




-
抗生素在生产和使用过程中会产生大量含抗生素废水[1],制药废水是抗生素的最大来源,通常含抗生素浓度高、盐分高、毒性大,其处理是水处理领域中的一项难题[2]。磺胺甲恶唑(Sulfamethoxazole, SMZ)是一类典型的磺胺类抗生素,其在水体中相对稳定,不易被降解[3]。根据一项针对中国七大典型河流水域抗生素赋存的研究,SMZ的检出浓度最高[4],而且有研究表明人的尿液中可检出高达10 mg·L−1的SMZ[5]。SMZ对动植物以及人体健康均会造成危害,因此,研发利用高效的处理技术迫在眉睫。
许多研究表明,高级氧化技术对抗生素废水具有较好去除效果。其中电芬顿(electro-Fenton, EF)技术仅消耗O2和电能,绿色清洁、倍受关注[6-8]。EF技术可通过两电子氧还原反应(2e− ORR)原位生成H2O2,随后H2O2进一步被活化生成活性氧物种(reactive oxygen species, ROS),其可进一步高效去除水中抗生素[9-11]。基于铁离子催化的均相EF技术需在酸性条件下才能有效运行,反应前后需要调节pH,为拓宽EF技术的pH适用范围,开发了基于固相催化剂的非均相EF技术[12-14]。然而受阴极催化剂过渡金属氧化还原电对循环速率慢、稳定性差等限制[15],非均相EF技术对抗生素的降解效率亟待提升,开发高效稳定的阴极催化剂是目前非均相EF技术的主要应用瓶颈。
近年来,金属有机框架材料(metal organic framework, MOFs)是催化领域的研究热点,其不仅具有发达的孔隙结构,且拓扑结构能保障金属位点的均匀分散[16-19]。为了进一步促进金属氧化还原电对在催化反应中的循环速率,构建更多活性位点,研究通过组合不同种类的金属开发出系列双金属MOFs,显著提高了MOFs的催化活性[20-21]。然而MOFs作为EF阴极催化剂时导电性能欠佳[22-23],且其有机配体容易被EF反应中生成的ROS氧化,造成催化剂多孔结构坍塌及失活[24],因此,同步提高电学性能和化学稳定性是关键需求。
本研究以双金属MOFs—FeCo-ZIF为前体,以三聚氰胺(melamine, MA)为碳源和氮源,将两者共混煅烧制备了氮掺杂碳纳米管封装铁钴合金阴极催化剂(N-CNT@FeCo),考察了MA对FeCo-ZIF衍生催化剂EF性能的影响规律,探究了体系中的主要活性物种和催化机理,考察了溶液初始pH、应用电位、共存阴离子及重复循环次数对SMZ降解效果的影响,以评价N-CNT@FeCo作为EF阴极催化剂的基础应用性能。
-
实验材料:六水合硝酸钴(Co(NO3)2·6H2O)、无水硫酸钠(Na2SO4)、无水乙醇(C2H6O)均为分析纯并购于南京化学试剂股份有限公司;七水合硫酸亚铁(FeSO4·7H2O)、氢氧化钠(NaOH)、浓硫酸(H2SO4)、浓硝酸(HNO3)均为分析纯并购于国药集团化学试剂有限公司;三聚氰胺(C3H6N6)为分析纯并购于上海麦克林生化科技股份有限公司;磺胺甲恶唑(C10H11N3O3S)、N,N-二甲基甲酰胺(C3H7NO, N,N-Dimethylformamide, DMF)、2-甲基咪唑(C4H6N2)、甲醇(CH3OH, Methanol, MeOH)、叔丁醇(C4H10O, Tert-Butanol, TBA)、糠醇(C5H6O2, Furfuryl alcohol, FFA)、对苯醌(C6H4O2, 1,4-Benzoquinone, p-BQ)、磷酸二氢钾(KH2PO4)、草酸钛钾(K2TiO(C2O4)2)均为分析纯并购于上海阿拉丁生化科技股份有限公司;5,5-二甲基-1-吡咯啉-N-氧化物(C6H11NO, 5,5-Dimethyl-1-pyrroline N-oxide, DMPO)购于和光纯药工业株式会社;4-氨基-2,2,6,6-四甲基哌啶(C9H20N2, 4-Amino-2,2,6,6-tetramethylpiperidine, TEMP)购于希恩思生化科技有限公司;碳布(W1S1009)购于台湾碳能科技有限公司;Nafion溶液(5 wt%)购于美国杜邦科技有限公司。
实验仪器:管式炉(TL1200,南京博蕴通仪器科技有限公司);电化学工作站(CHI760E,上海辰华仪器有限公司);高效液相色谱仪(LC-2040C,日本岛津公司,high performance liquid chromatography,HPLC);电感耦合等离子光谱发生仪(iCAP 8000,美国赛默飞世尔科技公司,inductive coupled plasma emission spectrometer,ICP);总有机碳分析仪(Multi N/C 3100,德国耶拿分析仪器公司,total organic carbon,TOC)扫描电子显微镜(S4800,日本日立公司,scanning electron microscope,SEM);透射电子显微镜(Tecnai G2 F30,美国FEI公司,transmission electron microscope,TEM);X射线衍射仪(D8 ADVANCE,德国布鲁克公司,X-ray diffractometer,XRD);X射线光电子能谱仪(K-Alpha,美国赛默飞世尔科技公司,X-ray photoelectron spectroscopy,XPS)。
-
1) N-CNT@FeCo的制备:通过典型水热法制备FeCo-ZIF[25],以FeCo-ZIF为前驱体,将FeCo-ZIF和MA按照质量比为1:M混合均匀后置于坩埚中并加盖放入管式炉内,使用氩气作为保护气体(300 mL·min−1),以5 ℃·min−1升温至800 ℃,煅烧2 h,待冷却至室温得到煅烧后的产物,在此条件下制备的催化剂命名为N-CNT@FeCo-M,其中N-CNT@FeCo-100简写为N-CNT@FeCo。此外,在不添加MA的条件下制备的催化剂命名为FeCo-N。
2)修饰阴极的制备:称取6.0 mg催化剂加入到100 μL去离子水、300 μL无水乙醇和5 μL Nafion溶液的混合液中,所配溶液超声30 min使催化剂分散均匀,然后将其滴涂到直径约为4 cm的碳布上,在室温下自然干燥得到负载催化剂的碳布修饰阴极。
-
采用XRD对催化剂的晶体结构进行表征。采用SEM和TEM对催化剂的微观形貌进行分析。采用XPS对催化剂进行元素价态分析。利用循环伏安法(cyclic voltammetry, CV)、电化学阻抗测试(electrochemical impedance spectroscopy, EIS)和旋转盘电极测试(rotating disk electrode, RDE)研究材料的电化学性能。
-
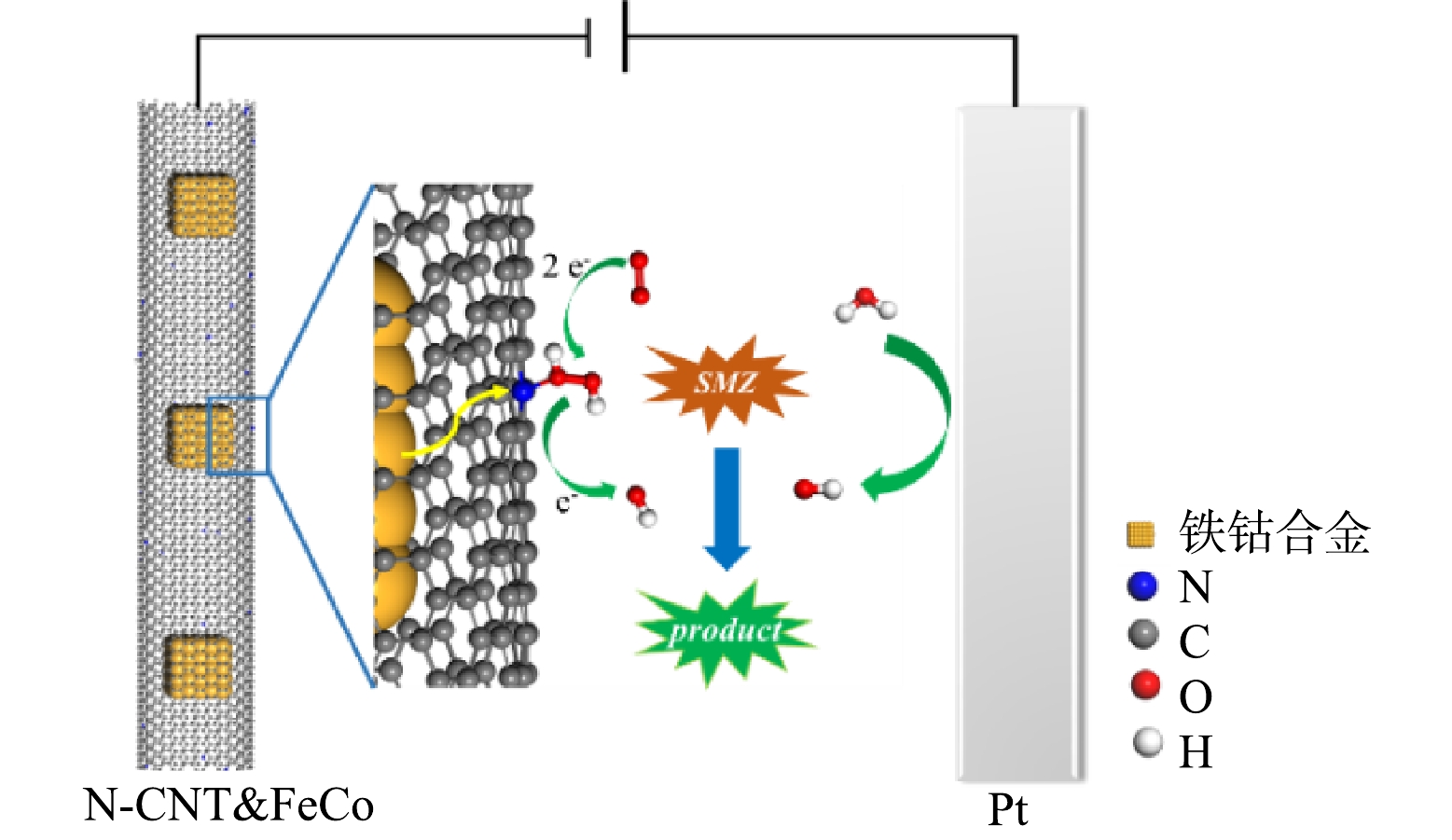
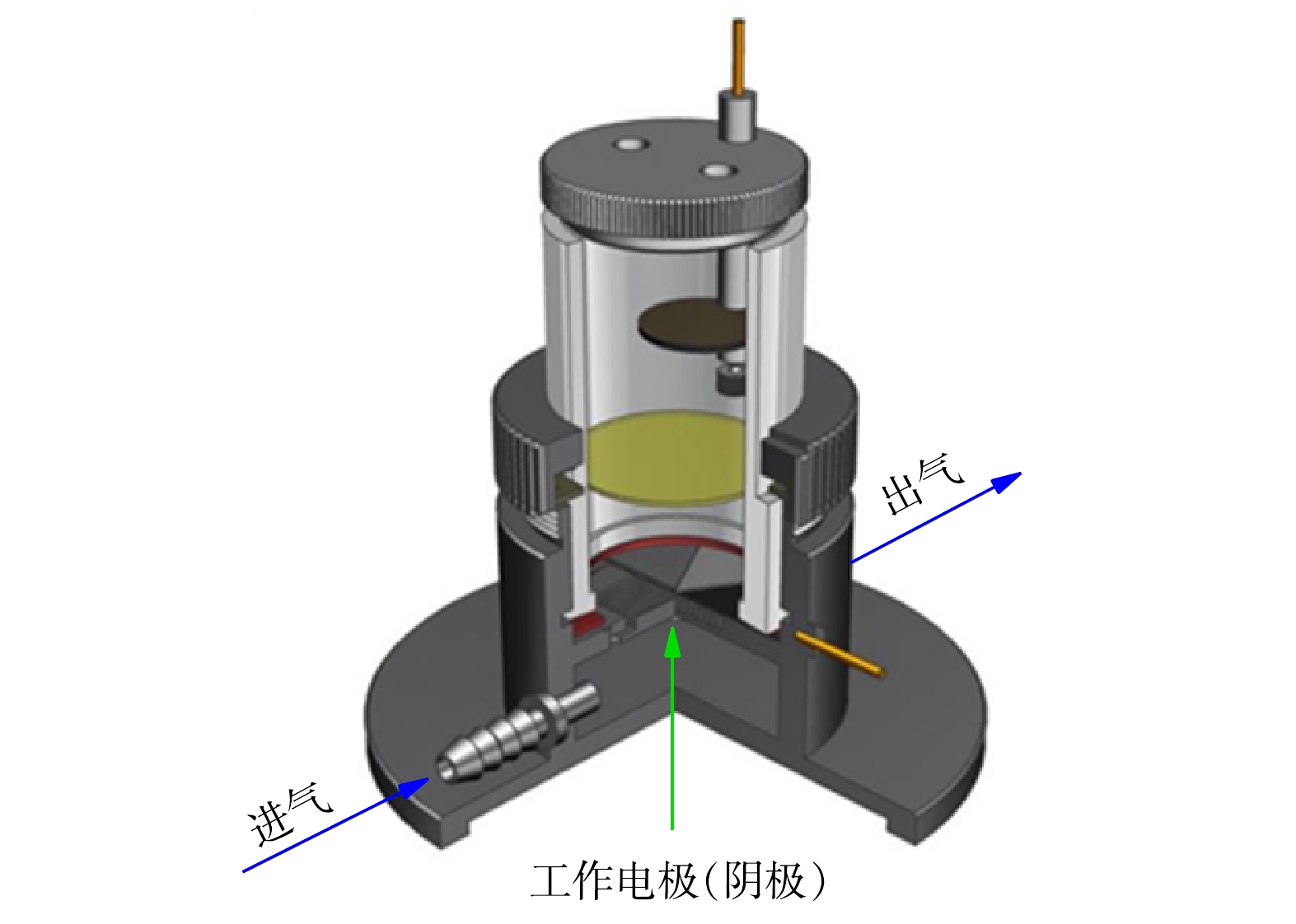
非均相EF降解实验:非均相EF反应在气体扩散反应器中(图1)进行,以均匀负载催化剂的碳布为工作电极,2.0 cm2的铂片和饱和甘汞电极分别为对电极和参比电极,反应通过CHI760E电化学工作站控制,氧气从进气口进入,为保证催化剂不易从碳布上被气流吹落,在装置另一侧设计了出气口。除非另作说明,降解实验均在−0.5 V的恒电位模式下进行。配置含20 mg·L−1 SMZ和50 mmol·L−1的Na2SO4的溶液为模拟制药废水,溶液pH使用NaOH和H2SO4调节,若未作说明,实验在初始pH下进行(pH = 5.2)。降解实验开始前,工作电极浸没在电解液中30 min达到吸附平衡。
SMZ浓度测定:SMZ浓度通过HPLC进行测定,样品通过C18色谱柱进行分离,在柱温30 ℃的条件下,用二极管阵列检测器进行检测,流动相为磷酸二氢钾/乙腈(60:40)混合液,流速为1 mL·min−1,检测波长为270 nm。金属浸出量测定:反应60 min取适量水样经聚四氟乙烯滤膜过滤后,使用ICP测定溶液中铁离子和钴离子浓度。H2O2浓度测定:将样品加入到去离子水、H2SO4与K2TiO(C2O4)2混合溶液中,摇匀后静置10 min显色,在波长386 nm处测定其吸光度[26]。
通过准一级动力学方程对非均相EF降解SMZ的过程进行拟合,SMZ降解速率常数根据式(1)计算。
式中:C0为SMZ初始质量浓度,mg·L−1;t为反应时间,min;C为反应t时的SMZ质量浓度,mg·L−1;k为降解速率常数,min−1。
-
如图2(a)所示,FeCo-ZIF的形貌为表面粗糙的球状微粒,粒径约为300~500 nm,而添加MA后FeCo-ZIF的煅烧产物形貌产生显著变化。如图2(b)所示,FeCo-ZIF的球状结构转变为N-CNT@FeCo相互缠绕的碳纳米管(carbon nanotubes, CNT)结构。
由图3(a)可以看出,N-CNT@FeCo由CNT封装深色的纳米颗粒组成,CNT的管径约为10~120 nm。在图3(b)中能明显观察到对应Fe0.3Co0.7合金中(110)晶面的晶格间距(0.201 nm)[27],结合XRD谱图(图3(c))中45.1°和65.5°处的Fe0.3Co0.7合金特征峰(PDF#50-0795)以及44.0°、51.2°和76.8°处的Fe0.28Co0.72合金特征峰(PDF#51-0740),证实深色纳米颗粒为铁钴合金。此外,在图3(b)观察到0.340 nm的晶格间距对应石墨碳的(002)晶面,XRD谱图中26.5°处也出现了明显的石墨碳特征峰(PDF#41-1487)。这种高结晶度的石墨碳具有优异的导电性能,有利于内层合金纳米颗粒在EF体系中ORR与类芬顿反应的电荷传输。
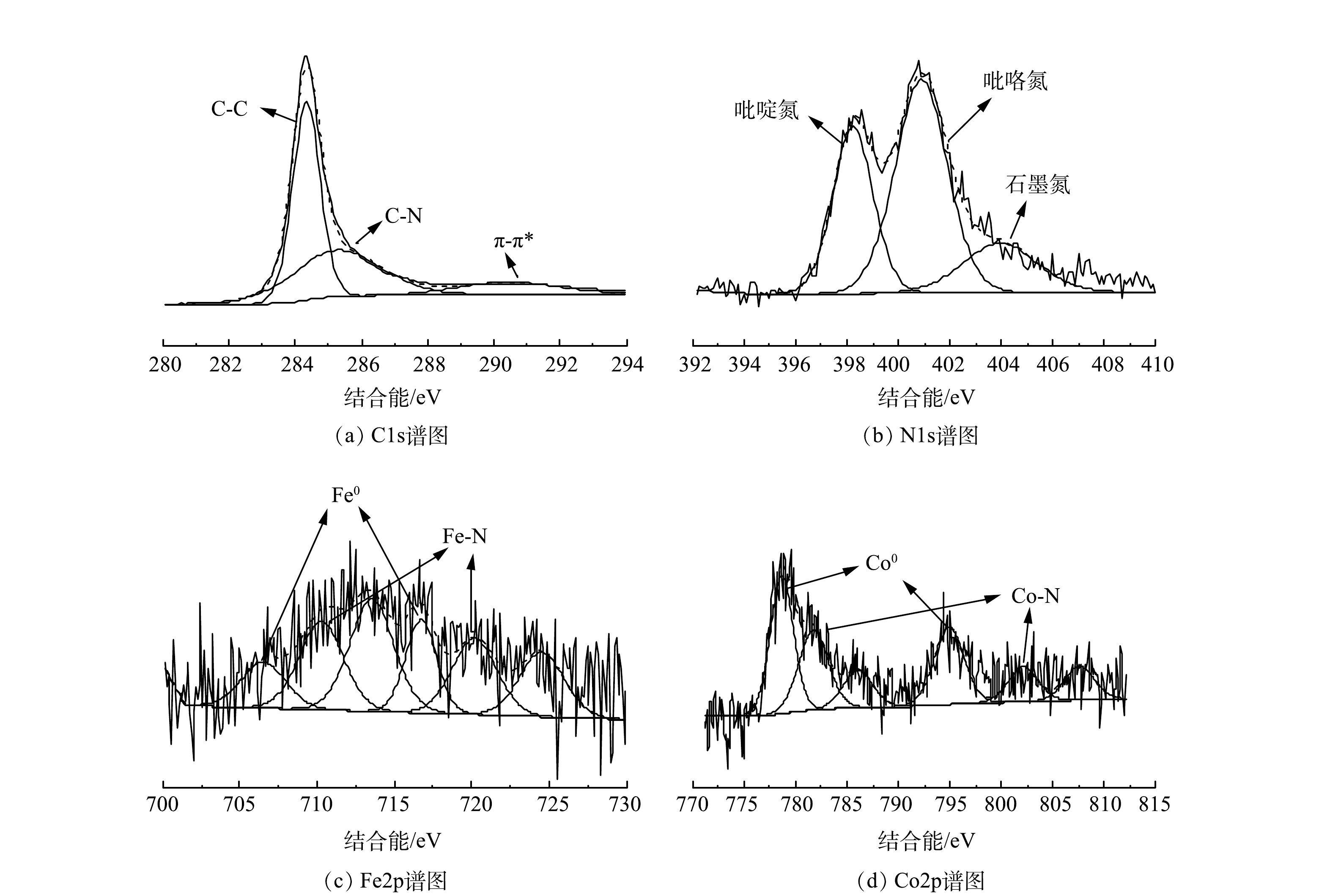
利用XPS分析了材料表面各元素组成及价态。由图4(a)的C1s精细谱中可以看出,位于284.3、285.3和290.4 eV处的3个峰,分别对应石墨碳、C-N和π-π*。这说明碳材料的石墨化程度较高,与TEM以及XRD分析结果一致[28]。在图4(b) 的N1s精细谱中位于398.2、400.9和403.9 eV处的特征峰分别对应吡啶氮、吡咯氮和石墨氮[29]。证明氮在CNT上的成功掺杂,这有利于提高催化剂对2e− ORR的选择性[30-31]。图4(c)和图4(d)中出现的Fe0特征峰(706.5 eV和716.7 eV处)及Co0特征峰(778.7 eV和794.9 eV)则进一步证明了铁钴合金的存在[32]。Co2p精细谱在781.8 eV和802.8 eV处还出现了对应Co-N的特征峰[33],表明铁钴合金可能通过金属-氮(M-N)结合键与外层N-CNT相连。
-
如图5(a)和图5(b)所示,N-CNT@FeCo对ORR的响应电流较FeCo-ZIF明显提升,ORR起始电位更正。表明N-CNT@FeCo的ORR活性高于FeCo-ZIF。如图5(c)所示,催化剂的EIS图均由一段高频区的圆弧和一条低频区的斜线组成。高频区中圆弧的直径代表电荷转移阻抗,直径越小则电荷传递速率越高,低频区斜线的斜率代表电解质和电极间的物质扩散[34-35]。煅烧后的N-CNT@FeCo圆弧直径明显小于FeCo-ZIF,表现出更低的电荷转移阻力(60.7 Ω<187.9 Ω),这也导致了更强的电化学活性,与CV测试结果一致,以上结果表明煅烧处理提高了催化剂的电子传递速率。
-
如图6(a)和图6(b)所示,FeCo-ZIF直接应用于EF降解SMZ,虽然60 min对SMZ的去除率可达91.7%,但铁离子和钴离子的浸出量高达1.22 mg·L−1和8.65 mg·L−1,均相EF的作用不容忽视。由FeCo-ZIF直接煅烧得到的FeCo-N在EF反应中催化氧化SMZ的活性及稳定性低,在60 min对SMZ的去除率仅为73.7%,且钴离子的浸出量仍有1.65 mg·L−1。图2(c)中看到,FeCo-N微球表面密集覆盖了大量金属团簇,在EF反应中直接暴露在强氧化环境下,导致材料稳定性较差。而FeCo-ZIF在添加不同比例的MA煅烧后得到的N-CNT@FeCo-M,反应60 min时对SMZ的去除率提升至88.2%~100%,钴的浸出量下降至0.05~0.50 mg·L−1,性能最佳的N-CNT@FeCo-100的k值高达0.057 min−1,约为FeCo-N的3倍(图6(c)),60 min可实现SMZ的完全降解,TOC去除率为45.3%。
EIS测试结果(图5(c))表明,N-CNT@FeCo-100(60.7 Ω)的电荷转移阻力远小于FeCo-N(145.9 Ω)。这可能得益于添加MA后衍生出的N-CNT能加速电荷传输[36-37]。结合N-CNT@FeCo的表征分析,MA作为碳源和氮源衍生出的N-CNT能通过促进内部铁钴合金之间的电子转移,提高EF催化活性,同时N-CNT作为金属纳米颗粒的保护层,还能有效提高内层合金在强氧化环境下的化学稳定性。综上所述,优选FeCo-ZIF与MA质量比为1:100进行后续研究。
SMZ的降解通常是电吸附、阳极氧化、均相EF以及非均相EF过程共同作用的结果,因此评估了反应40 min时各部分的贡献率,结果如图6(d)所示。电吸附、阳极氧化、均相EF以及非均相EF过程对SMZ降解的贡献率依次为5.5%、22.1%、16.7%和55.7%,说明非均相EF对SMZ的降解起主要作用。
-
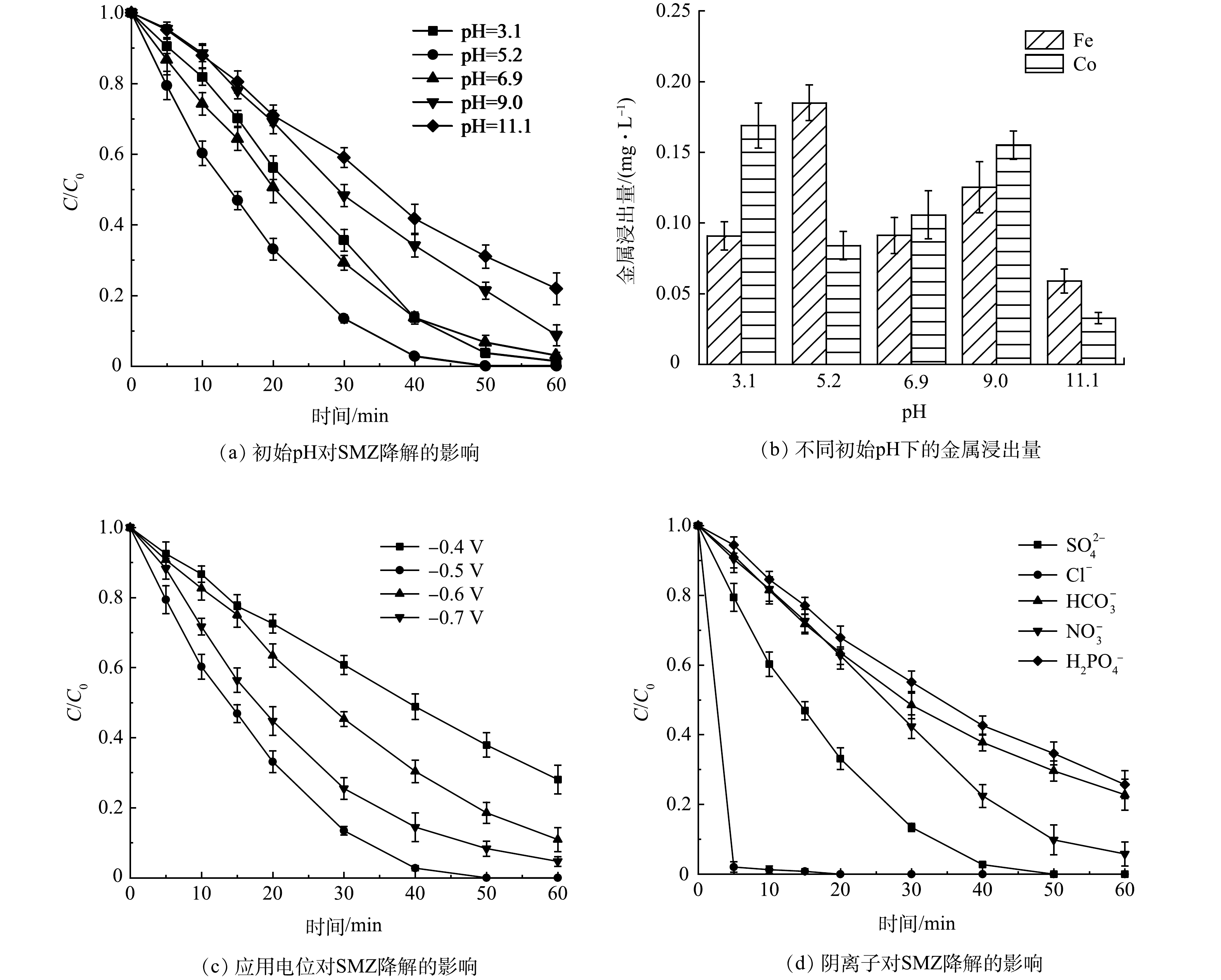
1) pH的影响。不同初始pH下SMZ的降解效率变化如图7(a)所示。EF性能随着初始pH的增大先升高后下降,无需调节pH时(pH=5.2)降解性能最佳,40 min对SMZ的去除率即可达到97.3%。由图7(b)可知,即使在pH=3.1时,催化剂仍保持高稳定性,铁和钴的浸出量分别仅有0.09 mg·L−1和0.17 mg·L−1。综上所述,后续实验选择在pH=5.2下进行。
2)应用电位的影响。如图7(c)所示,当应用电位为-0.5 V时,在60 min时SMZ的去除率为100%。将应用电位调节至-0.6 V时,SMZ的去除率下降了11.0%。这是由析氢反应以及H2O2被进一步还原为H2O所致,系列副反应抑制了SMZ降解。选择应用电位为-0.5 V进行后续实验。
3)阴离子的影响。由图7(d)可见,Cl−能够促进SMZ的降解。这是因为Cl−在阳极生成强氧化性的HClO[38]。而其余3种阴离子抑制了SMZ的降解,抑制作用从强到弱依次为H2PO4−≈HCO3−>NO3−,因为3种阴离子均会淬灭·OH生成氧化性更弱的自由基[39]。
-
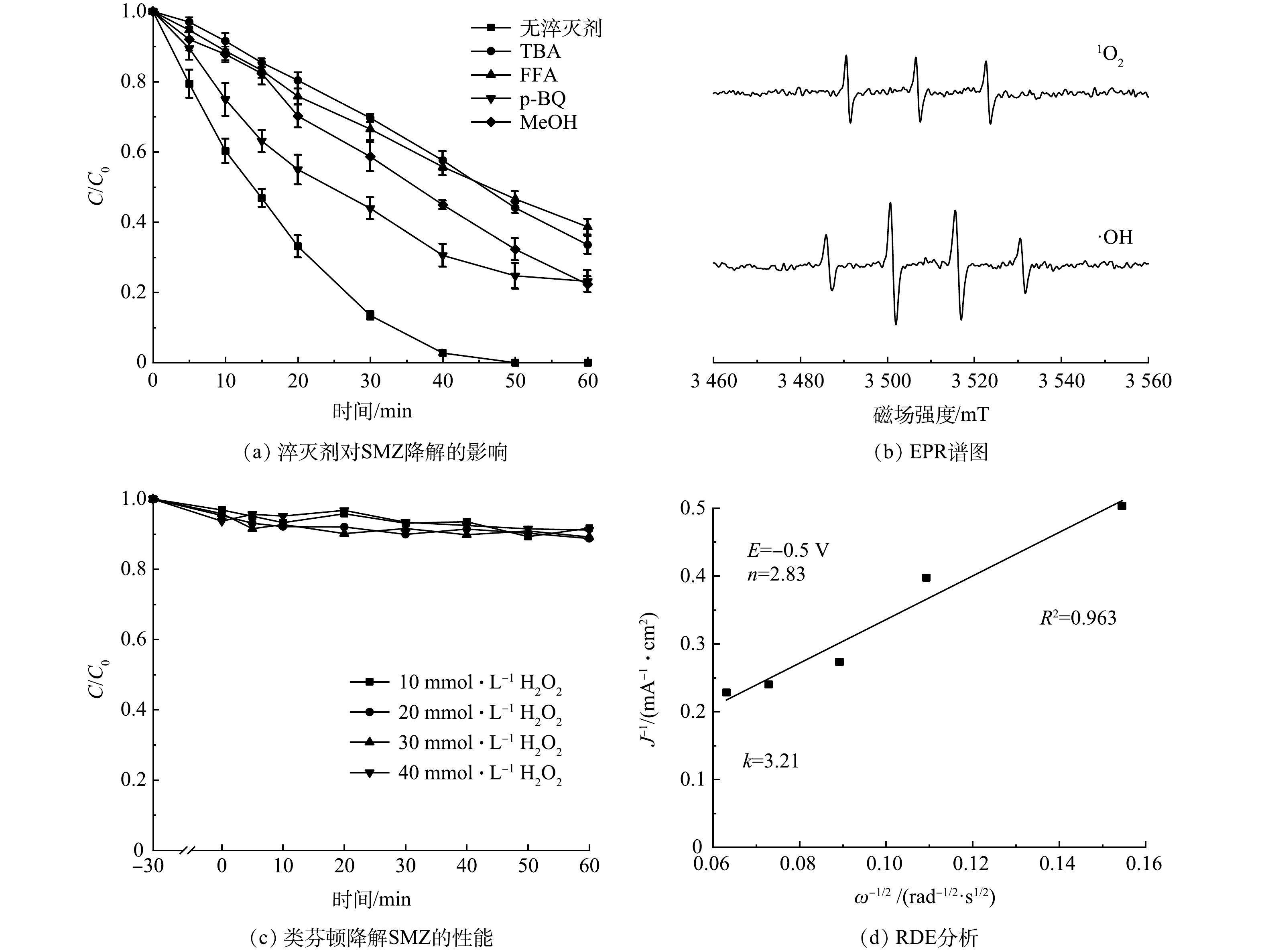
使用TBA(100 mmol·L−1)、FFA(5 mmol·L−1)、p-BQ(5 mmol·L−1)、MeOH(50 mmol·L−1)分别作为·OH、1O2、·O2−、·SO4−的淬灭剂[40],在此条件下TBA、FFA和MeOH对·OH的淬灭能力维持在同一水平。如图8(a)所示,TBA的加入对SMZ降解产生明显抑制,而加入FFA的抑制效果(38.7%)与TBA(33.6%)几乎一致。这说明·OH对污染物的降解起到了至关重要的作用,1O2虽然能通过链式反应生成,但其贡献较小。MeOH对SMZ降解的抑制作用弱于TBA,说明·SO4−的贡献可以忽略不计。淬灭·O2−不但会抑制H2O2的生成,同时也会抑制Fe(Ⅲ)还原为Fe(Ⅱ),降低了·OH的生成速率,进而削弱了SMZ的降解[41-42]。图8(b)为电子顺磁共振(EPR)测试结果。可以看出,基于N-CNT@FeCo的EF体系中出现了DMPO-OH和TEMP-1O2的特征峰,证实·OH和1O2的存在[24]。综上所述,污染物的降解是以·OH为主导,1O2和·O2−共同参与的过程。
此外,还考察了N-CNT@FeCo在类芬顿体系中直接活化H2O2的能力,发现N-CNT@FeCo(图8(c))无法有效地活化H2O2生成·OH降解水中的SMZ。这是因为碳材料自身活化H2O2的能力较差。同时铁钴合金被较厚的碳层(图2(a))包裹,在碳层层数较厚(> 3层)的情况下电子难以从铁钴合金中传递到碳材料表面[43],因此N-CNT@FeCo的非均相芬顿活性差,以上结果证实了电场不可或缺的作用。
N-CNT已经被证实是一种有效的2e− ORR催化剂,其中石墨氮是生成H2O2的主要活性位点,基于N-CNT@FeCo的EF技术可能的机理如图9所示。虽然被N-CNT封装的铁钴合金不直接参与H2O2的活化,但在电场作用下其具有强给电子能力,使N-CNT表面生成的H2O2在解吸前就得到电子被还原为·OH,O2通过如式(2)所示的3电子途径直接生成·OH[44],因此,溶液中H2O2浓度维持在较低水平(< 5.0 mg·L−1)。RDE测试结果如图8(d)所示,在−0.5 V的应用电位下N-CNT@FeCo的ORR电子转移数为2.83,近似于3的电子转移数也证明O2可能通过式(2)生成·OH,这种3电子氧还原途径能有效地避免传统EF中过渡金属氧化还原电对循环速率慢的问题。
-
N-CNT@FeCo的循环回用性能如表1所示,经过5次循环后60 min时SMZ的去除率仍有96.0%,且铁、钴离子最高浸出质量浓度分别仅为0.31 mg·L−1和0.30 mg·L−1,表明N-CNT@FeCo具有良好的稳定性能。
-
1)以MA为碳源和氮源,在铁钴合金外衍生的N-CNT可同时提高催化剂的活性和稳定性。在FeCo-ZIF与MA质量比为1:100条件下制备的N-CNT@FeCo具有最佳的催化性能,相较于FeCo-N,在60 min对SMZ的去除率提升了26.3%,而金属浸出量降低了84.9%。
2)在初始pH为5.2,应用电位为-0.5 V的最佳运行条件下,N-CNT@FeCo的k值可达0.057 min−1。H2PO4−、HCO3−和NO3−会抑制SMZ的降解,而Cl−会促进污染物的降解。N-CNT@FeCo循环回用5次,60 min对SMZ的去除率仍有96.0%。
3)·OH是主要的活性物种,1O2和·O2−也参与了SMZ的降解;结合H2O2累积浓度检测、非均相芬顿实验以及RDE检测,推测基于N-CNT@FeCo的EF体系中O2是通过3电子还原生成·OH。
4)反应40 min时电吸附、阳极氧化、均相EF以及非均相EF对SMZ降解的贡献率依次为5.5%、22.1%、16.7%和55.7%。
封装型双金属阴极催化剂强化电芬顿技术高效去除磺胺甲恶唑
Highly efficient removal of sulfamethoxazole by encapsulated bimetallic cathode catalyst enhanced electro-Fenton technology
-
摘要: 传统非均相电芬顿(EF)技术主要面临活性氧物种生成速率慢、催化剂稳定性差等不足。将FeCo-ZIF和三聚氰胺(MA)共混(质量比为1:100)煅烧,成功制备出氮掺杂碳纳米管封装铁钴合金阴极催化剂(N-CNT@FeCo),可强化EF高效去除水中磺胺甲恶唑(SMZ,初始质量浓度为20 mg·L−1),在近中性条件下50 min内即可完全去除,降解速率常数可达0.057 min−1,是单独煅烧FeCo-ZIF制备的裸露型双金属催化剂FeCo-N的3倍,且前者的金属浸出总量(0.27 mg·L−1)仅为后者(1.79 mg·L−1)的15.1%。循环回用5次后,60 min内N-CNT@FeCo对SMZ的去除率仍可达到96.0%。扫描电子显微镜表征与电化学阻抗测试结果表明,由MA诱导生成的N-CNT,不仅通过封装结构有效限制了内部铁钴合金受强氧化性环境腐蚀破坏,而且显著加速了内部铁钴合金的电子传递速率,N-CNT@FeCo的独特封装结构使其兼具高催化活性和高稳定性。本研究为高效稳定的阴极催化剂提供了稳定、可控、易放大的封装策略。Abstract: The conventional non-homogeneous electro-Fenton (EF) technology mainly faces the deficiencies of slow generation rate of active oxygen species and poor catalyst stability. A nitrogen-doped carbon nanotube-encapsulated iron-cobalt alloy cathode catalyst (N-CNT@FeCo) was successfully prepared by calcination of FeCo-ZIF and melamine (MA) in a co-blend with mass ratio of 1:100, which could enhance EF to efficiently remove sulfamethoxazole (SMZ, initial concentration set as 20 mg·L−1) from water, and SMZ could be completely removed within 50 min under near-neutral conditions with degradation. The degradation rate constant was up to 0.057 min−1, which was two times higher than that of the bare bimetallic catalyst FeCo-N prepared by calcination of FeCo-ZIF alone, and the total metal leaching from the former (0.27 mg·L−1) was only 15.1% of that from the latter (1.79 mg·L−1). 96.0% of SMZ removal was still achieved at 60 min when N-CNT@FeCo was recycled after five times. Scanning electron microscopy analysis and electrochemical impedance test results showed that the N-CNT induced by MA not only effectively limited the corrosion damage of the internal iron-cobalt alloy by the strong oxidizing environment through the encapsulation structure, but also significantly accelerated the electron transfer rate of the internal iron-cobalt alloy, and the unique encapsulation structure of N-CNT@FeCo made it both highly catalytic activity and highly stable. This study provides a stable, tunable and easy to enlarge encapsulation strategy for the preparation of efficient and stable cathode catalysts.
-
Key words:
- electro-Fenton /
- encapsulate /
- bimetallic /
- iron-cobalt alloy /
- sulfamethoxazole
-
随着我国城镇化步伐的加快,大量池塘受到污染或被填埋。这些池塘通过合理规划和利用不仅可以作为可观赏的景观塘,还可以兼用于污水的生化处理。本实验将景观设计和水体修复相结合,通过组合型生态浮岛原位修复技术改善水质,美化环境。
生态浮岛技术出现于20世纪50年代,直到20世纪80年代以后,生态浮岛技术在社会科技大发展的前提下才得以深入研究。DESTEFANI等[1]采用生态浮床净化某自然公园里的受污染的水体,浮岛植物选用香蒲、香根草、灯芯草,实验结果表明,浮床对水体中COD、TN、TP的去除率分别达到了66%、65%、13%。KANSIIME等[2]采用纸莎草浮床研究其对水体中N、P污染物的去除效果,实验结果表明,该浮床对TN、TP的去除率分别为80%~90%、70%~80%。李欲如等[3]在冬季采用生态浮床研究了多花黑麦草、大蒜、水芽对富营养化水体的处理效果,实验结果表明,3种植物对水体中COD去除率为49.2%~55.1%、总氮去除率为29.1%~58.9%、氨氮的去除率为39.7%~65.6%、总磷去除率为33.3%~54.9%。何成达[4]采用美人蕉浮床处理生活污水,实验结果表明,COD去除率达到90%以上,TN、TP去除率均达到80%以上。王郑等[5]通过将球形填料与美人蕉构建的组合型生态浮床处理农家乐废水,实验结果表明,该组合型生态浮床对COD、NH4+-N、TN、TP的去除率分别为79.71%、88.67%、73.88%、85.61%。
研究[6-7]表明,单一的生态浮岛由于浮岛植物本身的性质,对环境和水质都有一定的要求并对污染物的吸收效率各不相同,因此,需要对特定物理情况和环境因素制定相匹配的浮床和浮岛植物。本研究将浮岛植物黄花鸢尾与改良型火山石填料相结合,通过改良型火山石的强化作用,为黄花鸢尾提供一个较好的环境,从而达到治理重污染水体的效果。
1. 材料与方法
1.1 实验装置
该组合型生态浮岛由浮岛植物黄花鸢尾、塑料浮板、改良型火山石填料、活性炭网组成(如图1所示)。将花黄鸢尾固定安放在中心镂空的塑料浮板上,每块浮板上种植1株黄花鸢尾,通过浮板的浮力使其漂浮在水面上,黄花鸢尾根部和改良型火山石填料用活性炭网包裹,所使用的改良型火山石直径略大于活性炭网网格孔径,每块浮板下改良型火山石填料用量控制在1.2~1.5 kg。各实验组在100 cm(长)×100 cm(宽)×80 cm(高)塑料水箱中进行,塑料水箱中水深控制在70 cm。
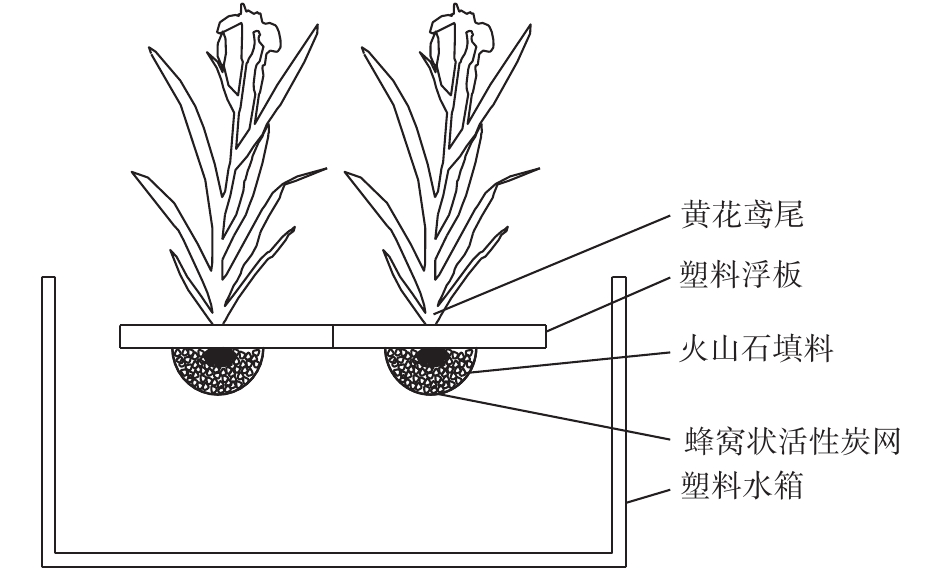 图 1 组合型生态浮岛装置示意图Figure 1. Schematic diagram of combined ecologicalfloating island device
图 1 组合型生态浮岛装置示意图Figure 1. Schematic diagram of combined ecologicalfloating island device1.2 实验方案
实验前,先将购买的黄花鸢尾幼苗放入实验水体中进行适应性培养1周,选取长势较好、株高基本一致的植株为实验所用,并将其根部做适当修剪,以保证实验植株的可对比性。将改良型火山石填料在实验室内用实验水体进行人工挂膜(水温控制在25 ℃左右),每隔6 h,通过曝气风机向水中曝气1次(水中溶解氧控制在2 mg·L−1),每隔3 d换1次水样,直至完全挂膜为止(开始挂膜时间约27 h;挂膜完全15 d)。实验共分为4组:空白对照组(0#)、黄花鸢尾处理组(1#)、火山石填料处理组(2#)、组合型生态浮岛处理组(3#)。实验从2018年4月25日开始,至2018年5月22日结束,实验期间平均气温在16 ℃左右。各组均放置在有阳光照射的池塘边,并做好防雨措施。每3 d取1次样,测定水体各指标,结束后测定植株高度和根部长度。本次实验水体取自某人工池塘,水体水质基本指标COD、TN、TP、NH4+-N、NO3−-N、DO分别为80.6~107.3、9.16~11.97、0.86~1.14、7.58~8.44、3.11~3.23、1.4~2.3 mg·L−1。
1.3 测试方法
各水质指标的测定方法参照文献中的方法[8]。COD采用重铬酸钾法测定,TN采用碱性过硫酸钾氧化法测定,TP采用钼锑抗分光光度法测定,NH4+-N采用纳氏试剂分光光度法测定,DO采用电极测量法测定,植物株高及根长采用标准卷尺测定。
2. 结果与讨论
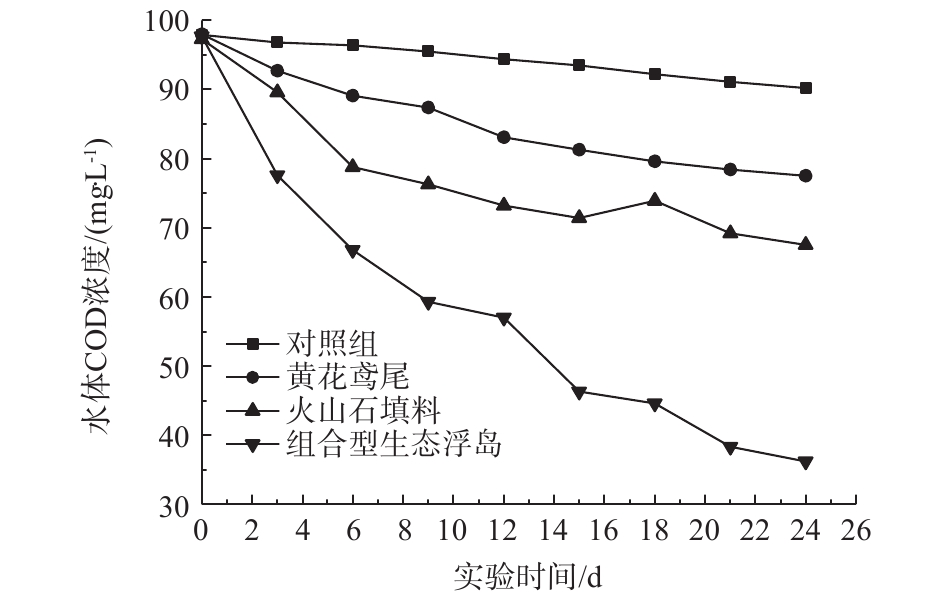
2.1 水体COD的变化
各处理组对水体COD的影响如图2所示。实验经过24 d后,0#、1#、2#、3#处理组对COD的去除率分别为7.86%、20.84%、30.63%、62.95%。由此可见,组合型生态浮岛对COD的去除率最高,且较单一的植物和填料处理组中COD去除效果明显,这与大量组合型生态浮岛处理污水的研究结果[9-11]一致。组合型生态浮岛对COD的去除率较高的原因主要包括3个方面。其一,浮岛下部的填料为改良型火山石。火山石自身具有对水流阻力小、不易堵塞、布水布气均匀、表面粗糙、挂膜速度快、反冲洗时微生物膜不易脱落、多孔性等物理特性。而且火山岩可以使水中的离子活跃(主要是增加了氧离子的含量)[12],从而促进了好氧微生物的生长,而好氧微生物生长过程需要消耗大量有机物。改良型火山石进一步扩大了其表面积,为微生物的生长提供了更大的空间,增加了微生物的数量。其二,黄花鸢尾为人工筛选后种植,其根系相对较为发达,为微生物繁殖提供了场所,且根系的泌氧作用为好氧微生物提供了有利的生存环境,增加了水体中有机物的消耗。其三,有研究[13]表明,活性炭网对水体和底泥中芳香族有机物有较好的去除效果。水体中的有机物一部分被活性炭网吸附,提高了有机物的去除率,并加速了水体中COD的去除。由图2也可以看出,前6 d内,2#、3#中COD降解速度较快。2#相较于1#处理组,处理效率高,这表明改良型火山石填料在COD的去除过程中起主要的作用。
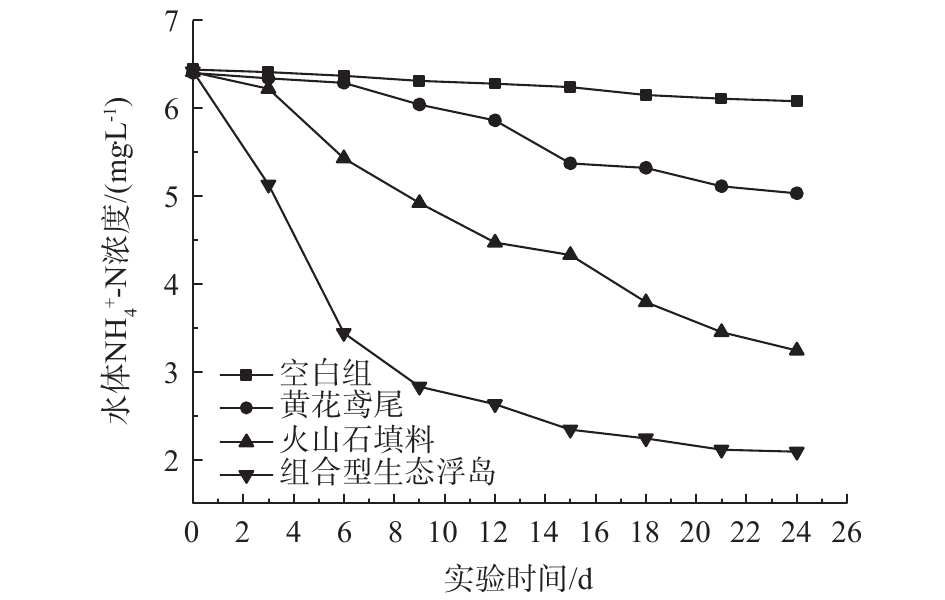
2.2 水体NH4+-N的变化
各处理组对水体NH4+-N的影响如图3所示。可以看出,在实验期内,各组NH4+-N的含量都呈下降的趋势,其中组合型生态浮岛(3#)对NH4+-N的去除率最高,达到67.45%。其余3个处理组0#、1#、2#去除率分别为5.59%、21.41%、49.45%。水体中NH4+-N的去除主要通过植物吸收、微生物的硝化作用及NH3自然挥发[14]。可以看出,改良型火山石填料组(2#)对NH4+-N的去除率相对于黄花鸢尾组(1#)较高,这说明对NH4+-N的去除,微生物的硝化作用高于黄花鸢尾的吸收作用。改良型火山石对NH4+-N的去除率较高的原因是由于它的多孔性产生的高表面积同样也是培养水中硝化细菌的良好温床,并且其表面带正电荷有利于微生物固着生长,亲水性强,把水中各种原因产生的NO2−-N和NH4+-N转化成毒性相对小的NO3−-N,从而降低了水体中NH4+-N的含量。组合型生态浮岛通过改良型火山石填料中丰富的微生物及植物根系的泌氧作用,为硝化细菌提供了一个良好的生长繁殖条件,从而大幅度提高了NH4+-N的去除率。
2.3 水体NO3−-N的变化
水体中的硝酸盐氮主要来源于受污染水体中自身含有的NO3−-N和由硝化作用产生的NO3−-N,它的主要去除形式有反硝化作用和植物吸收等[15]。
各处理组对水体NO3−-N的影响如图4所示。可以看出,空白对照组(0#)、改良型火山石填料处理组(2#)呈上升的趋势,组合型生态浮岛组(3#)呈先升后降的趋势,而黄花鸢尾处理组(1#)则呈下降趋势。由此推断出,黄花鸢尾对NO3−-N的吸收作用高于水体中微生物的硝化作用。改良型火山石填料处理组水体中NO3−-N大幅度升高,主要是由于其表面大量的硝化细菌将水体中的NH4+-N转化成了NO3−-N,这从NH4+-N的去除曲线中也能明显地看出。组合型生态浮岛处理组在9 d前NO3−-N处于上升阶段,这是由于整个水体中的硝化作用大于黄花鸢尾的吸收和反硝化作用。9 d后,NO3−-N呈下降趋势,这是由于水体中溶解氧减少,硝化作用减弱,植物的吸收能力和反硝化作用占主导地位。21 d后,水体中NO3−-N趋于平衡,这可能一方面由于植物的吸收达到饱和,另一方面从COD的变化曲线中可以推断,由于水体中碳源数量减少从而使反硝化作用降低。从图4中还可以看出,组合型生态浮岛处理组前期NO3−-N含量的上升速度高于改良型火山石填料组,这是由于改良型火山石和植物根系两者的协同作用使水体中的氧离子高于单纯的改良性火山石填料组。
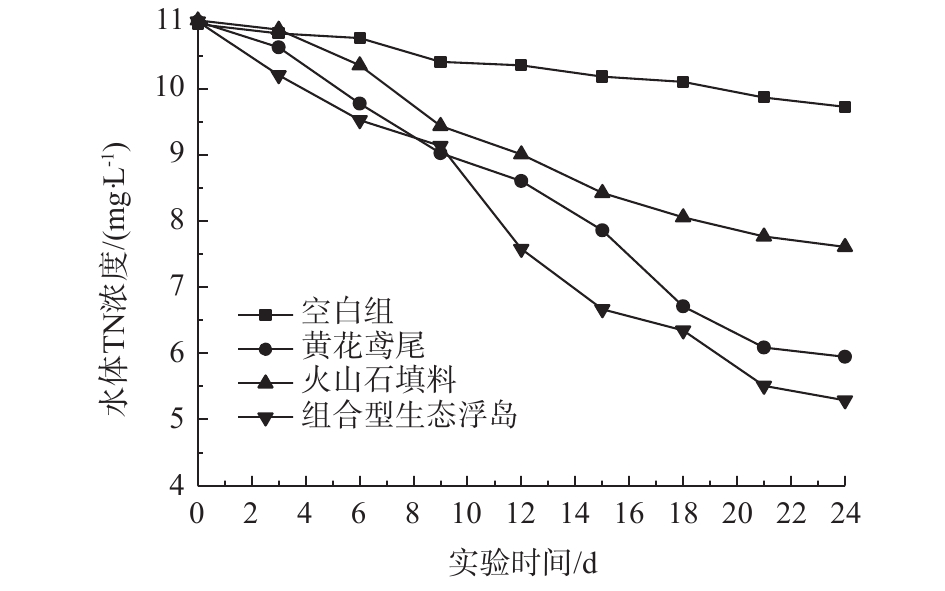
2.4 水体TN的变化
各处理组对水体TN的影响如图5所示。可以看出,4个处理组TN都呈下降趋势,0#、1#、2#、3#对TN的去除率分别为11.38%、46.01%、31.07%、51.99%,组合型生态浮岛(3#)处理效率高于其他实验组。组合型生态浮岛对水体中TN有较高的去除效果,这主要是由于改良型火山石填料为黄花鸢尾提供了一个较佳的生长环境。经过一段时间后,黄花鸢尾的根系植入改良型火山石填料内,有利于根系的保护,从而提高了黄花鸢尾根系对氮元素的吸收。同样,改良型火山石填料与植物根系的结合也为硝化及反硝化细菌提供了好氧与厌氧的生存环境,进一步加强了脱氮的效果。图5显示,黄花鸢尾处理组对TN的去除率高于填料处理组,而从空白组中可以看出,水体中分散的微生物对TN的自然去除率并不高,这表明黄花鸢尾处理组主要是靠植物的吸收作用和其根系微生物的硝化及反硝化作用达到去除目的。具体是由黄花鸢尾植物本身的吸收作用还是根系微生物的分解消耗起主导作用需要进一步研究。而改良型火山石填料处理组中TN去除则主要依靠微生物的作用。该组合型生态浮岛中微生物的脱氮作用应占主导地位,但其与花黄鸢尾吸收作用之间的差异还须做进一步研究。
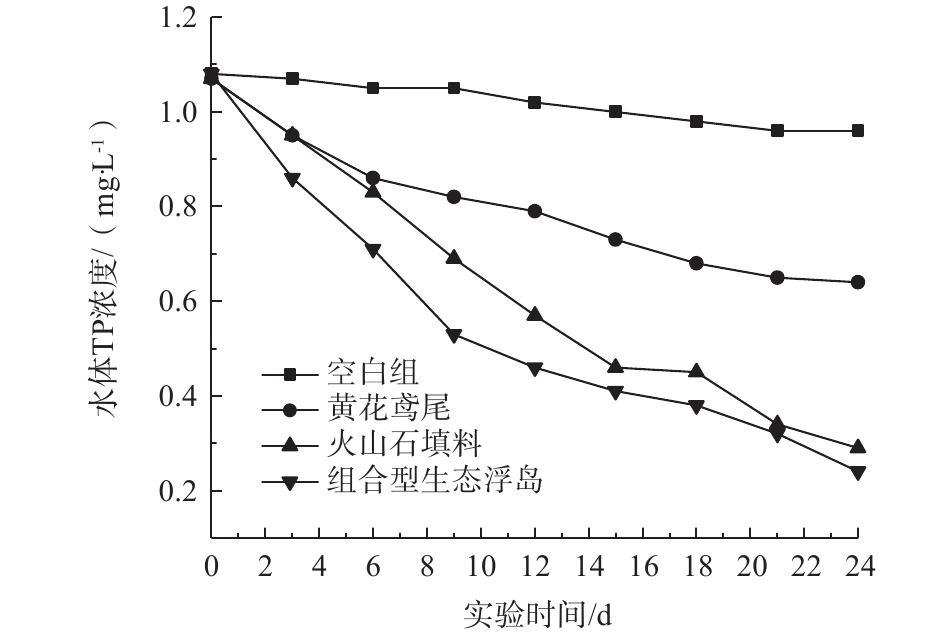
2.5 水体TP的变化
水体中的磷大体可分为颗粒型和溶解型2种形态。对TP的去除主要是靠植物吸收、微生物固定以及填料的吸附等[16]。各处理组对水体TP的影响如图6所示。可以看出,4个处理组TP都呈下降趋势,0#、1#、2#、3#对TP的去除率分别为11.11%、40.19%、72.90%、77.78%。可见组合型生态浮床对TP的去除效果较高,原因如下:1)黄花鸢尾根系对TP的吸收利用;2)改良型火山石填料中的聚磷菌在好氧条件下将部分TP作为自身生长繁殖所需的元素进行吸收并同化;3)通过填料自身的吸附作用将水体中颗粒型磷吸附到填料表面,从而降低水体中TP的含量。改良型火山石填料组对TP的去除速度及效率均高于黄花鸢尾处理组,这表明组合型生态浮岛中微生物的吸收同化及填料吸附作用是TP去除的主要机制。
2.6 黄花鸢尾生长状况
黄花鸢尾实验开始与结束的生长状况如表1所示。可以看出,组合型生态浮岛中黄花鸢尾的株高及根长均大于黄花鸢尾组,由此看来,改良型火山石填料中的微生物促进了黄花鸢尾的生长。研究[17-18]表明,植物的氮磷吸收及固定的量与植物生物量呈显著相关,植物生物量越大,对氮磷吸收越多,对水体净化能力越强,这与本研究结果一致。
表 1 黄花鸢尾生长状况Table 1. Growth status of Iris pseudoacorus组别 开始 结束 株高/cm 根长/cm 株高/cm 根长/cm 黄花鸢尾组 24.3 3.7 41.2 18.9 组合型生态浮岛组 24.1 3.6 55.7 32.6 | Show Table DownLoad:
CSV
DownLoad:
CSV
3. 结论
1)该组合型生态浮岛对COD、NH4+-N、TN、TP的去除率分别为62.95%、67.45%、51.99%、77.78%,且植物生物量高于单纯的植物处理组,填料中的微生物促进了植物的生长,从而提高了水质净化效果。
2)黄花鸢尾与改良型火山石、表面微生物具有很好的协同作用,共同促进了水体中COD、NH4+-N、TN、TP等污染物的去除。
3)针对不同的水质污染状况,合理搭配浮岛的组合方式可实现污染物的快速降解,达到净化水质的目的,适用于城镇水体尤其是居住区的静态水体的景观治理。
-

图 3 N-CNT@FeCo的TEM、HRTEM以及XRD图
Figure 3. TEM, HRTEM imagines and XRD patterns of N-CNT@FeCo
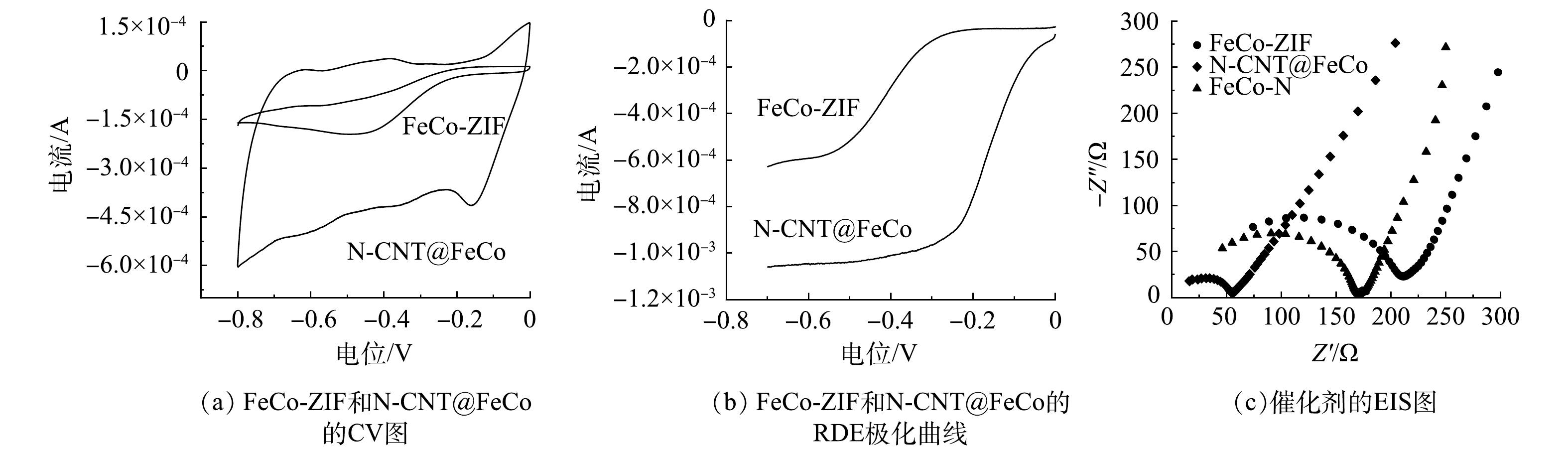
图 5 FeCo-ZIF、N-CNT@FeCo和FeCo-N的电化学表征
Figure 5. Electrochemical characterization of FeCo-ZIF, N-CNT@FeCo and FeCo-N
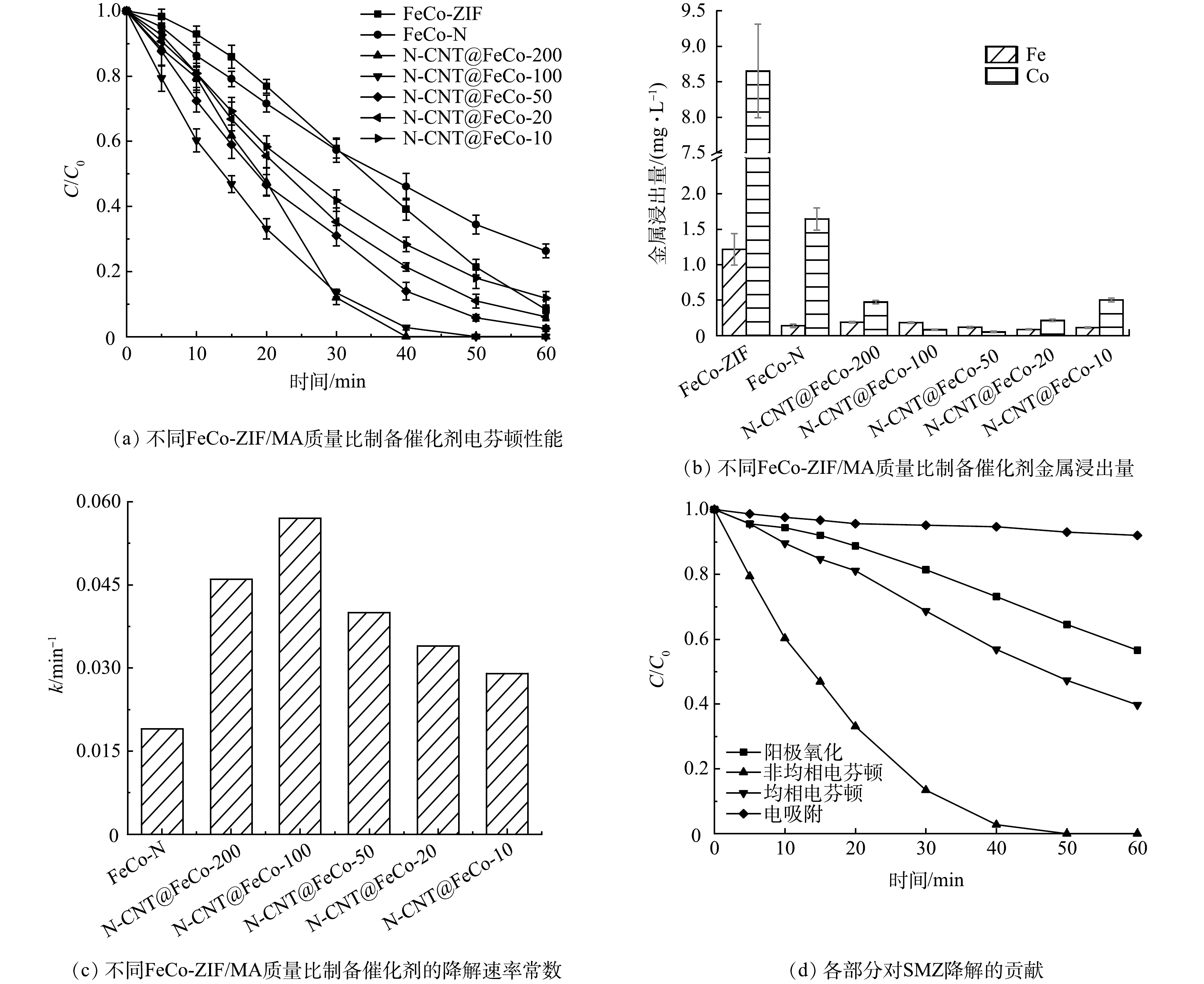
图 6 不同FeCo-ZIF和MA质量比制备催化剂的EF性能
Figure 6. EF performance of catalyst prepared with different FeCo-ZIF and MA mass ratios
表 1 N-CNT@FeCo的循环回用性能
Table 1. Cyclic reuse performance of N-CNT@FeCo
循环次数 去除率/% 浸出量/(mg·L-1) Fe Co 第1次 100 0.19 0.08 第2次 100 0.27 0.10 第3次 96.6 0.31 0.30 第4次 96.0 0.24 0.27 第5次 96.0 0.13 0.28
下载: 导出CSV
-
[1] 孟庆玲, 欧晓霞, 张梦然, 等. 抗生素污染废水处理技术研究进展[J]. 绿色科技, 2021, 23(2): 81-83. doi: 10.3969/j.issn.1674-9944.2021.02.029 [2] 齐亚兵, 张思敬, 孟晓荣, 等. 抗生素废水处理技术现状及研究进展[J]. 应用化工, 2021, 50(9): 2587-2593. doi: 10.3969/j.issn.1671-3206.2021.09.054 [3] CARVALHO I T, SANTOS L. Antibiotics in the aquatic environments: A review of the European scenario[J]. Environment International, 2016, 94: 736-757. doi: 10.1016/j.envint.2016.06.025 [4] 赵富强, 高会, 张克玉, 等. 中国典型河流水域抗生素的赋存状况及风险评估研究[J]. 环境污染与防治, 2021, 43(1): 94-102. doi: 10.15985/j.cnki.1001-3865.2021.01.018 [5] ZHANG J J, LIU X J, ZHU Y T, et al. Antibiotic exposure across three generations from Chinese families and cumulative health risk[J]. Ecotoxicology and Environmental Safety, 2020, 191: 110237. doi: 10.1016/j.ecoenv.2020.110237 [6] QIU S Y, WANG Y, WAN J Q, et al. Enhanced electro-Fenton catalytic performance with in-situ grown Ce/Fe@NPC-GF as self-standing cathode: Fabrication, influence factors and mechanism[J]. Chemosphere, 2021, 273: 130269. doi: 10.1016/j.chemosphere.2021.130269 [7] DU X, FU W, SU P, et al. Trace FeCu@PC derived from MOFs for ultraefficient heterogeneous electro-Fenton process: Enhanced electron transfer and bimetallic synergy[J]. ACS ES& T Engineering, 2021, 1(9): 1311-1322. [8] CHENG S, ZHENG H, SHEN C, et al. Hierarchical iron phosphides composite confined in ultrathin carbon layer as effective heterogeneous electro-Fenton catalyst with prominent stability and catalytic activity[J]. Advanced Functional Materials, 2021, 31(48): 2106311. doi: 10.1002/adfm.202106311 [9] 王奇, 潘家荣, 梅朋森, 等. 电Fenton及光电Fenton法废水处理技术研究进展[J]. 三峡大学学报(自然科学版), 2008(2): 89-94. [10] LIU X C, HE C S, SHEN Z Y, et al. Mechanistic study of Fe(III) chelate reduction in a neutral electro-Fenton process[J]. Applied Catalysis B:Environmental, 2020, 278: 119347. doi: 10.1016/j.apcatb.2020.119347 [11] WANG Y Z, ZHANG H M, LI B K, et al. γ-FeOOH graphene polyacrylamide carbonized aerogel as air-cathode in electro-Fenton process for enhanced degradation of sulfamethoxazole[J]. Chemical Engineering Journal, 2019, 359: 914-923. doi: 10.1016/j.cej.2018.11.096 [12] GANIYU S O, ZHOU M H, MARTÍNEZ-HUITLE C A. Heterogeneous electro-Fenton and photoelectro-Fenton processes: A critical review of fundamental principles and application for water/wastewater treatment[J]. Applied Catalysis B:Environmental, 2018, 235: 103-129. doi: 10.1016/j.apcatb.2018.04.044 [13] ZHANG J J, QIU S, FENG H P, et al. Efficient degradation of tetracycline using core–shell Fe@Fe2O3-CeO2 composite as novel heterogeneous electro-Fenton catalyst[J]. Chemical Engineering Journal, 2022, 428: 131403. doi: 10.1016/j.cej.2021.131403 [14] ZHAO K, QUAN X, CHEN S, et al. Enhanced electro-Fenton performance by fluorine-doped porous carbon for removal of organic pollutants in wastewater[J]. Chemical Engineering Journal, 2018, 354: 606-615. doi: 10.1016/j.cej.2018.08.051 [15] YE Z H, PADILLA J A, XURIGUERA E, et al. A highly stable metal–organic framework-engineered FeS2/C nanocatalyst for heterogeneous electro-Fenton treatment: Validation in wastewater at mild pH[J]. Environmental Science & Technology, 2020, 54(7): 4664-4674. [16] YANG T Y, YU D Y, WANG D, et al. Accelerating Fe(Ⅲ)/Fe(Ⅱ) cycle via Fe(Ⅱ) substitution for enhancing Fenton-like performance of Fe-MOFs[J]. Applied Catalysis B:Environmental, 2021, 286: 119859. doi: 10.1016/j.apcatb.2020.119859 [17] YIN Y, REN Y, LU J H, et al. The nature and catalytic reactivity of UiO-66 supported Fe3O4 nanoparticles provide new insights into Fe-Zr dual active centers in Fenton-like reactions[J]. Applied Catalysis B:Environmental, 2021, 286: 119943. doi: 10.1016/j.apcatb.2021.119943 [18] YUAN R R, QIU J L, YUE C L, et al. Self-assembled hierarchical and bifunctional MIL-88A(Fe)@ZnIn2S4 heterostructure as a reusable sunlight-driven photocatalyst for highly efficient water purification[J]. Chemical Engineering Journal, 2020, 401: 126020. doi: 10.1016/j.cej.2020.126020 [19] ZHOU L, LI N, OWENS G, et al. Simultaneous removal of mixed contaminants, copper and norfloxacin, from aqueous solution by ZIF-8[J]. Chemical Engineering Journal, 2019, 362: 628-637. doi: 10.1016/j.cej.2019.01.068 [20] LI H X, ZHANG J, YAO Y Z, et al. Nanoporous bimetallic metal-organic framework (FeCo-BDC) as a novel catalyst for efficient removal of organic contaminants[J]. Environmental Pollution, 2019, 255: 113337. doi: 10.1016/j.envpol.2019.113337 [21] LI H X, YANG Z X, LU S, et al. Nano-porous bimetallic CuCo-MOF-74 with coordinatively unsaturated metal sites for peroxymonosulfate activation to eliminate organic pollutants: Performance and mechanism[J]. Chemosphere, 2021, 273: 129643. doi: 10.1016/j.chemosphere.2021.129643 [22] DU J, LI F, SUN L C. Metal–organic frameworks and their derivatives as electrocatalysts for the oxygen evolution reaction[J]. Chemical Society Reviews, 2021, 50(4): 2663-2695. doi: 10.1039/D0CS01191F [23] SUN L, CAMPBELL M G, DINCĂ M. Electrically conductive porous metal–organic frameworks[J]. Angewandte Chemie International Edition, 2016, 55(11): 3566-3579. doi: 10.1002/anie.201506219 [24] CHENG S, SHEN C, ZHENG H, et al. OCNTs encapsulating Fe-Co PBA as efficient chainmail-like electrocatalyst for enhanced heterogeneous electro-Fenton reaction[J]. Applied Catalysis B:Environmental, 2020, 269: 118785. doi: 10.1016/j.apcatb.2020.118785 [25] MENG J S, NIU C J, XU L H, et al. General oriented formation of carbon nanotubes from metal–organic frameworks[J]. Journal of the American Chemical Society, 2017, 139(24): 8212-8221. doi: 10.1021/jacs.7b01942 [26] 陆平. 草酸钛钾分光光度法测定Fenton高级氧化系统中的过氧化氢[J]. 建筑工程技术与设计, 2014(8): 582-582,517. doi: 10.3969/j.issn.2095-6630.2014.08.553 [27] LI Y S, FENG Y, LI L, et al. PBA@PPy derived N-doped mesoporous carbon nanocages embedded with FeCo alloy nanoparticles for enhanced performance of oxygen reduction reaction[J]. Journal of Alloys and Compounds, 2020, 823: 153892. doi: 10.1016/j.jallcom.2020.153892 [28] AGO H, KUGLER T, CACIALLI F, et al. Work functions and surface functional groups of multiwall carbon nanotubes[J]. The Journal of Physical Chemistry B, 1999, 103(38): 8116-8121. doi: 10.1021/jp991659y [29] LIU Q T, LIU X F, ZHENG L R, et al. The solid-phase synthesis of an Fe-N-C electrocatalyst for high-power proton-exchange membrane fuel cells[J]. Angewandte Chemie International Edition, 2018, 57(5): 1204-1208. doi: 10.1002/anie.201709597 [30] SU P, ZHOU M H, LU X Y, et al. Electrochemical catalytic mechanism of N-doped graphene for enhanced H2O2 yield and in-situ degradation of organic pollutant[J]. Applied Catalysis B:Environmental, 2019, 245: 583-595. doi: 10.1016/j.apcatb.2018.12.075 [31] HAIDER M R, JIANG W L, HAN J L, et al. In-situ electrode fabrication from polyaniline derived N-doped carbon nanofibers for metal-free electro-Fenton degradation of organic contaminants[J]. Applied Catalysis B:Environmental, 2019, 256: 117774. doi: 10.1016/j.apcatb.2019.117774 [32] LIU X, WANG L, YU P, et al. A stable bifunctional catalyst for rechargeable zinc-air batteries: Iron-cobalt nanoparticles embedded in a nitrogen-doped 3D carbon matrix[J]. Angewandte Chemie International Edition, 2018, 57(49): 16166-16170. doi: 10.1002/anie.201809009 [33] LIANG H W, WEI W, WU Z S, et al. Mesoporous metal-nitrogen-doped carbon electrocatalysts for highly efficient oxygen reduction reaction[J]. Journal of the American Chemical Society, 2013, 135(43): 16002-5. doi: 10.1021/ja407552k [34] FAN L S, WU H X, WU X, et al. Fe-MOF derived jujube pit like Fe3O4/C composite as sulfur host for lithium-sulfur battery[J]. Electrochimica Acta, 2019, 295: 444-451. doi: 10.1016/j.electacta.2018.08.107 [35] SU P, ZHOU M H, REN G B, et al. A carbon nanotube-confined iron modified cathode with prominent stability and activity for heterogeneous electro-Fenton reactions[J]. Journal of Materials Chemistry A, 2019, 7(42): 24408-24419. doi: 10.1039/C9TA07491K [36] QIN Y X, ZHANG L Z, AN T C. Hydrothermal carbon-mediated Fenton-like reaction mechanism in the degradation of alachlor: Direct electron transfer from hydrothermal carbon to Fe(III)[J]. ACS Applied Materials & Interfaces, 2017, 9(20): 17115-17124. [37] YOO S H, JANG D, JOH H-I, et al. Iron oxide/porous carbon as a heterogeneous Fenton catalyst for fast decomposition of hydrogen peroxide and efficient removal of methylene blue[J]. Journal of Materials Chemistry A, 2017, 5(2): 748-755. doi: 10.1039/C6TA07457J [38] RAO X F, SHAO X L, XU J, et al. Efficient nitrate removal from water using selected cathodes and Ti/PbO2 anode: Experimental study and mechanism verification[J]. Separation and Purification Technology, 2019, 216: 158-165. doi: 10.1016/j.seppur.2019.02.009 [39] WANG J L, WANG S Z. Reactive species in advanced oxidation processes: Formation, identification and reaction mechanism[J]. Chemical Engineering Journal, 2020, 401: 126158. doi: 10.1016/j.cej.2020.126158 [40] LIU Z, DING H J, ZHAO C, et al. Electrochemical activation of peroxymonosulfate with ACF cathode: Kinetics, influencing factors, mechanism, and application potential[J]. Water Research, 2019, 159: 111-121. doi: 10.1016/j.watres.2019.04.052 [41] CAO P K, QUAN X, ZHAO K, et al. Selective electrochemical H2O2 generation and activation on a bifunctional catalyst for heterogeneous electro-Fenton catalysis[J]. Journal of Hazardous Materials, 2020, 382: 121102. doi: 10.1016/j.jhazmat.2019.121102 [42] YANG Z C, QIAN J S, YU A Q, et al. Singlet oxygen mediated iron-based Fenton-like catalysis under nanoconfinement[J]. Proceedings of the National Academy of Sciences, 2019, 116(14): 6659-6664. doi: 10.1073/pnas.1819382116 [43] DENG J, YU L, DENG D H, et al. Highly active reduction of oxygen on a FeCo alloy catalyst encapsulated in pod-like carbon nanotubes with fewer walls[J]. Journal of Materials Chemistry A, 2013, 1(47): 14868-14873. doi: 10.1039/c3ta13759g [44] XIAO F, WANG Z N, FAN J Q, et al. Selective electrocatalytic reduction of oxygen to hydroxyl radicals via 3-electron pathway with FeCo alloy encapsulated carbon aerogel for fast and complete removing pollutants[J]. Angewandte Chemie International Edition, 2021, 60(18): 10375-10383. doi: 10.1002/anie.202101804 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4354
- HTML全文浏览数: 4354
- PDF下载数: 92
- 施引文献: 0
