













-
芬顿(Fenton)工艺具有氧化性强、反应速率快等优点,广泛应用于造纸、纺织、制药和制油等行业难降解废水的处理。但芬顿工艺会产生大量成分复杂的芬顿污泥,需要进行处理以满足环境要求,填埋、焚烧等传统处理方式会造成二次污染并增加成本,芬顿污泥的处理难题已成为限制芬顿工艺推广应用的阻碍[1]。由于芬顿试剂含有大量亚铁盐,芬顿污泥中铁的质量分数可达30%~60%[2],具有较高的资源化利用潜力。目前,芬顿污泥的再利用方法主要包括回用于类芬顿工艺、制备催化剂、制备铁盐和亚铁盐等[3-6]。已有研究表明,主要成分为氢氧化铁和有机物的芬顿污泥可通过热处理转化为磁性铁氧化物和碳材料[7-8],铁氧化物和碳材料又可协同吸附重金属[9]。因此,通过热处理将芬顿污泥转化为磁性吸附材料具有一定可行性[10]。
常用的污泥热处理方法包括水热碳化法和热解法[11]。TONG等[1]通过水热碳化法将芬顿污泥转化为具有良好可回收性的磁性氨化水合炭,对Pb2+的吸附容量达359 mg∙g−1。WANG等[12]通过400 ℃热解将芬顿污泥转化为具有高电容和优异导电性的磁性生物炭,用于增强厌氧消化,可显著提高甲烷产量。陈丽群[13]以芬顿污泥和造纸黑液木质素为原料,热解制备出具有优异吸附和再生性能的磁性活性炭,对亚甲基蓝和苯酚的平衡吸附量分别为314 mg∙g−1和98 mg∙g−1,高于文献报道中的大部分磁性吸附剂。与水热碳化法相比,热解法无需高压环境,过程中不产生废液,有望作为制备新型芬顿污泥基磁性材料的方法。重金属锑(Sb)及其化合物对人体和环境生物具有毒性作用,被多国列为重点污染物。Sb在自然水体中主要以Sb(Ⅴ)价态存在,Sb(V)的迁移率和溶解度也均大于Sb(Ⅲ),研究去除水中Sb(V)的有效方法具有重要意义[14]。
本研究选择Sb(Ⅴ)作为目标污染物,通过煅烧焦化废水芬顿污泥制备磁性吸附剂,并采用酸改性优化对Sb(Ⅴ)的吸附性能,考察了pH、共存离子等因素对吸附效果的影响,对吸附实验数据进行了动力学和等温吸附拟合,结合材料表征,分析了磁性吸附剂吸附Sb(Ⅴ)的机理,此外,考察了吸附剂对实际废水的处理性能。
-
芬顿污泥取自江西省某焦化厂,产自采用芬顿工艺的焦化废水深度脱色处理工序,所采集的污泥已经过离心机脱水,含水量约为80%。芬顿污泥采集至实验室后,于鼓风干燥箱中65 ℃干燥至恒重,先后经粉碎机和球磨机进行粉碎和研磨,密封保存待用。
-
取适量研磨后的芬顿污泥,于管式炉中密闭煅烧,煅烧温度为300~800 ℃,煅烧时间为3 h,升温速率为20 ℃∙min−1,降温方式为炉内自然降温,煅烧产物以FS和煅烧温度命名(FS350)。取2.5 g芬顿污泥煅烧后产物投入100 mL一定浓度的硫酸溶液中,70 ℃水浴加热搅拌1 h,停止加热后继续搅拌冷却至室温,随后在磁铁辅助下分离出其中磁性材料,用水和无水乙醇清洗至pH呈中性,于真空干燥箱中65 ℃真空干燥,得到酸改性磁性材料,以AFS和煅烧温度命名(AFS350)。
-
管式炉(SKGL-1200,上海大恒光学精密机械有限公司);电子天平(ME206,Mettler Toledo);真空干燥箱(DZ-2BCIV,天津泰斯特仪器);电热鼓风干燥箱(GZX-9140MBE,上海博讯医疗生物仪器);粉碎机(YF6-2,浙江瑞安永历制药机械);行星球磨机(QM-QX1L,长沙米琪仪器);恒速电动搅拌器(JJ-4A,常州国宇仪器)、恒温水浴锅(DZKW-D-1,北京永光明医疗仪器);恒温振荡器(HZQ-X300C,上海一恒科学仪器);电感耦合等离子体质谱仪(iCAP-RQ,America Thermo);X射线多晶衍射仪(SmartLab/3KW,Janpan Rigaku)、振动样品磁强计(SQUID-VSM,America Quantum Design)、扫描电子显微镜(FEI Quanta 400 FEG,America FEI)、X射线透射电镜能谱仪(Edax appllo xl,America EDAX)、傅立叶变换红外吸收光谱仪(Nicolet iS50 FT-IR,American Thermo)、比表面积及孔径分析仪(3Flex,America Micromeritics)、纳米粒度电位仪(Zetasizer Nano ZS90,England Malvern)
氢氧化钠(NaOH)、硝酸(HNO3)、硫酸(NaOH)、无水乙醇(CH3CH2OH)、氯化钠(NaCl)、硝酸钾(KNO3)、无水氯化钙(CaCl2)、无水硫酸镁(MgSO4)、十八水硫酸铝(Al2(SO4)3∙18H2O)、十水碳酸钠(Na2CO3∙10H2O)和十二水磷酸三钠(Na3PO4∙12H2O)购于国药集团化学试剂有限公司,均为分析纯;砷标准溶液(1g∙L−1)和五价锑标准溶液(1g∙L−1)购于厦门海标科技有限公司;实验用水为超纯水。
-
以五价锑标准溶液为母液稀释配制不同浓度的Sb(Ⅴ)溶液,吸附和解吸实验均在恒温振荡器中进行,吸附转速为200 r·min−1,解吸转速为250 r·min−1,除等温吸附实验外,其他实验温度均为25 ℃。煅烧温度、酸改性和pH影响实验的Sb(Ⅴ)溶液初始质量浓度分别为0.474、0.683和0.883 mg∙L−1,吸附剂投加量分别为1.5、0.5和0.5 g∙L−1,反应时间为4 h。动力学、等温吸附、共存离子和再生利用实验的吸附剂投加量均为1 g∙L−1,pH均为4(通过NaOH和HNO3调节);动力学实验选择Sb(Ⅴ)溶液初始质量浓度为0~30 mg∙L−1内的3个浓度;等温吸附实验温度选择15、25和35 ℃,Sb(Ⅴ)溶液初始质量浓度选择0~100 mg∙L−1内的7个浓度;解吸实验振荡时间为3 h,其他实验反应时间为6 h。
-
使用电感耦合等离子体质谱仪(ICP-MS)检测溶液中的Sb和Fe的浓度。对AFS350等材料进行表面面积分析(BET)、X射线衍射分析(XRD)、傅里叶红外光谱分析(FTIR)、磁滞回线分析(VSM)、扫描电镜和X射线能谱分析(SEM+EDS)、X射线光电子能谱分析(XPS)、电动电势分析(Zeta电位),探究材料颗粒特性及对Sb(Ⅴ)的吸附机制。
-
去除率、吸附量和解吸率根据式(1)、式(2)和式(3)计算。
式中:
Rq 为去除率,%;Qe 为吸附量,mg∙g−1;Rd 为解吸率,%;C0 和Ct 分别为Sb(Ⅴ)溶液初始质量浓度和t时刻或吸附结束后Sb(Ⅴ)溶液质量浓度,mg∙L−1;V为溶液体积,L;m为吸附剂质量,g;Cd 为解吸液中Sb(Ⅴ)质量浓度,mg∙L−1;Vd 为为解吸液体积,L。吸附动力学拟合根据准一级动力学模型(式(4))、准二级动力学模型(式(5))、Elovich模型(式(6))和颗粒内扩散模型(式(7))计算。
式中:
qt 为t时刻的吸附容量,mg∙g−1;qe 为饱和吸附容量,mg∙g−1;t为吸附时间,min;k1 为准一级反应动力学常数,min−1;k2 为准二级反应动力学常数,g∙(mg∙min)−1;α为起始吸附速率,mg∙(g∙min)−1;β 为与表面覆盖度和化学吸附活化能有关的解吸常数,g∙mg−1;kp 为颗粒内扩散速率常数,mg·(g·min1/2)−1;C 为常数,mg∙g−1。吸附等温线拟合根据Langmuir模型(式(8)和式(9))、Freundlich模型(式(10))和Temkin模型(式(11)和式(12))计算。
式中:
qe和qm 分别为平衡吸附容量和理论最大吸附容量,mg∙g−1;Ce 为平衡质量浓度,mg∙L−1;KL 为Langmuir吸附系数,L∙mg−1;KF 和1/n为Freundlich常数;T 为绝对温度,K;bT 为与吸附热有关的常数,J∙mol−1;R为热力学常数,8.314 J∙(mol∙K)−1;BT 为Temkin常数;AT 为对应最大结合能的平衡结合常数,L∙mg−1。 -
煅烧温度直接影响制备材料的成分、形貌和稳定性。不同温度煅烧产物对Sb(Ⅴ)的去除情况见图1。同条件下FS350对Sb(Ⅴ)去除率最高。使用酸液对材料进行改性,可提高孔隙度,暴露出更多的活性基团和吸附位点。本研究选择硫酸溶液对煅烧材料进行改性,不同浓度硫酸溶液改性后的FS350对Sb(Ⅴ)的去除率变化见图2。0.2 mol∙L−1的硫酸溶液改性效果最佳,将其命名为AFS350且用于后续实验。
-
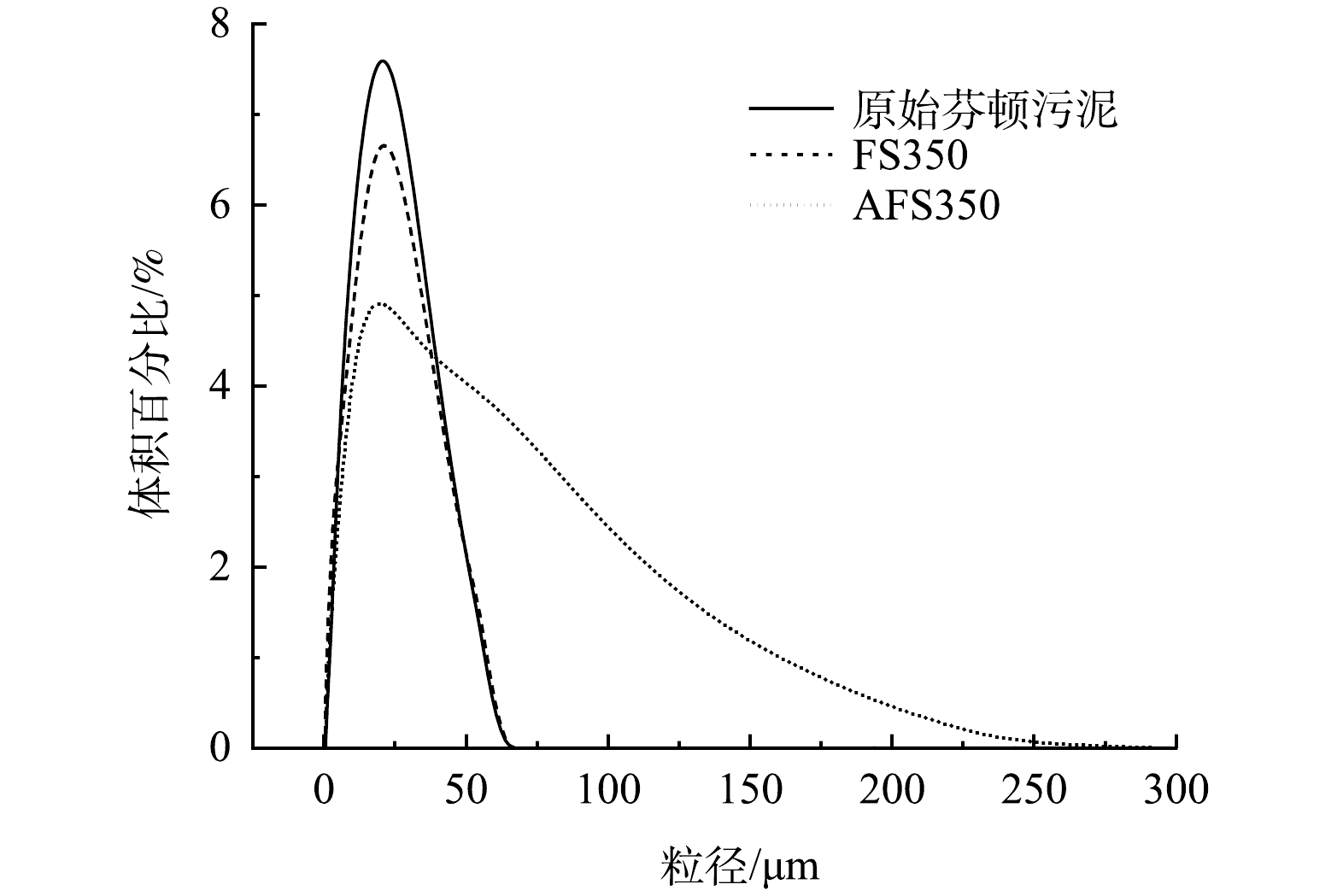
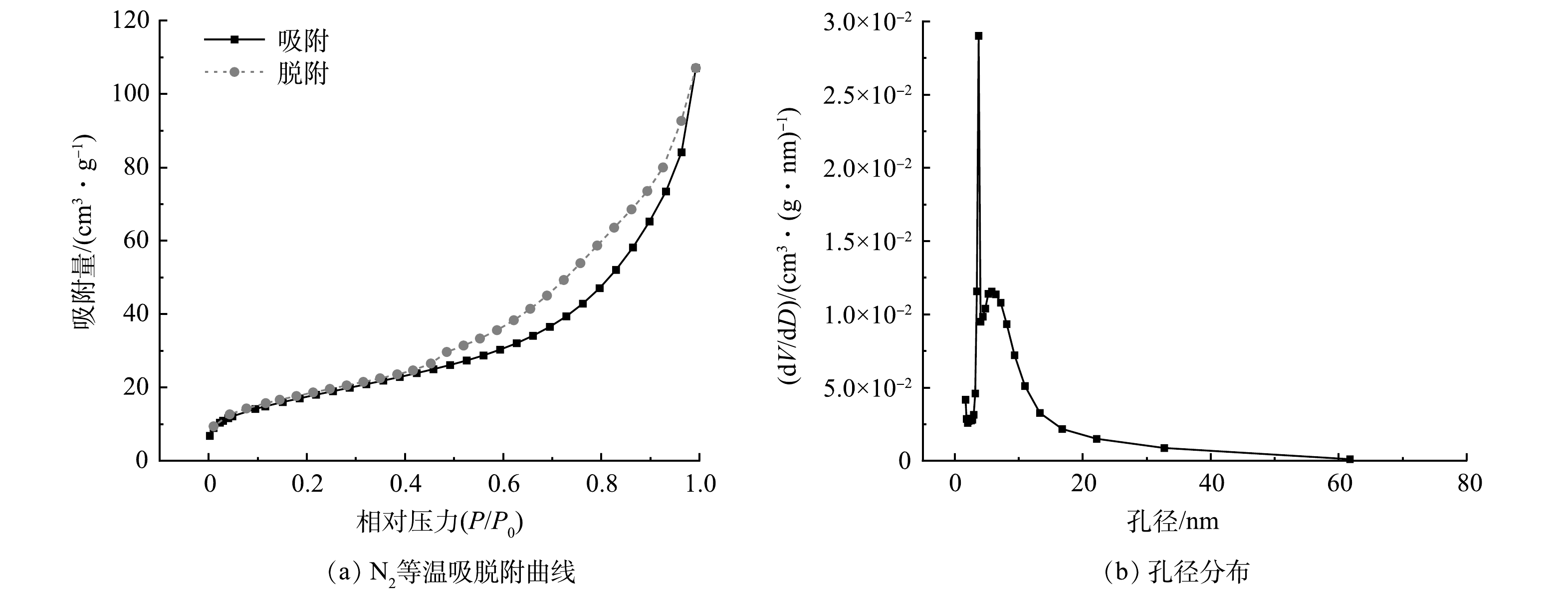
由图3可见,FS350与AFS350具有粗糙的多孔结构,存在不规则的孔洞,其中AFS350经酸液溶蚀后,表面更粗糙,孔洞更多。EDS表征显示,AFS350中Fe、O和C元素的质量占比分别为51.2%、24.6%和19.9%,Na、Si、Ca等其他元素共计占比约4.3%,Fe、O、C元素的原子比为1:1.68:1.79。由图4可见,原始芬顿污泥和FS350的颗粒粒径在65 μm以内,20 μm左右分布最多。AFS350的粒径分布在0~250 μm,由于存在略微的团聚,酸改性后的颗粒粒径有所增大。原始芬顿污泥、FS350和AFS350的比表面积分别为80.2、55.2和63.8 m2∙g−1,孔径体积分别为0.144、0.128和0.166 cm³∙g−1,平均孔径分别为6.42、7.99和8.88 nm。由图5可见,AFS350的N2吸脱附曲线符合Ⅳ型吸附等温线,存在明显的吸附回滞环,表明AFS350为裂隙孔材料,孔形可能为狭长裂口形,对N2的吸附是在介孔吸附剂上的物理吸附,对应有毛细凝聚现象的单层吸附体系[15]。
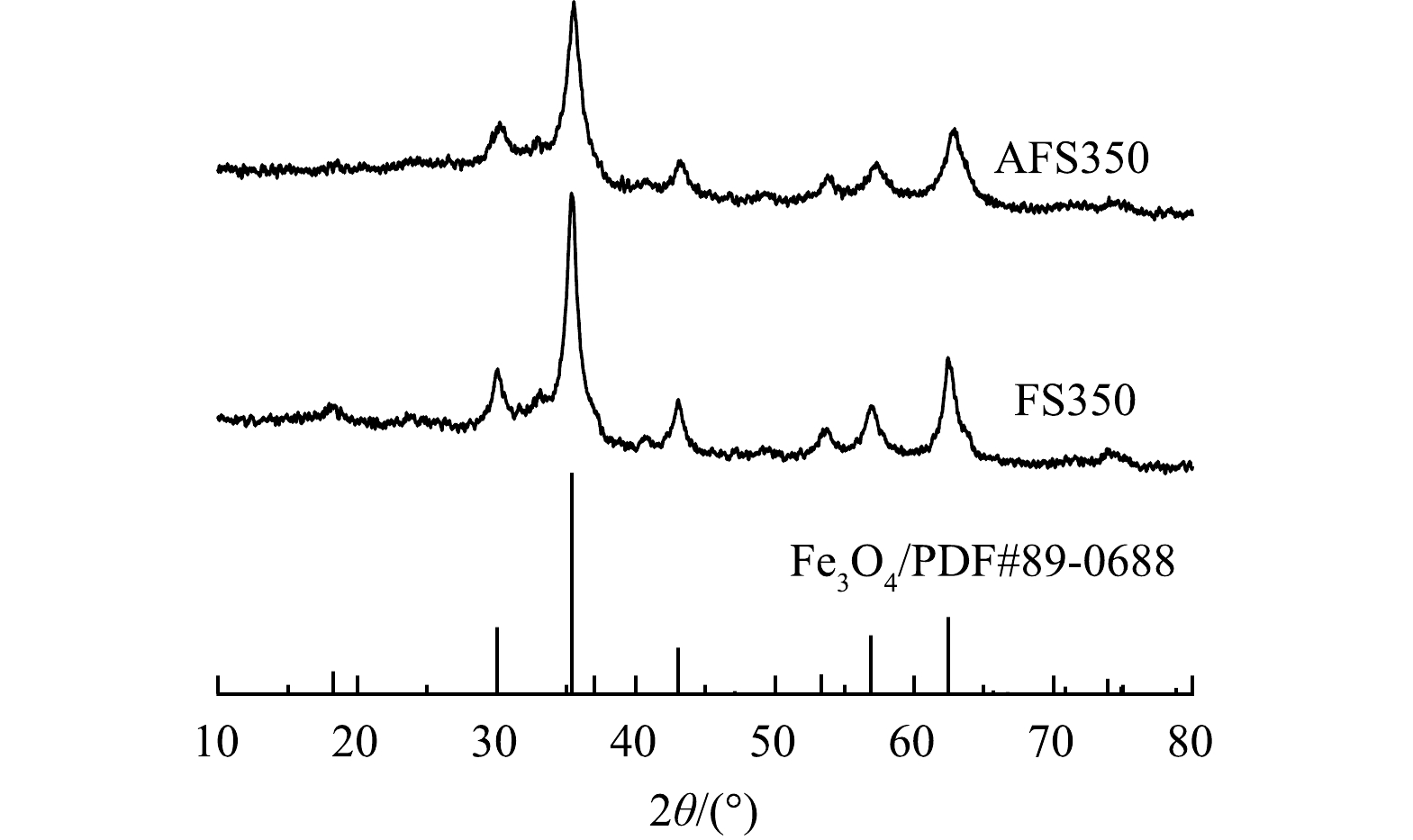
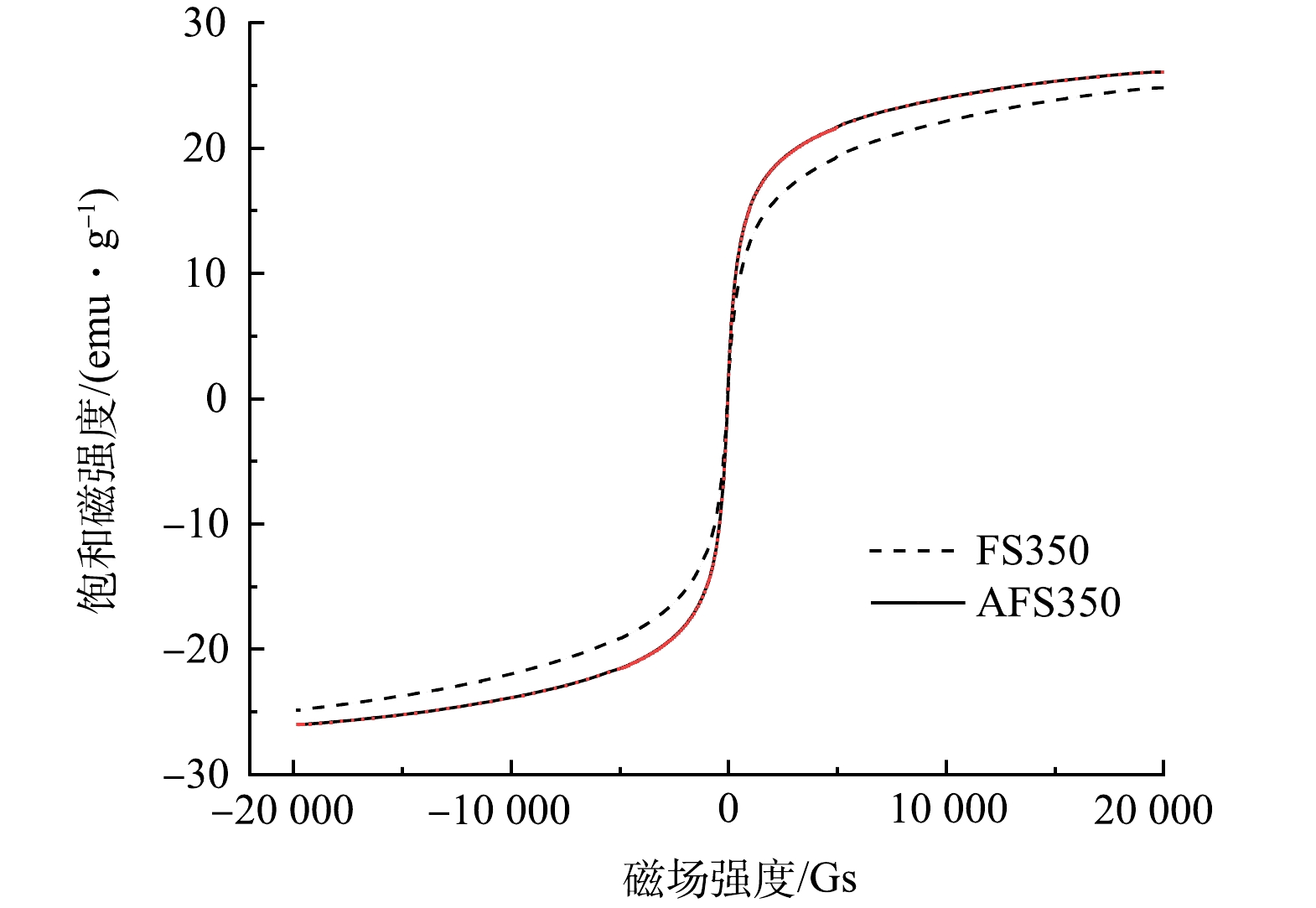
由图6可见,2θ为30.3°、35.7°、43.4°、53.9°、57.3°、63°的衍射峰与Fe3O4标准谱(PDF#89-0688)上(220)、(311)、(400)、(422)、(511)、(440)晶面对应良好。这表明2种材料的主要成分为Fe3O4。由图7可见,FS350和AFS350的饱和磁强度分别为24.8 emu∙g−1和26.1 emu∙g−1,在磁场作用下可快速聚集沉降,为其从溶液中快速分离提供基础。
由图8可见,3 419 cm−1强而宽的吸收峰归因于羟基(—OH)的伸缩振动。表明存在吸附水[16]。1 625 cm−1和1 035 cm−1的谱峰归因于COOH基团[16],1 385 cm−1处的峰归因于—OH的弯曲振动[17],1 120 cm−1处的峰可能与C—O的伸缩振动和—OH的弯曲振动有关[18],568 cm−1处谱峰归因于Fe—O官能团的伸缩振动,表明材料中存在铁氧化物[16]。以上结果表明,AFS350表面含有较丰富的羟基和羧基[1]。AFS350的羟基伸缩振动峰峰强和峰宽均较FS350显著增加。这是因为酸改性向FS350中引入了新的羟基,酸溶蚀作用也使其暴露出更多的功能基团。
-
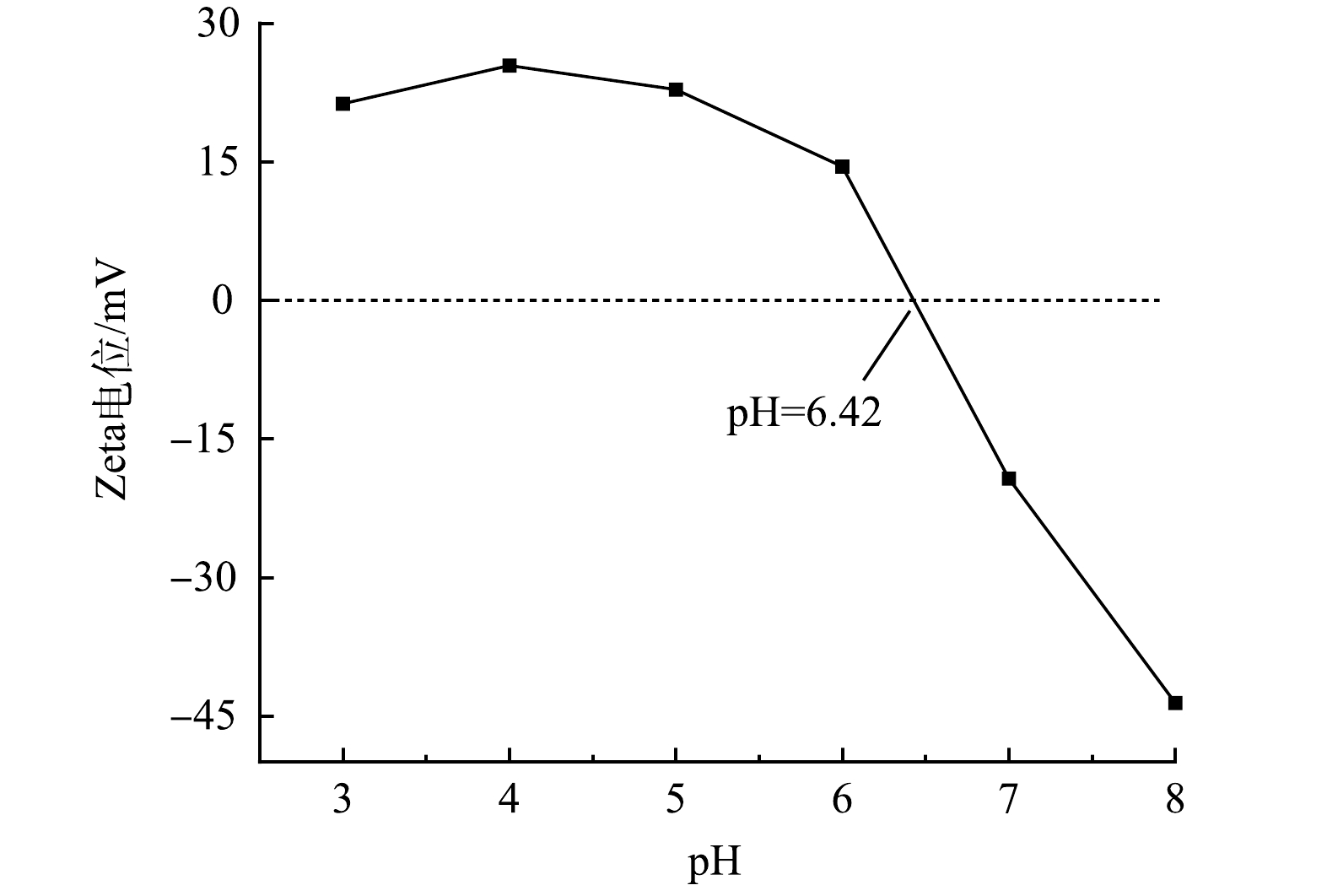
溶液pH会影响吸附剂与吸附质的表面带电情况,对Sb(Ⅴ)的吸附产生直接影响。由图9可见,在pH为3~10时,Sb(Ⅴ)的去除率随pH升高先缓慢后迅速降低。这是因为Sb(Ⅴ)在pH>3的溶液中主要以
Sb(OH)−6 形式存在[19],而图10中AFS350的等电点为6.42,当pH>6.42时AFS350表面带负电荷,因此当溶液pH呈碱性时,AFS350对Sb(OH)−6 的排斥力占主导,导致其对Sb(Ⅴ)的吸附量显著降低。根据不同pH下AFS350对Sb(Ⅴ)的去除率变化,确定最佳pH为4。结果表明,AFS350在pH为7时铁溶出量最低,酸性条件下的铁溶出量高于碱性条件。这是由于Fe3+和Fe2+在酸碱条件下不同存在形式的原因。集中式生活饮用水地表水源地的铁含量标准限值为0.3 mg∙L−1,与之相比,绝大多数情况下AFS350的铁溶出量是安全的。 -
AFS350对Sb(Ⅴ)的吸附量随时间的变化如图11所示,表现出随时间增加先快速吸附后缓慢吸附至平衡的曲线形态。动力学模型拟合参数见表1,结果表明,AFS350吸附Sb(Ⅴ)与准二级动力学模型更为符合,可决系数R2大于0.98,高于准一级动力学,这说明AFS350有饱和吸附点[20],对Sb(Ⅴ)的吸附主要为化学吸附[21],吸附速率与驱动力的平方成正比关系[22]。准一级动力学模型拟合的R2值(0.94~0.95)也较高,表明还存在着非主导的物理吸附过程,这与材料本身的物理特性有关。此外,AFS350对Sb(Ⅴ)的吸附还符合Elovich模型(R2>0.99),说明该吸附为非均质表面吸附[23-24],吸附中可能发生了共用电子对及价电子的转移,存在化学反应[20]。
颗粒内扩散模型可判定吸附过程中是否由颗粒内扩散起决定作用[25],
kp 越大,金属离子越容易在吸附材料表面进行内部扩散。粒子内扩散模型可分为2个阶段进行分别拟合,一段为外表面扩散拟合,另一段为内表面扩散拟合。由图12和表2可见,颗粒内扩散模型拟合的R2值高,拟合图由两段直线组成,将反应分为20 min前后2个阶段,一阶段表示吸附质扩散到吸附剂表面的过程,即膜扩散过程,二阶段为吸附质在吸附剂空隙内扩散的过程,即颗粒内扩散过程[22]。拟合结果中3个浓度的2个阶段拟合截距均不过零点,说明颗粒内扩散速率不是控制AFS350吸附Sb(Ⅴ)的唯一速率,吸附速率是由膜扩散和颗粒内扩散速率共同决定的[22]。
-
吸附等温线是在温度一定的条件下,表达反应平衡时吸附量同溶液浓度之间关系的数学式,可解释吸附现象的本质。由图13和表3的拟合结果可见,AFS350对Sb(Ⅴ)的吸附量随温度升高而增加。说明该吸附为吸热反应,提高温度有利于吸附进行。由拟合的可决系数可知,3种等温模型的拟合度顺序为Freundlich>Temkin>Langmuir,说明AFS350对Sb(Ⅴ)的吸附是多分子层吸附,材料表面可能不均匀,吸附位点异质性分布,存在各处吸附能力不相同的情况。这与SEM的表征结果一致[22,24,26-27]。从Freundlich模型中拟合得到3个温度的吸附浓度指数1/n均小于0.5,说明吸附容易进行[22]。当Sb(Ⅴ)溶液为不同初始质量浓度时,根据Langmuir方程定义的无量纲因子
RL 均在0~1,说明该吸附为优惠吸附,容易自发进行[22]。Temkin模型拟合的可决系数较高(>0.92),表明该吸附过程中吸附热随温度线性变化,为化学吸附[22]。 -
通过共存离子影响实验考察多种阴阳离子对Sb(Ⅴ)去除的影响,实验所用Sb(Ⅴ)溶液的质量浓度为17.32 mg∙L−1。共存离子中,
As3+(20) 和PO3−4(20) 的质量浓度为20 mg∙L−1,其他溶液的离子质量浓度为50 mg∙L−1。由图14可见,SO2−4 (硫酸钠配制)的存在对溶液中Sb(Ⅴ)的去除有一定影响,这与已有研究报道一致[27-28]。Al3+(硫酸铝配制)、As3+和PO3−4 对Sb(Ⅴ)去除影响较大,其中SO2−4 、As3+和PO3−4 影响Sb(Ⅴ)去除的原因相同,是由于SO2−4 、AsO−2和PO3−4 阴离子在铁氧化物材料表面形成了内层络合物,与溶液中的Sb(OH)−6 竞争吸附,其相似的吸附机理抑制了AFS350对Sb(Ⅴ)的吸附[27-29];Al3+对Sb(Ⅴ)的去除产生抑制是因为配制Al3+溶液时引入了浓度数倍于Al3+的SO2−4 。As、P由于和Sb为同一主族元素,又同是以三元酸形式存在,具有几乎相似的结构和化学性质,与Sb(Ⅴ)竞争相同吸附点位的作用更强,因此,更不利于Sb(Ⅴ)的去除,并且在共存溶液表现出较Sb(Ⅴ)优先被去除的特性[30]。Na+、K+等常见阳离子对Sb(Ⅴ)的去除几乎没有影响,说明静电吸引在吸附过程中不占主导作用。 -
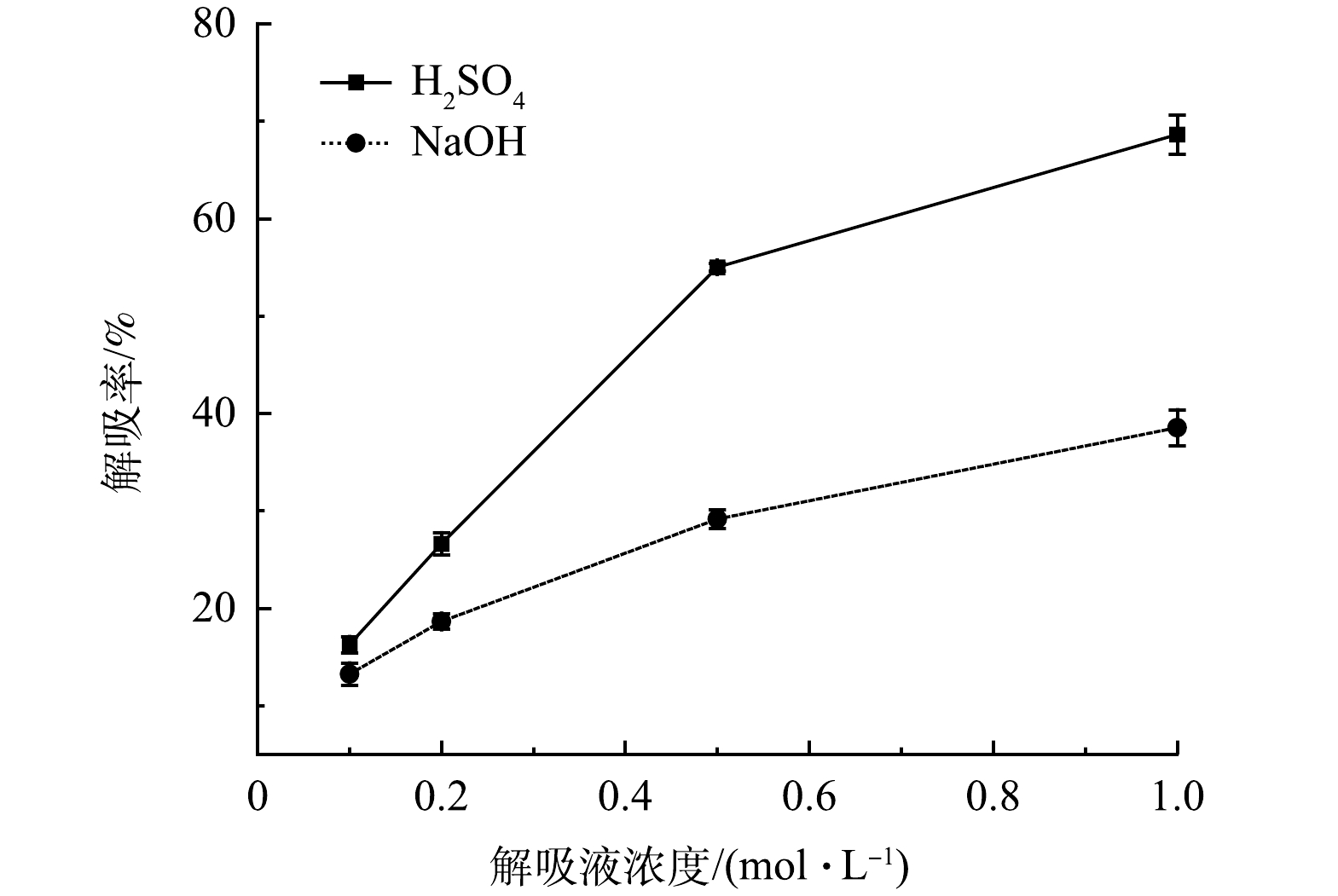
对吸附剂进行再生和再利用,可回收重金属,减少处理废物的产生,降低成本。实验中用于吸附的Sb(Ⅴ)溶液质量浓度为9.94 mg∙L−1,再生液选择体积为吸附溶液体积1/10的H2SO4和NaOH溶液,以解吸率作为评价标准,考察不同解吸液对AFS350的解吸再生能力。由图15可见,H2SO4溶液对Sb(Ⅴ)的解吸率比NaOH溶液更高,经过3 h的脱附,1 mol∙L−1的H2SO4溶液对AFS350的解吸率约为70%。根据不同解吸液的解吸表现,选择1 mol∙L−1的H2SO4溶液作为解吸液。
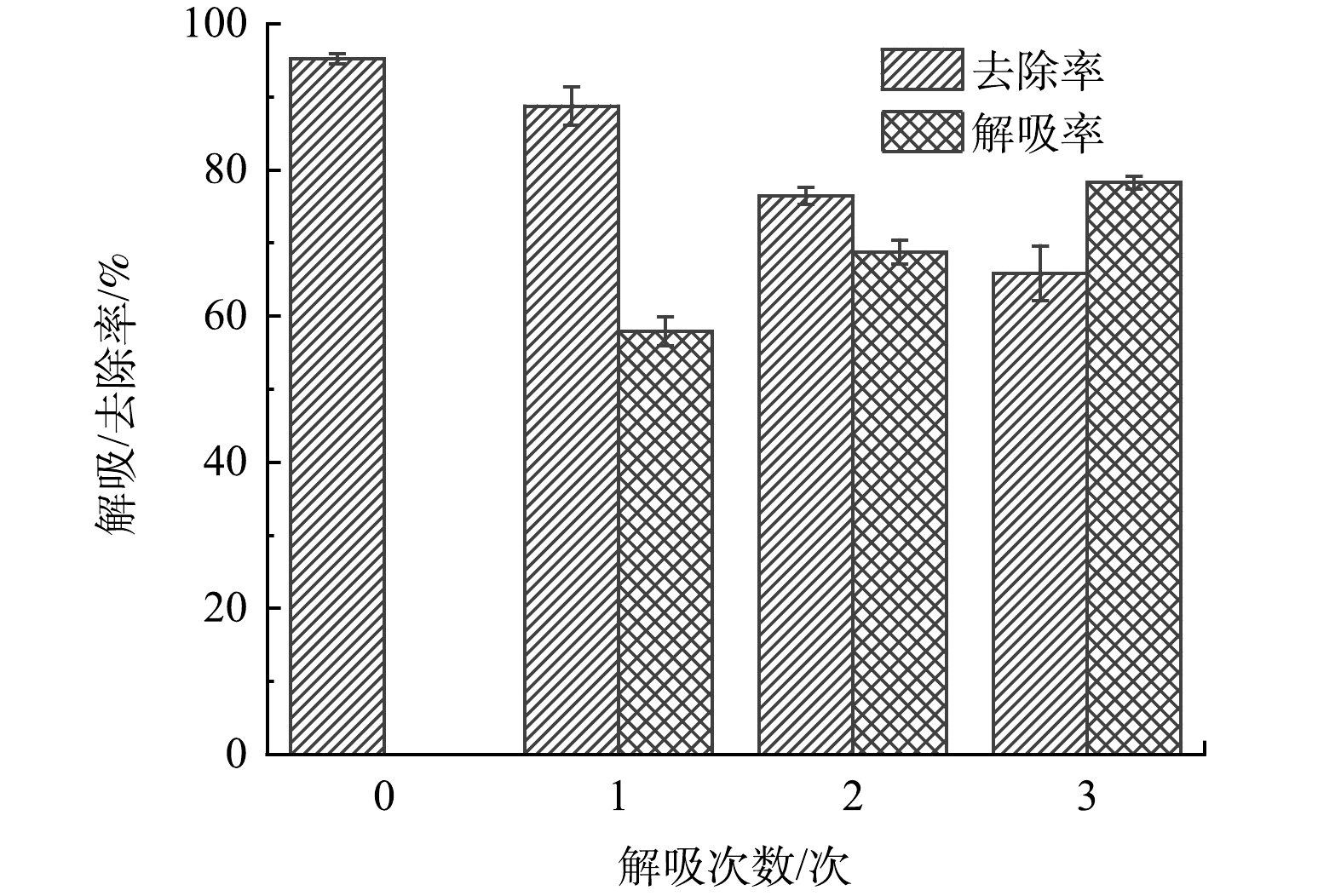
以1 mol∙L−1的H2SO4溶液对吸附后的AFS350解吸再生,再生后的吸附剂再次投入Sb(Ⅴ)溶液中进行吸附,依次循环。由图16可见,AFS350对溶液中Sb(Ⅴ)的初始去除率为95.2%,经3次再生后Sb(Ⅴ)的去除率降为65.9%。解吸率在多次再生中逐渐升高。这是因为每次脱附均不完全,吸附剂中积累的未脱附Sb(Ⅴ)的量逐渐增多,因此,每次的脱附量和解吸率逐渐增大。由于材料自身结晶度不高,又在解吸再生中被酸液溶蚀,多次再生后,AFS350的稳定性降低,根据材料稳定性和去除率表现,认为3次的再生次数较为合适。
-
为考察AFS350对实际含锑废水的处理效果,选择了2种含锑废水进行实验,处理目标为≤5 μg∙L−1。水样1为废弃矿洞涌水,pH为7.1,锑含量为3.33 mg∙L−1,以5.5 g∙L−1投加量投加AFS350,吸附后的锑含量为4.2 μg∙L−1;水样2为某锑制品厂洗地水,pH为7.3,锑含量为10.7 mg∙L−1。采用了2种方法进行处理:方法一为2次吸附法,第1次吸附以4 g∙L−1投加量吸附4 h,处理后的锑含量为0.17 mg∙L−1,第2次吸附以0.5 g∙L−1投加量再吸附4 h,处理后废水的锑含量为3.7 μg∙L−1;方法二为絮凝沉淀联合吸附法,先投加PFS(投加量为0.2 g∙L−1)和微量PAM溶液(质量浓度为3 g∙L−1)进行混凝沉淀,处理后上清液的锑含量为0.70 mg∙L−1,再以2 g∙L−1的投加量向上清液中投加AFS350进行吸附,吸附4 h后废水的锑含量为2.1 μg∙L−1。以上AFS350对实际废水的处理去除率均接近100%。实验还对AFS350进行了再生利用,经1、2次再生后吸附容量分别降低4%~8%和10%~16%。
-
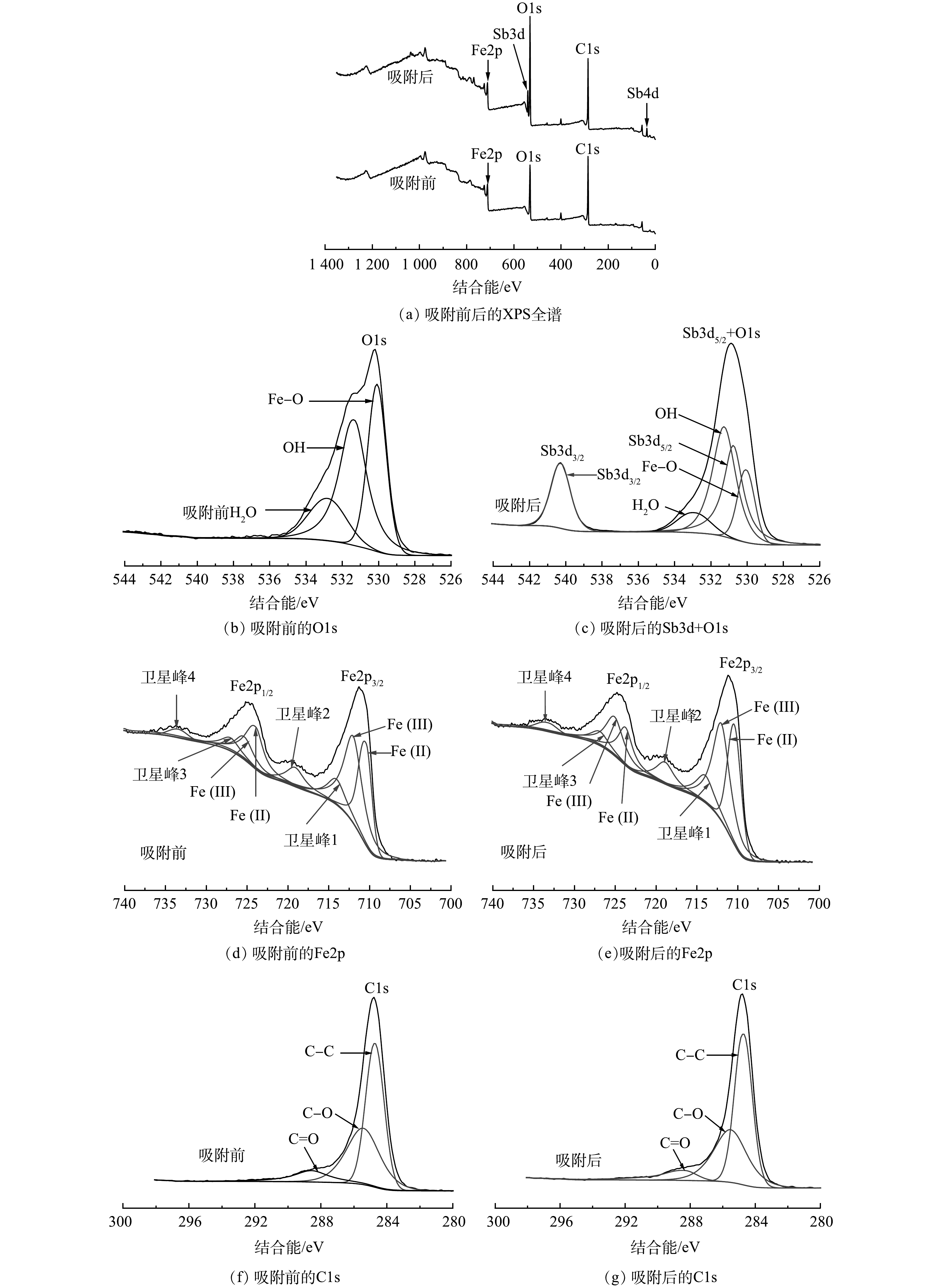
有研究[27]表明,铁氧化物表面的羟基在吸附Sb(Ⅴ)的过程中发挥重要作用。由图8可见,吸附后AFS350的羟基特征峰强度减弱。造成红外光谱谱带吸收损失的原因是AFS350中铁氧化物表面功能性羟基对锑的吸附[18]。Fe—O的峰强在吸附后减弱,是由于Sb的负载形成了Fe—O—Sb络合减弱了信号[31]。有研究[32-35]表明,锑酸盐、磷酸盐和砷酸盐都是以内层络合形式结合到针铁矿、磁铁矿等铁氧化物表面。内层结合阴离子的主要机制是配体交换,表面羟基与阴离子配体的交换过程中,由于表面羟基的质子化,较低的pH有利于表面络合物的形成[36]。这与本研究中溶液pH对Sb(Ⅴ)吸附影响的结果一致。因此,分析认为,AFS350对Sb(Ⅴ)的吸附主要为其中铁氧化物对Sb(Ⅴ)的化学吸附,主要作用是羟基的配体交换,形成了FeO—
Sb(OH)−5 形式的内层络合物[36-38]。由图17(b)可见,吸附前的O1s峰结合能为530.2 eV,可分为530.1、531.4和532.8 eV的3个子峰,结合能为530.1 eV处的第1个峰为Fe3O4晶格和金属氧化物键中存在的氧[39];531.4 eV处的峰对应于—OH基团的峰[40];532.8 eV处的峰表明材料表面存在吸附水[26,40]。吸附后,由于Sb的负载,图17(c)中出现了Sb的峰,由于Sb的峰与O的峰重叠,Sb3d5/2+O1s的结合能增加到530.9 eV,峰强明显增强。Sb3d3/2和Sb3d5/2的结合能分别为540.3 eV和530.8 eV(Δ=9.5 eV),对照数据库判断其价态为正五价,与吸附所用初始溶液中的Sb价态相同,表明Sb在吸附过程中未发生氧化还原反应。有研究[36,41]表明,铁氧化物吸附Sb(Ⅴ)后,由于Fe—OH和Sb—OH发生配位反应,—OH的特征峰面积及在O1s峰面积中的占比会降低。AFS350在吸附后,—OH峰的面积增加了约42%,面积和占比均未按照预期降低。这是因为吸附是在pH为4的酸性溶液中进行,向吸附剂中引入了新的羟基。吸附后,AFS350的O1s峰位置和强度变化表明含氧官能团参与了Sb(Ⅴ)的吸附,其中—OH官能团起到了重要作用,形成了Fe—O—Sb的内层络合结构[42-43]。
由图17(d)和图17(e)的Fe2p光谱中可见,吸附前后Fe2p1/2和Fe2p3/2自旋轨道能级的能量差分别为13.9 eV和13.6 eV。这与已有研究结果一致[44]。在吸附前后Fe2p3/2主峰中均检测到Fe(Ⅱ)(710.6 eV和710.5 eV)和Fe(Ⅲ)(712.1 eV和712.1 eV)的子峰,表明Fe(Ⅱ)和Fe(Ⅲ)氧化物的存在[45]。其中吸附前Fe2p3/2谱峰中Fe(Ⅲ)与Fe(Ⅱ)峰面积比为1.44:1,表明Fe(Ⅲ)的量多于Fe(Ⅱ)。这是由于制备时的煅烧温度不高,Fe(Ⅲ)转化为Fe3O4的量尚少的原因,因此,与一般磁铁矿中Fe(Ⅲ)和Fe(Ⅱ)的化学计量比存在差异[46]。吸附后Fe(Ⅲ)的峰面积占比略有降低,是因为在酸性溶液中被碳少量还原的原因。Fe2p和C1s光谱在吸附前后的形状和强度基本没有变化,表明其在吸附过程结构保持稳定[16]。
-
1)350 ℃煅烧热解并以0.2 mol∙L−1硫酸溶液改性制备的芬顿污泥基磁性吸附剂(AFS350)为介孔结构颗粒,具有粗糙的表面和较大的比表面积(63.8 m2∙g−1),主要成分为Fe3O4和碳,饱和磁强度为26.1 emu∙g−1,可在磁场作用下快速分离。
2)动力学和等温吸附拟合表明,AFS350吸附Sb(Ⅴ)更符合准二级动力学和Freundlich等温模型,Elovich动力学模型、颗粒内扩散模型和Temkin等温模型的可决系数也较高,说明AFS350对Sb(Ⅴ)的吸附是多分子层异质吸附,吸附过程以化学吸附为主导,膜扩散和颗粒内扩散速率共同决定吸附速率。
3)酸性条件有利于AFS350对Sb(Ⅴ)的吸附,
SO2−4 、AsO−2和PO3−4 会抑制Sb(Ⅴ)的去除;1 mol∙L−1硫酸适宜作为AFS350的再生液,3次再生后吸附容量降低了约29.3%。AFS350对Sb(Ⅴ)的吸附以形成Fe—O—Sb配位结构的内层络合作用为主,—OH的配体交换在其中发挥了重要作用。4) AFS350可将较高浓度的矿洞涌水和锑制品厂废水中的锑去除至5 μg∙L−1以下,去除率接近100%,经过2次再生后吸附容量降低10%~16%,再生利用性能较为稳定,通过煅烧芬顿污泥制备磁性吸附剂具有实际应用价值。
芬顿污泥制备磁性吸附剂去除水中Sb(Ⅴ)
Preparation of Fenton sludge-based magnetic adsorbent for Sb(Ⅴ) removal from water
-
摘要: 为实现芬顿污泥的有效处置,开发了一种基于煅烧芬顿污泥制备磁性吸附剂的回收利用方法,通过进一步酸改性优化吸附性能,用于去除水中的Sb(Ⅴ)。结果表明,350 ℃煅烧并以硫酸改性的磁性吸附剂(AFS350)具有粗糙的多介孔结构和较大的比表面积,主要成分为Fe3O4和碳;准二级动力学模型、Elovich动力学模型、颗粒内扩散模型、Freundlich和Temkin等温吸附模型对AFS350吸附Sb(Ⅴ)拟合效果好,表明吸附为多层异质化学吸附,吸附速率由膜扩散和颗粒内扩散速率共同控制;机理分析认为,AFS350对Sb(Ⅴ)的吸附以形成Fe—O—Sb配位结构的内层络合作用为主。Sb(Ⅴ)的吸附受多因素影响,酸性条件和升温有利于吸附进行,
SO2−4 、AsO−2和PO3−4 会抑制AFS350对Sb(Ⅴ)的吸附。AFS350可进行多次脱附再生,对实际废水处理去除率接近100%,表明AFS350是一种有效的磁性锑吸附剂,煅烧制备磁性吸附剂是实现芬顿污泥再利用的可行途径。Abstract: In order to realize the effective disposal of Fenton sludge, a recycling method based on calcined Fenton sludge to prepare magnetic adsorbent was developed, and the adsorption performance was further optimized by acid modification, which was used to remove Sb(V) in water. The results showed that the magnetic adsorbent (AFS350) calcined at 350 ℃ and modified with sulfuric acid had a rough mesoporous structure and a large specific surface area, and its main components were Fe3O4 and carbon. The pseudo-second-order kinetic model, Elovich kinetic model, intraparticle diffusion model, Freundlich and Temkin adsorption isotherm models fitted well for Sb(V) adsorption on AFS350, which indicated that the adsorption was a multilayer heterogeneous chemisorption, and the adsorption rate was controlled by both membrane diffusion and intraparticle diffusion rate. Mechanism analysis suggested that Sb(V) adsorption on AFS350 was mainly due to the formation of inner-layer complexation of Fe—O—Sb coordination structure. And multi-factors contributed to Sb(V) adsorption, acidic conditions and elevated temperature were conducive to Sb(V) adsorption,SO2−4 ,AsO−2andPO3−4 could inhibit Sb(V) adsorption. AFS350 could be desorbed and regenerated for many times, and the Sb(V) removal rate in actual wastewater was close to 100%. This indicated that AFS350 is an effective magnetic antimony adsorbent, and the preparation of magnetic adsorbent by calcination is a feasible way to realize the reuse of Fenton sludge.-
Key words:
- Fenton sludge /
- recycling /
- calcination /
- magnetic adsorbent /
- Sb(Ⅴ) in water
-
微生物污染问题作为全球饮用水供给安全中的重大挑战,在缺乏强效饮用水消毒措施的不发达国家和地区尤为严重[1–3]。传统的饮用水消毒手段存在一些局限,主要是消毒不彻底,极易生成有毒害副产物等,例如臭氧消毒不仅成本高、难储存,易腐蚀管道,而且会产生多余的消毒副产物(disinfection byproducts,DBPs)[3–5];紫外消毒存在着光穿透率低、易受水质状况影响、紫外灯耗电量大,以及微生物易光复活和暗复活等问题;加氯消毒则无法灭活两虫等抗氯微生物,而且会生成较多的卤代消毒副产物 [6–8]。为了应对传统消毒方法的局限性,近年来一些先进的消毒技术作为替代方案应运而生[9–11]。
在各种先进的消毒技术中,基于硫酸根自由基(SO4−•,通过活化过一硫酸盐(peroxymonosulfate , PMS)和过二硫酸盐(peroxo-disulfate,PDS))的高级氧化工艺(advanced oxidation processes, AOPs)具有较好的应用前景,主要基于以下几方面原因:(1)SO4•−具有宽pH适应范围,在pH = 3—11均具有较高的氧化电位(E0(SO4−•/SO42−) = +2.60 — +3.10 VNHE),能够氧化灭活多种微生物,并且能降解胞内基因来抑制微生物假死复活[12];(2)SO4−•的活化方式多样,如可以通过光、热、碱、超声或投加催化剂活化过硫酸盐来获得SO4−•,且过硫酸盐能够溶于水来渗透进土壤来进行修复[12–14];(3)与加氯和臭氧工艺相比,SO4−•消毒后产生的副产物数量较少[15];(4)SO4−•具有较高的选择性,在含溶解有机物、阴离子、阳离子等不同水环境中受影响较小,并能保持较高的消毒效果[12–13]。虽然基于SO4−•的AOPs用于消毒已经有较好的文献报道[16–20],但到目前为止仍没有相关的文章综述总结和比较基于SO4−•的AOPs用于灭活不同微生物的效率及其适用性。
当采用基于SO4−•的AOPs处理工艺对废水进行消毒时,不同水质环境和操作条件对于评估和优化该工艺至关重要。以往的研究评估了pH值、共存的溶解性有机物 (dissolved organic matter, DOM)阴离子、催化剂用量、温度和光强对消毒效率的影响[21–23],因此需要进行全面的概述来总结基于SO4−•的最佳消毒工艺条件。此外,目前对基于SO4−•的AOPs消毒工艺灭活微生物的机理仍存在一些争论:从过程角度来看,灭菌主要活性物种究竟是SO4−•,还是OH•、O2•−、1O2、激发态电子等仍然存在争议;从微生物的角度来看,有报告指出活性物种最先氧化微生物的壁或膜, 导致其不可逆转的损伤,最终影响膜的通透性和生理功能[17, 24-25];或有文献也强调活性物种还会攻击细胞内成分,甚至影响到微生物的酶与基因组[26–28]。因此,总结分析基于SO4−•的消毒工艺将提高对该技术的基础理解,并为指导其工程应用提供帮助。本文总结了各种基于SO4−•工艺的灭菌反应体系及其灭菌机理,同时讨论了水质环境的影响以及副产物的生成,进一步对该方向的研究需求和未来发展方向作出展望。
1. 不同SO4−•工艺对微生物的灭活(Microorganism inactivation by various SO4−• based processes)
1.1 铁基催化活化PS灭活微生物
PS可被铁、钴、铜、锰等多种金属活化,其中如溶解态铁、螯合铁和固态铁等铁基催化剂具有较为实际的应用前景[13, 29-33]。将铁类物种与PS结合以产生用于微污染物降解的自由基已在文献中得到充分证明[13]。这种组合还可用于对水中不同类型微生物的灭活[31-33]。表1总结了包括亚铁离子(Fe2+)、零价铁(ZVI = Zero valent iron)、铁矿物和氧化铁(Fe2O3)在内的不同铁基活化PS的消毒效率。Wordofa等研究了Fe2+/PDS系统对大肠杆菌O157:H7的灭活作用,在3 mmol·L−1 S2O82−+3 mmol·L−1 Fe2+投加量条件下,130 min后灭活率达到95%,灭活速率常数为0.025 min−1[16]。而Qi等对Fe2+/PDS系统进行优化,在投加量为40 mmol·L−1 S2O82− + 13.3 mmol·L−1 Fe2+,即 1:0.33的最佳条件下,120 s后(7.77 ± 0.57)lg CFU·mL−1的大肠杆菌O157:H7和(7.25 ± 0.36)lg CFU·mL−1的李斯特菌全部灭活[31]。Xia 等使用天然磁黄铁矿活化PDS使大肠杆菌K-12失活,达到了0.47 lg CFU·min−1的失活速率常数并在15 min内使7 lg CFU·mL−1细胞完全失活[33]。 RodriguezChueca等比较了使用Fe2+、Fe2O3、ZVI活化PDS和PMS以灭活大肠杆菌[34]。在很短的反应时间内PMS与Fe2+、Fe2O3和ZVI的偶联反应便使大肠杆菌完全失活,同时,PDS诱导的细菌灭活率低于PMS[34]。
表 1 铁基活化SO4−•致微生物失活Table 1. Microorganism inactivation by SO4−•-based processes activated by iron species体系Process 目标微生物Target microorganisms 实验条件Experimental conditions 消毒效率Disinfection efficiency 参考文献Reference Fe2+/PDS O157:H7大肠杆菌 3 mmol·L−1 PDS、3 mmol·L−1 FeSO4、3 mmol·L−1 NH2OH和pH7的饮用水 约130 min达到95%的灭活率 [16] Fe2+/PDS O157:H7大肠杆菌和李斯特菌 40 mmol·L−1 PDS 和 13.3 mmol·L−1 Fe2+ 120 s后(7.77 ± 0.57 )lg CFU·mL−1的大肠杆菌和(7.25 ± 0.36) lg CFU·mL−1的李斯特菌失活 [31] ZVI/PDS 大肠杆菌BL21和粪肠球菌 大肠杆菌测试中:3 mmol·L−1 PDS、 0.6 g·L−1 ZVI粪肠球菌测试中:1 mmol·L−1 PDS、0.2 g·L−1 ZVI以及 pH 为5.5 80 min后90%的大肠杆菌失活;12 min后75%的粪肠球菌失活 [11] 天然黄铁矿/PDS 大肠杆菌K-12 0.5 mmol·L−1 PDS、1g·L−1 NP 以及 pH 为5.0 15 min内7 lg CFU·mL−1大肠杆菌失活 [33] Fe2+, Fe2O3, nZVI/PDS和PMS 大肠杆菌 0.5 mmol·L−1 PDS、 0.5 mmol·L−1 PMS、1 mmol·L−1 Fe(Ⅱ)、1 mmol·L−1 Fe2O3以及50 mg·L−1 ZVI 5 min内PMS与Fe2+、Fe2O3和ZVI的偶联反应使大肠杆菌完全失活 [34] | Show Table DownLoad:
CSV
DownLoad:
CSV
上述结果表明,无论是溶解态还是固态,基元态还是复合态,铁基材料都能有效激活PDS或PMS生成活性物种用于微生物失活。将铁物种用于PS的活化和消毒的优点包括:(1)铁基材料种类丰富且易于获得;(2)与钴、铜等金属相比,使用铁基材料相对安全;(3)固态铁很容易从处理过的水中分离提取出来。与铁介导的活化PS灭菌机理相关的争论之一是除了活性物种直接攻击外,铁物种尤其是固态铁是否可以增强微生物的失活:在一些情况下观察到某些固体铁可通过界面正价铁来对带负电的微生物发生静电吸附作用[11, 34],吸附过程导致二价铁渗透到胞内会增强微生物的失活 [35]。
1.2 光介导活化PS灭活微生物
除了铁基介导的电子转移途径外,还可以通过紫外线、可见光等光介导的能量激活PS,如表2所示 [17, 24, 26]。Sun等报告了采用UV/PDS对大肠杆菌、MS2噬菌体和枯草芽孢杆菌孢子的灭活,当PDS剂量为0.3 mmol·L−1时,使4 lg CFU·mL−1的大肠杆菌、MS2噬菌体和枯草芽孢杆菌孢子失活所需紫外线剂量分别为8.8、30、30 mJ·cm−2 [36]。UV/PDS对大肠杆菌和MS2的灭活效率高于单独UV,但在灭活孢子作用不显著[36]。值得注意的是,消毒所需的紫外线剂量远低于降解有机污染物所需的剂量,这表明采用UV/PDS进行消毒是实际可行的。Wen等报道了使用UV/PMS工艺对真菌孢子(T. harzianum)进行灭活比单独使用UV的效果更好,当PMS剂量为0.1 mmol·L−1时,使2 lg CFU·mL−1的T. harzianum孢子失活所需的紫外线剂量为35 mL·cm−2[37]。除了能量密度相对较高的紫外光,可见光也被应用于活化PDS进行深度消毒。Wang等报道了在不投加催化剂条件下可见光活化PS灭活水中大肠杆菌 [17],当PDS剂量为2.0 mmol·L−1,将反应溶液暴露于带有420 nm截止滤光片的太阳模拟器中,在40 min内约6 lg CFU·mL−1大肠杆菌细胞被灭活[17];引入Fe3O4基的磁性水热炭在太阳光/PDS体系中可进一步增强该过程,在相同的实验条件下,灭活效率从6 lg CFU·mL−1提高到8 lg CFU·mL−1[24]。在大型塑料瓶中太阳能可以提高PMS和PMS/Fe2+体系对大肠杆菌的灭活率。当PMS和Fe2+剂量分别为36 μmol·L−1和18 μmol·L−1时,太阳光/PMS工艺灭活5 lg CFU·mL−1的大肠杆菌所需太阳能为650 Wh·m−2,而在添加了Fe2+后,太阳光能耗降低到450 Wh·m−2[38]。
表 2 光介导活化SO4−•致微生物失活Table 2. Microorganism inactivation by SO4−•-based processes activated by light体系Process 目标微生物Target microorganisms 实验条件Experimental conditions 消毒效率Disinfection disinfection 参考文献Reference UV/PDS 大肠杆菌、MS2噬菌体、枯草芽孢杆菌孢子 0.3 mmol·L−1 PDS,大肠杆菌、噬菌体MS2、枯草芽孢杆菌孢子紫外光剂量分别为8.8、30、30 mL·cm−2 3种微生物在30 min内均达到4 lg CFU·mL−1失活 [36] UV/PDS T. harzianum孢子 0.1 mmol·L−1 PMS, 紫外光剂量为35 mL·cm−2 达到2 lg CFU·mL−1失活 [37] 可见光/PDS 大肠杆菌 2 mmol·L−1 PDS, 30 °C, pH 6.0, λ ≥ 420 nm 40 min内6 lg CFU·mL−1大肠杆菌失活 [17] 可见光/ hydrochar /PS 大肠杆菌 200 mg·L−1 MHC; 2 mmol·L−1 PS; 25 °C, pH 6.0, λ ≥ 420 nm 40 min后8 lg CFU·mL−1大肠杆菌失活 [24] 太阳光/PMS;太阳光/Fe2+/PMS 大肠杆菌 装有蒸馏水的塑料瓶中加入18 μmol·L−1 Fe2+, 290 μmol·L−1 H2O2, 和 36 μmol·L−1 PMS 650 Wh·m−2太阳光/PMS体系和450 Wh·m−2太阳光/Fe2+/PMS体系达到5 lg CFU·mL−1失活 [38] | Show TableDownLoad:
CSV
以上结果表明,光介导活化PDS/PMS可有效杀灭多种微生物。使用光介导的过程有几个优点:例如,光辐射被认为是一种无化学物质的能量输入,它可以减少如在使用铁基催化剂时的铁污泥等副产物。实际上,可以较为容易地将紫外线或可见光灯安装到现有的水处理机组中,并且不会占用很多空间。此外,由于太阳能是一种自然资源而且容易获得,利用太阳能更具可持续性。太阳能介导的PDS/PMS活化在水的深度消毒方面很有前景,这在碳中和背景下发展绿色清洁技术有很深的意义,为发展中国家减少污染排放提供了帮助。
1.3 碱法活化PS灭活微生物
据报道,PS也能够被氢氧化钠(NaOH)活化,用于灭活大肠杆菌O157:H7[31]。研究进行了两组实验,当使用40 mmol·L−1 PS和30 mmol·L−1 NaOH时,120 s后使6.21 lg CFU·mL−1的大肠杆菌失活[31];当化学剂量增加至500 mmol·L−1 PS和350 mmol·L−1 NaOH时,120 s后8.64 lg CFU·mL−1的大肠杆菌失活[31]。与之前的铁介导或光辅助过程相比,碱基介导的研究较少,其主要原因可能是将溶液pH值调整为强碱性进行处理后需再将pH调整回来,这在实际运用中可能是不可行的。然而,该工艺可能会适用于某些特殊工业废水或某些特殊地区的地下水处理。
1.4 压电催化活化PS灭活微生物
普遍报道的铁介导法和光辅助法都有其固有的局限性,除此以外,近年来一些新型的PDS活化法也被报道用于水的深度消毒。Xia等报道了压电催化过硫酸盐活化体系进行消毒,他们合成了银改性的钛酸钡(Ag-BTO)并将其用作压电催化剂,在超声振动(US)下激活PDS以灭活大肠杆菌[39]。新开发的US/Ag-BTO/PDS工艺可以在5 min内使6.2 lg CFU·mL−1的大肠杆菌完全进入存活但不可培养(VBNC = viable but non-culturable)状态,并在20 min处理后彻底灭活VBNC状态细菌[39]。如图1所示,US/Ag-BTO/PDS体系的消毒性能比其他对照组有更好的消毒效果[39]。在该体系中,压电由超声驱动,可通过超声破坏细菌细胞膜而强化活性物种穿透,而且压电电子也能高效活化过硫酸盐,实现过硫酸盐的高效利用和催化灭菌性能。
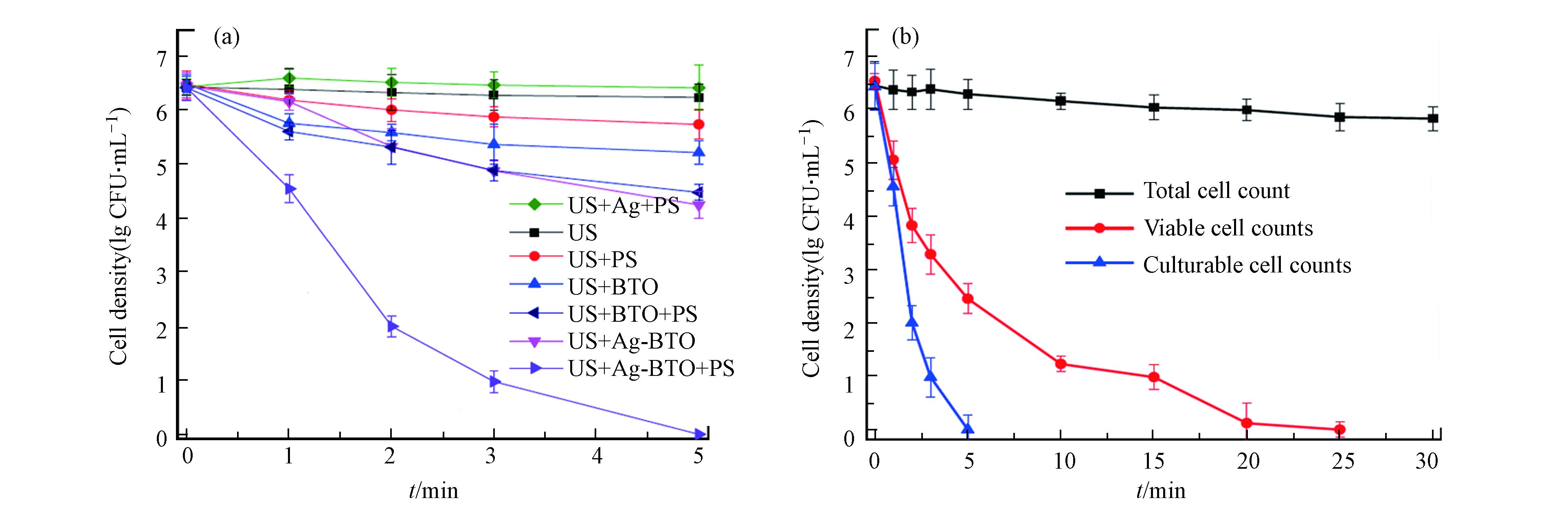
2. 操作条件与环境条件影响(Effects of operational andenvironmental conditions)
基于SO4−•的AOPs工艺消毒效率受催化剂和PDS/PMS的用量、接触时间、溶液pH、共存的阴离子或阳离子等操作和环境条件的影响。这些因素对消毒效率的影响在不同的工艺过程中大致呈相同的趋势,即随着剂量和接触时间的增加而增加,但在如碱基活化PDS体系较特殊的过程中会有一些例外。本节对上述提到的活化方法中相关因素的影响进行了总结。
2.1 CT值的影响
在Wordofa等用Fe2+/PDS体系对大肠杆菌进行灭活的研究中,灭活速率常数k几乎随着过硫酸盐的用量增加而呈线性增加,而诱导时间与过硫酸盐的用量增加呈反指数衰减关系[16]。速度常数k在稳定前也随Fe2+用量的增加而迅速增加,而诱导时间则随Fe2+浓度的增加而呈指数衰减[16]。在Xia等使用天然磁黄铁矿/PDS体系灭活大肠杆菌的研究中,随着PDS从0.2 mmol·L−1增加到1.0 mmol·L−1,NP剂量从0.15 g·L−1增加到1.0 g·L−1,灭活效率也得到提高。而由于过量的铁对活性物质的消耗,高浓度的铁会抑制消毒效率[33]。同样,在Sun等使用动力学模型模拟证明中,随着PDS剂量从0 mmol·L−1增加到10 mmol·L−1,MS2噬菌体的失活效率增加,然而大肠杆菌和杆菌孢子的失活效率受PDS剂量的影响较小[36]。与“CT值”类似的是在使用光辅助活化工艺时,灭活效率也随着紫外或可见光通量的增加而增加(光通量=光强度×辐照时间)。
2.2 pH值的影响
根据相关研究报道,消毒效率会受到pH值的显著影响。例如在Xia等报告中,天然磁黄铁矿/PDS系统对大肠杆菌的灭活效率随着pH值由3.0增加到9.0而逐渐降低[33];在Wang等的报告中,随着pH值从4.0增加到9.0,可见光/PDS体系的消毒效率逐渐降低[17];在Wordofa等的研究中,在Fe2+/PDS体系中的大肠杆菌灭活速率随着pH值从5.0增加到9.0而降低,这主要是因为碱性条件下Fe2+形成絮体发生沉降导致过硫酸盐活化效率降低。他们还发现溶液pH值对失活速率常数的影响比活化时长更加显著[16]。
pH值对消毒过程的影响有以下几个方面:(1)pH值影响溶液中铁离子的形态;(2)pH值影响PDS/PMS活化过程中活性物种的生成;(3)pH值也会影响微生物的表面电荷及其对活性物种的亲和力。大多数天然水和再生水的pH值一般在6.5—8.5之间,在这个范围内铁介导的过程可能无法很好地进行,而光辅助催化和新型压电催化在这个pH范围内能体现出其使用的优势,以及在某些碱性较强的工业废水或极端条件下,碱活化的PDS/PMS体系可能会成为深度消毒的良好选择。
2.3 水质的影响
溶解氧(DO = dissolved oxygen)、DOM(dissolved organic matter)、无机阴离子和阳离子以及悬浮固体(SS = suspended solid)等水基质会在不同程度上会影响SO4−•的消毒效率。如1—20 mg·L−1的碳酸氢盐能够降低对大肠杆菌的消毒效率,在降低溶解氧含量时会降低天然磁黄铁矿/PDS体系内的消毒效率[33]。在大多数研究报道中DOM会降低体系内的消毒效率,但在天然磁黄铁矿/PDS体系中DOM对大肠杆菌的消毒效果具有双重作用,其研究表明,在添加1 mg·L−1的DOM后,大肠杆菌的灭活率提高,但随着DOM进一步增加至5 mg·L−1和20 mg·L−1,大肠杆菌的灭活率显著降低[33]。低浓度DOM的增强效应归因于DOM中的对苯二酚和醌基团可以激活PDS生成SO4−•和HO•等活性物种[33]。与光催化相比,铁基催化对水基质成分相对更敏感,因为铁物种会活跃的参与到氧化还原反应和络合反应中,因此在实际应用中应当考虑到目标水质状况来选择合适的处理手段。
3. SO4−•致微生物灭活机理(Mechanisms of microorganism inactivation by SO4−• based processes)
目前已经有文献提出了SO4−•灭活微生物的机理,但现有文献中的观点仍存在有争议的部分。本章将从两个角度总结灭活机理:(1)从反应过程角度,重点介绍了活性物种是如何产生的,负责微生物灭活的主要物种以及主要途径是什么;(2)从微生物角度,重点介绍了微生物的哪些部位(如细胞膜、酶、基因组等)受到活性物种的攻击而导致失活。
3.1 活性物种的生成与贡献度
在紫外光、可见光活化的PDS或PMS的过程中,活性物种的生成是相对简单清晰的。如在UV/PDS体系中,Sun等计算了不同活性物质相对于大肠杆菌、MS2和杆菌孢子的CT值,结果表明消毒效果为HO• > SO4−• > CO3−• O2−•[36]。但与之相反的是,在Wang等的研究中,SO4−•对通过可见光/PDS体系使大肠杆菌失活的贡献程度远远大于HO•和O2−•[17]。这是因为与自由基相比,SO4−•具有相对较长的寿命和更强的氧化能力。不同研究中结果的不一致性是由于其对活性物质不同的量化方法,例如在Sun等的研究中使用动力学模型来预测每种活性物质的贡献[36],而在Wang等的研究中则使用自由基抑制剂达到同样的目的。
在铁介导的过程中,由于铁物种的参与,自由基的化学过程相对更加复杂。在Fe2+/PDS体系中,Wordofa等通过实验和构建模型的方法证明SO4−•导致大肠杆菌O157:H7细胞灭活的速度比HO•快5倍[16],且同时提出SO4−•的优势可能与其与大肠杆菌O157:H7细胞膜表面的富电子基团的高度选择性有关[16]。在天然磁黄铁矿/PDS体系中,根据抑制剂实验和原位ROS的测定,灭活大肠杆菌的活性物质贡献为SO4−• > HO• >H2O2[33],研究证明PDS能够有效地结合到天然磁黄铁矿表面,形成电荷转移复合物从而介导活性物种的产生。在Wang等使用可见光/PDS/hydro-char体系灭活大肠杆菌的工作中,SO4−•被认为是导致大肠杆菌灭活的主要活性物质[17]。在该体系中,SO4−•主要通过3种途径产生:(1)通过可见光激活PDS;(2)通过表面Fe(Ⅱ)激活PDS;(3)通过光催化产生的电子激活PDS。在关于铁介导的PDS/PMS活化体系的一个新的观点是,高价态铁物种可能参与到此类灭活过程,尤其是在接近中性的pH下。在对有机污染物的降解过程中这些高价态铁与自由基一样具有不可忽略的贡献[40-41]。例如,一些研究预测Fe(Ⅳ)可能会参与到微生物的去除过程[42],但迄今为止没有直接证据证明,因此这个问题值得进一步研究。
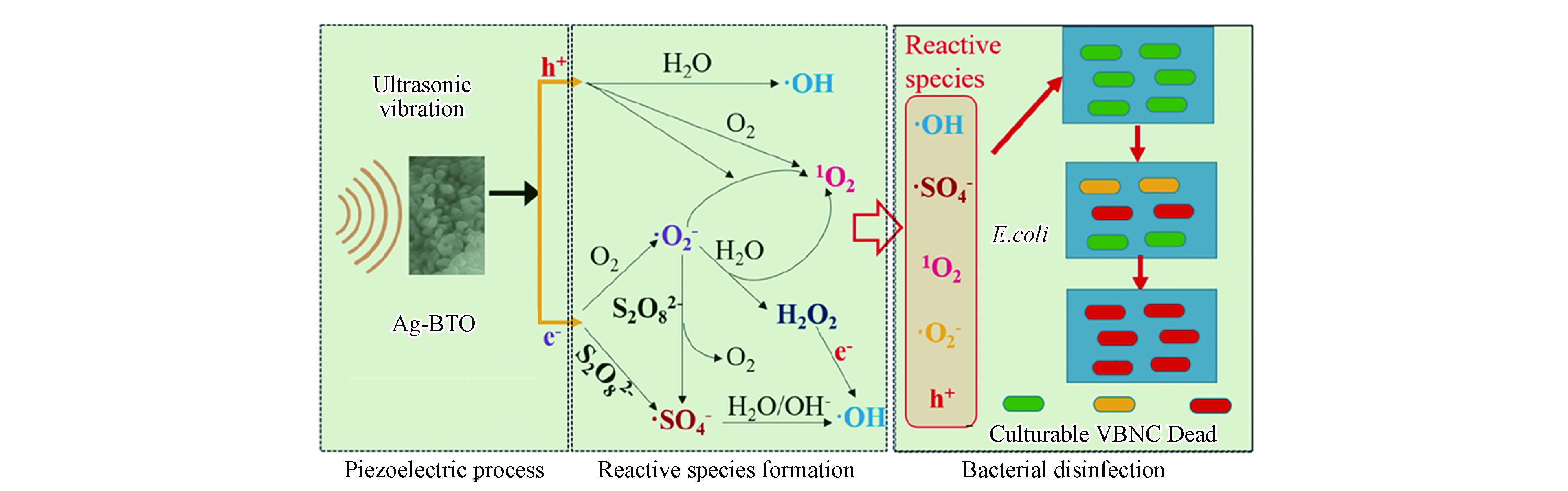
在新型压电催化系统中,Xia等报道了通过压电催化产生电子和超氧自由基活化PDS连续生成SO4−•和HO•对大肠杆菌进行高效灭活[39]。图2显示了活性物种的产生途径[39]。其中值得关注的是通过使用US/Ag-BTO/PS体系灭活大肠杆菌的过程中发现了超声波和自由基氧化之间的协同作用,如压电电子和超声本身可以帮助活化过硫酸盐来强化自由基生成。
3.2 微生物的损伤和灭活
过往的研究提供了一些关于微生物是如何被破坏和灭活的信息,它们是基于SO4−•的消毒工艺处理前后微生物从宏观到微观的特征而得出的。例如Wang等报道了大肠杆菌的细胞膜受到了被可见光激活PDS所产生的活性物质的破坏,随后CAT(catalase)和SOD(superoxide dismutase)等一些抗氧化酶被诱导进而使基因组DNA受损,导致细胞死亡[17]。在他们接下来的工作中使用水热炭增强了该过程,并得到了相似的机制,即细菌灭活过程被确定为从外膜开始最终到细胞内成分降解 [24]。Xiao等研究了用于大肠杆菌灭活的ZVI/PDS体系,通过扫描电子显微镜(SEM)拍摄的受试细菌的形态图像,表明SO4−•在细胞壁或细胞膜上引发氧化反应,影响膜的通透性和生理功能,最终导致其不可逆损伤[32]。 Xia等利用天然磁黄铁矿/PDS工艺系统地研究了大肠杆菌细胞破坏过程,由外膜、肽聚糖层和细胞质膜组成的细菌包膜首先受到系统中产生的活性物质的攻击[33]。利用BacLight kit显微荧光法直接观察天然磁黄铁矿/PS体系中细胞膜的通透性变化,同时由于ATP水平急剧下降,细胞几乎在瞬间因代谢停滞而失活。此外,他们的研究还提到反应性物种通过表现为质子载体或通过抑制呼吸链中的酶来抑制ATP的形成,活性物种在穿透细胞包膜后,通过与细胞质蛋白、基因组等多种生物分子发生反应损伤细胞,最后还观察到DNA条带的荧光强度逐渐减弱可知基因组DNA被破坏 [33]。但作者同时指出,处理后的细胞可能处于一种可存活但不可培养(VBNC)的状态,这种状态仍可能对水体造成健康风险[33]。Xia等提出了一种新的压电催化强化消毒工艺来处理VBNC状态下的微生物[39]。研究人员通过跟踪ATP(adenosine triphosphate)水平检测了可培养状态的大肠杆菌以及VBNC大肠杆菌细胞的代谢活性,并通过荧光光谱仪检测大肠杆菌的成分变化证明大肠杆菌在可培养状态和VBNC状态均被有效灭活 [39]。因此,基于SO4−•的AOPs能够通过氧化协同其他外加能量如光,超声等作用实现对微生物胞膜到胞内物质的裂解矿化。
4. 消毒副产物的形成(Formation of disinfection by-products)
基于SO4−•的工艺已被发展用于水的深度处理,但消毒过程中产生的消毒副产物却很少被提及。事实上在任何涉及消毒的化学过程中,都不可避免地会形成DBPs(disinfection by-products),虽然基于SO4−•的工艺手段中产生的DBPs可能与氯、氯胺或二氧化氯等消毒过程中形成的DBPs不同,但它们也同样会造成一定的健康风险。本节将对基于SO4−•的深度水消毒处理过程中形成的潜在DBPs进行总结。
4.1 无机副产物的形成
使用基于SO4−•的深度水处理过程中会形成无机副产物,典型的无机副产物是由PDS或PMS分解生成的硫酸盐。当使用PDS时由于1 mol的PDS可产生2 mol硫酸盐,无机副产物的形成尤其严重。由于摄入含有高硫酸盐含量的饮用水会对胃肠道产生影响,世卫组织建议向卫生当局通报硫酸盐浓度超过500 mg·L−1的饮用水源[42- 43]。然而硫酸盐在美国环保局中按二级最大污染物水平(secondary maximum contaminant level = SMCL)标准分类,饮用水中硫酸盐的SMCL为250 mg·L−1 [44]。但假设所有剂量的PDS或PMS最终全部分解为硫酸盐,并以250 mg·L−1倒推,则PDS和PMS的最大剂量仅为1.3 mmol·L−1和2.6 mmol·L−1。但在许多已报道的文献中,PDS/PMS的剂量均超过了该计算值,这表明在实际应用此类技术时,过量硫酸盐的形成可能会成为值得注意的问题。
而除了硫酸盐,用于自由基生成的催化剂也可能是这些过程的副产物。除了上文提到的铁泥外,其他金属或非金属材料也可能成为有害副产物。例如在PDS/PMS活化中,钴表现出比铁更优越的催化性能,但钴金属有毒,不能留在任何处理过的饮用水中。在实际应用时,金属或非金属材料的浸出始终是一个主要问题。
此外,当使用SO4−•进行水的深度处理时,氯酸盐的形成可能成为另一个问题。氯离子在水中普遍存在,而氯化物能够被SO4−•氧化成氯酸盐,这对饮用处理水的人群造成了极大的健康问题[45]。世卫组织和美国环境保护局将饮用水中氯酸盐的水质健康参考水平标准分别定为0.7 mg·L−1和0.21 mg·L−1 [46]。因此在使用基于SO4−•工艺处理含高浓度氯化物的水时需要对氯酸盐的形成应予以关注。
4.2 有机副产物的形成
在基于SO4−•的工艺用于水的深度消毒处理时还会产生有机副产物。SO4−•被认为是一种绿色环保的自由基,与如活性氯和活性溴等的卤素自由基相比,SO4−•产生的有机副产品数量有限。然而,最近的一项研究表明在用SO4−•处理水中有机污染物和灭菌的过程中会普遍产生有机硫酸盐[47]。其中一些有机硫酸盐,例如硫酸苯酯(phenyl sulfate)和硫酸对甲酚酯(p-cresyl sulfate)均是有害的尿毒症毒素。因此在使用基于SO4−•的水深度消毒处理的过程中,应进一步调查关注有机硫酸盐的形成、去向及其毒性。
同时,水中普遍存在的氯化物可通过SO4−•氧化为活性氯物种,如Cl•、Cl2•−和ClO•[46],这些活性氯物种通过取代反应和加成反应与水中溶解的有机物反应形成如三卤甲烷、卤乙酸、水合氯醛等的氯化副产物[48]。这些氯化副产品是会对人类健康构成严重威胁的致癌物[49]。而由于自由基转化,还可能会形成溴化和碘化副产物。因此在对水使用基于SO4−•的深度消毒处理时需要对卤化副产物的形成、迁移和毒性进行评估。
5. 总结与展望(Conclusions and future perspectives)
本文综述了基于SO4−•的AOPs应用于消毒的研究进展。本文首先回顾了过往研究中各种基于SO4−•工艺对不同微生物的灭活作用,这些工艺根据其驱动力主要分为三大类:铁介导、光辅助和新型过硫酸盐活化(例如压电催化),针对各类当前的研究进展和主要发现进行总结,并讨论了各类工艺的深度消毒处理的优缺点。接着本文概述了不同操作因素和环境条件对消毒性能的影响,包括过硫酸盐和催化剂的用量、接触时间、光照通量、pH值、溶解氧和其他水基质成分等,对上述不同因素造成影响的原因进行了讨论。进一步概述了基于SO4−•消毒的微生物灭活机理,并从两个角度对这些机理进行了综述,即从反应过程角度总结了活性物种是如何形成的,以及每种活性物种对微生物失活的贡献,并且从微生物的角度总结了微生物的损伤及灭活过程。最后,本文回顾了基于SO4−•工艺应用中无机和有机消毒副产物的形成,通过机理解释和讨论指出了与副产物形成有关的一些被忽视的问题。该综述还为基于SO4−•工艺的饮用水处理过程中工程设计和运行提供了建议。
(1)目前的研究仅局限于对少数微生物的失活,如大肠杆菌、MS2噬菌体和杆菌孢子,这对基于SO4−•的消毒方法在广谱致病微生物灭活方面的适用性和优点提供的信息较为有限。建议对更多与人类健康相关的微生物进行调查。此外对不同微生物进行的比较较少,考虑到SO4−•的选择性,建议在分子水平或借助一些微生物学工具对不同微生物的失活进行比较以供将来研究。
(2)在涉及铁物种活化的过程中,高价铁(如Fe(Ⅳ)和Fe(Ⅴ))对微生物失活的作用尚不清楚。因此,目前在铁介导活化方面对不同工艺过程下的SO4−•、HO−•以及H2O2、O2−•、和1O2等其他活性物质的相对贡献的判断可能不准确。建议对高价铁(如Fe(Ⅳ)和Fe(Ⅴ))等对微生物失活的贡献进行定性和定量的研究说明。
(3) 目前,基于SO4−•工艺中消毒副产物的形成往往被忽略,需要对微生物本身或共存的有机物产生的有机副产物和无机副产物进行更多的研究,以及分析探究更多关于基于SO4−•工艺处理前后水的毒理学变化的更基本的资料。
(4)基于SO4−•的AOP处理技术,虽然文献报道可以通过增加反应物浓度提升消毒效率,但如此也会引发一些技术应用问题,如吨水处理费用会远高于UV和氯,且大量硫酸盐会高于各类水质标准,甚至会导致饮用水后续管网腐蚀或污水排入河流中导致底泥生成H2S气体等。针对这些应用局限性,可以考虑引入微纳气泡或结合UV来协同提高过硫酸盐的活化效率及杀菌速率,一方面能有效降低反应物的投加浓度,另一方面还可以高效抑制微生物的复活。
(5)综合该工艺的优缺点,基于SO4−•的AOPs仍然在再生水、地下水或是应急场所中的消毒应用有发展前景。一是再生水,由于再生水中还含有较多持久性污染物,SO4−•可以同时降解某些污染物,并且可以灭活多种病原微生物;二是地下水,如紫外和臭氧无法穿透土壤,而过硫酸盐是可以溶解渗透的;且过硫酸盐是可以高效灭活本身具有抗氯性的真菌及阿米巴等微生物;三是某些特殊应急场所有可能处于复杂pH范围,而SO4−•具有广泛pH适应范围。
考虑到对过硫酸盐剂量和pH条件较严格的要求,在诸如使用点或入口水处理装置等一些小型处理装置中基于SO4−•的深度消毒水处理工艺的应用似乎比在大型水处理厂中的应用更有前景。在这种情况下,利用太阳能等自然资源加强基于SO4−•的消毒过程可能成为实现可持续发展目标——“清洁的水和卫生”要求的直接现实途径。
-
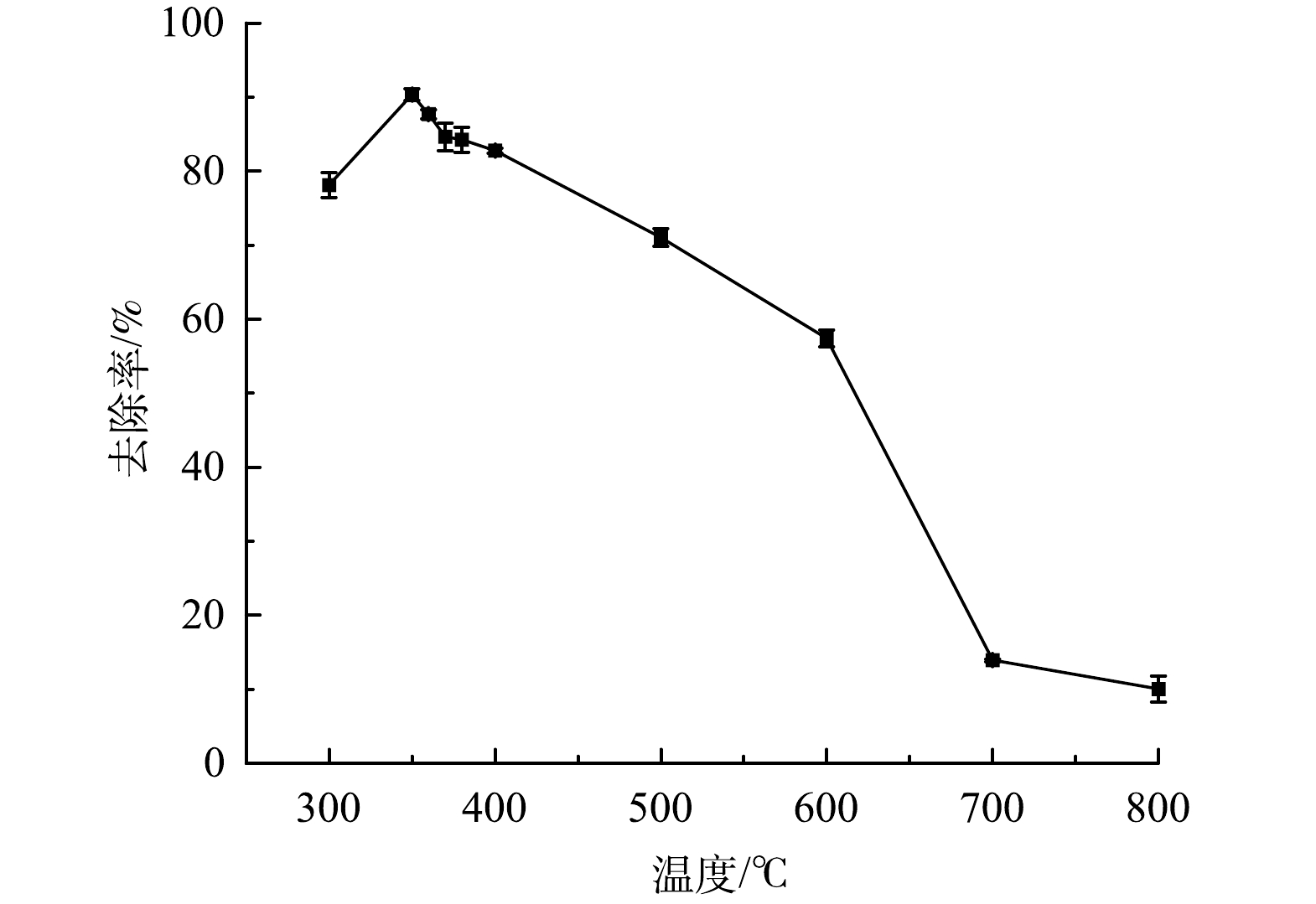
图 1 煅烧温度对Sb(Ⅴ)去除率的影响
Figure 1. Effect of calcination temperature on Sb(Ⅴ) removal rate
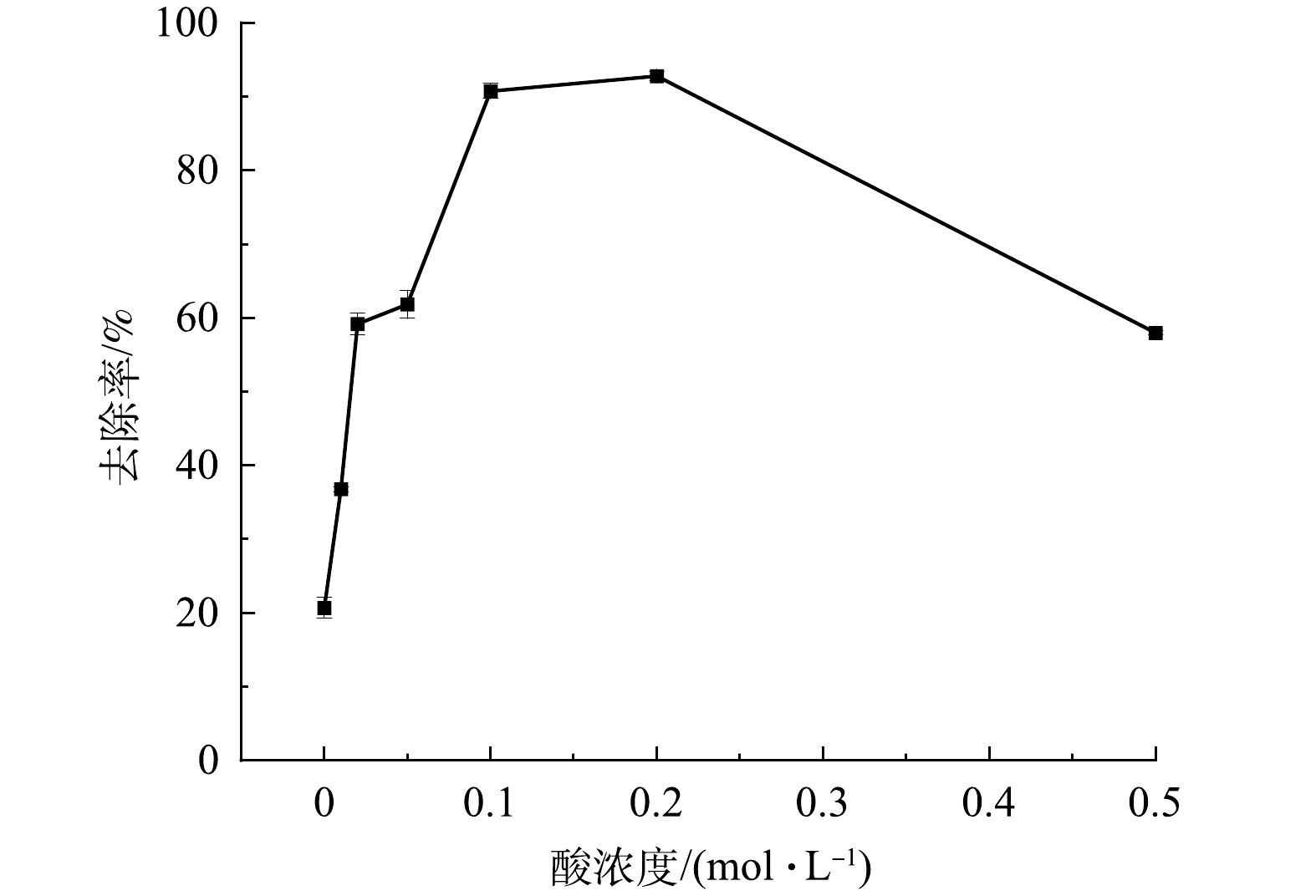
图 2 改性酸浓度对Sb(Ⅴ)去除率的影响
Figure 2. Effect of modified acid concentration on Sb(Ⅴ) removal rate
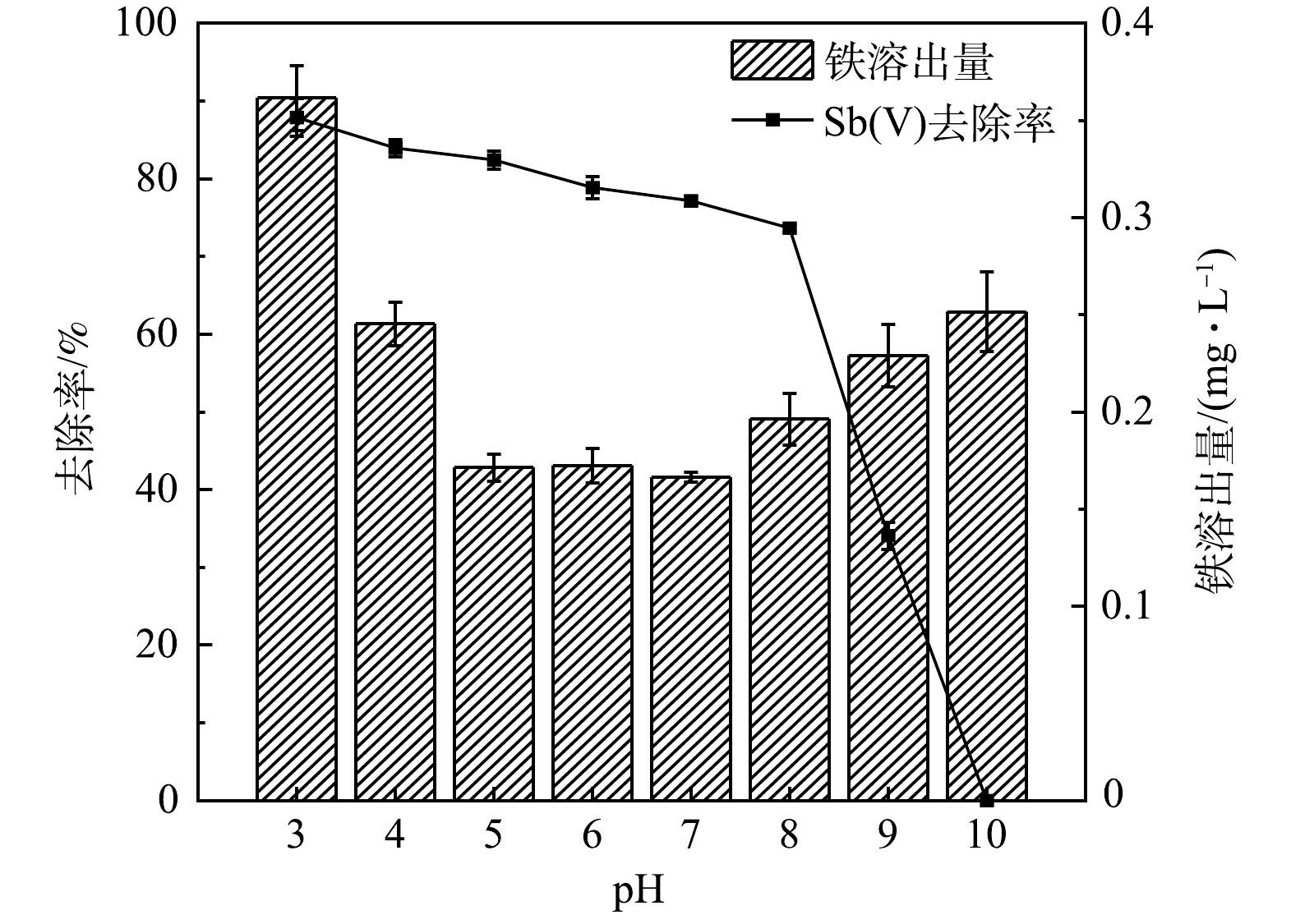
图 9 不同pH对Sb(Ⅴ)吸附和铁浸出量影响
Figure 9. Effects of different pH on Sb(Ⅴ) adsorption and iron leaching
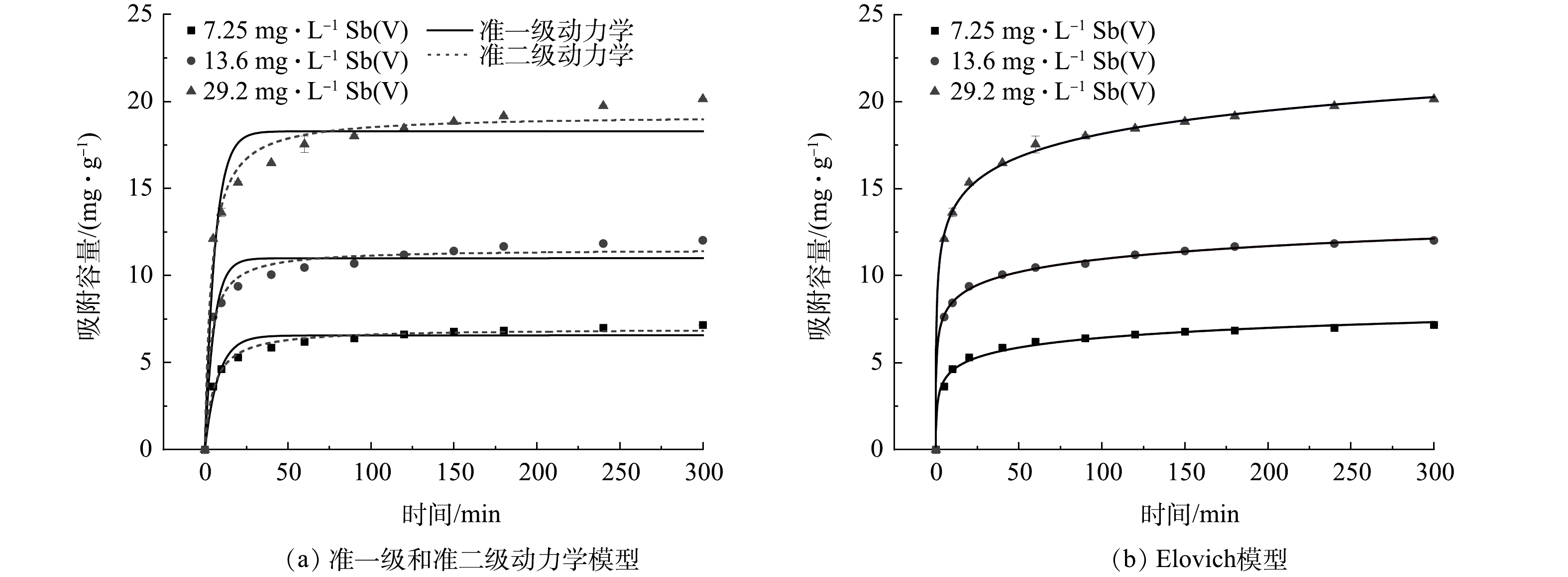
图 11 AFS350吸附Sb(Ⅴ)的吸附动力学模型
Figure 11. Adsorption kinetics of Sb(Ⅴ) adsorption by AFS350
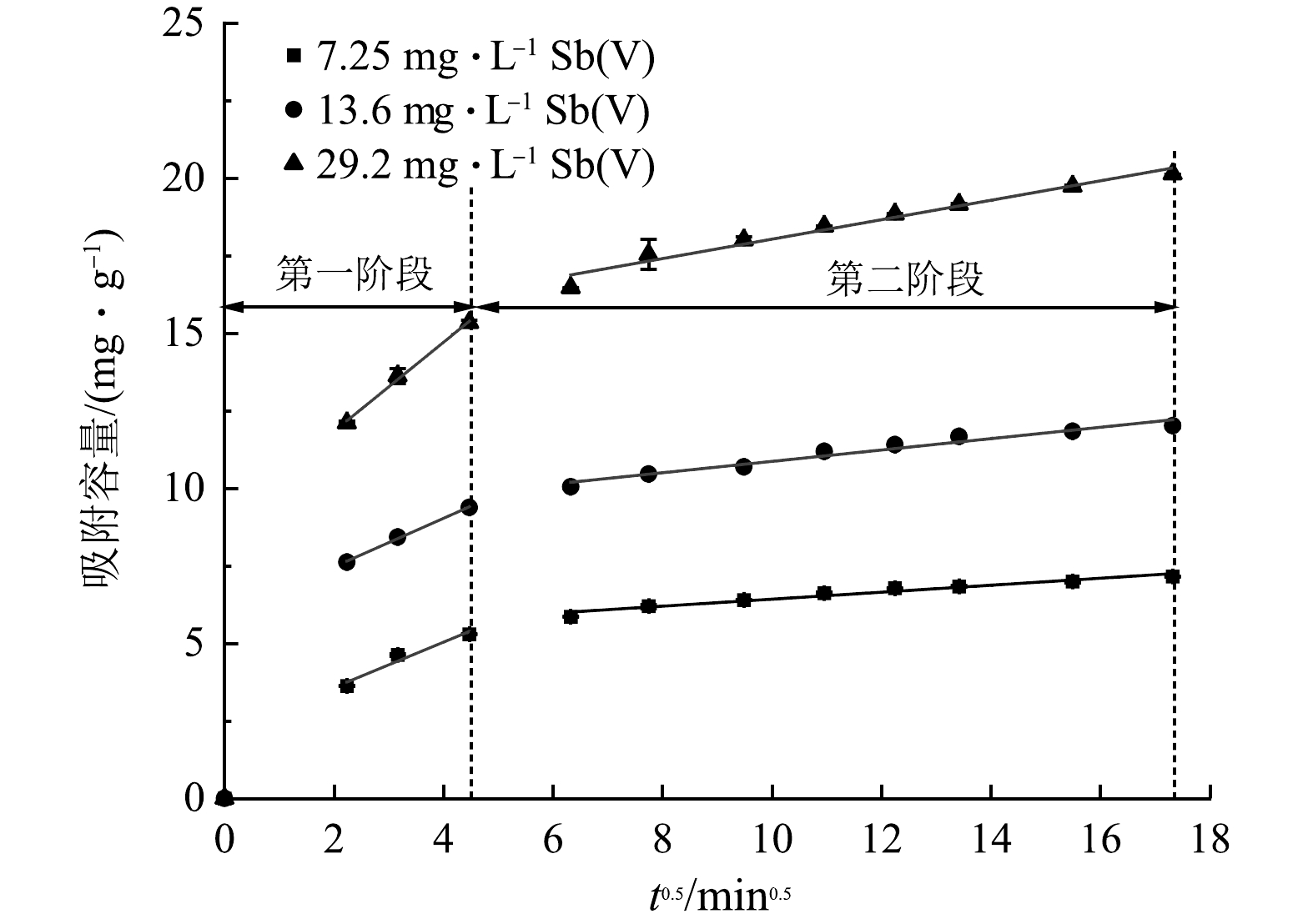
图 12 AFS350吸附Sb(Ⅴ)的颗粒内扩散模型
Figure 12. Intraparticle diffusion model of Sb(V) adsorption by AFS350
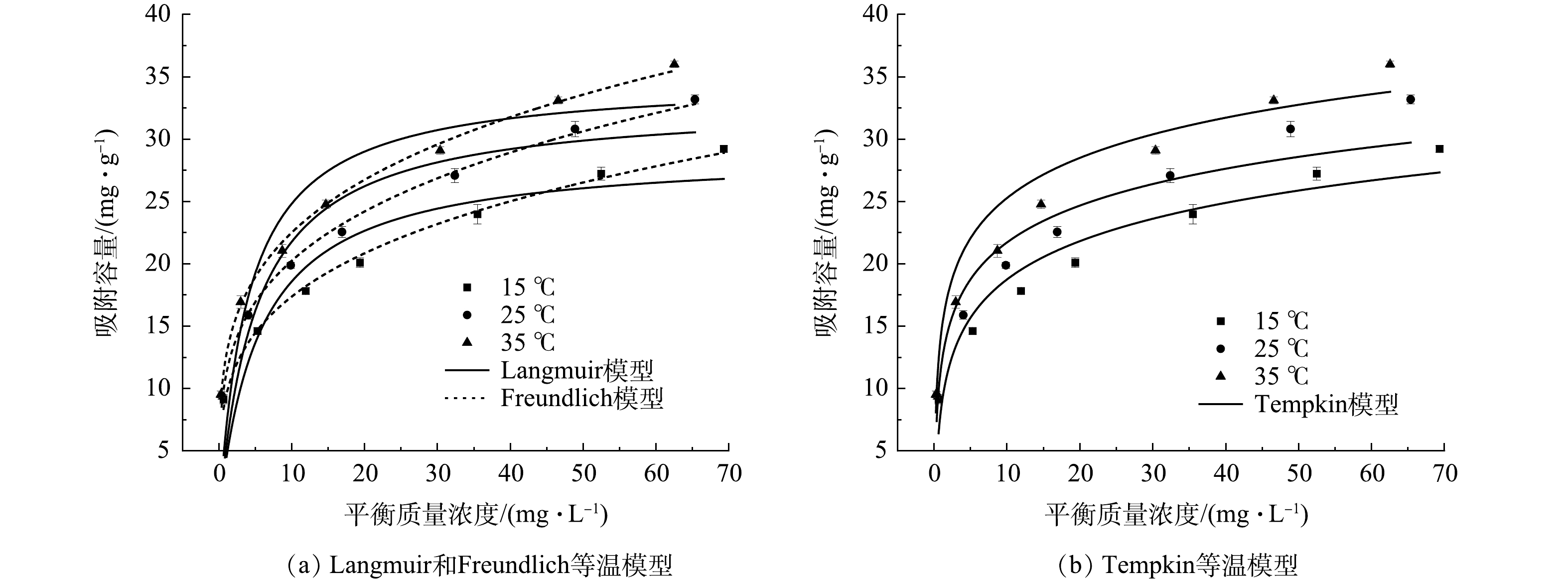
图 13 AFS350吸附Sb(Ⅴ)的吸附等温线模型
Figure 13. Adsorption isotherms of Sb(Ⅴ) adsorption by AFS350
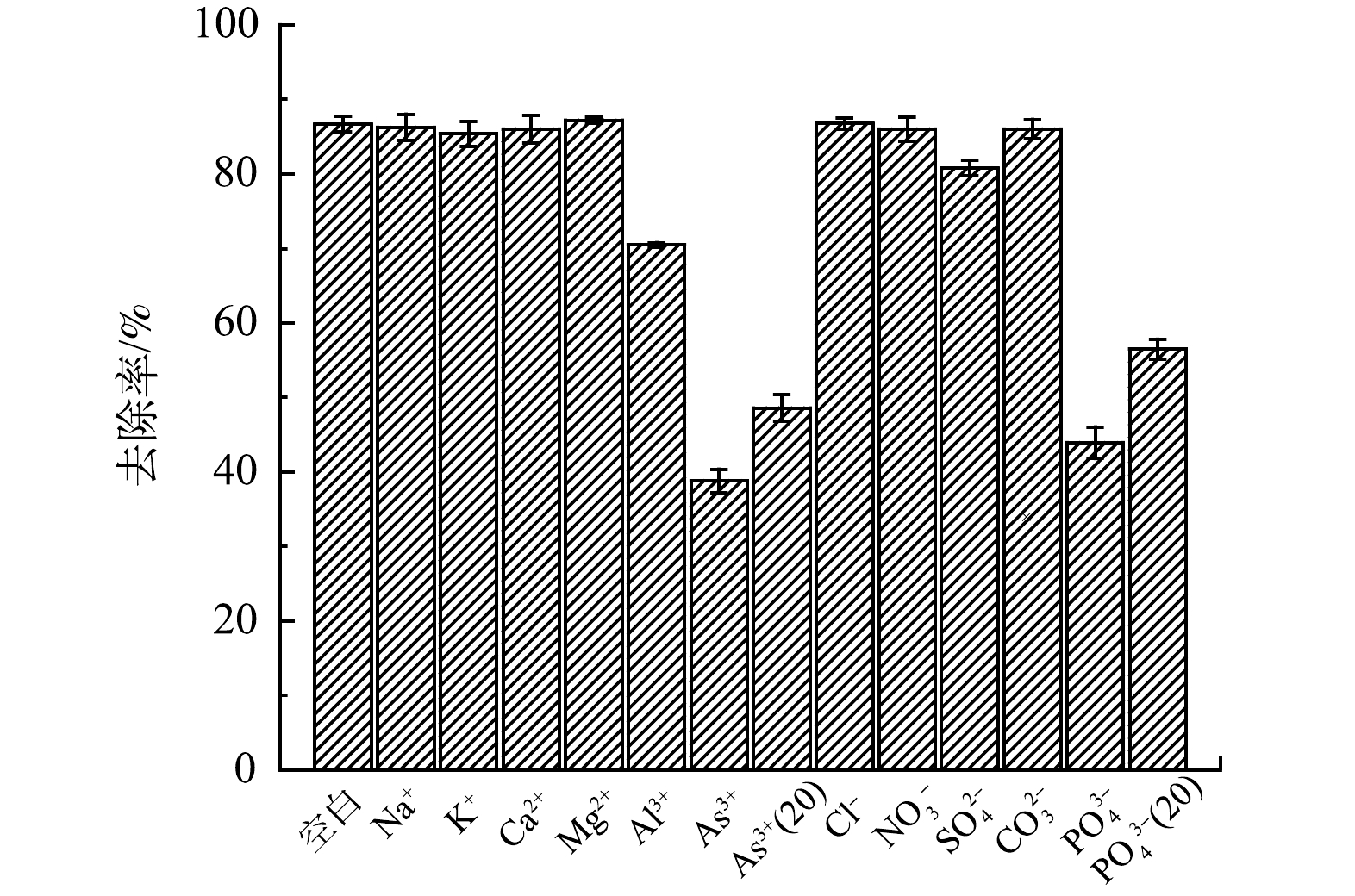
图 14 共存离子对AFS350吸附Sb(Ⅴ)的影响
Figure 14. Effects of coexisting ions on the adsorption of Sb(Ⅴ) by AFS350
表 1 AFS350吸附Sb(Ⅴ)的吸附动力学模型参数
Table 1. Adsorption kinetic fitting parameters of Sb(Ⅴ) adsorption by AFS350
初始质量浓度/(mg∙L−1) 准一级动力学模型 准二级动力学模型 Elovich模型 k1 qe R2 k2 qe R2 α β R2 7.25 0.125 6.56 0.953 0.027 6.96 0.991 21.7 1.23 0.994 13.6 0.184 11.0 0.941 0.026 11.5 0.981 287 0.931 0.999 29.2 0.165 18.3 0.940 0.014 19.2 0.982 242 0.520 0.999
下载: 导出CSV
表 2 AFS350吸附Sb(Ⅴ)的颗粒内扩散模型参数
Table 2. Intraparticle diffusion fitting parameters of Sb(V) adsorption by AFS350
初始质量浓度/(mg∙L−1) 第1阶段 第2阶段 kp1 C R2 kp2 C R2 7.25 0.731 2.11 0.956 0.113 5.30 0.959 13.6 0.783 5.90 0.997 0.183 9.03 0.961 29.2 1.44 8.97 0.996 0.314 14.9 0.970
下载: 导出CSV
表 3 AFS350吸附Sb(Ⅴ)的吸附等温线模型参数
Table 3. Adsorption isotherms fitting parameters of Sb(Ⅴ) adsorption by AFS350
温度/℃ Langmuir Freundlich Temkin KL qm R2 KF 1/n R2 AT BT R2 15 0.179 29.0 0.813 9.49 0.263 0.995 6.88 4.43 0.930 25 0.195 32.9 0.821 11.2 0.257 0.996 16.3 4.26 0.922 35 0.248 34.9 0.828 12.7 0.248 0.997 23.0 4.65 0.959
下载: 导出CSV
-
[1] TONG S, SHEN J, JIANG X, et al. Recycle of Fenton sludge through one-step synthesis of aminated magnetic hydrochar for Pb2+ removal from wastewater[J]. Journal of Hazardous Materials, 2021, 406: 124581. doi: 10.1016/j.jhazmat.2020.124581 [2] BABUPONNUSAMI A, MUTHUKUMAR K. A review on Fenton and improvements to the Fenton process for wastewater treatment[J]. Journal of Environmental Chemical Engineering, 2014, 2(1): 557-572. doi: 10.1016/j.jece.2013.10.011 [3] BOLOBAJEV J, KATTEL E, VIISIMAA M, et al. Reuse of ferric sludge as an iron source for the Fenton-based process in wastewater treatment[J]. Chemical Engineering Journal, 2014, 255: 8-13. doi: 10.1016/j.cej.2014.06.018 [4] ZHANG H, LIU J, OU C, et al. Reuse of Fenton sludge as an iron source for NiFe2O4 synthesis and its application in the Fenton-based process[J]. Journal of Environmental Sciences, 2017, 53: 1-8. doi: 10.1016/j.jes.2016.05.010 [5] MAHTAB M S, FAROOQI I H, KHURSHEED A. Zero Fenton sludge discharge: A review on reuse approach during wastewater treatment by the advanced oxidation process[J]. International Journal of Environmental Science and Technology, 2021: 1-14. [6] 于子扬, 于贺伟, 赵改菊, 等. 芬顿铁泥资源化利用研究进展[J/OL]. 无机盐工业: 1-12 2-03-05]. DOI:10.19964/j.issn.1006-4990.2021-0406.
[7] 郭慧. 氢氧化铁和羟基氧化铁的催化相转化机理研究[J]. 石家庄: 河北师范大学, 2006. [8] 王博, 刘燕, 张延宗. 两步煅烧法回收芬顿污泥[J]. 地球环境学报, 2020, 11(5): 554-561. doi: 10.7515/JEE192055 [9] LYU H, TANG J, CUI M, et al. Biochar/iron (BC/Fe) composites for soil and groundwater remediation: Synthesis, applications, and mechanisms[J]. Chemosphere, 2020, 246: 125609. doi: 10.1016/j.chemosphere.2019.125609 [10] YI Y, HUANG Z, LU B, et al. Magnetic biochar for environmental remediation: A review[J]. Bioresource Technology, 2020, 298: 122468. doi: 10.1016/j.biortech.2019.122468 [11] GU L, LI B, WEN H, et al. Co-hydrothermal treatment of fallen leaves with iron sludge to prepare magnetic iron product and solid fuel[J]. Bioresource Technology, 2018, 257: 229-237. doi: 10.1016/j.biortech.2018.02.113 [12] WANG M, ZHAO Z, ZHANG Y. Magnetite-contained biochar derived from fenton sludge modulated electron transfer of microorganisms in anaerobic digestion[J]. Journal of Hazardous Materials, 2021, 403: 123972. doi: 10.1016/j.jhazmat.2020.123972 [13] 陈丽群. 黑液木质素基磁性活性炭的制备及其吸附性能的研究[D]. 天津: 天津科技大学, 2020. [14] LIU R, XU W, HE Z, et al. Adsorption of antimony (V) onto Mn (II)-enriched surfaces of manganese-oxide and FeMn binary oxide[J]. Chemosphere, 2015, 138: 616-624. doi: 10.1016/j.chemosphere.2015.07.039 [15] 近藤精一, 石川达雄, 安部郁夫, 等. 吸附科学[J]. 李国希, 译. 北京:化学工业出版社, 2006: 65-70. [16] LI H, XIONG J, ZHANG G, et al. Enhanced thallium(I) removal from wastewater using hypochlorite oxidation coupled with magnetite-based biochar adsorption[J]. Science of the Total Environment, 2020, 698: 134166. doi: 10.1016/j.scitotenv.2019.134166 [17] BUTWONG N, KUNAWONG T, LUONG J H T. Simultaneous analysis of hydroquinone, arbutin, and ascorbyl glucoside using a nanocomposite of Ag@ AgCl nanoparticles, Ag2S nanoparticles, multiwall carbon nanotubes, and chitosan[J]. Nanomaterials, 2020, 10(8): 1583. doi: 10.3390/nano10081583 [18] WANG L, WANG J, WANG Z, et al. Enhanced antimonate (Sb(V)) removal from aqueous solution by La-doped magnetic biochars[J]. Chemical Engineering Journal, 2018, 354: 623-632. doi: 10.1016/j.cej.2018.08.074 [19] MITSUNOBU S, TAKAHASHI Y, TERADA Y, et al. Antimony(V) incorporation into synthetic ferrihydrite, goethite, and natural iron oxyhydroxides[J]. Environmental Science & Technology, 2010, 44(10): 3712-3718. [20] HO Y S, MCKAY G. Pseudo-second order model for sorption processes[J]. Process Biochemistry, 1999, 34(5): 451-465. doi: 10.1016/S0032-9592(98)00112-5 [21] SHEPHERD J G, JOSEPH S, SOHI S P, et al. Biochar and enhanced phosphate capture: Mapping mechanisms to functional properties[J]. Chemosphere, 2017, 179: 57-74. doi: 10.1016/j.chemosphere.2017.02.123 [22] 党志, 郑刘春, 卢桂宁. 矿区污染源头控制[J]. 北京:科学出版社, 2015: 54-56. [23] 张华. 柚皮基活性炭制备及吸附应用机理研究[D]. 南宁: 广西大学, 2013. [24] 廖路, 吴攀, 王兵, 等. 改性生物炭对高浓度锑废水中Sb(Ⅴ)的去除效果[J]. 环境工程学报, 2021, 15(2): 435-445. doi: 10.12030/j.cjee.202005006 [25] OGIWARA Y, KUBOTA H. Combination of cellulosic materials and metallic ions[J]. Journal of Polymer Science Part A-1:Polymer Chemistry, 1969, 7(8): 2087-2095. doi: 10.1002/pol.1969.150070807 [26] LI H, LI X, CHEN Y, et al. Removal and recovery of thallium from aqueous solutions via a magnetite-mediated reversible adsorption-desorption process[J]. Journal of Cleaner Production, 2018, 199: 705-715. doi: 10.1016/j.jclepro.2018.07.178 [27] WANG X, HE M, LIN C, et al. Antimony(Ⅲ) oxidation and antimony(V) adsorption reactions on synthetic manganite[J]. Geochemistry, 2012, 72: 41-47. doi: 10.1016/j.chemer.2012.02.002 [28] 周楚晨, 李成, 杨昆仑, 等. 铁氧化物对模拟印染废水中锑的去除性能研究[J]. 环境科学学报, 2022, 42(2): 96-107. doi: 10.13671/j.hjkxxb.2021.0155 [29] XI J, HE M, WANG K, et al. Adsorption of antimony(Ⅲ) on goethite in the presence of competitive anions[J]. Journal of Geochemical Exploration, 2013, 132: 201-208. doi: 10.1016/j.gexplo.2013.07.004 [30] 赵济金, 戚菁, 吉庆华, 等. 铁锰改性铜绿微囊藻对锑的吸附性能[J]. 环境工程学报, 2019, 13(7): 1573-1583. doi: 10.12030/j.cjee.201901071 [31] GUO X, WU Z, HE M, et al. Adsorption of antimony onto iron oxyhydroxides: adsorption behavior and surface structure[J]. Journal of Hazardous Materials, 2014, 276: 339-345. doi: 10.1016/j.jhazmat.2014.05.025 [32] HE Z, LIU R, LIU H, et al. Adsorption of Sb(Ⅲ) and Sb(V) on freshly prepared ferric hydroxide (FeOxHy)[J]. Environmental Engineering Science, 2015, 32(2): 95-102. doi: 10.1089/ees.2014.0155 [33] SHAN C, MA Z, TONG M. Efficient removal of trace antimony(Ⅲ) through adsorption by hematite modified magnetic nanoparticles[J]. Journal of Hazardous Materials, 2014, 268: 229-236. doi: 10.1016/j.jhazmat.2014.01.020 [34] AJMAL Z, MUHMOOD A, USMAN M, et al. Phosphate removal from aqueous solution using iron oxides: adsorption, desorption and regeneration characteristics[J]. Journal of Colloid and Interface Science, 2018, 528: 145-155. doi: 10.1016/j.jcis.2018.05.084 [35] LATA S, SAMADDER S R. Removal of arsenic from water using nano adsorbents and challenges: A review[J]. Journal of Environmental Management, 2016, 166: 387-406. doi: 10.1016/j.jenvman.2015.10.039 [36] LEUZ A K, MÖNCH H, JOHNSON C A. Sorption of Sb(Ⅲ) and Sb(V) to goethite: influence on Sb(Ⅲ) oxidation and mobilization[J]. Environmental Science & Technology, 2006, 40(23): 7277-7282. [37] MCCOMB K A, CRAW D, MCQUILLAN A J. ATR-IR spectroscopic study of antimonate adsorption to iron oxide[J]. Langmuir, 2007, 23(24): 12125-12130. doi: 10.1021/la7012667 [38] 缪阳洋. 载纳米水合氧化铁吸附剂去除水中Sb(V)的比较研究[D]. 南京: 南京大学, 2013. [39] CHAN C C P, GALLARD H, MAJEWSKI P. Fabrication of amine-functionalized magnetite nanoparticles for water treatment processes[J]. Nanotechnology for Sustainable Development. Springer, Cham, 2012: 137-147. [40] BHUVANESWARI S, PRATHEEKSHA P M, ANANDAN S, et al. Efficient reduced graphene oxide grafted porous Fe3O4 composite as a high performance anode material for Li-ion batteries[J]. Physical Chemistry Chemical Physics, 2014, 16(11): 5284-5294. doi: 10.1039/c3cp54778g [41] YANG K, ZHOU C, LI C, et al. Efficient removal of Sb(V) in textile wastewater through novel amorphous Si-doped Fe oxide composites: phase composition, stability and adsorption mechanism[J]. Chemical Engineering Journal, 2021, 407: 127217. doi: 10.1016/j.cej.2020.127217 [42] YANG K, ZHOU J, LOU Z, et al. Removal of Sb(V) from aqueous solutions using Fe-Mn binary oxides: the influence of iron oxides forms and the role of manganese oxides[J]. Chemical Engineering Journal, 2018, 354: 577-588. doi: 10.1016/j.cej.2018.08.069 [43] YU G, FU F. Exploration of different adsorption performance and mechanisms of core-shell Fe3O4@ Ce-Zr oxide composites for Cr(VI) and Sb(Ⅲ)[J]. Journal of Colloid and Interface Science, 2020, 576: 10-20. doi: 10.1016/j.jcis.2020.05.008 [44] LI H, CHEN Y, LONG J, et al. Removal of thallium from aqueous solutions using Fe-Mn binary oxides[J]. Journal of Hazardous Materials, 2017, 338: 296-305. doi: 10.1016/j.jhazmat.2017.05.033 [45] LIU J, REN S, CAO J, et al. Highly efficient removal of thallium in wastewater by MnFe2O4-biochar composite[J]. Journal of Hazardous Materials, 2021, 401: 123311. doi: 10.1016/j.jhazmat.2020.123311 [46] MISSANA T, MAFFIOTTE C, GARCı́A-GUTIÉRREZ M. Surface reactions kinetics between nanocrystalline magnetite and uranyl[J]. Journal of Colloid and Interface Science, 2003, 261(1): 154-160. doi: 10.1016/S0021-9797(02)00227-8 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4393
- HTML全文浏览数: 4393
- PDF下载数: 85
- 施引文献: 0







































