




-
近年来,药物和个人护理产品(pharmaceuticals and personal care products, PPCPs)在环境中的频繁出现引起了越来越多的关注。PPCPs可对水生生物和人类造成慢性毒性和内分泌紊乱,甚至引起致病菌耐药性的发生,对人体健康和生态系统构成潜在的严重威胁。布洛芬(ibuprofen, IBP)是目前世界上应用最多的一种非甾体抗炎药,也是在城镇污水处理厂二级出水中检出频率最高的一种PPCPs[1]。因此,探索从水环境中去除以布洛芬为代表的PPCPs技术已经成为环境领域的研究热点[2-3]。
目前, PPCPs的去除技术主要有生物技术[4-5]、臭氧氧化技术[6-8]、膜技术[9-11]、活性炭吸附技术[12-13]等。生物技术对PPCPs的降解效果并不稳定,这种现象与微生物的降解特性和PPCPs的结构有关[14]。膜技术与臭氧氧化技术对PPCPs的去除效果受PPCPs的种类和浓度影响[15]。活性炭可有效去除部分PPCPs,但对于极性化合物的去除效果有限,并且其他竞争物质的出现也会对活性炭的吸附能力产生影响[16]。
电催化强化技术在难降解废水的深度处理中表现出高效的污染物去除能力。CHEN等[17]通过O3+H2O2强化电催化体系对地表水中的氧氟沙星进行降解,发现O3单独和O3+H2O2强化电催化体系均可快速氧化氧氟沙星,与单独臭氧氧化(30%)相比,O3+H2O2体系显著提高了氧氟沙星的矿化率(55%)。BAVASSO等[18]采用O3强化电催化对利尿酮进行了去除,结果表明,在200 mA电流和酸性pH条件下,在2 h内可实现利尿酮的快速降解和矿化,这与羟基自由基的大量产生密不可分。SANTANA-MARTINEZ等[19]评价了H2O2强化电催化法的氧化效率,结果表明,在pH为3、电流密度为60 mA·cm−2、H2O2流速为4.7 L·min−1和电解质浓度为0.05 mol·L−1的条件下,苯酚的最高矿化率约为75%,其出水的原始毒性显著降低。综上所述,电催化强化对水体中的PPCPs类污染物具有高效的去除效率,然而电催化强化体系对布洛芬的深度去除效果和机制研究还很缺乏。因此,本研究设计了3种电催化强化处理体系,考察了H2O2浓度、O3投加量、腐殖酸等因素对IBP去除效果的影响,基于布洛芬的降解产物,阐明了IBP的降解机理。本研究以期为电催化强化体系去除以布洛芬为代表的PPCPs类污染物提供参考。
-
实验中所用到的布洛芬为分析纯,购于西格玛公司,实验中使用的其他试剂(如硫酸钠、磷酸氢二钠、硫酸、碳酸钙、过氧化氢等)均为分析纯购于国药公司,腐殖酸购买于西格玛公司,醋酸铵为色谱纯,购自于英国Alfa Aesar公司。实验中所需溶液均由密理博的高纯水系统产生的高纯水(阻抗18.2 MΩ)配制,实验中布洛芬溶液的质量浓度为10 mg·L−1。
-
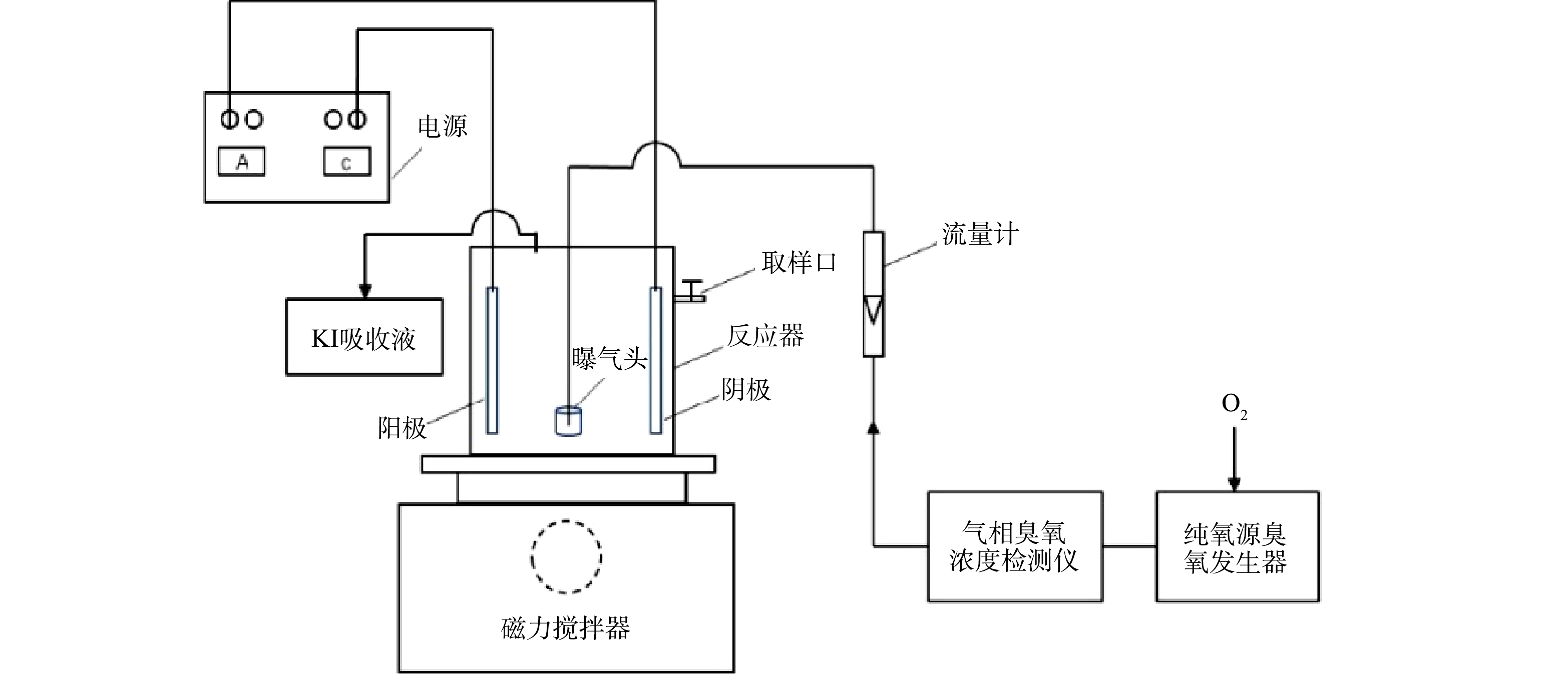
电催化及其强化的实验均在自行设计的玻璃容器中进行,容器有效容积为500 mL。实验中采用的阳极为钛镀钌铱电极,阴极为钛电极,电极间采用单极式连接方式,电极间距为4 mm,电极的有效面积为25 cm2(图1)。在处理中,每批次处理量为 200 mL,运行时间为0~30 min。电流由恒流恒压电源(大华,MC-100/5)控制,输出电压为0~50 V,输出电流为0~5 A,电流密度为30 mA·cm−2。所有实验均重复3次。对于电催化强化实验,则为在电催化反应基础上分别加入H2O2、O3和O3+H2O2,从而形成3种强化方式,单独电催化、H2O2强化电催化、O3强化电催化、O3+H2O2强化电催化以下分别简称为E0、E1,E2和E3。
-
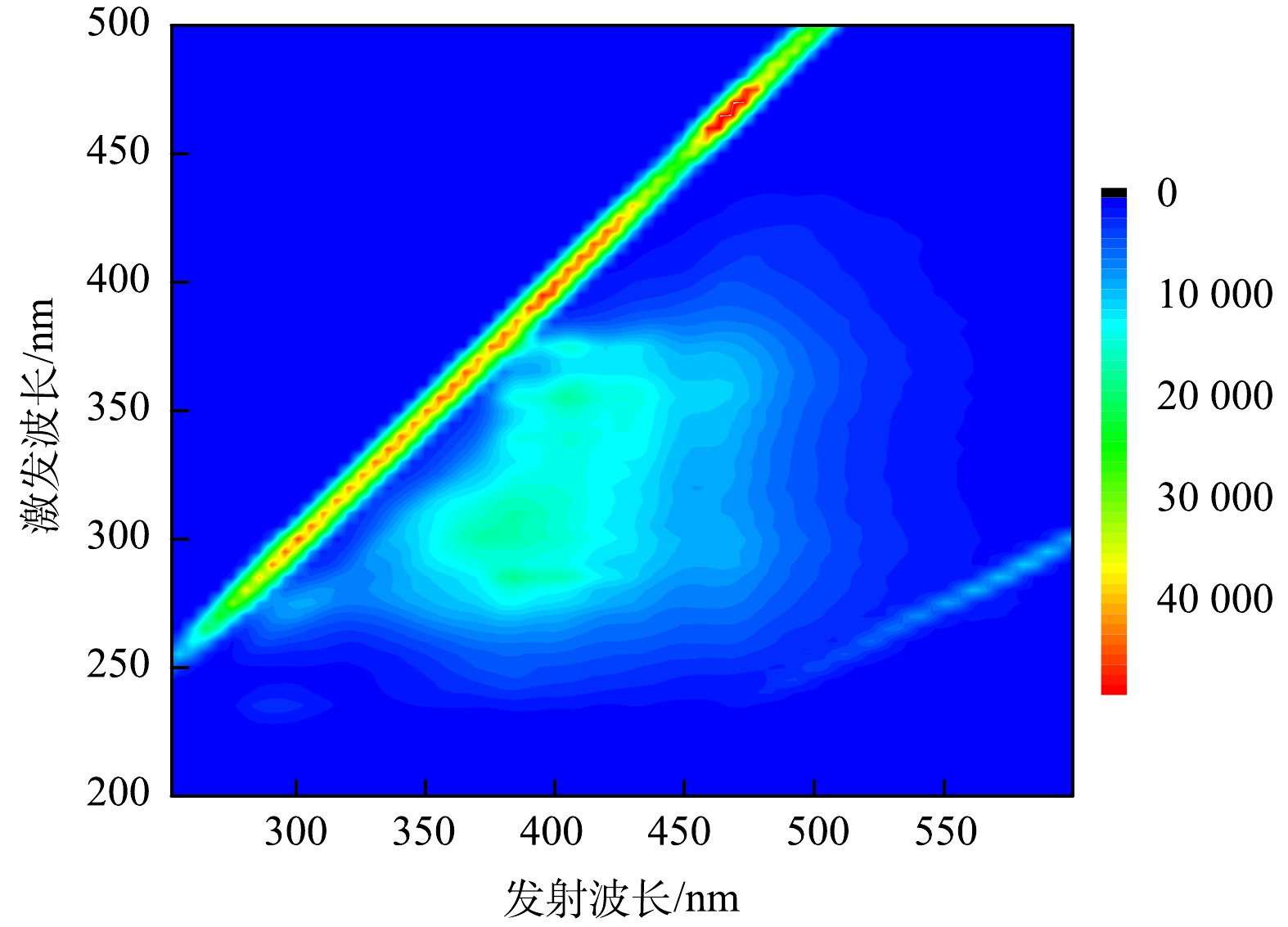
水样的荧光图谱采用Hitachi F-4600 (日本Hitachi公司)荧光色谱仪进行分析。分析仪激发光源为150 w氙弧灯,PMT电压为700 V,信噪比为110;激发波长为5 nm, 发射波长为5 nm; 响应时间设为自动,扫描速度为12 000 nm·min−1,激发波长为200~500 nm,发射波长为220~600 nm。布洛芬质量浓度通过Waters高效液相色谱仪测试,色谱柱为Agilent TC-C18,柱温为30 ℃,检测波长220 nm,流动相为75%甲醇和25%超纯水,流动相流速1 mL·min−1,进样体积50 μL。水样中有机物表观分子质量分布采用尺度排阻色谱-紫外分析仪测定(SEC-UV)。色谱柱采用 TSK-GEL G3000 PWxl型柱子(日本Tosoh Bioscience),流动相为1.2 g·L−1的磷酸二氢钠和2.5 g·L−1磷酸二氢钾的混合溶液,流速为1.0 mL·min−1。布洛芬在反应过程中的中间产物采用超高效液相色谱串联飞行时间质谱进行检测(UPLC/Q-TOF-MS)(UPLC,Ultimate 3200,Dionex,USA,micrOTOF III,Bruker,Germany),流动相为水(2 mmol·L−1醋酸铵)和乙腈,采用Waters Cortecs C18 色谱柱(1.7 μm,50 mm × 2.1 mm),流速为 0.4 mL· min−1,洗脱从10%的乙腈开始,持续2 min,然后在23 min内增加至60%的乙腈洗脱液,保持5 min。反应过程中产生的羟基自由基(·OH)采用Waters高效液相色谱仪测定,·OH的捕获剂为对-氯苯甲酸(0.5 μmol·L−1),色谱柱为Agilent TC-C18,检测波长为 254 nm,柱温为30 ℃;流动相为75%甲醇和25%超纯水,流速为1 mL· min−1。
-
为考察H2O2电催化强化体系对布洛芬的去除影响,分别投加3、6、9、12、15和17 mmol·L−1的H2O2,反应30 min结束后,计算布洛芬的去除率。由图2可见,随着H2O2投加量的提高,布洛芬的去除率显著升高。在H2O2为3 mmol·L−1时,布洛芬的去除率为34.1%,其去除率低于单独电催化时的去除率(46.2%);而在在H2O2为17 mmol·L−1时,布洛芬的去除率为54.1%,升高了58.6%。此外,从布洛芬的去除率变化趋势可以看出,H2O2添加并不能持续有效提高布洛芬的去除率。这是因为布洛芬在H2O2投加量为12、15和17 mmol·L−1条件下的去除率基本保持不变。添加H2O2有利于·OH的生成,但其生成效率受多方面影响。反应过程中H2O2的添加有利于·OH的生成速率增加, H2O2在反应中易分解生成·OH(式(1)),但随着H2O2浓度的增加,·OH的生成速率也会受到影响。这是因为H2O2会抑制·OH的生成(式(2)~式(4))。所以过量的H2O2不仅会增加运行成本,也不能高效去除布洛芬。结果表明,在单独电催化中投加3 mmol·L−1 H2O2所获得的布洛芬去除率相比单独电催化的去除率低,可能因为H2O2浓度的增加抑制了· OH的生成,从而影响了布洛芬的降解。
图3反映了在O3的质量浓度分别为5、10、20、30和40 mg·L−1、O3流量为0.8 mL·min−1,反应时间为30 min的条件下,O3电催化强化体系对布洛芬的去除效果。由图3可见,O3电催化强化体系对布洛芬去除效果较为显著。在O3加入量由5 mg·L−1增加到30 mg·L−1时,布洛芬的去除率由56%提高到84.5%。这是因为O3是氧的同素异形体,是一种较强的氧化剂,其氧化还原电位高达2.07 V,可通过断链、开环等一系列反应降解去除污染物;同时,在电催化过程中,O3的添加可促进反应过程中有效产生·OH,进而对布洛芬进行有效去除(式(5)~式(7))。
为了对比3种电催化强化体系对布洛芬的去除效果,分别考察了17 mmol·L−1 H2O2、30 mg·L−1 O3 (流量为0.8 mL·min−1)和17 mmol ·L−1H2O2+ 30 mg·L−1 O3 (流量为0.8 mL·min−1)3个体系对布洛芬的去除情况。如图4所示,随着反应时间的延长,布洛芬的去除率先快速上升,然后持续增加,3种强化方式在前10 min处理中的去除率上升较快,分别达到31.6% (E1)、45.1%(E2)和55.8%(E3)。在30 min后,对布洛芬的去除率顺序为为E3>E2>E1,其去除率分别为93.2%、84.5%和52.7%,高于电催化(E0)时46.2%的去除率。
图5所示的电催化及电催化强化体系中·OH的生成量,可以很好的解释上述布洛芬去除率的差异。由图5可知,E3在这几种电催化强化过程中的·OH生成量最高,并且其生成量随时间增加呈线性增长趋势,表明在该过程中·OH浓度基本保持稳定。其中,E0体系中·OH的生成量最低,在反应30 min后,·OH仅为0.13×10−9 mol·L−1。从电催化反应机理的角度来看,通常是通过电催化的电极材料表面直接氧化水中的有机污染物,同时电极材料通过电化学作用产生具有强氧化能力的自由基基团(羟基自由基(·OH)、超氧自由基(·O2)、H2O2等)间接氧化水中的有机污染物,最终达到降解去除污染物的目的。在O3和H2O2同时添加体系中,反应过程中H2O2与通入的 O3 发生反应(式(8))[20],可高效生成·OH (图5),对布洛芬进行深度降解。有研究[21]表明,O3 + H2O2强化电催化体系比单独的臭氧氧化和单独电催化体系对有机污染物具有更高效的去除效率。图4中E3对布洛芬的高效去除也进一步证实了这一点。
-
1)腐殖酸的质量浓度影响分析。污水处理厂的二级出水以及地表水环境中均包含大量的天然有机物质(natural organic matter, NOM),NOM不仅会影响电催化强化体系的催化效果,也会影响布洛芬的去除。以不同质量浓度的腐殖酸(humic acid, HA)为代表物质,考察HA对布洛芬去除效果的影响。电催化强化实验采用图4中的实验参数(17 mmol·L−1 H2O2、30 mg·L−1 O3 (流量为0.8 mL·min−1)和17 mmol ·L−1H2O2+ 30 mg·L−1 O3 (流量为0.8 mL·min−1)),反应时间为30 min。
由图6可见,HA的添加降低了电催化强化体系对布洛芬的去除效率。当HA的质量浓度为从1 mg·L−1增加到10 mg·L−1时,E1~E3中布洛芬的去除率均低于不添加HA的溶液,例如,E1在HA质量浓度为1 mg·L−1时的去除率为33.3%,而在10 mg·L−1质量浓度下的去除率为25.4%。这是由于反应过程中HA的添加会限制·OH的生成,减弱H2O2和O3的强化作用,从而抑制布洛芬的去除[22]。此外,布洛芬的去除率在HA质量浓度为10 mg·L−1时相比1 mg·L−1有显著的升高。这是因为HA会促进臭氧的自由基链式反应从而强化了布洛芬的去除[23]。布洛芬的去除受到以上2种因素共同作用。
2)分子质量分析。作为一个重要的水处理参数,有机污染物的分子量分布对研究有机污染物的特性以及布洛芬在电催化强化过程中的降解及去除机理具有重要作用。根据尺度排阻色谱-紫外分析结果,本研究的水样中主要含有2种分子质量的有机污染物。由图7可见,这2种有机物的保留时间分别为7.1 min和8.6 min,参照聚乙二醇和聚氧化乙烯标准物质的出峰时间,2种污染物的名义分子质量分别为28 800 Da和2 900 Da。结果表明,电催化强化体系对这2种有机污染物去除效果不同,其中单独电催化体系对2 900 Da的污染物没有去除作用,而对28 800Da的污染物可进行有效降解。在7.7 min 处有新的紫外吸收峰出现,所对应的化合物分子质量为11 150 Da。该结果表明,单独的电催化体系(E0)可使得28 800 Da的有机污染物发生部分降解,同时也产生了新的污染物。相比单独电催化(E0)的去除,电催化强化对这2种污染物均可有效去除。E3、E2和E1体系可完全去除28 800 Da的污染物,E3和E2对2 900 Da的污染物的去除率则为53.1%和41.2%,而E1对此污染物并没有去除作用。由此可见,电催化强化体系对大分子和小分子污染物均有较好的去除作用,并且E3的强化作用有利于小分子污染物的去除。该结果也进一步证明E3在3种电催化强化体系中的氧化作用最强,不仅能高效去除布洛芬(图4),对其他有机污染物也具有高效的降解;同时结合图7结果,该部分研究说明腐殖酸中分子质量为28 800 Da和2 900 Da的污染物降低了布洛芬的去除效率。
3)荧光光谱分析。荧光光谱分析对于研究布洛芬在电催化强化过程中的去除效果及降解机制具有重要作用。由图8可以发现,水样中主要包含2个特征荧光峰,对应的是溶解性微生物副产物和类腐殖酸化合物。这2种有机物也是限制布洛芬去除的主要原因。根据图中荧光强度的变化分析得出,溶解性微生物副产物和类腐殖酸化合物被有效去除,而且类腐殖酸化合物比溶解性微生物副产物更容易被去除。由图9可以看出,经30 min 处理后,E1、E2和 E3对类腐殖酸化合物的去除率分别为47.1%、69.2%和84.1%。这与氧化作用对有机污染物的选择性降解有直接的联系,在类似研究[24]中也证实了电化学氧化对类腐殖酸的去除率要高于溶解性微生物副产物。
-
硬度普遍存在于地表水和废水中,而过高的硬度会降低电催化体系对布洛芬的去除效率,并且增加运行成本[25]。硬度对电催化强化布洛芬去除率的影响结果如图10所示。硬度对布洛芬的去除具有显著的抑制作用,随着硬度的增加,3种电催化强化体系的去除率都受到影响。在没有硬度的干扰下,E3对布洛芬的去除率为93.2%,而在15 mg·L −1 CaCO3的质量浓度下,布洛芬的去除率为68.4%,下降了26.6%;而E2和E1的去除率也相应的下降了30.6%和40.2%。基于电催化过程的反应原理,水中的钙离子会在外加电场的作用下向阴极迁移,使得阴极溶液中碳酸钙的浓度达到过饱和,容易在极板附着形成沉淀,从而降低了反应的催化效率[26-27],最终导致布洛芬去除率的下降。
-
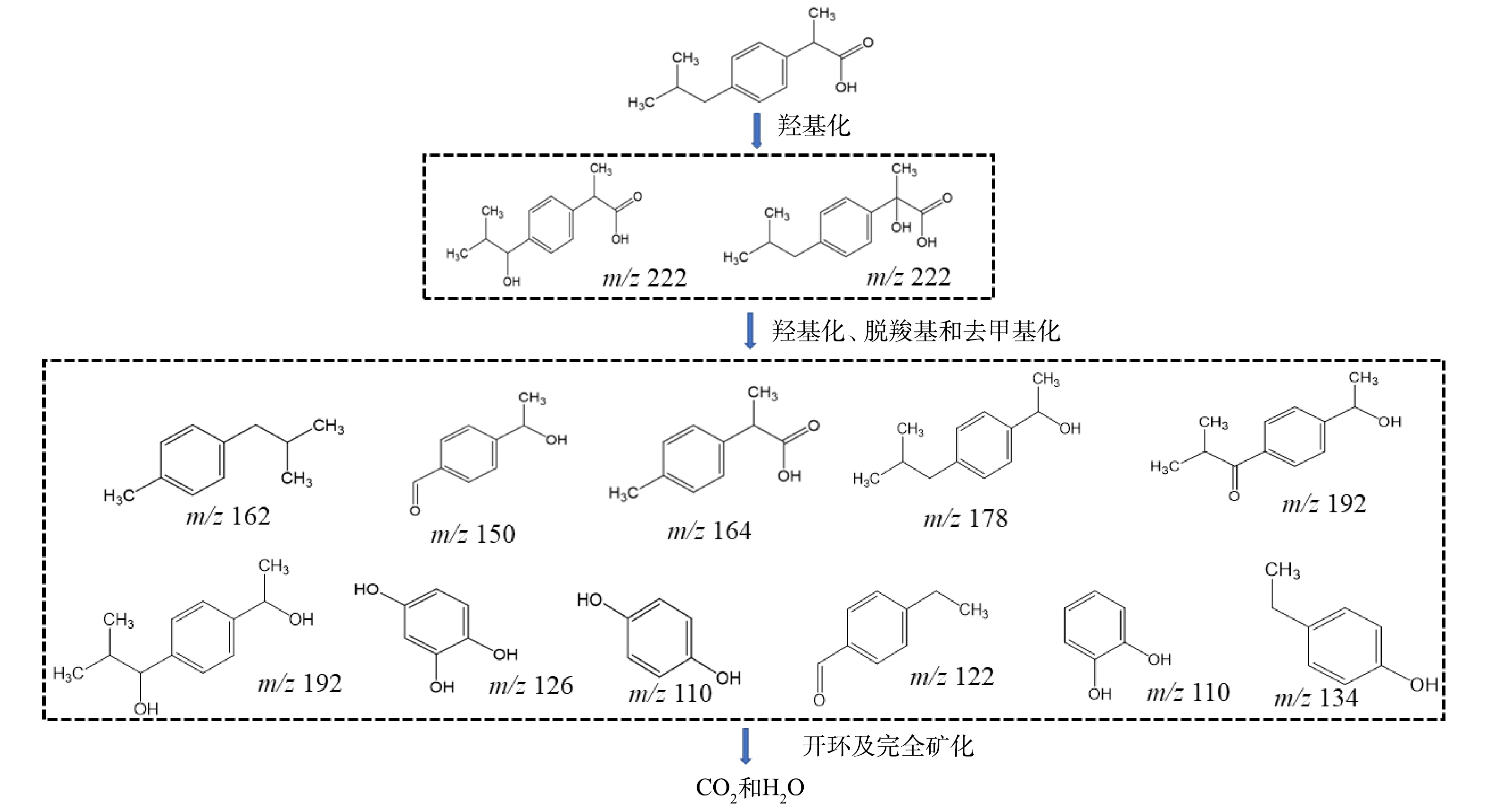
采用高效液相色谱串联飞行时间质谱(UPLC/Q-TOF-MS)分析了电催化强化反应过程中布洛芬的降解物。由表1可知,布洛芬在降解过程中共产生了13种中间产物,其他研究也证实了这些降解产物是电催化降解过程中的主要产物[28]。根据以上的降解产物提出了布洛芬的降解路径(图11)。由于反应过程中存在大量的中间体以及反应机制的复杂性,因此,该降解路径分析仅提供可能的理论参考。首先,在电催化强化反应过程中产生的·OH在不同位置攻击布洛芬,最终形成羟基化布洛芬异构体。然后羟基化的布洛芬进一步通过脱羧、脱甲基等一系列反应,生成了不同的小分子降解产物,反应过程中生成的·OH最终对苯环进行开环,氧化污染物为CO2和H2O,完成布洛芬的矿化作用[29]。
-
1)电催化强化可显著提高对布洛芬的去除率,在30 min的反应过程中,布洛芬的去除率从高到低依次为O3 + H2O2强化电催化> O3强化电催化> H2O2强化电催化,其去除率分别达到93.2%、84.5%和52.7%,高于单独电催化时的去除率。
2)有机物和硬度的存在均降低了电催化强化对布洛芬的去除率,其中类腐殖酸和溶解性微生物副产物是限制布洛芬去除的主要原因。3种电催化强化体系可彻底去除分子质量为28 800 Da的污染物,O3+H2O2体系和O3体系对分子质量为2 900 Da的污染物的去除率分别为53.1%和41.2%,而H2O2强化电催化对该有机污染物并没有去除效果。
3)布洛芬在降解过程中产生了13种中间产物,反应过程中产生的·OH从不同位置攻击布洛芬,形成羟基化布洛芬异构体,然后羟基化的布洛芬进一步通过脱羧、脱甲基等一系列反应,最终将污染物氧化为CO2和H2O。
电催化强化对布洛芬的去除效果及机制
Removal effect and mechanism of ibuprofen by enhanced electrocatalysis
-
摘要: 对比研究了O3 + H2O2电催化、 O3电催化和H2O2电催化这3种电催化强化体系对水中难降解污染物布洛芬的去除效果,并且进一步阐明了布洛芬的降解机制。结果表明:经30 min反应后,3种电催化强化体系对布洛芬的去除率分别达到93.2%、84.5%和52.7%,均高于在单独电催化条件下的去除率(46.2%)。尺度排阻色谱-紫外分析结果表明3种电催化强化体系对大分子(28 800 Da)和小分子(2 900 Da)有机污染物均有较好的去除作用。腐殖酸的存在会显著降低布洛芬的去除率。荧光光谱分析结果表明,类腐殖酸和溶解性微生物副产物是限制布洛芬去除的主要原因。硬度离子的存在对布洛芬去除影响较大,在CaCO3的质量浓度为15 mg·L −1时,O3 + H2O2体系对水中布洛芬的去除率相比去离子水溶液时下降了26.6%。研究阐明了布洛芬的降解路径,布洛芬在降解过程中产生了13种中间产物,反应中通过脱羧、脱甲基等一系列反应,最终将污染物氧化为CO2和H2O。Abstract: In this study, the performance of O3+H2O2 enhanced electrocatalysis, O3 enhanced electrocatalysis and H2O2 enhanced electrocatalysis on ibuprofen removal from water was compared, and the degradation mechanism was analyzed. The results showed that after 30 min reaction, the removal rates of ibuprofen by these three types of enhanced electrocatalysis were 93.2%, 84.5% and 52.7%, respectively, which were higher than 46.2% by electrocatalysis alone. The three enhanced electrocatalysis systems had good removal effects on macromolecular (28 800 Da) and small molecular (2 900 Da) pollutants detected by size exclusion chromatography-UV. Humic acid in water significantly reduced the removal effect of ibuprofen. Fluorescence spectrum analysis showed that the humic acid like and soluble microbial by-products were the main reason for restricting ibuprofen removal. The hardness of water significantly reduced the removal efficiency of ibuprofen. The removal rate of ibuprofen by O3+H2O2 enhanced electrocatalysis decreased by 26.6% at the 15 mg·L−1 CaCO3 in comparison with that in deionized aqueous background solution. The degradation path of ibuprofen was clarified, 13 intermediate products were produced during the degradation process of ibuprofen. In the reaction, the ibuprofen was finally oxidized to CO2 and H2O through a series of reactions such as decarboxylation and demethylation.
-
Key words:
- electrocatalysis /
- ibuprofen /
- organic matters /
- degradation /
- molecular weight
-
煤矸石是煤炭开采过程中的大宗工业固废,目前处置方式以堆存为主,存在占用土地、生态破坏、地下水污染等问题。煤矿瓦斯在经燃烧利用所产中低温烟气余热未得到有效利用,造成能源的大量浪费。
煤矸石在矿物组成上与土壤具有高度的相似性,但因其粒径粗大、保水性能差等原因,煤矸石大规模生态化利用受限。魏忠义和王秋兵[1]研究认为微粒含量上升即持水率提高是煤矸石复垦的先决条件,胡振琪等[2]认为矿区复垦重点在于土壤性能综合提升。煤矸石颗粒粉化可提升基质持水能力,实现基质成土的初期演替。自然环境中,煤矸石等岩石颗粒的风化粉化主要由温度和水分驱动,但自然扰动强度小、频率低,煤矸石风化进程缓慢。蔡毅等[3]对淮南矿区煤矸石进行采样分析,结果表明风化2~12年的煤矸石持水率才具有较为明显的提升。张清峰等[4]研究表明煤矸石经36个月风化后含水率趋于稳定并超过10%。针对煤矸石风化粉化,梁冰等[5]利用木霉菌对煤矸石进行分解改良,作用30 d后,0.5 mm以下微粒上升了5%左右,时间成本较高;尚志等[6]、ZHANG等[7]采用冻融循环提高煤矸石黏粒细砂含量,研究表明冻融对土壤矿物化学性能改变不大,但对其粒径粉化效果显著,但冻融循环受环境制约难以大规模推广。张素等[8]认为干湿/冷热扰动有利于基质微粒含量的快速提升。通过瓦斯烟气余热利用对煤矸石进行强化风化处理,增强环境扰动的强度和频率,有望实现煤矸石的快速粉化及矿物转化,均衡其土壤学要素,达到综合性能的提升,开辟煤矸石原位风化成土的新途径。所以将煤矸石与瓦斯烟气余热进行协同处理处置,实现资源与能源的高效利用,是煤炭企业绿色可持续发展的重要课题。
针对煤矸石粒径粗大、持水能力薄弱的问题,本研究利用瓦斯烟气余热,强化煤矸石干湿/冷热的交替循环,加速煤矸石结构风化粉化,促进煤矸石的矿物分解与重构,提升风化产物的透气保墒性能,并通过植物生长考核其风化产物的生态化应用效果,以实现煤矸石与瓦斯烟气余热的资源能源的高效利用。
1. 材料与方法
1.1 实验原料
实验所用煤矸石来自云南恩洪煤矿,岩性以页岩、砂岩、泥岩为主,矿物成分主要为钙长石、伊利石、方解石等,以及少量黄铁矿。实验设置3组样品,初始持水率为6.3%±0.5%,每组样品重量为1 kg,样品粒径分布如表1所示。
表 1 煤矸石样品初始粒径分布Table 1. The initial particle size distribution of coal gangue samples5~10 mm 2~5 mm 1~2 mm 0~1 mm 68%±5% 6%±2% 12%±2% 14%±2% | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 实验方法
1) 干湿交替实验。对煤矸石进行高频干湿/冷热交替的扰动模拟,将煤矸石用滤布包裹后放入直径15 cm的PVC反应器,向容器中注入1 000 mL超纯水对煤矸石浸泡30 min并记录容器总重;提起滤布取出样品,沥干30 min,然后在45~50 ℃下烘干8 h,此为1个循环。样品分别经历10、20、30次循环,样品命名为CG1、CG2、CG3,干湿交替过程中堆浸液循环使用,每次浸泡时补充超纯水至初始记录总重,堆浸液命名为R1、R2、R3。反应完成后底部煤矸石微粒采用真空抽滤进行分离。
2) 紫花苜蓿生长试验。处理后的样品各取80 g,混匀后放入种植盆,每盆播种20颗紫花苜蓿种子,观察紫花苜蓿发芽及生长状况。每组样品设计3组平行样。
1.3 分析方法
1) 堆浸液。堆浸液静置沉淀30 min,样品过0.45 μm滤膜,使用电导率仪 (上海雷磁,DDS-11A) 检测其电导率,为减小实验误差,相邻2次电导率取平均值作图。样品分别循环处理完成后,30次循环的堆浸液上清液过0.45 μm滤膜烘干得到堆浸液盐分结晶,测定浸出液盐分的元素种类。样品粒径检测采用干筛法,筛分时间t (
m样品50g×20 2) 风化产物。利用XRD (日本 Rigaku Ultima IV) 检测颗粒矿物种类;将干湿交替循环结束的样品经105 ℃干燥,利用体式显微镜 (江西凤凰 XTL-165-VB) 表征循环处理风化产物表面形貌,用XRF (美国 Thermo Scientific™ Niton™ XL3t 600) 表征结晶盐元素含量,使用SEM-EDS (捷克 TESCAN MIRA LMS) 表征20次循环颗粒表面结晶盐形貌及元素种类;紫花苜蓿发芽后记录发芽时间,待其继续生长7 d后测定其发芽率、用直尺测量茎长。
2. 结果与讨论
2.1 煤矸石干湿/冷热交替盐分溶出规律
如图1所示,干湿/冷热交替过程中,R1、R2、R3堆浸液电导率呈现上升、下降、再次上升的趋势,将0~6次循环划分为干湿交替初期。将7~22次循环划分为干湿交替中期,将22次以后的循环划分为后期。溶液电导率与其离子质量浓度呈正比,说明干湿交替初期煤矸石盐分快速溶出,造成堆浸液电导率上升,中期时堆浸液内盐分下降,而后煤矸石颗粒再次发生溶出。
 图 1 循环过程堆浸液电导率变化情况Figure 1. Changes in conductivity of heap leaching solution during circulation
图 1 循环过程堆浸液电导率变化情况Figure 1. Changes in conductivity of heap leaching solution during circulation干湿/冷热交替初期盐分大量溶出,电导率持续上升说明除颗粒表面游离盐分外,煤矸石可溶性组分也不断溶解。干湿交替过程提高了水分的迁移速率,增加了与可溶性组分的接触面积,对其进行溶解,而后在干燥的条件下水分受热蒸发,向颗粒表面移动,盐分随着水流一起向外转移,并进入溶液。这与SHEILA[9]、ZHANG等[10]研究结果相符,干湿交替促进水分不断侵入新区域,还萌生、连接了微裂隙,提高了岩石颗粒孔隙度。可溶组分的持续溶解和溶出通道的形成加速了干湿交替初期煤矸石所含盐分向溶液转移的过程,导致盐分质量浓度的显著上升。
干湿/冷热交替中期堆浸液电导率下降可能的原因是煤矸石颗粒经历了初期的快速溶出,颗粒表面活性组分含量下降,盐分溶出作用受限。而溶液中盐分在干燥作用下发生重结晶,不但堵塞盐分溶出通道还粘附在容器上,从溶液中脱除;除重结晶作用,盐分还被煤矸石中微粒吸附、固定,当盐分脱除速度高于盐分的溶出速度时,溶液盐分质量浓度下降。ZHAO等[11]对砂岩的研究结果表明,随着干湿交替进行,颗粒中低溶性组分相对含量增加,盐分溶出速率下降。HAYNES等[12]、曹勇等[13]认为非晶铝硅酸盐、页硅酸盐以及采矿伴生的黏土对溶液离子有较强吸附作用。本试验初期的盐分溶出使矿物向非晶格形式或次生黏土矿物转变,溶液中离子的吸附固定作用增强,总体而言,溶液盐分含量变化受溶出作用与多方面的固定作用影响,盐分的固定作用强于溶出作用时,溶液盐分质量浓度下降。
干湿/冷热交替后期溶液电导率上升说明颗粒表面可溶组分再次增加,其可能的原因是煤矸石颗粒形成了大量的活性界面,水的溶出作用会降低表面活性,所以这部分活性界面的形成应来源于干湿交替过程中颗粒的物理性损伤。ZHU等[14]研究煤矸石粉改良膨润土裂隙发育的机理,结果表明干湿交替过程中颗粒内部会形成不与外界相连的裂隙,随着循环次数的增加,裂隙不断发育、连接,最终形成宏观裂隙或断面,与外界联系。HUANG等[15]的研究结果也表示风化导致煤矸石中粗孔、大裂隙发育明显,提升了煤矸石元素的溶出速率。MA等[16]研究了干湿交替对固废再生絮凝膏体的破坏作用,发现样品宏观裂隙是逐渐发育的过程,颗粒受到周期性损伤,在中途的4~5个循环时变质程度显著加大。随着干湿/冷热交替的不断进行,煤矸石内微裂隙逐渐发育,形成宏观断面,暴露出新鲜界面,使后期溶液盐分质量浓度再次上升。
2.2 干湿/冷热交替作用下煤矸石矿物溶出与重构过程分析
如图2所示,煤矸石颗粒表面出现盐分重结晶,能谱表征显示其为硫酸盐,结果表明干湿/冷热交替过程中煤矸石发生了涉硫组分的溶出与重构。煤矸石中涉硫组分以黄铁矿为主,占总硫组分90%以上,所以认为颗粒表面硫酸盐的来源是黄铁矿氧化溶解。硫酸盐随水分向颗粒表面转移,而后水分蒸发,盐分再结晶并附在颗粒表面。黄铁矿氧化过程如式(1)所示。黄铁矿在氧气和水分作用下氧化,干湿交替增大了煤矸石的孔隙度,从而加速氧气与水分的扩散,提高了黄铁矿的氧化速度。
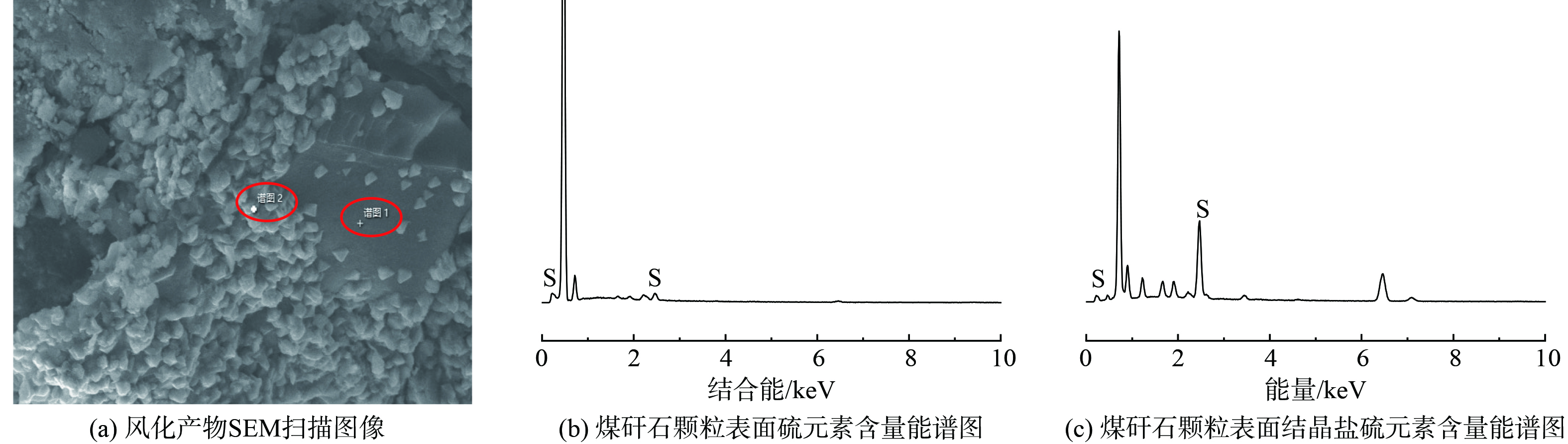 图 2 煤矸石颗粒表面结晶盐显微及能谱分析Figure 2. Microscopic and energy spectrum analysis of crystallized salt on the surface of coal gangue particles
图 2 煤矸石颗粒表面结晶盐显微及能谱分析Figure 2. Microscopic and energy spectrum analysis of crystallized salt on the surface of coal gangue particlesstringUtils.convertMath(!{formula.content}) (1) 干湿/冷热交替处理30次的风化产物进行XRD表征,其结果与原煤矸石结果对比如图3所示。处理后煤矸石所含黄铁矿、钙长石、方解石等衍射强度显著降低,黄铁矿峰趋于消失。但煤矸石为复杂矿物混合物,杂峰较多,且本试验未外加溶蚀药剂对矸石浸出,所以图3显示的前后样品结果变化幅度较小,难以直接说明煤矸石矿物溶出重构过程。CHETTY[17]认为矿渣脉石矿物所含长石、伊利石在表面溶蚀作用下向高岭石转变,YUAN等[18]认为中性条件下,长石向高岭石转变并生成正硅酸。基于图3显示的煤矸石黄铁矿、钙长石、方解石发生溶出,在H+存在及中性条件下的条件下,对含钾含钙矿物进行热力学分析,反应如式(2)~(7)所示。
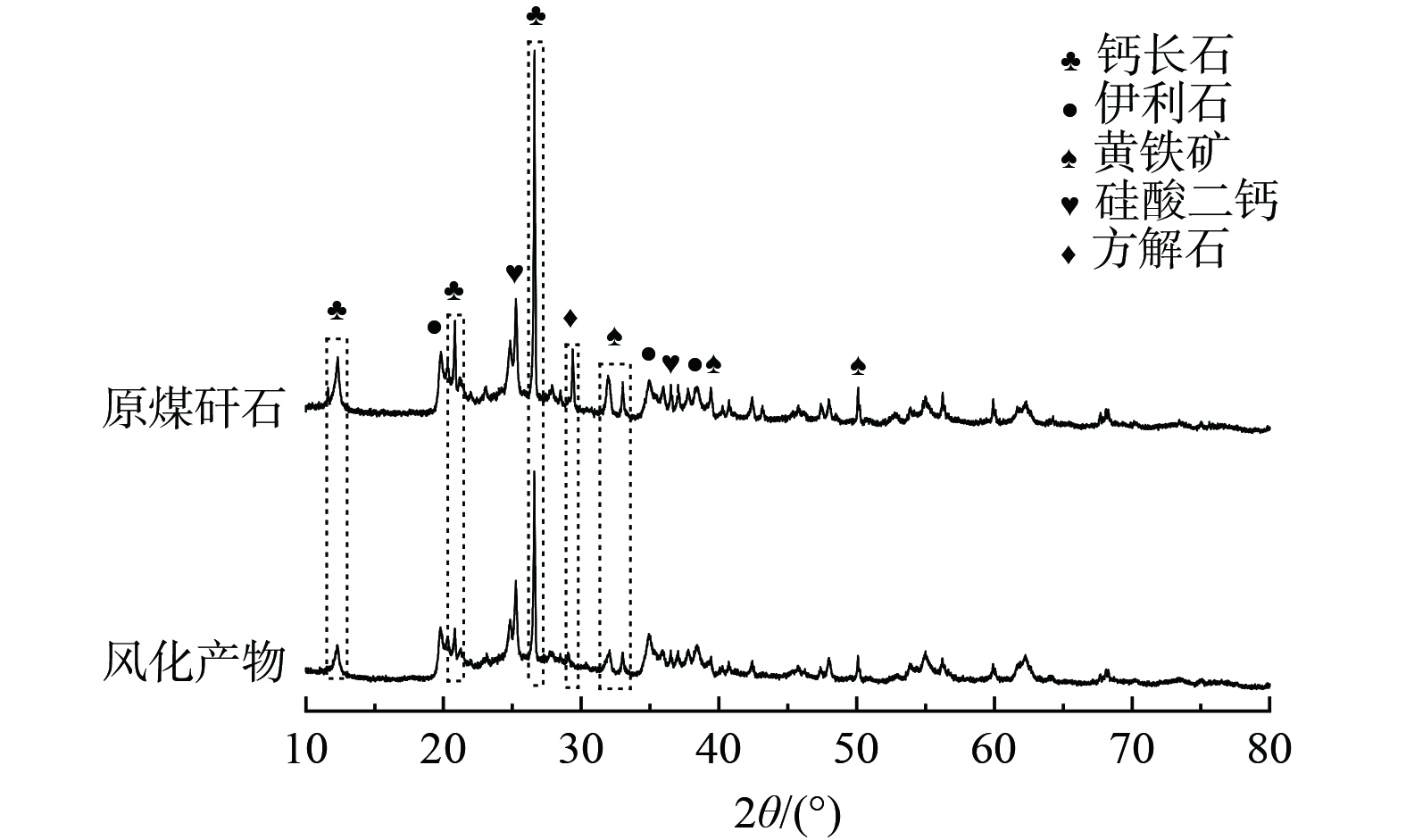 图 3 30次循环处理产物与原煤矸石XRD结果对比图Figure 3. Comparison chart of XRD results between 30 cycles of processing products and raw coal gangue
图 3 30次循环处理产物与原煤矸石XRD结果对比图Figure 3. Comparison chart of XRD results between 30 cycles of processing products and raw coal ganguestringUtils.convertMath(!{formula.content}) (2) stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) stringUtils.convertMath(!{formula.content}) (5) stringUtils.convertMath(!{formula.content}) (6) stringUtils.convertMath(!{formula.content}) (7) 公式(2)~(7)说明钙长石、伊利石等含钾含钙矿物在H+存在时具有自发反应趋势,而在无H+时反应难度较大。煤矸石中含有的黄铁矿在干湿/冷热交替下氧化溶解速率提升,H+产率上升,从而加快长石等硅酸盐矿物溶解速率。NORDSTROM等[19]研究认为硅酸盐矿物、碳酸盐矿物在高浓度酸性矿山废水中起到显著的中和作用,说明黄铁矿的大量氧化形成的H+可与伴生矿物快速反应并导致钾、钙组分的溶出。
样品处理完成后,将30次处理的浸出液烘干得到盐结晶,进行XRD表征,结果如图4所示。堆浸液结晶盐主要成分为硫酸钙、硫酸钠、硫酸钾,说明溶出的盐分中SO42−、K+、Ca2+、Na+含量最高。其中SO42−、K+、Ca2+来源于矿物的溶出,Na+在原煤矸石XRD表征中没有对应的晶型含钠矿物,所以认为钠元素为煤矸石自身所含的可溶性组分,在处理过程中直接溶解在水中,在水分蒸发后与溶液中硫酸根结合,形成硫酸钠。
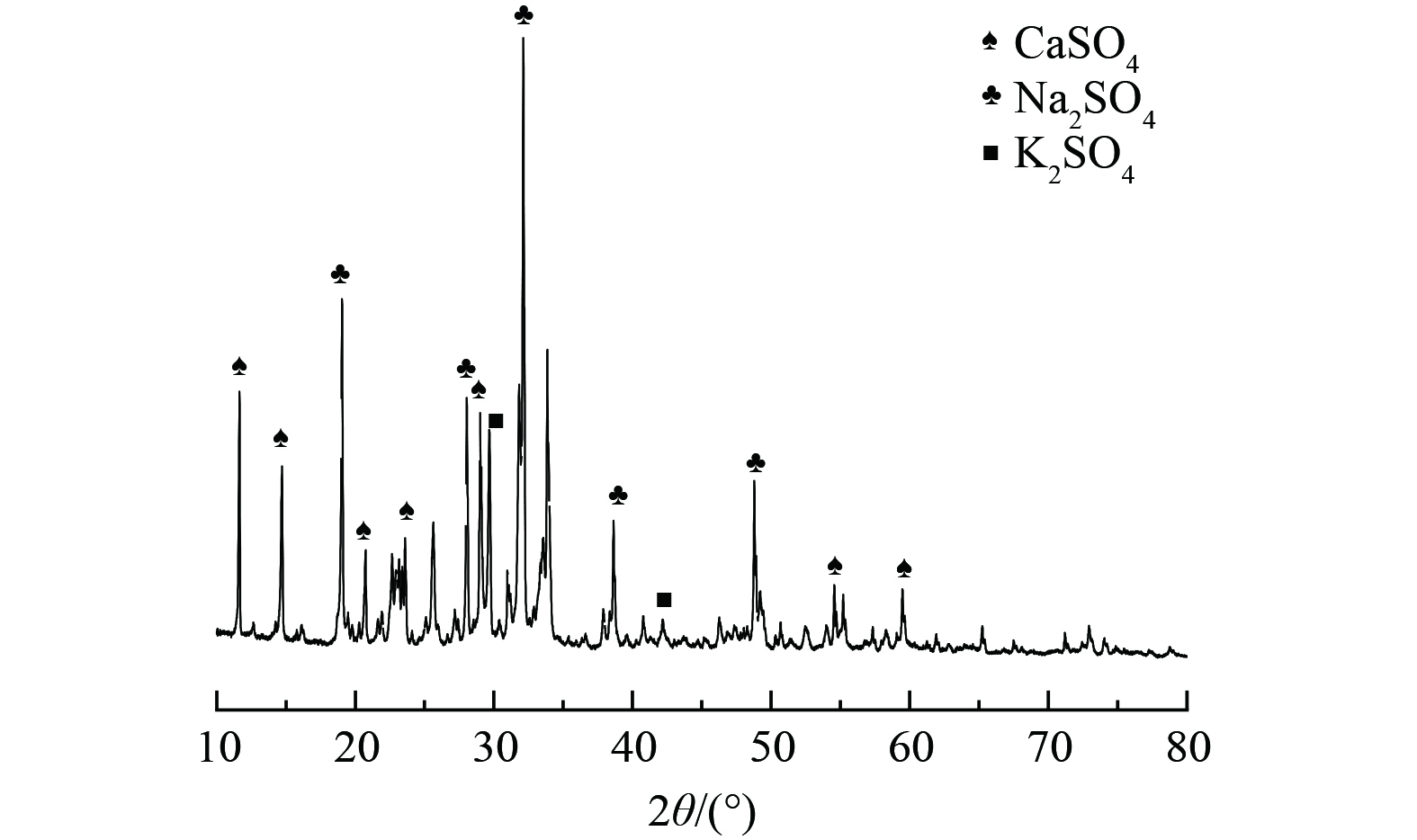 图 4 30次循环浸出液结晶盐XRD结果Figure 4. XRD results of crystallized salt in leaching solution after 30 cycles
图 4 30次循环浸出液结晶盐XRD结果Figure 4. XRD results of crystallized salt in leaching solution after 30 cycles使用XRF测定堆浸液烘干后的盐结晶,结果如表2所示。除硅酸盐矿物组分外,Fe含量也较高,根据前文结果,认为这部分Fe来自黄铁矿的氧化溶出。干湿/冷热交替过程导致岩石矿物碎裂,进而生成大量高反应活性界面,热力学不平衡也会导致矿物化学键的减弱,所以也存在含钾含钙矿物直接与表面水膜发生物质交换的可能。
表 2 堆浸液结晶盐XRF检测结果Table 2. XRF detection results of heap leach liquid crystallization saltmg·kg−1 样品编号 K Ca S Fe R1 2 371 9 557 160.4×103 1 734 R2 4 203 7 396 142.3×103 1 458 R3 820 18.1·103 96.5×103 1 487 | Show TableDownLoad:
CSV
2.3 煤矸石粉化过程分析
如图5所示,干湿/冷热交替处理完成后观察20次处理的颗粒表面形貌,图(a)、(b)、(c)、(d)表明10~30次处理后产物颗粒都出现盐分重结晶现象。所以煤矸石颗粒的粉化除2.2所述可溶矿物的溶解外,还受盐分结晶的挤压作用。高润东等[20]研究认为干湿交替使含硫混凝土盐分溶出,随着蒸发进行,盐分发生重结晶,而结晶作用可对颗粒产生挤压应力,促使裂隙的进一步发育。CELIK和SERT[21]对安山岩进行冻融、热冲击和盐结晶处理,试验表明空隙的重结晶作用有助于裂隙和孔洞的扩大。AN等[22]对长石砂岩进行干湿交替处理,试验发现盐的结晶作用对砂岩的劣化效果最为显著。干湿/冷热交替加速颗粒组分的结晶频率,从而促使颗粒裂隙发育,侵蚀离子、水分子进入颗粒内部,与新鲜活化界面反应、溶出,使煤矸石粉化反应不断进行。
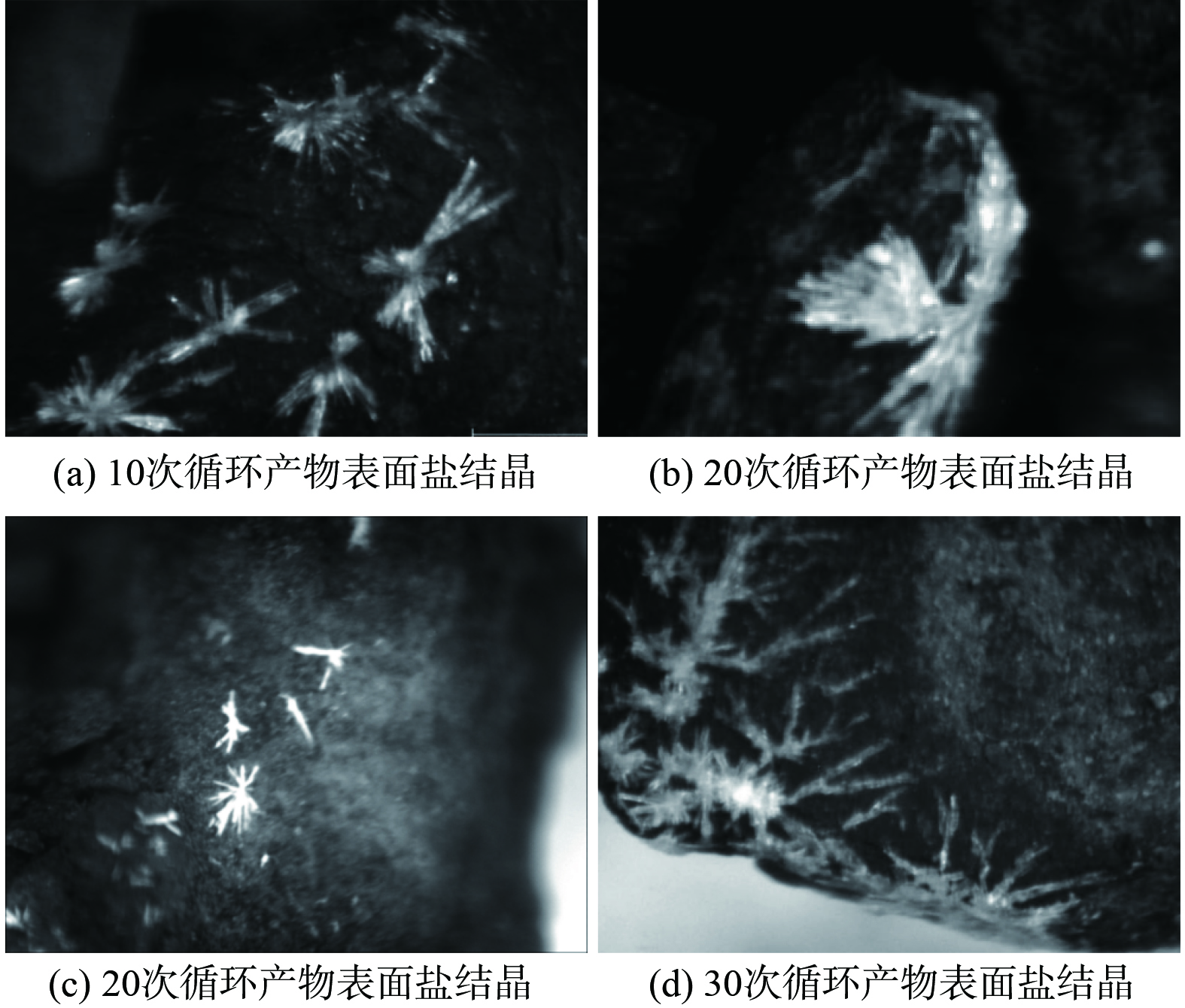 图 5 20次循环处理产物表面盐结晶Figure 5. Salt crystallization on the surface of the product after 20 cycles
图 5 20次循环处理产物表面盐结晶Figure 5. Salt crystallization on the surface of the product after 20 cycles如图6所示,图(a)、(b)、(c)、(d)表明10~30次处理后煤矸石颗粒都受到损伤,出现裂隙,根据煤矸石颗粒岩性,认为这部分裂隙除盐结晶作用外,还可能是黏土矿物不均匀性膨胀、收缩造成的。煤矸石中伊利石、长石等具有不同的膨胀性能,梁冰等[23]对弱崩解泥岩开展干湿循环试验,结果表明含伊利石、高岭石较多的泥岩在干湿交替过程中,界面结合处发生力学破坏能,加速促进裂隙、微裂隙的扩展。YANG等 [24]对泥岩进行干湿交替处理,发现水的侵入导致黏土矿物膨胀,并产生拉伸应力,失水导致的收缩使裂隙不断发展,进而有利于下一次水分的侵入,展示了混合矿物颗粒裂隙发育原理。因此,煤矸石在干湿交替过程中会发生物理性损伤而粉化。
煤矸石颗粒在干湿/冷热交替过程中发生盐分溶解作用、盐结晶挤压作用、黏土矿物崩解作用,3类作用促使煤矸石颗粒不断粉化,形成微粒,过程如图7所示。
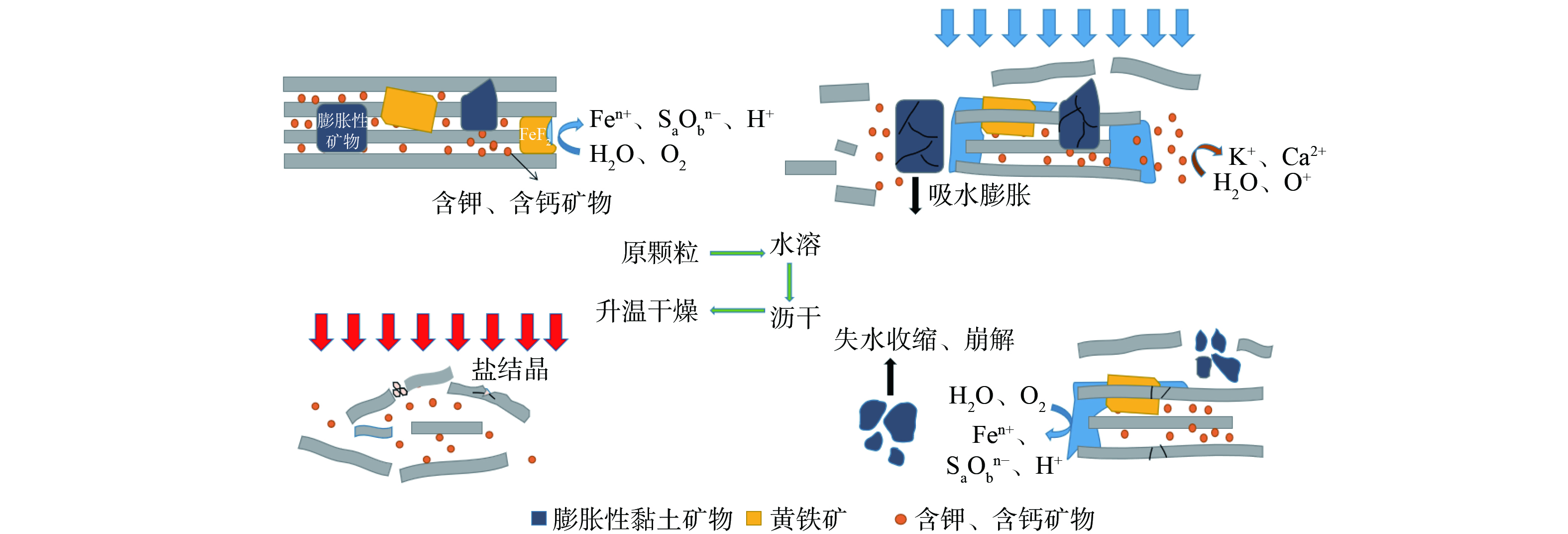 图 7 干湿交替作用下煤矸石风化粉化机理图Figure 7. Mechanism of coal gangue weathering and pulverization under the alternating action of dry and wet
图 7 干湿交替作用下煤矸石风化粉化机理图Figure 7. Mechanism of coal gangue weathering and pulverization under the alternating action of dry and wet2.4 煤矸石风化产物土壤性能
干湿/冷热交替处理后检测煤矸石粉化效果,测定各粒径范围内煤矸石颗粒的含量变化,结果如表3、图8所示。干湿交替对煤矸石颗粒粉化具有显著效果,3组样品中,5~10 mm颗粒显著下降,1 mm以下微粒显著上升,20和30次循环后1 mm以下细砂含量上升30%以上,可达35.4%,较低扰动强度 (10次循环) 对1 mm以下微粒提升也有较好的效果。ZHANG等[25]实验表明干湿交替处理过程中,10次循环时,岩石颗粒微观结构已发生较大变化,20~30个循环后,颗粒劣化速度趋于稳定。1~5 mm颗粒含量略微上升,说明在此粒径范围内集中的主要是结构较为稳定的矿物或岩石颗粒,粉化作用较5~10 mm颗粒弱。
表 3 干湿/冷热交替处理前后煤矸石样品粉化粒径分布Table 3. Pulverized particle size distribution of coal gangue samples before and after dry-wet/cold-heat alternate treatment粒径分布/mm CG3 CG2 CG1 处理前/% 处理后/% 处理前/% 处理后/% 处理前/% 处理后/% 5~10 71.5 28 66.1 24.5 63.1 34.2 2~5 4.8 7.2 9.5 15 7.1 9.3 1~2 12.2 18 12.2 15.6 15 18.5 0~1 11.5 46.9 12.4 44.9 15.2 38 | Show TableDownLoad:
CSV
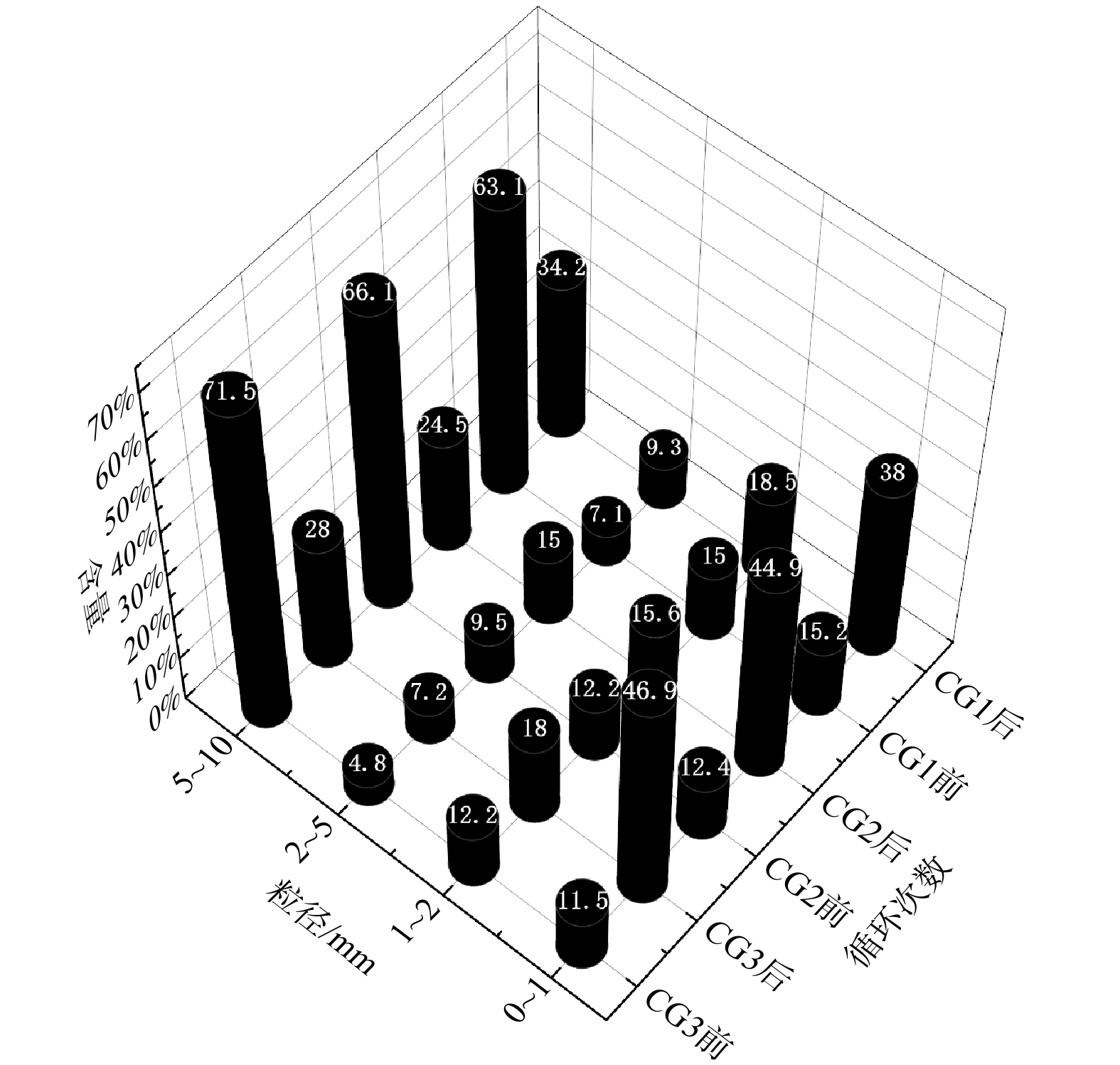 图 8 干湿交替前后煤矸石粒径分布Figure 8. Particle size distribution of coal gangue before and after dry-wet alternation
图 8 干湿交替前后煤矸石粒径分布Figure 8. Particle size distribution of coal gangue before and after dry-wet alternation煤矸石经历干湿/冷热交替后,其粒度显著下降,风化产物的持水率与孔隙度上升,与循环次数呈正比,如图9所示。持水率和空隙度是土壤物理性质的重要指标,两者的提高有利于风化基质的生态化利用。风化产物持水率在30次循环后最大,达到12.82%,该指标满足砂土持水标准。毛管空隙度上升可提高风化产物透气性能,未经处理的煤矸石粒径粗大,颗粒间缝隙大,因此宏观上较为透气,但颗粒本身致密,无法满足土壤基质缓冲、透气的要求。本试验中煤矸石毛管孔隙度随循环次数增加而不断上升,与DEHESTANI等[26]研究结果一致,即岩石颗粒的有效孔隙度与干湿循环次数呈正比。
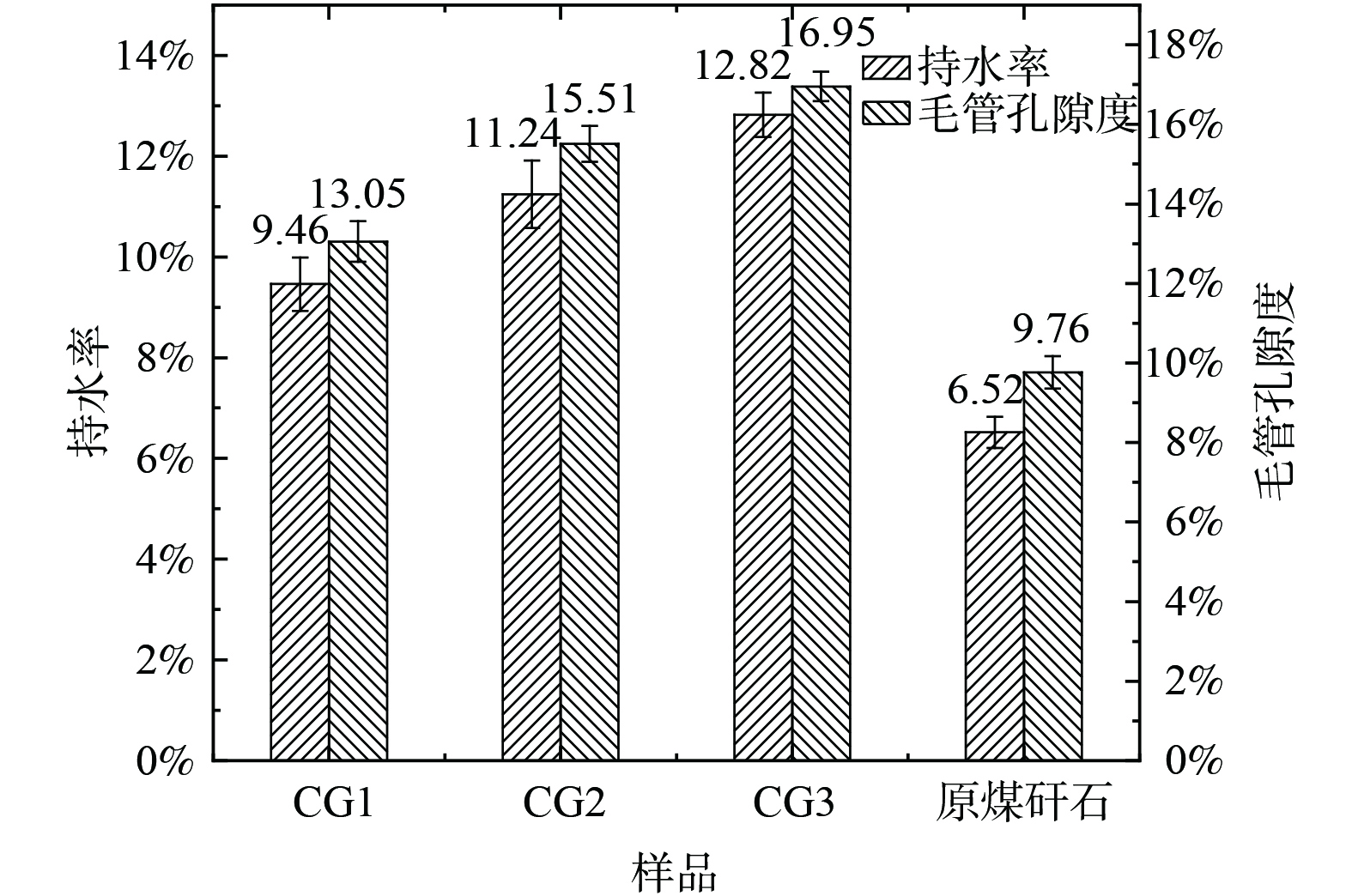 图 9 干湿交替前后煤矸石持水率及孔隙度Figure 9. Water holding capacity and porosity of coal gangue before and after dry-wet alternation
图 9 干湿交替前后煤矸石持水率及孔隙度Figure 9. Water holding capacity and porosity of coal gangue before and after dry-wet alternation20与30次循环后,煤矸石1 mm以下微粒增长量相似,但30次循环后风化基质毛管孔隙度提升较多,说明30次循环后样品含有较多的微裂隙或小孔,此结果与前文所述煤矸石电导率变化情况相符合,证明煤矸石颗粒的崩解存在一个缓慢发育的周期,由微裂隙/微孔逐渐扩大,最终形成断面,微粒含量提高。
紫花苜蓿是常见的牧草,是牧业种植的重要经济作物。风化产物进行紫花苜蓿种植,其考核结果如表4所示,经过干湿交替处理的煤矸石基质发芽率、同生长时间茎长等均优于未处理的对照组,图10为3组样品经干湿交替后种植效果展示。
表 4 紫花苜蓿生长指标Table 4. Growth index of alfalfa样品 发芽率/% 茎长/cm 发芽时间/d 原煤矸石 20 0.97 20 10次循环产物 40 1.76 20 20次循环产物 60 2.21 21 30次处理产物 85 2.64 18 | Show TableDownLoad:
CSV
3. 结论
1) 干湿/冷热交替可显著降低煤矸石的结构强度,加速煤矸石的风化粉化,风化产物中0~1 mm微粒含量上升30%以上,风化产物粒度级配得到优化,显著提升了煤矸石风化产物的透气保墒性能。
2) 干湿/冷热交替促进煤矸石发生矿物分解与重构,加速盐分的溶出与重结晶,产生挤压应力加速煤矸石碎裂及矿物稳定化;硫化矿物发生氧化溶出,并轰击硅酸盐矿物,导致K、Ca、S、Si等元素的溶出,提升煤矸石风化产物的肥力性能。
3) 紫花苜蓿种植结果表明,基于余热利用的干湿循环可显著加速煤矸石风化成土过程,显著提升风化产物的土壤学性能,有效促进苜蓿等绿肥植物的生长,进而为煤矸石的大规模生态化利用提供可靠路径。
-
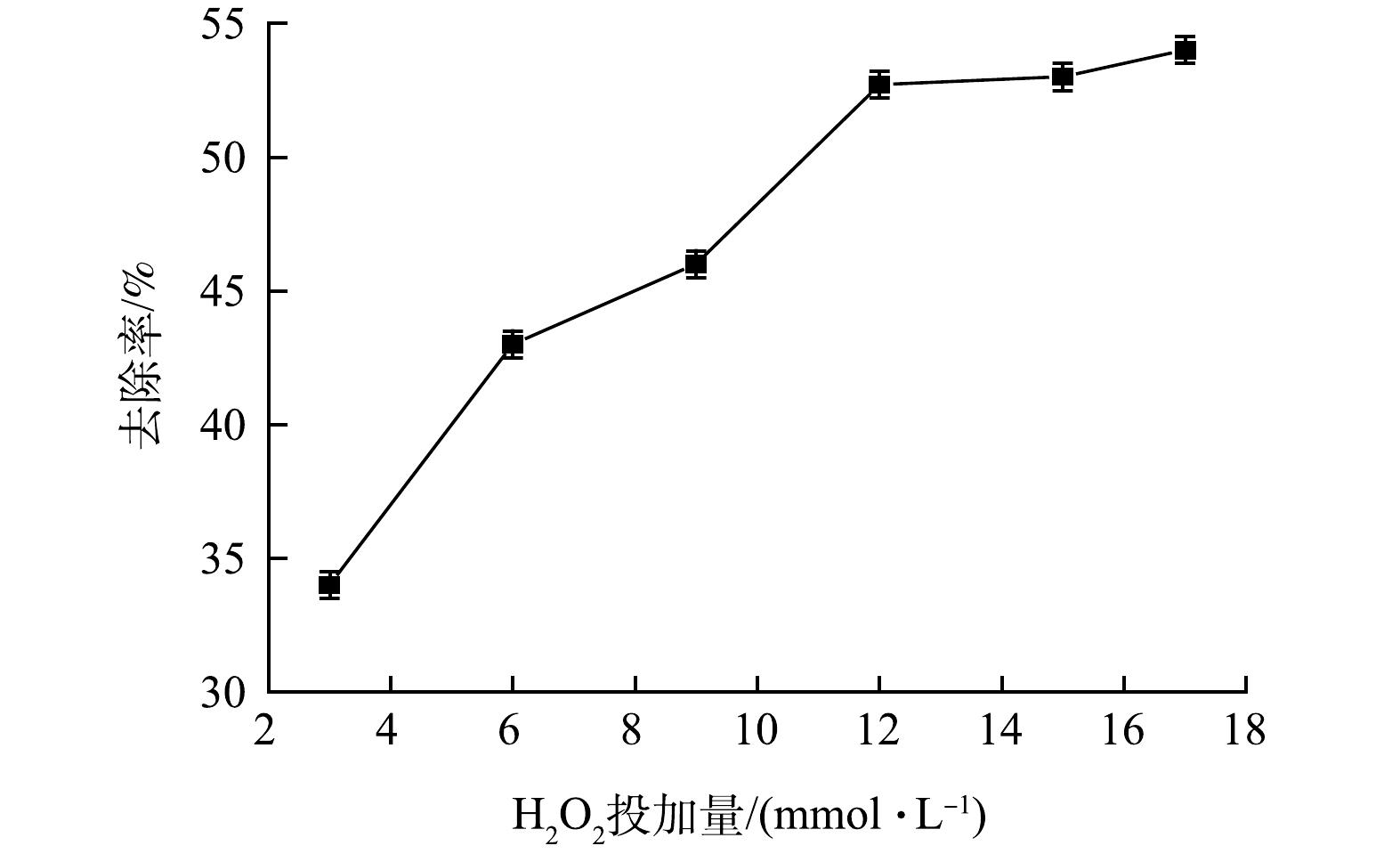
图 2 H2O2电催化强化体系对布洛芬去除率的影响
Figure 2. Effect of H2O2 enhanced electrocatalysis system on ibuprofen removal rate
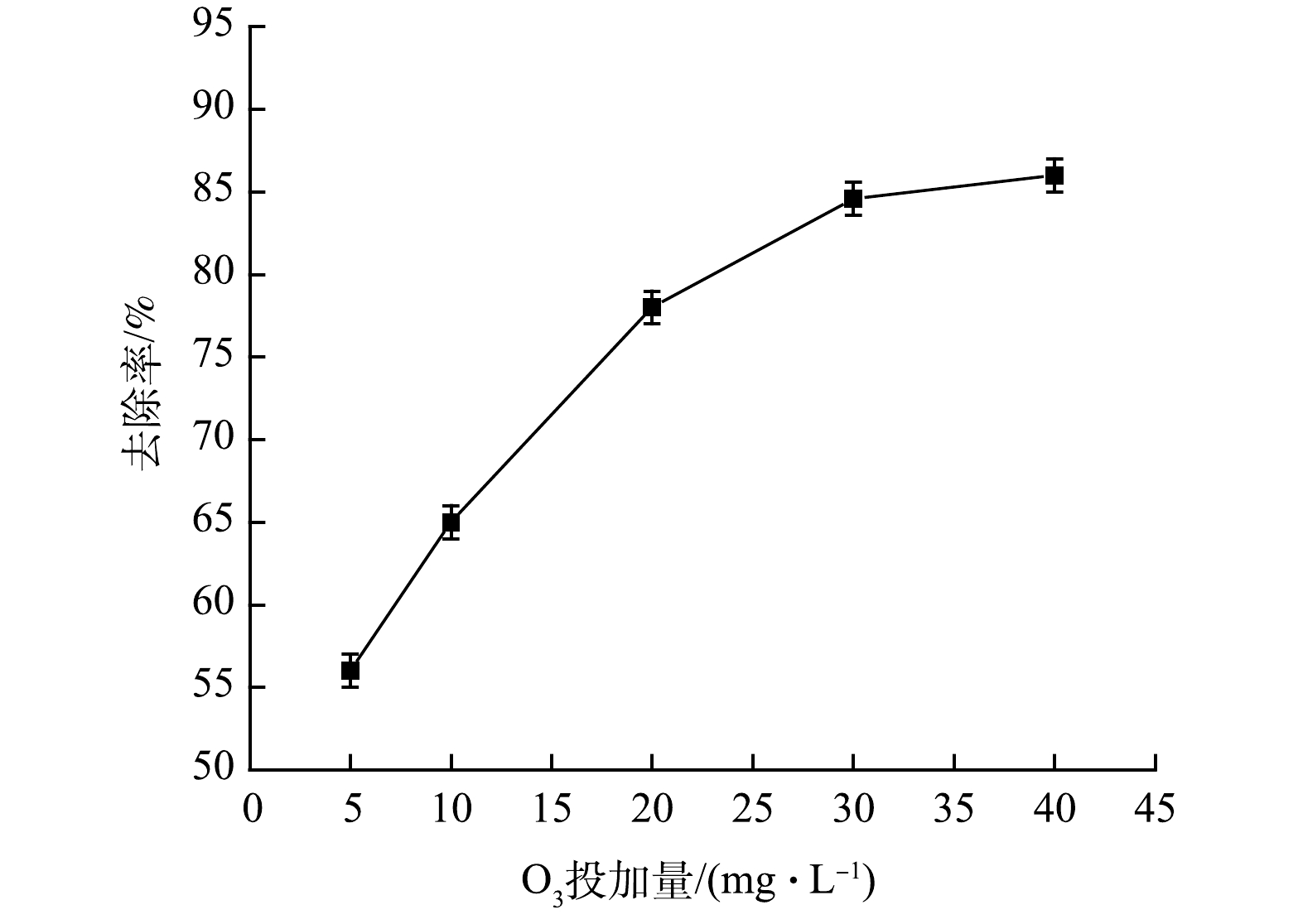
图 3 O3电催化强化体系对布洛芬去除率的影响
Figure 3. Effect of O3 enhanced electrocatalysis system on ibuprofen removal rate

图 4 电催化及电催化强化体系对布洛芬去除率的影响
Figure 4. Effect of electrocatalysis and enhanced electrocatalysis system on ibuprofen removal rate
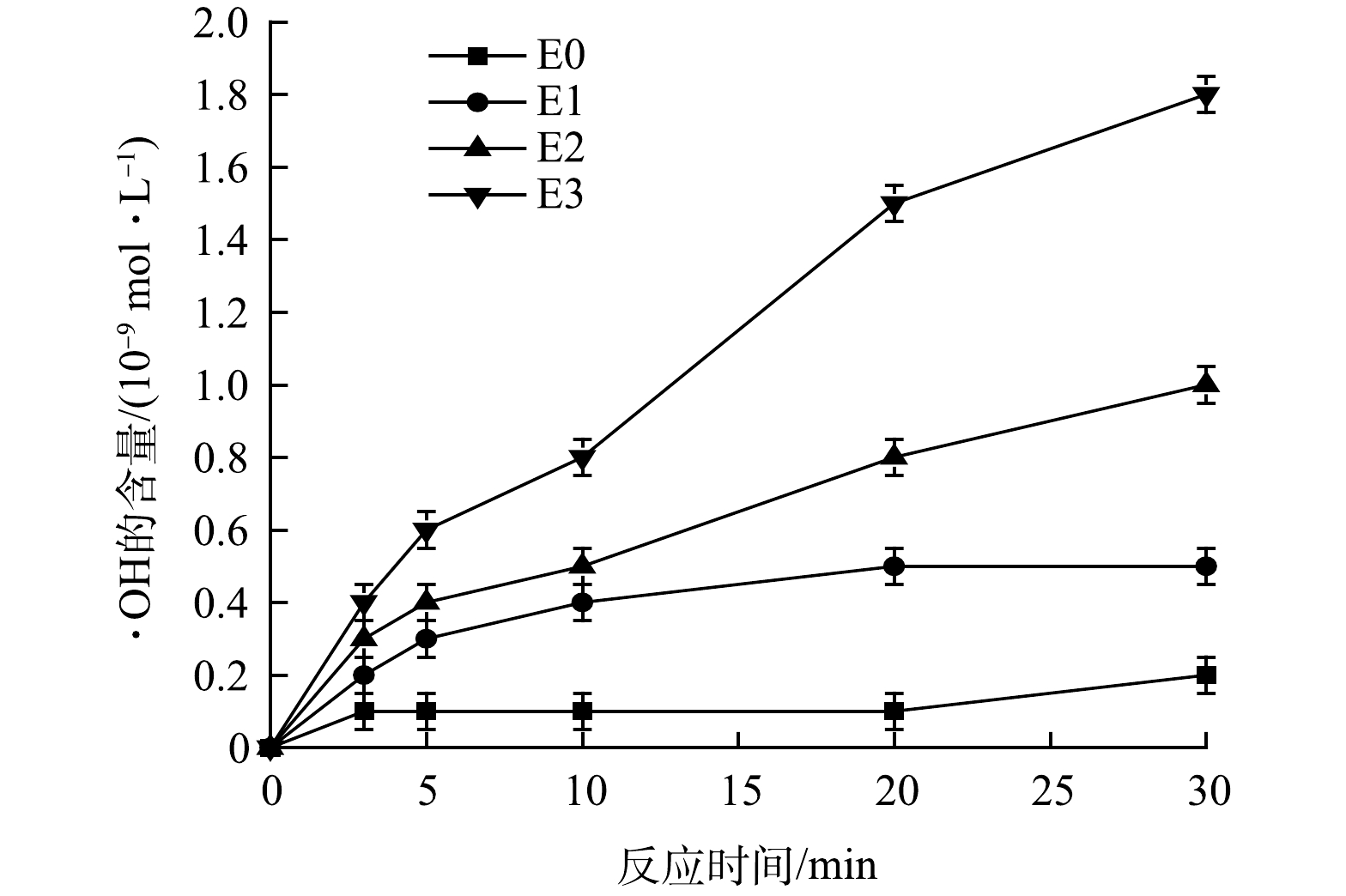
图 5 电催化及电催化强化体系中·OH的生成量
Figure 5. ·OH formation in electrocatalysis and its enhanced systems
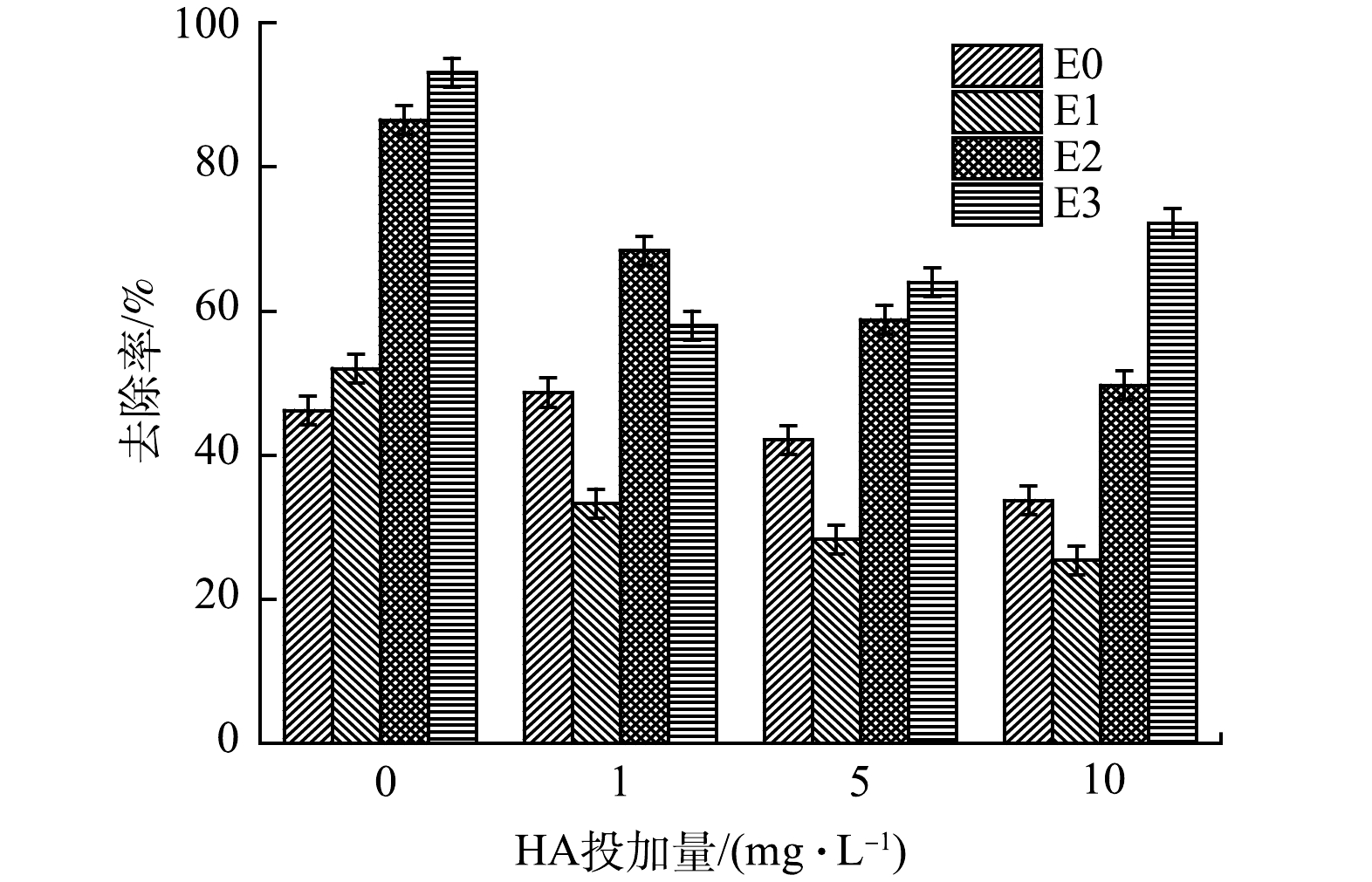
图 6 腐殖酸质量浓度对布洛芬去除率的影响
Figure 6. Effect of humic acid concentration on ibuprofen removal rate
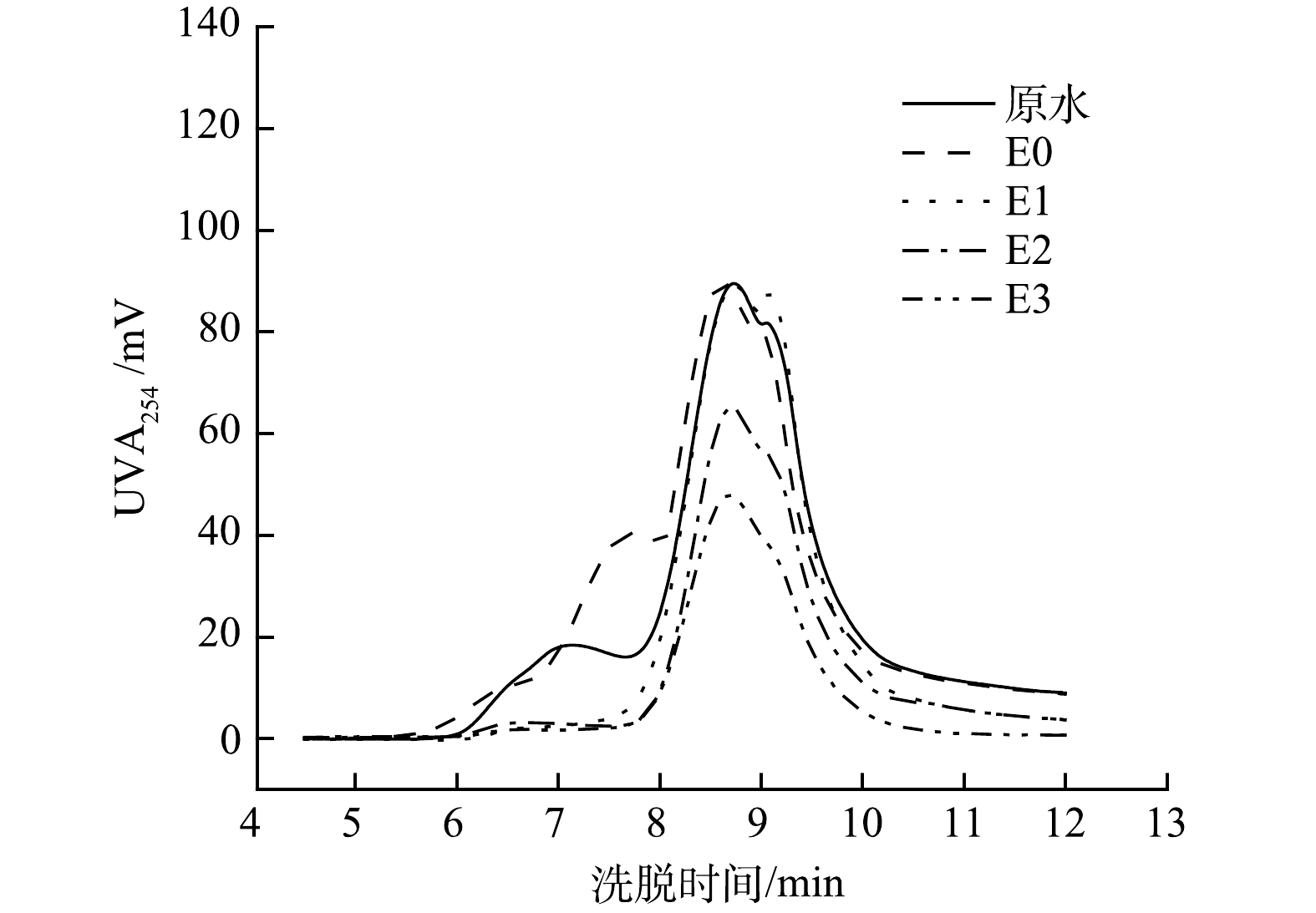
图 7 电催化强化体系对不同分子量污染物的去除
Figure 7. Removal of pollutants with different molecular weights by enhanced electrocatalysis system
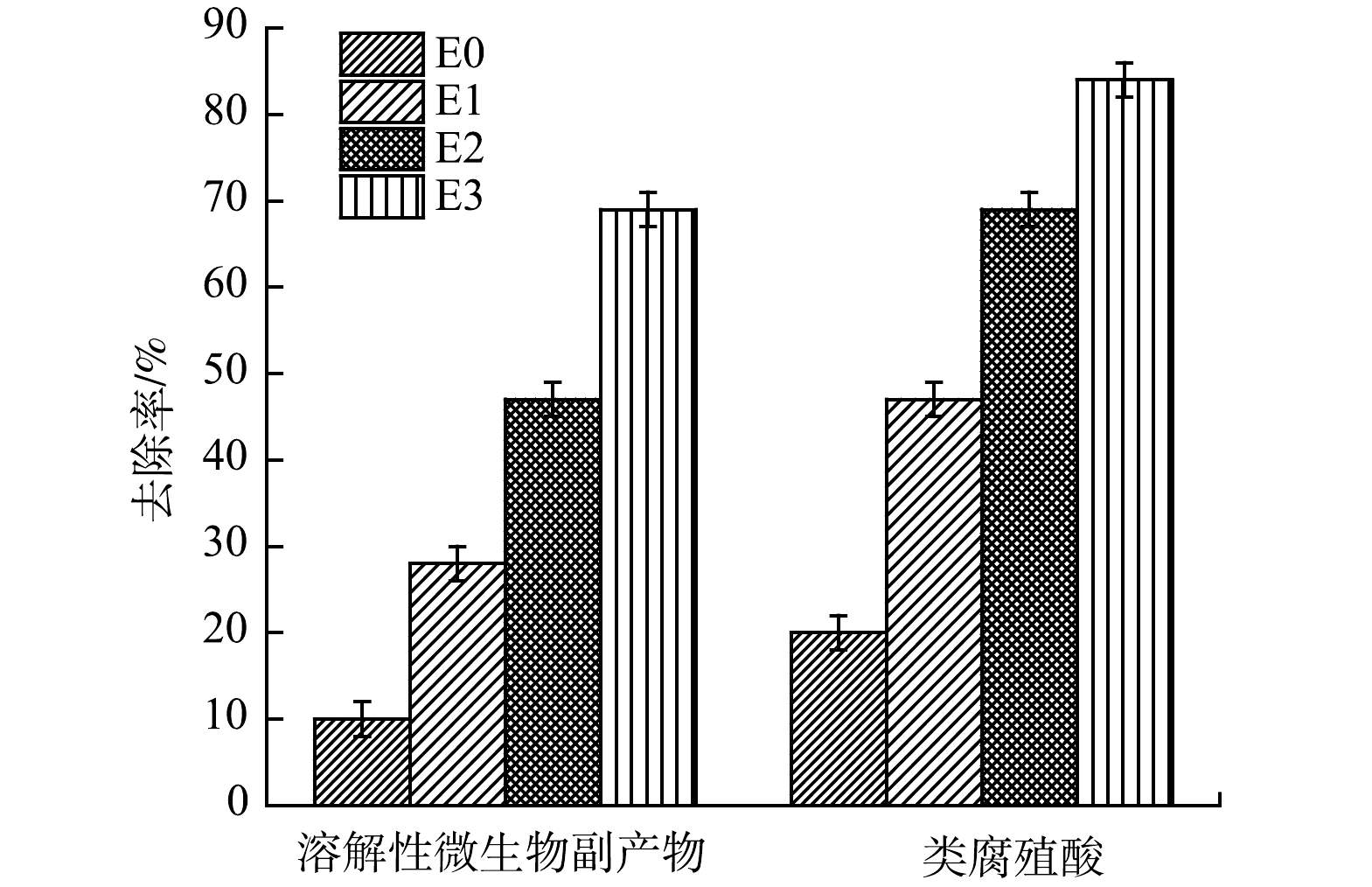
图 9 电催化强化体系对类腐殖酸和溶解性微生物副产物的去除
Figure 9. Removal of humic acids and soluble microbial by-products by enhanced electrocatalysis system
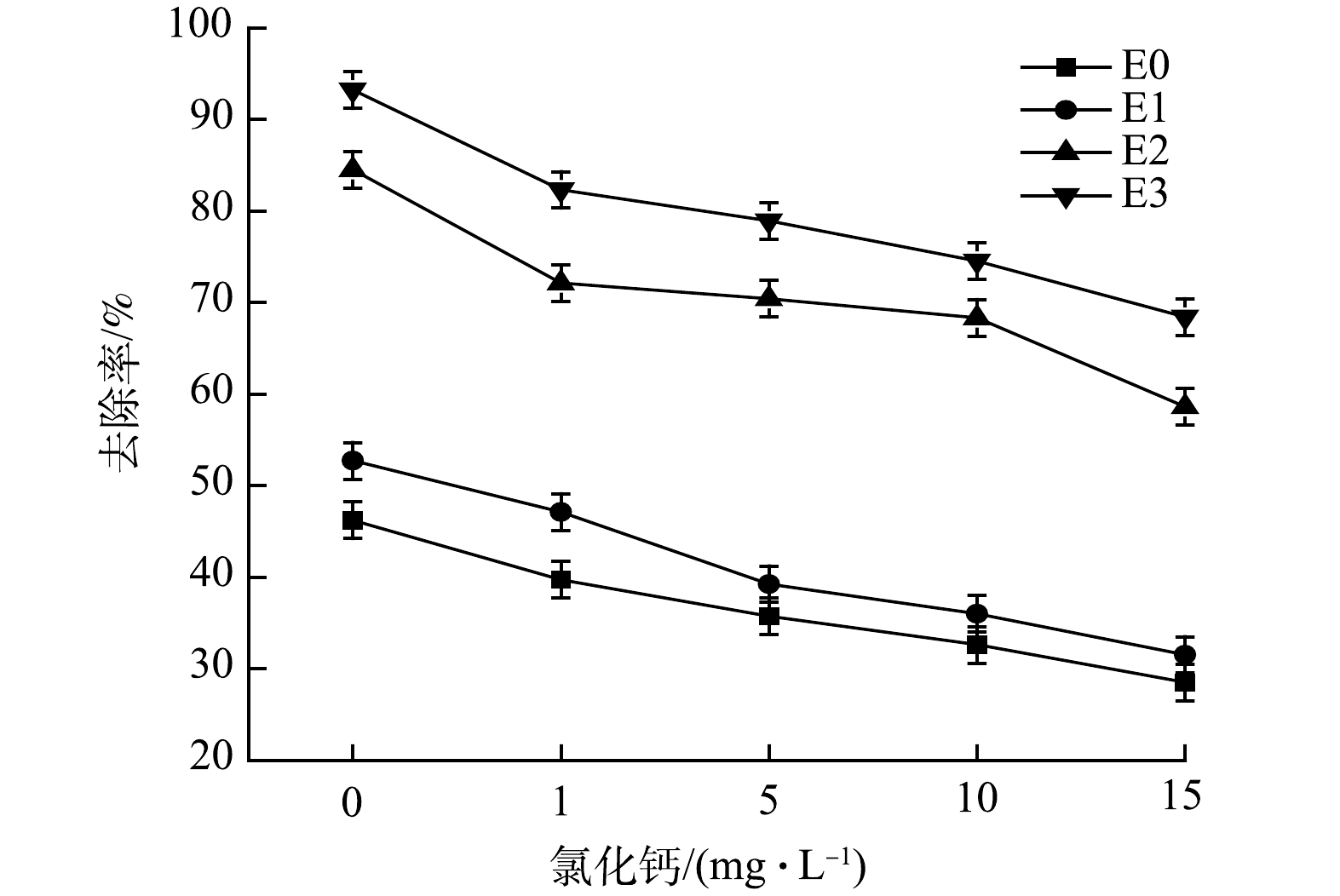
图 10 硬度对布洛芬在电催化强化体系中的去除影响
Figure 10. Effect of hardness on the removal of ibuprofen by enhanced electrocatalysis system
表 1 高效液相色谱串联飞行时间质谱检测到布洛芬在反应过程的中间产物
Table 1. Intermediate products of ibuprofen in the reaction process detected by high performance liquid chromatography-time-of-flight mass spectrometry
序号 名称 化学式 m/z 保留时间/min 1 2-(3-羟基-4-异丁基苯基)丙酸 C13H18O3 222 2.5 2 2-羟基-2-(4-异丁基苯基)丙酸 C13H18O3 222 6.6 3 4-(1-羟乙基)苯甲醛 C9H10O2 150 13.9 4 2-(4-甲基苯基)丙酸 C9H8O3 164 8.2 5 1-(4-异丁基苯基)乙醇 C11H13O2 178 11.7 6 1-(4-(1-羟乙基)苯基)-2-甲基丙烷-1-酮 C12H16O2 192 2.7 7 4-(1-羟基-2-甲基丙基)苯乙酮 C12H16O2 192 14 8 1,2,4-苯三酚 C6H6O3 126 1.7 9 邻苯二酚 C6H6O2 110 2.1 10 4-乙基苯酚 C8H10O 122 2.4 11 对苯二酚 C6H6O2 110 1.8 12 4-乙基苯甲醛 C9H10O 134 3.5 13 1-乙基-4-(2-甲基丙基)苯 C12H18 162 3.9
下载: 导出CSV
-
[1] FALAHI O, ABDULLAH S, HASAN H, et al. Simultaneous removal of ibuprofen, organic material, and nutrients from domestic wastewater through a pilot-scale vertical sub-surface flow constructed wetland with aeration system[J]. Journal of Water Process Engineering, 2021, 43: 102214. doi: 10.1016/j.jwpe.2021.102214 [2] OBA S, IGHALO J, ANIAGOR C, et al. Removal of ibuprofen from aqueous media by adsorption: A comprehensive review[J]. Science of the Total Environment, 2021, 780: 146608. doi: 10.1016/j.scitotenv.2021.146608 [3] KRISHNAN R, MANIKANDAN S, SUBBAIYA R, et al. Removal of emerging micropollutants originating from pharmaceuticals and personal care products (PPCPs) in water and wastewater by advanced oxidation processes: A review[J]. Environmental Technology & Innovation, 2021, 23: 101757. [4] GUO M, FENG Y, LI X, et al. Enhanced degradation of pharmaceuticals and personal care products (PPCPs) by three-dimensional electrocatalysis coupled biological aerated filter[J]. Journal of Environmental Chemical Engineering, 2021, 9(5): 106035. doi: 10.1016/j.jece.2021.106035 [5] LI J, HAN X, BRANDT B, et al. Physico-chemical and biological aspects of a serially connected lab-scale constructed wetland-stabilization tank-GAC slow sand filtration system during removal of selected PPCPs[J]. Chemical Engineering Journal, 2019, 369: 1109-1118. doi: 10.1016/j.cej.2019.03.105 [6] LEE C, HOWE K, THOMSON B. Ozone and biofiltration as an alternative to reverse osmosis for removing PPCPs and micropollutants from treated wastewater[J]. Water Research, 2012, 46(4): 1005-1014. doi: 10.1016/j.watres.2011.11.069 [7] ESPLUGAS S, BILA D, KRAUSE L, et al. Ozonation and advanced oxidation technologies to remove endocrine disrupting chemicals (EDCs) and pharmaceuticals and personal care products (PPCPs) in water effluents[J]. Journal of Hazardous Materials, 2007, 149(3): 631-642. doi: 10.1016/j.jhazmat.2007.07.073 [8] QUERO-PASTOR M, GARRIDO-PEREZ M, QUIROGA J. Ozonation of ibuprofen: A degradation and toxicity study[J]. Science of the Total Environment, 2014, 466-467: 957-964. doi: 10.1016/j.scitotenv.2013.07.067 [9] XU R, ZHANG P, WANG Q, et al. Influences of multi influent matrices on the retention of PPCPs by nanofiltration membranes[J]. Separation and Purification Technology, 2019, 212: 299-306. doi: 10.1016/j.seppur.2018.11.040 [10] ZHANG L, SHA J, SUN G, et al. Vacancy engineering and constructing built-in electric field in Z-scheme full-spectrum-Response 0D/3D BiOI/MoSe2 heterojunction modified PVDF membrane for PPCPs degradation and anti-biofouling[J]. Chemical Engineering Journal, 2021, 414: 128867. doi: 10.1016/j.cej.2021.128867 [11] YANG Y, OK Y, KIM K, et al. Occurrences and removal of pharmaceuticals and personal care products (PPCPs) in drinking water and water/sewage treatment plants: A review[J]. Science of the Total Environment, 2017, 596-597: 303-320. doi: 10.1016/j.scitotenv.2017.04.102 [12] GERRITY D, SNYDER S. Review of Ozone for Water Reuse Applications: Toxicity, Regulations, and Trace Organic Contaminant Oxidation[J]. Ozone-Science & Engineering, 2011, 33(4): 253-266. [13] YI C, QIN W, WEN X. Renovated filter filled with poly-3-hydroxybutyrateco-hydroxyvalerate and granular activated carbon for simultaneous removal of nitrate and PPCPs from the secondary effluent[J]. Science of the Total Environment, 2020, 749: 141494. doi: 10.1016/j.scitotenv.2020.141494 [14] BOYD G R, ZHANG S Y, GRIMM D A. Naproxen removal from water by chlorination and biofilm processes[J]. Water Research, 2005, 39(4): 668-676. doi: 10.1016/j.watres.2004.11.013 [15] KUJAWSKA A, KIELKOWSKA U, ATISHA A, et al. Comparative analysis of separation methods used for the elimination of pharmaceuticals and personal care products (PPCPs) from water: A critical review[J]. Separation and Purification Technology, 2022, 290: 120797. doi: 10.1016/j.seppur.2022.120797 [16] VIENI N M, HARKKI H, TUHKANEN T, et al. Occurrence of pharmaceuticals in river water and their elimination a pilot-scale drinking water treatment plant[J]. Environmental Science & Technology, 2007, 41(14): 5077-5084. doi: 10.1021/es062720x [17] CHEN H, WANG J. Degradation and mineralization of ofloxacin by ozonation and peroxone (O3/H2O2) process[J]. Chemosphere, 2021, 269: 128775. doi: 10.1016/j.chemosphere.2020.128775 [18] BAVASSO I, MONTANARO D, PALMA L, et al. Electrochemically assisted decomposition of ozone for degradation and mineralization of Diuron[J]. Electrochimica Acta, 2020, 331: 135423. doi: 10.1016/j.electacta.2019.135423 [19] SANTANA-MARTINEZ G, ROA-MORALES G, GOMEZ-OLIVAN L, et al. Downflow bubble column electrochemical reactor (DBCER): In-situ production of H2O2 and O3 to conduct electroperoxone process[J]. Journal of Environmental Chemical Engineering, 2021, 9(4): 105148. doi: 10.1016/j.jece.2021.105148 [20] SLJUKIC B, BANKS C E, COMPTON R G. An overview of the electrochemical reduction of oxygen at carbon-based modified electrodes[J]. Journal of the Iranian Chemical Society, 2005, 2: 1-25. doi: 10.1007/BF03245775 [21] BAKHEET B, QIU C, YUAN S, et al. Inhibition of polymer formation in electrochemical degradation of p-nitrophenol by combining electrolysis with ozonation[J]. Chemical Engineering Journal, 2014, 252: 17-21. doi: 10.1016/j.cej.2014.04.103 [22] YONG E, LIN Y. Kinetics of natural organic matter as the initiator, promoter, and inhibitor and their influences on the removal of ibuprofen in ozonation[J]. Ozone-Science & Engineering, 2013, 35: 472-481. [23] LAJEUNESSE A, BLAIS M, BARBEAU B, et al. Ozone oxidation of antidepressants in wastewater treatment evaluation and characterization of new by-products by LC-Q-TOF-MS[J]. Chemistry Central Journal, 2013, 15: 7. [24] CHANG C, CHEN T, CHIN C, et al. Enhanced electrochemical degradation of ibuprofen in aqueous solution by PtRu alloy catalyst[J]. Chemosphere, 2017, 175: 76-84. doi: 10.1016/j.chemosphere.2017.02.021 [25] RAPHAEL R, ADISA A. Life cycle environmental impacts of advanced wastewater treatment techniques for removal of pharmaceuticals and personal care products (PPCPs)[J]. Journal of Environmental Management, 2018, 215: 258-272. [26] AMARASOORIYA A, KAWAKAMI T. Removal of fluoride, hardness and alkalinity from groundwater by electrolysis[J]. Groundwater for Sustainable Development, 2019, 9: 100231. doi: 10.1016/j.gsd.2019.100231 [27] 张霄磊. 电化学法在钢铁企业循环冷却水处理中的研究[J]. 给水排水, 2014, 50(1): 256-261. doi: 10.3969/j.issn.1002-8471.2014.z1.079 [28] SOLTANI R, MASHAYEKHI M. Decomposition of ibuprofen in water via an electrochemical process with nano-sized carbon black-coated carbon cloth as oxygen-permeable cathode integrated with ultrasound[J]. Chemosphere, 2018, 194: 471-480. doi: 10.1016/j.chemosphere.2017.12.033 [29] AMBULUDI S, PANIZZA M, OTURAN N, et al. Kinetic behavior of anti-inflammatory drug ibuprofen in aqueous medium during its degradation by electrochemical advanced oxidation[J]. Environmental Science and Pollution Research, 2013, 20(4): 2381-2389. doi: 10.1007/s11356-012-1123-6 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4404
- HTML全文浏览数: 4404
- PDF下载数: 98
- 施引文献: 0
