


-
石油烃 (PHC) 是目前环境中广泛存在的有机污染物,是多种烃类 (正烷烃、支链烷烃、环烷烃、芳烃) 和少量其他有机物的混合物[1]。PHC进入土壤后,不仅会破坏土壤结构,与无机氮、磷结合并限制硝化作用和脱磷酸作用,减少土壤中有效氮、磷等养分的含量,还会通过挥发或食物链富集等途径对人群健康构成潜在风险[2]。因此,PHC污染土壤的修复治理迫在眉睫[3]。化学氧化剂过硫酸盐 (PS) 因其在环境中存在时间长、适用pH范围广、氧化性强等优势被广泛应用于有机污染土壤修复领域。PS可以在水中分解产生强氧化剂S2O82−,经活化后会产生氧化性更强的SO4−·,能够在广泛的环境条件下转化各种环境污染物[4]。
有机污染物进入土壤后往往以“快”“慢”“极慢”等解吸组分形式存在[5],由于“快”解吸组分易被氧化,氧化后污染物往往以“慢”“极慢”等解吸组分形式存在[6],难以进一步去除,化学氧化末期往往会出现拖尾现象[7-8]。虽然投加过量药剂可以提高PHC降解效率,但也会增加修复成本。因此,化学氧化和其他修复技术的联合应用备受关注[9],如已有研究表明化学氧化与微生物联合处理能够更高效地去除土壤中的PHC[10-12]。由于缺氧条件下微生物对有机污染物的降解效率较低,目前与微生物联合降解有机污染物的研究多在好氧条件下进行[13],然而,通过降解参数优化 (如营养物质、电子受体等) ,微生物的缺氧降解效率会明显提升[14-16]。此外,深层有机污染土壤往往处于缺氧环境[17],因此探究缺氧条件下化学氧化与微生物联合修复PHC污染土壤具有重要意义。
由于PS氧化后土壤中残留的PHC以“慢”“极慢”等解吸组分形式存在,生物有效性更低,限制了其进一步生物降解[11]。加入表面活性剂可以增强土壤中PHC的解吸能力,提高其生物有效性[18-19]。常见的表面活性剂增效修复技术使用单一阴离子或非离子表面活性剂,单一阴离子表面活性剂会发生沉淀作用,非离子表面活性剂则易被土壤吸附,而混合表面活性剂体系既能减少表面活性剂在土壤表面的吸附,提高解吸效率,又能使得洗脱液中污染物浓度和表面活性剂浓度对微生物降解产生正效应,是一种更有效的修复方式[20]。但表面活性剂对PS氧化后土壤中微生物以及PHC缺氧生物降解的影响尚不清楚。因此,本研究将通过加入不同质量浓度和质量比的SDBS和Tween 80,探究其对PS氧化后土壤中PHC解吸、土著微生物群落结构和丰度、PHC缺氧降解的影响,为深层石油污染土壤的微生物修复提供参考。
-
实验所用土壤采自华北地区某加油站地下2~5 m处,在采样现场过2 mm筛,运回实验室用四分法混匀。部分土壤−20 ℃保存用于测定PHC质量分数和微生物指标,其余土壤4 ℃保存用于分析理化性质和后续实验处理。土壤样品理化性质:pH为7.12;含水率为8.13%;TOC为1.52%;总铁为2.17%;SO42−和NO3−质量分数分别为122.29和60.83 mg·kg−1;粒径分布为黏土15.70%,粉土84.13%,砂土0.17%。
实验所用阴离子表面活性剂SDBS为化学纯;非离子表面活性剂Tween 80、过硫酸钠 (Na2S2O8) 均为分析纯;正己烷 (C6H14) 、二氯甲烷 (CH2Cl2) 均为色谱纯;高纯氮气 (N2,99.999%) ;石油烃 (C10~C40) 标准溶液 (1 mg·L−1,美国AccuStandard公司) 。
-
称取15 g新鲜土壤样品置于50 mL血清瓶中,在水土比0.4∶1的条件下[5]加入6 mL PS溶液作为氧化处理组PS1 (使土壤中PS质量分数约为1%) ,将未经处理的土壤样品记为原土US,同时设置加入等量去离子水的空白对照组CK1,每个处理设置3组重复。将处理完成的样品在手套箱中完成缺氧处理后用带橡胶垫的铝盖密封[21],转移至恒温培养箱,在30 ℃下缺氧培养30 d (此时土样中的PS基本消耗殆尽[5]) 。同时设置灭菌对照组 (灭菌后原土组记为SS,氧化处理组记为PS2,空白对照组记为CK2) ,灭菌处理参考文献方法[5],使用高压蒸汽灭菌锅,灭菌温度为121 ℃,时间为30 min,灭菌重复3次。
使用经PS氧化的土样进行解吸实验,将样品转移至50 mL离心管,在水土比为1∶1的条件下分别加入不同质量浓度和质量比的SDBS和Tween 80溶液,同时设置空白对照组CK3,放入25 ℃恒温振荡器内150 r·min−1振荡36 h。根据不同质量浓度及类型的表面活性剂对多环芳烃的解吸效果研究可知[22],当部分表面活性剂的质量浓度达到800 mg·L−1时,便开始有解吸效果;质量浓度为3 000 mg·L−1时,解吸效果开始变得更加明显,但已经对土著微生物丰度产生不利影响。而阴-非离子混合表面活性剂对有机污染物的解吸效果强于单一表面活性剂[20],但混合体系质量比对PHC解吸效果的影响尚不清楚,因此解吸实验选用表面活性剂的质量浓度为800和3 000 mg·L−1,质量比为SDBS∶Tween 80=0∶1、1∶3、1∶2、1∶1、2∶1、3∶1和1∶0。另取一部分PS氧化后的土样,在水土比为1∶1的条件下向血清瓶中加入与上述质量浓度和质量比相同的表面活性剂溶液进行缺氧微生物降解实验,同时设置空白对照CK4,在手套箱中完成缺氧处理后密封血清瓶,转移至30 ℃恒温培养箱继续培养120 d。
-
土壤中PHC的提取采用超声萃取法[16],上清液中PHC的提取采用液液萃取法[23]。PHC检测仪器为气相色谱仪 (7890B,美国Agilent公司) ,色谱柱为HP-5MS (30 m×0.25 mm×0.25 μm) ,测定条件:进样口温度300 ℃,不分流进样,进样量1.0 μL;柱箱初始温度为50 ℃,保持2 min,以40 ℃·min−1升至230 ℃,再以20 ℃·min−1升至320 ℃,保持20 min;气体流量:氮气1.5 mL·min−1,氢气30 mL·min−1,空气300 mL·min−1。
-
土壤DNA采用MOBIO Power Soil DNA Isolation Kit试剂盒提取,细菌丰度的测定采用实时定量PCR扩增技术[24]。以16S rDNA作为靶基因对细菌丰度进行检测。细菌引物为338F (5'-ACTCCTACGGGAGGCAGCAG-3') 和806R (5'-GGACTACHVGGGTWTCTAAT-3') ,片段大小为460。反应条件为:95 ℃预变性3 min,95 ℃变性30 s,58 ℃退火30 s,72 ℃延伸1 min,35个循环。完成上述操作后,将待测样品放在荧光定量PCR仪中进行反应,实时定量PCR扩增效率为97.44%。
微生物群落结构分析测试参照FREY等的方法[25],使用Fastp软件对原始测序序列进行质控,使用Flash软件进行拼接 (最小重叠长度为10 bp,重叠区允许的最大错配比为0.2) 。使用UPARSE软件在97%的相似度对序列进行OTU聚类分析。使用RDP classifier分类器对每条序列进行物种分类注释,比对Silva 16S rRNA数据库进行物种注释分析 (比对阈值为80%) ,统计各样品的细菌群落组成。
-
本研究土样中PHC质量分数见表1。由于原土中C31~C40组分质量分数较低 (仅占C10~C40的2.59%) ,对此组分不做进一步讨论。本研究主要探讨碳链长度为C10~C16 (F1) 、C17~C23 (F2) 、C24~C30 (F3) 的组分,其在原土 (C10~C30)中的占比分别为29.83%、62.19%、7.98%,即本研究原土中石油烃污染物以F2组分为主,其次为F1组分。与原土相比,灭菌后的原土中F1组分的损失率最大 (80.47%) ,其次是F2组分 (37.85%) ,F3组分损失率最小 (1.85%) 。
-
由图1 (a) 可以看出,灭菌空白对照组 (CK2) 与灭菌后的原土 (SS) 中ΣPHC无明显差异 (p>0.05) ,但原土空白对照组 (CK1) 中ΣPHC相对于原土 (US) 有明显减少 (p<0.05) ,表明30 d缺氧培养期间原土中PHC发生了缺氧微生物降解,去除了15.06%的ΣPHC。与SS相比,灭菌土壤的氧化处理组 (PS2) 中ΣPHC明显减少 (p<0.05) ,在仅PS氧化降解作用下去除了20.86%的ΣPHC。与US相比,原土的氧化处理组 (PS1) 中ΣPHC明显减少 (p<0.05) ,在此过程中同时发生了PS氧化和缺氧微生物降解,二者联合作用去除了30.84%的ΣPHC。这表明缺氧条件下PS氧化和缺氧微生物降解均能去除土壤中的PHC,但2者联合作用的效果更加显著。ZHANG等[12]利用PS和微生物联合在好氧条件下去除土壤中PHC,其结果也表明联合修复效果优于单一处理效果。此外,从碳链长度角度来看,缺氧微生物降解对F1、F2、F3组分的去除率分别为30.24%、9.21%、3.88%,PS氧化对F1、F2、F3组分的去除率分别为34.83%、20.69%、11.30%,联合作用对F1、F2、F3组分的去除率分别为40.43%、29.21%、7.78% (图1 (b)~图1 (d) ) ,表明土壤中PHC的去除率与其碳链长度成反比,这在其他研究中也得到了相似的结果[10,26]。
-
表面活性剂对PHC解吸的影响实验以原土经PS氧化30 d后PHC质量分数为初始值。如图2 (a) 所示,空白对照组 (CK3) 上清液中ΣPHC质量浓度为5.47 mg·L−1。与CK3相比,800 mg·L−1表面活性剂处理组上清液中ΣPHC增加了25.11%~65.50%,3 000 mg·L−1表面活性剂处理组上清液中ΣPHC增加了45.04%~251.51%,高浓度表面活性剂解吸效果更强,2种处理均在SDBS∶Tween 80=1∶3时解吸效果最好 (分别为65.50%和251.51%) ,但在高浓度处理组中SDBS∶Tween 80=1∶3时解吸效果更显著 (p<0.05) 。表面活性剂对F1、F2、F3组分的解吸效果如图2 (b)~图2 (d) 所示,当表面活性剂质量比相同时,高浓度表面活性剂对各组分的解吸效果更强;而浓度相同时,混合体系对各组分解吸效果强于单一表面活性剂,且随着体系中Tween 80的比例增加而增加。陈巧超等[20]利用SDBS-Tween 80混合表面活性剂对土壤中的芘解吸时发现,Tween 80占比提高时,混合体系解吸能力相应提高,且质量比相同时高浓度混合表面活性剂解吸效果更好。
-
各处理土壤中的细菌丰度如图3所示。US中总细菌基因拷贝数对数值为7.16,PS1中对数值为5.66,细菌数量较US下降了1.50个数量级;空白对照组 (CK4) 中对数值为7.36,较US上升了0.20个数量级。这表明缺氧条件下加入PS对土壤中细菌造成了负面影响,但经过一段时间的培养后细菌数量得以恢复,在其他研究中也观察到了这种现象[27]。在本研究中,氧化后加入表面活性剂继续缺氧培养120 d的土壤与CK4相比,800 mg·L−1处理组土壤中除SDBS∶Tween 80=3∶1和1∶0之外 (较CK4分别增加了0.19和0.49个数量级) ,细菌数量明显增加 (p<0.05) ,增加了0.61~0.81个数量级;3 000 mg·L−1处理组土壤中细菌数量与CK4相比出现显著下降 (p<0.05) ,下降了1.98~2.46个数量级。这表明缺氧条件下低浓度表面活性剂对PS氧化后土壤中细菌生长起到了促进作用,而高浓度则表现出抑制作用。TIAN等[28]在好氧条件下使用表面活性剂对原油处理后发现,表面活性剂对微生物的毒性会随着其剂量的增加而增加,在高浓度下抑制微生物增殖,而在低浓度时有促进增殖的作用。
如图4所示,US中优势菌种主要为放线菌门 (Actinobacteria) 、变形菌门 (Proteobacteria) 和厚壁菌门 (Firmicutes) 。PS1中3种细菌的相对丰度与US相比均有所下降。已有研究表明,PHC降解菌主要属于Proteobacteria和Firmicutes等[29]。GRAY等[30]探究了厌氧条件下PHC的降解机制,调查表明96%的可用细菌序列与15个不同的门有关,且以Firmicutes和所有类别的Proteobacteria为主。US中Firmicutes和Proteobacteria的基因拷贝数对数值为6.87,在CK4中,Firmicutes和Proteobacteria的对数值为7.27,与US相比增加了0.40个数量级。氧化后土壤中加入表面活性剂继续培养120 d,800 mg·L−1处理组土壤中除SDBS∶Tween 80=3∶1和1∶0之外 (较CK4分别增加了0.17和0.49个数量级) ,Firmicutes和Proteobacteria的总数量较CK4增加了0.61~0.81个数量级,土壤中PHC降解菌数量明显增加 (p<0.05) 。而3 000 mg·L−1时土壤中Firmicutes和Proteobacteria的总数量较CK4下降了2.13~2.58个数量级,土壤中PHC降解菌的数量显著减少 (p<0.05) 。
-
表面活性剂对PS氧化后土壤中PHC缺氧微生物降解的影响实验以原土经PS氧化30 d后PHC质量分数为初始值。向氧化后土壤中加入表面活性剂继续缺氧培养120 d后,如图5 (a) 所示,CK4中ΣPHC残留率为69.71%。3 000 mg·L−1表面活性剂处理组土壤中,除SDBS∶Tween 80=0∶1和1∶3外,其余组别ΣPHC的残留率均高于CK4 (增加了1.82%~11.08%) 。虽然高浓度表面活性剂对土壤中PHC解吸能力更强 (图2) ,但显著降低了土壤中细菌的丰度,抑制了PHC降解菌的数量 (较CK4下降了2.13~2.58个数量级,图3) ,因此加入3 000 mg·L−1表面活性剂抑制了PS氧化后土壤中PHC的缺氧微生物降解。
张秀霞等[31]考察了好氧条件下SDBS添加量对微生物修复石油污染土壤效果的影响,结果表明土壤中PHC的降解率会随着表面活性剂质量浓度的增加而降低,修复体系中残留高浓度的SDBS对微生物修复石油污染土壤具有抑制作用,而低浓度的SDBS有促进作用。在本研究中,缺氧条件下800 mg·L−1表面活性剂处理组土壤中ΣPHC的残留率与CK4相比出现明显降低 (p<0.05) ,下降了5.93%~15.80%,在SDBS∶Tween 80=1∶3时残留率最低 (53.91%) 。低浓度表面活性剂促进了PS氧化后土壤中ΣPHC的进一步降解,可能是因为其不仅促进了土壤中ΣPHC的解吸 (如图2,在SDBS∶Tween 80=1∶3时解吸效果最好) ,同时还提高了土壤中的细菌丰度,尤其是对PHC降解起重要作用的Firmicutes和Proteobacteria的数量 (较CK4增加了0.17~0.81个数量级,图3) 。
短链石油烃的分子量小、疏水性弱,容易被生物降解,而长链石油烃因为疏水性强,生物有效性较低[32-33],在土壤中的残留率更高。如图5 (b)~图5 (d) 所示,向PS氧化后的土壤中加入800 mg·L−1表面活性剂继续培养120 d后,F1、F2、F3组分的残留率均低于对照组CK4,对氧化后土壤中残留的中短碳链PHC的强化降解效果更强,这与表面活性剂对土壤中PHC的解吸效果相符。
-
1) 缺氧条件下PS氧化和缺氧微生物降解联合作用对土壤中的PHC有较好的处理效果,去除了30.84%的ΣPHC (C10~C30) 。
2) 表面活性剂SDBS和Tween 80混合体系对土壤中PHC的解吸效果强于单一表面活性剂,解吸效果随着表面活性剂浓度和混合体系中Tween 80比例的增加而增加。
3) 缺氧条件下向PS氧化后土壤中加入低浓度表面活性剂不仅促进了土壤中PHC的解吸,对微生物生长也起到了促进作用,除SDBS∶Tween 80=3∶1和1∶0之外 (较对照组分别增加了0.17和0.49个数量级) ,使土壤中Firmicutes和Proteobacteria的总数量较对照组明显增加了0.61~0.81个数量级,进而降低了土壤中PHC的残留率。降解效果的提升和碳链长度成反比,且随着混合体系中Tween 80的比例增加而增加,在SDBS∶Tween 80=1∶3时残留率最低 (53.91%) 。
4) 尽管高浓度表面活性剂对土壤中PHC的解吸效果更强,但处理后土壤中PHC降解菌总数量较对照组下降了2.13~2.58个数量级,抑制了PHC的缺氧微生物降解。
表面活性剂SDBS和Tween 80强化过硫酸盐氧化后土壤中石油烃的缺氧生物降解
Surfactants SDBS and Tween 80 enhance anoxic biodegradation of petroleum hydrocarbons (PHC) in soil pretreated with persulfate
-
摘要: 化学氧化和微生物联合修复是去除土壤中石油烃 (PHC) 的有效技术,但氧化后土壤中残留PHC的生物有效性较低,难以进一步生物降解。向过硫酸盐 (PS) 氧化后的土壤中加入不同质量浓度和质量比的表面活性剂十二烷基苯磺酸钠 (SDBS) 和聚氧乙烯山梨醇酐单油酸酯 (Tween 80) ,探究其对PS氧化后土壤中PHC解吸、土著微生物群落结构和丰度、PHC缺氧降解的影响。结果表明,缺氧条件下PS氧化和微生物联合降解去除了土壤中30.84%的ΣPHC (C10~C30) 。向PS氧化后土壤中加入SDBS和Tween 80能够有效促进PHC解吸,解吸效果随表面活性剂质量浓度和混合体系中Tween 80比例的增加而增加。加入3 000 mg·L−1表面活性剂继续缺氧培养120 d后,氧化后土壤中Firmicutes和Proteobacteria的总数量较对照组减少了2.13~2.58个数量级,抑制了土壤中PHC的缺氧降解。加入800 mg·L−1表面活性剂后,土壤中Firmicutes和Proteobacteria的总数量较对照组增加了0.17~0.81个数量级,促进了PHC的缺氧降解,在SDBS∶Tween 80=1∶3时ΣPHC残留率最低 (较对照组降低了15.80%) 。本研究结果可为深层石油污染土壤的微生物修复提供参考。Abstract: Chemical oxidation combined with biodegradation is an effective technology for removing petroleum hydrocarbons (PHC) from soil. However, the residual PHC in soil after oxidation is difficult to be further biodegraded due to their low bioavailability. The surfactants of sodium dodecyl benzene sulfonate (SDBS) and polyoxyethylene sorbitan monooleate (Tween 80) with different doses and mass ratios were added to the soil pretreated with persulfate (PS), to investigate their effect on PHC desorption, indigenous microbial community composition and abundance, PHC anoxic biodegradation. The results showed that 30.84% of ΣPHC(C10~C30) in soil were removed by PS oxidation combined with anoxic biodegradation. SDBS and Tween 80 were added to the soil pretreated with PS had effectively promoted the desorption of PHC, the effect increased with the increase of surfactant’s concentration and the proportion of Tween 80 in the mixed system. After 120 d of continued anoxic incubation, the total amount of Firmicutes and Proteobacteria in soil added surfactants with the concentration of 3 000 mg·L−1 decreased by 2.13~2.58 orders of magnitude compared with the control treatment, which inhibited the anoxic biodegradation of PHC. The total amount of Firmicutes and Proteobacteria in soil added surfactants with the concentration of 800 mg·L−1 increased by 0.17~0.81 orders of magnitude compared with the control treatment, which promoted the anoxic biodegradation of PHC, the residual rate of ΣPHC was lowest at SDBS:Tween 80=1:3 (15.80% lower than the control group). This study can provide a reference for the microbial remediation of subsurface soil contaminated with PHC.
-
Key words:
- surfactants /
- petroleum hydrocarbons /
- anoxic biodegradation /
- chemical oxidation
-
石油烃 (PHC) 是目前环境中广泛存在的有机污染物,是多种烃类 (正烷烃、支链烷烃、环烷烃、芳烃) 和少量其他有机物的混合物[1]。PHC进入土壤后,不仅会破坏土壤结构,与无机氮、磷结合并限制硝化作用和脱磷酸作用,减少土壤中有效氮、磷等养分的含量,还会通过挥发或食物链富集等途径对人群健康构成潜在风险[2]。因此,PHC污染土壤的修复治理迫在眉睫[3]。化学氧化剂过硫酸盐 (PS) 因其在环境中存在时间长、适用pH范围广、氧化性强等优势被广泛应用于有机污染土壤修复领域。PS可以在水中分解产生强氧化剂S2O82−,经活化后会产生氧化性更强的SO4−·,能够在广泛的环境条件下转化各种环境污染物[4]。
有机污染物进入土壤后往往以“快”“慢”“极慢”等解吸组分形式存在[5],由于“快”解吸组分易被氧化,氧化后污染物往往以“慢”“极慢”等解吸组分形式存在[6],难以进一步去除,化学氧化末期往往会出现拖尾现象[7-8]。虽然投加过量药剂可以提高PHC降解效率,但也会增加修复成本。因此,化学氧化和其他修复技术的联合应用备受关注[9],如已有研究表明化学氧化与微生物联合处理能够更高效地去除土壤中的PHC[10-12]。由于缺氧条件下微生物对有机污染物的降解效率较低,目前与微生物联合降解有机污染物的研究多在好氧条件下进行[13],然而,通过降解参数优化 (如营养物质、电子受体等) ,微生物的缺氧降解效率会明显提升[14-16]。此外,深层有机污染土壤往往处于缺氧环境[17],因此探究缺氧条件下化学氧化与微生物联合修复PHC污染土壤具有重要意义。
由于PS氧化后土壤中残留的PHC以“慢”“极慢”等解吸组分形式存在,生物有效性更低,限制了其进一步生物降解[11]。加入表面活性剂可以增强土壤中PHC的解吸能力,提高其生物有效性[18-19]。常见的表面活性剂增效修复技术使用单一阴离子或非离子表面活性剂,单一阴离子表面活性剂会发生沉淀作用,非离子表面活性剂则易被土壤吸附,而混合表面活性剂体系既能减少表面活性剂在土壤表面的吸附,提高解吸效率,又能使得洗脱液中污染物浓度和表面活性剂浓度对微生物降解产生正效应,是一种更有效的修复方式[20]。但表面活性剂对PS氧化后土壤中微生物以及PHC缺氧生物降解的影响尚不清楚。因此,本研究将通过加入不同质量浓度和质量比的SDBS和Tween 80,探究其对PS氧化后土壤中PHC解吸、土著微生物群落结构和丰度、PHC缺氧降解的影响,为深层石油污染土壤的微生物修复提供参考。
1. 材料与方法
1.1 实验材料
实验所用土壤采自华北地区某加油站地下2~5 m处,在采样现场过2 mm筛,运回实验室用四分法混匀。部分土壤−20 ℃保存用于测定PHC质量分数和微生物指标,其余土壤4 ℃保存用于分析理化性质和后续实验处理。土壤样品理化性质:pH为7.12;含水率为8.13%;TOC为1.52%;总铁为2.17%;SO42−和NO3−质量分数分别为122.29和60.83 mg·kg−1;粒径分布为黏土15.70%,粉土84.13%,砂土0.17%。
实验所用阴离子表面活性剂SDBS为化学纯;非离子表面活性剂Tween 80、过硫酸钠 (Na2S2O8) 均为分析纯;正己烷 (C6H14) 、二氯甲烷 (CH2Cl2) 均为色谱纯;高纯氮气 (N2,99.999%) ;石油烃 (C10~C40) 标准溶液 (1 mg·L−1,美国AccuStandard公司) 。
1.2 实验方法
称取15 g新鲜土壤样品置于50 mL血清瓶中,在水土比0.4∶1的条件下[5]加入6 mL PS溶液作为氧化处理组PS1 (使土壤中PS质量分数约为1%) ,将未经处理的土壤样品记为原土US,同时设置加入等量去离子水的空白对照组CK1,每个处理设置3组重复。将处理完成的样品在手套箱中完成缺氧处理后用带橡胶垫的铝盖密封[21],转移至恒温培养箱,在30 ℃下缺氧培养30 d (此时土样中的PS基本消耗殆尽[5]) 。同时设置灭菌对照组 (灭菌后原土组记为SS,氧化处理组记为PS2,空白对照组记为CK2) ,灭菌处理参考文献方法[5],使用高压蒸汽灭菌锅,灭菌温度为121 ℃,时间为30 min,灭菌重复3次。
使用经PS氧化的土样进行解吸实验,将样品转移至50 mL离心管,在水土比为1∶1的条件下分别加入不同质量浓度和质量比的SDBS和Tween 80溶液,同时设置空白对照组CK3,放入25 ℃恒温振荡器内150 r·min−1振荡36 h。根据不同质量浓度及类型的表面活性剂对多环芳烃的解吸效果研究可知[22],当部分表面活性剂的质量浓度达到800 mg·L−1时,便开始有解吸效果;质量浓度为3 000 mg·L−1时,解吸效果开始变得更加明显,但已经对土著微生物丰度产生不利影响。而阴-非离子混合表面活性剂对有机污染物的解吸效果强于单一表面活性剂[20],但混合体系质量比对PHC解吸效果的影响尚不清楚,因此解吸实验选用表面活性剂的质量浓度为800和3 000 mg·L−1,质量比为SDBS∶Tween 80=0∶1、1∶3、1∶2、1∶1、2∶1、3∶1和1∶0。另取一部分PS氧化后的土样,在水土比为1∶1的条件下向血清瓶中加入与上述质量浓度和质量比相同的表面活性剂溶液进行缺氧微生物降解实验,同时设置空白对照CK4,在手套箱中完成缺氧处理后密封血清瓶,转移至30 ℃恒温培养箱继续培养120 d。
1.3 PHC的提取与测定
土壤中PHC的提取采用超声萃取法[16],上清液中PHC的提取采用液液萃取法[23]。PHC检测仪器为气相色谱仪 (7890B,美国Agilent公司) ,色谱柱为HP-5MS (30 m×0.25 mm×0.25 μm) ,测定条件:进样口温度300 ℃,不分流进样,进样量1.0 μL;柱箱初始温度为50 ℃,保持2 min,以40 ℃·min−1升至230 ℃,再以20 ℃·min−1升至320 ℃,保持20 min;气体流量:氮气1.5 mL·min−1,氢气30 mL·min−1,空气300 mL·min−1。
1.4 分析方法
土壤DNA采用MOBIO Power Soil DNA Isolation Kit试剂盒提取,细菌丰度的测定采用实时定量PCR扩增技术[24]。以16S rDNA作为靶基因对细菌丰度进行检测。细菌引物为338F (5'-ACTCCTACGGGAGGCAGCAG-3') 和806R (5'-GGACTACHVGGGTWTCTAAT-3') ,片段大小为460。反应条件为:95 ℃预变性3 min,95 ℃变性30 s,58 ℃退火30 s,72 ℃延伸1 min,35个循环。完成上述操作后,将待测样品放在荧光定量PCR仪中进行反应,实时定量PCR扩增效率为97.44%。
微生物群落结构分析测试参照FREY等的方法[25],使用Fastp软件对原始测序序列进行质控,使用Flash软件进行拼接 (最小重叠长度为10 bp,重叠区允许的最大错配比为0.2) 。使用UPARSE软件在97%的相似度对序列进行OTU聚类分析。使用RDP classifier分类器对每条序列进行物种分类注释,比对Silva 16S rRNA数据库进行物种注释分析 (比对阈值为80%) ,统计各样品的细菌群落组成。
2. 结果与讨论
本研究土样中PHC质量分数见表1。由于原土中C31~C40组分质量分数较低 (仅占C10~C40的2.59%) ,对此组分不做进一步讨论。本研究主要探讨碳链长度为C10~C16 (F1) 、C17~C23 (F2) 、C24~C30 (F3) 的组分,其在原土 (C10~C30)中的占比分别为29.83%、62.19%、7.98%,即本研究原土中石油烃污染物以F2组分为主,其次为F1组分。与原土相比,灭菌后的原土中F1组分的损失率最大 (80.47%) ,其次是F2组分 (37.85%) ,F3组分损失率最小 (1.85%) 。
表 1 土壤中PHC质量分数Table 1. Mass fraction of PHC in soilPHC组分 组名 原土PHC质量分数/mg·kg−1 原土灭菌后PHC质量分数/mg·kg−1 原土经PS氧化30 d后PHC质量分数/mg·kg−1 灭菌土经PS氧化30 d后PHC质量分数/mg·kg−1 C10~C16 F1 251.64±3.94 49.14±7.72 149.90±22.45 32.03±1.17 C17~C23 F2 524.74±34.97 326.13±33.77 371.47±41.07 258.65±3.89 C24~C30 F3 67.37±0.21 66.12±0.74 62.14±3.95 58.65±0.28 C31~C40 — 22.41±6.47 20.66±5.38 — — ΣPHC(C10~C30) — 843.75±30.82 441.39±26.79 583.51±59.89 349.33±5.34 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.1 PS氧化和缺氧微生物降解对PHC去除的影响
由图1 (a) 可以看出,灭菌空白对照组 (CK2) 与灭菌后的原土 (SS) 中ΣPHC无明显差异 (p>0.05) ,但原土空白对照组 (CK1) 中ΣPHC相对于原土 (US) 有明显减少 (p<0.05) ,表明30 d缺氧培养期间原土中PHC发生了缺氧微生物降解,去除了15.06%的ΣPHC。与SS相比,灭菌土壤的氧化处理组 (PS2) 中ΣPHC明显减少 (p<0.05) ,在仅PS氧化降解作用下去除了20.86%的ΣPHC。与US相比,原土的氧化处理组 (PS1) 中ΣPHC明显减少 (p<0.05) ,在此过程中同时发生了PS氧化和缺氧微生物降解,二者联合作用去除了30.84%的ΣPHC。这表明缺氧条件下PS氧化和缺氧微生物降解均能去除土壤中的PHC,但2者联合作用的效果更加显著。ZHANG等[12]利用PS和微生物联合在好氧条件下去除土壤中PHC,其结果也表明联合修复效果优于单一处理效果。此外,从碳链长度角度来看,缺氧微生物降解对F1、F2、F3组分的去除率分别为30.24%、9.21%、3.88%,PS氧化对F1、F2、F3组分的去除率分别为34.83%、20.69%、11.30%,联合作用对F1、F2、F3组分的去除率分别为40.43%、29.21%、7.78% (图1 (b)~图1 (d) ) ,表明土壤中PHC的去除率与其碳链长度成反比,这在其他研究中也得到了相似的结果[10,26]。
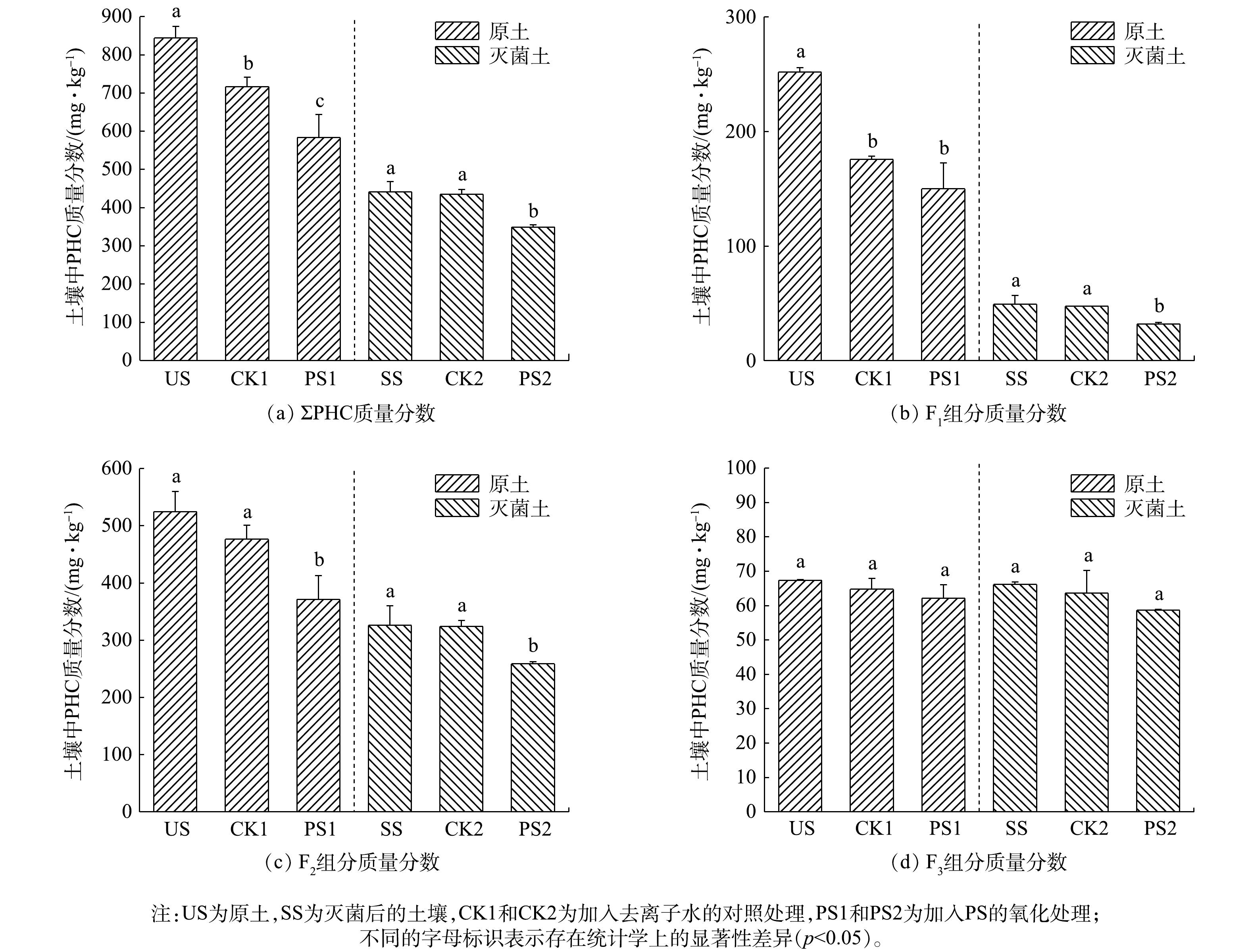 图 1 缺氧条件下PS氧化和微生物降解对PHC去除的影响Figure 1. Effect of PS oxidation and biodegradation on removal of PHC under anoxic condition
图 1 缺氧条件下PS氧化和微生物降解对PHC去除的影响Figure 1. Effect of PS oxidation and biodegradation on removal of PHC under anoxic condition2.2 表面活性剂对PHC解吸的影响
表面活性剂对PHC解吸的影响实验以原土经PS氧化30 d后PHC质量分数为初始值。如图2 (a) 所示,空白对照组 (CK3) 上清液中ΣPHC质量浓度为5.47 mg·L−1。与CK3相比,800 mg·L−1表面活性剂处理组上清液中ΣPHC增加了25.11%~65.50%,3 000 mg·L−1表面活性剂处理组上清液中ΣPHC增加了45.04%~251.51%,高浓度表面活性剂解吸效果更强,2种处理均在SDBS∶Tween 80=1∶3时解吸效果最好 (分别为65.50%和251.51%) ,但在高浓度处理组中SDBS∶Tween 80=1∶3时解吸效果更显著 (p<0.05) 。表面活性剂对F1、F2、F3组分的解吸效果如图2 (b)~图2 (d) 所示,当表面活性剂质量比相同时,高浓度表面活性剂对各组分的解吸效果更强;而浓度相同时,混合体系对各组分解吸效果强于单一表面活性剂,且随着体系中Tween 80的比例增加而增加。陈巧超等[20]利用SDBS-Tween 80混合表面活性剂对土壤中的芘解吸时发现,Tween 80占比提高时,混合体系解吸能力相应提高,且质量比相同时高浓度混合表面活性剂解吸效果更好。
 图 2 表面活性剂对PS氧化后土壤中PHC解吸效果的影响Figure 2. Effect of surfactants on desorption of PHC in soil pretreated with PS
图 2 表面活性剂对PS氧化后土壤中PHC解吸效果的影响Figure 2. Effect of surfactants on desorption of PHC in soil pretreated with PS2.3 表面活性剂对细菌丰度及群落结构的影响
各处理土壤中的细菌丰度如图3所示。US中总细菌基因拷贝数对数值为7.16,PS1中对数值为5.66,细菌数量较US下降了1.50个数量级;空白对照组 (CK4) 中对数值为7.36,较US上升了0.20个数量级。这表明缺氧条件下加入PS对土壤中细菌造成了负面影响,但经过一段时间的培养后细菌数量得以恢复,在其他研究中也观察到了这种现象[27]。在本研究中,氧化后加入表面活性剂继续缺氧培养120 d的土壤与CK4相比,800 mg·L−1处理组土壤中除SDBS∶Tween 80=3∶1和1∶0之外 (较CK4分别增加了0.19和0.49个数量级) ,细菌数量明显增加 (p<0.05) ,增加了0.61~0.81个数量级;3 000 mg·L−1处理组土壤中细菌数量与CK4相比出现显著下降 (p<0.05) ,下降了1.98~2.46个数量级。这表明缺氧条件下低浓度表面活性剂对PS氧化后土壤中细菌生长起到了促进作用,而高浓度则表现出抑制作用。TIAN等[28]在好氧条件下使用表面活性剂对原油处理后发现,表面活性剂对微生物的毒性会随着其剂量的增加而增加,在高浓度下抑制微生物增殖,而在低浓度时有促进增殖的作用。
 图 3 表面活性剂对PS氧化后土壤中细菌丰度的影响Figure 3. Effect of surfactants on bacterial abundance in soil pretreated with PS
图 3 表面活性剂对PS氧化后土壤中细菌丰度的影响Figure 3. Effect of surfactants on bacterial abundance in soil pretreated with PS如图4所示,US中优势菌种主要为放线菌门 (Actinobacteria) 、变形菌门 (Proteobacteria) 和厚壁菌门 (Firmicutes) 。PS1中3种细菌的相对丰度与US相比均有所下降。已有研究表明,PHC降解菌主要属于Proteobacteria和Firmicutes等[29]。GRAY等[30]探究了厌氧条件下PHC的降解机制,调查表明96%的可用细菌序列与15个不同的门有关,且以Firmicutes和所有类别的Proteobacteria为主。US中Firmicutes和Proteobacteria的基因拷贝数对数值为6.87,在CK4中,Firmicutes和Proteobacteria的对数值为7.27,与US相比增加了0.40个数量级。氧化后土壤中加入表面活性剂继续培养120 d,800 mg·L−1处理组土壤中除SDBS∶Tween 80=3∶1和1∶0之外 (较CK4分别增加了0.17和0.49个数量级) ,Firmicutes和Proteobacteria的总数量较CK4增加了0.61~0.81个数量级,土壤中PHC降解菌数量明显增加 (p<0.05) 。而3 000 mg·L−1时土壤中Firmicutes和Proteobacteria的总数量较CK4下降了2.13~2.58个数量级,土壤中PHC降解菌的数量显著减少 (p<0.05) 。
 图 4 表面活性剂对PS氧化后土壤中细菌群落组成的影响 (门水平)Figure 4. Effect of surfactants on bacterial community composition in soil pretreated with PS (phylum levels)
图 4 表面活性剂对PS氧化后土壤中细菌群落组成的影响 (门水平)Figure 4. Effect of surfactants on bacterial community composition in soil pretreated with PS (phylum levels)2.4 表面活性剂对PS氧化后土壤中PHC缺氧微生物降解的影响
表面活性剂对PS氧化后土壤中PHC缺氧微生物降解的影响实验以原土经PS氧化30 d后PHC质量分数为初始值。向氧化后土壤中加入表面活性剂继续缺氧培养120 d后,如图5 (a) 所示,CK4中ΣPHC残留率为69.71%。3 000 mg·L−1表面活性剂处理组土壤中,除SDBS∶Tween 80=0∶1和1∶3外,其余组别ΣPHC的残留率均高于CK4 (增加了1.82%~11.08%) 。虽然高浓度表面活性剂对土壤中PHC解吸能力更强 (图2) ,但显著降低了土壤中细菌的丰度,抑制了PHC降解菌的数量 (较CK4下降了2.13~2.58个数量级,图3) ,因此加入3 000 mg·L−1表面活性剂抑制了PS氧化后土壤中PHC的缺氧微生物降解。
 图 5 表面活性剂对PS氧化后土壤中石油烃缺氧生物降解的影响Figure 5. Effect of surfactants on anoxic biodegradation of PHC in soil pretreated with PS
图 5 表面活性剂对PS氧化后土壤中石油烃缺氧生物降解的影响Figure 5. Effect of surfactants on anoxic biodegradation of PHC in soil pretreated with PS张秀霞等[31]考察了好氧条件下SDBS添加量对微生物修复石油污染土壤效果的影响,结果表明土壤中PHC的降解率会随着表面活性剂质量浓度的增加而降低,修复体系中残留高浓度的SDBS对微生物修复石油污染土壤具有抑制作用,而低浓度的SDBS有促进作用。在本研究中,缺氧条件下800 mg·L−1表面活性剂处理组土壤中ΣPHC的残留率与CK4相比出现明显降低 (p<0.05) ,下降了5.93%~15.80%,在SDBS∶Tween 80=1∶3时残留率最低 (53.91%) 。低浓度表面活性剂促进了PS氧化后土壤中ΣPHC的进一步降解,可能是因为其不仅促进了土壤中ΣPHC的解吸 (如图2,在SDBS∶Tween 80=1∶3时解吸效果最好) ,同时还提高了土壤中的细菌丰度,尤其是对PHC降解起重要作用的Firmicutes和Proteobacteria的数量 (较CK4增加了0.17~0.81个数量级,图3) 。
短链石油烃的分子量小、疏水性弱,容易被生物降解,而长链石油烃因为疏水性强,生物有效性较低[32-33],在土壤中的残留率更高。如图5 (b)~图5 (d) 所示,向PS氧化后的土壤中加入800 mg·L−1表面活性剂继续培养120 d后,F1、F2、F3组分的残留率均低于对照组CK4,对氧化后土壤中残留的中短碳链PHC的强化降解效果更强,这与表面活性剂对土壤中PHC的解吸效果相符。
3. 结论
1) 缺氧条件下PS氧化和缺氧微生物降解联合作用对土壤中的PHC有较好的处理效果,去除了30.84%的ΣPHC (C10~C30) 。
2) 表面活性剂SDBS和Tween 80混合体系对土壤中PHC的解吸效果强于单一表面活性剂,解吸效果随着表面活性剂浓度和混合体系中Tween 80比例的增加而增加。
3) 缺氧条件下向PS氧化后土壤中加入低浓度表面活性剂不仅促进了土壤中PHC的解吸,对微生物生长也起到了促进作用,除SDBS∶Tween 80=3∶1和1∶0之外 (较对照组分别增加了0.17和0.49个数量级) ,使土壤中Firmicutes和Proteobacteria的总数量较对照组明显增加了0.61~0.81个数量级,进而降低了土壤中PHC的残留率。降解效果的提升和碳链长度成反比,且随着混合体系中Tween 80的比例增加而增加,在SDBS∶Tween 80=1∶3时残留率最低 (53.91%) 。
4) 尽管高浓度表面活性剂对土壤中PHC的解吸效果更强,但处理后土壤中PHC降解菌总数量较对照组下降了2.13~2.58个数量级,抑制了PHC的缺氧微生物降解。
-
图 1 缺氧条件下PS氧化和微生物降解对PHC去除的影响
Figure 1. Effect of PS oxidation and biodegradation on removal of PHC under anoxic condition
图 2 表面活性剂对PS氧化后土壤中PHC解吸效果的影响
Figure 2. Effect of surfactants on desorption of PHC in soil pretreated with PS
图 3 表面活性剂对PS氧化后土壤中细菌丰度的影响
Figure 3. Effect of surfactants on bacterial abundance in soil pretreated with PS
图 4 表面活性剂对PS氧化后土壤中细菌群落组成的影响 (门水平)
Figure 4. Effect of surfactants on bacterial community composition in soil pretreated with PS (phylum levels)
图 5 表面活性剂对PS氧化后土壤中石油烃缺氧生物降解的影响
Figure 5. Effect of surfactants on anoxic biodegradation of PHC in soil pretreated with PS
表 1 土壤中PHC质量分数
Table 1. Mass fraction of PHC in soil
PHC组分 组名 原土PHC质量分数/mg·kg−1 原土灭菌后PHC质量分数/mg·kg−1 原土经PS氧化30 d后PHC质量分数/mg·kg−1 灭菌土经PS氧化30 d后PHC质量分数/mg·kg−1 C10~C16 F1 251.64±3.94 49.14±7.72 149.90±22.45 32.03±1.17 C17~C23 F2 524.74±34.97 326.13±33.77 371.47±41.07 258.65±3.89 C24~C30 F3 67.37±0.21 66.12±0.74 62.14±3.95 58.65±0.28 C31~C40 — 22.41±6.47 20.66±5.38 — — ΣPHC(C10~C30) — 843.75±30.82 441.39±26.79 583.51±59.89 349.33±5.34
下载: 导出CSV
-
[1] 刘自力, 王红旗, 孔德康, 等. 不同植物-微生物联合修复体系下石油烃的降解[J]. 环境工程学报, 2018, 12(1): 190-197. doi: 10.12030/j.cjee.201704016 [2] 周启星, 展海银. 石油及石化污染场地生物修复技术进展与展望[J]. 应用技术学报, 2021, 21(4): 285-292. [3] 李晓晶, 赵倩, 张月勇, 等. 微生物燃料电池修复石油污染盐碱土壤[J]. 环境工程学报, 2017, 11(2): 1185-1191. doi: 10.12030/j.cjee.201509031 [4] TSITONAKI A, PETRI B, CRIMI M, et al. In situ chemical oxidation of contaminated soil and groundwater using persulfate: A review[J]. Critical Reviews in Environmental Science and Technology, 2010, 40(1): 55-91. doi: 10.1080/10643380802039303 [5] GOU Y L, ZHAO Q Y, YANG S C, et al. Enhanced degradation of polycyclic aromatic hydrocarbons in aged subsurface soil using integrated persulfate oxidation and anoxic biodegradation[J]. Chemical Engineering Journal, 2020, 394: 125040. doi: 10.1016/j.cej.2020.125040 [6] HATZINGER P B, ALEXANDER M. Effect of aging of chemicals in soil on their biodegradability and extractability[J]. Environmental Science & Technology, 1995, 29(2): 537-545. [7] CASSIDY D, NORTHUP A, HAMPTON D. The effect of three chemical oxidants on subsequent biodegradation of 2, 4-dinitrotoluene (DNT) in batch slurry reactors[J]. Journal of Chemical Technology and Biotechnology, 2009, 84(6): 820-826. doi: 10.1002/jctb.2140 [8] CHEN K F, CHANG Y C, CHIOU W T. Remediation of diesel-contaminated soil using in situ chemical oxidation (ISCO) and the effects of common oxidants on the indigenous microbial community: a comparison study[J]. Journal of Chemical Technology and Biotechnology, 2016, 91(6): 1877-1888. doi: 10.1002/jctb.4781 [9] SAHL J, MUNAKATA-MARR J. The effects of in situ chemical oxidation on microbiological processes: A review[J]. Remediation Journal:The Journal of Environmental Cleanup Costs, Technologies & Techniques, 2006, 16(3): 57-70. [10] LI Y T, LI D, LAI L J, et al. Remediation of petroleum hydrocarbon contaminated soil by using activated persulfate with ultrasound and ultrasound/Fe[J]. Chemosphere, 2020, 238: 124657. doi: 10.1016/j.chemosphere.2019.124657 [11] LOMINCHAR M A, SANTOS A, MIGUEL E, et al. Remediation of aged diesel contaminated soil by alkaline activated persulfate[J]. Science of the Total Environment, 2018, 622-623: 41-48. doi: 10.1016/j.scitotenv.2017.11.263 [12] ZHANG B W, GUO Y, HUO J Y, et al. Combining chemical oxidation and bioremediation for petroleum polluted soil remediation by BC-nZVI activated persulfate[J]. Chemical Engineering Journal, 2020, 382: 123055. doi: 10.1016/j.cej.2019.123055 [13] GHATTAS A K, FISCHER F, WICK A, et al. Anaerobic biodegradation of (emerging) organic contaminants in the aquatic environment[J]. Water Research, 2017, 116: 268-295. doi: 10.1016/j.watres.2017.02.001 [14] LU X Y, LI B, ZHANG T, et al. Enhanced anoxic bioremediation of PAHs-contaminated sediment[J]. Bioresource Technology, 2012, 104: 51-58. doi: 10.1016/j.biortech.2011.10.011 [15] MEDINA R, GARA P M D, FERNÁNDEZ-GONZÁLEZ A J, et al. Remediation of a soil chronically contaminated with hydrocarbons through persulfate oxidation and bioremediation[J]. Science of the Total Environment, 2018, 618: 518-530. doi: 10.1016/j.scitotenv.2017.10.326 [16] YANG S C, GOU Y L, SONG Y, et al. Enhanced anoxic biodegradation of polycyclic aromatic hydrocarbons (PAHs) in a highly contaminated aged soil using nitrate and soil microbes[J]. Environmental Earth Sciences, 2018, 77: 432. doi: 10.1007/s12665-018-7629-6 [17] FIERER N, SCHIMEL J P, HOLDEN P A. Variations in microbial community composition through two soil depth profiles[J]. Soil Biology and Biochemistry, 2003, 35(1): 167-176. doi: 10.1016/S0038-0717(02)00251-1 [18] 马浩, 刘元元, 肖文燕, 等. 表面活性剂CMC对石油烃污染土壤的增溶[J]. 环境工程学报, 2016, 10(12): 7333-7338. doi: 10.12030/j.cjee.201507133 [19] SEMPLE K T, MORRISS A W J, PATON G I. Bioavailability of hydrophobic organic contaminants in soils: fundamental concepts and techniques for analysis[J]. European Journal of Soil Science, 2003, 54(4): 809-818. doi: 10.1046/j.1351-0754.2003.0564.x [20] 陈巧超, 王一宁, 董旭斌. 阴-非混合表面活性剂增效修复有机污染土壤的影响因素研究[J]. 科学技术创新, 2021(20): 52-53. doi: 10.3969/j.issn.1673-1328.2021.20.022 [21] 赵倩云, 苟雅玲, 杨苏才, 等. 硫酸盐对老化土壤中多环芳烃厌氧降解的影响[J]. 环境科学与技术, 2018, 41(12): 200-205. doi: 10.19672/j.cnki.1003-6504.2018.12.029 [22] 刘志号. 缺氧条件下温度和表面活性剂对土壤中多环芳烃微生物降解的影响[D]. 北京: 轻工业环境保护研究所, 2021. [23] 李佳斌. 土壤中石油烃的芬顿氧化实验研究[D]. 北京: 轻工业环境保护研究所, 2016. [24] GOU Y L, YANG S C, CHENG Y J, et al. Enhanced anoxic biodegradation of polycyclic aromatic hydrocarbons (PAHs) in aged soil pretreated by hydrogen peroxide[J]. Chemical Engineering Journal, 2019, 356: 524-533. doi: 10.1016/j.cej.2018.09.059 [25] FREY B, RIME T, PHILLIPS M, et al. Microbial diversity in European alpine permafrost and active layers[J]. FEMS Microbiology Ecology, 2016, 92(3): fiw018. doi: 10.1093/femsec/fiw018 [26] LEI Y J, ZHANG J, TIAN Y, et al. Enhanced degradation of total petroleum hydrocarbons in real soil by dual-frequency ultrasound-activated persulfate[J]. Science of the Total Environment, 2020, 748: 141414. doi: 10.1016/j.scitotenv.2020.141414 [27] BARTLETT C K, SLAWSON R M, THOMSON N R. Response of sulfate-reducing bacteria and supporting microbial community to persulfate exposure in a continuous flow system[J]. Environmental Science:Processes & Impacts, 2019, 21(7): 1193-1203. [28] TIAN W, YAO J, LIU R P, et al. Effect of natural and synthetic surfactants on crude oil biodegradation by indigenous strains[J]. Ecotoxicology and Environmental Safety, 2016, 129: 171-179. doi: 10.1016/j.ecoenv.2016.03.027 [29] WU M L, WU J L, ZHANG X H, et al. Effect of bioaugmentation and biostimulation on hydrocarbon degradation and microbial community composition in petroleum-contaminated loessal soil[J]. Chemosphere, 2019, 237: 124456. doi: 10.1016/j.chemosphere.2019.124456 [30] GRAY N D, SHERRY A, HUBERT C, et al. Chapter 5- Methanogenic degradation of petroleum hydrocarbons in subsurface environments: remediation, heavy oil formation, and energy recovery[J]. Advances in Applied Microbiology, 2010, 72: 137-161. [31] 张秀霞, 熊鑫, 郭鹏, 等. SDBS残留对微生物修复石油污染土壤的影响[J]. 石油学报(石油加工), 2021, 37(5): 1167-1173. [32] KOSTKA J E, PRAKASH O, OVERHOLT W A, et al. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the deepwater horizon oil spill[J]. Applied and Environmental Microbiology, 2011, 77(22): 7962-7974. doi: 10.1128/AEM.05402-11 [33] CHEN F, LI X X, ZHU Q L, et al. Bioremediation of petroleum-contaminated soil enhanced by aged refuse[J]. Chemosphere, 2019, 222: 98-105. doi: 10.1016/j.chemosphere.2019.01.122 期刊类型引用(5)
1. 朱西,李昕妍,孙晓春. 基于POEC策略的真实问题解决的高中化学教学设计——以“如何科学使用补铁剂”为例. 云南化工. 2024(04): 173-177 .  百度学术
百度学术
2. 何梦婷,肖荣波,黄飞,王鹏,王韵泽,罗树华,杨洁鑫,徐美丽. 抗坏血酸改性纳米硫化亚铁对水体Cr(Ⅵ)的还原特性及机制. 环境科学学报. 2023(02): 61-72 . 百度学术
3. 方政,陈平,刘洋,余前锋,郑铱汛,杨慧敏,张立,吕文英,刘国光. Co~(2+)/CaO_2/草酸高级氧化体系降解水中SMT的机理研究. 环境科学学报. 2023(03): 40-51 . 百度学术
4. 张玉连,李幸,葛志强. CaO_2-硅藻土对尿液中头孢克肟的去除研究. 环境科学与技术. 2022(10): 159-167 . 百度学术
5. 邵子纯,卢建,高仕谦,徐劼,张占恩. 共价三嗪骨架活化过一硫酸盐降解磺胺甲恶唑. 中国环境科学. 2020(01): 135-142 . 百度学术
其他类型引用(4)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 4580
- HTML全文浏览数: 4580
- PDF下载数: 130
- 施引文献: 9
