


 下载:
下载:


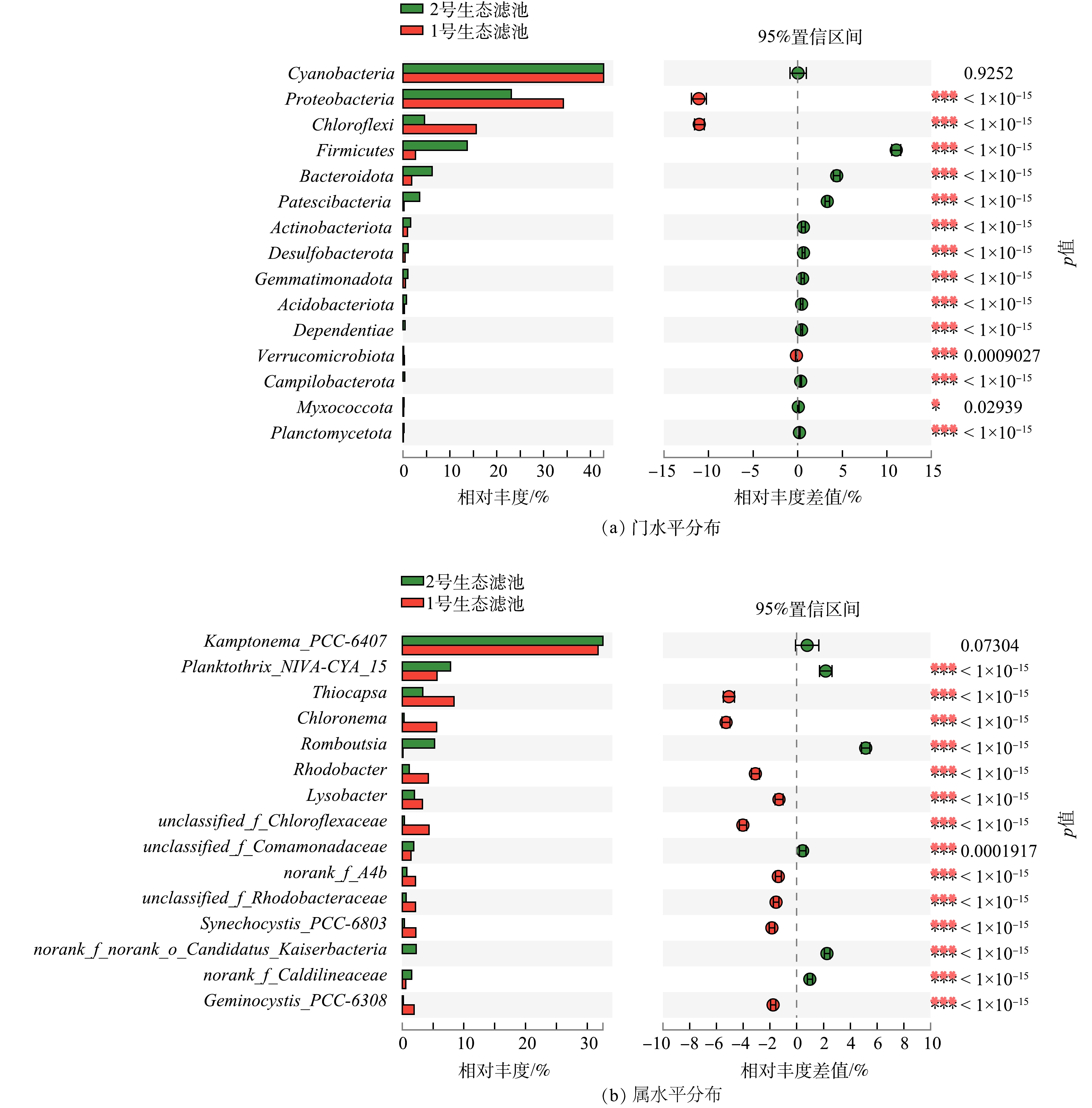
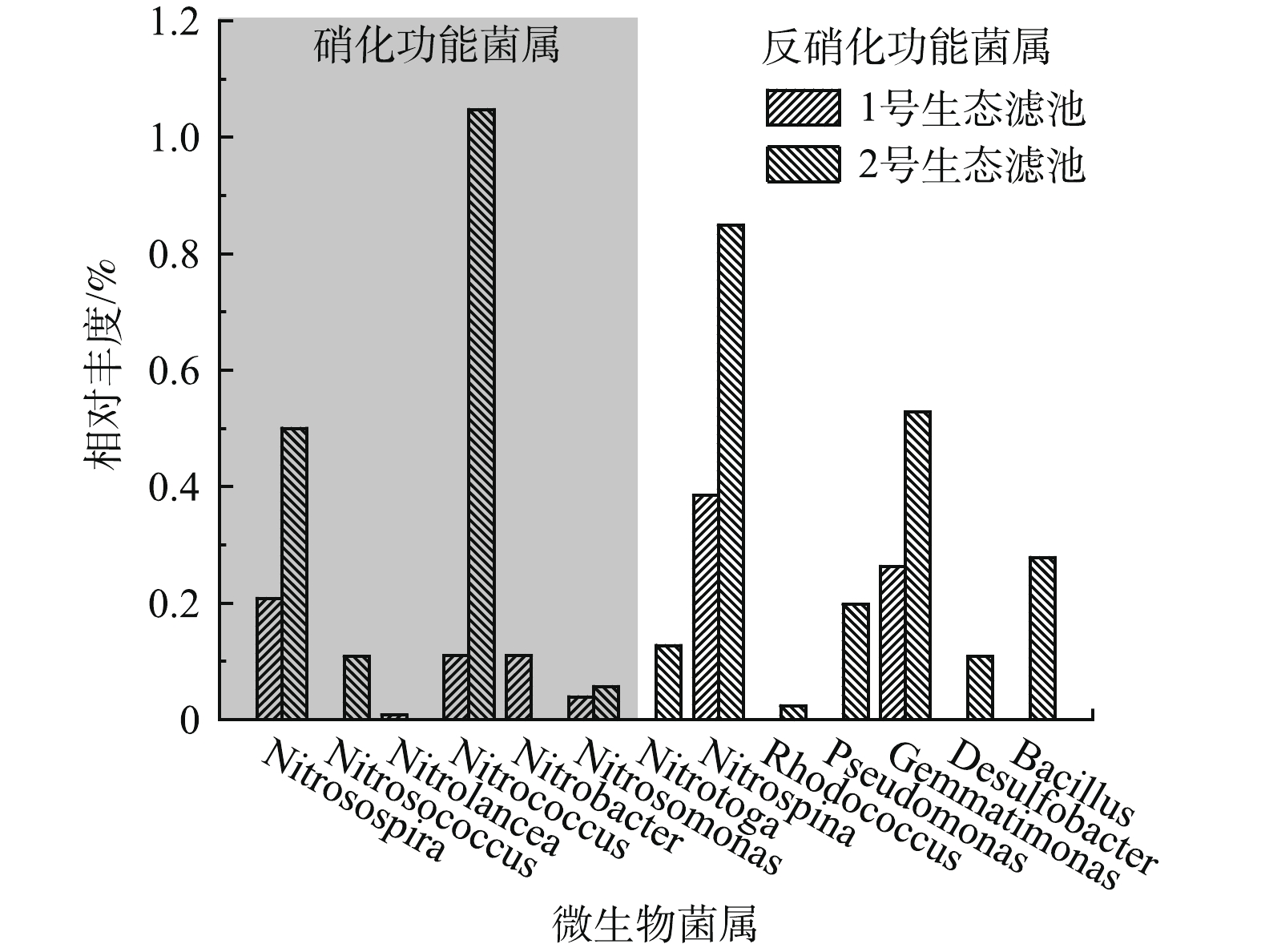


-
随着新型冠状病毒(corona virus disease 2019, COVID-19)在全球持续肆虐,治疗和预防COVID-19的药物使用量越来越大,但人体可以吸收利用的药物剂量较少。大部分未被利用的药物会通过人体排泄进入生态环境,经过不断的生物富集作用在食物链中逐步转移、积累和放大,对人类的健康造成了威胁[1]。其中,布洛芬(ibuprofen,IBP)是典型的抗炎和退热药物,是英国药品机构推荐治疗COVID-19症状的药物之一[2-3]。有研究表明,欧洲、美洲和亚洲国家水体中可检测到的IBP质量浓度分别为95 μg·L−1、208 ng·L−1和92 μg·L−1[4]。由于传统水处理工艺无法有效去除IBP,国内外学者对IBP降解进行了大量的研究,包括臭氧氧化法[5]、光催化氧化法[6]、电化学氧化法[7]、类芬顿法[8]等,其中非均相催化臭氧化技术具有污染物去除彻底、催化剂易回收和无二次污染等优点受到广大研究者的青睐。
目前,金属氧化物、碳材料、天然矿物以及硅基介孔分子筛等材料被广泛应用于非均相催化臭氧化领域[9-10]。由于非均相催化臭氧化以界面反应为主[10],具有较高比表面积、长程有序孔道结构以及优异稳定性的硅基介孔分子筛(包括MCM-41、MCM-48和SBA-15等)受到了广泛关注。纯的硅基介孔分子筛缺少酸性位点,常需与金属活性组分复合才具备活化臭氧(O3)的能力[11]。在金属改性硅基介孔分子筛上,金属阳离子等路易斯(Lewis)酸位点可以吸附O3并将其转化为氧化能力更强的羟基自由基(·OH)[12]。铁(Fe)作为地壳中含量第二高的金属元素,具有价态多、氧化还原能力强、价廉易得和无毒性等优点,因此,在催化领域被大量使用[13]。其中,Fe-MCM-41是应用比较广泛的O3催化剂[9-10]。但Fe-MCM-41表面酸性较弱,对O3利用率低;同时Fe-MCM-41表面对·OH束缚能力差,·OH等短寿命组分向溶液中扩散损耗较大。现有研究表明,解决这2个问题的关键是提高催化剂表面Lewis酸性,这不仅可以提升O3向催化剂表面传质的效率,而且可以加速O3转化为·OH的过程[9-10]。非均相催化臭氧化反应是由溶液反应和界面反应共同参与的体系,FENG等[14]认为强Lewis酸性位点会减少催化剂表面的·OH向溶液中扩散,有利于污染物的界面氧化;而中酸性Lewis酸位点产生的·OH会迅速脱离催化剂表面参与溶液反应。FENG等构建了具有中酸位点的Fe-Mn/MCM-41,提高了溶液中甲基橙的去除效率。而现有研究关注于构建具有强酸位点的催化剂,提高界面反应效率[9-10]。但这些催化剂无法同时增强界面反应和溶液反应的效率,对污染物的单一降解途径会极大限制催化剂的应用。因此,如何构建一种同时具有强酸和中酸位点的催化剂是亟待解决的难题。
近年来,许多研究发现电负性差异较大的2种金属可以引起催化剂内部电子重排,从而构建拥有贫富电子双中心的催化剂。张帆等通过电负性差异较大的Fe和Ti在Al2O3表面构建了Fe富电子中心和Ti贫电子中心[15]。富电子中心可以活化O3产生·OH,加快IBP降解,同时IBP作为电子供体在贫电子中心被氧化去除,大大提高了对IBP的去除效率。XIE等合成了具有贫电子Cu中心和富电子Bi中心的新型类芬顿催化剂γ-Cu-Al2O3-Bi12O15Cl6,H2O2可以同时在贫富电子中心通过不同途径被活化为·OH,加快了酚类化合物的选择性降解[16]。有研究表明,锌(Zn)是一种活性比Fe更强的金属[17],它在进入二氧化硅骨架后可以表现出比Fe更强的酸性[18]。同时,Zn的电负性为1.65,而Fe的电负性为1.83。因此,在Fe-MCM-41的骨架中引入Zn后,理论上可形成Fe富电子中心和Zn贫电子中心。FENG等研究发现,贫电子位点的强正电性会表现出强酸性[14]。因此,可以实现具有中等酸性的Fe位点和强酸性的Zn位点催化剂的目标。
基于此,本文通过一步水热合成方法研制了具有双酸性中心Fe和Zn的Fe-Zn-MCM-41催化剂。使用XRD、N2吸附-脱附等温线、TEM、XPS等手段对Fe-Zn-MCM-41进行表征;通过对比不同臭氧化体系中IBP去除率和O3利用率,探究Fe-Zn-MCM-41的活性;通过pyridine-FTIR、自由基淬灭实验、ATR-FTIR以及电化学实验揭示Fe-Zn-MCM-41催化臭氧化去除IBP机理;同时评价了Fe-Zn-MCM-41的稳定性和重复使用性;最后利用GC-MS检测了IBP降解的中间产物,并推导出Fe-Zn-MCM-41/O3降解IBP路径。
-
试剂:九水硅酸钠(Na2SiO3·9H2O)、十六烷基三甲基溴化铵(CTAB)、硝酸铁(Fe(NO3)3·9H2O)、硫酸锌(ZnSO4·7H2O)、叔丁醇(TBA)、二甲基亚砜(DMSO)、硫代硫酸钠(Na2S2O3)、布洛芬、强氧化钠(NaOH)、盐酸(HCl)均为分析纯;甲醇(CH3OH)、乙腈(CH3CN)均为色谱纯。
仪器:高效液相色谱(high performance liquid chromatography,HPLC,LC-10AT vp,Shimadzu,Japan);有机碳分析仪(TOC-Vwp,Shimadzu,Japan);X射线衍射仪(X-ray diffraction,XRD,D-MAX 2200 VPC,Rigaku,Japan);透射电子显微镜(transmission electron microscope,TEM,JEM-2100,JEOL,Japan);比表面积分析仪(BET,ASAP-2020,Micromeritics,USA);X射线光电子能谱(X-ray photoelectron spectroscopy,XPS,AXIS SUPRA,Shimadzu,Japan);吡啶红外光谱(pyridine-FTIR)和傅里叶变换衰减全反射红外光谱(attenuated total reflectance Flourier transformed infrared,ATR-FTIR)(Nicolet 6700,美国热电尼高力)。
-
称取28.4 g Na2SiO3·9H2O、0.17 g Fe(NO3)3·9H2O和0.12 g ZnSO4·7H2O分别溶于55、10和10 mL去离子水中,随后将其混合溶液在35 ℃恒温水浴锅中搅拌30 min,用2 mol·L−1的硫酸调节pH直至形成凝胶。充分搅拌后,向凝胶溶液中加入25 mL溶有7.28 g CTAB溶液并继续搅拌混合。然后将所得溶液置于200 mL聚四氟乙烯内衬的高压反应釜中,在145 ℃下水热晶化48 h。所得白色固体经过洗涤、干燥后再马弗炉中550 ℃下煅烧6 h,可得到Fe-Zn-MCM-41(摩尔比Si/Fe=Si/Zn=240)。MCM-41、Fe-MCM-41和Zn-MCM-41的制备方法与上述方法相同,且Fe-MCM-41和Zn-MCM-41中摩尔比Si/Fe=Si/Zn=120,制备过程中不添加Fe或/和Zn盐。
-
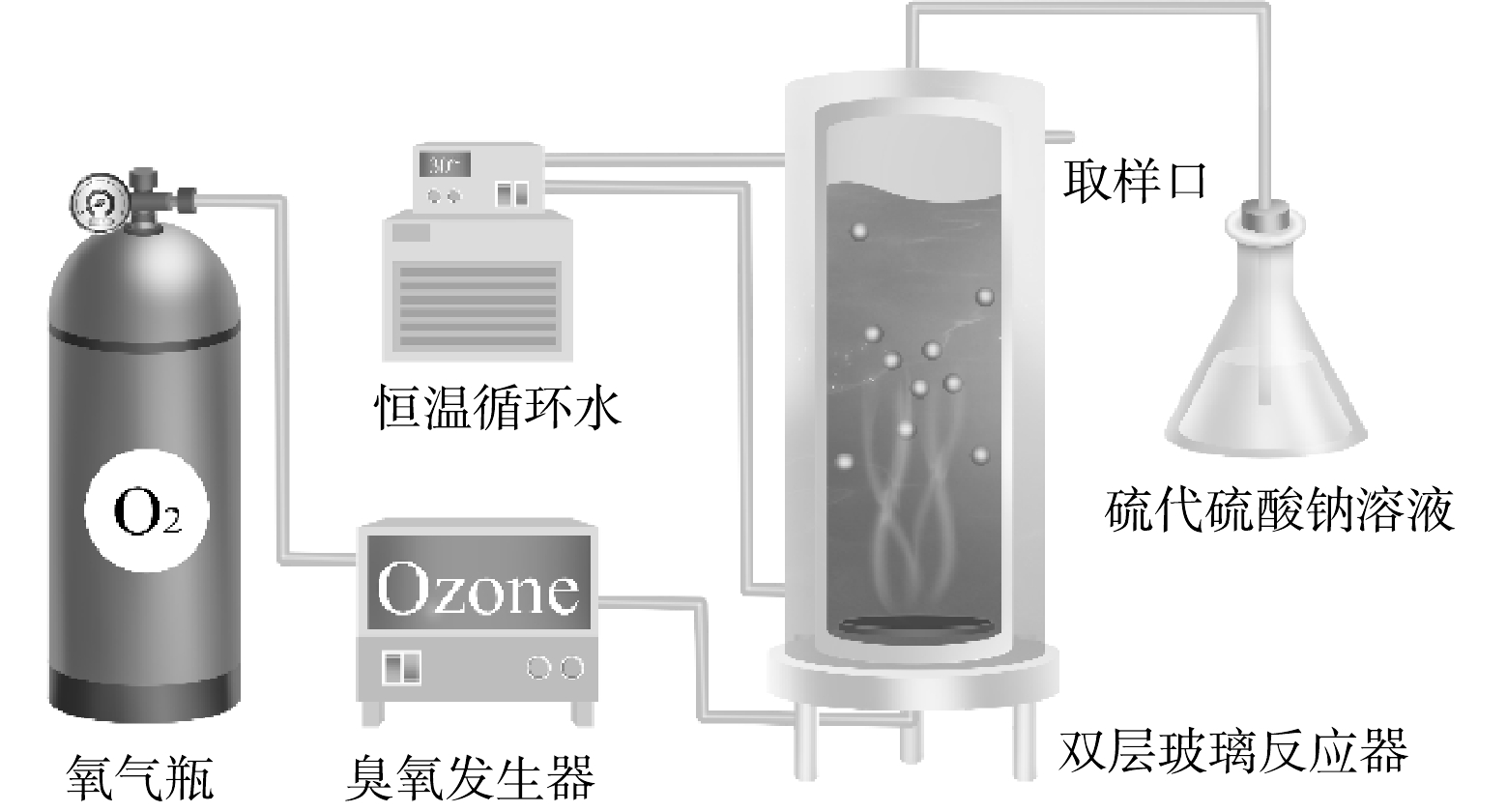
如图1所示,本研究使用内径80 mm、外径100 mm和高300 mm的双层玻璃反应器,内层进行臭氧化反应,外层通循环水保持恒温(30 ℃),内层底部为砂芯布气板。实验开始前先打开臭氧发生器预热30 min,并打开恒温水槽,使反应器外循环水温度达到实验条件。臭氧发生器预热稳定后,称取0.5 g催化剂与1.0 L、pH为5且质量浓度为10 mg·L−1的IBP溶液混合后倒入反应器中。O3由氧气经过臭氧发生器产生,气体流量为1.0 L·min−1,O3混合气体经反应器底部多孔砂板进入反应器,均匀曝气,使溶液、催化剂和气体三相均匀接触。尾气用5%的Na2S2O3溶液吸收处理。所有样品均用0.45 μm有机滤膜过滤后进行分析。IBP浓度通过HPLC分析,色谱条件:紫外检测器波长为220 nm;色谱柱为5U C18柱(250 mm×4.6 mm);流动相为20 mmol,pH=2.5磷酸盐缓冲溶液-乙腈(40∶60),流速为1.0 mL·min−1。
-
采用扫描范围为0.5°~10°的小角XRD对催化剂晶型结构进行表征,测试过程中用Cu靶Kα射线(λ=0.154 18 nm)为激发源、管电压为40 kV,管电流为30 mA,扫描速度为0.5°min−1;利用低温N2吸附比表面分析仪测得催化剂的N2吸附-脱附等温曲线,采用BET法计算催化剂比表面积,采用BJH法分析催化剂孔径分布和孔容;采用TEM观察催化剂的微观结构特征和构造;采用XPS对表面信息进行收集,用C1s的结合能284.6 eV进行校准;采用pyridine-FTIR测定不同催化剂的酸性和酸量;在0.1 mol·L−1的Na2SO4电解质溶液中的三电极电池系统(Ag/AgCl为参比电极、铂丝为对电极、催化剂为工作电极)上进行线性扫描伏安法(linear sweep voltammetry,LSV)和电化学阻抗谱(electrochemical impedance spectroscopy,EIS)实验。
-
利用小角XRD表征改性前后的MCM-41晶体结构变化,结果如图2(a)所示。MCM-41在2θ=2.4°、3.8°和4.4°附近均出现特征衍射峰,分别对应(100)、(110)和(200)晶面,这与具有高度有序的二维六方介孔结构相一致[14]。在掺杂了Fe和/或Zn后,金属改性MCM-41的(100)晶面峰强度降低且向较小角度移动,特征衍射峰强度降低是由于Fe和Zn成功进入MCM-41骨架后导致MCM-41有序度降低所引起的[12]。因为Fe3+(0.063 nm)和Zn2+(0.060 nm)的半径均大于Si4+(0.040 nm),Fe和Zn原子取代Si原子会导致晶胞膨胀,所以Fe-MCM-41、Zn-MCM-41和Fe-Zn-MCM-41衍射峰均向小角度移动[12]。XRD结果表明Fe和Zn成功进入MCM-41骨架,Fe-Zn-MCM-41保留了MCM-41有序的介孔结构。
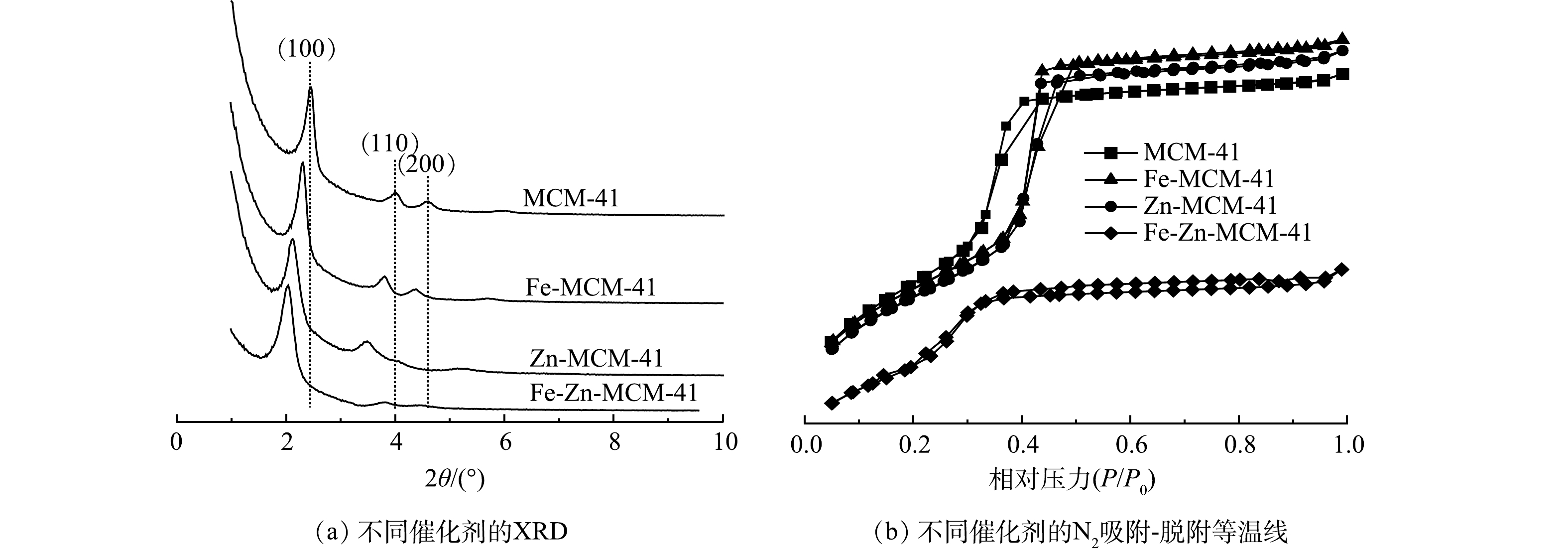
如图2(b)所示,Fe-MCM-41、Zn-MCM-41和Fe-Zn-MCM-41的N2吸附-脱附等温线与MCM-41相同,呈现典型的Ⅳ型吸附-脱附等温线。这表明改性后的MCM-41均保持介孔结构[12]。Fe-MCM-41和Zn-MCM-41具有与MCM-41相同的H1型回滞环,说明单金属改性后MCM-41保持了较好的有序介孔结构;而Fe-Zn-MCM-41呈现出H4型回滞环,这意味着Fe和Zn共掺杂破坏了MCM-41部分骨架结构,从而导致介孔结构的有序性降低[19],这与XRD结果一致。从表1中可以发现,MCM-41具有较大的比表面积,数值可达1 171.62 m2·g−1。向骨架中引入Fe和Zn后,Fe-MCM-41、Zn-MCM-41和Fe-Zn-MCM-41的比表面积分别减小为1 136.27、1 112.34和831.60 m2·g−1。这一变化归因于Fe和Zn进入MCM-41骨架后引起的结构变化[12]。除了Fe和Zn具有较大的原子半径外,Fe-O和Zn-O键均比Si-O键更长[12],因此,Fe-MCM-41、Zn-MCM-41和Fe-Zn-MCM-41具有更大的晶胞参数,孔径由3.33 nm分别增加到3.36、3.40和3.62 nm。由于在水热反应过程中,部分Fe和Zn未进入骨架而在催化剂孔道内沉积[14],MCM-41的孔容从0.98 cm3·g−1分别减小到0.82、0.57和0.50 cm3·g−1。
由图3(a)和图3(b)中可以明显看出Fe-Zn-MCM-41拥有长程有序的孔道结构,这表明Fe-Zn-MCM-41仍保持了较规则的孔道结构。这与XRD和N2吸附-脱附结果相一致。由图3(c)~(f)可以看出,O、Si、Zn和Fe是Fe-Zn-MCM-41的主要组成元素,且Fe和Zn均匀分布在Fe-Zn-MCM-41骨架中。

如图4(a)所示,Fe-Zn-MCM-41表面主要含Si、O、Fe和Zn元素,这与TEM的元素分布图结果相对应。C元素的存在可能是催化剂表面或仪器表面受污染所致。图4(b)为Fe-MCM-41和Fe-Zn-MCM-41的Fe2p精细谱。Fe2p精细谱可以被拟合为Fe2p3/2(~711.5 eV)和Fe2p1/2(~725.0 eV)2个自旋轨道峰。2种催化剂Fe2p3/2和Fe2p1/2的能级差均大于13.0 eV,该结果表明Fe-MCM-41和Fe-Zn-MCM-41中的Fe均以Fe3+形式存在[12]。与Fe-MCM-41相比,Fe-Zn-MCM-41中Fe2p3/2和Fe2p1/2特征峰的结合能分别变小了0.3 eV和0.4 eV,这是Zn进入骨架后与Fe相互作用的结果[20]。Fe和Zn的电负性分别为1.83和1.65,Fe可以吸引Zn原子外层电子,因此,Fe的电子云密度变大,结合能相应变小[14, 21]。图4(c)中Zn2p精细谱分析结果显示,Zn2p精细谱可以拟合为Zn2p3/2(~1 022.2 eV)和Zn2p1/2(~1 045.5 eV)2个自旋轨道峰,2个特征峰的能级差均为23.3 eV,表明Fe-MCM-41和Fe-Zn-MCM-41中的Zn均以Zn2+形式存在[22]。与Zn-MCM-41相比,Fe-Zn-MCM-41中Zn2p3/2和Zn2p1/2 2个特征峰均变大了0.4 eV,这与Fe2p分析结果相一致。
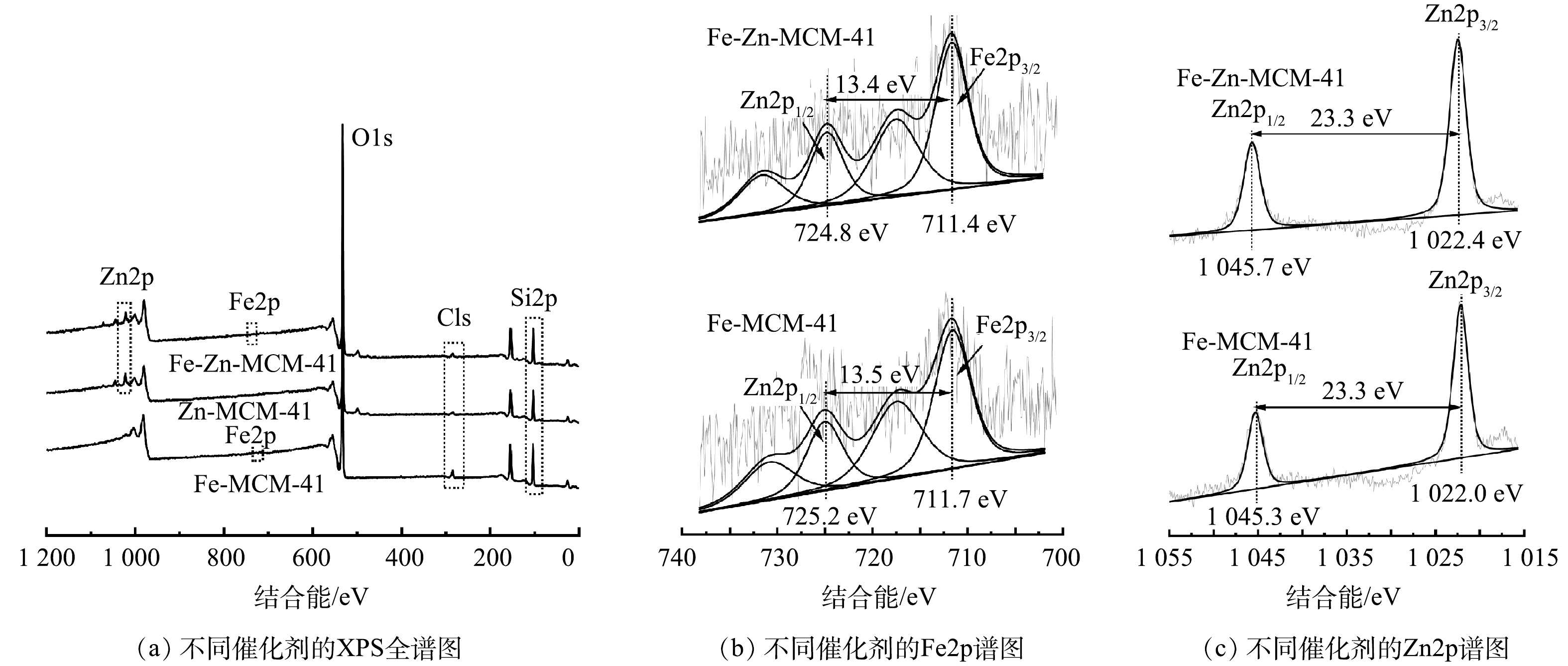
综合以上表征结果,可以确定Fe和Zn进入MCM-41骨架内并分别形成了富电子和贫电子中心。由于贫、富电子位点分别表现出强酸和中酸性[14, 21],因此成功构建了拥有中酸性Fe位点和强酸性Zn位点的Fe-Zn-MCM-41催化剂。
-
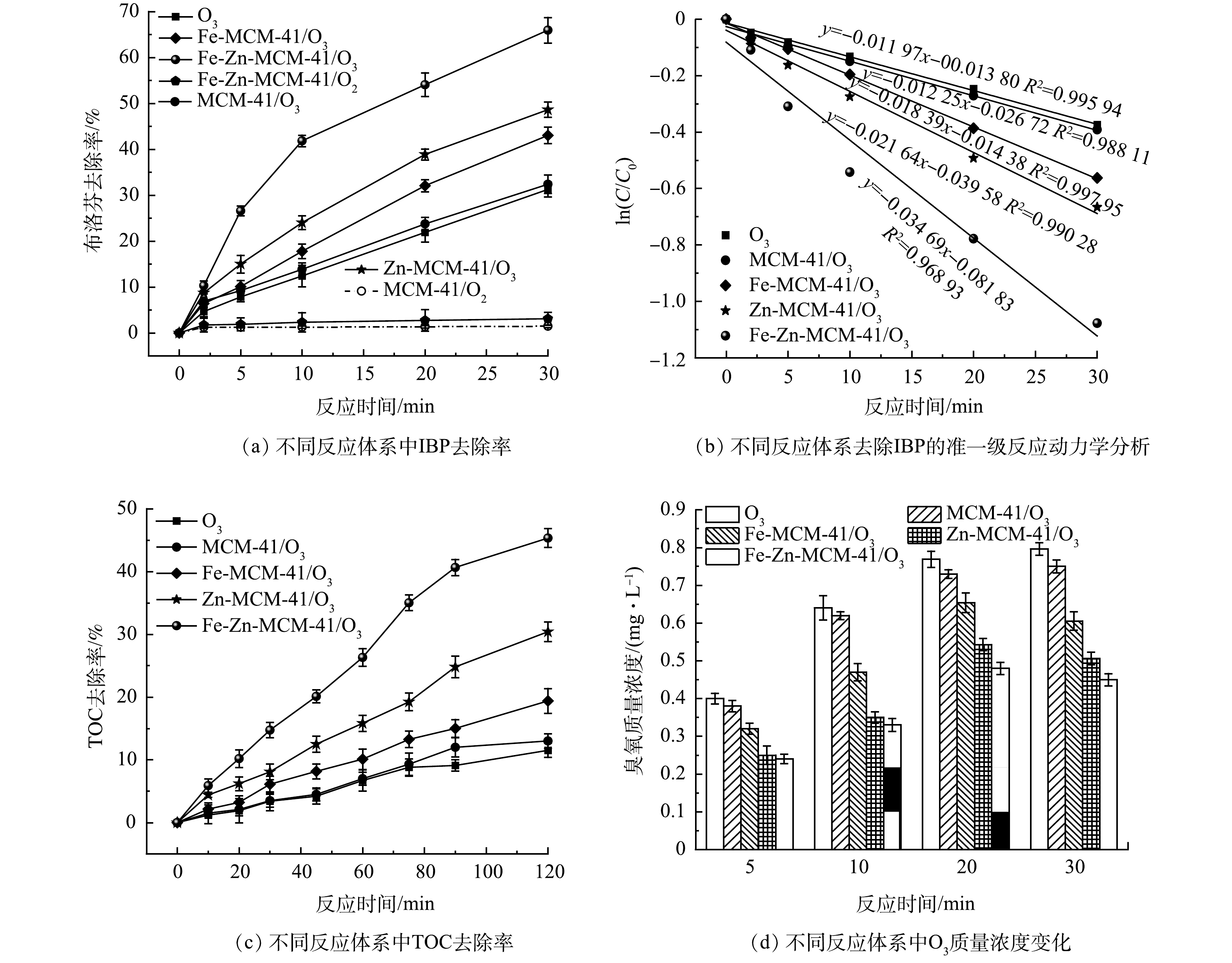
如图5(a)所示,反应30 min后,单独O3对IBP去除率为31.3%;催化剂的加入可以提高O3对IBP的去除率。加入MCM-41、Fe-MCM-41和Zn-MCM-41后,IBP的去除率分别提高到32.3%、43.1%和48.6%,说明Fe和Zn改性MCM-41具有较好的活性,且Zn-MCM-41比Fe-MCM-41表现出更强的O3活化能力。Fe-Zn-MCM-41/O3对IBP的去除效果可达到65.9%,分别是单独O3、MCM-41、Fe-MCM-41/O3和Zn-MCM-41/O3的2.1、2.0、1.5和1.4倍。单独MCM-41和Fe-Zn-MCM-41对IBP的去除分别为1.5%和3.1%,这是因为Fe和Zn进入MCM-41骨架后减少了疏水Si-OH官能团的数量,提高了催化剂表面疏水性,从而增加了对疏水性IBP的亲和力,这有助于IBP向Fe-Zn-MCM-41表面传质,加快降解IBP的效率[12, 23]。为了进一步探究不同臭氧化工艺降解IBP的能力,对不同臭氧化体系进行准一级动力学分析。如图5(b)所示,单独O3和MCM-41/O3降解IBP的表观速率常数(k)均为0.012 min−1,在单独O3体系中加入Fe-MCM-41、Zn-MCM-41和Fe-Zn-MCM-41后,k分别增大为0.018、0.022和0.035 min−1,该结果表明Fe-Zn-MCM-41对IBP降解具有更大的反应速率。除此之外,如图5(c)所示,Fe-Zn-MCM-41对IBP具有优异的矿化能力,反应120 min后IBP的矿化率为45.3%,分别是单独O3、MCM-41/O3、Fe-MCM-41/O3和Zn-MCM-41/O3的3.9、3.5、2.3和1.5倍。如图5(d)所示,不同臭氧化体系中O3质量浓度变化可以间接反映该体系中O3利用率。随着反应时间延长,进入溶液中的O3量逐渐增加,单独O3体系中溶液残留的O3量持续增加,但催化臭氧化体系中残留的O3量均出现了先增加后降低的现象。Fe-Zn-MCM-41/O3中O3质量浓度均低于Fe-MCM-41/O3、Zn-MCM-41/O3、MCM-41/O3和单独O3体系,这表明Fe-Zn-MCM-41可以更高效的活化水溶液中的O3。这是因为一方面Fe-Zn-MCM-41拥有更多可以吸附和活化O3的活性位点;另一方面Fe-Zn-MCM-41较大的孔径结构有助于O3传质。因此,Fe-Zn-MCM-41具有优异的催化活性,可以有效提高水溶液中O3的利用率。
-
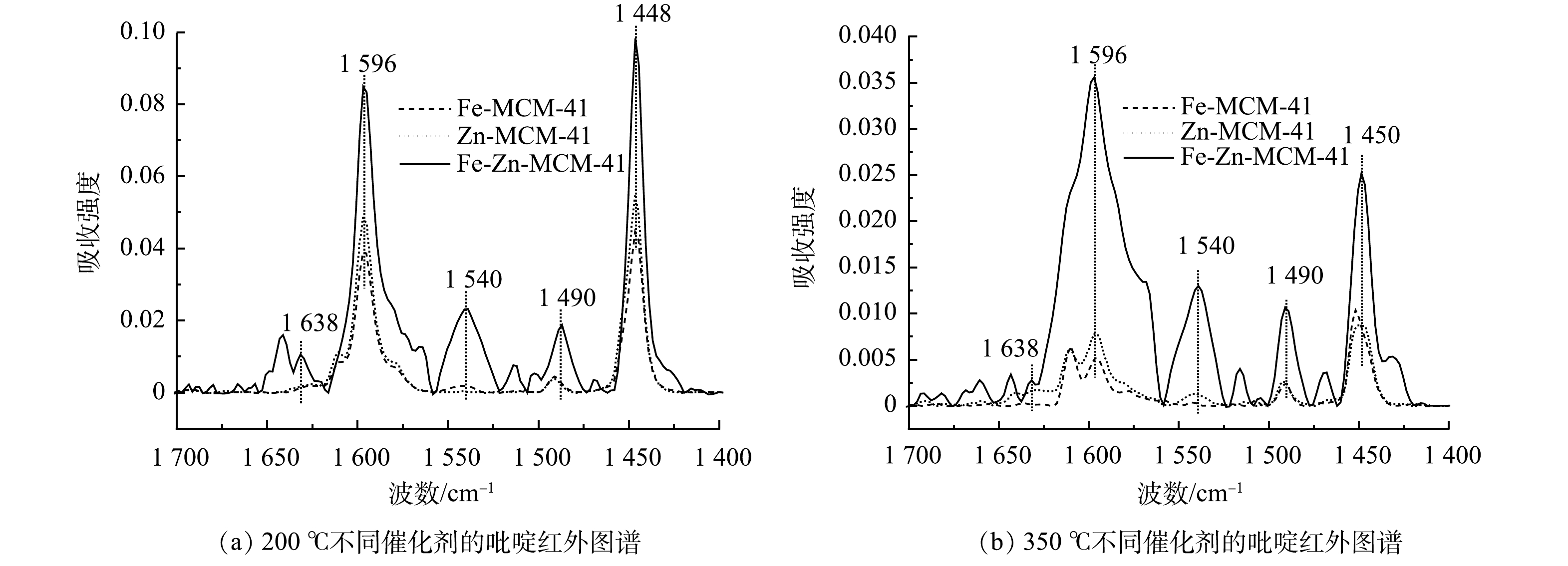
为了探究双金属改性后Fe-Zn-MCM-41的表面双酸性位点,使用pyridine-FTIR技术对比分析单金属和双金属掺杂后MCM-41表面酸性位点的类型和酸量变化。催化剂在50、200和350 ℃的pyridine-FTIR脱附峰分别代表催化剂的总酸、中酸和强酸特征[24]。如图6所示,在1 596和1 448 cm−1处是吡啶吸附在Lewis酸位点上形成的特征峰,在1 638和1 540 cm−1处的特征峰对应于布朗斯特(Brønsted)酸位点上的吡啶吸附峰,1 490 cm−1处特征峰是由吡啶吸附在Lewis酸和Brønsted酸位点共同形成的[12]。由图6可以看出,Zn-MCM-41比Fe-MCM-41拥有更多的强酸性位点,这与已有研究结果相一致[18]。在Fe和Zn共掺杂后,Fe-Zn-MCM-41同时拥有更多的中酸和强酸位点。由图6和表2可知,随着温度升高所有酸性位点特征峰的强度降低,但Lewis酸位点的峰强度和峰面积始终高于Brønsted酸位点。这表明Lewis酸位点是催化剂的主要酸性位点。表2中酸量结果表明,Fe-Zn-MCM-41的中Lewis酸和强Lewis酸的量分别为33.55 μmol·g−1和7.97 μmol·g−1,均高于Fe-MCM-41和Zn-MCM-41。因此,Fe-Zn-MCM-41表现出优异的催化臭氧化活性。
-
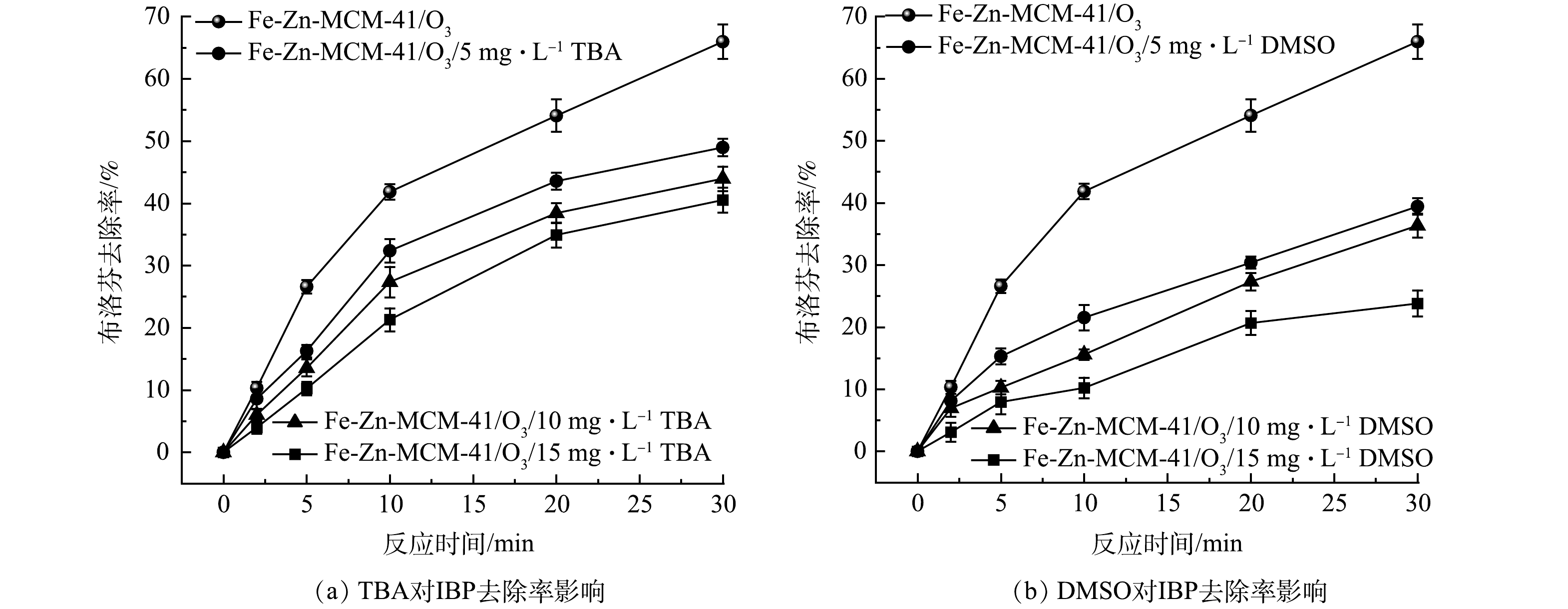
中酸位点产生的·OH(·OHfree)会迅速脱离催化剂表面进入溶液,而强酸位点产生的·OH(·OHad)键合在催化剂表面,它们在非均相催化臭氧化反应中去除污染物的途径不同[14, 21]。TBA亲水性强,它主要淬灭·OHfree;而DMSO具有疏水性,它可以淬灭·OHad[25]。因此,TBA和DMSO两种·OH淬灭剂被用来探究Fe-Zn-MCM-41/O3对IBP去除的机理。如图7(a)所示,随着TBA质量浓度从5 mg·L−1增大到15 mg·L−1,IBP的去除率从65.9%分别降低为48.6%、43.9%和40.5%,说明溶液中的·OHfree对IBP的去除有较大贡献。DMSO对Fe-Zn-MCM-41/O3表现出更明显的抑制作用。如图7(b)所示,在加入质量浓度为5~15 mg·L−1的DMSO后,IBP去除率分别下降到39.4%、36.3%和23.8%,这表明·OHad对IBP的去除起主导作用。结合XPS和pyridine-FTIR分析结果,Fe-Zn-MCM-41表面的Fe位点是中Lewis酸位点,可以产生能快速脱离Fe-Zn-MCM-41表面的·OH,它们迅速扩散到溶液中加快体相反应进程;Zn位点是强Lewis酸位点,其吸附O3后产生的·OH键合在Fe-Zn-MCM-41表面,促进了界面反应的发生。
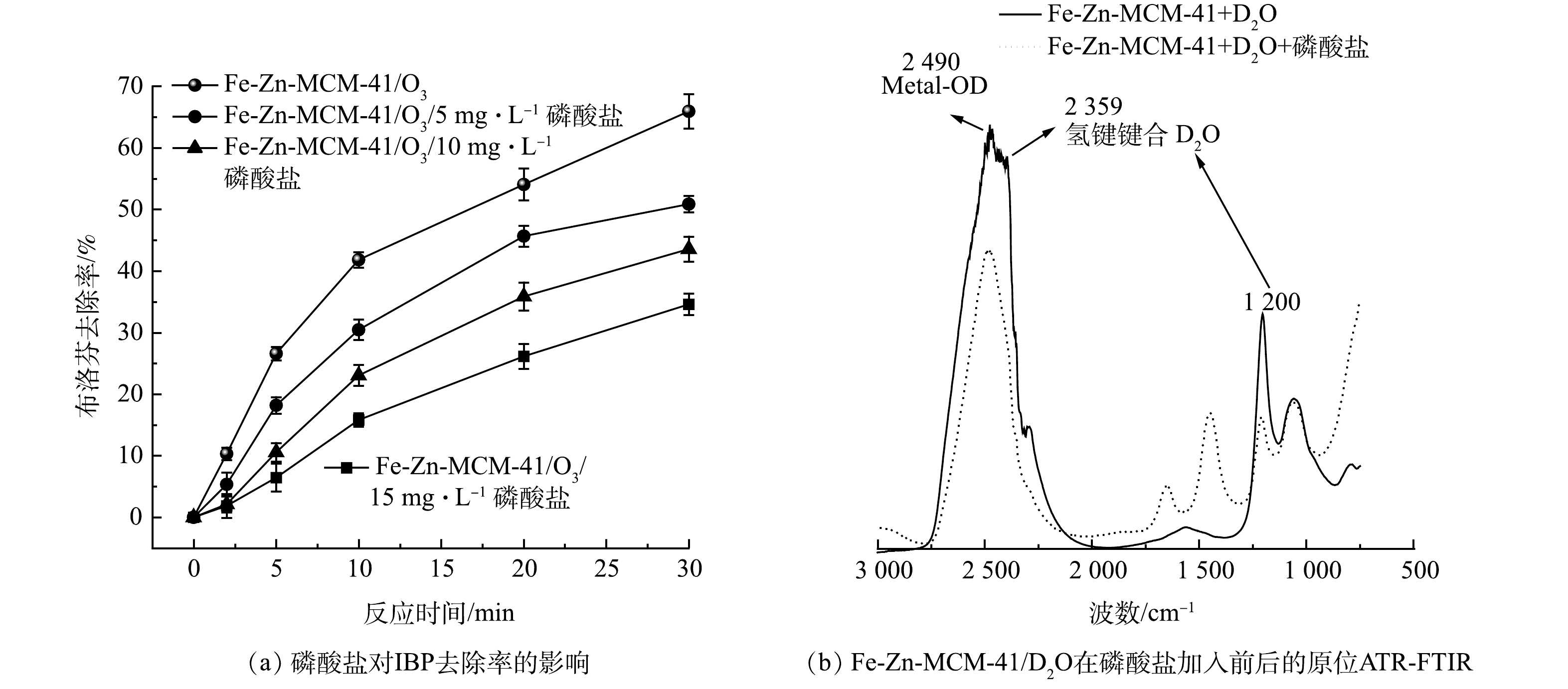
催化剂表面Lewis酸位点与溶剂水会形成金属羟基(Metal-OH),通常认为,Metal-OH活化O3是产生·OH的关键步骤[10]。磷酸盐(phosphate)作为比水和O3更强的Lewis碱,可以占据Lewis酸位点阻碍Metal-OH的形成,从而抑制O3的活化过程[26]。如图8(a)所示,加入5~15 mg·L−1磷酸盐后,Fe-Zn-MCM-41/O3对IBP去除率从65.9%分别降低为50.9%、43.6%和34.2%。随着磷酸盐量增加,Fe-Zn-MCM-41表面更多的Fe和Zn位点被占据,阻碍了O3在Lewis酸位点的活化和·OH的产生。Metal-OH的作用可以更直观的通过原位ATR-FTIR来呈现。如图8(b)所示,为了区分溶剂在Fe-Zn-MCM-41表面形成的Metal-OH和Fe-Zn-MCM-41自带的羟基基团,使用重水(D2O)代替水作为溶剂,可以更清晰的揭示Fe-Zn-MCM-41表面Metal-OH在加入磷酸盐前后的作用机理。Lewis酸位点金属与D2O形成的Metal-OD特征峰位于2 490 cm−1处。2 359和1 200 cm−1处是氢键键合的D2O所形成的特征峰[27]。当加入磷酸盐后,Metal-OD特征峰和氢键键合的D2O的特征峰均明显降低,并且在1 656和1 452 cm−1处新出现了磷酸根特征峰。原位ATR-FTIR结果表明磷酸根占据了Lewis酸位点,从而抑制O3与Lewis酸位点的相互作用,阻碍了·OH的产生。因此,Fe-Zn-MCM-41表面Lewis酸位点对O3活化和·OH的产生至关重要。
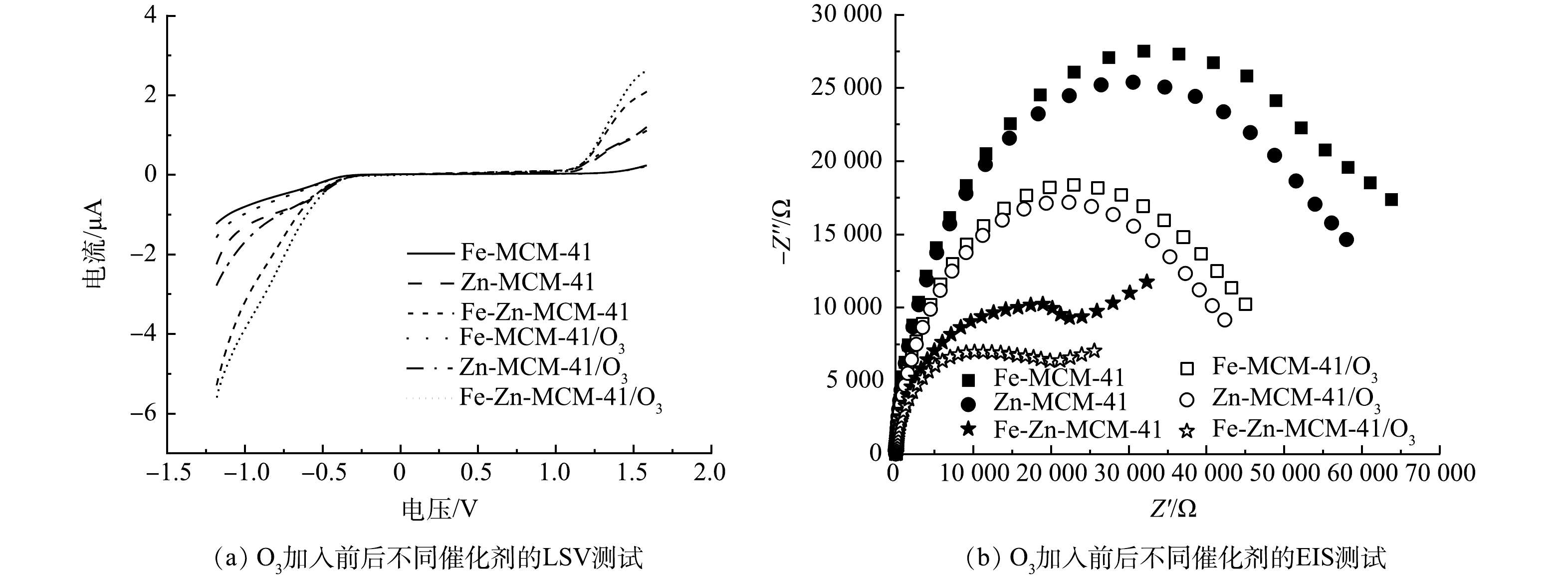
在催化臭氧化体系中,催化剂与O3之间的电子传递过程是O3活化产生·OH的关键[28],因此,使用原位LSV和EIS探究不同催化剂与O3之间电子传递效率有助于进一步揭示Fe-Zn-MCM-41/O3降解IBP的机理。如图9(a)所示,在未加入O3时,Fe-Zn-MCM-41表现出比Fe-MCM-41和Zn-MCM-41更大的起始电流信号,这说明双金属的协同作用可以提高催化剂的电子传递能力[24]。加入O3后,所有催化剂的电流信号强度明显增大,其中Fe-Zn-MCM-41/O3表现出最强的起始电流信号。该结果表明,Fe-Zn-MCM-41不仅具有较强的电子传递能力,而且拥有较强的O3亲和力[12]。为了进一步验证Fe-Zn-MCM-41的强电子传递能力,使用EIS对不同催化剂的阻抗进行测定。一般来说,催化剂电子传递阻力越小则EIS的电弧半径越小,较低的阻抗意味着较高的电子传递能力[29]。如图9(b)所示,Fe-Zn-MCM-41具有最小的阻抗,特别是存在O3的时候表现出更低的阻抗。由此可知,Fe和Zn进入MCM-41骨架后,提高了Fe-Zn-MCM-41的电子传递能力。Fe-Zn-MCM-41具有优异的电子传递能力和O3亲和力。
-
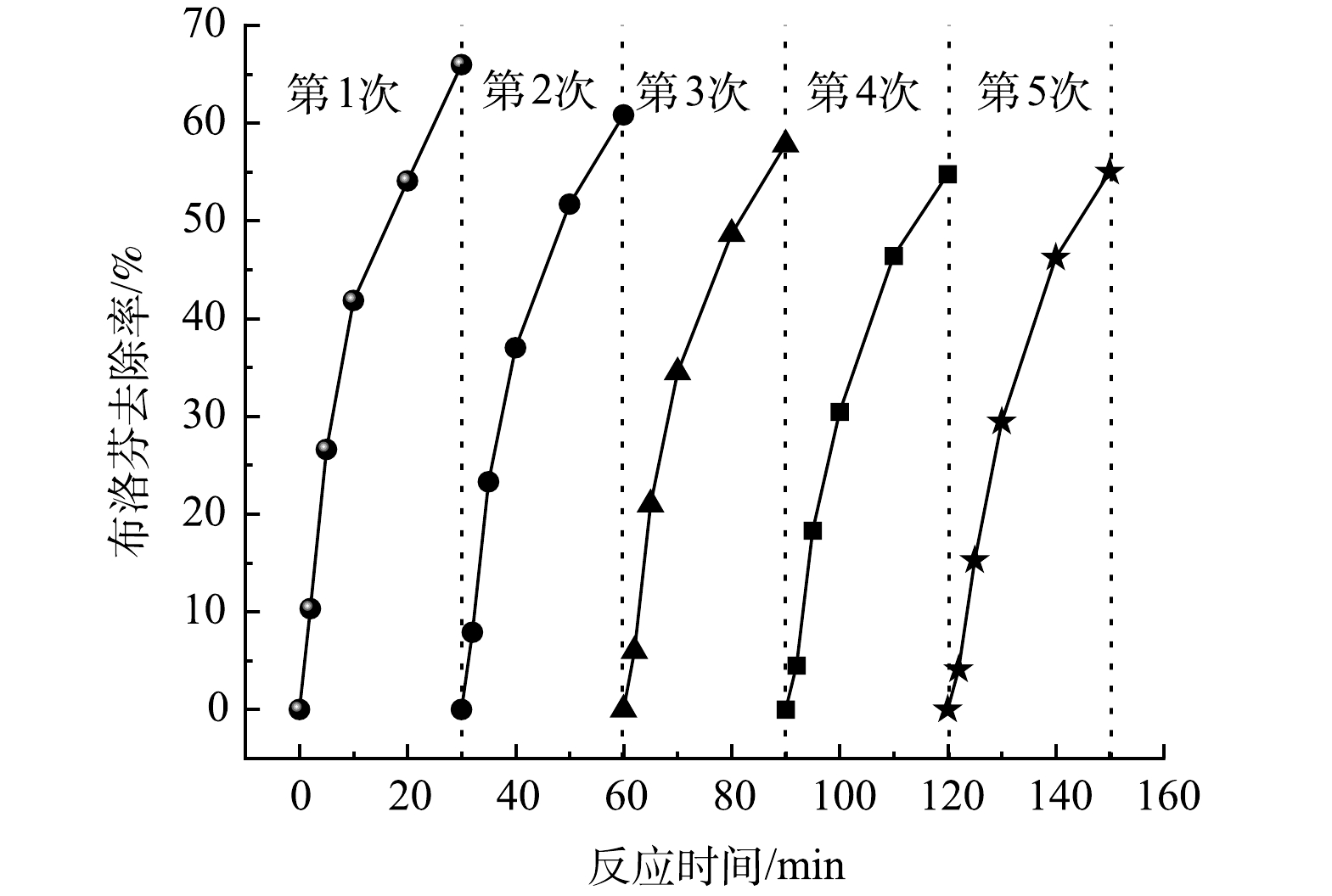
催化剂的稳定性评价包括反应过程中Fe和Zn的溶出量、催化剂重复使用活性两方面。反应30 min后溶液中Fe和Zn的质量浓度分别为0.13 mg·L−1和0.28 mg·L−1,而我国现行《生活饮用水卫生标准》(GB5746-2006)中Fe和Zn的质量浓度限值分别为0.3 mg·L−1和1.0 mg·L−1,这说明在催化臭氧化过程中Fe-Zn-MCM-41具有较好的稳定性,不会引起水体二次污染。Fe-Zn-MCM-41的重复利用性能如图10所示,经过5次重复使用后Fe-Zn-MCM-41的活性逐渐降低,对IBP的去除率由65.9%分别下降为60.8%、57.3%、54.9%和55.1%。尽管Fe-Zn-MCM-41重复使用5次后活性降低,但对IBP的催化臭氧化去除效率仍然高于单独O3(31.3%)、MCM-41/O3(32.3%)、Fe-MCM-41/O3 (43.1%)和Zn-MCM-41/O3(48.6%)。以上结果表明Fe-Zn-MCM-41具有较好的稳定性和可循环使用性。如表3所示,对回收后的Fe-Zn-MCM-41进行BET测试可以发现,回用Fe-Zn-MCM-41的比表面积、孔径和孔容均随着使用次数增多而变小。这可能是Fe-Zn-MCM-41循环使用后活性降低的原因。
-
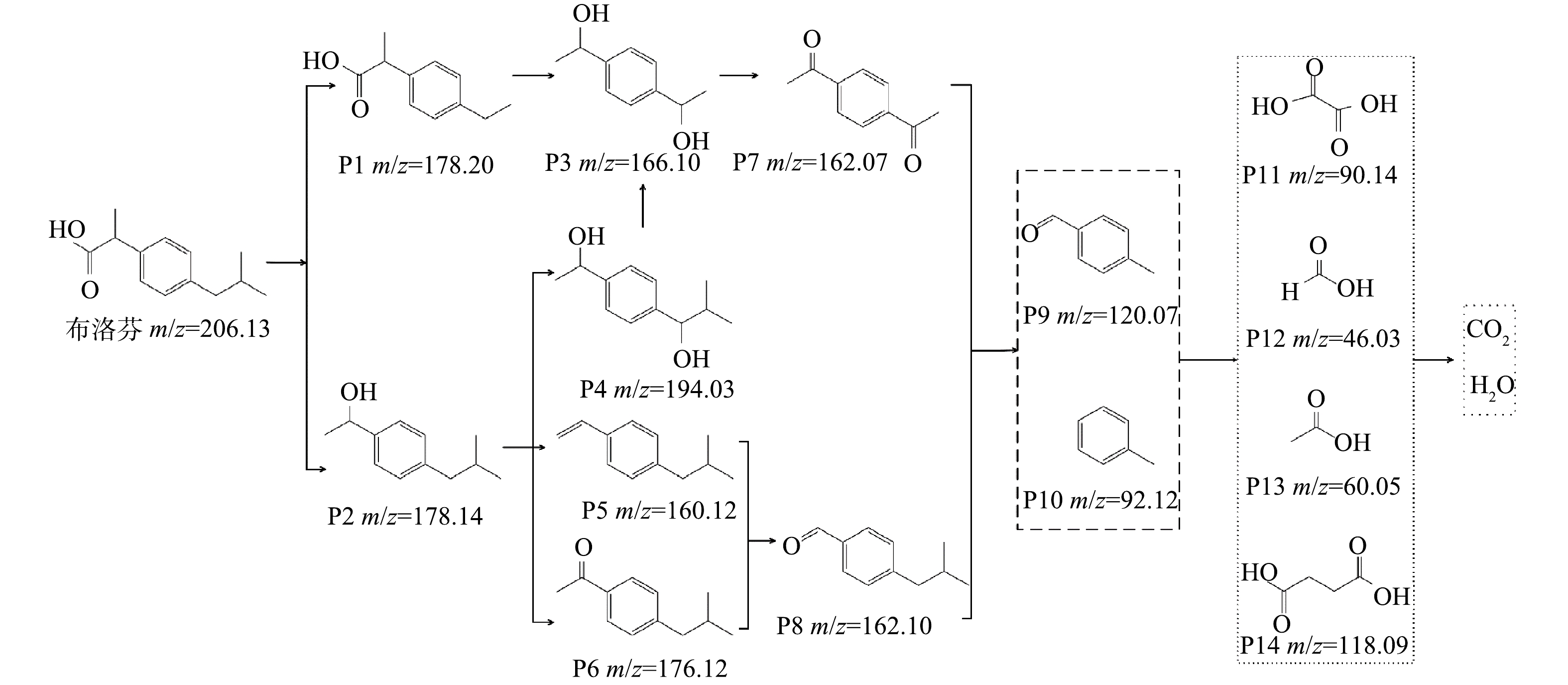
结合相关研究[12, 30-31]和GC-MS检测结果,确定了14种Fe-Zn-MCM-41/O3降解IBP的中间产物,并以此推测IBP的降解路径。如图11所示,由于IBP结构中异丁基和丙酸基的位阻作用[12],·OH优先进攻IBP的异丁基和丙酸基后分别生成了P1和P2,随着·OH对P1进一步进攻,在经过羟基的取代和加成反应后转化为P3,P3通过·OH的抽氢作用产生P7。同时,·OH进攻P2通过羟基加成、脱水和抽氢反应后分别转化为P4、P5和P6,P4可以通过脱甲基反应产生P3,P5和P6分别通过加成反应和脱甲基反应产生P8。P7和P8在O3和·OH的持续攻击下产生简单芳香族物质P9和P10,随着O3和·OH的进一步氧化转化为草酸(P11)、甲酸(P12)、乙酸(P13)和琥珀酸(P14)等脂肪酸,部分脂肪酸被·OH矿化为CO2和H2O。
-
1)通过一步水热合成方法成功制备了Fe和Zn共掺杂的MCM-41(Fe-Zn-MCM-41),Fe-Zn-MCM-41保留了纯MCM-41较好的介孔结构和较大的比表面积。
2) Fe-Zn-MCM-41/O3对IBP有较好的降解效果,30 min内对10 mg·L−1的IBP去除率可达到65.9%,分别是单独O3、MCM-41/O3、Fe-MCM-41/O3和Zn-MCM-41/O3的2.1、2.0、1.5和1.4倍。同时,Fe-Zn-MCM-41具有较好的稳定性,循环使用5次后对IBP去除率仍可达到55.1%。
3) Fe和Zn是Fe-Zn-MCM-41的活性组分,其在Fe-Zn-MCM-41表面构建了双酸性中心;基于Fe的中等Lewis酸位点有助于产生·OHfree,基于Zn的强Lewis酸位点产生·OHad。·OHfree和·OHad共同作用加快了IBP的降解,但·OHad参与的界面反应贡献更大。
4) Fe-Zn-MCM-41不仅具有较好的电子传递能力,而且具有较强的O3亲和力。
图 1
生态滤池装置
Figure 1.
Ecological filter device
