


 DownLoad:
DownLoad:

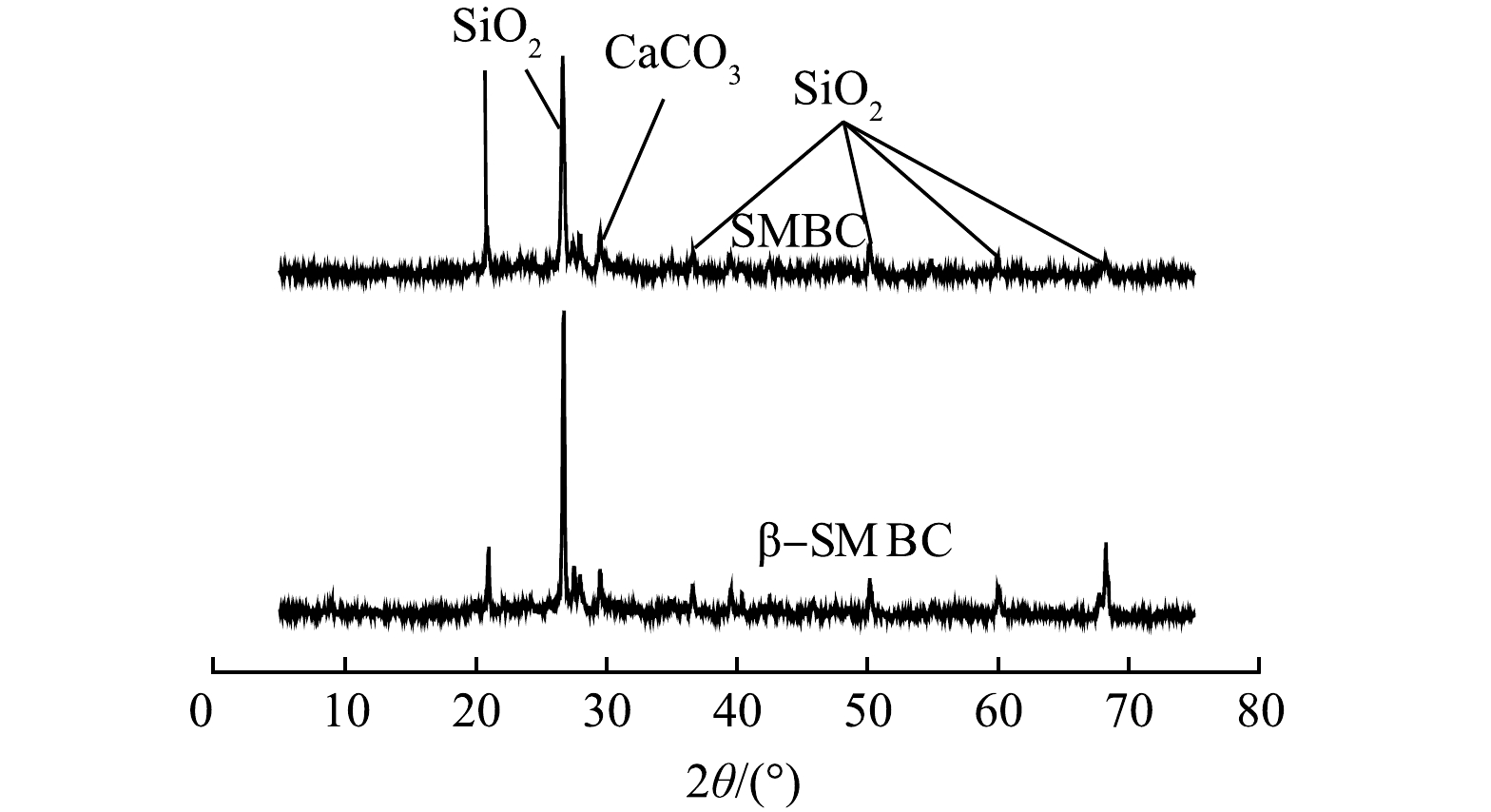
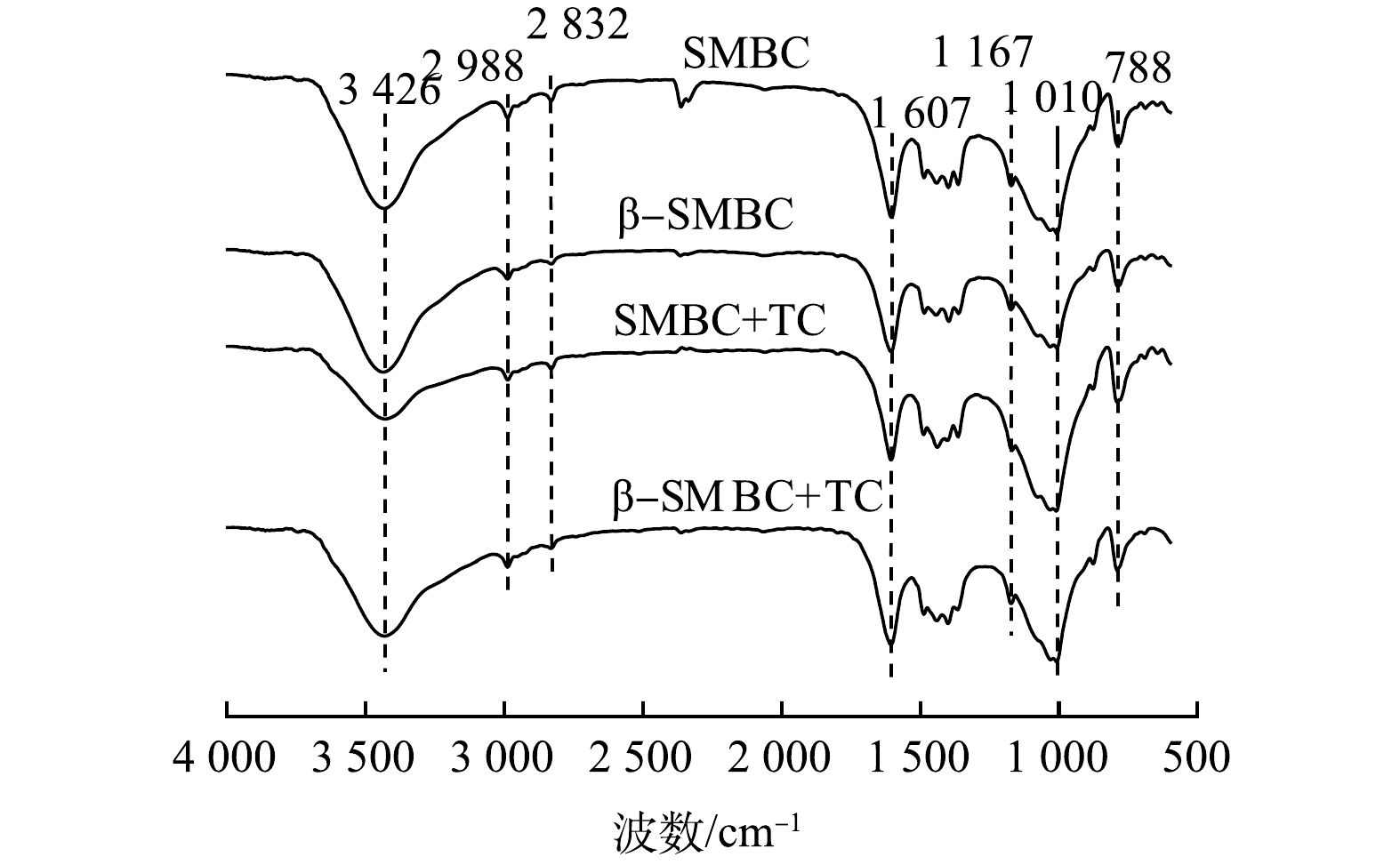


-
TESSIER等[1]于1979年提出连续提取法,以用于分析金属在土壤或固废中的结合形态。该法将土壤金属的结合态分为5种,即可交换态、碳酸盐结合态、铁锰氧化物结合态、有机物结合态、残渣态;各结合态的提取难度依次增大,其所对应的生物有效性依次降低。Tessier连续提取法作为一种操作性定义,以特定提取剂和一定的提取条件来区分元素与土壤特定组分的结合态,有其相对合理性,也存在着一定的局限性,但现阶段还未找到更好的替代方法。Tessier连续提取法最初只对8种土壤金属(Cd、Co、Cu、Ni、Pb、Zn、Fe、Mn)进行了分析,但其中并不包含铬。近年来,随着土壤铬污染问题及其修复技术研发的需要,一些研究开始将Tessier连续提取法拓展到土壤铬的结合态分析上来,有些研究仅针对总铬(TCr)[2-4],有些也包括六价铬[Cr(Ⅵ)][5-7],该方法的应用场景越来越广泛[8-9]。
土壤TCr以三价铬[Cr(Ⅲ)]和Cr(Ⅵ) 2种价态存在。其中,Cr(Ⅵ)的可迁移性和毒性远远高于Cr(Ⅲ)。因此,铬污染土壤修复通常是指清除土壤中的Cr(Ⅵ),或将其转化成低毒性的Cr(Ⅲ)[10]。Tessier连续提取法在铬结合态分析中的应用主要出于3个目的:1)了解自然环境下未污染土壤中原生铬结合态的分布;2)了解铬污染土壤中Cr(Ⅲ)、Cr(Ⅵ)和TCr结合态的分布[11];3)比较修复前、后土壤Cr(Ⅲ)、Cr(Ⅵ)或TCr结合态分布的变化,从结合态的角度评估修复技术的有效性[12-13]。然而,Tessier连续提取法在其第3步铁锰氧化物结合态的提取中使用了还原剂盐酸羟胺(NH2OH·HCl),存在将Cr(Ⅵ)还原成Cr(Ⅲ)的可能性。在第4步有机结合态的提取中使用的双氧水(H2O2)也可能导致Cr(Ⅵ)-Cr(Ⅲ)的转化。此外,对于经还原修复的铬污染土壤,土壤中残留的还原剂也可能在提取过程中将Cr(Ⅵ)还原成Cr(Ⅲ),从而影响修复效果评估的准确性。但关于以上条件对铬结合态分析的影响至今仍缺少相关研究报道。目前,Tessier连续提取法在未加评估的情况下,被直接用于铬污染土壤和含铬固废的结合态分析。
本研究针对当前Tessier连续提取法使用的3种场景,即:1)未污染土壤的原生铬结合态;2)铬污染土壤中的铬结合态;3)铬污染土壤还原修复后残留铬的结合态(还原剂为亚铁[4]和硫化钠[14])。通过液相机理研究和土壤相验证研究相结合的方式,探究提取液自身组分和残留还原剂导致的各提取步骤中Cr(Ⅵ)与Cr(Ⅲ)的转化及机理。本研究结果可为利用Tessier连续提取法准确评估铬污染土壤修复效果提供参考。
HTML
-
实验试剂。重铬酸钾(K2Cr2O7)、七水硫酸亚铁(FeSO4·7H2O)、九水硫化钠(Na2S·9H2O)、六水合氯化镁(MgCl2·6H2O)、乙酸钠(NaCOOH)、乙酸(HCOOH)、盐酸羟胺(NH2OH·HCl)、30%过氧化氢(H2O2)、乙酸铵(NH4COOH)、硝酸(HNO3)均为分析纯。
硫化物溶液。为模拟经硫化钠处理后的土壤中残留硫化物的状态,残留还原剂的影响实验中使用的硫化物溶液取自K2Cr2O7与Na2S溶液密闭反应7 d后的上清液,主要成分为硫离子、多硫化物、硫代硫酸钠等[15]。
本研究采用4种土壤样品进行土壤相实验。
土样1。未污染土壤,取自重庆大学校园内挖出的未经污染的原生黏土。采集后风干、过1 mm孔径的筛网备用。
土样2。铬污染土壤,采自重庆某铬渣堆场。该渣场铬渣已被清理,土壤样品取自该渣场底部。
土样3。经硫酸亚铁稳定化处理后的Cr(Ⅵ)污染土壤。以硫酸亚铁为还原剂,对土样2进行稳定化处理,用量为与Cr(Ⅵ)按化学计量比反应所需剂量的5倍。期间密封保存,稳定化处理时间为7 d,然后对其进行Tessier连续提取分析。
土样4。经硫化钠稳定化处理后的Cr(Ⅵ)污染土壤。以硫化钠为还原剂,用量为与Cr(Ⅵ)按化学计量比反应所需剂量的5倍。稳定化处理方法同土样3,然后对其进行Tessier连续提取分析。
-
Tessier连续提取法引起的铬结合态分析误差主要来自于2个方面,一是其提取液自身组分在第3、4步提取过程中对Cr(Ⅵ)的还原;二是土壤中残留还原剂在第1、2步提取过程中对Cr(Ⅵ)的还原。
1)第3、4步中提取液组分的影响。本实验采用的Tessier连续提取法[1]操作步骤详见表1,其在第3步和第4步分别使用了NH2OH·HCl和H2O2。在酸性条件下,NH2OH·HCl和H2O2均可能将释放到提取液中的Cr(Ⅵ)还原成Cr(Ⅲ)[16-17]。为避免土壤中铬提取不完全的误差和土壤中其他离子的干扰,本实验不加入土壤基质,以Cr(Ⅵ)标准液替代铬污染土壤,进行溶液相反应机理研究。向150 mL锥形瓶中加入0.5 mL不同浓度的Cr(Ⅵ)标准液,具体加入量见表2中“Cr(Ⅵ)初始量”,实验编号分别L3-1、L3-2和L4-1、L4-2。再分别按照Tessier连续提取法第3步、第4步进行操作,测定溶液中残留的Cr(Ⅵ)量。
2)第1、2步中残留还原剂的影响。对于修复后的铬污染土壤,残留还原剂和剩余Cr(Ⅵ)都会释放到提取液中,2者可能在第1、2步的操作过程中发生反应,导致Cr(Ⅵ)的还原。在本实验中,首先向第1步和第2步的提取液中添加Cr(Ⅵ)标准液,再加入还原剂FeSO4溶液或硫化物溶液,按表1中步骤操作完成后,检测溶液中剩余的Cr(Ⅵ)量。
3)土壤铬的Tessier连续提取实验。按照表1中步骤分析4种土壤样品中Cr(Ⅵ)和TCr结合态。每步提取完成后,使用离心机进行固液分离(4 000 r·min−1,10 min)。上清液经0.45 µm滤膜过滤后测定Cr(Ⅵ)和TCr含量,离心管中的土壤继续用于下一步的提取分析。
以上所有液相和固相土壤实验均设置3个平行。
-
硫化物溶液浓度(以S2-计)的测定采用碘量法(HJ/T 60-2000)[18]。水溶液中Cr(Ⅵ)的测定采用二苯碳酰二肼分光光度法(EPA Method 7196a)[19],水溶液中TCr的测定采用高锰酸钾氧化-二苯碳酰二肼分光光度法(GB 7466-1987)[20]。土壤Cr(Ⅵ)的测定采用碱消解(Method 3060a)[21]联合二苯碳酰二肼分光光度法(Method 7196a);土壤TCr的测定采用微波消解[22]联合高锰酸钾氧化-二苯碳酰二肼分光光度法(GB 7466-1987)。以上检测采用空白样、实验室控制样和加标样作为质控措施。
在残留还原剂的影响实验中,为保证剩余Cr(Ⅵ)浓度测定的准确性,需采取一定方法减小检测误差:为避免残留Fe2+对Cr(Ⅵ)测定的干扰[23],实验完成后要先将溶液pH调至11以上,曝气50 min,放置1 d左右;然后,滤去Fe(OH)3沉淀,测定滤液Cr(Ⅵ)浓度,在此操作下,Fe(OH)3沉淀的吸附不影响Cr(Ⅵ)检测[24]。为减小残留硫化物对Cr(Ⅵ)测定的干扰,实验完成后,对溶液中残留的Cr(Ⅵ)同时采用二苯碳酰二肼显色法(EPA Method 7196a)和UV-VIS扫描测定[25]。如果发现残留硫化物导致显色法测定结果出现显著负偏差,而UV-VIS扫描测定的结果在其检出限以上,则采用UV-VIS扫描的测定结果;如果2种方法的检测结果均在其检出限以下,则认为残留Cr(Ⅵ)含量未检出,记为“ND”。
1.1. 实验原料
1.2. 实验方法
1.3. 分析方法
-
1) NH2OH·HCl对Cr(Ⅵ)的还原。实验结果表明(表2),在Tessier提取的第3步,Cr(Ⅵ)被提取液组分中的还原剂NH2OH·HCl所还原,其还原Cr(Ⅵ)的量可高达约50.88 mg(L3-2),换算成1.0 g土样中的Cr(Ⅵ)含量为50 880 mg·kg−1。该量远超常见铬污染土壤中的Cr(Ⅵ)浓度[26-27],这意味着该步骤提取的Cr(Ⅵ)可被全部还原成Cr(Ⅲ),导致误判。在真实铬污染土壤提取中,NH2OH·HCl不仅会还原Cr(Ⅵ),还会还原土壤中铁锰所氧化物。因此,其实际用于还原Cr(Ⅵ)的量随土壤组分而变化。
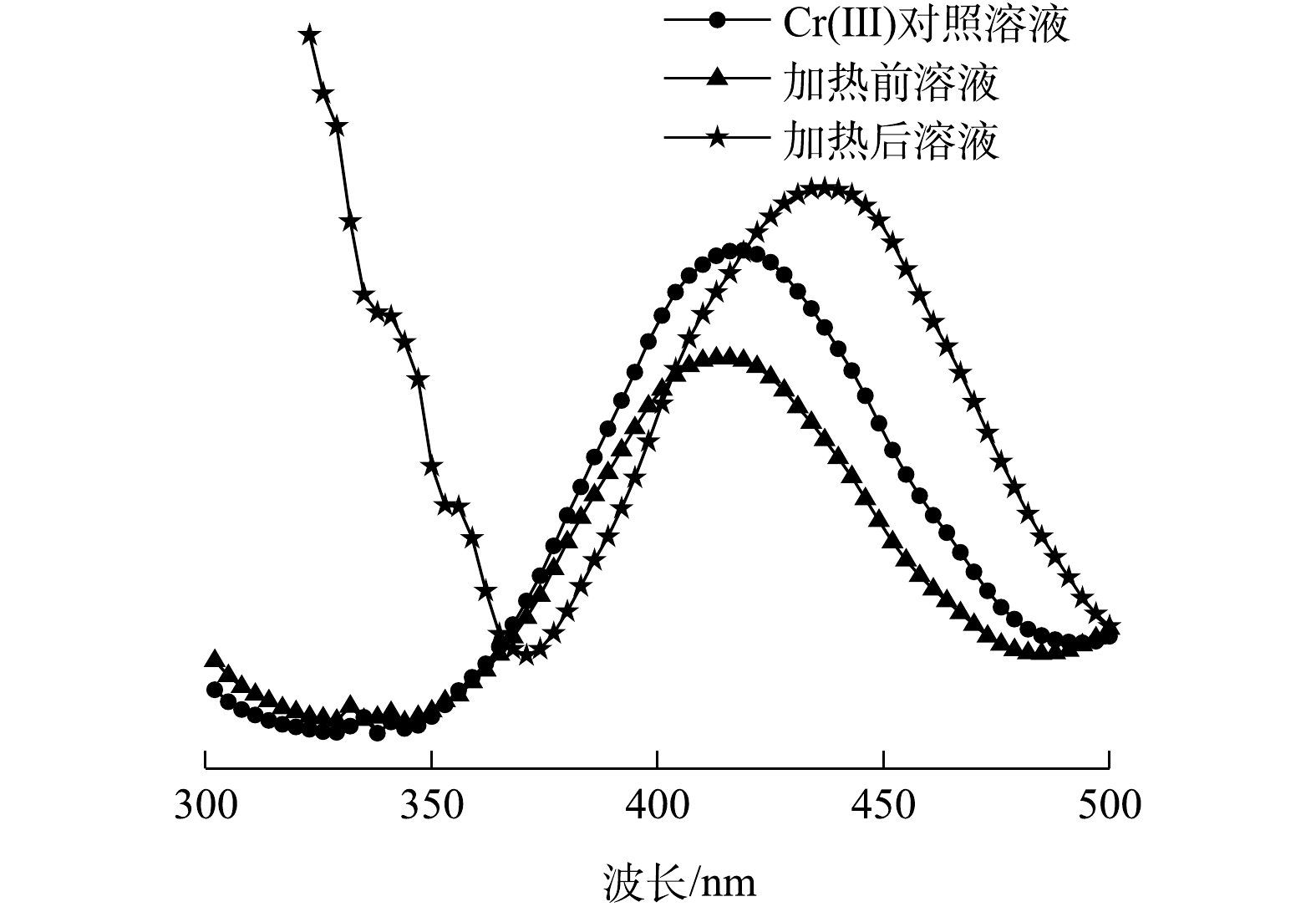
在第3步实验中观察到,Cr(Ⅵ)与NH2OH·HCl提取液混合后,有气泡产生(N2和N2O);溶液迅速变成蓝色(Cr(Ⅲ)及其配合离子的混合色),加热后转变为绿色(图1)。加热前、后溶液的UV-VIS扫描结果显示(图2),溶液在375 nm处均无Cr(Ⅵ)吸收峰[25],这说明Cr(Ⅵ)已被全部还原。在加热前,溶液在415 nm处有吸收峰,与含Cr(Ⅲ)对照溶液[Cr2(SO4)3·6H2O配制]吸收峰位置基本相同;加热后,溶液吸收峰位置右移至437 nm处,且吸光度增大,说明有Cr(Ⅲ)配合物产生。

第3步铁锰氧化物结合态的提取原理是,在酸性条件下,以NH2OH·HCl为还原剂,将土壤中以固相存在三价铁还原为易溶于水的二价铁[28-29],使得附着其上的其它金属离子失去附着基质而被释放到提取液中,反应方程式如式(1)~式(10)所示。
当有Cr(Ⅵ)存在时,还会发生式(7)~式(10)的反应。
在液相实验中,由于没有土壤,因此不发生与Fe(Ⅲ)有关的反应,NH2OH·HCl被全部用于还原Cr(Ⅵ)。
2) H2O2对Cr(Ⅵ)的还原。Tessier连续提取法第4步的操作pH为2.0,在酸性条件下,H2O2相对于土壤有机物为强氧化剂,但相对于Cr(Ⅵ)却是还原剂(式11)。值得注意的是,在碱性条件下,H2O2可以反过来将Cr(Ⅲ)氧化成Cr(Ⅵ)(式12)[30-31]。
根据表2可知,实验L4-1中Cr(Ⅵ)因量太低被全部还原,而实验L4-2中有0.54 mg(0.01 mmol)的Cr(Ⅵ)被H2O2还原。在H2O2加入到Cr(Ⅵ)溶液中(L4-2)的瞬间,溶液变成紫色(Cr(Ⅵ)离子的颜色)[17, 32],反应后紫色褪去恢复黄色。提取液中H2O2的加入量为1.5 g(0.044 mol),若按式(11)与Cr(Ⅵ)完全反应,则可还原的Cr(Ⅵ)总量为1.529 g(0.029 mol),远大于0.54 mg。这表明在第4步中,H2O2对Cr(Ⅵ)的还原能力较弱。但其还原的Cr(Ⅵ)量对应到土壤中高达540 mg·kg−1,因此不可忽视其影响。在实际的土壤提取操作中,H2O2会被土壤中的其他物质消耗,实际还原的Cr(Ⅵ)量小于该值。
-
1)亚铁离子的影响。Fe2+与Cr(Ⅵ)的氧化还原反应(式10)在弱碱性和酸性条件下均可进行。Tessier连续提取法的第1步未对pH加以控制,第2步要求pH控制在5.0,而要阻止Fe2+与Cr(Ⅵ)的氧化还原反应,溶液的pH通常需要大于10[23, 33]。实验结果(表3)表明,当Fe2+过量时(L1-1-Fe、L1-2-Fe、L2-1-Fe和L2-2-Fe),Cr(Ⅵ)会被Fe2+全部还原。
本实验发现,L1-3-Fe的Cr(Ⅵ)反应量大于L2-3-Fe;对比发现,L1-3-Fe反应后pH明显低于L2-3-Fe。这是因为,L1-3-Fe产生的Fe3+在生成Fe(OH)3的过程中释放出H+。在L2-3-Fe实验中,由于NaAc-HOAc提取液具有较强的缓冲能力,从而能维持在pH=5.0。当有土壤存在时,由于土壤具有较强的缓冲能力,通常不会出现L1-3-Fe中的低pH情况。
2)硫化物的影响。Na2S还原处理后的铬污染土壤中,残留在土壤中的含硫物质并非S2-这1种形式,还包含零价硫(S0)、多硫化物(
S2−n S2O2−3 SO2−3 SO2−4 SO2−4 实验结果表明(表4),在第1步提取实验中,Cr(Ⅵ)的绝对反应量随其初始量的增加而增加,但占初始量的比重却逐渐降低,依次为35%、6%和3%,整体还原效率远低于Fe2+。
在第2步提取实验中,Cr(Ⅵ)的绝对反应量随其初始量的增加而增加,占初始量的比重分别为55%、32%和27%,与第1步有相同规律。但Cr(Ⅵ)的绝对反应量相较于第1步明显增加,这主要归因于第1步提取液pH呈弱碱性,Cr(Ⅵ)与S2−、
S2O2−3 SO2−3 S2O2−3 SO2−3 在第2 d对溶液中的剩余Cr(Ⅵ)浓度进行复检时,Cr(Ⅵ)浓度下降。这说明该反应过程仍在缓慢进行,进一步影响了该提取步骤检测结果的可信度和重现性。
-
根据表5可知,未污染土壤(土样1)中不含有Cr(Ⅵ)。TCr即为Cr(Ⅲ),其可交换态的检测结果低于检出限,这与自然条件下未污染土壤中Cr(Ⅲ)溶解度极低、通常不检出的情况一致。TCr的5种结合态加和值大于直接检测值,这是实验的系统误差所致。操作中发现,由于Tessier连续提取分析时土壤量少,导致数据重现性下降,数据偏差较大。
在各步提取液中均未检测到Cr(Ⅵ),说明不存在Cr(Ⅲ)转化成Cr(Ⅵ)的情况,即在实际土壤的Tessier连续提取过程中,有机结合态提取步骤中使用的H2O2只能氧化土壤有机物,不能氧化Cr(Ⅲ),与液相实验结果一致。这表明,当土壤中只有Cr(Ⅲ)时,如农田中少量的铬污染,其存在形态通常只有Cr(Ⅲ)这一种形态,在土壤中的结合态分析可以采用Tessier连续提取法。
铬污染土壤(土样2)的Tessier连续提取结果表明(表6),其含有较高的Cr(Ⅵ)和TCr。在TCr的可交换态和碳酸盐结合态中,Cr(Ⅵ)占比分别高达91%和78%。与土样1相比,可交换态检测到Cr(Ⅲ)的存在,这是由于该铬污染土壤中的绝大部分Cr(Ⅲ)以Cr(OH)3沉淀存在,仅有少量Cr3+和无定性Cr(OH)3吸附在土壤颗粒表面,这部分Cr(Ⅲ)易被提取出来。
Cr(Ⅵ)的铁锰氧化物结合态和有机结合态的检测结果低于检出限,且这2步的加标回收率分别为0和11%~12%,相比于可交换态和碳酸盐结合态的加标回收率在103%~109%之间(每步2个加标回收平行样,RPD = 0~1%),这说明在铁锰氧化物结合态和有机结合态的提取过程中存在严重的Cr(Ⅵ)还原现象。该结果与溶液相实验结果一致,即提取液中的还原性组分会将提取到溶液中的Cr(Ⅵ)还原。此外,在有机结合态提取步骤中,还可能存在原本不能直接与Cr(Ⅵ)反应的土壤有机质,经H2O2氧化降解后与Cr(Ⅵ)发生反应的情况[35]。
TCr的铁锰氧化物结合态和有机结合态检测结果的可信度受多重因素的影响,如:Cr(Ⅵ)被还原成Cr(Ⅲ)后,是否一部分会被重新吸附或沉淀到土壤中?属于铁锰氧化物结合态的Cr(Ⅵ)被还原成Cr(Ⅲ)后,是否会与土壤有机质结合转化成有机结合态[36]?这些影响因素对TCr结合态的检测结果影响是否达到不可接受的水平?需要根据土壤样品的具体成分进行评估研究。
表7表明,经Na2S还原处理后的铬污染土壤(土样4),其Cr(Ⅵ)的可交换态、碳酸盐结合态、铁锰氧化物结合态和有机结合态均未检出。经FeSO4还原处理后的铬污染土壤(土样3),除了可交换态少量检出外,其他结合态均未检出,以上实验结果与溶液相实验结果(表3和表4)一致。苏长青[37]的研究中也出现了类似的可交换态、碳酸盐结合态、铁锰氧化态和有机态为零或少量检出的情况;但更多报道是5种结合态都有检出,这与土壤Cr(Ⅵ)含量、氧化性物质含量等诸多因素有关。土样4中,TCr的可交换态低于检出限,这是由于Na2S的强碱性导致土样4的pH较高(pH=9),Cr(Ⅲ)因生成Cr(OH)3而未被提取。在碳酸盐结合态的检测中可发现,当提取液pH被控制在5时,Cr(Ⅲ)开始大量溶出。其他研究中也存在还原修复后土壤TCr的可交换态低于检出限的情况[2]。
另外,由于在碳酸盐结合态提取步骤中依然存在Cr(Ⅵ)还原现象,说明土样3、土样4中残留还原剂在可交换态提取步骤未被完全去除。而还原剂在碳酸盐结合态提取步骤之后是否有残留,继续影响第3、4步的检测结果;由于提取液自身还原性组分的影响,此处无法进一步判断。土样3和土样4中Cr(Ⅵ)的5种结合态检测结果之和显著小于直接检测值,也说明Tessier连续提取过程从整体上存在Cr(Ⅵ)的还原现象。
目前的研究通常未提供Cr(Ⅵ)检测结果的关键质控数据(如加标回收率),或者未通过直接检测值与5种结合态检测加和值的比较来判断数据的可信度[38],残渣态通常根据直接检测值减去前4种结合态求得,这种算法掩盖了提取过程中的Cr(Ⅵ)还原问题。此外,大部分研究仅将Tessier连续提取法用于TCr的分析[4, 39],只有少数用于Cr(Ⅵ)[6, 37, 40],但由于Cr(Ⅵ)毒性远大于Cr(Ⅲ),对于以评价土壤修复效果为目的的研究,仅对比还原修复前、后TCr结合态分布的变化,存在一定的不合理性。
此外,BCR提取法中也使用了NH2OH·HCl作为还原剂(pH = 2)[12, 38],在常温下提取可还原结合态。而本研究结果表明,即使在室温条件下,Cr(Ⅵ)也会被还原成Cr(Ⅲ)(图1、图2),这说明BCR提取法可能也面临同样的Cr(Ⅵ)-Cr(Ⅲ)转化问题。
陈英旭等[41]提出了一种专门针对土壤铬的5步提取方案:水溶态、交换态(1 mol·L−1 CH3COONH4)、沉淀态(2 mol·L−1 HCl)、有机结合态(5% H2O2 -2 mol·L−1 HCl)、残渣态。其沉淀态包含了Tessier连续提取法的碳酸盐结合态和铁锰氧化物结合态,因其沉淀态提取液中没有使用还原剂,该步骤不会发生提取液组分还原Cr(Ⅵ)的问题。其有机结合态的分析原理与Tessier连续提取法相同,使用H2O2在酸性条件下氧化有机物,同样可能导致Cr(Ⅵ)还原问题。但该方法对Cr(Ⅵ)的整体还原程度远低于Tessier连续提取法。因此可以认为,对于铬污染土壤的结合态分析,陈英旭等[41]提出的方法使用面相对较广,可适用于未经修复的铬污染土壤中Cr(Ⅵ)的结合态分析,但对于修复后残留大量还原剂的铬污染土壤依然不适用。
2.1. 第3、4步中提取液组分的影响
2.2. 第1、2步提取操作中残留还原剂的影响
2.3. 土壤铬的Tessier连续提取实验
-
1)Tessier连续提取法的提取液自身还原性组分会在铁锰氧化物结合态和有机结合态的检测中导致Cr(Ⅵ)被还原为Cr(Ⅲ),可还原的Cr(Ⅵ)最大量分别为50.88 mg、0.54 mg。经还原修复的铬污染土壤,土壤残留还原剂会在可交换态和碳酸盐结合态的检测中导致Cr(Ⅵ)的还原。
2)当用于了解未污染土壤中原生铬结合态分布时,使用Tessier连续提取法无不利影响。
3)当用于了解铬污染土壤中TCr和Cr(Ⅵ)的结合态分布时,Cr(Ⅵ)铁锰氧化物结合态和有机结合态的检测结果明显低于真实值,TCr结合态的检测结果重现性可能较差,但不一定出现不可接受的偏差。
4)当用于评价铬污染土壤修复前、后Cr(Ⅵ)结合态分布变化时,修复后土壤中Cr(Ⅵ)可交换态、碳酸盐结合态、铁锰氧化物结合态和有机结合态的检测结果均可能明显低于真实值。
