



















-
纳米材料是指在三维空间尺度中,至少有一维的尺度在1—100 nm之间的材料[1]。随着纳米技术的不断发展,各种各样的纳米材料已经广泛应用于生物医药、航空航天、环境保护等很多领域,并发挥着巨大的作用[2-5]。纳米二氧化钛,又称纳米钛白粉,是目前使用较为广泛的无机纳米材料之一[6]。由于纳米二氧化钛具有良好的光催化特性以及紫外线屏蔽能力,其被广泛地应用于化妆品、涂料、油漆和光催化剂等产品中[7-9]。在纳米二氧化钛广泛应用的同时,其对生态环境的负面影响也受到了研究人员的关注[10]。在大量生产、运输、使用和废弃过程中,纳米二氧化钛颗粒会被不可避免地释放到环境中去[11-13]。当纳米二氧化钛被释放到环境介质中时,巨大的比表面积以及超强吸附性能使得它极易吸附环境介质中存在的有毒物质,如持久性有机污染物、重金属等,它可以充当这些有毒有害物质在环境介质中转移的载体或流动相,促进这些污染物在环境介质中的迁移,从而导致污染距离的增大和污染范围的扩大,最终会威胁生态环境安全和人体健康[12,14]。迁移过程是纳米二氧化钛环境地球化学行为中的重要步骤,研究纳米二氧化钛在自然界多孔介质中的迁移行为是揭示其在“固-液”和“固-液-气”界面迁移机制及对其在实际环境介质中的迁移行为做出正确评估的关键。因此,有必要进行纳米二氧化钛在环境介质中的迁移的研究。
近年来,国内外研究工作者关于探究纳米二氧化钛在多孔介质中的迁移规律做了大量的研究。研究表明,影响纳米二氧化钛在多孔介质中迁移行为的因素有很多,如纳米颗粒粒径、浓度、表面性质等纳米材料的性质[15-17],介质种类、孔隙结构、粒径分布等多孔介质的性质[18-19],以及pH 值、离子强度、离子组成、表面活性剂等背景溶液的性质[12,19-24]都会影响纳米二氧化钛在多孔介质中的迁移。目前,虽然有关纳米二氧化钛在多孔介质中迁移行为的研究较多,但这些研究大多限于均质的石英砂介质[25]。相比于石英砂,双孔隙介质更为复杂,与真实土壤介质更为接近。双孔隙介质这一概念最早由PIRSON[26]提出用于解释裂缝性油藏原油采收率低的问题,他表示裂缝性储集岩的孔隙度可以认为是由占储集层大部分孔隙空间的颗粒间孔隙度和占小部分孔隙空间的裂缝和裂隙空间组成的两个以上的孔隙-渗透系统组成。之后,PHILIP[27]提出了另一种双孔隙介质类型——聚集质介质(aggregated media)的概念,他表示这种类型介质的孔隙空间由两种不同类型的孔隙度组成:大孔隙 (颗粒间孔隙度)和微孔隙率(颗粒内孔隙率),并表示聚集体介质构成了聚集成岩土壤的真实表现。纳米二氧化钛在相同环境条件下在石英砂和双孔介质中的迁移规律是否一致,是有待研究的一个问题。目前,关于纳米二氧化钛在双孔隙介质中迁移行为的研究较少。因此,系统研究纳米二氧化钛在双孔隙介质中的迁移过程,对于理解和评估纳米二氧化钛在实际土壤及地下水中的环境风险是非常有必要的。
本研究选取石英砂和双孔隙介质两种多孔介质,探究pH、离子强度(IS)和十二烷基苯磺酸钠(SDBS)的3种环境因素对纳米二氧化钛迁移行为的影响,并通过HYDRUS-1D软件模拟实验结果,预测纳米二氧化钛在两种多孔介质中的迁移机理,为评价纳米二氧化钛的环境风险提供更为全面的理论依据。
-
实验试剂:纳米二氧化钛(99.8%,锐钛矿,亲水),南京先丰纳米材料科技有限公司;氯化钠(NaCl),AR,天津市科密欧化学试剂有限公司;盐酸(HCl,AR);氢氧化钠(NaOH,AR),天津市天力化学试剂有限公司;十二烷基苯磺酸钠(SDBS C18H29NaO3S,CP),天津市科密欧化学试剂有限公司;石英砂 ( 98% SiO2 ; Fe, Al < 2 %);双孔隙介质(80% SiO2 ; 18% Ca ; Mg, K, Na < 1 %);实验用水为超纯水。
实验仪器:TU-1810型紫外可见分光光度计;ISO 9001型蠕动泵;HZT-A2000型电子天平;101 型电热恒温鼓风干燥箱;KQ-50型超声波清洗器;JY92型超声波细胞粉碎机;ME204分析天平。
-
用分析天平称取1 g纳米二氧化钛于500 mL烧杯中,加去离子水后,用超声波细胞粉碎机,325 W功率下超声2 min,倒入1 L的容量瓶中定容,配成1 g·L−1的纳米二氧化钛溶液(储备液A)并用紫外分光光度计测量A0。从储备液A中取出25 mL于1 L容量瓶中,加去离子水稀释定容,配成25 mg·L−1的纳米二氧化钛溶液(储备液B)。
-
用稀释后的盐酸和氢氧化钠溶液调节去离子水的pH,分别配制pH = 2(结合文献[28],为了探究低pH环境因素对纳米二氧化钛在实验介质中迁移行为的影响)、pH = 6.9和pH = 12(由于石英砂柱实验中pH=12条件下的流出液浑浊,所以纳米二氧化钛数据无法用分光光度法测量,故此文对其不再做讨论)的背景溶液。
-
用电子天平称取58.44 g NaCl晶体于1 L容量瓶中,加去离子水至刻度线定容,配成1 g·L−1的NaCl溶液备用。用移液管移取配置好的NaCl溶液,稀释成1 mmol·L−1、50 mmol·L−1的NaCl背景溶液和50 mmol·L−1的NaCl示踪剂溶液。
-
SDBS的临界胶束浓度(cmc)为1.2 mmol·L−1。首先,称取2.0909 g SDBS固体粉末于500 mL容量瓶内,加去离子水至刻度线定容,配制12 mmol·L−1的SDBS储备液;分别移取0.05 mL、0.5 mL SDBS储备液和1.25 mL纳米二氧化钛储备液A于50 mL容量瓶定容,配制成含SDBS 0.01 cmc、0.1 cmc的纳米二氧化钛悬浊液;取1.25 mL纳米二氧化钛储备液A于50 mL容量瓶定容配制成含0 cmc SDBS的纳米二氧化钛溶液作参照实验。
-
首先对石英砂反复冲洗;第二步用pH = 3—4的盐酸浸泡24 h之后,用去离子水反复冲洗;第三步用pH = 10—11的氢氧化钠浸泡清洗,继续用去离子水清洗至pH呈中性,以除去挥发性有机物;双孔隙介质用去离子水彻底清洗消除其表面的微粒,两种介质都放在烘箱中在105 ℃烘干,放在带盖的桶里储藏,以待实验[29]。
-
采用激光粒度仪(Malvern 2000)来确定两种多孔介质的粒径分布;使用注汞式孔隙度仪(AutoPoreIV9500V1.07)来确定两种多孔介质的孔径分布。利用Philip XL-30环境扫描电子显微镜(ESEM)对多孔介质进行表征。
-
首先将多孔介质颗粒碾碎成接近胶体的细小颗粒,然后将小颗粒悬浮在离子强度为0.1 mmol·L−1 NaCl的溶液中,并在小瓶中剧烈摇动。然后收集一份上清液,采用纳米粒度电位仪Malvern ZS3000 HAS测定两种多孔介质的表面Zeta电位,结果表明,石英砂的表面Zeta电位为 (–39.6 ± 1.8) mV;双孔隙介质的表面Zeta电位为 (–12.5 ± 1.8) mV。
-
装柱:迁移实验使用高度15 cm,直径3.5 cm的玻璃高压层析柱,空柱质量为399.49 g,空柱体积为144.32 cm3。在柱下端放置一张尼龙网以防止多孔介质堵塞出口,将已经洗净、烘干的多孔介质缓慢倒入柱中,轻敲柱壁夯实,重复上述操作至满柱。称取装满多孔介质的柱重,计算出柱中多孔介质质量[30]。
洗柱:连接柱实验装置,从左至右分别为背景溶液、蠕动泵、层析柱、比色皿。调整蠕动泵转速在10—11,恒定流速在3.3—3.7(mL∙min−1)范围,自下而上泵入背景溶液饱和柱子,饱和时间2 h,饱和后拆下柱子称取其质量,计算柱体中含水质量,进而求出水的体积,也即柱的孔隙体积,最后由孔隙体积算出多孔介质的孔隙率。实验测得石英砂柱的孔隙率在0.4—0.5范围内,而双孔隙介质柱的孔隙率在0.8—0.85的范围内。
进样:待柱子饱和后,用移液管移取15 mL的示踪剂或纳米二氧化钛悬浊液于小烧杯中,开始进样的同时用秒表计时,以进样时背景溶液的体积为初始体积,与此同时收集流出液在比色皿中;进样结束时,记录进样时间,继续泵入背景溶液。记录每一次接样时的时间和背景溶液体积,直到收集液中检测不到纳米二氧化钛。
测量:首先,预热紫外-可见分光光度计,之后对光度计进行暗电流矫正;然后在200—400 nm波长范围内,对纳米二氧化钛悬浊液样品进行光谱扫描,得出该浓度悬浊液的紫外-可见扫描光谱图,确定最大波长;在最大波长下,以去离子水为基准线,先测量纳米悬浊液的初始吸光度A1。进样后以收集的第一个样为基准,测量收集流出液样品的吸光度记录数据,利用吸光度与浓度关系计算出相应纳米二氧化钛颗粒浓度。
流出曲线:以孔隙体积数(V/V0)为横坐标,纳米二氧化钛流出浓度与初始浓度比值(C/C0)为纵坐标绘制纳米二氧化钛流出曲线图(BTC)。
-
对纳米二氧化钛在多孔介质中的穿透曲线采用动力学截留模型进行拟合,相应的质量平衡用一维对流-弥散方程来描述:
式中,θ 为水饱和度;C为水相中纳米二氧化钛的浓度;t为时间;ρ为装柱密度;x为纵向坐标;D为水力扩散系数;q为达西流速;s为截留位的固相颗粒浓度。
物质在固液间转移的质量平衡可由下式来表示:
式中,katt和kd分别是吸附和解吸系数;s为固相颗粒浓度;ψ是沉积作用有关的一个功能函数,其动力学可表示为下列公式:
式中,smax为截留位上的最大固相颗粒浓度。dc为介质颗粒的直径;x为与柱子深度有关的截留发生的起点坐标;β为控制颗粒空间分布的一个经验参数。Bradford等发现对于与柱子深度有关的截留作用取0.432最优[15]。
-
颗粒之间的相互作用力一般采用DLVO 理论来描述,该理论将颗粒间的总势能Φtot表示为范德华力ΦVDW和静电作用力ΦEL之和:
通常情况下,总势能Φtot、范德华力ΦVDW和静电作用力ΦEL均除以玻尔兹曼常数(k = 1.38 ×10−23 J·K−1)和绝对温度(T),即用无量纲形式来表示,故Φ以kT为度量。
粒子之间的静电作用可用下式来表示:
式中,ε = 78.5,为水的相对介电常数;ε0 = 8.854×10−12 C2·N−1·m2为真空绝对介电常数;rp是二氧化钛的平均粒径,ψ01和ψ02分别是二氧化钛和多孔介质的表面Zeta电势;h是二氧化钛和多孔介质颗粒表面之间的距离,κ是德拜参数。
粒子间的范德华作用力可由下式表示:
式中,λ为特征波长,一般取100 nm;A = 1×10−20为Hamaker常数。
-
实验用石英砂的密度为2.65 g·cm−3。由图1a可知,石英砂的粒径分布在0.16—0.79 mm之间,其平均粒径为0.36 mm;由图1b可知,石英砂的孔径分布为5—200 μm,孔径分布图只有1个峰,且峰值为55 μm。
实验用双孔隙介质的密度为2.49 g·cm−3。由图1a可知,双孔隙介质的粒径在0.4—5.0 mm之间,平均粒径为1.5 mm;由图1b可以看出,双孔隙介质孔隙分布复杂,孔隙分布图有两个孔径范围,1个在0.005—5 μm,峰值为0.035 μm;1个是孔隙范围在5—360 μm,有两个峰值,15 μm和200 μm。
从孔径分布图可以看出,相较于石英砂的单峰孔径分布图,双孔隙介质的孔径分布图,呈多范围、多峰值的特点,说明双孔隙介质内部孔道更为发达,内部结构更为复杂。图1c、图1d分别是石英砂和双孔隙介质的扫描电镜图。从图像中可以清晰地看到,石英砂颗粒表面均匀,颗粒内部无孔隙;而双孔隙介质表面粗糙,颗粒内部有很多大小不一的小孔。
-
采用50 mmol·L−1的NaCl作为示踪剂,分别对石英砂和双孔隙介质两种不同多孔介质中的水流特征进行表征。示踪剂在石英砂和多孔介质中的实验条件如表1所示,石英砂在柱子里的堆积密度为1.42 g·cm−3,而双孔隙介质的堆积密度是0.44 g·cm−3. 石英砂在柱子中的孔隙度为0.47,而双孔隙介质在柱子中的孔隙度为0.83。相比于石英砂,双孔隙介质的堆积密度很小,孔隙体积很大,这与前文介质表征时所表明的双孔隙介质内部由复杂的孔隙结构密切相关。
图2是示踪剂在两种多孔介质中的穿透曲线。由图2可以发现,示踪剂穿过石英砂的时候,穿透曲线比较对称,这说明水流经过石英砂的时候流动状态比较均匀;而示踪剂穿过双孔隙介质的时候,穿透曲线是不对称的,有明显拖尾现象,这说明水流经过双孔隙介质的时候,由于双孔隙介质内部孔隙复杂不均,导致流动状态是不均匀的。表2是拟合参数表,示踪剂实验中的实验现象与HYDRUS模拟软件得出的拟合参数相一致:示踪剂穿过装有石英砂的柱中,水流扩散系数是0.19 cm,而穿过装有双孔隙介质的柱中,水流扩散系数为1.95 cm。
-
图3a是不同pH条件下,实验所得纳米二氧化钛在饱和石英砂中的穿透曲线及模拟曲线。从图3可以看出,不同pH条件下,纳米二氧化钛在石英砂中的穿透曲线都有峰,表明两种条件下均有二氧化钛从饱和石英砂中流出;然而,相比于pH = 6.9时的穿透曲线,pH = 2时的穿透曲线峰值很低,与横坐标轴围成的图形较小,表明pH = 2时饱和石英砂中纳米二氧化钛流出较少。
由实验条件表表3可知,在饱和石英砂介质中,pH = 2时,纳米二氧化钛的回收率为1.7%;而在pH = 6.9时,回收率为30%。
通过HYDRUS-1D软件,对实验进行了多次模拟,得到实验模拟结果参数表(表4)。由表4可知,参数katt的值在pH = 2时远大于pH = 6.9时的值。结合两点动力学理论分析,在两点动力学理论中,katt越大说明介质表面对胶体颗粒的吸附能力越强,因此在实验条件下,pH = 2时石英砂对纳米二氧化钛的吸附能力要远大于pH = 6.9时的吸附能力,所以导致纳米二氧化钛大量滞留不易流出。
图4a为不同pH条件下石英砂与纳米二氧化钛之间的能势图。结合DLVO理论分析,由图4可知pH = 2时,纳米二氧化钛颗粒与石英砂表面之间的总势能Φtot小于0,且随着纳米二氧化钛颗粒与石英砂表面的距离增加,总势能Φtot的值不断趋近于0 KT。Φtot小于0,表明石英砂表面与纳米二氧化钛颗粒之间成吸附作用,与模拟结果相一致。相反地,pH = 6.9时,在纳米二氧化钛颗粒距离石英砂介质表面0—20 nm范围内,能势曲线存在一个峰,峰值为200 KT,表明纳米二氧化钛与石英砂之间表现为静电斥力作用,能垒较大,导致纳米二氧化钛不易吸附在石英砂表面,从而导致pH = 6.9时纳米二氧化钛从饱和石英砂中的流出能力高于pH = 2。据文献表述[21],纳米二氧化钛的等电点为4.4—6.2,当pH小于等电点时,未被包被的二氧化钛会在带负电的介质中大量滞留,实验结论与其相一致。
图3b为不同pH条件下,纳米二氧化钛在双孔隙介质中的穿透曲线图,与石英砂相似,pH = 2时纳米二氧化钛在双孔隙介质中的穿透曲线与坐标轴围成的图形要小于pH = 6.9时的穿透曲线与坐标轴围成的图形。由表3可知,与在石英砂中相似,pH = 2时纳米二氧化钛在双孔隙介质中的回收率为2.6%,远小于pH = 6.9时的50.8%。由表4可知,与在石英砂中模拟结果相似,pH = 2时的katt为19.4,远大于pH = 6.9时的0.753。图4b是纳米二氧化钛与双孔隙介质的能势图,与在石英砂中情况类似,pH = 2时的纳米二氧化钛与双孔隙介质表面的总势能Φtot小于0 kT,使得纳米二氧化钛与双孔隙介质表面成吸附作用;而pH = 6.9时的Φtot则大于0 kT,且存在一个能量壁垒,使得纳米二氧化钛与双孔隙介质表面成静电斥力作用,导致pH = 2时纳米二氧化钛在双孔隙介质中穿透能力小于pH = 6.9时。
结论虽相似,然细节有不同。相比于纳米二氧化钛在石英砂中的穿透曲线,所有pH条件下,纳米二氧化钛在双孔隙介质中的穿透曲线对称度较低且有明显的拖尾现象;相比于饱和石英砂中纳米二氧化钛的迁移回收率,饱和双孔隙介质中纳米二氧化钛的迁移回收率较大。从表4参数对比中可以看出,相同pH条件对比下,饱和石英砂中的katt值均大于饱和双孔隙介质中的katt值,表示实验中相同pH条件下石英砂对纳米化钛的吸附能力要大于双孔隙介质,造成纳米二氧化钛在饱和双孔隙介质中的迁移能力较强,回收率较大。从能势图对比可以看出,pH = 2的条件下,纳米二氧化钛与双孔隙介质的初级能阱绝对值为60 kT,小于纳米二氧化钛与石英砂的初级能阱的100 KT,表示pH = 2时,纳米二氧化钛与石英砂的吸附作用大于纳米二氧化钛与双孔隙介质的吸附作用,与前述模拟结果相符;然而pH = 6.9的条件下,纳米二氧化钛与石英砂表面的能量势垒峰值为200 kT,大于与双孔隙介质的能量壁垒峰值20 kT,表示pH = 6.9条件下,纳米二氧化钛与石英砂表面的静电斥力作用要大于与双孔隙介质表面的静电斥力作用,以此推测,纳米二氧化钛在石英砂中的迁移能力要大于在饱和双孔隙介质中的迁移能力,与前述结论不符。纳米颗粒的团聚作用是造成pH = 6.9条件下与结论不符的原因,由于静电斥力作用,纳米二氧化钛颗粒会发生团聚作用,导致纳米二氧化钛颗粒粒径增大,静电斥力作用越大,团聚作用越剧烈。纳米二氧化钛与石英砂表面的静电斥力作用远大于与双孔隙介质间的静电斥力作用,导致纳米二氧化钛在石英砂中的团聚作用较强,从而迁移能力减弱;另一方面,有文献报道,纳米颗粒在孔径比较大的多孔介质中的回收率要大于孔径小的回收率[31],由介质表征可知双孔隙介质的孔径要大于石英砂介质的孔径,从而,相比之下,双孔隙介质中本就较弱的团聚作用的影响也不明显。
综上所述,pH对纳米二氧化钛在双孔隙介质中迁移行为的影响类似于纳米二氧化钛子在饱和石英砂中的迁移行为的影响,然而由于双孔隙介质自身孔隙较大的特性,相同pH条件下的纳米二氧化钛在双孔隙介质中的回收率要大于纳米二氧化钛在石英砂中的回收率。
-
图5a是在不同离子强度下,纳米二氧化钛在石英砂中的穿透曲线。可以看出,纳米二氧化钛均可穿透饱和石英砂柱,且低离子强度(1 mmol∙L−1)流出曲线在上,高离子强度(50 mmol∙L−1)流出曲线在下。由表3可知高离子强度下纳米二氧化钛的回收率为64.2%,小于低离子强度下的67.6%。由表4可知,高离子强度的katt参数为8.73,而低离子强度的katt值是0.316。高离子强度的katt值比低离子强度大了一个数量级,结合两点动力学理论可知高离子强度下石英砂对纳米二氧化钛的吸附能力较强,因而造成纳米二氧化钛的不易迁移,回收率较低。
同时,图6a是石英砂表面与纳米颗粒间的能势图,高离子强度条件下的能势图在下,总势能值始终为负值;低离子强度条件下的势能图在上,存在能垒,总势能值始终为正值;结合DLVO理论可知,高离子强度下石英砂表面与纳米二氧化钛颗粒之间呈吸附作用,与前述结论一致。
造成这一现象的原因是,外来离子会对纳米颗粒表面的双电层产生影响,高离子条件下,纳米二氧化钛双电层被压缩,石英砂与纳米二氧化钛颗粒之间的静电斥力作用减弱,吸附作用增强,从而抑制了纳米二氧化钛的迁移,造成高离子强度下纳米二氧化钛的回收率较低。
在饱和双孔隙介质中,不同离子强度下,纳米二氧化钛的穿透曲线如图5b。相比于饱和石英砂,不同离子条件下双孔隙介质中纳米二氧化钛的穿透曲线有明显的拖尾现象,并且,双孔隙中纳米二氧化钛的初始流出时间要早于石英砂中纳米二氧化钛的流出时间。由表3可知,高离子强度下纳米二氧化钛的回收率为42.4%,低离子强度条件下的回收率为47.1%。与饱和石英砂条件下类似,饱和双孔隙介质中,高离子强度条件下纳米二氧化钛的回收率小于低离子强度条件下的回收率。然而,与石英砂相比,在所有离子强度下,双孔隙介质条件下的纳米二氧化钛的回收率要小于石英砂中纳米二氧化钛的回收率。
由表4可知,与石英砂条件下类似,双孔隙介质中,高离子强度下的katt值大于低离子强度下的katt值,表明高离子强度下双孔隙介质对纳米二氧化钛颗粒的吸附作用大于低离子强度的吸附作用从而与穿透曲线结果相符。但是,相较于石英砂低离子强度条件下的katt值,双孔隙介质低离子强度条件下的katt值较大,但相差不大;同时,相比于石英砂高离子强度条件下的katt值,双孔隙介质高离子强度下的katt值较小。以此结合两点动力学推测,不同离子条件下应是石英砂条件下的回收率较低或者两者差别不大。但是,相同离子条件下,石英砂中的kdet值远大于双孔隙介质中的kdet值,结合两点动力学理论,kdet越大表示纳米颗粒的脱附能力越强。上述表明,不同离子条件下,纳米二氧化钛在石英砂中的脱附能力远大于在双孔隙介质中的脱附能力,从而使得其在石英砂中的回收率远大于在双孔隙介质中的回收率;而此时,纳米二氧化钛在不同介质对的吸附能力大小对纳米颗粒的迁移影响甚微。
图6b是双孔隙介质与纳米二氧化钛颗粒间的能势图,与石英砂中相似,低离子强度条件下,双孔隙介质表面对纳米二氧化钛呈静电斥力作用,且存在能量壁垒;而相反,高离子强度下的能势曲线在下,且小于0 kT,表示此时双孔隙介质对纳米二氧化钛成吸附作用。但是,相比于石英砂,双孔隙介质低离子强度下最大势能绝对值较小,表示此时双孔隙介质对纳米二氧化钛的静电斥力作用小于石英砂对纳米二氧化钛的静电斥力作用,导致相比之下,纳米二氧化钛更易在双孔隙介质中滞留,从而使得低离子强度下双孔隙介质中的回收率较低。纳米颗粒与介质表面同一距离下,双孔隙介质高离子强度下的总势能绝对值与石英砂中相似,表明此时两者对纳米二氧化钛颗粒的吸附能力相同,结合实验模拟分析,在吸附能力相近的情况下,纳米二氧化钛在石英砂中的脱附能力较大,故而纳米二氧化钛在石英砂中的迁移能力较强,回收率高。
综上所述,离子强度对两种介质中纳米二氧化钛迁移行为的影响是相似的,都是高离子强度抑制,低离子强度促进纳米二氧化钛的迁移。但是,由于纳米二氧化钛在两种介质中的吸附和脱附能力的不同,导致相同离子强度条件下,双孔隙介质表现为抑制纳米二氧化钛的迁移。
-
图7a是纳米二氧化钛在饱和石英砂中的穿透曲线。
由图7可知,实验条件下不同浓度的SDBS、纳米二氧化钛均能从石英砂中穿透,并且从3种SDBS条件下纳米二氧化钛均是从0.5个孔隙体积左右开始流出,之后在1.25孔隙体积流出浓度达到峰值。在开始的1.25个孔隙体积数内相对流出浓度从高到低分别对应SDBS的含量为 0.1 cmc、0.01 cmc、0 cmc,但是在1.25个孔隙体积数后,从高到低的顺序变为了0.01 cmc、0.1 cmc、0 cmc。介质表征可知石英砂的表面光滑,所以其对SDBS的吸附能力较弱,所以在最初1.25个孔隙体积数时,表面活性剂对的纳米二氧化钛流出曲线的作用主要体现在纳米二氧化钛和介质之间的静电排斥力,体现到曲线上就是1 cmc的体系中纳米二氧化钛的相对流出浓度较高。
由表3可知,随着SDBS含量的增加,纳米二氧化钛的回收率也越来越大,表明随着SDBS的增加,纳米二氧化钛在石英砂中的迁移能力越来越强。SDBS是一种典型的阴离子表面活性剂,它可以吸附于纳米二氧化钛表面或者吸附于石英砂表面,从而改变石英砂与二氧化钛之间的空间结构及相互作用力,从而影响纳米二氧化钛在介质中的迁移。从表4可以看出,随着SDBS浓度的增大,对应的katt值不断减小,结合两点动力学理论,katt值减小说明纳米二氧化钛的吸附能力减弱,从而其迁移能力不断增强,导致回收率不断增加。Godinez [23]研究表明,SDBS的存在和浓度的增加增强了纳米二氧化钛在饱和多孔介质中的迁移,表面活性剂对空间斥力的影响使纳米二氧化钛保持稳定或分散,从而增加了迁移率。
图7b是双孔隙介质中不同SDBS存在条件下的纳米二氧化钛的穿透曲线图。由图7可知,相比于石英砂的流出曲线,同一SDBS浓度下,双孔隙介质中纳米二氧化钛的迁移曲线具有较为明显的拖尾现象,同时随着表面活性剂浓度的升高拖尾越严重。由图7可知,双孔隙介质中,纳米二氧化钛从0.1孔隙体积开始流出,比石英砂的0.5孔隙体积流出较快。在开始的0.5个孔隙体积数内,纳米二氧化钛在双孔隙介质柱的相对流出浓度从高到低分别对应的含SDBS浓度为 0.01 cmc、0 cmc、0.1 cmc,在0.5个孔隙体积数后,从高到低的顺序变为了0.1cmc、0.01cmc、0 cmc。这是由于在最初的0.5个孔隙体积数内,SDBS的桥键作用起了主导优势,SDBS浓度越高,桥键作用越大,因此0.1 cmc体系中纳米二氧化钛的相对流出浓度最低;随着时间的推移,淋溶液中阴离子表面活性剂SDBS占据了介质表面的正电荷位点,随着SDBS在介质上的吸附量逐渐增加,纳米二氧化钛和介质之间的静电排斥力增加且随着SDBS浓度的增加更加明显。因此在0.5个孔隙体积数后,静电排斥力的优势更明显,使含高浓度SDBS体系中的纳米二氧化钛更不容易被多孔介质截留,所以最后0.1 cmc SDBS的体系中纳米二氧化钛的相对流出浓度最高。由表3可知与石英砂中相似,纳米二氧化钛的回收率随着SDBS浓度的增加而增加。然而,相比于石英砂中纳米二氧化钛的流出率,同一SDBS浓度下,双孔隙介质中纳米二氧化钛的回收率较小。从表4可以看出随着SDBS的浓度增加,katt值逐渐降低,表示纳米二氧化钛的吸附能力逐渐降低。
结合表3、表4分析,SDBS存在条件下,双孔隙介质实验参数模拟结果的katt值与石英砂实验参数模拟结果相比差距不大,结合两点里动力学分析,此时两种介质表面对纳米二氧化钛颗粒的吸附能力相当;然而相对应的双孔隙介质实验参数模拟的的kdet值要比石英砂的小几个数量级,表明此时虽然吸附能力相当,但是纳米二氧化钛颗粒在石英砂中的脱附能力要远大于在双孔隙介质中的脱附能力,从而导致同一SDBS浓度下,纳米二氧化钛在石英砂中的滞留较少,回收率较高。
综上所述,无论多孔介质是均质的石英砂还是非均质的双孔隙介质,SDBS的加入,都会大幅度提高纳米二氧化钛的迁移能力。然而,相比于在石英砂中而言,纳米二氧化钛在双孔隙介质中的回收率较小。
-
(1) pH对纳米二氧化钛在两种多孔介质中迁移行为的影响与纳米二氧化钛等电点有关,当pH低于等电点时,纳米二氧化钛会在石英砂和双孔隙中大量滞留。
(2) 由于双孔隙介质对纳米二氧化钛的吸附能力较弱以及本身孔隙较多的综合原因,使得相同pH条件下,纳米二氧化钛在双孔隙介质中比在细砂中的迁移能力较强,回收率较高。
(3) IS对纳米二氧化钛在两种介质中迁移行为的影响相似,均为:高离子强度抑制、低离子强度促进纳米二氧化钛的迁移。
(4) 由于纳米二氧化钛在双孔隙介质中比在细砂介质中的脱附能力弱,导致相比于细砂介质,在相同离子强度条件下,双孔隙介质表现为抑制纳米二氧化钛的迁移。
(5) SDBS的存在促进了纳米二氧化钛在两种多孔介质中的迁移,且随着SDBS浓度的增加,纳米二氧化钛的迁移能力增强,回收率增加。
(6) 由于纳米二氧化钛颗粒在石英砂中的脱附能力要远大于在双孔隙介质中的脱附能力,从而导致相较于细砂介质,在同一SDBS浓度下,纳米二氧化钛在双孔隙介质中的滞留较多,回收率较低。
不同环境因素对纳米二氧化钛在双孔隙介质中迁移行为的影响
TiO2 nanoparticles transport in aggregated porous media with intra- and interaggregate porosities: Effects of pH, ionic strength and sodium dodecylbenzene sulfonate
-
摘要: 选取石英砂和双孔隙介质两种多孔介质,通过柱实验探究pH、离子强度(IS)和阴离子表面活性剂十二烷基苯磺酸钠(SDBS)的3种环境因素对纳米二氧化钛在饱和多孔介质中迁移行为的影响。通过HYDRUS-1D用两点动力学模型对实验结果进行模拟,并结合DLVO理论进行分析。结果表明,pH对纳米二氧化钛在两种多孔介质中迁移行为的影响与纳米二氧化钛等电点有关,当pH低于等电点时,纳米二氧化钛会在石英砂和双孔隙中大量滞留;由于双孔隙介质孔隙较多,且对纳米二氧化钛的吸附能力较弱,因而在相同pH下,纳米二氧化钛在双孔隙介质中比在细砂中的迁移能力较强,回收率较高;IS对纳米二氧化钛在两种介质中迁移行为的影响相似,均为高离子强度抑制、低离子强度促进纳米二氧化钛的迁移;由于纳米二氧化钛在双孔隙介质中比在细砂介质中的脱附能力弱,导致相比于细砂介质,在相同离子强度条件下,双孔隙介质表现为抑制纳米二氧化钛的迁移;SDBS的存在促进了纳米二氧化钛在两种多孔介质中的迁移,且随着SDBS浓度的增加,纳米二氧化钛的迁移能力增强,回收率增加;由于纳米二氧化钛颗粒在石英砂中的脱附能力要远大于在双孔隙介质中的脱附能力,从而导致相较于细砂介质,在同一SDBS浓度下,纳米二氧化钛在双孔隙介质中的滞留较多,回收率较低。Abstract: The influences of pH, ionic strength (IS) and sodium dodecylbenzene sulfonate (SDBS) on TiO2 nanoparticles transport behavior in saturated porous media were investigated by column experiment, and by selecting two kinds of porous media, i.e. quartz sand and aggregated porous media with intra- and interaggregate porosities (dual-porosity media). Experimental results were simulated using two-site model by HYDRUS-1D code and analyzed theoretically according to DLVO theory. The results showed that the effect of pH on the transport of TiO2 nanoparticles in both porous media was related to the isoelectric point of TiO2 nanoparticles. When pH was lower than the isoelectric point, TiO2 nanoparticles would be largely retained in both media. Due to the large number of pores and the weak adsorption capacity of the dual-porosity media for TiO2 nanoparticles and the large number of pores, the TiO2 nanoparticles in the dual-porosity media had a stronger transport capacity and a higher recovery rate under the same pH, compared to fine sand. Given to the effect of IS, high IS inhibited the transport of TiO2 nanoparticles, and low IS promoted the transport of TiO2 nanoparticles for both porous media. However, the transport of TiO2 nanoparticles was inhibited in the dual-porosity media under the same ionic strength compared to fine sand, because the desorption ability of TiO2 nanoparticles from the dual-porosity media was weak. The presence of SDBS promoted the transport of TiO2 nanoparticles in both porous media. Furthermore, with the increase of SDBS concentration, the transport ability of TiO2 nanoparticles was enhanced, and the recovery rate increased correspondingly. Because the desorption ability of TiO2 nanoparticles in fine sand was much greater than that in dual-porosity media, TiO2 nanoparticles remained more in dual-porosity media with the same SDBS concentration, and the recovery rate was correspondingly lower.
-
Key words:
- TiO2 nanoparticle /
- saturated porous media /
- transport
-
循环活性污泥法(cyclic activated sludge technology, CAST)作为一种基于序批式反应器(sequencing batch reactor, SBR)工艺改良的新技术,因其运行简便、结构紧凑及运行费用低等优点而被广泛应用于城镇污水处理厂中[1-2]。CAST反应池通常由选择池、缺氧池和好氧池3部分构成。在生物选择池内,将原水和混合液按体积比为5∶1的比例混合,使回流污泥处于高负荷条件下,这能有效抑制丝状菌的增殖,避免污泥膨胀[3-4]。由于污泥回流过程中会带有一定量的硝态氮(NO3−-N),反硝化菌在选择池和缺氧池中利用有机物将NO3−-N还原为氮气(N2)的同时,也将原水中的有机物储存为细胞内碳源[5],这为好氧池内的同步硝化-反硝化脱氮(simultaneous nitrification and denitrification, SND)创造了条件。BIAN等[6]同时研究CAST工艺的脱碳与脱氮性能,结果表明,TN主要通过以亚硝酸盐为间歇产物的同步硝化-反硝化机制去除,曝气期间同步硝化-反硝化的发生是由于反应器悬浮载体中存在微缺氧环境所致。WANG等[7]指出通过改变关键操作条件(温度,DO等)可以控制脱氮路径,控制同步硝化-反硝化成为主要脱氮路径,并在不同温度下通过调整有机负荷改善脱氮效率。WANG等[8]在对CAST系统进行脱氮性能的评估中,同样证实同步硝化-反硝化脱氮贡献与操作条件有关,通过优化操作条件可提高同步硝化-反硝化过程的脱氮性能,推测DO在1 mg·L−1时亚硝酸盐氧化菌的抑制作用会影响亚硝酸盐的积累。然而,目前关于CAST工艺的脱氮路径及脱氮特性的报道不多。基于此,对CAST工艺的脱氮性能进行评价十分必要。
为此,本研究以某污水处理厂CAST工艺为研究对象,通过对典型周期内氮组分、污泥浓度等进行测定,结合污泥活性、同步硝化-反硝化速率和饱食-饥饿(feast-famine)等批次实验,评价了该工艺的脱氮性能,以期为我国城市污水处理厂CAST工艺的设计、运行及水厂提质增效提供参考。
1. 材料与方法
1.1 污水处理厂简介
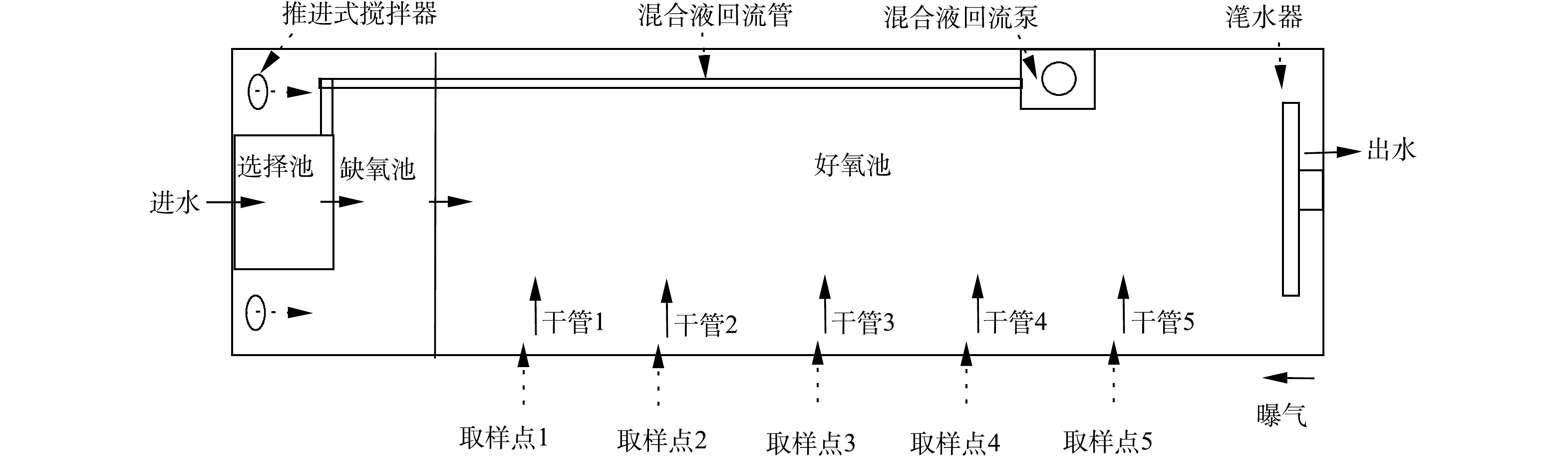
该污水处理厂处理规模为3.5×104 m3·d−1,其中一期工程2.0×104 m3·d−1,采用CAST工艺;二期工程1.5×104 m3·d−1,采用A2/O工艺。CAST工艺由4组反应池构成,每一组反应池的尺寸为76 m×11.6 m×6 m,选择池、缺氧池和好氧池体积比为1∶4∶25。该工艺水力停留时间(hydraulic retention time, HRT)为18.8 h,污泥龄(sludge retention time, SRT)为14.7 d,污泥回流比为20%。CAST工艺包括4个运行步骤:进水曝气、曝气、沉淀和出水,每步骤时长为1 h。缺氧池中安装潜水混合器,保证缺氧池内活性污泥处于悬浮状态;好氧池内安装微孔曝气器,以维持所需溶解氧(dissolved oxygen, DO)浓度及搅拌。在好氧池末端设有混合液回流泵,每运行周期前2 h进行回流,流量为80 m3·d−1。厌氧池和缺氧池设有氧化还原电位(oxidation-reduction potential, ORP)测定仪,在线显示氧化还原电位。好氧池中设有溶解氧仪和污泥浓度仪,在线检测DO浓度和污泥浓度,并反馈至中控室,随时调节鼓风机送风量和排泥量。
1.2 常规指标的分析方法
氨氮(NH4+-N)、亚硝氮(NO2−-N)和硝氮(NO3−-N)等的测定参照标准分析方法[9]。NH4+-N的测定采用纳氏试剂分光光度法,NO2−-N的测定采用N-(1-萘基)-乙二胺分光光度法,NO3−-N的测定采用紫外分光光度法,MLSS和MLVSS的测定采用重量法。DO由便携式溶氧仪(SevenGo pro SG6)测定。本研究中以乙酸钠作为批次实验的碳源,其测定方法为气象色谱法,测定仪器为SP-3420A型气象色谱仪。
1.3 CAST工艺取样点及脱氮贡献计算
为研究CAST工艺好氧池同步硝化-反硝化脱氮特性,沿池长设定5个取样点(图1),在进水曝气和曝气段每间隔15 min取样且测定氮组分浓度和记录DO浓度。
CAST反应池的脱氮主要由3部分贡献:缺氧池反硝化脱氮(DEN)、同步硝化-反硝化脱氮(SND)、沉淀阶段的内源反硝化脱氮(ED)。总脱氮量根据式(1)计算,同步硝化-反硝化脱氮量根据式(2)计算,内源反硝化脱氮量根据式(3)计算,选择池及缺氧池脱氮量根据式(4)计算,每周期开始时池内总氮量根据式(5)计算,每周期进水总氮量根据式(6)计算,每周期出水总氮量根据式(7)计算,曝气结束时池内总氮量根据式(8)计算。
M=Minf−Meff (1) Msnd=Mst+Minf−Mend−Mhd (2) Med=TNend−TNeff−TNst (3) Mhd=1000V1TC1 (4) Mst=1000V2C2 (5) Minf=1000V3C3 (6) Meff=1000V3C4 (7) Meff=1000(V2+V3)C5 (8) 式中:M 为每周期总脱氮量,mg(以N计);Minf为每周期进水总氮量,mg;Meff为每周期出水总氮量,mg;Msnd为每周期同步硝化-反硝化脱氮量,mg;Mst为每周期起始时CAST池内总氮量,mg;Mhd为每周期缺氧池脱氮量,mg;Med为每周期内源反硝化脱氮量,mg;Mend为每周期曝气结束时CAST池总氮量,mg; V1为混合液回流量,80 m3·h−1;T为回流时间,2 h;C1为回流混合液平均NO3−-N浓度,mg·L−1;V2为每周期开始时池内混合液体积,m3;C2为每周期开始时池内混合液总氮浓度,mg·L−1;V3为进水体积,m3;C3为进水总氮浓度,mg·L−1;C4为出水总氮浓度,mg·L−1;C5为曝气结束时池内混合液总氮浓度,mg·L−1。
1.4 同步硝化-反硝化速率测定
为研究CAST反应池内不同DO浓度下的同步硝化-反硝化脱氮特性,本研究在CAST工艺进水结束时取1号采样点的泥水混合物于5 L试验装置内,在DO浓度分别为1.0、1.5、2.0和2.5 mg·L−1下进行同步硝化-反硝化率测定。试验装置内设有搅拌器,在反应器顶端设有取样口。测定过程中,每隔15 min取样并测定NH4+-N和NO3−-N浓度,随后根据浓度变化拟合得出NH4+-N消耗速率和NO3−-N生成速率,最终根据两速率计算得出同步硝化-反硝化率(式(9))。
R=(1−RNO−3−NRNH+4−N)⋅100% (9) 式中:
R RNH+4-N RNH+4-N 1.5 饥饿时长对微生物吸收乙酸的测定
为确定CAST工艺中饥饿时长对微生物吸收乙酸的影响,本研究将活性污泥等分为5份,置于1 L血清瓶中,活性污泥(SS)浓度为3 608 mg·L−1,VSS·SS−1为0.55。向每一份污泥加入足量的NO3−-N使其处于饥饿状态,饥饿时长依次为5、24、36、48和72 h。饥饿结束后加入足量的乙酸、NO3−-N和NH4+-N,间隔15 min进行取样,测定基质浓度变化,根据式(10)~式(12)计算最大乙酸吸收量。
ΔMHAc1=2.67(ΔMNO−3−ΔMNO−2)+1.07ΔMNO−2 (10) ΔMHAc2=ΔMHAc0−ΔMHAc1 (11) ΔMHAc=ΔMHAc2×0.001CVSS (12) 式中:
ΔMHAc1 ΔMNO−3 ΔMNO−2 ΔMHAc0 ΔMHAc2 CVSS ΔMHAc 1.6 内源反硝化活性测定
活性污泥的内源反硝化活性采用基质消耗速率法测定,具体操作如下:CAST工艺每周期曝气结束时,在好氧池末端取一定量的活性污泥,用水淘洗去除残余基质,将淘洗过的活性污泥加入密闭的反应器内,向其中加入硝酸钠,控制NO3−-N为20 mg·L−1,每隔30 min取泥水混合物进行过滤,之后测定滤液中的NO3−-N、NO2−-N和NH4+-N的浓度,活性通过基质浓度变化线性拟合得出。
2. 结果与讨论
2.1 CAST工艺的脱氮效果
CAST工艺自2013年投入运行以来,各项水质指标均满足《城镇污水处理厂污染物排放标准》(GB 18918-2002)一级B标准。2021年10月—2022年5月,CAST工艺的主要进出水水质见表1。可以看出,CAST工艺出水COD、TN和PO43--P浓度分别小于30、15和0.3 mg·L−1,均达到A标准的水质要求。而对于出水NH4+-N,尽管略高于A标准所规定的1.5 mg·L−1,但在二期A2/O工艺出水稀释下,污水厂总出水可达标排放。
表 1 进出水水质Table 1. Quality of influent and effluentmg·L−1 水样 COD NH4+-N NO3−- N TN PO43--P 进水 188±22 32.7±7.60 — 40.50±9.50 2.98±1.15 出水 20±3 1.60±1.30 6.74±3.59 11.30±2.20 0.07±0.06 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.2 CAST工艺脱氮途径及原理分析
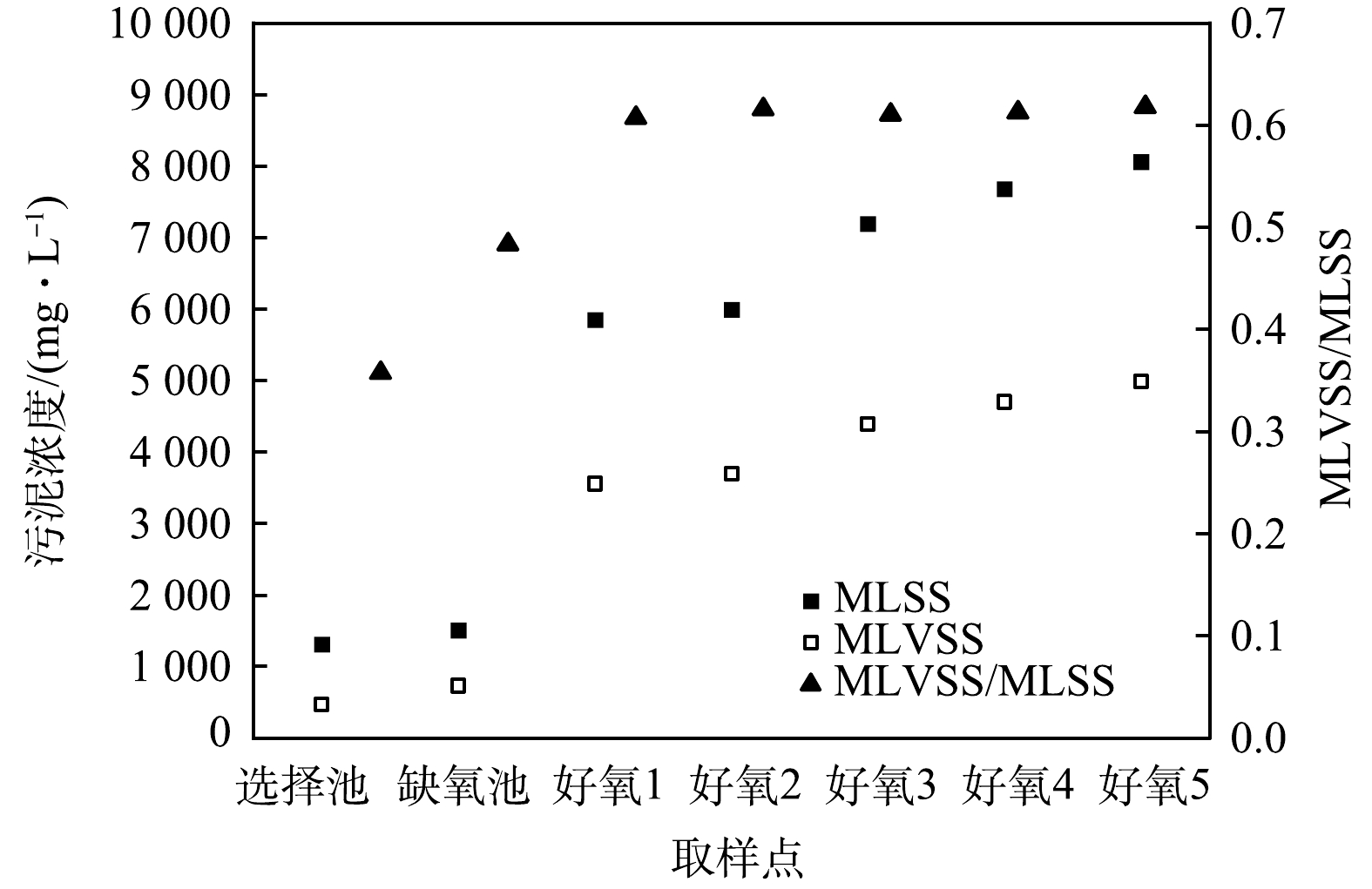
CAST工艺属于SBR工艺的一种演变,其运行方式也与SBR工艺相同,但CAST的推流式进水方式使得好氧池的反应并非完全混合式。考虑到CAST工艺的脱氮主要在好氧池。因此,首先对好氧池流态进行分析,结合选择池、缺氧池和好氧池的污泥浓度测定,判断各反应区的流体力学特性。由测定结果可知(图2),各反应区的污泥浓度分布并不均匀,在选择池和缺氧池内污泥浓度基本不变,说明这2个反应区的流态呈完全混合式;而好氧池进水端污泥浓度最低,约6 000 mg·L−1;出水端最高,约8 000 mg·L−1,说明回流混合液和原水混合进入好氧池后,以推流式方式前进,由于混合液回流比较低,使得污泥浓度沿程增加。
CAST工艺序批式的运行方式及推流式进水方式使其具有独特的脱氮特性。在周期开始时,回流混合液和原水在选择池内混合,此时混合液中的NO3−-N被反硝化菌利用原水中的有机物还原成N2(反硝化脱氮)。值得注意的是,CAST工艺的回流比仅为20%,使得回流至缺氧池的NO3−-N含量有限,继而选择池和缺氧池对TN脱除的贡献率较低;其次,回流混合液中的活性污泥在选择池和缺氧池中将原水中的有机物以胞内聚合物的形式储存,之后进入好氧池内,进而作为同步硝化-反硝化脱氮的碳源;最后,当曝气结束后,好氧池内活性污泥开始沉淀,池中剩余DO首先被消耗殆尽,此时好氧池部分区域进入缺氧状态,反硝化菌开始利用胞内聚合物及内碳源进行内源反硝化脱氮。综上所述,CAST工艺脱氮途径包括选择池及缺氧池反硝化脱氮、好氧池同步硝化-反硝化脱氮及内源反硝化脱氮。
为明确3种脱氮途径对TN脱除的贡献率,结合典型周期水质指标,利用式(1)~(8)对其进行计算,结果见表2。可以看出,选择池和缺氧池中反硝化脱氮贡献率最低,仅为(1.64±0.05)%,这一结果主要和混合液回流比较低(20%)有关,其可通过增大回流比提升,但需对回流泵进行更换;沉淀出水阶段的内源反硝化脱氮贡献最大,可达(62.86±4.13)%,类似结果在二沉池中也有观察到[10-11],但受限于沉淀时间短等因素,其脱氮贡献率难以进一步提升;除去反硝化和内源反硝化脱氮,TN的脱除全由好氧池同步硝化-反硝化途径完成,贡献率可达(35.50±4.15)%,对其进行优化,或成为进一步提升CAST工艺脱氮性能的突破口。基于此,本研究从如何优化同步硝化-反硝化过程入手,通过探究DO浓度和饥饿时长对同步硝化-反硝化脱氮性能的影响机制,从而确定适宜CAST工艺的运行策略。
表 2 CAST工艺典型周期各途径脱氮贡献占比Table 2. Contribution of each pathway to nitrogen removal脱氮方式 脱氮量/g 占比/% 反硝化脱氮 526.01±19.91 1.64±0.05 内源反硝化脱氮 20 093.13±1 291.55 62.86±4.13 同步硝化-反硝化脱氮 11 366.27±1 553.06 35.50±4.15 总脱氮 31 985.40±1 212.71 100 | Show TableDownLoad:
CSV
2.3 DO浓度对同步硝化-反硝化脱氮性能的影响
为分析DO浓度对同步硝化-反硝化过程的影响特性,测定了不同DO浓度下好氧池曝气阶段DO、NH4+-N及NO3−-N浓度随时间的变化情况,结果如图3所示。图3(a)~(b)为低DO浓度(0.5~1 mg·L−1)下同步硝化-反硝化的脱氮特性。由图3(a)可知,反应过程中NH4+-N仅很少一部分被硝化,曝气结束时仍有约12 mg·L−1NH4+-N,而NO3−-N浓度几乎为零,不能为同步硝化-反硝化过程提供足够的电子受体,进而导致脱氮效率极低。
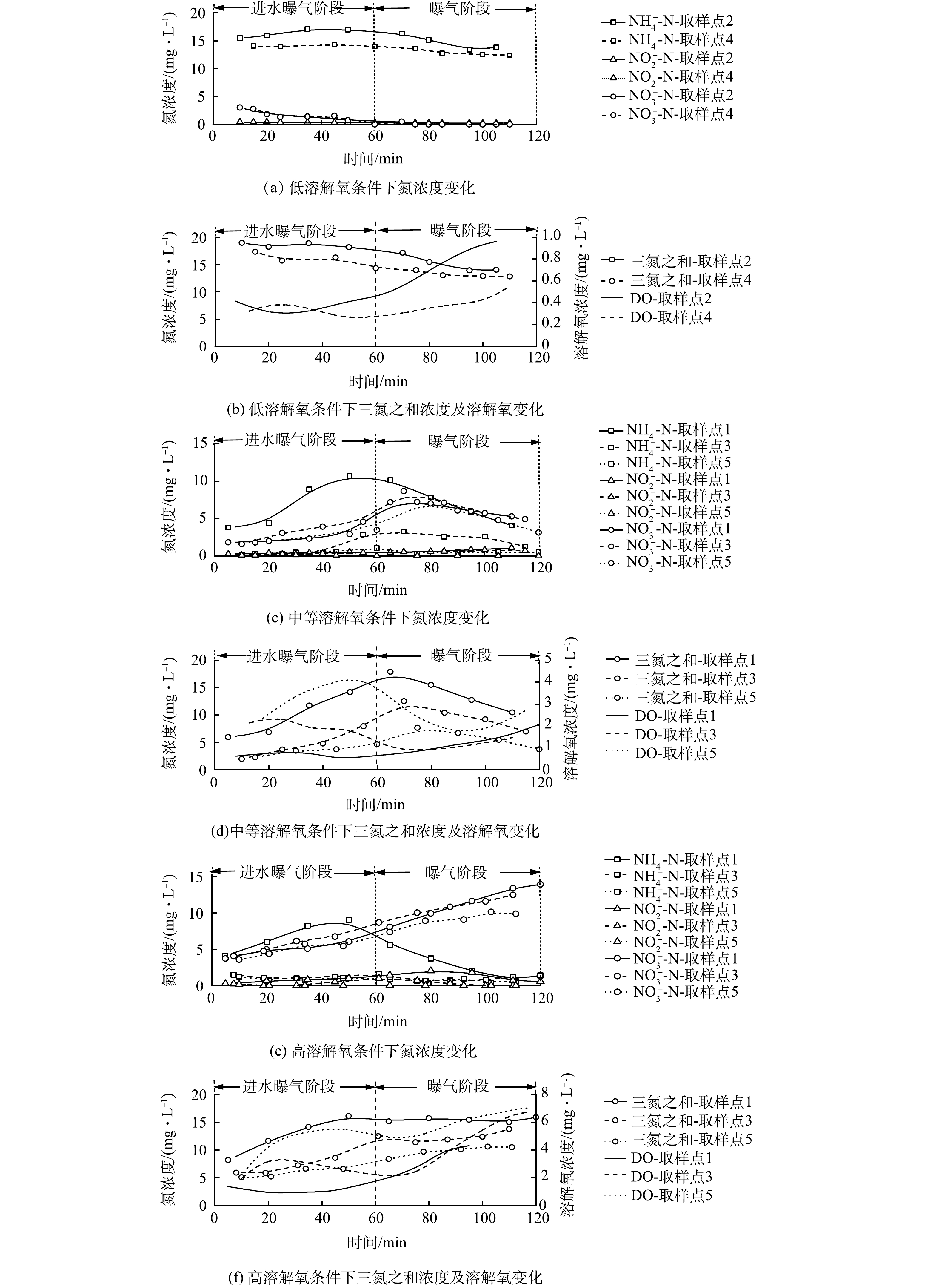 图 3 不同DO浓度下各取样点基质浓度变化Figure 3. Variation of nitrogen concentration at each sampling point at different DO concentrations
图 3 不同DO浓度下各取样点基质浓度变化Figure 3. Variation of nitrogen concentration at each sampling point at different DO concentrations图3(c)~(d)为中等DO浓度(1~4 mg·L−1)下同步硝化-反硝化脱氮特性。由图3(c)可知,取样点1、3和5的NH4+-N和NO3−-N浓度随反应的进行呈现先升后降的趋势。在进水曝气阶段,取样点1 NH4+-N浓度由3.80 mg·L−1升至10.72 mg·L−1,而NO3−-N浓度仅由1.85 mg·L−1升至2.94 mg·L−1,这一结果由原水和硝化反应联合贡献。原水会带入一定量的NH4+-N,但此时硝化能力有限,无法及时将NH4+-N氧化为NO3−-N,于是NH4+-N浓度不断升高。对NO3−-N而言,原水会对其进行稀释,但伴随着硝化反应的进行,NO3−-N会不断积累,因而NO3−-N浓度略有增加,但会明显低于NH4+-N浓度的增加;在曝气阶段,若仅存在硝化反应,NH4+-N的下降将同NO3−-N的升高相匹配。但实际情况是,NH4+-N和 NO3−-N浓度同时下降,在曝气结束时分别降至4.09 mg·L−1和5.29 mg·L−1,此结果证实了同步硝化-反硝化脱氮的存在。由于取样点3和5距进水口有一定距离,推流式进水方式使得其起始NH4+-N和COD较低,进而DO浓度快速升高。而当进水逐渐推移到这两点时(取样点3为20~40 min;取样点5为40~60 min),由于COD降低和硝化反应的进行,DO浓度开始下降,之后随着NH4+-N和COD的降低,DO浓度开始缓慢回升。依据计算可以得出,同步硝化-反硝化、内源反硝化和反硝化途径对TN的脱除贡献率分别为65.7%、32.5%和1.80%。依据计算可得,同步硝化-反硝化、内源反硝化和反硝化途径对TN的脱除贡献分别为65.7%、32.5%和1.80%(进水和出水TN分别为38.80 mg·L−1和6.01 mg·L−1, TN去除率为84.51%)。
图3(e)~(f)为高DO浓度(1~6 mg·L−1)下同步硝化-反硝化脱氮特性。由图3(e)和图3(f)可知,3个取样点的 DO 浓度随时间变化规律与中 DO 水平下的变化规律基本一致,但其浓度均高于后者,取样点1、3、5的DO平均浓度分别为3.41、4.48和6.07 mg·L−1。在高DO浓度条件下,氧扩散能力增强,活性污泥絮体内部难以维持缺氧环境,同步硝化-反硝化脱氮过程所需环境遭到破坏,致使NO3−-N不断积累,曝气结束时3个取样点的NH4+-N和NO3−-N浓度依次达到1.41、1.27、0.58 mg·L−1和13.92、12.48、9.88 mg·L−1。依据计算可得,同步硝化-反硝化、内源反硝化和反硝化途径对TN的脱除贡献分别为39.2%、58.6%和2.20%(进水和出水TN分别为40.01和10.20 mg·L−1,TN去除率为74.51%)。
综上所述,中DO浓度下可观察到明显的同步硝化-反硝化过程,同步硝化-反硝化脱氮效果最理想,而高DO和低DO浓度下无法观察到明显的同步硝化-反硝化过程,脱氮效果较差。因此,CAST工艺好氧池DO应控制为中等浓度,这对同步硝化-反硝化过程进行更有利,从而提高TN去除率。
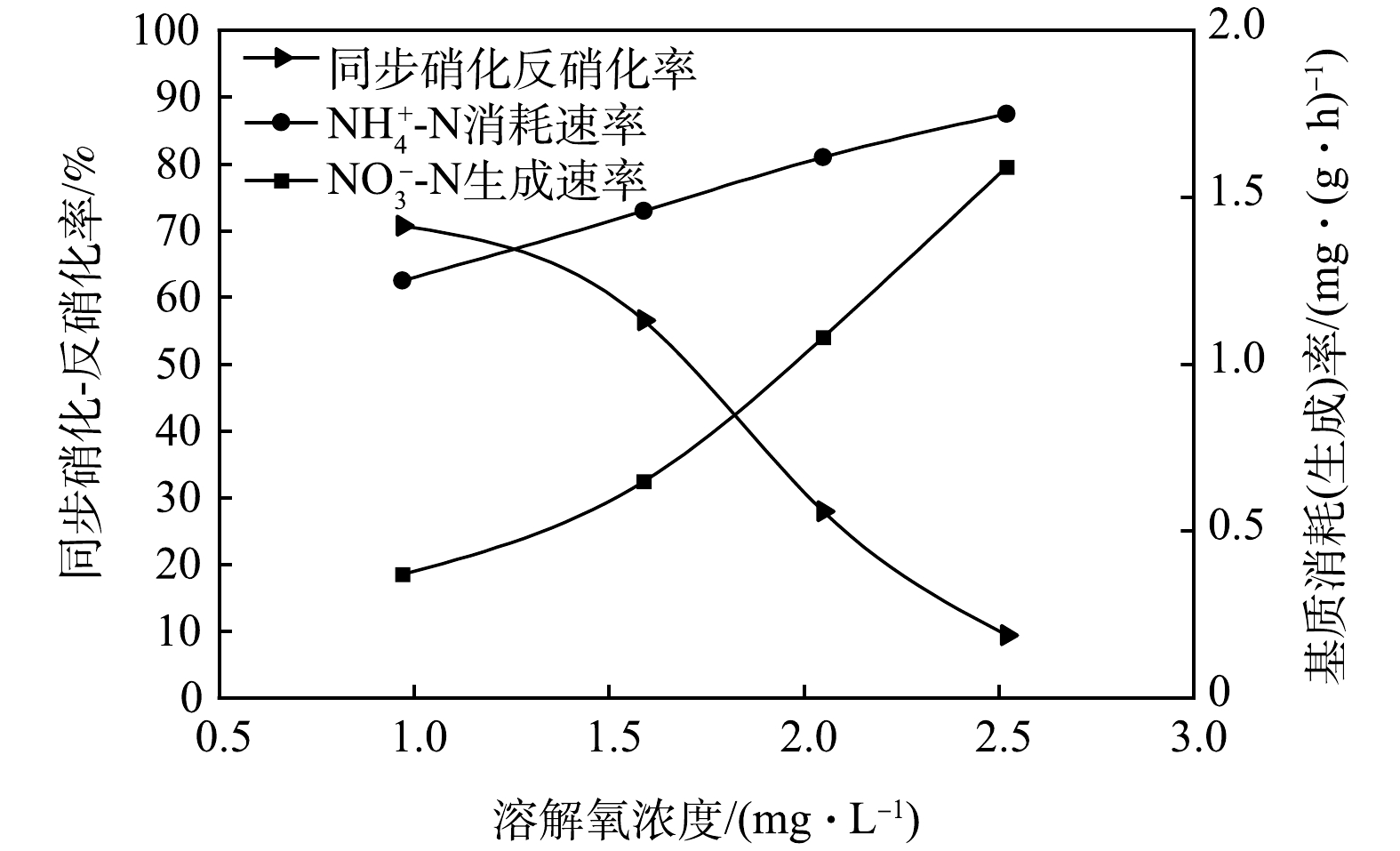
基于不同DO浓度下典型周期内同步硝化-反硝化对TN脱除的贡献变化,特进行同步硝化-反硝化率测定,以确定同步硝化-反硝化脱氮的最适宜DO浓度。在CAST反应池进水结束后取取样点1的混合液,利用序批式反应器对不同DO浓度(1.0~2.5 mg·L−1)下的同步硝化-反硝化率进行测定,结果如图4所示。由图4可知,随着DO浓度升高,同步硝化-反硝化率逐渐下降。当DO浓度控制在 1 mg·L−1 时,同步硝化-反硝化率达到最高值(70.66%),NH4+-N消耗速率和NO3−-N生成速率分别为1.25 mg·(g·h)−1和0.37 mg·(g·h)−1。而当DO浓度控制在2.52 mg·L−1时,同步硝化-反硝化率降为9.34%,NH4+-N消耗速率和NO3−-N生成速率分别为1.75 mg·(g·h)−1和1.59 mg·(g·h)−1。相比较而言,随着DO浓度的升高,NH4+-N消耗速率增幅并不明显,仅为40%,但NO3−-N生成速率增幅明显,达到329.73%。这表明在DO浓度为2.52 mg·L−1时同步硝化-反硝化过程已严重受限。因此,CAST工艺DO浓度应控制在1.0~1.5 mg·L−1,此时同步硝化-反硝化性能最强,这一结果与实际运行过程内中等DO浓度下所得结果一致,同样符合徐凤等[12]认为的最佳溶解氧浓度(0~2mg·L−1)的观点。
2.4 饥饿时长对微生物吸收HAc的影响
CAST工艺中20%的混合液回流比使得回流至CAST反应池前端的污泥有限,且污泥在好氧池内以推流式方式前进,污泥从好氧池末端回流至前端理论需要3~5 d,于是微生物将长期处于饥饿状态,即CAST工艺运行具有饱食-饥饿的特性,即非平衡增长[13-14]。饱食-饥饿理论[15]认为污泥所处饥饿时长会影响胞内聚合物的储存量,而同步硝化-反硝化脱氮能力又取决于胞内聚合物的量。因此,确定合适的饥饿时长,进而明确合适的污泥回流比对提高CAST工艺脱氮效率尤为重要。
为探究饥饿时长对微生物吸收有机物的影响,进行批次实验,结果见表3。由表3可知,随着饥饿时长增加,活性污泥对HAc的吸收量呈现出先上升后下降的趋势。在饥饿时长为5 h时
ΔMHAc ΔMHAc ΔMHAc 表 3 不同饥饿时长下活性污泥对HAc的吸收量Table 3. Uptake of HAc by activated sludge at different famine durations饥饿时长/h ΔMHAc0 ΔMNO−3 ΔMNO−2 ΔMHAc1 ΔMHAc2 5 335.16 79.35 74.87 91.31 243.84 24 394.68 101.39 88.34 128.47 266.2 36 468.91 103.59 94.3 124.77 344.14 48 407.25 102.71 93.03 124.46 282,79 72 376.14 96.54 84.47 121.76 254.38 | Show TableDownLoad:
CSV
2.5 CAST工艺内源反硝化脱氮特性分析
为了解CAST工艺好氧池内源反硝化脱氮特性,在曝气结束时取1、3和5取样点的泥水混合物,对其内源反硝化活性进行了测定,结果见图5。由图5可知,取样点1泥水混合物内源反硝化活性最大,达到1.48 mg·(g·h)−1,明显高于取样点3(1.08 mg·(g·h)−1)和取样点5 (1.05 mg·(g·h)−1)。造成这一结果的原因是取样点1靠近进水端,活性污泥所吸附的有机物及微生物储存的胞内聚合物较多,所以可用于内源反硝化反应的电子供体较多。而对于取样点3和取样点5的活性污泥,较长的污泥龄导致外源有机物和胞内聚合物已被大量消耗,内源反硝化反应只能以细胞作为碳源进行,致使内源反硝化速率较低。内源反硝化主要是利用内碳源(细胞体)进行反硝化,细胞体的化学结构通式为C5H7NO2,化学计量反应如式(13)所示。
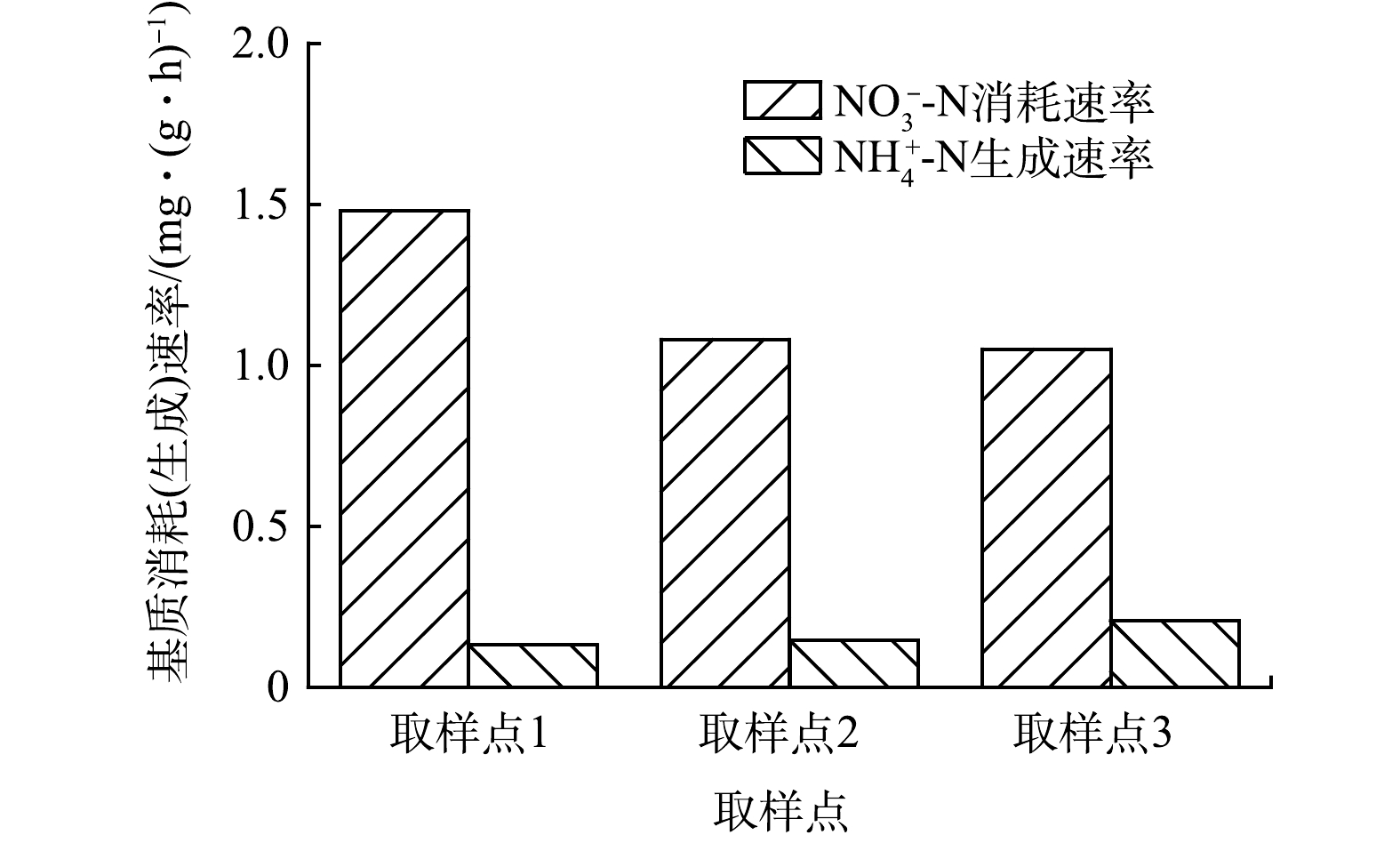 图 5 不同取样点内源反硝化过程中氮消耗(生成)速率Figure 5. Rate of nitrogen consumption (production) during endogenous denitrification at different sampling points
图 5 不同取样点内源反硝化过程中氮消耗(生成)速率Figure 5. Rate of nitrogen consumption (production) during endogenous denitrification at different sampling pointsC5H7NO2+4NO−3→2N2+5CO2+NH3+4OH− (13) 由式(13)可知,每去除1 g的NO3−-N理论上会产生0.25 g的NH4+-N。而在CAST工艺中,取样点1、3和5每去除1 g的NO3−-N依次产生0.09、0.13和0.20 g NH4+-N,均低于利用细胞体进行反硝化的理论值,但沿着推流方向逐渐接近理论值。此结果证实内源反硝化过程不单以细胞体作为碳源,还包括污泥所吸附的有机物及微生物储存的胞内聚合为碳源。
2.6 CAST工艺脱氮性能评价与运行调控建议
表4列举了CAST、A2/O和氧化沟工艺的脱氮指标。由表4可知,3种工艺出水COD、NH4+-N和TN浓度相当,均满足相关排放要求。而对于出水TN,CAST、A2/O和氧化沟工艺的去除量分别为29.1、36.06和39.03 mg,每去除1 mg TN所需COD依次为5.77、6.41和15.94 mg,此结果说明CAST工艺更具脱氮潜力,但考虑到CAST工艺出水TN明显高于A2/O和氧化沟工艺,因而对其运行提出以下调控建议:1)DO浓度调控:依据现场测定和同步硝化-反硝化率测定实验,为获得良好的同步硝化-反硝化脱氮性能(较高同步硝化-反硝化率)及降低运行费用(减少供氧量),在CAST工艺好氧池内实行阶梯曝气,沿泥水混合物流动方向不断升增加曝气量,控制好氧池前段、中段和后段的DO质量浓度依次处于0.6~1.0、1.0~2.0和2~3 mg·L−1。2)污泥回流比调控:依据饱食-饥饿实验,为保证微生物将进水有机物尽可能多的以内碳源的形式储存,从而增强同步硝化-反硝化脱氮性能,将CAST工艺的回流比由当下的20%(饥饿时长为80 h)提升至45%(饥饿时长为36 h)。
表 4 CAST、A2/O和氧化沟工艺脱氮指标Table 4. Denitrification indicators of CAST, A2/O and oxidation ditch processesmg·L−1 工艺 进水COD 进水NH4+-N 进水TN 出水COD 出水NH4+-N 出水TN CAST 188±23 32.70±7.60 40.50±9.50 20±3 1.60±1.30 11.40±2.20 A2/O 249±38 36.12±5.38 44.36±4.87 18±3 0.32±0.26 8.30±1.26 氧化沟 643±136 35.12±4.89 47.50±5.33 21±3 1.07±0.57 8.47±1.01 | Show TableDownLoad:
CSV
3. 结论
1) CAST工艺好氧池内同步硝化-反硝化和沉淀过程中内源反硝化对总氮去除的贡献占据主导作用,贡献率分别为 (35.50±4.15)%和(62.86±4.13)%,而缺氧池反硝化脱氮贡献仅为(1. 64±0.05)%。
2) DO浓度对同步硝化-反硝化脱氮性能有重要影响,控制DO浓度在1.0~1.5 mg·L−1时,可获得最大同步硝化-反硝化脱氮效果。
3)饥饿时长对微生物吸收HAc有重要影响,当饥饿时长为36 h时,可获得最大HAc吸收量,达到每1 g VSS消耗0.173 g HAc,可为同步硝化-反硝化脱氮提供更多碳源。
-

图 1 两种多孔介质的粒径分布(a),孔径分布(b)和扫描电镜图(c:石英砂;d:双孔隙介质)
Figure 1. Particle size distribution (a), pore size distribution (b) and Scanning electron microscopy (c: quartz sand; d: dual-porosity media)
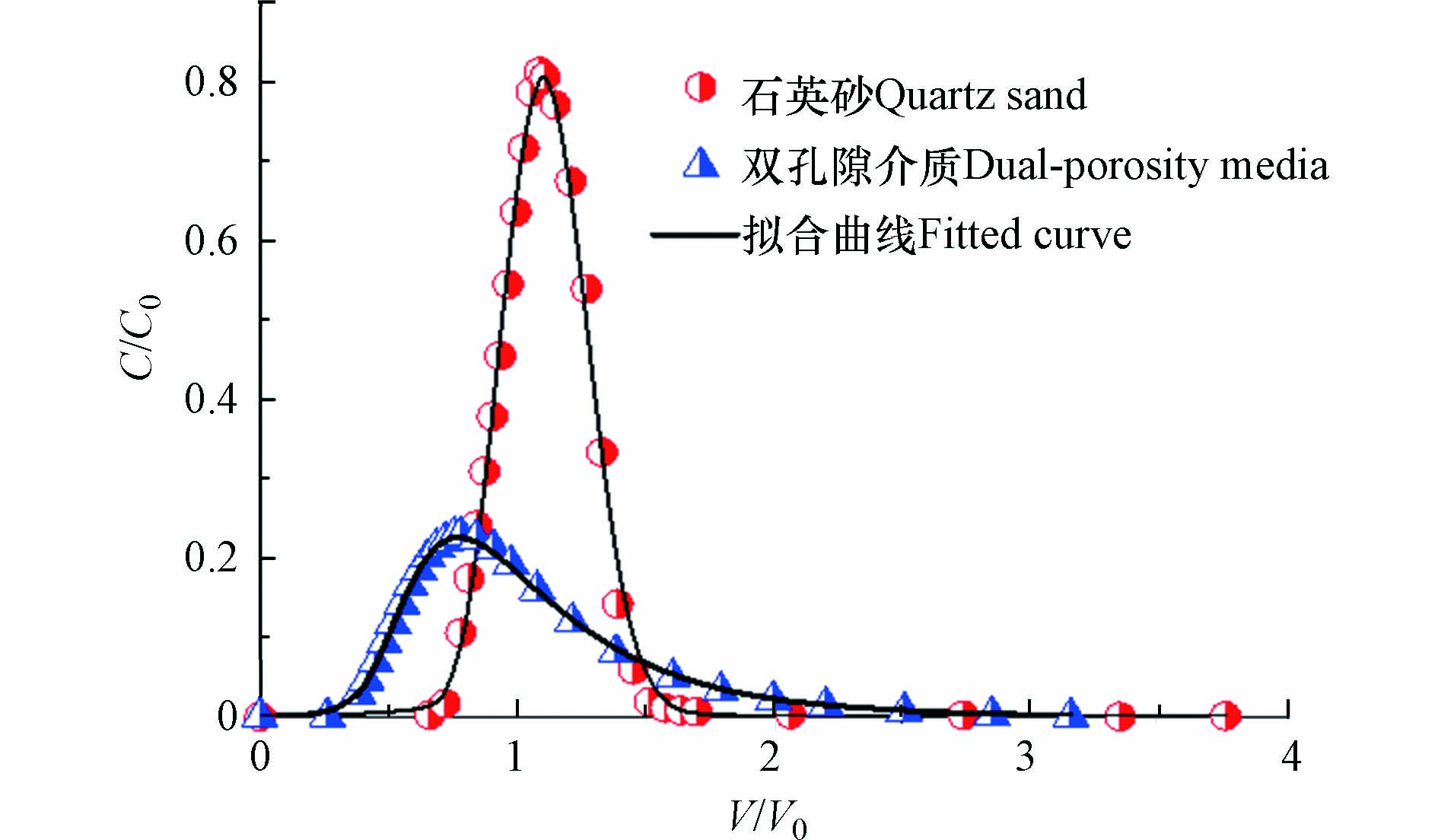
图 2 示踪剂在石英砂和双孔隙介质中穿透曲线和拟合曲线
Figure 2. Penetration curves and fitting curves of the tracer in quartz sand and dual-porosity media

图 3 不同pH下纳米二氧化钛在石英砂(a)和双孔隙介质(b)中的穿透曲线
Figure 3. Penetration curves of TiO2 nanoparticles in quartz sand (a) and dual-porosity media (b) at different pH
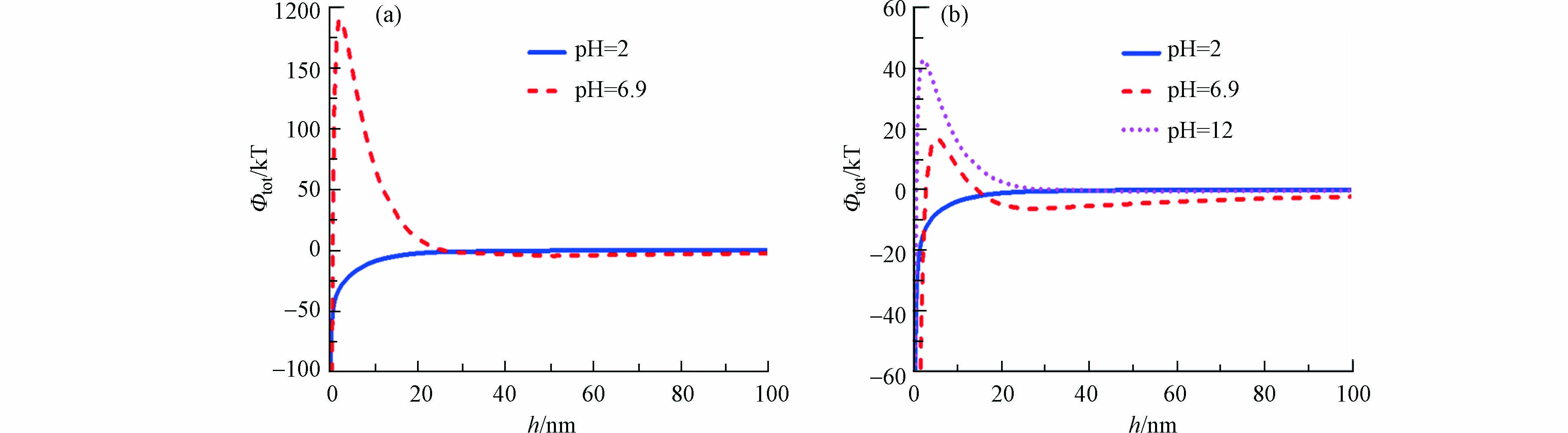
图 4 pH对纳米颗粒与石英砂(a)和双孔隙介质(b)间势能的影响
Figure 4. Influence of pH on potential energy between nanoparticles and quartz sand (a) and dual-porosity media (b)
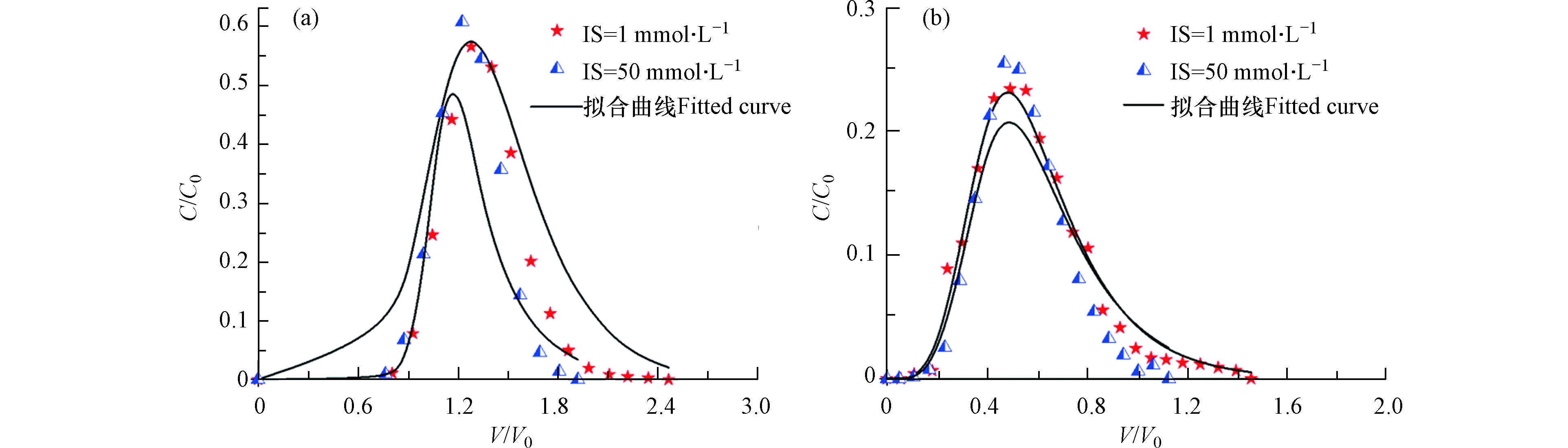
图 5 不同IS下纳米二氧化钛在石英砂(a)和双孔隙介质(b)中的穿透曲线
Figure 5. Penetration curves of TiO2 nanoparticles in quartz sand (a) and dual-porosity media (b) under different IS
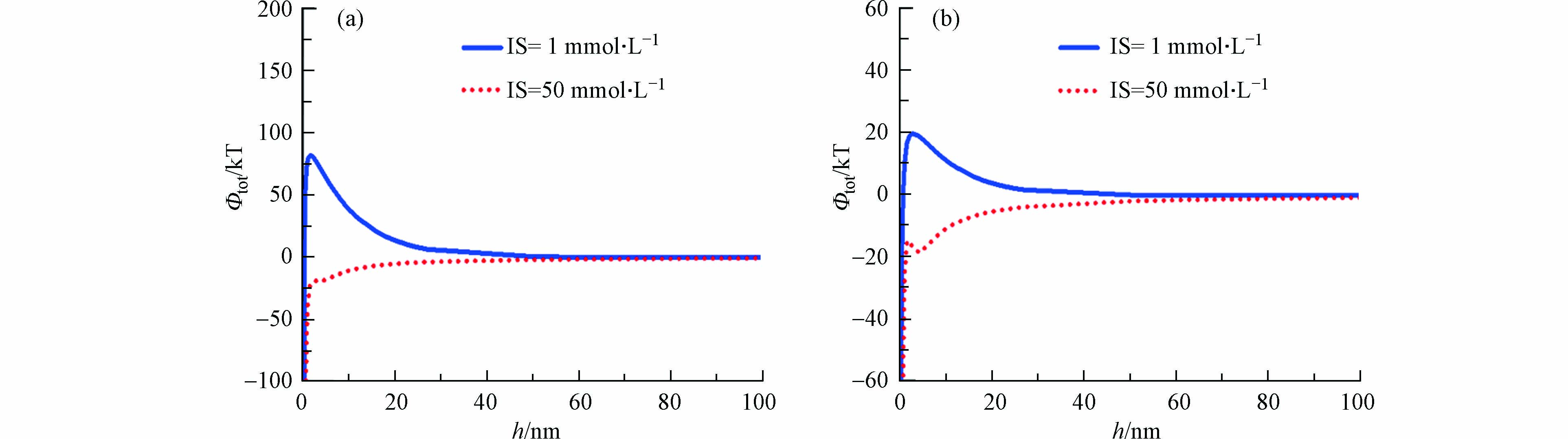
图 6 IS对纳米颗粒与石英砂(a)和双孔隙介质(b)间势能的影响
Figure 6. Effect of IS on potential energy between nanoparticles and quartz sand (a) and dual-porosity media (b)
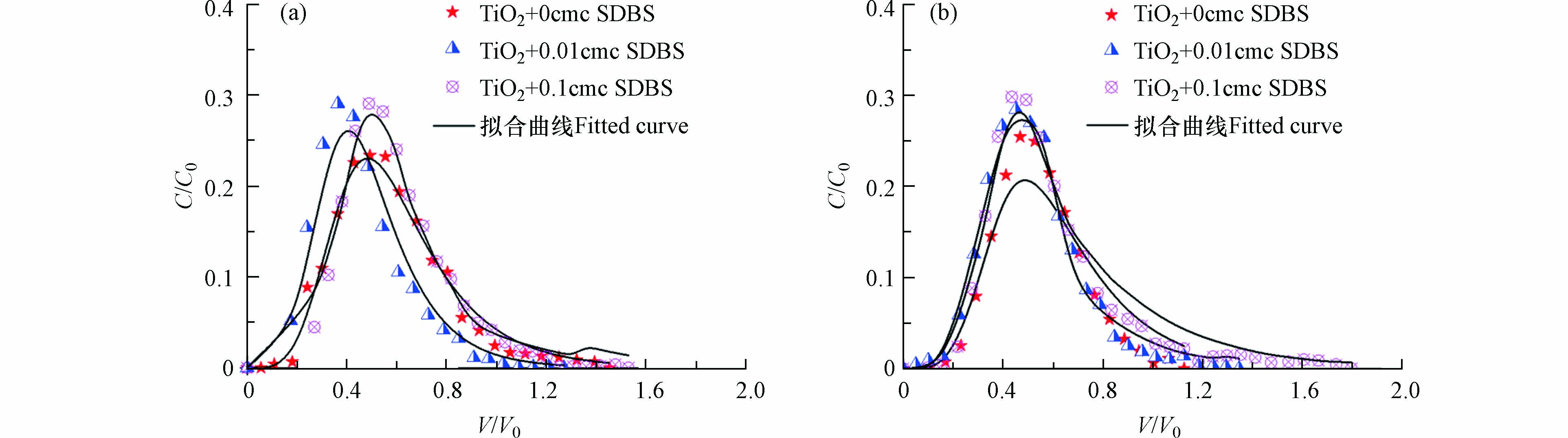
图 7 不同浓度SDBS下纳米二氧化钛在石英砂(a)和双孔隙介质(b)中的穿透曲线
Figure 7. Penetration curves of TiO2 nanoparticles in quartz sand (a) and dual-porosity media (b) at different concentrations of SDBS
表 1 示踪剂实验条件表
Table 1. Tracer experimental conditions
多孔介质Porous media 介质堆积密度/(g·cm−3)Bulk density 孔隙度Porosity 流速/(cm·min−1)Darcy velocity 注入时间/minInjection time 回收率/%Recovery 石英砂Quartz sand 1.42 0.47 0.37 4.28 99.4 双孔隙介质Dual-porosity media 0.44 0.83 0.36 4.32 99.9
下载: 导出CSV
表 2 示踪剂实验参数模拟结果表
Table 2. Simulation results of tracer experimental parameters
多孔介质Porous media 扩散系数/cmDiffusion coefficient 扩散速率/min−1Diffusion rate R2 石英砂Quartz sand 0.19 1.03×10−4 0.9961 双孔隙介质Dual-porosity media 1.95 3.4×10−1 0.9973
下载: 导出CSV
表 3 柱实验条件表
Table 3. Column experimental conditions
多孔介质Porous media 环境因素Environmental conditions 装柱密度/(g·cm−3)Bulk density 孔隙度/%Porosity 达西流速/(cm·min−1 )Darcy velocity 注入时间/minInjection time 回收率/%Recovery 滞留率/% Detention 石英砂Quartz sand pH 2 1.33 0.5 0.37 4.28 1.7 98.3 6.9 1.35 0.49 0.35 4.4 30 70 IS 1 1.42 0.47 0.38 4.12 67.6 32.4 50 1.49 0.44 0.36 4.33 64.2 35.8 SDBS 0 1.35 0.49 0.35 4.4 30 70 0.01 1.4 0.47 0.43 3.97 74.2 25.8 0.1 1.41 0.47 0.39 4.32 97.5 2.5 双孔隙介质Dual-porosity media pH 2 0.42 0.83 0.32 4.82 2.6 97.4 6.9 0.43 0.83 0.36 4.32 50.8 49.2 IS 1 0.45 0.82 0.38 4.12 47.1 52.9 50 0.46 0.82 0.35 4.55 42.4 57.6 SDBS 0 0.43 0.83 0.36 4.32 50.8 49.2 0.01 0.44 0.82 0.36 4.37 67 33 0.1 0.45 0.82 0.35 4.48 78.1 21.9
下载: 导出CSV
表 4 柱实验模拟结果参数表
Table 4. Parameters of column experiment simulation results
多孔介质 Porous media 环境因素 Environmental conditions 模拟结果Simulation results katt kdet R2 石英砂Quartz sand pH 2 1.31×102 1.23×10−2 0.7872 6.9 1.28 3.95×10−2 0.7493 IS 1 0.316 1.34 0.9457 50 8.73 1.42 0.8506 SDBS 0 1.28 3.95×10−2 0.7493 0.01 0.228 0.194 0.9685 石英砂Quartz sand SDBS 0.1 0.122 7.40×10−3 0.9939 双孔隙介质Dual-porosity media pH 2 19.4 3.50×10−4 0.9515 6.9 0.753 2.36×10−6 0.9467 IS 1 0.999 1.41×10−6 0.9828 50 1.77 3.92×10−5 0.9609 SDBS 0 0.753 2.36×10−6 0.9467 0.01 0.319 5.37×10−5 0.9647 0.1 0.261 1.39×10−5 0.965
下载: 导出CSV
-
[1] 沈钟, 赵振国, 康万利. 胶体与表面化学[M]. 4版. 北京: 化学工业出版社, 2012: 329. SHEN Z, ZHAO Z G, KANG W L. Colloid and surface chemistry[M]. Beijing: Chemical Industry Press, 2012: 329(in Chinese).
[2] 于荣丽, 孙铁珩, 胡晓钧. 纳米材料的生物负效应研究进展 [J]. 安全与环境学报, 2009, 9(2): 121-124. YU R L, SUN T H, HU X J. Research progress on the negative effects of nanomaterials on biological systems [J]. Journal of Safety and Environment, 2009, 9(2): 121-124(in Chinese).
[3] KHAN S, NAUSHAD M, AL-GHEETHI A, et al. Engineered nanoparticles for removal of pollutants from wastewater: Current status and future prospects of nanotechnology for remediation strategies [J]. Journal of Environmental Chemical Engineering, 2021, 9(5): 106160. doi: 10.1016/j.jece.2021.106160 [4] 李冉, 吴霜, 付鹏辉. 浅谈纳米技术在生活生产中的应用 [J]. 信息记录材料, 2019, 20(5): 14-15. LI R, WU S, FU P H. Application of nanotechnology in life production [J]. Information Recording Materials, 2019, 20(5): 14-15(in Chinese).
[5] 冯辰昀, 李旭东, 郑妤婕, 等. 纳米材料的毒理学研究进展[J/OL]. 中国科学: 化学: 2021, 51,doi: 10.1360/SSC-2021-0138. FENG C Y, LI X D, ZHENG Y J, et al. Recent progress in nanotoxicology of nanomaterials[J/OL]. Sci Sin Chim, 2021, 51,doi: 10.1360/SSC-2021-0138 (in Chinese).
[6] CHEN X B, MAO S S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications [J]. Chemical Reviews, 2007, 107(7): 2891-2959. doi: 10.1021/cr0500535 [7] 刁润丽, 赵世伟. 纳米二氧化钛的应用研究进展 [J]. 山西化工, 2021, 41(3): 25-26,31. DIAO R L, ZHAO S W. Application research progress of Nano-titanium dioxide [J]. Shanxi Chemical Industry, 2021, 41(3): 25-26,31(in Chinese).
[8] 张海丰, 张鹏宇, 赵贵龙, 等. 纳米二氧化钛的制备及其应用研究进展 [J]. 东北电力大学学报, 2014, 34(2): 52-56. doi: 10.3969/j.issn.1005-2992.2014.02.011 ZHANG H F, ZHANG P Y, ZHAO G L, et al. Advances on preparation, modification and application of nanosized titanium dioxide [J]. Journal of Northeast Dianli University, 2014, 34(2): 52-56(in Chinese). doi: 10.3969/j.issn.1005-2992.2014.02.011
[9] 刁润丽. 纳米二氧化钛的光催化性及其应用研究进展 [J]. 佛山陶瓷, 2021, 31(6): 5-7. DIAO R L. Research progress on photocatalytic activity and application of nano-titanium dioxide [J]. Foshan Ceramics, 2021, 31(6): 5-7(in Chinese).
[10] 李润虎. 纳米技术的风险问题及对策研究 [J]. 工程研究-跨学科视野中的工程, 2020, 12(4): 380-387. LI R H. Research on risks and countermeasures of nanotechnology [J]. Journal of Engineering Studies, 2020, 12(4): 380-387(in Chinese).
[11] 刘诚, 孙强, 张刚, 等. 磷酸盐对纳米TiO2在土壤中迁移的影响 [J]. 苏州科技大学学报(自然科学版), 2018, 35(3): 44-50. LIU C, SUN Q, ZHANG G, et al. Influence of phosphate on the transport of nTiO2 in soil column [J]. Journal of Suzhou University of Science and Technology (Natural Science Edition), 2018, 35(3): 44-50(in Chinese).
[12] 韩鹏, 王雪婷. 不同环境因子对纳米二氧化钛在饱和多孔介质中迁移持留行为的影响 [J]. 应用基础与工程科学学报, 2013, 21(3): 512-521. doi: 10.3969/j.issn.1005-0930.2013.03.013 HAN P, WANG X T. Transport and retention behaviors of TiO2 nanoparticles in saturated porous media: Effects of ionic strength, iron(hydr)oxide and humic acid [J]. Journal of Basic Science and Engineering, 2013, 21(3): 512-521(in Chinese). doi: 10.3969/j.issn.1005-0930.2013.03.013
[13] 彭爱夏, 占敬敬, 吴明火. 纳米粒子在多孔介质中迁移模型的优化 [J]. 化工学报, 2021, 72(10): 5114-5122. PENG A X, ZHAN J J, WU M H. Optimization of nanoparticles transport model in porous media [J]. CIESC Journal, 2021, 72(10): 5114-5122(in Chinese).
[14] 方婧, 余博阳. 3种金属氧化物纳米材料在不同土壤中运移行为研究 [J]. 环境科学, 2013, 34(10): 4050-4057. FANG J, YU B Y. Transport behaviors of metal oxide nanomaterials in various soils [J]. Environmental Science, 2013, 34(10): 4050-4057(in Chinese).
[15] 张柯柯. 纳米TiO2在土壤中迁移及其与Pb共迁移机制研究[D]. 杭州: 浙江工商大学, 2015: 72. ZHANG K K. Transport of nano-TiO2 in soil and the CO-transport mechanisms of Pb with nano-TiO2[D]. Hangzhou: Zhejiang Gongshang University, 2015: 72(in Chinese).
[16] FANG J, SHAN X Q, WEN B, et al. Stability of titania nanoparticles in soil suspensions and transport in saturated homogeneous soil columns [J]. Environmental Pollution, 2009, 157(4): 1101-1109. doi: 10.1016/j.envpol.2008.11.006 [17] DUNPHY GUZMAN K A, FINNEGAN M P, BANFIELD J F. Influence of surface potential on aggregation and transport of titania nanoparticles [J]. Environmental Science & Technology, 2006, 40(24): 7688-7693. [18] CAI L, PENG S N, WU D, et al. Effect of different-sized colloids on the transport and deposition of titanium dioxide nanoparticles in quartz sand [J]. Environmental Pollution, 2016, 208: 637-644. doi: 10.1016/j.envpol.2015.10.040 [19] 罗小廷, 吴丹, 梁嘉良, 等. 典型阴离子对纳米二氧化钛在载铁石英砂多孔介质中迁移行为的影响 [J]. 北京大学学报(自然科学版), 2017, 53(4): 749-757. LUO X T, WU D, LIANG J L, et al. Influence of typical anions on the transport of titanium dioxide nanoparticles in iron oxide-coated porous media [J]. Acta Scientiarum Naturalium Universitatis Pekinensis, 2017, 53(4): 749-757(in Chinese).
[20] 吴其圣, 杨琛, 胡秀敏, 等. 环境因素对纳米二氧化钛颗粒在水体中沉降性能的影响 [J]. 环境科学学报, 2012, 32(7): 1596-1603. WU Q S, YANG C, HU X M, et al. Influences of environmental factors on aggregation of titanium dioxide nanoparticles [J]. Acta Scientiae Circumstantiae, 2012, 32(7): 1596-1603(in Chinese).
[21] 张瑞昌, 章海波, 涂晨, 等. 腐殖酸作用下酸性多孔介质中纳米TiO2的迁移与滞留机制 [J]. 中国环境科学, 2018, 38(9): 3542-3551. doi: 10.3969/j.issn.1000-6923.2018.09.041 ZHANG R C, ZHANG H B, TU C, et al. Mechanisms of mobility and retention of nano-TiO2 in acidic porous media in the presence of humus acids [J]. China Environmental Science, 2018, 38(9): 3542-3551(in Chinese). doi: 10.3969/j.issn.1000-6923.2018.09.041
[22] SUN P D, ZHANG K K, FANG J, et al. Transport of TiO2 nanoparticles in soil in the presence of surfactants [J]. Science of the Total Environment, 2015, 527/528: 420-428. doi: 10.1016/j.scitotenv.2015.05.031 [23] GODINEZ I G, DARNAULT C J G. Aggregation and transport of nano-TiO2 in saturated porous media: Effects of pH, surfactants and flow velocity [J]. Water Research, 2011, 45(2): 839-851. doi: 10.1016/j.watres.2010.09.013 [24] 张柯柯, 汪敏浩. 离子强度和富里酸对纳米TiO2在土壤中迁移的影响 [J]. 广州化工, 2014, 42(23): 110-111. ZHANG K K, WANG M H. Effect of ionic strength and fulvic acid on the transport of TiO2 nanoparticles in soil [J]. Guangzhou Chemical Industry, 2014, 42(23): 110-111(in Chinese).
[25] 杜宜春, 梁志卿. 多孔介质中纳米颗粒及其携带重金属污染物迁移研究进展 [J]. 河南科技, 2019(32): 133-136. doi: 10.3969/j.issn.1003-5168.2019.32.047 DU Y C, LIANG Z Q. Review of migration of nanoparticle and its facilitated heavy metal contaminants in porous media [J]. Henan Science and Technology, 2019(32): 133-136(in Chinese). doi: 10.3969/j.issn.1003-5168.2019.32.047
[26] PIRSON S J. Performance of fractured oil reservoirs [J]. AAPG Bulletin, 1953, 377(2): 232-244. doi: 10.1306/5ceadc4a-16bb-11d7-8645000102c1865d [27] PHILIP J R. The theory of absorption in aggregated media [J]. Soil Research, 1968, 6(1): 1. doi: 10.1071/SR9680001 [28] FANG J, XU M J, WANG D J, et al. Modeling the transport of TiO2 nanoparticle aggregates in saturated and unsaturated granular media: Effects of ionic strength and pH [J]. Water Research, 2013, 47(3): 1399-1408. doi: 10.1016/j.watres.2012.12.005 [29] PACKMAN A I, BROOKS N H, MORGAN J J. A physicochemical model for colloid exchange between a stream and a sand streambed with bed forms [J]. Water Resources Research, 2000, 36(8): 2351-2361. doi: 10.1029/2000WR900059 [30] WANG D J, CHU L Y, PARADELO M, et al. Transport behavior of humic acid-modified nano-hydroxyapatite in saturated packed column: Effects of Cu, ionic strength, and ionic composition [J]. Journal of Colloid and Interface Science, 2011, 360(2): 398-407. doi: 10.1016/j.jcis.2011.04.064 [31] 方华祥. 溶液性质对金属纳米颗粒在多孔介质中迁移持留行为的影响机制研究[D]. 杭州: 浙江大学, 2017: 87. FANG H X. The influence mechanisms of solutions properties on the transport and retention behaviors of metal-based nanoparticles in porous media[D]. Hangzhou: Zhejiang University, 2017: 87(in Chinese).
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2917
- HTML全文浏览数: 2917
- PDF下载数: 100
- 施引文献: 0
