

-
石油类污染物的意外泄漏事故频繁发生,导致一定数量的非水相液体 (NAPL) 通过包气带进入地下造成污染[1]。在包气带内挥发性有机物 (VOCs) 会通过挥发作用进入土壤气体,VOCs气体在迁移过程中发生生物降解,源区轻非水相液体 (LNAPL) 由生物降解作用导致的衰减占LNAPL总质量损失的90%~99% [2-3]。因此,挥发性石油烃在包气带中的气相自然衰减和生物降解动力学规律的研究对VOCs蒸气入侵风险评估和石油场地监测自然衰减应用都具有重要意义[4-5],气相生物降解速率是蒸气入侵建模和定量风险评估的关键输入参数[6-7]。
当VOCs在污染源挥发成为气态在包气带扩散迁移过程中,吸附、生物降解等机制会导致显著的质量衰减,这被称为气相自然衰减[8-9]。在某汽油污染场地,从地下水源挥发的总碳氢化合物质量的68%在毛细管层被生物降解,石油烃蒸气潜水面上1 m内包气带内被降解殆尽[10]。HÖHENER等[11]通过微宇宙实验、柱实验及现场研究测定了包气带中13种VOCs的气相生物降解规律。VOCs气相自然衰减受到土壤类型、温度、含水率、营养水平等多种因素影响。土壤类型,特别是土壤孔隙度、含水量、土壤渗透性、有效扩散性和有机碳吸附能力是影响VOCs气相迁移归趋的重要因素[12]。YAO等[13]报道,土壤质地对VOCs浓度衰减的影响,其中土壤粒径的对底板下土壤气衰减因子的影响平均约为0.4个数量级。BEKELE等[14]研究了三氯乙烯 (TCE) 在5种不同的土壤中的气相吸附,粘土质量分数增加1倍会导致TCE蒸气吸附量增加11倍。但是,目前关于包气带VOCs气相自然衰减的研究仍然很不充分,对于不同种类石油烃蒸气在不同土壤中的吸附与生物降解规律以及我国不同区域土壤的气相自然衰减潜力尚不清楚。
本研究采用微宇宙实验,选取4种正构烷烃 (正戊烷、正己烷、正庚烷、正辛烷) 、4种环烷烃 (环戊烷、环己烷、环庚烷、环辛烷) 及4种苯系物 (苯、甲苯、乙苯、对二甲苯) ,对其在黑土、黄土、红土及石英砂等4种土壤中的气相自然衰减规律和生物降解速率进行了系统研究。
-
实验试剂采用色谱纯的正戊烷、正己烷、正庚烷、正辛烷、环戊烷、环己烷、环庚烷、环辛烷、苯、甲苯、乙苯、对二甲苯。本研究所用的黄土、黑土、红土和石英砂分别取自河北省、黑龙江省、云南省和江西省,土壤经自然风干后过2 mm标准方孔筛备用,其基本理化性质如表1所示。
-
微宇宙实验在50 mL的玻璃旋盖小瓶中进行,分别取20 g土壤及20 μL混合污染物于瓶中,使用特氟龙微型阀瓶盖密封。为了研究非生物作用对气相自然衰减的贡献,同时做灭菌对照组,使用120 ℃高压蒸汽灭菌30 min。每隔一段时间进行气体取样,采用250 μL气密性微量取样针取100 μL气体,用气相色谱仪测定污染物蒸气的质量浓度 (g·m−3) 。
-
污染物气体质量浓度采用安捷伦7890B 气相色谱仪,检测器为火焰离子化检测器(FID),温度250 ℃,HP-5毛细管柱( 30 m×0. 32 mm×0. 25 μm),进样口温度设置为200 ℃,分流比10∶1。测试正构烷烃时,柱箱升温程序为初始温度55 ℃保持时间2.5 min,升温至140 ℃,保持时间0.2 min,升温速率为30 ℃·min−1。测试环烷烃时,柱箱温度为50 ℃保持时间2 min,升温至75 ℃,保持时间0.2 min,升温速率为10 ℃·min−1。测试苯系物时色谱柱箱恒温120 ℃。
自然衰减通常用一级动力学模型描述。气态烃的表观一级衰减速率是通过质量浓度数据明显下降阶段的ln (Ct/C0) 与时间 (t) 线性回归来确定,其中C0是污染蒸气的初始质量浓度,Ct是t时刻污染蒸气质量浓度[15]。非灭菌土壤中的表观一级衰减速率是生物降解和非生物自然衰减速率的总和,而灭菌土壤中的污染物质量浓度下降仅由非生物机制引起,因此标记为“非生物衰减速率”。对于同一种气态烃,非灭菌土壤中的“表观一级衰减速率”减去灭菌土壤中的“非生物衰减速率”是生物降解的真正贡献,标记为“生物降解速率”[16]。
-
4种正构烷烃 (正戊烷、正己烷、正庚烷、正辛烷) 在灭菌组和非灭菌组的衰减去除率均表现为黑土>黄土>红土>石英砂 (表2) 。4种正构烷烃的生物降解速率表现为黑土>黄土>红土,石英砂未发生明显的生物降解,因此无法拟合石英砂的降解速率 (图1、表3) 。灭菌组中气态烃的自然衰减机制主要是吸附,而土壤有机质质量分数是4种土壤气相吸附能力差异大的主要原因。4种土壤的有机质的质量分数顺序:黑土 (36.70%) >黄土 (0.66%) >红土 (0.33%) >石英砂 (0.32%) 。这与灭菌组中的气态烃衰减去除率一致。其他研究[14,17]也发现,土壤有机质质量分数是土壤对气态VOCs吸附的主要因素,高有机质质量分数会增加土壤对VOCs的吸附。BEKELE等[14]报道,土壤有机质质量分数增加1倍导致总三氯乙烯蒸气吸附量增加7倍。UGWOHA和ANDRESEN [17]发现,土壤有机质质量分数从0增加至5%后气态烷烃的吸附质量增加了2倍,土壤有机质量分数的增加会增强汽油烃蒸气的吸附速度和吸附量。气态VOCs需要首先被土壤吸附固定,然后才能被土壤微生物代谢降解[18],较高的有机质质量分数会提高土壤吸附固定VOCs的能力,从而会提高其生物降解能力。因此,土壤有机质质量分数是4种土壤气相吸附能力和生物降解能力差异大的主要原因。除有机质质量分数外,土壤的生物降解能力还与土壤土著微生物数量密切相关,经测试发现,本研究所用的黑土 (5.2×106 cfu·g−1) 和黄土 (2.1×106 cfu·g−1) 中的微生物数量远大于红土 (1.2×105 cfu·g−1) 和石英砂 (2.0×103 cfu·g−1) 。综上所述,较高有机质质量分数和微生物数量使得黑土对于气态烃具有较强的自然衰减能力。
-
4种环烷烃 (环戊烷、环己烷、环庚烷、环辛烷) 在不同土壤中的衰减规律与正构烷烃相似,在灭菌组和非灭菌组的衰减去除率整体上符合黑土>黄土>红土>石英砂的规律 (表2、图2) 。环烷烃在黑土、黄土和红土中的生物降解速率均很小 (<0.06 d−1) ,而石英砂中几乎不存在生物降解。这说明环烷烃蒸气的生物降解性极低 (表3) 。HöHENER等[11]也发现,气相环烷烃的自然衰减速率很低,实验和场地等不同方式获得的气相一级衰减速率常数均低于0.31 d−1。这可能由于环烷烃的溶解度较低,导致土壤气体和水之间的界面传质速率较低[19]。而且短链烷烃 (<C9) 对许多微生物都有毒性,中间链长的正构烷烃 (C10~C24) 降解速度最快,短链烷烃通过其溶剂作用来抑制微生物活性,它们在作为溶剂存在时会破坏脂质膜[20-21]。大量有机地球化学研究证实环烷烃的生物降解性低于正构烷烃[22-23],且生物降解随着环数的增加而降低[24]。环烷烃通过氧化酶的作用降解为环醇,进一步脱氢为酮,环烷烃代谢的主要产物是环酮和环烷烃-羧酸[20,24]。
-
4种苯系物 (苯、甲苯、乙苯、对二甲苯) 在4种土壤在灭菌组和非灭菌组中的自然衰减去除率均表现为黑土>黄土>红土>石英砂 (表2、图3) 。4种苯系物的生物降解速率均符合黑土 (3.90~9.76 d−1) >黄土 (1.18~8.24 d−1) >红土 (0.90~2.76 d−1) 的规律,而石英砂几乎不存在生物降解 (表3) 。因为黑土中的高有机质质量分数会促进苯系物蒸气的吸附,进而促进生物降解。UGWOHA和ANDRESEN[17]发现土壤有机质的质量分数增加5%导致E20乙醇汽油中苯的吸附量增加76%。在不同的土壤中苯系物的一级生物降解速率均为甲苯>苯>乙苯>对二甲苯。苯系物的生物降解速率依赖于分子化学结构,甲苯通常认为是苯系物中最容易被生物降解的。因为环上存在取代基提供了攻击侧链或氧化芳香环的替代途径[25]。苯氧化的第一步是由双加氧酶催化的羟基化,苯环上取代基的存在允许两种可能的机制:攻击侧链或氧化芳环,所有这些途径汇聚形成邻苯二酚中间体。苯主要中间产物是邻苯二酚,甲苯和乙苯在单独的途径上降解,产生各自的主要中间体3甲基邻苯二酚和3-乙基邻苯二酚,二甲苯均代谢为单甲基化儿茶酚[25]。BTEX化合物之间存在复杂的底物相互作用以及其生物降解过程中的竞争性抑制效应[26];有研究报道[27],在混合物中苯和甲苯根据竞争性抑制动力学被去除,而对二甲苯在苯和甲苯的存在下通过共代谢过程部分去除,甲苯、苯或乙苯的存在对二甲苯的降解速率有负面影响。尽管芳环上没有官能团可能使苯难以被生物转化,但苯在水中的溶解度 (1 791.00 mg·L−1) 在4种苯系物中最高(甲苯、乙苯及对二甲苯溶解度分别为535.00、161.00、156.00 mg·L−1),也促进了苯的生物降解[25]。
-
苯系物的自然衰减去除率和生物降解速率远高于正构烷烃和环烷烃,环烷烃最难被生物降解。4种苯系物中一级生物降解速率均为甲苯>苯>乙苯>对二甲苯,甲苯最容易被生物降解。苯、甲苯、乙苯、对二甲苯生物降解速率0.90~9.76 d−1,正戊烷、正己烷、正庚烷、正辛烷生物降解速率小于0.28 d−1,环戊烷、环己烷、环庚烷、环辛烷生物降解速率小于0.06 d−1。苯系物的表观一级衰减速率和生物降解速率都远高于正构烷烃和环烷烃,可能因为苯系物的溶解度远高于正构烷烃和环烷烃[22]。PASTERIS等[21]也报道过,柱实验数据拟合的甲苯的一级生物降解速率常数为3.2 d−1,而短链烷烃如戊烷和己烷、环烷烃和的生物降解速率较慢,估计为0.1~1.2 d−1。脂肪族烃缺乏官能团且水溶性极低,微生物对脂肪烃都表现出较低的化学反应性和生物利用度,烷烃的微生物降解的敏感性:正构烷烃>异构烷烃>环烷烃[20]。
-
1) 正构烷烃、环烷烃和苯系物蒸气在4种土壤中的自然衰减,整体自然衰减去除率黑土>黄土>红土>石英砂,黑土生物降解速率高于黄土,红土和石英砂的生物降解能力很低。
2) 苯系物的气相自然衰减速率和生物降解速率远高于正构烷烃和环烷烃。包气带中的气相生物降解是石油污染场地一种重要但被忽视的自然衰减机制,该过程对于挥发性石油烃蒸气入侵暴露以及污染物自然衰减均具有重要的影响。
3) 气相自然衰减和气相生物降解动力学数据可以为蒸气入侵定量风险评估和石油污染场地自然衰减速率定量评估模型提供关键输入参数。
石油烃在不同土壤中的气相自然衰减规律
Natural attenuation of petroleum hydrocarbon vapors in different soils
-
摘要: 挥发性有机物 (VOCs) 在包气带中的迁移扩散是土壤和地下水中可挥发污染物自然衰减的重要机制,也与蒸气入侵暴露和风险评估密切相关。采用微宇宙实验对12种挥发性石油烃 (正戊烷、正己烷、正庚烷、正辛烷、环戊烷、环己烷、环庚烷、环辛烷、苯、甲苯、乙苯、对二甲苯) 在4种土壤 (黑土、黄土、红土、石英砂) 中的气相自然衰减机制和气态生物降解动力学规律进行了研究。结果表明,正构烷烃、环烷烃和苯系物蒸气在4种土壤中的气相自然衰减去除率都遵循黑土>黄土>红土>石英砂的规律;黑土中生物降解对污染物去除率的贡献高于黄土,而红土和石英砂中的生物降解速率极低;4种苯系物的自然衰减和生物降解潜力远高于正构烷烃和环烷烃;苯系物气相生物降解速率排序为:甲苯>苯>乙苯>对二甲苯。本研究结果可为蒸气入侵定量风险评估和石油污染场地自然衰减速率定量评估提供参考。Abstract: The transport of volatile organic compounds (VOCs) in the vadose zone is an important mechanism for the natural attenuation of volatile pollutants in soil and groundwater and is also closely related to vapor intrusion and risk assessment. The natural attenuation mechanism and biodegradation kinetics of 12 volatile petroleum hydrocarbons (n-pentane, n-hexane, n-heptane, n-octane, cyclopentane, cyclohexane, cycloheptane, cyclooctane, benzene, toluene, ethylbenzene, p-xylene) in four soils (black soil, yellow earth, lateritic red earth, and quartz sand) were studied by microcosm experiments in detail. The results showed that the natural attenuation removal rates of n-alkane, cycloalkane and monoaromatic hydrocarbon vapors in four soils followed the order: black soil > yellow earth > lateritic red earth > quartz sand; the biodegradation contribution in black soil was higher compared to yellow earth, and the biodegradation rates of petroleum hydrocarbon vapors in lateritic red earth and quartz sand were very low; the natural attenuation removal rates and biodegradation contribution of the four monoaromatic hydrocarbons were much higher than n-alkanes and cycloalkane; the biodegradation rates of four monoaromatic hydrocarbons were in the order of toluene > benzene> ethylbenzene > p-xylene. This study provided valuable insights into vapor intrusion risk assessment and natural attenuation rate quantification at petroleum contaminated sites.
-
Key words:
- contaminated site /
- soil remediation /
- groundwater remediation /
- petroleum /
- natural attenuation /
- biodegradation /
- unsaturated zone
-
石油类污染物的意外泄漏事故频繁发生,导致一定数量的非水相液体 (NAPL) 通过包气带进入地下造成污染[1]。在包气带内挥发性有机物 (VOCs) 会通过挥发作用进入土壤气体,VOCs气体在迁移过程中发生生物降解,源区轻非水相液体 (LNAPL) 由生物降解作用导致的衰减占LNAPL总质量损失的90%~99% [2-3]。因此,挥发性石油烃在包气带中的气相自然衰减和生物降解动力学规律的研究对VOCs蒸气入侵风险评估和石油场地监测自然衰减应用都具有重要意义[4-5],气相生物降解速率是蒸气入侵建模和定量风险评估的关键输入参数[6-7]。
当VOCs在污染源挥发成为气态在包气带扩散迁移过程中,吸附、生物降解等机制会导致显著的质量衰减,这被称为气相自然衰减[8-9]。在某汽油污染场地,从地下水源挥发的总碳氢化合物质量的68%在毛细管层被生物降解,石油烃蒸气潜水面上1 m内包气带内被降解殆尽[10]。HÖHENER等[11]通过微宇宙实验、柱实验及现场研究测定了包气带中13种VOCs的气相生物降解规律。VOCs气相自然衰减受到土壤类型、温度、含水率、营养水平等多种因素影响。土壤类型,特别是土壤孔隙度、含水量、土壤渗透性、有效扩散性和有机碳吸附能力是影响VOCs气相迁移归趋的重要因素[12]。YAO等[13]报道,土壤质地对VOCs浓度衰减的影响,其中土壤粒径的对底板下土壤气衰减因子的影响平均约为0.4个数量级。BEKELE等[14]研究了三氯乙烯 (TCE) 在5种不同的土壤中的气相吸附,粘土质量分数增加1倍会导致TCE蒸气吸附量增加11倍。但是,目前关于包气带VOCs气相自然衰减的研究仍然很不充分,对于不同种类石油烃蒸气在不同土壤中的吸附与生物降解规律以及我国不同区域土壤的气相自然衰减潜力尚不清楚。
本研究采用微宇宙实验,选取4种正构烷烃 (正戊烷、正己烷、正庚烷、正辛烷) 、4种环烷烃 (环戊烷、环己烷、环庚烷、环辛烷) 及4种苯系物 (苯、甲苯、乙苯、对二甲苯) ,对其在黑土、黄土、红土及石英砂等4种土壤中的气相自然衰减规律和生物降解速率进行了系统研究。
1. 材料与方法
1.1 实验材料
实验试剂采用色谱纯的正戊烷、正己烷、正庚烷、正辛烷、环戊烷、环己烷、环庚烷、环辛烷、苯、甲苯、乙苯、对二甲苯。本研究所用的黄土、黑土、红土和石英砂分别取自河北省、黑龙江省、云南省和江西省,土壤经自然风干后过2 mm标准方孔筛备用,其基本理化性质如表1所示。
表 1 实验土壤的基本理化性质Table 1. Basic physical-chemical characteristics of experimental soils土壤类型 pH 孔隙度 含水率 有效扩散系数 (20 ℃)/ (正戊烷,m2·d−1) 密度/ (g·cm−1) 有机质质量分数/% 土壤质地 机械组成/% 砂粒 粉粒 黏粒 黄土 7.8 0.5 0.02 0.28 1.3 0.66 壤土 49.6 39.6 10.7 黑土 6.1 0.8 0.09 0.80 0.6 36.70 壤质砂土 74.6 19.6 5.8 红土 5.7 0.6 0.02 0.36 1.0 0.33 粘土 11.2 5.4 83.4 石英砂 7.6 0.4 0.01 0.19 1.5 0.32 砂土 97.4 1.8 0.9 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 实验方法
微宇宙实验在50 mL的玻璃旋盖小瓶中进行,分别取20 g土壤及20 μL混合污染物于瓶中,使用特氟龙微型阀瓶盖密封。为了研究非生物作用对气相自然衰减的贡献,同时做灭菌对照组,使用120 ℃高压蒸汽灭菌30 min。每隔一段时间进行气体取样,采用250 μL气密性微量取样针取100 μL气体,用气相色谱仪测定污染物蒸气的质量浓度 (g·m−3) 。
1.3 分析方法
污染物气体质量浓度采用安捷伦7890B 气相色谱仪,检测器为火焰离子化检测器(FID),温度250 ℃,HP-5毛细管柱( 30 m×0. 32 mm×0. 25 μm),进样口温度设置为200 ℃,分流比10∶1。测试正构烷烃时,柱箱升温程序为初始温度55 ℃保持时间2.5 min,升温至140 ℃,保持时间0.2 min,升温速率为30 ℃·min−1。测试环烷烃时,柱箱温度为50 ℃保持时间2 min,升温至75 ℃,保持时间0.2 min,升温速率为10 ℃·min−1。测试苯系物时色谱柱箱恒温120 ℃。
自然衰减通常用一级动力学模型描述。气态烃的表观一级衰减速率是通过质量浓度数据明显下降阶段的ln (Ct/C0) 与时间 (t) 线性回归来确定,其中C0是污染蒸气的初始质量浓度,Ct是t时刻污染蒸气质量浓度[15]。非灭菌土壤中的表观一级衰减速率是生物降解和非生物自然衰减速率的总和,而灭菌土壤中的污染物质量浓度下降仅由非生物机制引起,因此标记为“非生物衰减速率”。对于同一种气态烃,非灭菌土壤中的“表观一级衰减速率”减去灭菌土壤中的“非生物衰减速率”是生物降解的真正贡献,标记为“生物降解速率”[16]。
2. 结果与讨论
2.1 正构烷烃在4种土壤中的气相自然衰减
4种正构烷烃 (正戊烷、正己烷、正庚烷、正辛烷) 在灭菌组和非灭菌组的衰减去除率均表现为黑土>黄土>红土>石英砂 (表2) 。4种正构烷烃的生物降解速率表现为黑土>黄土>红土,石英砂未发生明显的生物降解,因此无法拟合石英砂的降解速率 (图1、表3) 。灭菌组中气态烃的自然衰减机制主要是吸附,而土壤有机质质量分数是4种土壤气相吸附能力差异大的主要原因。4种土壤的有机质的质量分数顺序:黑土 (36.70%) >黄土 (0.66%) >红土 (0.33%) >石英砂 (0.32%) 。这与灭菌组中的气态烃衰减去除率一致。其他研究[14,17]也发现,土壤有机质质量分数是土壤对气态VOCs吸附的主要因素,高有机质质量分数会增加土壤对VOCs的吸附。BEKELE等[14]报道,土壤有机质质量分数增加1倍导致总三氯乙烯蒸气吸附量增加7倍。UGWOHA和ANDRESEN [17]发现,土壤有机质质量分数从0增加至5%后气态烷烃的吸附质量增加了2倍,土壤有机质量分数的增加会增强汽油烃蒸气的吸附速度和吸附量。气态VOCs需要首先被土壤吸附固定,然后才能被土壤微生物代谢降解[18],较高的有机质质量分数会提高土壤吸附固定VOCs的能力,从而会提高其生物降解能力。因此,土壤有机质质量分数是4种土壤气相吸附能力和生物降解能力差异大的主要原因。除有机质质量分数外,土壤的生物降解能力还与土壤土著微生物数量密切相关,经测试发现,本研究所用的黑土 (5.2×106 cfu·g−1) 和黄土 (2.1×106 cfu·g−1) 中的微生物数量远大于红土 (1.2×105 cfu·g−1) 和石英砂 (2.0×103 cfu·g−1) 。综上所述,较高有机质质量分数和微生物数量使得黑土对于气态烃具有较强的自然衰减能力。
表 2 石油烃在不同土壤中自然衰减去除率Table 2. The remove rates of petroleum hydrocarbons in different soils% 化合物 黄土 黑土 红土 石英砂 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 正戊烷 24.61 27.82 3.21 42.91 68.18 25.27 21.33 23.06 1.73 3.56 5.08 1.52 正己烷 35.35 45.64 10.29 64.67 87.24 22.57 32.89 36.05 3.16 2.18 3.03 0.86 正庚烷 51.17 74.77 23.61 74.62 93.20 18.58 40.25 45.29 5.04 0.61 1.39 0.78 正辛烷 63.14 78.43 15.28 78.12 96.55 18.44 49.37 46.77 2.60 5.63 7.87 2.24 环戊烷 13.30 33.68 20.38 20.90 43.04 22.14 5.29 6.29 1.00 5.58 9.62 4.05 环己烷 12.96 28.24 15.28 46.68 70.96 24.28 1.75 7.09 5.34 4.29 9.91 5.62 环庚烷 8.23 45.91 37.68 61.22 84.85 23.63 9.88 12.89 3.01 12.67 14.29 1.62 环辛烷 57.60 66.28 8.67 71.90 85.35 13.45 9.14 17.37 8.23 12.56 21.14 8.58 苯 69.62 81.55 11.93 77.66 92.32 14.66 64.88 75.69 10.81 27.15 27.51 0.36 甲苯 72.54 80.57 8.03 82.64 91.98 9.34 67.44 75.63 8.19 26.43 27.00 0.57 乙苯 72.31 79.12 6.81 80.98 85.64 4.66 53.64 56.86 3.23 19.38 19.88 0.50 对二甲苯 75.80 82.81 7.01 77.57 83.36 5.80 51.45 55.65 4.20 19.46 20.00 0.54 注:“*”表示生物降解贡献的去除率为非灭菌组与灭菌组去除率之差。 | Show TableDownLoad:
CSV
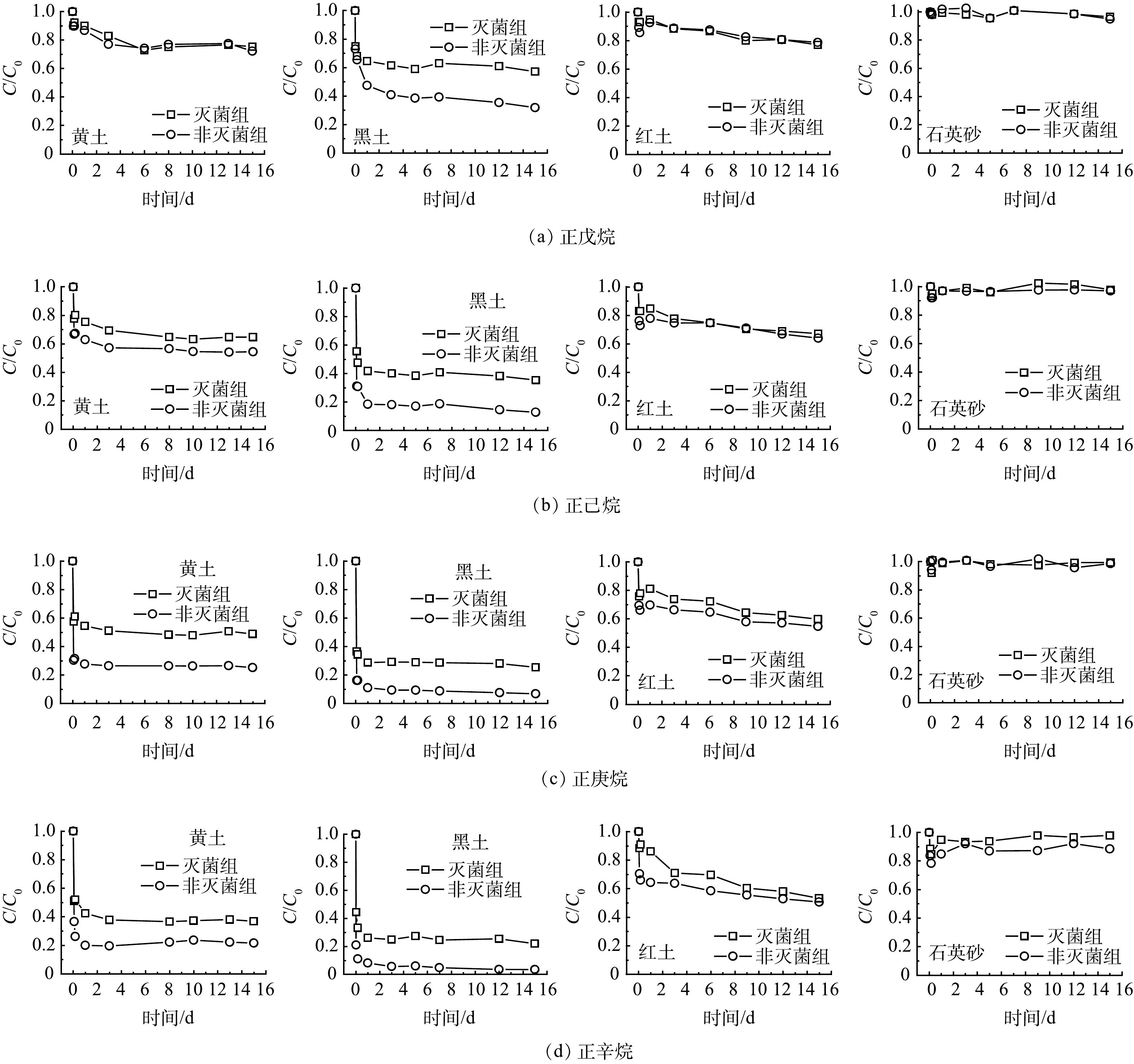 图 1 正构烷烃在4种土壤中的质量浓度变化曲线Figure 1. N-alkanes concentration attenuation curve in four soils表 3 石油烃在不同土壤中的一级衰减速率常数Table 3. The first order attenuation rate constants of petroleum hydrocarbons in different soils
图 1 正构烷烃在4种土壤中的质量浓度变化曲线Figure 1. N-alkanes concentration attenuation curve in four soils表 3 石油烃在不同土壤中的一级衰减速率常数Table 3. The first order attenuation rate constants of petroleum hydrocarbons in different soilsd−1 化合物 黄土 黑土 红土 石英砂 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 正戊烷 0.03 0.04 0.01 0.03 0.06 0.03 0.02 0.02 — — — — 正己烷 0.03 0.06 0.03 0.05 0.18 0.13 0.016 0.02 0.004 — — — 正庚烷 0.05 0.18 0.13 0.26 0.54 0.28 0.02 0.05 0.03 — — — 正辛烷 0.06 0.15 0.09 0.02 0.12 0.10 0.03 0.05 0.02 — — — 环戊烷 0.01 0.02 0.01 0.04 0.07 0.03 0.01 0.01 — — — — 环己烷 0.01 0.03 0.02 0.20 0.26 0.06 0.01 0.01 — — — — 环庚烷 0.02 0.05 0.03 0.28 0.32 0.04 0.01 0.01 — — — — 环辛烷 0.07 0.09 0.02 0.33 0.39 0.06 0.01 0.01 — — — — 苯 25.68 27.84 2.16 28.24 235.48 7.24 24.24 25.78 1.54 — — — 甲苯 30.72 38.96 8.24 29.36 39.12 9.76 25.68 28.44 2.76 — — — 乙苯 15.6 17.08 1.48 324.98 30.82 5.84 11.76 12.72 0.96 — — — 对二甲苯 14.4 15.58 1.18 23.76 27.66 3.90 11.04 11.94 0.90 — — — | Show TableDownLoad:
CSV
2.2 环烷烃在4种土壤中的气相自然衰减
4种环烷烃 (环戊烷、环己烷、环庚烷、环辛烷) 在不同土壤中的衰减规律与正构烷烃相似,在灭菌组和非灭菌组的衰减去除率整体上符合黑土>黄土>红土>石英砂的规律 (表2、图2) 。环烷烃在黑土、黄土和红土中的生物降解速率均很小 (<0.06 d−1) ,而石英砂中几乎不存在生物降解。这说明环烷烃蒸气的生物降解性极低 (表3) 。HöHENER等[11]也发现,气相环烷烃的自然衰减速率很低,实验和场地等不同方式获得的气相一级衰减速率常数均低于0.31 d−1。这可能由于环烷烃的溶解度较低,导致土壤气体和水之间的界面传质速率较低[19]。而且短链烷烃 (<C9) 对许多微生物都有毒性,中间链长的正构烷烃 (C10~C24) 降解速度最快,短链烷烃通过其溶剂作用来抑制微生物活性,它们在作为溶剂存在时会破坏脂质膜[20-21]。大量有机地球化学研究证实环烷烃的生物降解性低于正构烷烃[22-23],且生物降解随着环数的增加而降低[24]。环烷烃通过氧化酶的作用降解为环醇,进一步脱氢为酮,环烷烃代谢的主要产物是环酮和环烷烃-羧酸[20,24]。
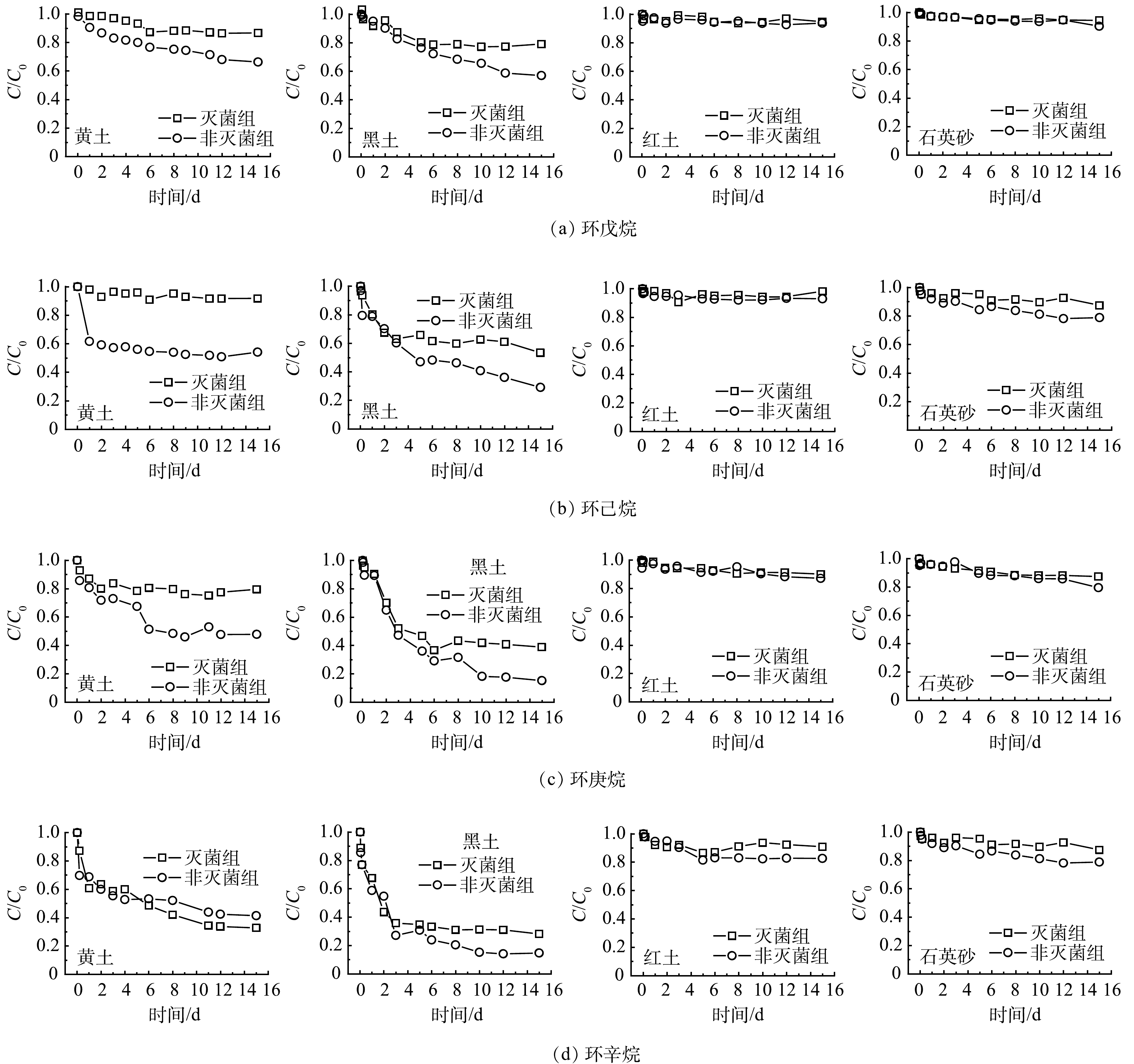 图 2 环烷烃在4种土壤中的质量浓度变化曲线Figure 2. Cyclic alkanes concentration attenuation curve in four soils
图 2 环烷烃在4种土壤中的质量浓度变化曲线Figure 2. Cyclic alkanes concentration attenuation curve in four soils2.3 苯系物在4种土壤中的气相自然衰减
4种苯系物 (苯、甲苯、乙苯、对二甲苯) 在4种土壤在灭菌组和非灭菌组中的自然衰减去除率均表现为黑土>黄土>红土>石英砂 (表2、图3) 。4种苯系物的生物降解速率均符合黑土 (3.90~9.76 d−1) >黄土 (1.18~8.24 d−1) >红土 (0.90~2.76 d−1) 的规律,而石英砂几乎不存在生物降解 (表3) 。因为黑土中的高有机质质量分数会促进苯系物蒸气的吸附,进而促进生物降解。UGWOHA和ANDRESEN[17]发现土壤有机质的质量分数增加5%导致E20乙醇汽油中苯的吸附量增加76%。在不同的土壤中苯系物的一级生物降解速率均为甲苯>苯>乙苯>对二甲苯。苯系物的生物降解速率依赖于分子化学结构,甲苯通常认为是苯系物中最容易被生物降解的。因为环上存在取代基提供了攻击侧链或氧化芳香环的替代途径[25]。苯氧化的第一步是由双加氧酶催化的羟基化,苯环上取代基的存在允许两种可能的机制:攻击侧链或氧化芳环,所有这些途径汇聚形成邻苯二酚中间体。苯主要中间产物是邻苯二酚,甲苯和乙苯在单独的途径上降解,产生各自的主要中间体3甲基邻苯二酚和3-乙基邻苯二酚,二甲苯均代谢为单甲基化儿茶酚[25]。BTEX化合物之间存在复杂的底物相互作用以及其生物降解过程中的竞争性抑制效应[26];有研究报道[27],在混合物中苯和甲苯根据竞争性抑制动力学被去除,而对二甲苯在苯和甲苯的存在下通过共代谢过程部分去除,甲苯、苯或乙苯的存在对二甲苯的降解速率有负面影响。尽管芳环上没有官能团可能使苯难以被生物转化,但苯在水中的溶解度 (1 791.00 mg·L−1) 在4种苯系物中最高(甲苯、乙苯及对二甲苯溶解度分别为535.00、161.00、156.00 mg·L−1),也促进了苯的生物降解[25]。
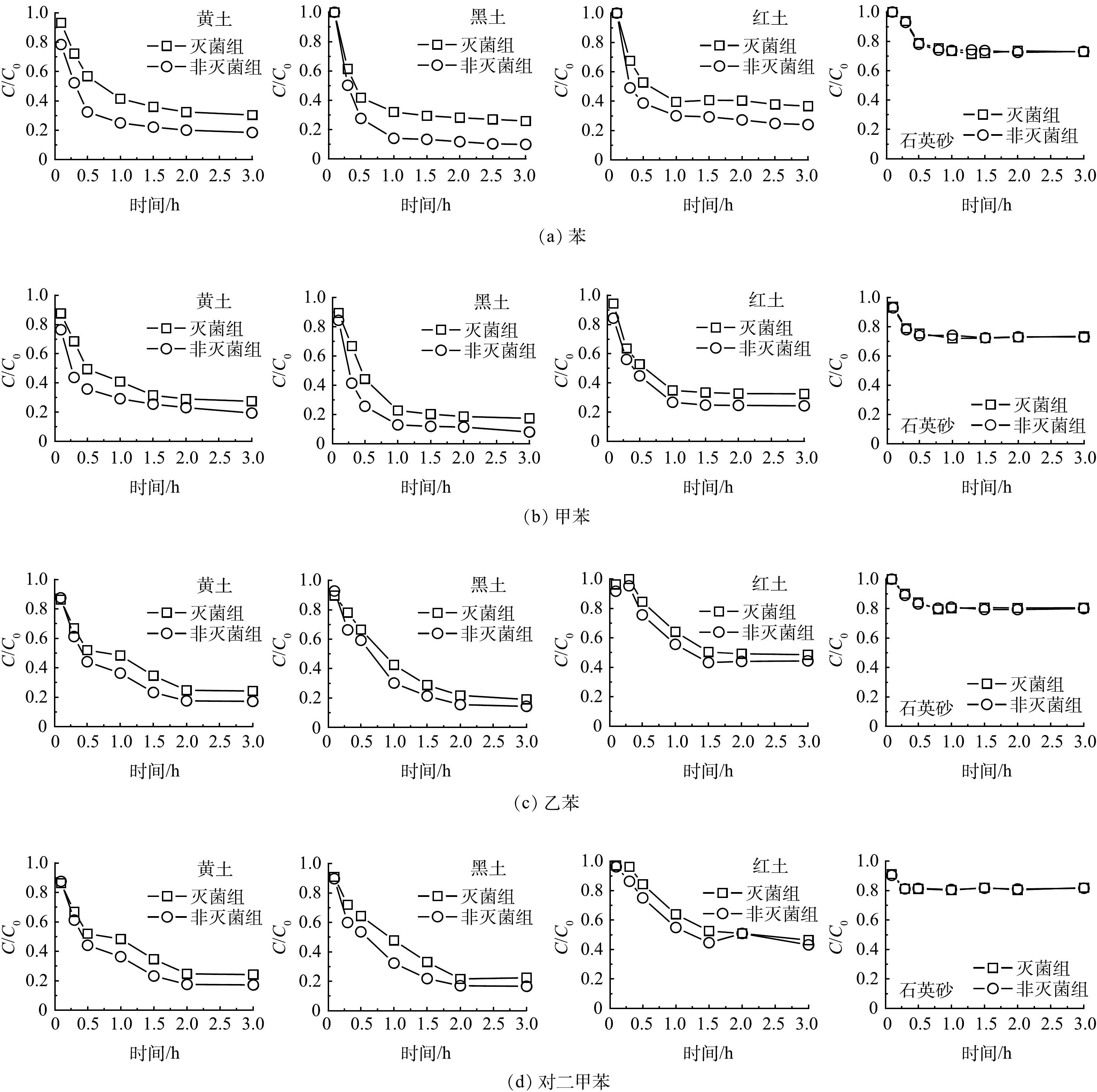 图 3 苯系物在4种土壤中的质量浓度变化曲线Figure 3. Monoaromatic hydrocarbons concentration attenuation curve in four soils
图 3 苯系物在4种土壤中的质量浓度变化曲线Figure 3. Monoaromatic hydrocarbons concentration attenuation curve in four soils2.4 不同种类石油烃的气相自然衰减潜力对比
苯系物的自然衰减去除率和生物降解速率远高于正构烷烃和环烷烃,环烷烃最难被生物降解。4种苯系物中一级生物降解速率均为甲苯>苯>乙苯>对二甲苯,甲苯最容易被生物降解。苯、甲苯、乙苯、对二甲苯生物降解速率0.90~9.76 d−1,正戊烷、正己烷、正庚烷、正辛烷生物降解速率小于0.28 d−1,环戊烷、环己烷、环庚烷、环辛烷生物降解速率小于0.06 d−1。苯系物的表观一级衰减速率和生物降解速率都远高于正构烷烃和环烷烃,可能因为苯系物的溶解度远高于正构烷烃和环烷烃[22]。PASTERIS等[21]也报道过,柱实验数据拟合的甲苯的一级生物降解速率常数为3.2 d−1,而短链烷烃如戊烷和己烷、环烷烃和的生物降解速率较慢,估计为0.1~1.2 d−1。脂肪族烃缺乏官能团且水溶性极低,微生物对脂肪烃都表现出较低的化学反应性和生物利用度,烷烃的微生物降解的敏感性:正构烷烃>异构烷烃>环烷烃[20]。
3. 结论
1) 正构烷烃、环烷烃和苯系物蒸气在4种土壤中的自然衰减,整体自然衰减去除率黑土>黄土>红土>石英砂,黑土生物降解速率高于黄土,红土和石英砂的生物降解能力很低。
2) 苯系物的气相自然衰减速率和生物降解速率远高于正构烷烃和环烷烃。包气带中的气相生物降解是石油污染场地一种重要但被忽视的自然衰减机制,该过程对于挥发性石油烃蒸气入侵暴露以及污染物自然衰减均具有重要的影响。
3) 气相自然衰减和气相生物降解动力学数据可以为蒸气入侵定量风险评估和石油污染场地自然衰减速率定量评估模型提供关键输入参数。
-
图 1 正构烷烃在4种土壤中的质量浓度变化曲线
Figure 1. N-alkanes concentration attenuation curve in four soils
图 2 环烷烃在4种土壤中的质量浓度变化曲线
Figure 2. Cyclic alkanes concentration attenuation curve in four soils
图 3 苯系物在4种土壤中的质量浓度变化曲线
Figure 3. Monoaromatic hydrocarbons concentration attenuation curve in four soils
表 1 实验土壤的基本理化性质
Table 1. Basic physical-chemical characteristics of experimental soils
土壤类型 pH 孔隙度 含水率 有效扩散系数 (20 ℃)/ (正戊烷,m2·d−1) 密度/ (g·cm−1) 有机质质量分数/% 土壤质地 机械组成/% 砂粒 粉粒 黏粒 黄土 7.8 0.5 0.02 0.28 1.3 0.66 壤土 49.6 39.6 10.7 黑土 6.1 0.8 0.09 0.80 0.6 36.70 壤质砂土 74.6 19.6 5.8 红土 5.7 0.6 0.02 0.36 1.0 0.33 粘土 11.2 5.4 83.4 石英砂 7.6 0.4 0.01 0.19 1.5 0.32 砂土 97.4 1.8 0.9
下载: 导出CSV
表 2 石油烃在不同土壤中自然衰减去除率
Table 2. The remove rates of petroleum hydrocarbons in different soils
% 化合物 黄土 黑土 红土 石英砂 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 正戊烷 24.61 27.82 3.21 42.91 68.18 25.27 21.33 23.06 1.73 3.56 5.08 1.52 正己烷 35.35 45.64 10.29 64.67 87.24 22.57 32.89 36.05 3.16 2.18 3.03 0.86 正庚烷 51.17 74.77 23.61 74.62 93.20 18.58 40.25 45.29 5.04 0.61 1.39 0.78 正辛烷 63.14 78.43 15.28 78.12 96.55 18.44 49.37 46.77 2.60 5.63 7.87 2.24 环戊烷 13.30 33.68 20.38 20.90 43.04 22.14 5.29 6.29 1.00 5.58 9.62 4.05 环己烷 12.96 28.24 15.28 46.68 70.96 24.28 1.75 7.09 5.34 4.29 9.91 5.62 环庚烷 8.23 45.91 37.68 61.22 84.85 23.63 9.88 12.89 3.01 12.67 14.29 1.62 环辛烷 57.60 66.28 8.67 71.90 85.35 13.45 9.14 17.37 8.23 12.56 21.14 8.58 苯 69.62 81.55 11.93 77.66 92.32 14.66 64.88 75.69 10.81 27.15 27.51 0.36 甲苯 72.54 80.57 8.03 82.64 91.98 9.34 67.44 75.63 8.19 26.43 27.00 0.57 乙苯 72.31 79.12 6.81 80.98 85.64 4.66 53.64 56.86 3.23 19.38 19.88 0.50 对二甲苯 75.80 82.81 7.01 77.57 83.36 5.80 51.45 55.65 4.20 19.46 20.00 0.54 注:“*”表示生物降解贡献的去除率为非灭菌组与灭菌组去除率之差。
下载: 导出CSV
表 3 石油烃在不同土壤中的一级衰减速率常数
Table 3. The first order attenuation rate constants of petroleum hydrocarbons in different soils
d−1 化合物 黄土 黑土 红土 石英砂 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 正戊烷 0.03 0.04 0.01 0.03 0.06 0.03 0.02 0.02 — — — — 正己烷 0.03 0.06 0.03 0.05 0.18 0.13 0.016 0.02 0.004 — — — 正庚烷 0.05 0.18 0.13 0.26 0.54 0.28 0.02 0.05 0.03 — — — 正辛烷 0.06 0.15 0.09 0.02 0.12 0.10 0.03 0.05 0.02 — — — 环戊烷 0.01 0.02 0.01 0.04 0.07 0.03 0.01 0.01 — — — — 环己烷 0.01 0.03 0.02 0.20 0.26 0.06 0.01 0.01 — — — — 环庚烷 0.02 0.05 0.03 0.28 0.32 0.04 0.01 0.01 — — — — 环辛烷 0.07 0.09 0.02 0.33 0.39 0.06 0.01 0.01 — — — — 苯 25.68 27.84 2.16 28.24 235.48 7.24 24.24 25.78 1.54 — — — 甲苯 30.72 38.96 8.24 29.36 39.12 9.76 25.68 28.44 2.76 — — — 乙苯 15.6 17.08 1.48 324.98 30.82 5.84 11.76 12.72 0.96 — — — 对二甲苯 14.4 15.58 1.18 23.76 27.66 3.90 11.04 11.94 0.90 — — —
下载: 导出CSV
-
[1] UNNITHAN A, BEKELE D N, CHADALAVADA S, et al. Insights into vapour intrusion phenomena: Current outlook and preferential pathway scenario[J]. Science of the Total Environment, 2021, 796: 148885. doi: 10.1016/j.scitotenv.2021.148885 [2] FENG S J, ZHU Z W, CHEN H X, et al. Two-dimensional analytical solution for subsurface volatile organic compounds vapor diffusion from a point source in layered unsaturated zone[J]. Journal of Contaminant Hydrology, 2021, 243: 103916. doi: 10.1016/j.jconhyd.2021.103916 [3] 孙琳, 张敏, 郭彩娟, 等. 非水相液体污染场地源区自然消除研究进展[J]. 岩矿测试, 2022, 41(5): 704-716. doi: 10.15898/j.cnki.11-2131/td.202110110145 [4] MA J, MCHUGH T, BECKLEY L, et al. Vapor Intrusion Investigations and Decision-Making: A Critical Review[J]. Environmental Science & Technology, 2020, 54(12): 7050-69. [5] YAO Y, MAO F, XIAO Y, et al. Modeling capillary fringe effect on petroleum vapor intrusion from groundwater contamination[J]. Water Research, 2019, 150: 111-119. doi: 10.1016/j.watres.2018.11.038 [6] 龚亚炬, 宋云, 李培中, 等. 粉砂介质包气带中甲苯蒸气挥发的迁移转化[J]. 环境工程学报, 2018, 12(1): 206-212. doi: 10.12030/j.cjee.201703080 [7] 王梦杰, 范婷婷, 王祥, 等. 典型农药污染场地地下水中氯代脂肪烃自然衰减研究[J]. 环境科学学报, 2022, 42(6): 155-166. doi: 10.13671/j.hjkxxb.2021.0505 [8] SOOKHAK LARI K, DAVIS G B, RAYNER J L, et al. Natural source zone depletion of LNAPL: A critical review supporting modelling approaches[J]. Water Research, 2019, 157: 630-646. doi: 10.1016/j.watres.2019.04.001 [9] RIVETT M O, WEALTHALL G P, DEARDEN R A, et al. Review of unsaturated-zone transport and attenuation of volatile organic compound (VOC) plumes leached from shallow source zones[J]. Journal of Contaminant Hydrology, 2011, 123(3/4): 130-156. doi: 10.1016/j.jconhyd.2010.12.013 [10] LAHVIS M A, BAEHR A L, BAKER R J. Quantification of aerobic biodegradation and volatilization rates of gasoline hydrocarbons near the water table under natural attenuation conditions[J]. Water Resources Research, 1999, 35(3): 753-765. doi: 10.1029/1998WR900087 [11] HöHENER P, DAKHEL N, CHRISTOPHERSEN M, et al. Biodegradation of hydrocarbons vapors: Comparison of laboratory studies and field investigations in the vadose zone at the emplaced fuel source experiment, Airbase Værløse, Denmark[J]. Journal of Contaminant Hydrology, 2006, 88(3/4): 337-358. doi: 10.1016/j.jconhyd.2006.07.007 [12] GARG S, NEWELL C J, KULKARNI P R, et al. Overview of Natural Source Zone Depletion: Processes, Controlling Factors, and Composition Change[J]. Groundwater Monitoring & Remediation, 2017, 37(3): 62-81. [13] YAO Y, WANG Y, ZHONG Z, et al. Investigating the Role of Soil Texture in Vapor Intrusion from Groundwater Sources[J]. Journal Of Environmental Quality, 2017, 46(4): 776-784. doi: 10.2134/jeq2017.01.0011 [14] BEKELE D N, NAIDU R, CHADALAVADA S. Influence of soil properties on vapor-phase sorption of trichloroethylene[J]. Journal of Hazardous Materials, 2016, 306: 34-40. doi: 10.1016/j.jhazmat.2015.12.002 [15] BUSHNAF K M, PURICELLI S, SAPONARO S, et al. Effect of biochar on the fate of volatile petroleum hydrocarbons in an aerobic sandy soil[J]. Journal of Contaminant Hydrology, 2011, 126(3/4): 208-215. doi: 10.1016/j.jconhyd.2011.08.008 [16] HöHENER P, DUWIG C, PASTERIS G, et al. Biodegradation of petroleum hydrocarbon vapors: laboratory studies on rates and kinetics in unsaturated alluvial sand[J]. Journal of Contaminant Hydrology, 2003, 66(1/2): 93-115. doi: 10.1016/S0169-7722(03)00005-6 [17] UGWOHA E, ANDRESEN J M. Sorption and phase distribution of ethanol and butanol blended gasoline vapours in the vadose zone after release[J]. Journal of Environmental Sciences, 2014, 26(3): 608-616. doi: 10.1016/S1001-0742(13)60436-2 [18] ENGLISH C W, LOEHR R C. Degradation of organic vapors in unsaturated soils[J]. Journal of Hazardous Materials, 1991, 28(1): 55-64. [19] LEE E H, KIM J, CHO K S, et al. Degradation of hexane and other recalcitrant hydrocarbons by a novel isolate, Rhodococcus sp. EH831[J]. Environmental Science and Pollution Research, 2010, 17(1): 64-77. doi: 10.1007/s11356-009-0238-x [20] SINGH S N, KUMARI B, MISHRA S. Microbial Degradation of Alkanes[M]. Microbial Degradation of Xenobiotics. 2012: 439-469. [21] PASTERIS G, WERNER D, KAUFMANN K, et al. Vapor phase transport and biodegradation of volatile fuel compounds in the unsaturated zone: A large scale lysimeter experiment[J]. Environmental Science & Technology, 2002, 36(1): 30-39. [22] MENG Q, WANG X, WANG X, et al. Biodegradation of light hydrocarbon(C5-C8) in shale gases from the Triassic Yanchang Formation, Ordos basin, China[J]. Journal of Natural Gas Science and Engineering, 2018, 51: 183-194. doi: 10.1016/j.jngse.2018.01.002 [23] CHIKERE C B, OKPOKWASILI G C, CHIKERE B O. Monitoring of microbial hydrocarbon remediation in the soil[J]. 3 Biotech, 2011, 1(3): 117-138. doi: 10.1007/s13205-011-0014-8 [24] VARJANI S J. Microbial degradation of petroleum hydrocarbons [J]. Bioresource Technology, 2017, 223: 277-286. [25] EL-NAAS M H, ACIO J A, EL TELIB A E. Aerobic biodegradation of BTEX: Progresses and Prospects[J]. Journal of Environmental Chemical Engineering, 2014, 2(2): 1104-1122. doi: 10.1016/j.jece.2014.04.009 [26] ALVAREZ P J, VOGEL T M. Substrate interactions of benzene, toluene, and para-xylene during microbial degradation by pure cultures and mixed culture aquifer slurries[J]. Applied and environmental microbiology, 1991, 57(10): 2981-2985. doi: 10.1128/aem.57.10.2981-2985.1991 [27] DEEB R A, ALVAREZ-COHEN L. Temperature effects and substrate interactions during the aerobic biotransformation of BTEX mixtures by toluene-enriched consortia and Rhodococcus rhodochrous[J]. Biotechnology and Bioengineering, 1999, 62(5): 526-536. doi: 10.1002/(SICI)1097-0290(19990305)62:5<526::AID-BIT4>3.0.CO;2-8 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4484
- HTML全文浏览数: 4484
- PDF下载数: 155
- 施引文献: 0
