


 下载:
下载:


-
抗生素在生产和使用过程中会产生大量含抗生素废水[1],制药废水是抗生素的最大来源,通常含抗生素浓度高、盐分高、毒性大,其处理是水处理领域中的一项难题[2]。磺胺甲恶唑(Sulfamethoxazole, SMZ)是一类典型的磺胺类抗生素,其在水体中相对稳定,不易被降解[3]。根据一项针对中国七大典型河流水域抗生素赋存的研究,SMZ的检出浓度最高[4],而且有研究表明人的尿液中可检出高达10 mg·L−1的SMZ[5]。SMZ对动植物以及人体健康均会造成危害,因此,研发利用高效的处理技术迫在眉睫。
许多研究表明,高级氧化技术对抗生素废水具有较好去除效果。其中电芬顿(electro-Fenton, EF)技术仅消耗O2和电能,绿色清洁、倍受关注[6-8]。EF技术可通过两电子氧还原反应(2e− ORR)原位生成H2O2,随后H2O2进一步被活化生成活性氧物种(reactive oxygen species, ROS),其可进一步高效去除水中抗生素[9-11]。基于铁离子催化的均相EF技术需在酸性条件下才能有效运行,反应前后需要调节pH,为拓宽EF技术的pH适用范围,开发了基于固相催化剂的非均相EF技术[12-14]。然而受阴极催化剂过渡金属氧化还原电对循环速率慢、稳定性差等限制[15],非均相EF技术对抗生素的降解效率亟待提升,开发高效稳定的阴极催化剂是目前非均相EF技术的主要应用瓶颈。
近年来,金属有机框架材料(metal organic framework, MOFs)是催化领域的研究热点,其不仅具有发达的孔隙结构,且拓扑结构能保障金属位点的均匀分散[16-19]。为了进一步促进金属氧化还原电对在催化反应中的循环速率,构建更多活性位点,研究通过组合不同种类的金属开发出系列双金属MOFs,显著提高了MOFs的催化活性[20-21]。然而MOFs作为EF阴极催化剂时导电性能欠佳[22-23],且其有机配体容易被EF反应中生成的ROS氧化,造成催化剂多孔结构坍塌及失活[24],因此,同步提高电学性能和化学稳定性是关键需求。
本研究以双金属MOFs—FeCo-ZIF为前体,以三聚氰胺(melamine, MA)为碳源和氮源,将两者共混煅烧制备了氮掺杂碳纳米管封装铁钴合金阴极催化剂(N-CNT@FeCo),考察了MA对FeCo-ZIF衍生催化剂EF性能的影响规律,探究了体系中的主要活性物种和催化机理,考察了溶液初始pH、应用电位、共存阴离子及重复循环次数对SMZ降解效果的影响,以评价N-CNT@FeCo作为EF阴极催化剂的基础应用性能。
-
实验材料:六水合硝酸钴(Co(NO3)2·6H2O)、无水硫酸钠(Na2SO4)、无水乙醇(C2H6O)均为分析纯并购于南京化学试剂股份有限公司;七水合硫酸亚铁(FeSO4·7H2O)、氢氧化钠(NaOH)、浓硫酸(H2SO4)、浓硝酸(HNO3)均为分析纯并购于国药集团化学试剂有限公司;三聚氰胺(C3H6N6)为分析纯并购于上海麦克林生化科技股份有限公司;磺胺甲恶唑(C10H11N3O3S)、N,N-二甲基甲酰胺(C3H7NO, N,N-Dimethylformamide, DMF)、2-甲基咪唑(C4H6N2)、甲醇(CH3OH, Methanol, MeOH)、叔丁醇(C4H10O, Tert-Butanol, TBA)、糠醇(C5H6O2, Furfuryl alcohol, FFA)、对苯醌(C6H4O2, 1,4-Benzoquinone, p-BQ)、磷酸二氢钾(KH2PO4)、草酸钛钾(K2TiO(C2O4)2)均为分析纯并购于上海阿拉丁生化科技股份有限公司;5,5-二甲基-1-吡咯啉-N-氧化物(C6H11NO, 5,5-Dimethyl-1-pyrroline N-oxide, DMPO)购于和光纯药工业株式会社;4-氨基-2,2,6,6-四甲基哌啶(C9H20N2, 4-Amino-2,2,6,6-tetramethylpiperidine, TEMP)购于希恩思生化科技有限公司;碳布(W1S1009)购于台湾碳能科技有限公司;Nafion溶液(5 wt%)购于美国杜邦科技有限公司。
实验仪器:管式炉(TL1200,南京博蕴通仪器科技有限公司);电化学工作站(CHI760E,上海辰华仪器有限公司);高效液相色谱仪(LC-2040C,日本岛津公司,high performance liquid chromatography,HPLC);电感耦合等离子光谱发生仪(iCAP 8000,美国赛默飞世尔科技公司,inductive coupled plasma emission spectrometer,ICP);总有机碳分析仪(Multi N/C 3100,德国耶拿分析仪器公司,total organic carbon,TOC)扫描电子显微镜(S4800,日本日立公司,scanning electron microscope,SEM);透射电子显微镜(Tecnai G2 F30,美国FEI公司,transmission electron microscope,TEM);X射线衍射仪(D8 ADVANCE,德国布鲁克公司,X-ray diffractometer,XRD);X射线光电子能谱仪(K-Alpha,美国赛默飞世尔科技公司,X-ray photoelectron spectroscopy,XPS)。
-
1) N-CNT@FeCo的制备:通过典型水热法制备FeCo-ZIF[25],以FeCo-ZIF为前驱体,将FeCo-ZIF和MA按照质量比为1:M混合均匀后置于坩埚中并加盖放入管式炉内,使用氩气作为保护气体(300 mL·min−1),以5 ℃·min−1升温至800 ℃,煅烧2 h,待冷却至室温得到煅烧后的产物,在此条件下制备的催化剂命名为N-CNT@FeCo-M,其中N-CNT@FeCo-100简写为N-CNT@FeCo。此外,在不添加MA的条件下制备的催化剂命名为FeCo-N。
2)修饰阴极的制备:称取6.0 mg催化剂加入到100 μL去离子水、300 μL无水乙醇和5 μL Nafion溶液的混合液中,所配溶液超声30 min使催化剂分散均匀,然后将其滴涂到直径约为4 cm的碳布上,在室温下自然干燥得到负载催化剂的碳布修饰阴极。
-
采用XRD对催化剂的晶体结构进行表征。采用SEM和TEM对催化剂的微观形貌进行分析。采用XPS对催化剂进行元素价态分析。利用循环伏安法(cyclic voltammetry, CV)、电化学阻抗测试(electrochemical impedance spectroscopy, EIS)和旋转盘电极测试(rotating disk electrode, RDE)研究材料的电化学性能。
-
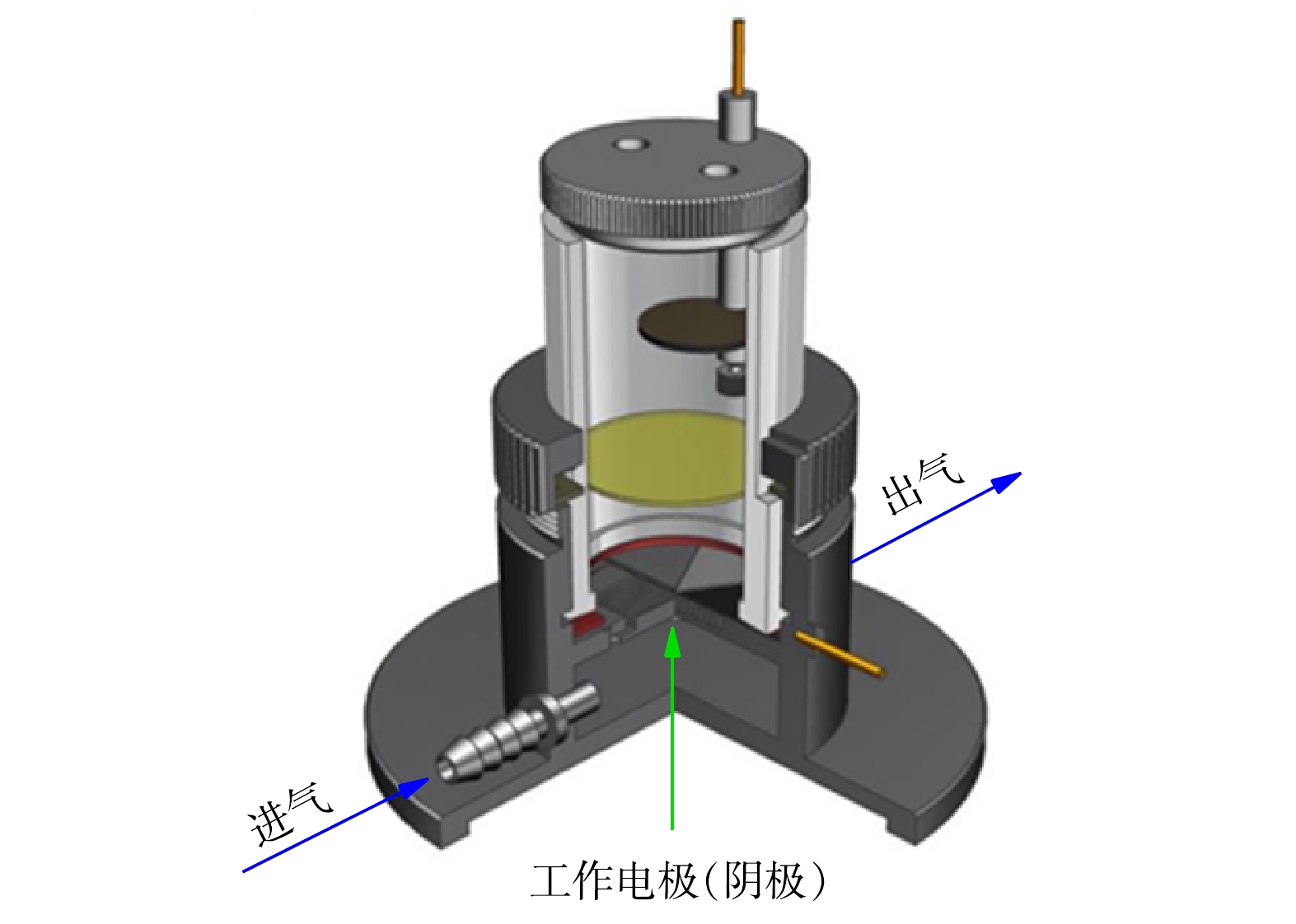
非均相EF降解实验:非均相EF反应在气体扩散反应器中(图1)进行,以均匀负载催化剂的碳布为工作电极,2.0 cm2的铂片和饱和甘汞电极分别为对电极和参比电极,反应通过CHI760E电化学工作站控制,氧气从进气口进入,为保证催化剂不易从碳布上被气流吹落,在装置另一侧设计了出气口。除非另作说明,降解实验均在−0.5 V的恒电位模式下进行。配置含20 mg·L−1 SMZ和50 mmol·L−1的Na2SO4的溶液为模拟制药废水,溶液pH使用NaOH和H2SO4调节,若未作说明,实验在初始pH下进行(pH = 5.2)。降解实验开始前,工作电极浸没在电解液中30 min达到吸附平衡。
SMZ浓度测定:SMZ浓度通过HPLC进行测定,样品通过C18色谱柱进行分离,在柱温30 ℃的条件下,用二极管阵列检测器进行检测,流动相为磷酸二氢钾/乙腈(60:40)混合液,流速为1 mL·min−1,检测波长为270 nm。金属浸出量测定:反应60 min取适量水样经聚四氟乙烯滤膜过滤后,使用ICP测定溶液中铁离子和钴离子浓度。H2O2浓度测定:将样品加入到去离子水、H2SO4与K2TiO(C2O4)2混合溶液中,摇匀后静置10 min显色,在波长386 nm处测定其吸光度[26]。
通过准一级动力学方程对非均相EF降解SMZ的过程进行拟合,SMZ降解速率常数根据式(1)计算。
式中:C0为SMZ初始质量浓度,mg·L−1;t为反应时间,min;C为反应t时的SMZ质量浓度,mg·L−1;k为降解速率常数,min−1。
-
如图2(a)所示,FeCo-ZIF的形貌为表面粗糙的球状微粒,粒径约为300~500 nm,而添加MA后FeCo-ZIF的煅烧产物形貌产生显著变化。如图2(b)所示,FeCo-ZIF的球状结构转变为N-CNT@FeCo相互缠绕的碳纳米管(carbon nanotubes, CNT)结构。

由图3(a)可以看出,N-CNT@FeCo由CNT封装深色的纳米颗粒组成,CNT的管径约为10~120 nm。在图3(b)中能明显观察到对应Fe0.3Co0.7合金中(110)晶面的晶格间距(0.201 nm)[27],结合XRD谱图(图3(c))中45.1°和65.5°处的Fe0.3Co0.7合金特征峰(PDF#50-0795)以及44.0°、51.2°和76.8°处的Fe0.28Co0.72合金特征峰(PDF#51-0740),证实深色纳米颗粒为铁钴合金。此外,在图3(b)观察到0.340 nm的晶格间距对应石墨碳的(002)晶面,XRD谱图中26.5°处也出现了明显的石墨碳特征峰(PDF#41-1487)。这种高结晶度的石墨碳具有优异的导电性能,有利于内层合金纳米颗粒在EF体系中ORR与类芬顿反应的电荷传输。

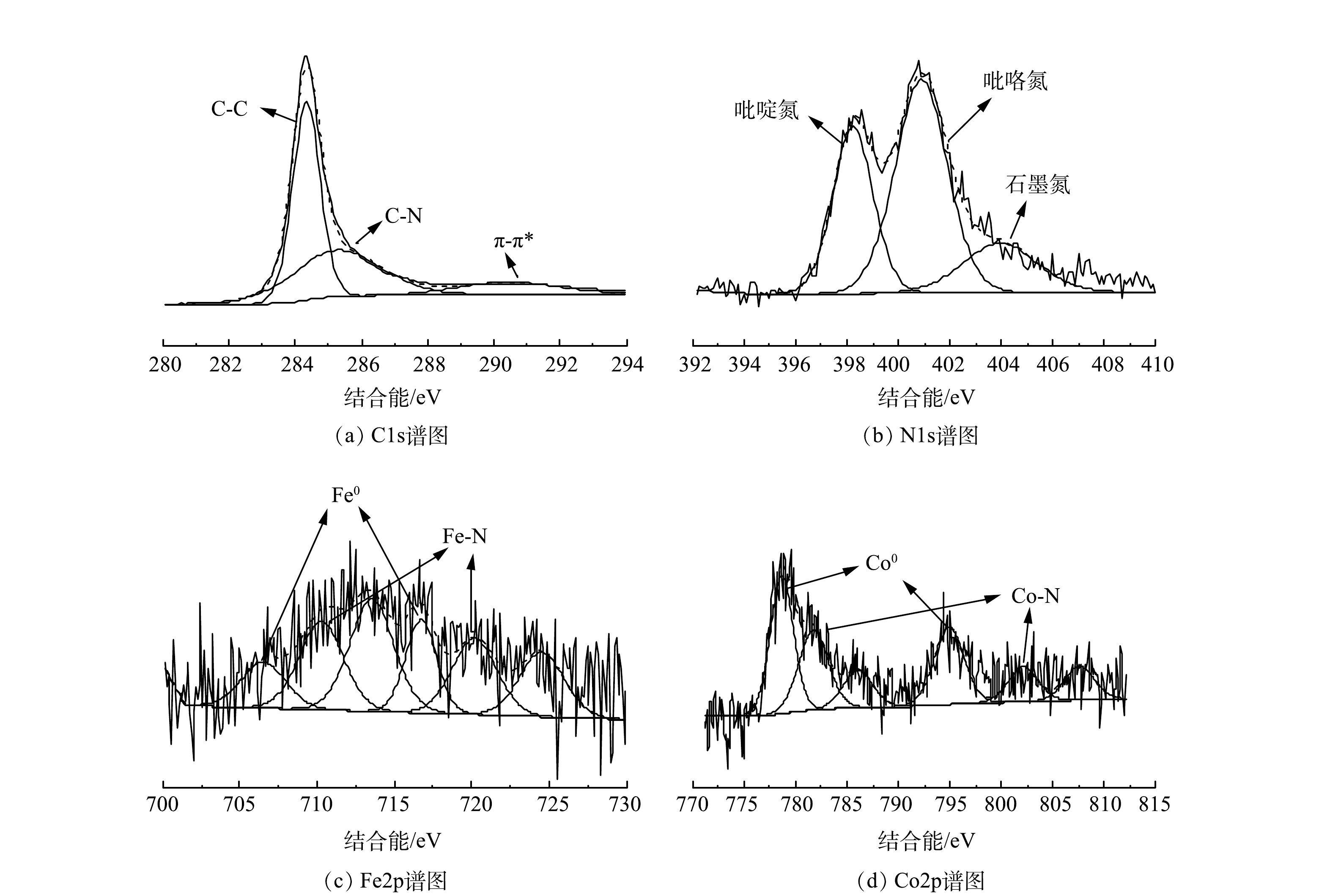
利用XPS分析了材料表面各元素组成及价态。由图4(a)的C1s精细谱中可以看出,位于284.3、285.3和290.4 eV处的3个峰,分别对应石墨碳、C-N和π-π*。这说明碳材料的石墨化程度较高,与TEM以及XRD分析结果一致[28]。在图4(b) 的N1s精细谱中位于398.2、400.9和403.9 eV处的特征峰分别对应吡啶氮、吡咯氮和石墨氮[29]。证明氮在CNT上的成功掺杂,这有利于提高催化剂对2e− ORR的选择性[30-31]。图4(c)和图4(d)中出现的Fe0特征峰(706.5 eV和716.7 eV处)及Co0特征峰(778.7 eV和794.9 eV)则进一步证明了铁钴合金的存在[32]。Co2p精细谱在781.8 eV和802.8 eV处还出现了对应Co-N的特征峰[33],表明铁钴合金可能通过金属-氮(M-N)结合键与外层N-CNT相连。
-
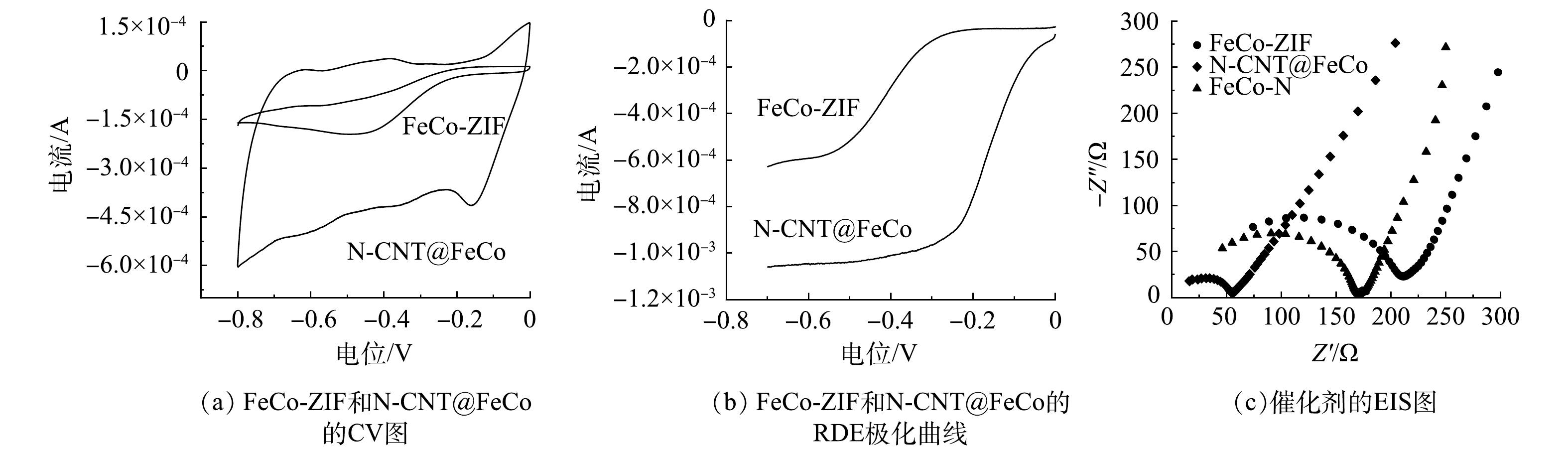
如图5(a)和图5(b)所示,N-CNT@FeCo对ORR的响应电流较FeCo-ZIF明显提升,ORR起始电位更正。表明N-CNT@FeCo的ORR活性高于FeCo-ZIF。如图5(c)所示,催化剂的EIS图均由一段高频区的圆弧和一条低频区的斜线组成。高频区中圆弧的直径代表电荷转移阻抗,直径越小则电荷传递速率越高,低频区斜线的斜率代表电解质和电极间的物质扩散[34-35]。煅烧后的N-CNT@FeCo圆弧直径明显小于FeCo-ZIF,表现出更低的电荷转移阻力(60.7 Ω<187.9 Ω),这也导致了更强的电化学活性,与CV测试结果一致,以上结果表明煅烧处理提高了催化剂的电子传递速率。
-
如图6(a)和图6(b)所示,FeCo-ZIF直接应用于EF降解SMZ,虽然60 min对SMZ的去除率可达91.7%,但铁离子和钴离子的浸出量高达1.22 mg·L−1和8.65 mg·L−1,均相EF的作用不容忽视。由FeCo-ZIF直接煅烧得到的FeCo-N在EF反应中催化氧化SMZ的活性及稳定性低,在60 min对SMZ的去除率仅为73.7%,且钴离子的浸出量仍有1.65 mg·L−1。图2(c)中看到,FeCo-N微球表面密集覆盖了大量金属团簇,在EF反应中直接暴露在强氧化环境下,导致材料稳定性较差。而FeCo-ZIF在添加不同比例的MA煅烧后得到的N-CNT@FeCo-M,反应60 min时对SMZ的去除率提升至88.2%~100%,钴的浸出量下降至0.05~0.50 mg·L−1,性能最佳的N-CNT@FeCo-100的k值高达0.057 min−1,约为FeCo-N的3倍(图6(c)),60 min可实现SMZ的完全降解,TOC去除率为45.3%。
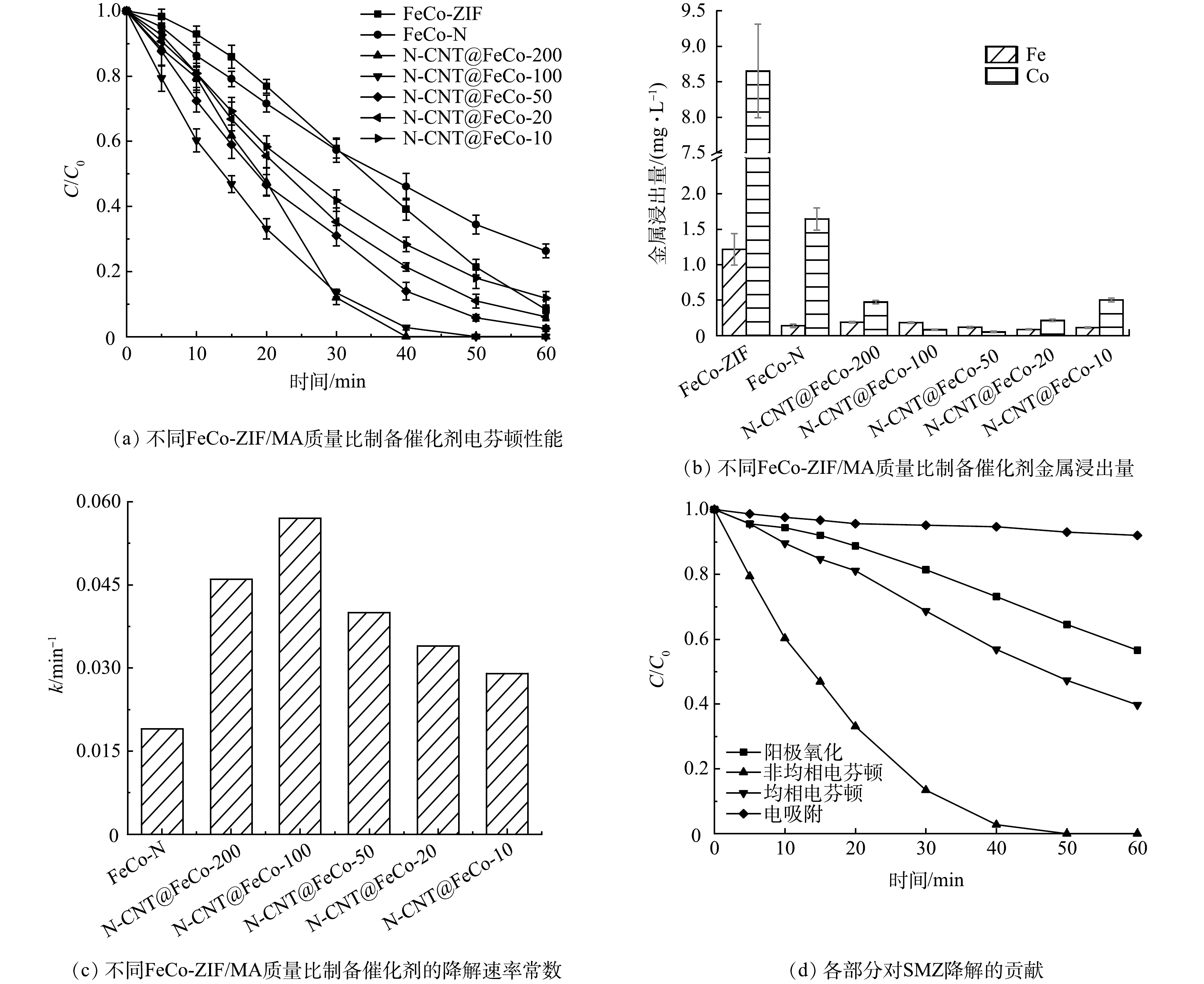
EIS测试结果(图5(c))表明,N-CNT@FeCo-100(60.7 Ω)的电荷转移阻力远小于FeCo-N(145.9 Ω)。这可能得益于添加MA后衍生出的N-CNT能加速电荷传输[36-37]。结合N-CNT@FeCo的表征分析,MA作为碳源和氮源衍生出的N-CNT能通过促进内部铁钴合金之间的电子转移,提高EF催化活性,同时N-CNT作为金属纳米颗粒的保护层,还能有效提高内层合金在强氧化环境下的化学稳定性。综上所述,优选FeCo-ZIF与MA质量比为1:100进行后续研究。
SMZ的降解通常是电吸附、阳极氧化、均相EF以及非均相EF过程共同作用的结果,因此评估了反应40 min时各部分的贡献率,结果如图6(d)所示。电吸附、阳极氧化、均相EF以及非均相EF过程对SMZ降解的贡献率依次为5.5%、22.1%、16.7%和55.7%,说明非均相EF对SMZ的降解起主要作用。
-
1) pH的影响。不同初始pH下SMZ的降解效率变化如图7(a)所示。EF性能随着初始pH的增大先升高后下降,无需调节pH时(pH=5.2)降解性能最佳,40 min对SMZ的去除率即可达到97.3%。由图7(b)可知,即使在pH=3.1时,催化剂仍保持高稳定性,铁和钴的浸出量分别仅有0.09 mg·L−1和0.17 mg·L−1。综上所述,后续实验选择在pH=5.2下进行。
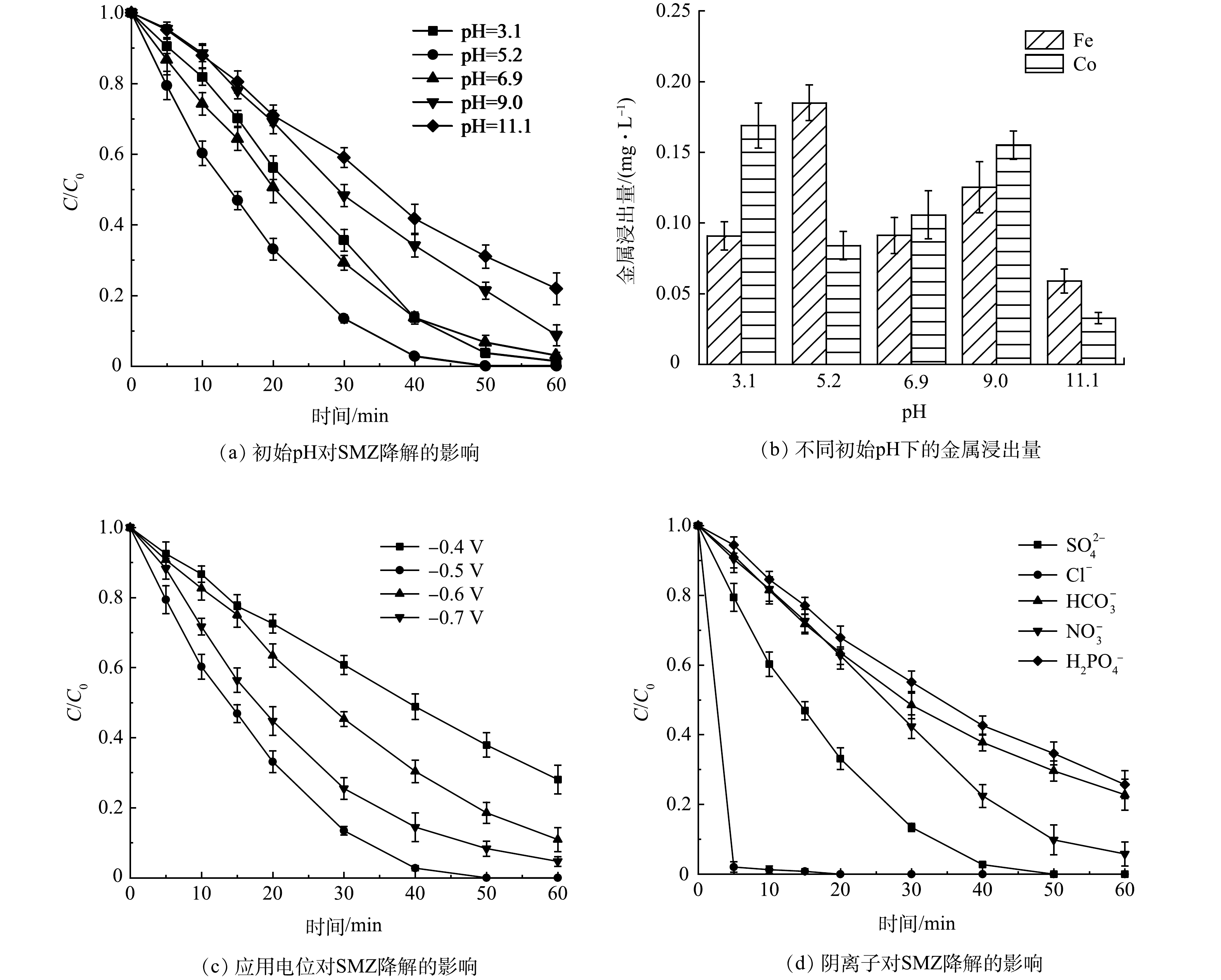
2)应用电位的影响。如图7(c)所示,当应用电位为-0.5 V时,在60 min时SMZ的去除率为100%。将应用电位调节至-0.6 V时,SMZ的去除率下降了11.0%。这是由析氢反应以及H2O2被进一步还原为H2O所致,系列副反应抑制了SMZ降解。选择应用电位为-0.5 V进行后续实验。
3)阴离子的影响。由图7(d)可见,Cl−能够促进SMZ的降解。这是因为Cl−在阳极生成强氧化性的HClO[38]。而其余3种阴离子抑制了SMZ的降解,抑制作用从强到弱依次为H2PO4−≈HCO3−>NO3−,因为3种阴离子均会淬灭·OH生成氧化性更弱的自由基[39]。
-
使用TBA(100 mmol·L−1)、FFA(5 mmol·L−1)、p-BQ(5 mmol·L−1)、MeOH(50 mmol·L−1)分别作为·OH、1O2、·O2−、·SO4−的淬灭剂[40],在此条件下TBA、FFA和MeOH对·OH的淬灭能力维持在同一水平。如图8(a)所示,TBA的加入对SMZ降解产生明显抑制,而加入FFA的抑制效果(38.7%)与TBA(33.6%)几乎一致。这说明·OH对污染物的降解起到了至关重要的作用,1O2虽然能通过链式反应生成,但其贡献较小。MeOH对SMZ降解的抑制作用弱于TBA,说明·SO4−的贡献可以忽略不计。淬灭·O2−不但会抑制H2O2的生成,同时也会抑制Fe(Ⅲ)还原为Fe(Ⅱ),降低了·OH的生成速率,进而削弱了SMZ的降解[41-42]。图8(b)为电子顺磁共振(EPR)测试结果。可以看出,基于N-CNT@FeCo的EF体系中出现了DMPO-OH和TEMP-1O2的特征峰,证实·OH和1O2的存在[24]。综上所述,污染物的降解是以·OH为主导,1O2和·O2−共同参与的过程。
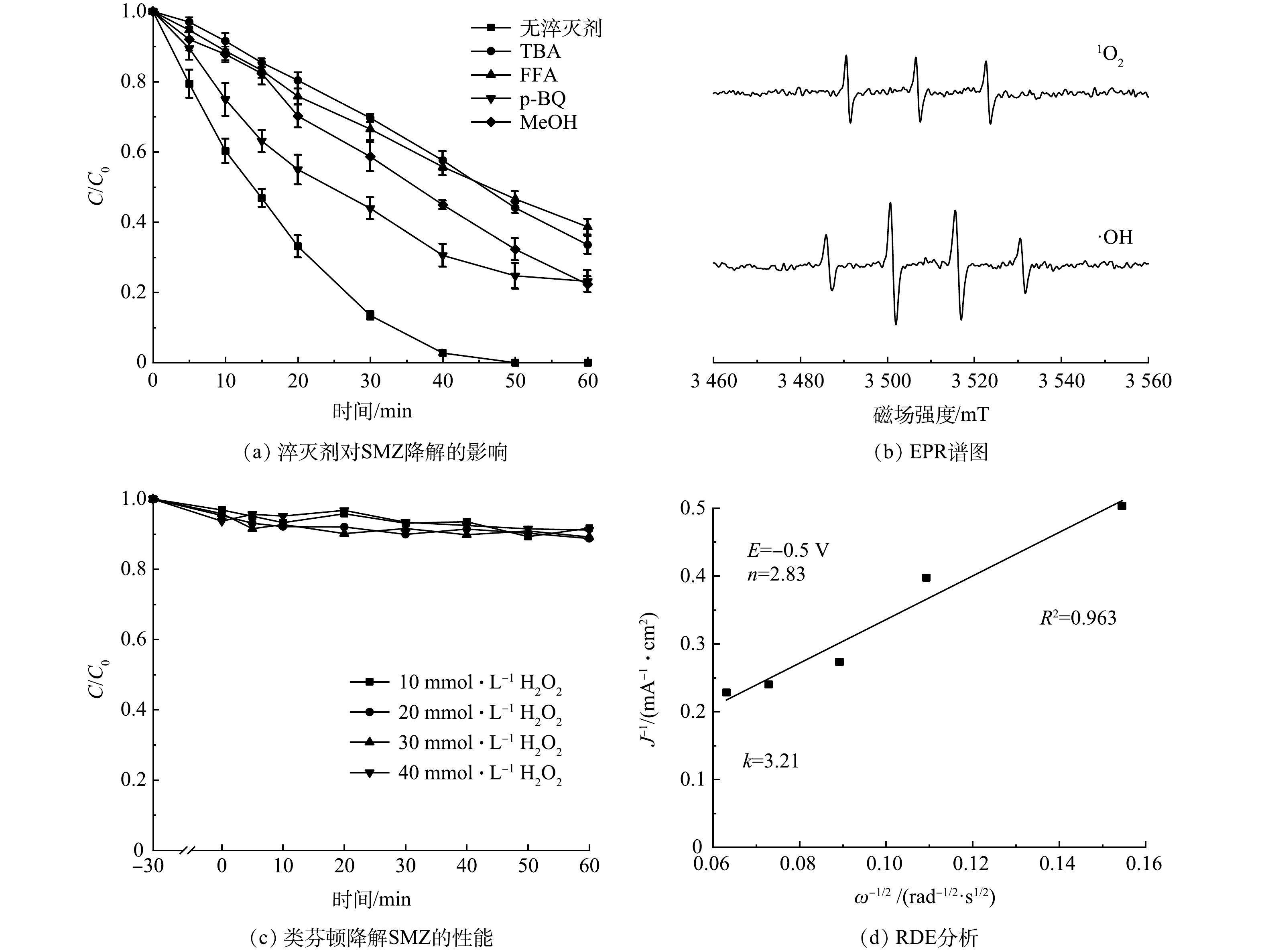
此外,还考察了N-CNT@FeCo在类芬顿体系中直接活化H2O2的能力,发现N-CNT@FeCo(图8(c))无法有效地活化H2O2生成·OH降解水中的SMZ。这是因为碳材料自身活化H2O2的能力较差。同时铁钴合金被较厚的碳层(图2(a))包裹,在碳层层数较厚(> 3层)的情况下电子难以从铁钴合金中传递到碳材料表面[43],因此N-CNT@FeCo的非均相芬顿活性差,以上结果证实了电场不可或缺的作用。
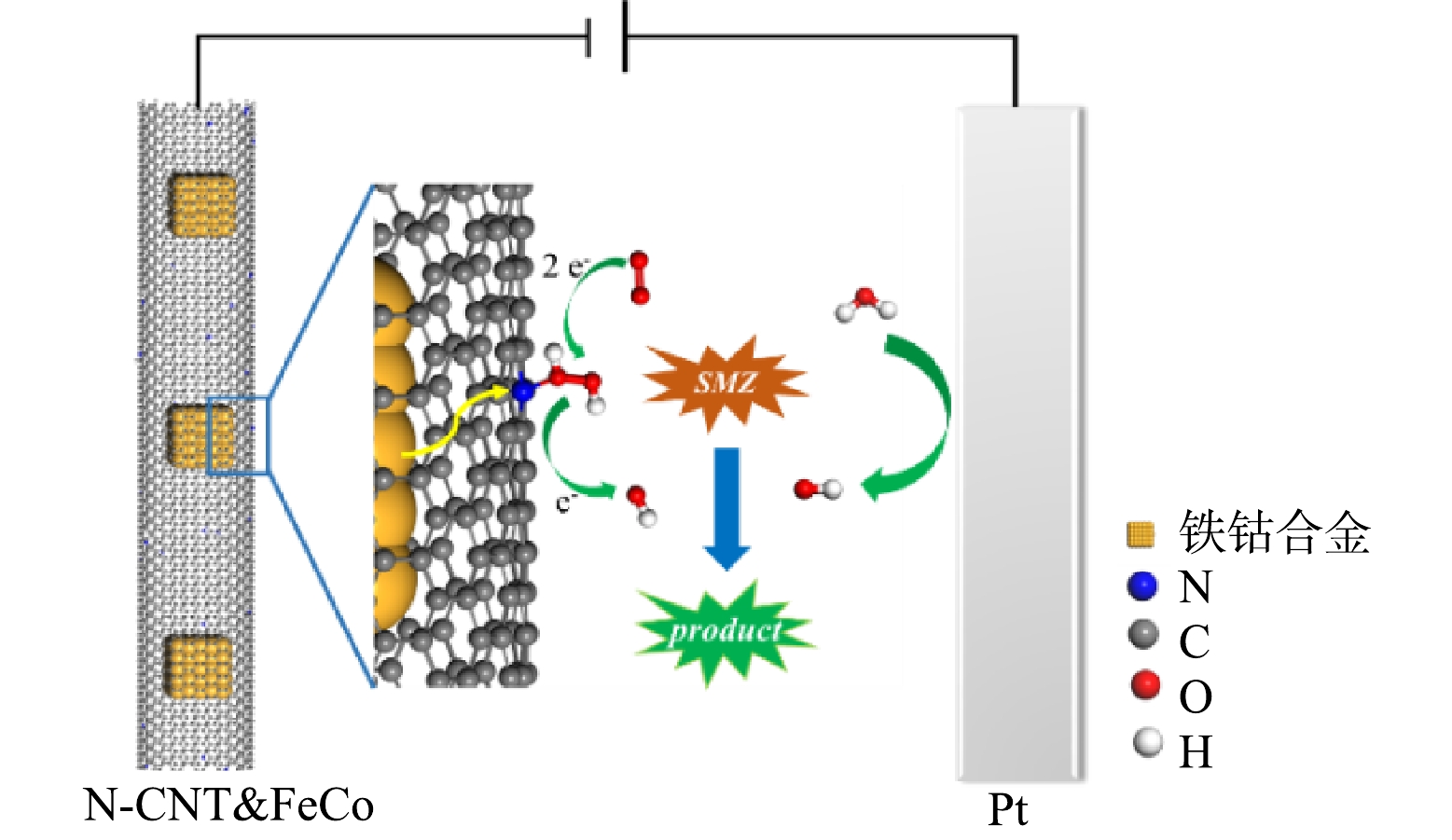
N-CNT已经被证实是一种有效的2e− ORR催化剂,其中石墨氮是生成H2O2的主要活性位点,基于N-CNT@FeCo的EF技术可能的机理如图9所示。虽然被N-CNT封装的铁钴合金不直接参与H2O2的活化,但在电场作用下其具有强给电子能力,使N-CNT表面生成的H2O2在解吸前就得到电子被还原为·OH,O2通过如式(2)所示的3电子途径直接生成·OH[44],因此,溶液中H2O2浓度维持在较低水平(< 5.0 mg·L−1)。RDE测试结果如图8(d)所示,在−0.5 V的应用电位下N-CNT@FeCo的ORR电子转移数为2.83,近似于3的电子转移数也证明O2可能通过式(2)生成·OH,这种3电子氧还原途径能有效地避免传统EF中过渡金属氧化还原电对循环速率慢的问题。
-
N-CNT@FeCo的循环回用性能如表1所示,经过5次循环后60 min时SMZ的去除率仍有96.0%,且铁、钴离子最高浸出质量浓度分别仅为0.31 mg·L−1和0.30 mg·L−1,表明N-CNT@FeCo具有良好的稳定性能。
-
1)以MA为碳源和氮源,在铁钴合金外衍生的N-CNT可同时提高催化剂的活性和稳定性。在FeCo-ZIF与MA质量比为1:100条件下制备的N-CNT@FeCo具有最佳的催化性能,相较于FeCo-N,在60 min对SMZ的去除率提升了26.3%,而金属浸出量降低了84.9%。
2)在初始pH为5.2,应用电位为-0.5 V的最佳运行条件下,N-CNT@FeCo的k值可达0.057 min−1。H2PO4−、HCO3−和NO3−会抑制SMZ的降解,而Cl−会促进污染物的降解。N-CNT@FeCo循环回用5次,60 min对SMZ的去除率仍有96.0%。
3)·OH是主要的活性物种,1O2和·O2−也参与了SMZ的降解;结合H2O2累积浓度检测、非均相芬顿实验以及RDE检测,推测基于N-CNT@FeCo的EF体系中O2是通过3电子还原生成·OH。
4)反应40 min时电吸附、阳极氧化、均相EF以及非均相EF对SMZ降解的贡献率依次为5.5%、22.1%、16.7%和55.7%。
图 1
反应器构造
Figure 1.
Illustration of reactor construction
