

-
重金属铬(Cr)及其化合物是我国工业用地中最常见的重金属污染物之一。土壤中铬的化合价主要有三价和六价,其他二价、四价和五价的铬化合物性质极其不稳定[1-2]。Cr(Ⅲ)和Cr(Ⅵ)具有完全不同的化学性质和生物毒性。Cr(Ⅲ)易水解形成沉淀吸附于土壤矿物和有机质中,毒性较低;而Cr(Ⅵ)具有剧毒性、强致癌和易迁移等特点[3],被列为我国当前亟需治理的重金属污染物之一[4]。
化学修复是Cr(Ⅵ)污染土最常用的修复技术之一,具有效果好、速度快、成本低和二次污染小等特点[5-6]。硫酸亚铁(FeSO4)和多硫化钙(CaSx)是修复Cr(Ⅵ)污染土常用的药剂,已在Cr(Ⅵ)污染修复方面得到大量应用[7-9]。任学昌等[7]采用FeSO4联合多种固化剂修复铬污染土壤,土壤中Cr(Ⅵ)浸出浓度可由527 mg·L−1降至1.5 mg·L−1。CHRYSOCHOOU等[9]采用CaS5修复电镀污染场地Cr(Ⅵ)污染土,实验结果表明:CaS5可显著降低污染土中的Cr(Ⅵ)质量浓度,当CaS5/Cr(Ⅵ)摩尔比为2时,60 d养护龄期的稳定土中Cr(Ⅵ)的浸出浓度低于1 mg·L−1。事实上,化学修复并不是完全去除污染物,而是将重金属污染物的价态或形态改变后稳定于土壤基质中,降低重金属向周围环境运移的能力。多孔介质溶质运移理论表明,土体的渗透系数是衡量重金属污染物运移的首要指标[10]。邵俐等[11]对复合重金属铜镉污染底泥进行固化处理,研究了不同污染物掺量对渗透系数及污染物渗出总量的影响。冯亚松[12]通过柔性壁渗透实验测试复合镍锌污染土和固化土的渗透系数,并进一步分析渗透液中重金属浓度和pH随渗透液体积的变化规律。由此可见,重金属污染土的渗透系数及渗出液中重金属浓度是评估污染土修复有效性和重金属运移特征的重要指标。
然而,前人的研究主要采用浸出实验和消解实验来评价Cr(Ⅵ)污染土的修复效果,有关Cr(Ⅵ)污染土修复后对环境的实际影响研究较少;此外,仅用散状土进行实验研究并不能很好地模拟污染场地土层的实际力学特性及重金属运移特征。本研究以某铁合金场地为研究背景,拟采用不同投加摩尔比的FeSO4和CaSx对Cr(Ⅵ)污染土壤进行修复处理,制备压实试样,通过渗透淋滤实验研究不同还原剂对Cr(Ⅵ)污染土渗透淋滤特性的影响规律,评估FeSO4和CaSx修复Cr(Ⅵ)污染土的实际效果;拟基于BCR形态提取法和X射线衍射法,从微观的角度对比分析不同还原剂掺量对土壤中物理化学反应产物的影响。本研究结果可为Cr(Ⅵ)污染土的化学稳定修复及安全再利用提供参考。
-
1) 实验用土。实验用土取自湖南某铁合金铬渣场地,粉质黏土广泛分布于场区,场地原有金属铬湿法冶炼生产线,长期生产过程产生的铬渣随意堆放于铬渣场,导致场区周边粉质黏土层Cr(Ⅵ)严重污染。为保证实验用土中Cr(Ⅵ)分布均匀且污染浓度恒定,实验用土取自该场地上游无污染的粉质黏土,人工制备Cr(Ⅵ)污染土。实验前将土在60 ℃下烘干24 h至恒重,粉碎过1 mm筛备用。粉质黏土主要物理力学特性指标见表1,土的物理力学实验方法参考《土工试验方法标准》 (GB/T50123-2019) [13],击实实验采用轻型击实法。原始土样中Cr(Ⅵ)质量浓度为12 mg·kg−1,在第一类用地管制值以下,为未污染土[14]。
2) 铬污染土制备。向烘干土样中加入重铬酸钾(K2Cr2O7)至Cr(Ⅵ)的质量浓度为200 mg·kg−1。其中,200 mg·kg−1为污染场地Cr(Ⅵ)平均浓度。再向污染土中均匀喷洒去离子水使土的含水率为20.5%,拌合均匀后密封,在 (20±2) ℃、湿度95%下养护7 d,使K2Cr2O7与土壤充分反应。
3) 实验试剂。重铬酸钾(K2Cr2O7)、盐酸(HCl)、硝酸(HNO3)、过氧化氢(H2O2)、硫酸亚铁(FeSO4)、质量分数为50%的多硫化钙(CaSx)等。以上材料除特殊说明外均为分析纯。实验过程中所用水均为去离子水。
-
按照目标干密度 (根据场地粉质黏土层干密度选定) 称取含一定质量添加药剂的养护土样填入不锈钢筒状模具中,利用电子万能试验机,以位移控制法压制试样。保持0.2 mm·min−1的垂直加载速率,将散状土样均匀压实至设计位置,在恒体积条件下静置0.5 h,防止试样回弹。最后,卸载后得到直径为50 mm、高度为5 mm、初始干密度为1.5 g·cm−3的压实试样,具体试样如图1所示。
-
1) 土壤修复实验。设计修复药剂不同投加摩尔比,开展污染土壤修复实验,实验具体方案见表2。将称量好的还原剂加入Cr(Ⅵ)污染土中,充分拌和至混合物无明显颜色差异,然后利用NJ-160台式电动搅拌机强化搅拌5 min,以提高其均匀性。搅拌结束后,将混合均匀的土样转移至容器中并用保鲜膜覆盖防止水分蒸发,在恒温条件下养护7 d。
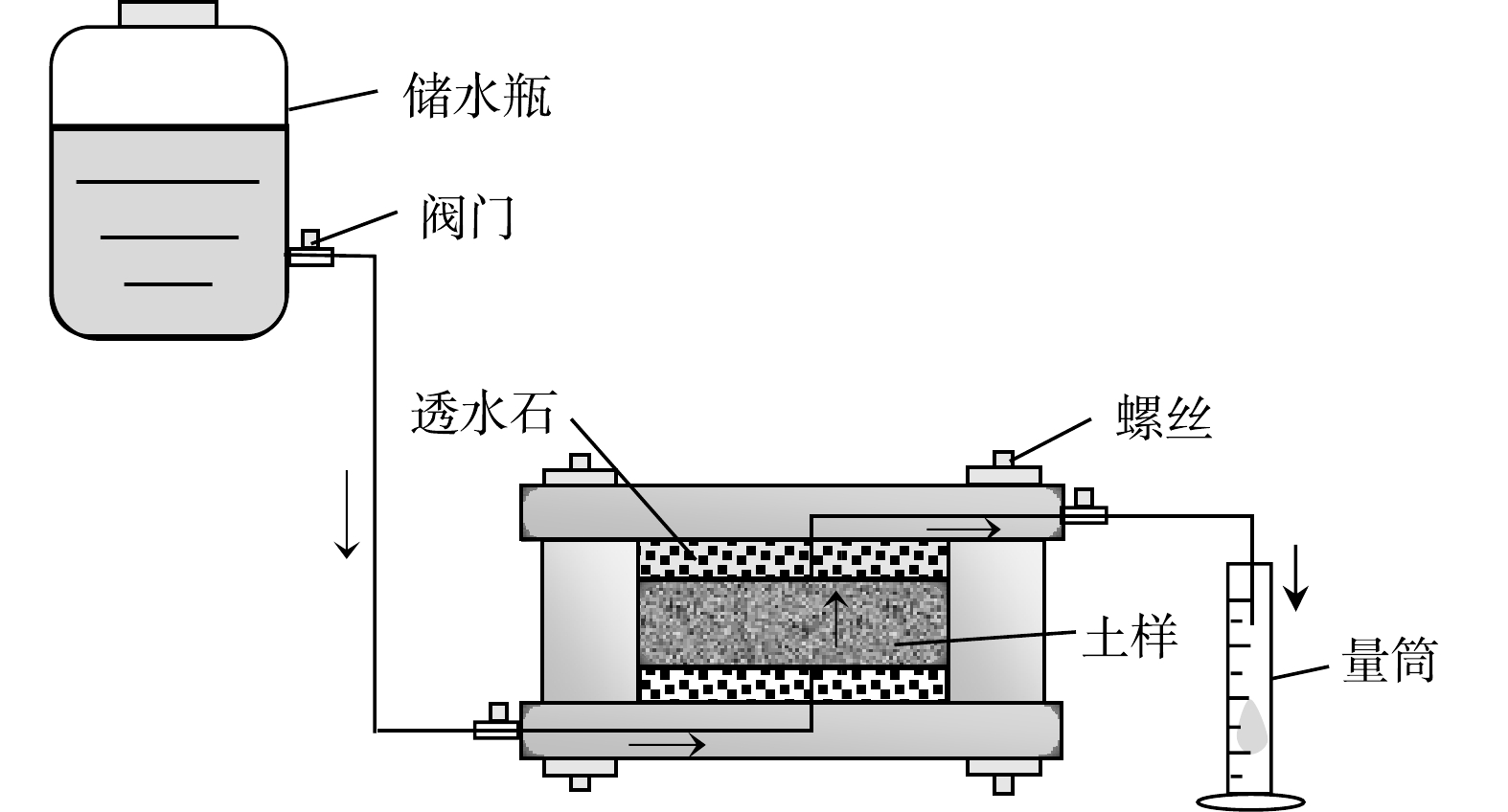
2) 渗透淋滤实验。渗透淋滤实验装置示意图如图2所示。实验过程中定期收集渗出液并测量体积,根据达西定律计算渗透系数,渗透系数计算公式如式1所示。根据计算得到的污染土和修复土的渗透系数,以及测定的渗出液中重金属离子浓度,进一步分析渗出液中金属浓度随渗出液体积的变化规律。
式中:K为渗透系数,cm·s−1;ΔQ为一定时间内出流水量,mL;L为土样高度,cm;Δh为水头差,cm;A为土样横截面积,cm2;Δt为渗流时间,s。
-
采用二苯碳酰二肼分光光度法测定土样渗出液中Cr(Ⅵ)浓度;采用XRD实验来判定修复前后Cr(Ⅵ)污染土的物相组成,XRD表征条件:CuKa靶,电压为40 kV,电流为40 mA,扫描速度为 2 °·min−1,光源波长为1.54 nm,角度为5°~90°;铬赋存形态采用改进BCR方法[19]。
-
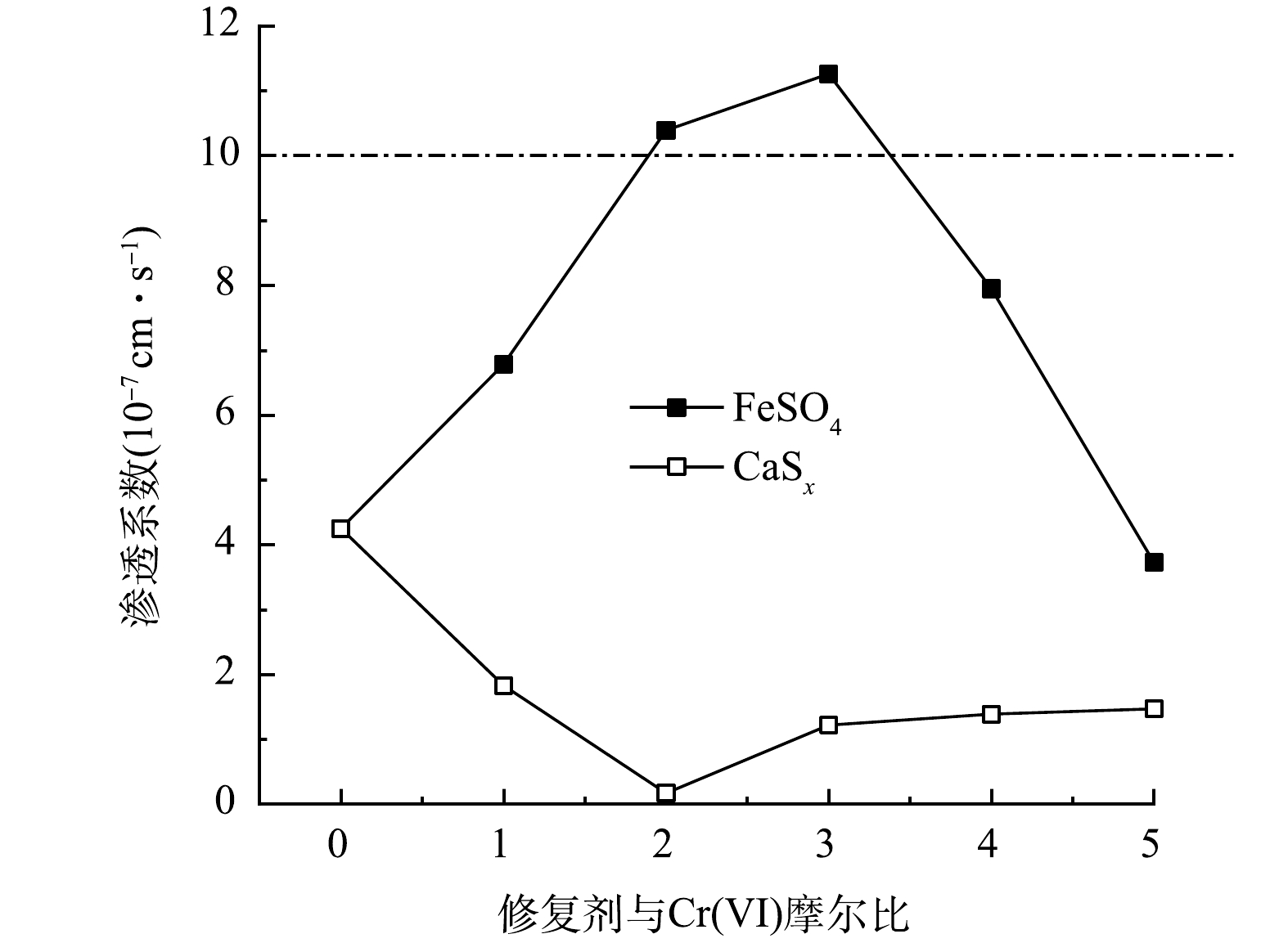
FeSO4和CaSx修复Cr(Ⅵ)污染粉质黏土的渗透系数随投加摩尔比变化的曲线如图3所示。由图可以看出,使用CaSx修复后的土样渗透系数明显低于使用FeSO4修复后土样的渗透系数,且使用CaSx修复后土样的渗透系数小于原污染土 (FeSO4/Cr(Ⅵ)摩尔比为0) 。
原污染土的渗透系数为4.25×10−7 cm·s−1,FeSO4/Cr(Ⅵ)摩尔比由1增加到5时,土样的渗透系数分别为6.78×10−7、1.04×10−6、1.13×10−6、7.95×10−7、3.73×10−7 cm·s−1,FeSO4修复Cr(Ⅵ)污染粉质黏土渗透系数随摩尔比的增加呈先增大后减小,在摩尔比为3时渗透系数最大。由表2反应机理式可知,FeSO4/Cr(Ⅵ)摩尔比为3时,Fe2+和Cr2O72-反应刚好能完全反应生成Cr3+和Fe3+。在FeSO4加入量较少时,Fe3+和Cr3+反应生成的(Fex,Cr1-x)(OH)3是难溶于水的金属化合物,但由于土壤环境为酸性,反应会向减少沉淀的方向进行;在FeSO4/Cr(Ⅵ)摩尔比超过3时,Fe2+投入过量,可生成大量Fe(OH)3沉淀,土壤的渗透系数降低。此外,通常将渗透系数小于1×10−6 cm·s−1作为土壤修复的控制标准之一[12],见图3虚线。通过对比Cr(Ⅵ)污染土修复前后渗透系数变化,可以发现:FeSO4处理后的污染土的渗透系数在FeSO4/ Cr(Ⅵ)摩尔比为2和3时要略高于该控制标准,而FeSO4/Cr(Ⅵ)摩尔比为1、4和5的土样渗透系数明显低于该控制标准。
使用CaSx修复时,CaSx/Cr(Ⅵ)摩尔比由1增加到5,土样的渗透系数分别为1.83×10−7、1.72×10−8、1.22×10−7、1.39×10−7、1.47×10−7 cm·s−1,实验在摩尔比为2时渗透系数最小,CaSx修复Cr(Ⅵ)污染粉质黏土渗透系数随摩尔比的增加呈先减小后缓慢增大的趋势,这与FeSO4修复后土壤渗透系数的变化相反。由表2反应机理公式可知,在CaS5/Cr(Ⅵ)摩尔比为1.5时,理想状态下,CaS5和Cr2O72-反应刚好能完全反应生成Cr(OH)3和单质S;在CaSx加入量较少时,CaSx和Cr2O72-反应不会生成过多的沉淀物质;在CaSx/Cr(Ⅵ)摩尔比超过2时,随着CaSx投入量的增加,土壤中产生的单质S和Ca2+也随之增加,Ca2+可生成微溶于水的CaSO4,使土壤的渗透系数增大。CaSx/Cr(Ⅵ)摩尔比为2时试样渗透系数最小,可能是由于下述原因。CaSx是含有多种硫化物的混合体系,本实验所用不为纯CaS5,其中含有CaS5及其他杂质,在铬污染土壤修复方面还没有比较完善的反应机理,尚需深入地研究[8,20]。
对比Cr(Ⅵ)污染土修复前后渗透系数的变化可以发现:CaSx修复的污染土渗透系数均明显低于修复土控制标准1×10−6 cm·s−1,总体比FeSO4处理后的土壤渗透系数要小。此前多见固化土渗透系数的研究,但使用还原剂稳定化后的土壤进行渗透实验的相关研究较少,可由本次实验得出稳定化Cr(Ⅵ)土壤渗透系数随还原剂种类及掺量的变化规律。
-
原污染土、FeSO4/ Cr(Ⅵ)摩尔比为1到5的修复土样渗出液Cr(Ⅵ)浓度如图4所示。由图可知:随渗出液体积增大,污染土样渗出液中重金属Cr(Ⅵ)浓度不断降低。在实验开始阶段,土样的孔隙水中溶解的Cr(Ⅵ)随渗出液被冲出,浸出液中重金属浓度较大;随着实验的进行,土样中Cr(Ⅵ)的总量逐渐减少,因此由固相 (黏土矿物吸附态) 进入液相 (孔隙水溶解态) 的重金属也逐渐减少,重金属的浓度曲线趋于平缓[12]。原污染土中Cr(Ⅵ)渗出浓度由一开始的21.96 mg·L−1降至9.05 mg·L−1,FeSO4/Cr(Ⅵ)摩尔比为1时,Cr(Ⅵ)渗出浓度由1.72 mg·L−1降至0.22 mg·L−1,均大于IV类地下水标准Cr(Ⅵ)浓度限值(0.10 mg·L−1)和III类地下水标准Cr(Ⅵ)浓度限值(0.05 mg·L−1)[21]。当FeSO4/Cr(Ⅵ)摩尔比大于3时,渗出液中Cr(Ⅵ)的浓度始终低于III类地下水标准。该现象进一步说明,用FeSO4修复Cr(Ⅵ)污染土的最优摩尔比为3,污染土中大量Cr(Ⅵ)被还原为Cr(Ⅲ)稳定在土壤中,有效减缓了重金属运移过程。
图5给出了原污染土、CaSx/Cr(Ⅵ)摩尔比为由1到5的修复土样渗出液Cr(Ⅵ)浓度。由图5可知:当渗出液体积随时间增大时,污染土样渗出液中重金属Cr(Ⅵ)浓度稳定降低。CaSx/Cr(Ⅵ)摩尔比为1时,土中的Cr(Ⅵ)渗出浓度由最初的9.42 mg·L−1降至5.41 mg·L−1,CaSx/Cr(Ⅵ)摩尔比为2时,土中的Cr(Ⅵ)渗出浓度由7.77 mg·L−1降至0.42 mg·L−1,均大于IV类地下水标准Cr(Ⅵ)浓度限度(0.10 mg·L−1)。当CaSx/Cr(Ⅵ)摩尔比大于3时,在实验过程后期,渗出液中Cr(Ⅵ)的浓度会降至低于III类地下水标准Cr(Ⅵ)浓度限值。该现象说明,使用CaSx修复Cr(Ⅵ)污染土,其反应的最优摩尔比与理论值 (1.5,见表2) 存在差异,需视具体反应的情况而定。
由图4和图5可知:随着渗出液体积逐渐增大,修复后土样渗出液中Cr(Ⅵ)浓度逐渐减小。与原Cr(Ⅵ)污染土样渗出液中Cr(Ⅵ)浓度相比,修复后土样渗出液中Cr(Ⅵ)浓度显著降低,而且随着掺入还原剂摩尔比的增加,渗出液中Cr(Ⅵ)浓度进一步降低。总体来说,污染土经修复处理后,孔隙水中Cr(Ⅵ)浓度显著降低。该现象说明:修复土中Cr(Ⅵ)通过渗透作用向周围环境运移达到稳定状态的时间,较未修复时得到了显著缩短。上述现象均说明在土壤修复后,土中Cr(Ⅵ)的化学稳定性得到明显提升、运移性显著降低。其原因是,CaSx和FeSO4与Cr(Ⅵ)通过一系列氧化还原反应、离子交换反应及氢氧化物沉淀反应[22-24],改变了铬的价态与形态,降低了铬的毒性,将铬稳定于土壤基质中,土样孔隙水中的Cr(Ⅵ)浓度降低,有效减弱了固相重金属向液相环境转化,因此在渗透实验中渗出液重金属浓度明显降低[25]。
-
FeSO4/Cr(Ⅵ)摩尔比对铬形态分布的影响如图6所示。从图6中可以发现,未修复的Cr(Ⅵ)污染土中弱酸提取态铬质量分数最高,达86%,这是因为采用室内人工配制的Cr(Ⅵ)污染土,污染周期较短,Cr(Ⅵ)主要以弱酸提取态赋存于土壤中[26],可还原态和可氧化态的铬质量分数次之,分别为6%和7%,残渣态铬最低,质量分数约为1%。随着FeSO4/Cr(Ⅵ)的摩尔比的增加,修复土中弱酸提取态铬质量分数明显降低,可还原态和可氧化态铬质量分数显著增大,残渣态的铬质量分数有细微增加。当摩尔比从0增加到 5时,修复土中弱酸提取态的铬质量分数从86%降低至23%,而可还原态和可氧化态的铬质量分数从6%、7%增加到27%、47%。实验结果表明:FeSO4可促使Cr从活性态向较稳定态转化,提高了修复土中Cr稳定性。
CaSx/Cr(Ⅵ)摩尔比对铬形态分布的影响如图7所示,与FeSO4效果类似。分析其原因,Fe2+将Cr(Ⅵ)还原成Cr(Ⅲ),一方面Cr(Ⅲ)与土壤中的氢氧化物、氧化物及黏土矿物发生离子交换生成Cr(OH)3等沉淀;另一方面Cr(OH)3与Fe3+发生离子交换生成较难溶的沉淀物 (Fex,Cr1-x)(OH)3,且在摩尔比较小时,土壤中的Cr(Ⅲ)主要以Cr(OH)3存在,随着摩尔比的增大,Cr(OH)3会逐步转化成(Fex,Cr1-x)(OH)3[27],铬逐渐从活性态向稳定态转化,降低了铬的迁移能力。CaSx将Cr(Ⅵ)还原成Cr(Ⅲ),由于CaSx能提高修复土的pH 值,一方面促使Cr(Ⅲ)与土壤中的氢氧化物、氧化物发生离子交换生成Cr(OH)3等沉淀;另一方面CaSx被Cr(Ⅵ)氧化成单质S,部分Cr(Ⅲ)被单质S络合或包裹;此外,CaSx将部分Cr(Ⅵ)还原,生成了Cr(OH)3沉淀,降低了渗出风险[28]。
-
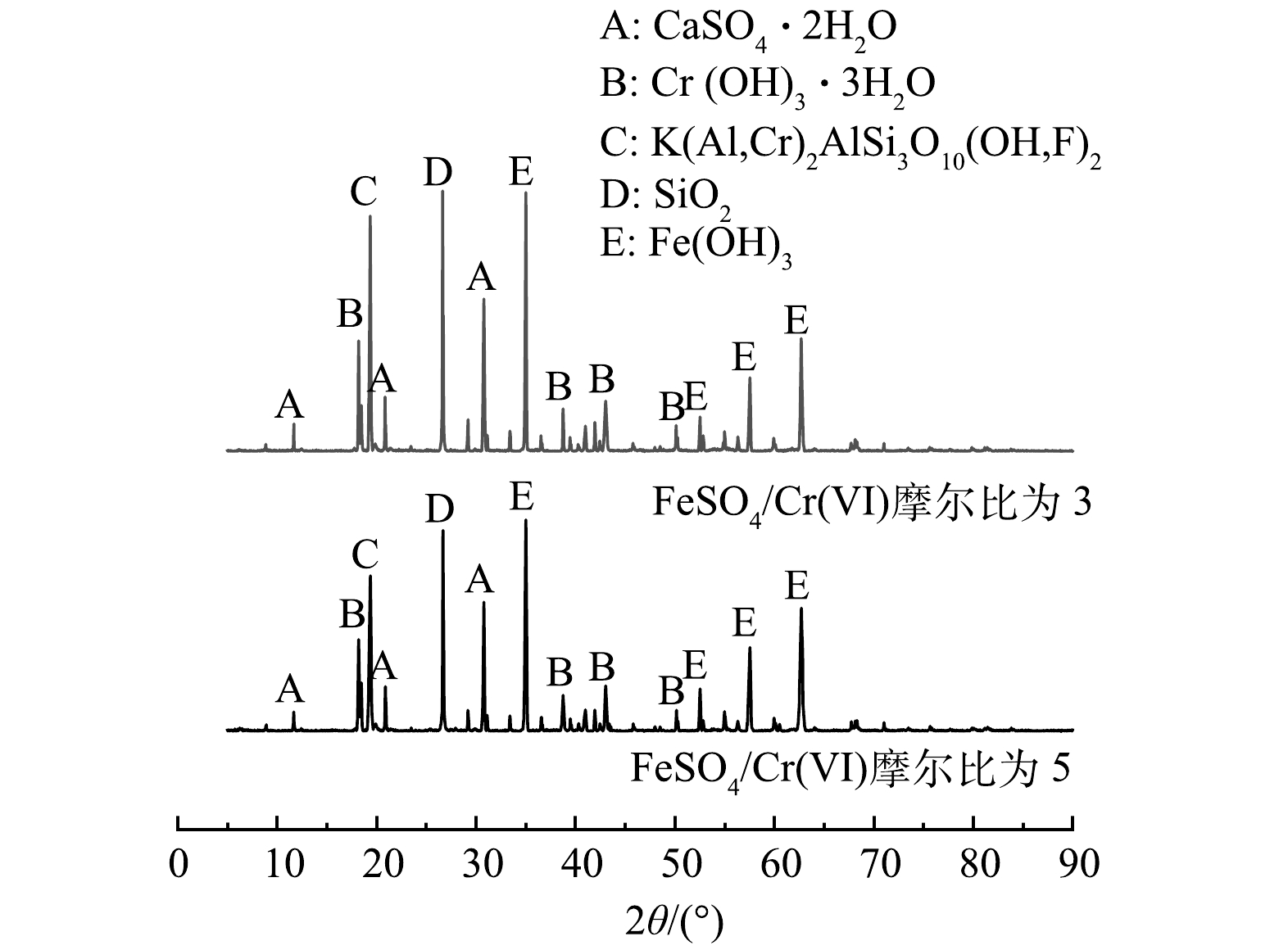
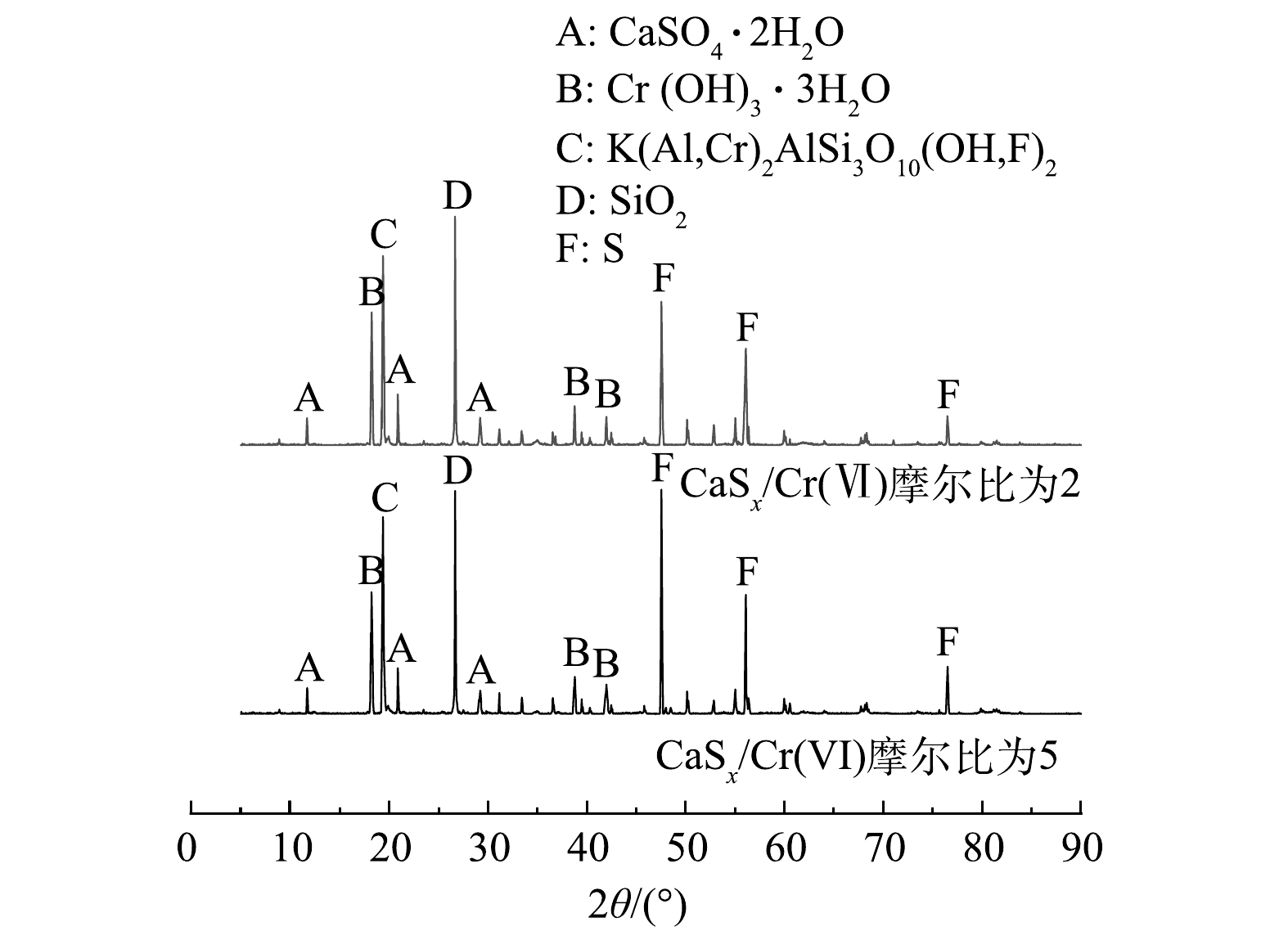
分别选取渗透实验中最具代表性的2组修复实验: FeSO4/Cr(Ⅵ)摩尔比为3和5、CaSx/Cr(Ⅵ)摩尔比为2和5的供试土壤,采用X射线衍射实验对土壤中的物质、元素、相对大颗粒进行物相分析,研究2种还原剂FeSO4和CaSx对Cr(Ⅵ)污染土的微观修复机理。
通过X射线衍射实验得到FeSO4和CaSx修复后土的衍射曲线如图8和9所示,在衍射角为5°~90°时,修复后土壤中主要存在石英(SiO2)、铬云母(KAl2[Si3AlO10](OH,F)2)和三水合氢氧化铬(Cr(OH)3·3H2O)的衍射峰。由图8中可知,用FeSO4修复后的Cr(Ⅵ)污染土中含有铁元素的衍射峰,当衍射角为35.24°时,FeSO4/Cr(Ⅵ)摩尔比为3和5的污染土中,氢氧化铁的衍射强度由1 003增至1 209 s−1,当衍射角为18.20°时,三水合氢氧化铬的衍射强度分别由3 901降至3 227 s−1。在图9中可知,用CaSx修复后的Cr(Ⅵ)污染土中含有硫单质的衍射峰,当衍射角为56.08°时,CaSx/Cr(Ⅵ)摩尔比为2和5的污染土中,单质S的衍射强度由3 452增至4 253 s−1,当衍射角为18.20°时,三水合氢氧化铬的衍射强度分别由4 744降至3 992 s−1,土样的其他主要物相峰值基本不变。以上现象证明了 FeSO4和CaSx在土中发生了一系列物理化学反应,使得土壤中的Cr(Ⅵ)质量分数降低,进一步验证了不同还原剂作用下,六价铬污染粉质黏土的渗透淋滤特性及组成成分变化。
-
1) 试样的渗透系数K随着还原剂的种类和摩尔比的不同有着非线性的关系,FeSO4处理后的土壤渗透系数随着渗透的进行先变大后减小,而CaSx处理后的土壤渗透系数随着渗透的进行先减小后变大。其中,FeSO4/Cr(Ⅵ)摩尔比为3时渗透系数最大;CaSx/Cr(Ⅵ)摩尔比为2时渗透系数最小。
2) 修复后土中Cr(Ⅵ)的渗出量随着时间的增长持续减小,当FeSO4或CaSx与Cr(Ⅵ)的摩尔比大于3时,最终渗出液质量浓度低于III类地下水标准,FeSO4处理后土壤Cr(Ⅵ)的渗出液质量浓度总体比CaSx处理后的土壤低。
3) 通过BCR分布提取法和XRD物相分析可知,重金属Cr(Ⅵ)与还原剂发生一系列物理化学反应,改变了修复土中各物质成分及铬的赋存形态。还原剂可促使铬由弱酸提取态向可还原态、可氧化态转化,而对残渣态的铬影响不大。
还原剂作用下Cr(Ⅵ)污染土渗透淋滤特性及成分分析
Percolation leaching characteristics and composition analysis of hexavalent chromium contaminated soil under the remediation of reductant
-
摘要: 以湖南某铬污染场地中典型污染土壤为对象,采用硫酸亚铁(FeSO4)和多硫化钙(CaSx)2种还原剂,开展土壤修复实验和渗透淋滤实验,探讨不同还原剂对铬污染土渗透淋滤特性的影响规律,利用BCR形态提取和X射线衍射分析方法,评估不同还原剂对铬污染土中Cr(VI)的还原效果。结果表明,FeSO4修复试样的渗透系数随FeSO4/Cr(Ⅵ)摩尔比的增加呈先增加后降低的趋势,CaSx修复试样的渗透系数变化呈现相反规律。相同条件下,FeSO4修复试样的渗透系数较CaSx修复试样的渗透系数更大,表明CaSx修复试样的抗渗效果更好。随着还原剂掺量增加,修复试样渗出液中Cr(Ⅵ)浓度随之降低,当FeSO4/Cr(Ⅵ)摩尔比大于3时,渗出液中Cr(Ⅵ)浓度低于Ⅲ类地下水标准Cr(Ⅵ)浓度基准值0.05mg·L−1。类似地,当CaSx/Cr(Ⅵ)摩尔比大于3时,淋滤实验后期渗出液Cr(Ⅵ)浓度也降至0.05 mg·L−1以下。修复土中铬的赋存形态随还原剂掺量的增大而发生变化,铬由弱酸提取态向可还原态和可氧化态转化,且还原剂摩尔比也会对修复土壤组成成分产生影响。本研究结果可为Cr(Ⅵ)污染场地土壤修复工程提供参考。Abstract: Taking the typical contaminated soil in a chromium contaminated site in Hunan Province as an object, the effects of different reductants on the percolation and leaching characteristics of chromium contaminated soil were studied through soil remediation tests and percolation leaching tests using ferrous sulfate (FeSO4) and calcium polysulfide (CaSx) as reductants. The effects of different reductants on the reduction and remediation of Cr(Ⅵ) in chromium contaminated soil were analyzed by BCR morphology extraction technology and X-ray diffraction analysis method. The results showed that the permeability coefficient of FeSO4 repair sample first increased and decreased with the increase of FeSO4/Cr(Ⅵ) molar ratio, which was contrary to the change rule of permeability coefficient of CaSx repair sample. Under the same conditions, the permeability coefficient of FeSO4 repair sample was higher than that of CaSx repair sample, indicating that CaSx had better impermeability. With the increase of the amount of reducing agent, the concentration of Cr(Ⅵ) in the exudate of the reducing agent repair sample decreased. When the FeSO4/Cr(Ⅵ) molar ratio was greater than 3, the concentration of Cr(Ⅵ) in the exudate was lower than the concentration limit of Cr(Ⅵ) in the Class Ⅲ groundwater standard. Similarly, when the molar ratio of CaSx/Cr(Ⅵ) was greater than 3, the concentration of Cr(Ⅵ) in leachate at the later stage of leaching test also droped below 0.05 mg·L−1. The occurrence form of chromium in stabilized soil changed with the increase of the amount of reducing agent. Chromium was transformed from the weak acid extraction state to the reducible and oxidable state, and different molar ratios of reducing agent would also affect the composition of the repaired soil. The research results can provide a theoretical reference for the application of soil remediation in Cr(Ⅵ) contaminated sites.
-
重金属铬(Cr)及其化合物是我国工业用地中最常见的重金属污染物之一。土壤中铬的化合价主要有三价和六价,其他二价、四价和五价的铬化合物性质极其不稳定[1-2]。Cr(Ⅲ)和Cr(Ⅵ)具有完全不同的化学性质和生物毒性。Cr(Ⅲ)易水解形成沉淀吸附于土壤矿物和有机质中,毒性较低;而Cr(Ⅵ)具有剧毒性、强致癌和易迁移等特点[3],被列为我国当前亟需治理的重金属污染物之一[4]。
化学修复是Cr(Ⅵ)污染土最常用的修复技术之一,具有效果好、速度快、成本低和二次污染小等特点[5-6]。硫酸亚铁(FeSO4)和多硫化钙(CaSx)是修复Cr(Ⅵ)污染土常用的药剂,已在Cr(Ⅵ)污染修复方面得到大量应用[7-9]。任学昌等[7]采用FeSO4联合多种固化剂修复铬污染土壤,土壤中Cr(Ⅵ)浸出浓度可由527 mg·L−1降至1.5 mg·L−1。CHRYSOCHOOU等[9]采用CaS5修复电镀污染场地Cr(Ⅵ)污染土,实验结果表明:CaS5可显著降低污染土中的Cr(Ⅵ)质量浓度,当CaS5/Cr(Ⅵ)摩尔比为2时,60 d养护龄期的稳定土中Cr(Ⅵ)的浸出浓度低于1 mg·L−1。事实上,化学修复并不是完全去除污染物,而是将重金属污染物的价态或形态改变后稳定于土壤基质中,降低重金属向周围环境运移的能力。多孔介质溶质运移理论表明,土体的渗透系数是衡量重金属污染物运移的首要指标[10]。邵俐等[11]对复合重金属铜镉污染底泥进行固化处理,研究了不同污染物掺量对渗透系数及污染物渗出总量的影响。冯亚松[12]通过柔性壁渗透实验测试复合镍锌污染土和固化土的渗透系数,并进一步分析渗透液中重金属浓度和pH随渗透液体积的变化规律。由此可见,重金属污染土的渗透系数及渗出液中重金属浓度是评估污染土修复有效性和重金属运移特征的重要指标。
然而,前人的研究主要采用浸出实验和消解实验来评价Cr(Ⅵ)污染土的修复效果,有关Cr(Ⅵ)污染土修复后对环境的实际影响研究较少;此外,仅用散状土进行实验研究并不能很好地模拟污染场地土层的实际力学特性及重金属运移特征。本研究以某铁合金场地为研究背景,拟采用不同投加摩尔比的FeSO4和CaSx对Cr(Ⅵ)污染土壤进行修复处理,制备压实试样,通过渗透淋滤实验研究不同还原剂对Cr(Ⅵ)污染土渗透淋滤特性的影响规律,评估FeSO4和CaSx修复Cr(Ⅵ)污染土的实际效果;拟基于BCR形态提取法和X射线衍射法,从微观的角度对比分析不同还原剂掺量对土壤中物理化学反应产物的影响。本研究结果可为Cr(Ⅵ)污染土的化学稳定修复及安全再利用提供参考。
1. 材料与方法
1.1 实验原料
1) 实验用土。实验用土取自湖南某铁合金铬渣场地,粉质黏土广泛分布于场区,场地原有金属铬湿法冶炼生产线,长期生产过程产生的铬渣随意堆放于铬渣场,导致场区周边粉质黏土层Cr(Ⅵ)严重污染。为保证实验用土中Cr(Ⅵ)分布均匀且污染浓度恒定,实验用土取自该场地上游无污染的粉质黏土,人工制备Cr(Ⅵ)污染土。实验前将土在60 ℃下烘干24 h至恒重,粉碎过1 mm筛备用。粉质黏土主要物理力学特性指标见表1,土的物理力学实验方法参考《土工试验方法标准》 (GB/T50123-2019) [13],击实实验采用轻型击实法。原始土样中Cr(Ⅵ)质量浓度为12 mg·kg−1,在第一类用地管制值以下,为未污染土[14]。
表 1 实验用土壤基本物理力学参数Table 1. Basic physical and mechanical parameters of soil天然含水率/% 天然干密度/(g·cm−3) 最优含水率/% 液限/% 塑限/% 比重 6.11 1.54 20.5 37.3 22.4 2.72 | Show Table DownLoad:
CSV
DownLoad:
CSV
2) 铬污染土制备。向烘干土样中加入重铬酸钾(K2Cr2O7)至Cr(Ⅵ)的质量浓度为200 mg·kg−1。其中,200 mg·kg−1为污染场地Cr(Ⅵ)平均浓度。再向污染土中均匀喷洒去离子水使土的含水率为20.5%,拌合均匀后密封,在 (20±2) ℃、湿度95%下养护7 d,使K2Cr2O7与土壤充分反应。
3) 实验试剂。重铬酸钾(K2Cr2O7)、盐酸(HCl)、硝酸(HNO3)、过氧化氢(H2O2)、硫酸亚铁(FeSO4)、质量分数为50%的多硫化钙(CaSx)等。以上材料除特殊说明外均为分析纯。实验过程中所用水均为去离子水。
1.2 土样制备
按照目标干密度 (根据场地粉质黏土层干密度选定) 称取含一定质量添加药剂的养护土样填入不锈钢筒状模具中,利用电子万能试验机,以位移控制法压制试样。保持0.2 mm·min−1的垂直加载速率,将散状土样均匀压实至设计位置,在恒体积条件下静置0.5 h,防止试样回弹。最后,卸载后得到直径为50 mm、高度为5 mm、初始干密度为1.5 g·cm−3的压实试样,具体试样如图1所示。
1.3 实验方法
1) 土壤修复实验。设计修复药剂不同投加摩尔比,开展污染土壤修复实验,实验具体方案见表2。将称量好的还原剂加入Cr(Ⅵ)污染土中,充分拌和至混合物无明显颜色差异,然后利用NJ-160台式电动搅拌机强化搅拌5 min,以提高其均匀性。搅拌结束后,将混合均匀的土样转移至容器中并用保鲜膜覆盖防止水分蒸发,在恒温条件下养护7 d。
表 2 土壤修复实验方案与反应机理Table 2. Stirring test designs and reaction mechanism between Cr(Ⅵ) and reductants序号 还原剂种类 还原剂与Cr(Ⅵ)摩尔比 反应机理 参考文献 1 FeSO4 1 6Fe2++Cr2O72-+14H+=2Cr3++6Fe3++7H2OxFe3++(1-x)Cr3++3H2O→(Fex,Cr1-x)(OH)3+3H+ [15-16] 2 2 3 3 4 4 5 5 6 CaSx 1 3CaS5+Cr2O72-+8H+=2Cr(OH)3+15S+3Ca2++H2O [5,16-18] 7 2 8 3 9 4 10 5 11 无 — — — | Show TableDownLoad:
CSV
2) 渗透淋滤实验。渗透淋滤实验装置示意图如图2所示。实验过程中定期收集渗出液并测量体积,根据达西定律计算渗透系数,渗透系数计算公式如式1所示。根据计算得到的污染土和修复土的渗透系数,以及测定的渗出液中重金属离子浓度,进一步分析渗出液中金属浓度随渗出液体积的变化规律。
K=ΔQ⋅LA⋅Δh⋅Δt (1) 式中:K为渗透系数,cm·s−1;ΔQ为一定时间内出流水量,mL;L为土样高度,cm;Δh为水头差,cm;A为土样横截面积,cm2;Δt为渗流时间,s。
1.4 测试方法
采用二苯碳酰二肼分光光度法测定土样渗出液中Cr(Ⅵ)浓度;采用XRD实验来判定修复前后Cr(Ⅵ)污染土的物相组成,XRD表征条件:CuKa靶,电压为40 kV,电流为40 mA,扫描速度为 2 °·min−1,光源波长为1.54 nm,角度为5°~90°;铬赋存形态采用改进BCR方法[19]。
2. 结果与讨论
2.1 还原剂掺量对渗透系数的影响
FeSO4和CaSx修复Cr(Ⅵ)污染粉质黏土的渗透系数随投加摩尔比变化的曲线如图3所示。由图可以看出,使用CaSx修复后的土样渗透系数明显低于使用FeSO4修复后土样的渗透系数,且使用CaSx修复后土样的渗透系数小于原污染土 (FeSO4/Cr(Ⅵ)摩尔比为0) 。
原污染土的渗透系数为4.25×10−7 cm·s−1,FeSO4/Cr(Ⅵ)摩尔比由1增加到5时,土样的渗透系数分别为6.78×10−7、1.04×10−6、1.13×10−6、7.95×10−7、3.73×10−7 cm·s−1,FeSO4修复Cr(Ⅵ)污染粉质黏土渗透系数随摩尔比的增加呈先增大后减小,在摩尔比为3时渗透系数最大。由表2反应机理式可知,FeSO4/Cr(Ⅵ)摩尔比为3时,Fe2+和Cr2O72-反应刚好能完全反应生成Cr3+和Fe3+。在FeSO4加入量较少时,Fe3+和Cr3+反应生成的(Fex,Cr1-x)(OH)3是难溶于水的金属化合物,但由于土壤环境为酸性,反应会向减少沉淀的方向进行;在FeSO4/Cr(Ⅵ)摩尔比超过3时,Fe2+投入过量,可生成大量Fe(OH)3沉淀,土壤的渗透系数降低。此外,通常将渗透系数小于1×10−6 cm·s−1作为土壤修复的控制标准之一[12],见图3虚线。通过对比Cr(Ⅵ)污染土修复前后渗透系数变化,可以发现:FeSO4处理后的污染土的渗透系数在FeSO4/ Cr(Ⅵ)摩尔比为2和3时要略高于该控制标准,而FeSO4/Cr(Ⅵ)摩尔比为1、4和5的土样渗透系数明显低于该控制标准。
使用CaSx修复时,CaSx/Cr(Ⅵ)摩尔比由1增加到5,土样的渗透系数分别为1.83×10−7、1.72×10−8、1.22×10−7、1.39×10−7、1.47×10−7 cm·s−1,实验在摩尔比为2时渗透系数最小,CaSx修复Cr(Ⅵ)污染粉质黏土渗透系数随摩尔比的增加呈先减小后缓慢增大的趋势,这与FeSO4修复后土壤渗透系数的变化相反。由表2反应机理公式可知,在CaS5/Cr(Ⅵ)摩尔比为1.5时,理想状态下,CaS5和Cr2O72-反应刚好能完全反应生成Cr(OH)3和单质S;在CaSx加入量较少时,CaSx和Cr2O72-反应不会生成过多的沉淀物质;在CaSx/Cr(Ⅵ)摩尔比超过2时,随着CaSx投入量的增加,土壤中产生的单质S和Ca2+也随之增加,Ca2+可生成微溶于水的CaSO4,使土壤的渗透系数增大。CaSx/Cr(Ⅵ)摩尔比为2时试样渗透系数最小,可能是由于下述原因。CaSx是含有多种硫化物的混合体系,本实验所用不为纯CaS5,其中含有CaS5及其他杂质,在铬污染土壤修复方面还没有比较完善的反应机理,尚需深入地研究[8,20]。
对比Cr(Ⅵ)污染土修复前后渗透系数的变化可以发现:CaSx修复的污染土渗透系数均明显低于修复土控制标准1×10−6 cm·s−1,总体比FeSO4处理后的土壤渗透系数要小。此前多见固化土渗透系数的研究,但使用还原剂稳定化后的土壤进行渗透实验的相关研究较少,可由本次实验得出稳定化Cr(Ⅵ)土壤渗透系数随还原剂种类及掺量的变化规律。
2.2 还原剂掺量对渗出液Cr(Ⅵ)浓度的影响
原污染土、FeSO4/ Cr(Ⅵ)摩尔比为1到5的修复土样渗出液Cr(Ⅵ)浓度如图4所示。由图可知:随渗出液体积增大,污染土样渗出液中重金属Cr(Ⅵ)浓度不断降低。在实验开始阶段,土样的孔隙水中溶解的Cr(Ⅵ)随渗出液被冲出,浸出液中重金属浓度较大;随着实验的进行,土样中Cr(Ⅵ)的总量逐渐减少,因此由固相 (黏土矿物吸附态) 进入液相 (孔隙水溶解态) 的重金属也逐渐减少,重金属的浓度曲线趋于平缓[12]。原污染土中Cr(Ⅵ)渗出浓度由一开始的21.96 mg·L−1降至9.05 mg·L−1,FeSO4/Cr(Ⅵ)摩尔比为1时,Cr(Ⅵ)渗出浓度由1.72 mg·L−1降至0.22 mg·L−1,均大于IV类地下水标准Cr(Ⅵ)浓度限值(0.10 mg·L−1)和III类地下水标准Cr(Ⅵ)浓度限值(0.05 mg·L−1)[21]。当FeSO4/Cr(Ⅵ)摩尔比大于3时,渗出液中Cr(Ⅵ)的浓度始终低于III类地下水标准。该现象进一步说明,用FeSO4修复Cr(Ⅵ)污染土的最优摩尔比为3,污染土中大量Cr(Ⅵ)被还原为Cr(Ⅲ)稳定在土壤中,有效减缓了重金属运移过程。
 图 4 FeSO4修复后土样的渗出液中Cr(Ⅵ)浓度Figure 4. Changes of Cr(VI) concentration in exudate of soil after FeSO4 repair
图 4 FeSO4修复后土样的渗出液中Cr(Ⅵ)浓度Figure 4. Changes of Cr(VI) concentration in exudate of soil after FeSO4 repair图5给出了原污染土、CaSx/Cr(Ⅵ)摩尔比为由1到5的修复土样渗出液Cr(Ⅵ)浓度。由图5可知:当渗出液体积随时间增大时,污染土样渗出液中重金属Cr(Ⅵ)浓度稳定降低。CaSx/Cr(Ⅵ)摩尔比为1时,土中的Cr(Ⅵ)渗出浓度由最初的9.42 mg·L−1降至5.41 mg·L−1,CaSx/Cr(Ⅵ)摩尔比为2时,土中的Cr(Ⅵ)渗出浓度由7.77 mg·L−1降至0.42 mg·L−1,均大于IV类地下水标准Cr(Ⅵ)浓度限度(0.10 mg·L−1)。当CaSx/Cr(Ⅵ)摩尔比大于3时,在实验过程后期,渗出液中Cr(Ⅵ)的浓度会降至低于III类地下水标准Cr(Ⅵ)浓度限值。该现象说明,使用CaSx修复Cr(Ⅵ)污染土,其反应的最优摩尔比与理论值 (1.5,见表2) 存在差异,需视具体反应的情况而定。
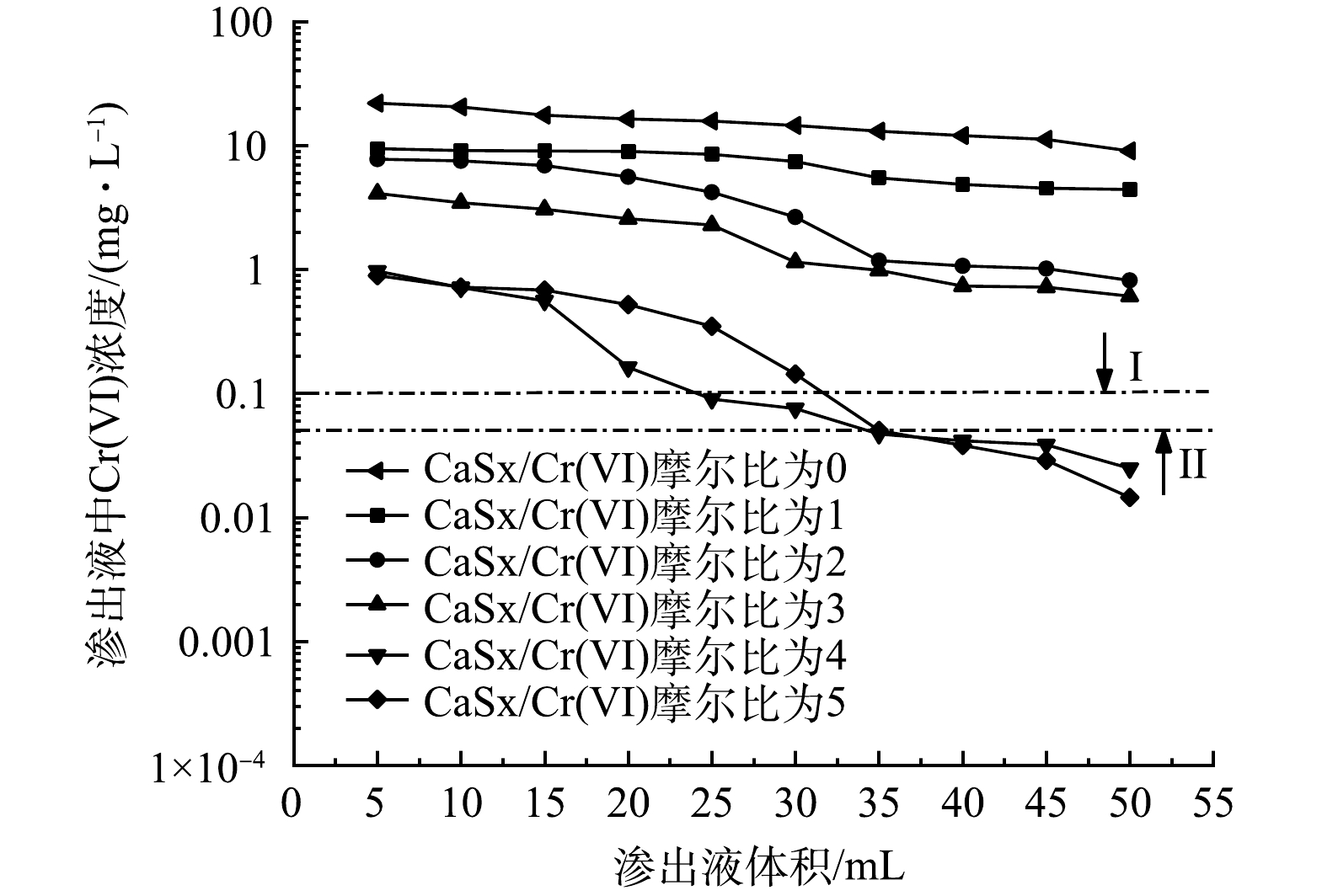 图 5 CaSx修复后土样的渗出液中Cr(Ⅵ)浓度Figure 5. Changes of Cr(Ⅵ) concentration in exudate of soil after CaSx repair
图 5 CaSx修复后土样的渗出液中Cr(Ⅵ)浓度Figure 5. Changes of Cr(Ⅵ) concentration in exudate of soil after CaSx repair由图4和图5可知:随着渗出液体积逐渐增大,修复后土样渗出液中Cr(Ⅵ)浓度逐渐减小。与原Cr(Ⅵ)污染土样渗出液中Cr(Ⅵ)浓度相比,修复后土样渗出液中Cr(Ⅵ)浓度显著降低,而且随着掺入还原剂摩尔比的增加,渗出液中Cr(Ⅵ)浓度进一步降低。总体来说,污染土经修复处理后,孔隙水中Cr(Ⅵ)浓度显著降低。该现象说明:修复土中Cr(Ⅵ)通过渗透作用向周围环境运移达到稳定状态的时间,较未修复时得到了显著缩短。上述现象均说明在土壤修复后,土中Cr(Ⅵ)的化学稳定性得到明显提升、运移性显著降低。其原因是,CaSx和FeSO4与Cr(Ⅵ)通过一系列氧化还原反应、离子交换反应及氢氧化物沉淀反应[22-24],改变了铬的价态与形态,降低了铬的毒性,将铬稳定于土壤基质中,土样孔隙水中的Cr(Ⅵ)浓度降低,有效减弱了固相重金属向液相环境转化,因此在渗透实验中渗出液重金属浓度明显降低[25]。
2.3 铬形态分布
FeSO4/Cr(Ⅵ)摩尔比对铬形态分布的影响如图6所示。从图6中可以发现,未修复的Cr(Ⅵ)污染土中弱酸提取态铬质量分数最高,达86%,这是因为采用室内人工配制的Cr(Ⅵ)污染土,污染周期较短,Cr(Ⅵ)主要以弱酸提取态赋存于土壤中[26],可还原态和可氧化态的铬质量分数次之,分别为6%和7%,残渣态铬最低,质量分数约为1%。随着FeSO4/Cr(Ⅵ)的摩尔比的增加,修复土中弱酸提取态铬质量分数明显降低,可还原态和可氧化态铬质量分数显著增大,残渣态的铬质量分数有细微增加。当摩尔比从0增加到 5时,修复土中弱酸提取态的铬质量分数从86%降低至23%,而可还原态和可氧化态的铬质量分数从6%、7%增加到27%、47%。实验结果表明:FeSO4可促使Cr从活性态向较稳定态转化,提高了修复土中Cr稳定性。
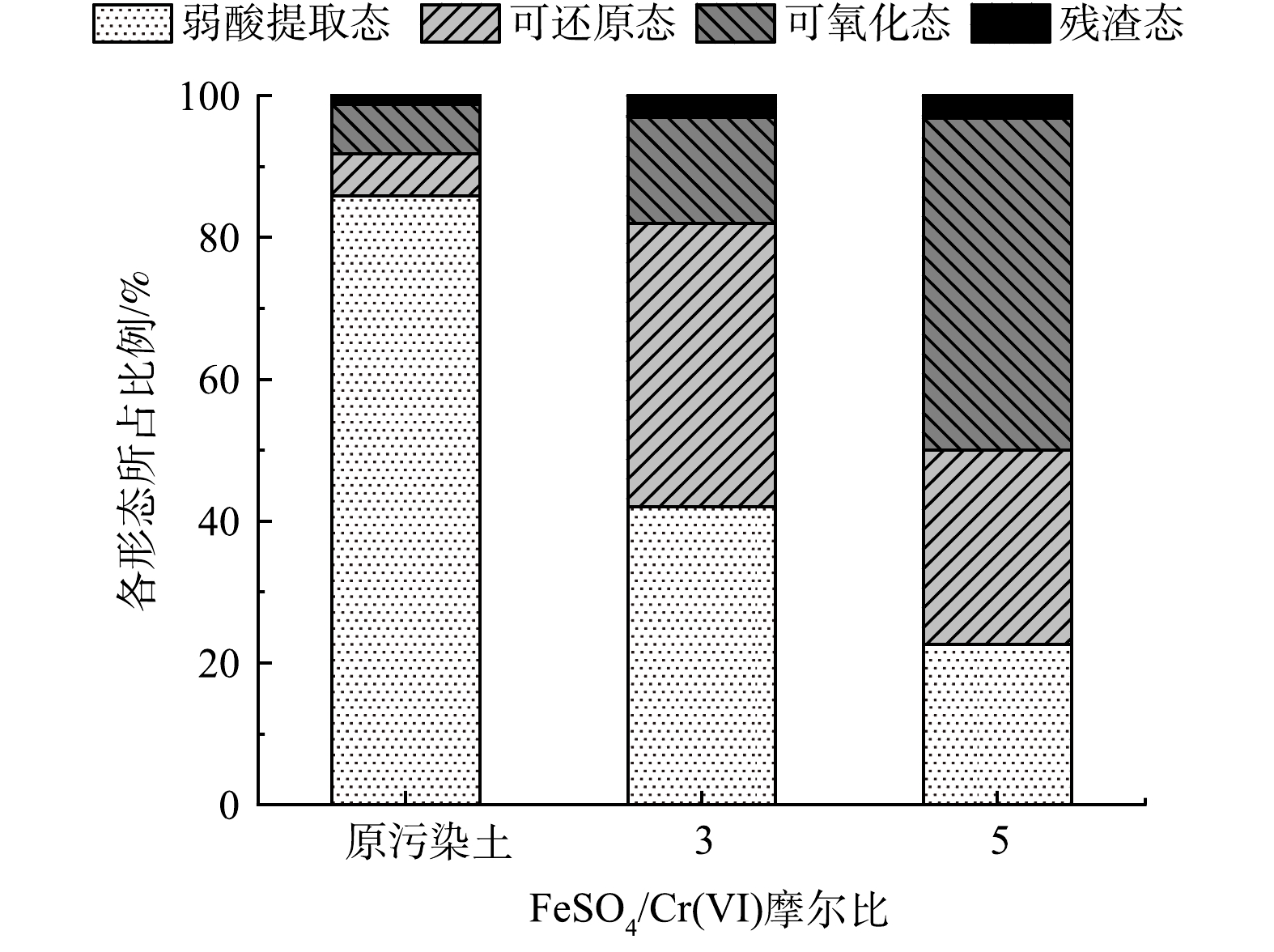 图 6 FeSO4/Cr(VI)摩尔比对铬形态分布的影响Figure 6. Effect of FeSO4 on chromium speciation in the soil
图 6 FeSO4/Cr(VI)摩尔比对铬形态分布的影响Figure 6. Effect of FeSO4 on chromium speciation in the soilCaSx/Cr(Ⅵ)摩尔比对铬形态分布的影响如图7所示,与FeSO4效果类似。分析其原因,Fe2+将Cr(Ⅵ)还原成Cr(Ⅲ),一方面Cr(Ⅲ)与土壤中的氢氧化物、氧化物及黏土矿物发生离子交换生成Cr(OH)3等沉淀;另一方面Cr(OH)3与Fe3+发生离子交换生成较难溶的沉淀物 (Fex,Cr1-x)(OH)3,且在摩尔比较小时,土壤中的Cr(Ⅲ)主要以Cr(OH)3存在,随着摩尔比的增大,Cr(OH)3会逐步转化成(Fex,Cr1-x)(OH)3[27],铬逐渐从活性态向稳定态转化,降低了铬的迁移能力。CaSx将Cr(Ⅵ)还原成Cr(Ⅲ),由于CaSx能提高修复土的pH 值,一方面促使Cr(Ⅲ)与土壤中的氢氧化物、氧化物发生离子交换生成Cr(OH)3等沉淀;另一方面CaSx被Cr(Ⅵ)氧化成单质S,部分Cr(Ⅲ)被单质S络合或包裹;此外,CaSx将部分Cr(Ⅵ)还原,生成了Cr(OH)3沉淀,降低了渗出风险[28]。
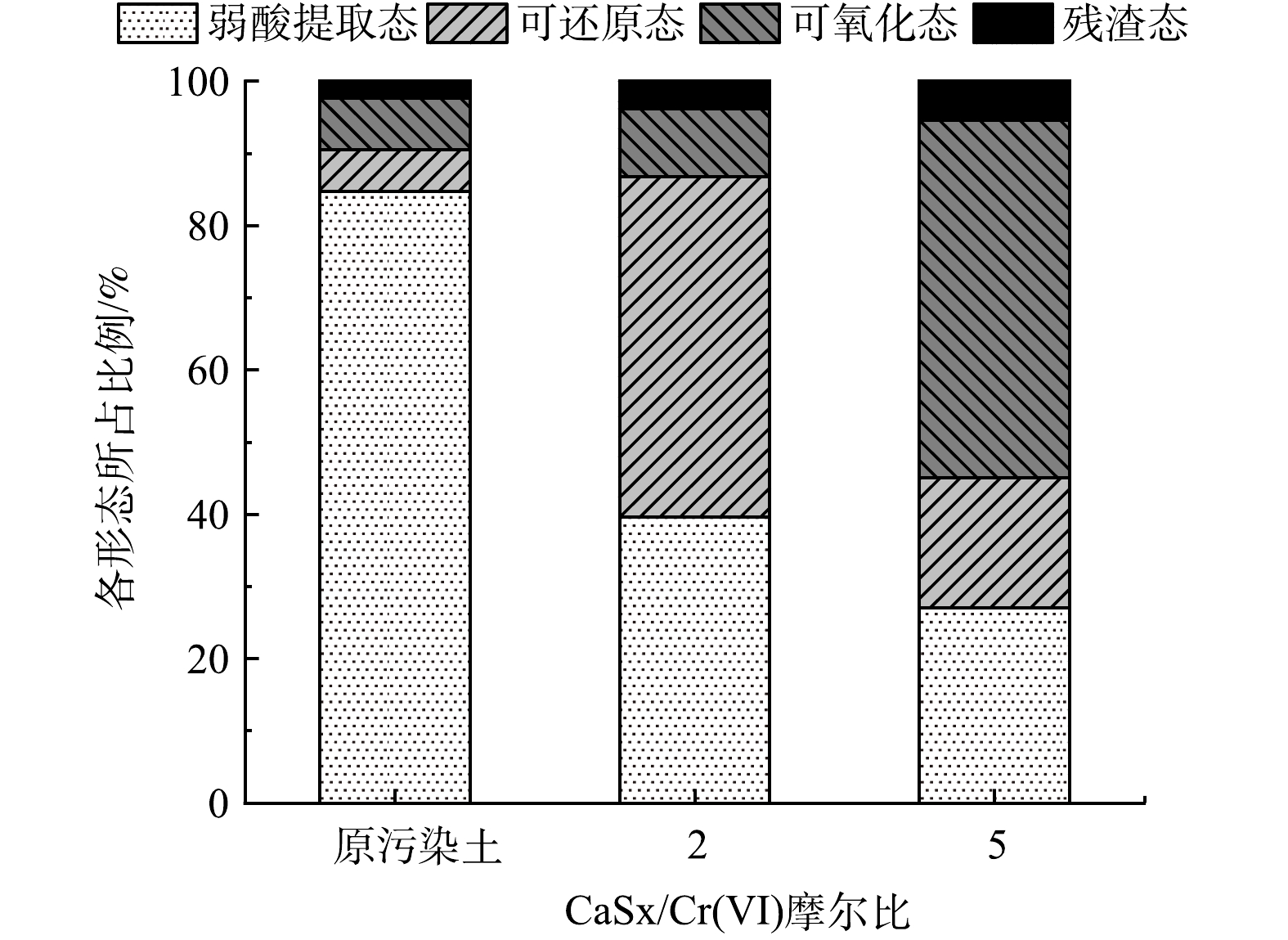 图 7 CaSx/Cr(VI)摩尔比对铬形态分布的影响Figure 7. Effect of CaSx on chromium speciation in the soil
图 7 CaSx/Cr(VI)摩尔比对铬形态分布的影响Figure 7. Effect of CaSx on chromium speciation in the soil2.4 成分分析
分别选取渗透实验中最具代表性的2组修复实验: FeSO4/Cr(Ⅵ)摩尔比为3和5、CaSx/Cr(Ⅵ)摩尔比为2和5的供试土壤,采用X射线衍射实验对土壤中的物质、元素、相对大颗粒进行物相分析,研究2种还原剂FeSO4和CaSx对Cr(Ⅵ)污染土的微观修复机理。
通过X射线衍射实验得到FeSO4和CaSx修复后土的衍射曲线如图8和9所示,在衍射角为5°~90°时,修复后土壤中主要存在石英(SiO2)、铬云母(KAl2[Si3AlO10](OH,F)2)和三水合氢氧化铬(Cr(OH)3·3H2O)的衍射峰。由图8中可知,用FeSO4修复后的Cr(Ⅵ)污染土中含有铁元素的衍射峰,当衍射角为35.24°时,FeSO4/Cr(Ⅵ)摩尔比为3和5的污染土中,氢氧化铁的衍射强度由1 003增至1 209 s−1,当衍射角为18.20°时,三水合氢氧化铬的衍射强度分别由3 901降至3 227 s−1。在图9中可知,用CaSx修复后的Cr(Ⅵ)污染土中含有硫单质的衍射峰,当衍射角为56.08°时,CaSx/Cr(Ⅵ)摩尔比为2和5的污染土中,单质S的衍射强度由3 452增至4 253 s−1,当衍射角为18.20°时,三水合氢氧化铬的衍射强度分别由4 744降至3 992 s−1,土样的其他主要物相峰值基本不变。以上现象证明了 FeSO4和CaSx在土中发生了一系列物理化学反应,使得土壤中的Cr(Ⅵ)质量分数降低,进一步验证了不同还原剂作用下,六价铬污染粉质黏土的渗透淋滤特性及组成成分变化。
3. 结论
1) 试样的渗透系数K随着还原剂的种类和摩尔比的不同有着非线性的关系,FeSO4处理后的土壤渗透系数随着渗透的进行先变大后减小,而CaSx处理后的土壤渗透系数随着渗透的进行先减小后变大。其中,FeSO4/Cr(Ⅵ)摩尔比为3时渗透系数最大;CaSx/Cr(Ⅵ)摩尔比为2时渗透系数最小。
2) 修复后土中Cr(Ⅵ)的渗出量随着时间的增长持续减小,当FeSO4或CaSx与Cr(Ⅵ)的摩尔比大于3时,最终渗出液质量浓度低于III类地下水标准,FeSO4处理后土壤Cr(Ⅵ)的渗出液质量浓度总体比CaSx处理后的土壤低。
3) 通过BCR分布提取法和XRD物相分析可知,重金属Cr(Ⅵ)与还原剂发生一系列物理化学反应,改变了修复土中各物质成分及铬的赋存形态。还原剂可促使铬由弱酸提取态向可还原态、可氧化态转化,而对残渣态的铬影响不大。
-
图 4 FeSO4修复后土样的渗出液中Cr(Ⅵ)浓度
Figure 4. Changes of Cr(VI) concentration in exudate of soil after FeSO4 repair
图 5 CaSx修复后土样的渗出液中Cr(Ⅵ)浓度
Figure 5. Changes of Cr(Ⅵ) concentration in exudate of soil after CaSx repair
图 6 FeSO4/Cr(VI)摩尔比对铬形态分布的影响
Figure 6. Effect of FeSO4 on chromium speciation in the soil
图 7 CaSx/Cr(VI)摩尔比对铬形态分布的影响
Figure 7. Effect of CaSx on chromium speciation in the soil
表 1 实验用土壤基本物理力学参数
Table 1. Basic physical and mechanical parameters of soil
天然含水率/% 天然干密度/(g·cm−3) 最优含水率/% 液限/% 塑限/% 比重 6.11 1.54 20.5 37.3 22.4 2.72
下载: 导出CSV
表 2 土壤修复实验方案与反应机理
Table 2. Stirring test designs and reaction mechanism between Cr(Ⅵ) and reductants
下载: 导出CSV
-
[1] 白俊宾, 江晓玲, 杨红健. 几种常用还原剂在铬污染土壤中的应用探讨[J]. 化学试剂, 2021, 43(2): 127-135. doi: 10.13822/j.cnki.hxsj.2021007819 [2] 王兴润, 李磊, 颜湘华, 等. 铬污染场地修复技术进展[J]. 环境工程, 2020, 38(6): 1-8. doi: 10.13205/j.hjgc.202006001 [3] CHOPPALA G, KUNHIKRISHNAN A, SESHADRI B, et al. Comparative sorption of chromium species as influenced by pH, surface charge and organic matter content in contaminated soils[J]. Journal of Geochemical Exploration, 2018, 184: 255-260. [4] PRADO C, PONCE S C, PAGANO E, et al. Differential physiological responses of two Salvinia species to hexavalent chromium at a glance[J]. Aquatic Toxicology (Amsterdam, Netherlands), 2016: 213-221. [5] 卢鑫, 罗启仕, 刘馥雯, 等. 硫化物对电镀厂铬污染土壤的稳定化效果及其机理研究[J]. 环境科学学报, 2017, 37(6): 2315-2321. doi: 10.13671/j.hjkxxb.2017.0028 [6] 史开宇, 王兴润, 范琴, 等. 不同还原药剂修复Cr(Ⅵ)污染土壤的稳定性评估[J]. 环境工程学报, 2020, 14(2): 473-479. [7] 任学昌, 张曦, 时秋红, 等. FeSO4联合固化剂修复铬污染土壤的稳定化研究[J]. 安全与环境学报, 2022, 22(4): 2231-2240. [8] 张亭亭, 魏明俐, 熊欢, 等. 多硫化钙对铬污染土的稳定性能及铬赋存形态试验研究[J]. 岩石力学与工程学报, 2017, 36(S2): 4282-4289. doi: 10.13722/j.cnki.jrme.2017.1166 [9] CHRYSOCHOOU M, JOHNSTON C P, DAHAL G. A comparative evaluation of hexavalent chromium treatment in contaminated soil by calcium polysulfide and green-tea nanoscale zero-valent iron[J]. Journal of Hazardous Materials, 2012, 201: 33-42. [10] SHACKELFORD C D, GLADE M J. Analytical mass leaching model for contaminated soil and soil stabilized waste[J]. Ground Water, 1997, 35(2): 233-242. doi: 10.1111/j.1745-6584.1997.tb00080.x [11] 邵俐, 施倩芸, 蒙强. 固化重金属铜镉污染底泥渗透特性试验研究[J]. 上海理工大学学报, 2018, 40(4): 403-408. doi: 10.13255/j.cnki.jusst.2018.04.015 [12] 冯亚松. 镍锌复合重金属污染黏土固化稳定化研究[D].南京: 东南大学, 2021. [13] 国家市场监督管理总局, 中华人民共和国住房和城乡建设部. 土工试验方法标准: GB/T 50123-2019[S]. 北京: 中国计划出版社, 2019. [14] 国家市场监督管理总局, 生态环境部. 土壤环境质量 建设用地土壤污染风险管控标准: GB 36600-2018[S]. 北京: 中国环境科学出版社, 2018. [15] 王宇峰, 杨强, 刘磊, 等. 不同还原剂对某铬渣污染场地修复效果的实验研究[J]. 环境污染与防治, 2017, 39(4): 384-387+391. doi: 10.15985/j.cnki.1001-3865.2017.04.008 [16] 黄莹, 徐民民, 李书鹏, 等. 还原稳定化法修复六价铬污染土壤的中试研究[J]. 环境工程学报, 2015, 9(2): 951-958. doi: 10.12030/j.cjee.20150274 [17] 王婉玉, 刘元元, 黄思敏, 等. 利用CPS还原稳定化修复Cr(Ⅵ)污染土壤[J]. 环境工程学报, 2017, 11(6): 3853-3860. doi: 10.12030/j.cjee.201604039 [18] 龚亚龙, 范敏, 高晓梅, 等. 铬污染土壤还原固化稳定化药剂的筛选[J]. 土木与环境工程学报(中英文), 2021, 43(5): 187-195. [19] RAURET G, LÓPEZ-SÁNCHEZ J F, SAHUQUILLO A, et al. Improvement of the BCR three step sequential extraction procedure prior to the certification of new sediment and soil reference materials[J]. Journal of Environmental Monitoring, 1999, 1(1): 57-61. doi: 10.1039/a807854h [20] 刘馥雯, 罗启仕, 卢鑫, 等. 多硫化钙对铬污染土壤处理效果的长期稳定性研究[J]. 环境科学学报, 2018, 38(5): 1999-2007. [21] 中华人民共和国国家质量监督检验检疫总局, 中国国家标准化管理委员会. 地下水质量标准: GB/T 14848-2017[S]. 北京: 中国环境科学出版社, 2018. [22] YUAN W Y, XU W T, ZHANG Z W, et al. Rapid Cr(VI) reduction and immobilization in contaminated soil by mechanochemical treatment with calcium polysulfide[J]. Chemosphere, 2019, 227: 657-661. doi: 10.1016/j.chemosphere.2019.04.108 [23] ZHANG W J, LIN M F. Influence of redox potential on leaching behavior of a solidified chromium contaminated soil[J]. Science of the Total Environment, 2020, 733: 139410. doi: 10.1016/j.scitotenv.2020.139410 [24] 贺勇, 胡广, 张召, 等. 污染场地六价铬迁移转化机制与数值模拟研究[J]. 岩土力学, 2022, 43(2): 528-538. [25] 李磊, 朱伟, 屈阳, 等. 低渗透污染土水动力弥散参数试验研究[J]. 岩土工程学报, 2011, 33(8): 1308-1312. [26] 赵虎彪. 铁系物还原稳定技术在铬污染土壤修复中的应用研究[D]. 杭州: 浙江大学, 2019. [27] 张亭亭, 李江山, 薛强, 等. FeSO4稳定铬污染土的铬赋存形态及浸出特性研究[J]. 岩土力学, 2019, 40(12): 4652-4658. [28] 赵祥, 姜春露, 陈星, 等. 多硫化钙与亚铁盐联用去除工业废水中的Cr(Ⅵ)[J]. 环境工程学报, 2021, 15(12): 3854-3864. doi: 10.12030/j.cjee.202108155 -

 点击查看大图
点击查看大图

计量
- 文章访问数: 4266
- HTML全文浏览数: 4266
- PDF下载数: 126
- 施引文献: 0
