












-
石油化工企业在炼油和化工生产过程中,会排放出大量含有高浓度硫化物(主要为Na2S,质量百分比0.5%~5%[1])、高COD的强碱性废液。此类废碱液具有强烈的恶臭[2]、较大的毒性且不易被处理,同时会造成严重的二次污染[3]。因此,必须将此类废水中有毒的低价硫化物转化成无毒、无二次污染的高价态硫酸盐后才能进入后续的处理过程。
目前国内外含硫废碱液处理主要有中和法、综合利用法、氧化法、沉淀法和生物强化法[4]。中和法即使用酸调整溶液pH至中性或酸性,使硫化氢释放,而后将废气燃烧或用作他用,但存在废气处理和高含盐量的酸性废水处理问题。综合利用即结晶回收硫化钠,再将硫化钠综合利用,但废水中的有机物和其他物质会影响硫化钠的结晶纯度。氧化法即使用空气、氧气、过氧化氢等作为氧化剂,在催化剂或无催化剂作用下将低价硫氧化。沉淀法即采用过渡金属盐与硫化钠生成沉淀,而后将硫化物沉淀分离,沉淀法同样会受废水中其他无机盐的影响使沉淀的盐不纯。生物法需要大量水(6倍以上)稀释,只适用于低浓度废碱液处理,故存在局限性。氧化法相比其他方法适用范围广、氧化效率高、无二次污染。
氧化法处理废碱液中最常用的是湿式空气氧化法。湿式空气氧化工艺是在一定温度和压力条件下,以空气中的氧作为氧化剂对污水中的污染物进行水相氧化的处理技术[5]。根据处理温度和是否加入催化剂分为低温湿式氧化、中温湿式氧化、高温湿式氧化、催化湿式氧化4类,具体如表1所示。
上述工艺均需要加压,并且反应温度超过了100 ℃,在工程中,加温加压将会提高对设备的压力等级要求、降低操作的安全性、增加能源的消耗。为了降低能耗和设备的压力等级,近期,有催化空气氧化工艺在小于100 ℃下处理废碱液的报道,详见表2。
但上述研究的重点均在废碱液中硫化钠的初步氧化去除上,并没有对废碱液中本身存在以及初步氧化后生成的高浓度硫代硫酸钠的去除进行讨论。在实际废碱液处理过程中,硫代硫酸钠的存在很大程度影响了后续盐分离的处理效果,从而影响后续生化。因此,针对含硫废碱液中硫代硫酸钠氧化困难的实际问题,本研究的重点为开发一种小于100 ℃常压催化湿式空气氧化硫代硫酸钠的脱硫工艺,通过高效脱硫催化剂提高低价硫的氧化效率和氧化深度,实现高效、经济的氧化硫代硫酸钠。
TiO2作为载体被广泛应用于催化湿式氧化及其他工业催化过程,具有高强度及耐酸碱等特点;Mn具有+2、+3、+4、+7等多种价态,可以形成不同性质的稳定氧化物,在催化反应中能够通过不同价态Mn的催化循环表现出优异的氧化还原能力[9-10],锰基催化剂常被用作氧化还原反应的催化剂,如NH3-SCR(selctive catalytic reduction)[11-12]。
基于上述研究结果,本研究应用MnOx@TiO2作为催化剂,实现常压条件下对硫代硫酸钠的催化氧化,使硫代硫酸钠在较短时间内氧化生成易于后续盐-水分离的亚硫酸钠及硫酸钠。同时考察气量、反应温度、反应时间等反应条件对硫代硫酸钠转化率的影响。
-
测试载体TiO2的吸水率,称取一定量硫酸锰(AR,上海吉至生化科技有限公司)溶解于一定量去离子水中,等体积浸渍一晚,得到中间体A。将A放入120 ℃烘箱中完全烘干,所得样品放入马弗炉内在450 ℃空气气氛中焙烧4 h,最终得到Mn质量分数为2%的MnOx@TiO2。
-
载体及使用前后的催化剂研磨成粉末作为待测样品。采用荷兰PANAlytical公司生产的Zetium型X射线荧光光谱仪(XRF)进行元素分析。X-射线衍射测试采用荷兰PANAlytical公司生产的Empyrean-100型X射线粉末衍射仪(XRD),陶瓷X光管Cu辐射源(Kα2/Kα1=0.5),扫描角度2θ为10°~90°,扫描时间为10 min,扫描步长为0.026°。采用美国ThermoFisher公司生产的Thermo ScientificTM K-AlphaTM XPS能谱仪进行X-射线光电子能谱(XPS)表征。采用Aventage软件对曲线进行分峰拟合,背景扣除方式为Shirley背景。采用NH3-TPD研究催化剂表面酸性活性位量,使用浙江泛泰仪器有限公司FINESORB-3010型装置,取100 mg左右样品粉末,150 ℃下Ar气氛预处理2 h,使用NH3体积分数为1%的NH3/Ar(20 mL·min−1)混合气常温吸附2 h至饱和,于Ar气氛下以10 ℃·min−1升温至500 ℃脱附,记录NH3-TPD图谱。采用O2-TPD研究催化剂表面对氧气的吸附性能,使用浙江泛泰FINESORB-3010型装置,取100 mg左右样品粉末,150 ℃下Ar气氛预处理2 h,使用O2体积分数为10%的O2/Ar(20 mL·min−1)混合气在75 ℃进行2 h吸附,于Ar气氛下10 ℃·min−1升温至800 ℃脱附,记录O2-TPD图谱。采用H2-TPR(浙江泛泰FINESORB-3010)研究催化剂氧化还原性能,取50 mg左右样品粉末,常温Ar气氛下吹扫100 min,使用H2体积分数为10%的H2/Ar (20 mL·min−1)混合气以10 ℃·min−1升温至800 ℃进行还原,记录H2-TPR图谱。比表面积的测试使用浙江泛泰FINESORB-3010型装置,采用单点法进行测试,取约500 mg样品粉末,通过N2低温吸附常温脱附和BET方程计算得到比表面积。
-
常压催化湿式氧化硫代硫酸钠的活性测试在鼓泡床反应器中进行,反应装置如图1所示。该装置主要分为3部分:气路系统、反应系统及冷却系统(防止溶液挥发)。反应器中装入50 mL催化剂和35 mL模型水,使用单独的硫代硫酸钠溶液作为模型底物,模型水中
S2O32− 质量浓度为10 000 mg·L−1,待空气流量与温度稳定后开始反应计为反应零点。每次反应结束后,出水使用0.45 μm滤头进行过滤,采用间接碘量法[13]测定每次出水水样中
S2O32− 和SO32− 质量浓度。使用体积比1∶1甲醛溶液屏蔽水样中SO32− ,在酸性条件下使用稀释50倍的碘标准溶液(C(1/2I2)=0.1 mol·L−1)滴定一定量稀释水样由无色变为浅蓝色计为反应终点,计算出S2O32− 的质量浓度。在一定量稀释水样中加入过量碘标准溶液,在酸性条件下使用硫代硫酸钠标准溶液(c(Na2S2O3)=0.1 mol·L−1)滴定剩余碘至溶液无色计为反应终点,计算得S2O32− 和SO32− 质量浓度之和,差值为SO32− 质量浓度。滴定过程发生的主要化学反应如式(1)~式(2)所示。 -
本研究所有反应均使用同一批次催化剂,每次使用后清洗干燥进行再生。催化剂使用过程中Mn的溶出量采用美国PerkinElmer公司生产的Optima 7300DV型电感耦合等离子体发射光谱仪(ICP)进行分析。
-
1) 空气气量的影响。在反应温度65 ℃、反应时间50 h的条件下,在5~50 mL·min−1内不同气量条件下硫代硫酸钠转化率(以
S2O32− 计)见图2。由图2可见,通入空气中氧气含量过量(经计算至少过量7.8倍),硫代硫酸钠的转化率可达96%,但不随气量的增加而升高,说明通过加大气量提高的传质水平已达上限。同时,MnOx@TiO2催化剂对亚硫酸钠的氧化也表现出了较好的催化效果,在通入气量为50 mL·min−1时氧化产物中的SO32− 质量浓度均在500 mg·L−1以下,说明大部分低价硫转化生成了硫酸钠。可见,当空气量在5~50 mL·min−1内,即空气过量的条件下气量对MnOx@TiO2活性的影响不大,通入气量为50 mL·min−1时最有利于硫代硫酸钠的深度氧化。2) 反应时间的影响。图3为反应温度65 ℃、空气气量50 mL·min−1的条件下,在25~75 h内反应时间对硫代硫酸钠转化率(以
S2O32− 计)的影响。由图3可知,反应时间对硫代硫酸钠的转化有较大影响,硫代硫酸钠的转化率随反应时间的增加而升高,反应50 h后硫代硫酸钠的转化率即可达到96%,同时出水中SO32− 的质量浓度为518 mg·L−1。当反应时间延长至75 h时,硫代硫酸钠基本完全转化,出水中还存在少量的SO32− ,说明反应时间由50 h延长到75 h时会发生硫代硫酸钠氧化和亚硫酸钠氧化的竞争反应。由此研究结果可知,反应时间对MnOx@TiO2的活性影响较大,在此系统中最佳反应时间为50 h。3) 反应温度的影响。在反应时间为25 h、空气气量为50 mL·min−1的条件下,反应温度对硫代硫酸钠(以
S2O32− 计)转化率的影响结果见图4。由图4可见,硫代硫酸钠的催化氧化反应对温度敏感,在65~85 ℃内转化率首先随着温度的升高而升高,在75 ℃转化率达到最高(96%),当温度升高至85 ℃时转化率反而下降。温度升高有利于提高氧化效率,但是温度升高也会导致氧气溶解度降低,在温度和溶解氧的共同作用下,反应温度为75 ℃时氧化效率最高。 -
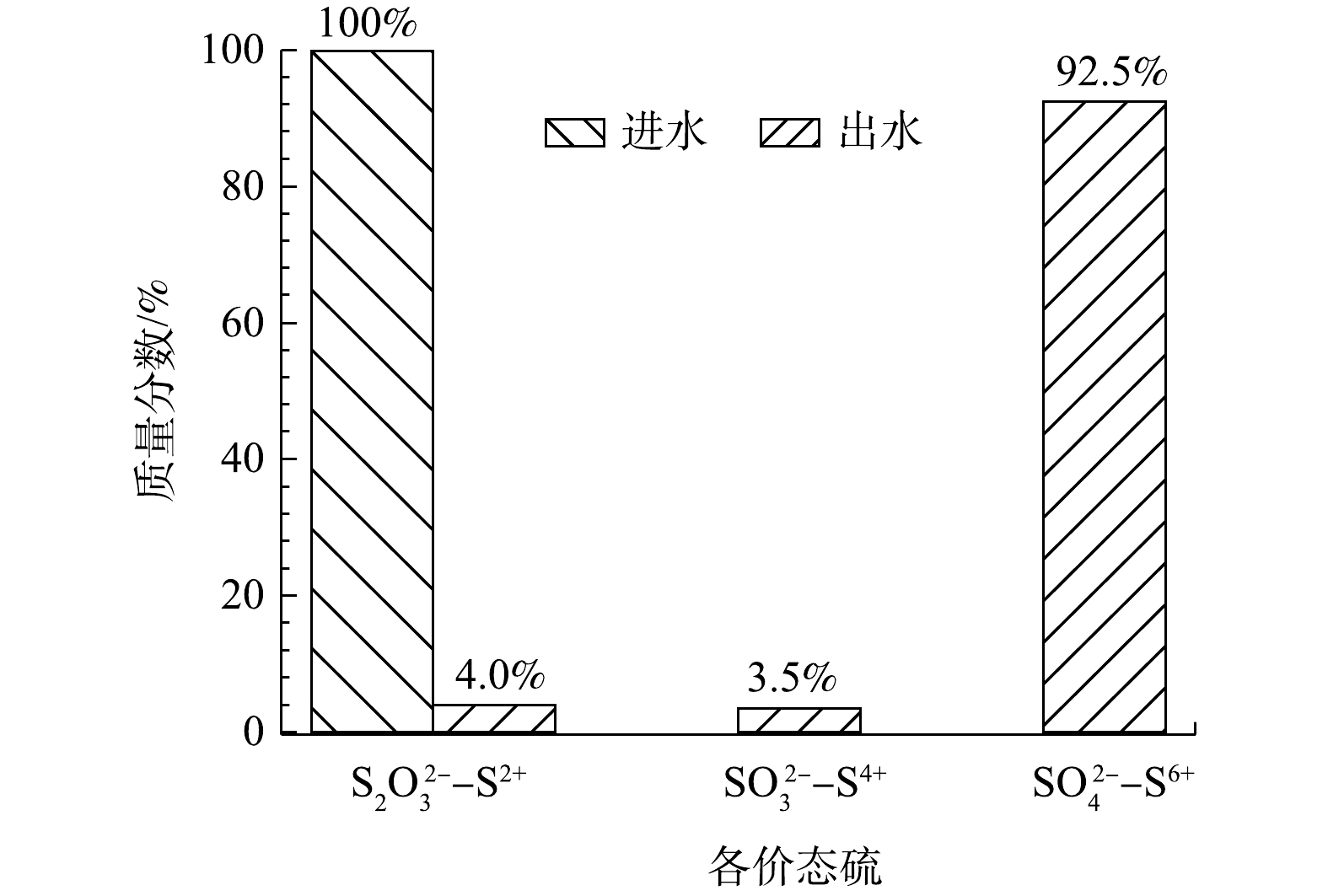
对最佳反应条件下(反应温度75 ℃、反应时间25 h、空气气量50 mL·min−1)不同因素对硫代硫酸钠转化的作用进行了讨论。通过空白反应(玻璃珠替代催化剂)考察空气氧化作用,此时硫代硫酸钠转化率(以
S2O32− 计)仅为4%;加入载体对硫代硫酸钠转化率的提高了1%;使用ICP分析MnOx@TiO2在最佳反应条件下Mn的溶出量为190.5 mg·L−1,在Mn2+(使用MnSO4为前驱体)质量浓度为190.5 mg·L−1的均相催化系统中,硫代硫酸钠的转化未见提高。硫代硫酸钠在最佳反应条件下转化率为96%。其中,空气氧化对硫代硫酸钠转化的贡献率为4.17%;吸附剂载体对硫代硫酸钠的吸附率为1.04%;而溶出Mn2+对硫代硫酸钠的转化没有影响;多相催化湿式空气氧化对硫代硫酸钠转化的贡献比重占94.79%。当硫代硫酸钠模型废水
S2O32− 质量浓度为10 000 mg·L−1,硫代硫酸钠转化率在最佳条件下为96%。由图5可知,在最优条件下92.5%的低价态硫均被氧化为最高价态的硫酸根。 -
1) XRF表征。由表3的XRF元素分析结果可见,TiO2载体本身不含Mn,载体在经过浸泡-烘干-煅烧后负载Mn,其质量分数1.9%,基本符合理论值2%。催化剂在经过共300 h的使用后,Mn质量分数仅剩0.2%,较使用前流失1.7%,但Mn的流失并未影响催化剂对硫代硫酸钠催化氧化的活性,同图5中硫代硫酸钠转化率分析结果一致,即均相Mn2+不起作用,主要起催化作用的是多相催化剂MnOx@TiO2。由此可见,Mn质量分数为0.2%的MnOx@TiO2催化剂活性就足以在此半连续反应系统中处理
S2O32− 质量浓度高达10 000 mg·L−1的硫代硫酸钠废液。使用前MnOx@TiO2主要杂质元素为S,其质量分数为0.7%,使用后主要杂质为Na,其质量分数为0.5%。但杂质元素未对催化剂活性产生影响。2) XRD表征。TiO2及MnOx@TiO2使用前后的XRD衍射图谱如图6所示。由图6可以看出,载体及使用前后的催化剂均有明显的锐钛矿型TiO2的特征峰。MnOx@TiO2使用前后的衍射图谱基本重叠,可以说明MnOx@TiO2在使用过程中未发生晶体结构上的改变,表现出良好的催化剂稳定性。使用前后的MnOx@TiO2衍射图谱中并未出现锰氧化物的特征峰,这可能是因为MnOx质量分数较低或MnOx以无固定形态在载体表面均匀分散。为进一步确定催化剂表面Mn价态和催化剂表面组成,对样品进行XPS表征。
3) XPS分析。催化剂表面元素种类、价态及氧物种是影响催化剂活性的重要因素。图7为MnOx@TiO2使用前后的XPS谱图,图7(a)为MnOx@TiO2使用前后的全谱图。可以看出,使用前的MnOx@TiO2表面主要出现Ti2p、O1s、Mn2p和S2p峰,使用后S2P峰基本消失,出现Na1s峰,其结果与XPF结果(表3)相符。因Mn负载量较小,扫描出的Mn3s谱图信噪比较差,因此,不在此列出。
为确定催化剂表面Mn价态,对Mn2p谱图进行分峰拟合(图7(b))。Mn2p图谱因能级分裂产生2个分裂峰:Mn2p3/2和Mn2p1/2。使用前后MnOx@TiO2的Mn2p3/2可拟合出3个峰,说明催化剂表面存在多种价态的Mn,分别对应Mn2+(640.4~641.0 eV)、Mn3+(642.4~642.7 eV)以及Mn4+(644.1~644.6 eV),在646.6 eV处出现伴峰。Mn3+和Mn4+的结合能明显高于理论值(641.7±0.2) eV和(643.0±0.2) eV[14-15],这种结合能的偏移是由MnOx与TiO2载体的相互作用导致的。为了对催化剂表面Mn价态和组成的比例进行分析,对拟合峰进行积分,结果汇总于表4。催化剂使用前后Mn4+原子百分比在8%~9%保持不变,Mn2+原子百分比由46%升到54%,而Mn3+原子百分比由23%降到16%。以上结果说明,在此催化系统中催化剂活性主要依赖于表面Mn2+与Mn3+之间的催化循环,催化剂中的Mn3+在反应中被还原,起到了氧化位点作用。
图7(c)为MnOx@TiO2使用前后O1s的XPS谱图。O1s的峰可分为2个峰[16],在低结合能处的峰(529.4~529.7 eV)可归属于表面晶格氧Oα,在高结合能处的峰则归属于化学吸附氧Oβ(531.3~531.5 eV),主要以氧缺陷的形式存在(
O22− 和O−[17])。结合表4可见,MnOx@TiO2在使用后,Oβ峰占比降低,Oα峰占比升高,说明催化剂表面化学吸附氧物种在硫代硫酸钠的氧化过程中可能转化为晶格氧。图7(d)为MnOx@TiO2使用前后S2p的XPS谱图。S2p的峰可分为4个峰[18],结合能由小到大分别为S4+2p3/2、S6+2p3/2、S4+2p1/2、S6+2p1/2。使用前MnOx@TiO2表面主要检测到S6+2p3/2峰及S6+2p1/2峰,说明S在使用前的MnOx@TiO2表面以硫氧化物的形态存在。反应后的MnOx@TiO2表面几乎检测不到S2p峰,可以推断,经MnSO4溶液浸泡、450 ℃空气气氛焙烧后的MnOx@TiO2表面仍有未完全转化为MnOx的MnSO4存在,而这些MnSO4随反应过程中的反复冲洗洗涤脱落,证明起到催化活性的不是MnSO4而是MnOx,这与硫代硫酸钠转化率分析结果一致。
4) O2-/NH3-TPD分析。催化剂表面氧气吸附性能和表面酸性是影响催化剂活性的2个重要因素。采用O2-TPD研究MnOx@TiO2表面对氧气的吸附性能。一般来说,在O2-TPD测试中,低于400 ℃的脱附峰可归属为表面吸附氧(Oβ)的脱附峰;而高于400 ℃的脱附峰可归属于晶格氧(Oα)的脱附峰[19]。由图8(a)可见,TiO2的O2-TPD图谱的脱附峰信号非常弱,这说明TiO2表面吸附氧物种量非常少可忽略。通过负载MnOx,低温区出现O2脱附峰,MnOx@TiO2表面可以吸附氧物种。使用后MnOx@TiO2具有更高的氧吸附量,说明MnOx@TiO2在催化硫代硫酸钠的氧化过程中形成了更多的氧缺陷更容易吸附氧气,所以反应后催化剂仍能保持较高的催化活性。
催化剂表面酸性通过NH3-TPD的检测进行分析。由图8(b)可见,载体本身并没有出现明显的NH3脱附峰,说明载体表面本身酸性位点量非常少。使用前的MnOx@TiO2在100~250 ℃以及使用后的MnOx@TiO2在100~250 ℃和250~500 ℃有明显的NH3脱附峰。100~250 ℃的低温NH3脱附峰属于弱酸性位点的NH3脱附峰,250~500 ℃的高温NH3脱附峰则属于中强和强酸性位点的NH3脱附峰[14, 17]。新鲜制备的催化剂出现较强的NH3低温脱附峰,说明在负载MnOx后催化剂表面主要形成弱酸性位点,而这些酸性位点主要以Brønsted酸的形式存在[20]。使用后的催化剂出现了更强的低温NH3脱附峰,并且出现了多个较强的高温NH3脱附峰,说明MnOx@TiO2在使用后酸性位点增加,结合O2-TPD的结果,使用后MnOx@TiO2表面酸性位点的增加可能与反应过程中MnOx@TiO2表面形成的氧缺陷有关[21]。
5) H2-TPR分析。图9为TiO2和MnOx@TiO2使用前后在150~750 ℃的H2-TPR图谱。TiO2没有耗氢峰,说明TiO2在这个范围内没有被H2还原。使用前后MnOx@TiO2在489~505 ℃出现的低温还原峰可归属为表面Mn4+氧化物还原为Mn3+氧化物的还原[22],结合XPS结果可见,MnOx@TiO2在使用前后Mn4+氧化物含量较少且保持不变。564~591 ℃内出现的还原峰可归属于Mn3+氧化物到Mn2+氧化物的的还原,此还原峰的显著降低可以再一次证明Mn3+氧化物在MnOx@TiO2催化氧化硫代硫酸钠过程中起到了重要的作用,上述结果同XPS结果一致。而600 ℃以上高温区出现的2个峰则可归属为表面氧及晶格氧的还原[23],使用后的MnOx@TiO2在此范围内只出现非常微弱的还原峰信号,说明表面活性氧及晶格氧在反应过程中的脱落,这与O2-/NH3-TPD的结果相符。
6) BET表征。BET测试结果表明,TiO2比表面积只有8 m2·g−1,通过浸渍-烘干-煅烧后MnOx@TiO2比表面积较载体增大至55 m2·g−1,通过负载煅烧Mn盐和TiO2载体相互作用改变了材料的孔结构,比表面积增加。MnOx@TiO2在每次反应过后都进行清洗干燥,在共计使用300 h后比表面积较使用前没有变化,表明MnOx@TiO2在使用后结构稳定。
-
结合2.2中的分析和催化表征结果可知,常压催化湿式空气氧化硫代硫酸钠中起主要催化作用的是多相催化湿式空气氧化,对硫代硫酸钠转化的贡献比重占94.79%,可能的反应机制如图10所示。催化剂活性位点为Mn2+/Mn3+催化循环和催化剂表面氧缺陷。一方面,催化剂中的Mn3+作为氧化位点在反应中被还原生成Mn2+,而后Mn2+会被氧气氧化回Mn3+状态,从而实现催化循环;另一方面,MnOx@TiO2中的氧缺陷更容易吸附氧气形成活性氧物种或晶格氧,实现硫代硫酸钠的氧化。
为进一步验证此反应机理,使用Mn2O3与硫代硫酸钠直接进行氧化反应。在最佳反应条件 (反应温度75 ℃、反应时间25 h、空气气量50 mL·min−1)下,加入与MnOx@TiO2等物质量的Mn的Mn2O3,硫代硫酸钠转化率为35%,说明Mn2O3作为活性物种参与了硫代硫酸钠的氧化过程。最佳反应条件下使用MnOx@TiO2的硫代硫酸钠转化率为96%,较使用Mn2O3的硫代硫酸钠转化率高出61%,此结果可以说明,MnOx@TiO2催化氧化硫代硫酸钠是Mn2+/Mn3+氧化位点的催化循环以及催化剂表面形成的氧缺陷的共同作用。
-
1) 在常压系统下,通过浸渍-煅烧制备的MnOx@TiO2对硫代硫酸钠氧化有较好的催化效果,其催化活性受反应温度的影响,较低的温度不利于氧化反应的进行,而较高的温度不利于常压下氧气在水中的溶解,最佳反应温度为75 ℃。
2) 硫代硫酸钠的转化率随反应时间的延长而提高,反应25 h转化率可达到90%以上;继续延长反应时间可以提升硫代硫酸钠的转化率,但不利于硫代硫酸钠的深度氧化。
3) 当硫代硫酸钠模拟废水中
S2O32− 质量浓度为10 000 mg·L−1时,最佳反应条件为反应温度75 ℃、反应时间25 h、空气气量50 mL·min−1;硫代硫酸钠转化率在最佳条件下为96%,92.5%的低价态硫都被氧化为最高价态的硫酸根。4) MnOx@TiO2的主要活性成分为Mn2O3,反应过程中,在MnOx@TiO2表面主要发生由Mn2O3到MnO的转化。同时,MnOx@TiO2表面形成的氧缺陷为O2与
S2O32− 之间的电子转移提供了更多位点,从而可提高MnOx@TiO2对硫代硫酸钠的催化氧化效率。
MnOx@TiO2常压催化湿式空气氧化硫代硫酸钠
Catalytic wet air oxidation of sodium thiosulfate under atmospheric pressure with MnOx@TiO2
-
摘要: 应用MnOx@TiO2催化剂在常压条件下催化湿式空气氧化高浓度硫代硫酸钠,分别考察了空气气量、反应温度和反应时间对高浓度硫代硫酸钠转化率的影响,并采用XRF、XRD、XPS、NH3-/O2-TPD、H2-TPR、BET对使用前后的催化剂进行了分析和表征。结果表明,通入空气中的氧气含量在过量的情况下,气量对硫代硫酸钠的转化影响不大。硫代硫酸钠的转化率随着反应时间的增加而升高;在65~85 ℃内,硫代硫酸钠的转化率先增大后减小。使用Mn负载质量分数为2%的催化剂,使用硫代硫酸钠为模型底物,当
S2O32− 质量浓度为10 000 mg·L−1时,最佳反应条件为气量50 mL·min−1、反应时间25 h、反应温度75 ℃,硫代硫酸钠转化率可达到96%。催化剂表征结果表明,MnOx@TiO2表面存在多种形态的锰氧化物,其中主要活性成分为Mn2O3,硫代硫酸钠的氧化过程伴随着催化剂表面Mn3+到Mn2+的电子转移。同时,MnOx@TiO2表面的各种锰氧化物为S2O32− 与O2之间的电子转移提供了更多位点,而这种电子转移也导致催化剂表面氧缺陷的增加。这些结果证明了MnOx@TiO2常压催化湿式空气氧化硫代硫酸钠的可行性,为含硫废碱液高效、经济的预处理技术的开发提供了可能性。Abstract: This work reported the effects of the airflow, the temperature, the reaction time on sodium thiosulfate conversion. In this process, MnOx@TiO2 catalyst was prepared for the catalytic wet air oxidation of sodium thiosulfate in high concentration under atmospheric pressure. X-ray fluorescence (XRF), X-ray diffraction (XRD), X-Ray Photoelectron Spectroscopy (XPS), temperature-programmed desorption (TPD) with NH3 and O2, temperature-programmed reduction with H2 (H2-TPR) and BET were used to characterize the physicochemical properties of MnOx@TiO2 catalysts before and after the reactions. The results showed that airflow had an insignificant effect on sodium thiosulfate oxidation under the absolute excess of oxygen. Reaction time and reaction temperature were two important influence factors of the catalytic activities. Reaction time increasing showed a positive influence on the conversion of sodium thiosulfate. The catalytic activities first increased then decreased with the increase of temperature from 65 ℃ to 85 ℃. The optimal experimental conditions were airflow of 50 mL·min−1, the reaction time of 25 h, and reaction temperature of 75 ℃, respectively. Under the optimal experimental conditions, the conversion of sodium thiosulfate reached 96% by MnOx@TiO2 with Mn loading amount of 2% when theS2O32− concentration was 10 000 mg·L−1. The result of MnOx@TiO2 characterization showed that there existed various forms of Mn oxides on the surface of the catalyst, of which Mn2O3 was the main active component. The sodium thiosulfate oxidation process was accompanied by the valence conversion of Mn3+ to Mn2+ and electron transfer. At the same time, various Mn oxides on MnOx@TiO2 surface provided more active sites for electron exchange betweenS2O32− and O2, which led to an increase of oxygen defects on the catalyst surface. All these results proved the feasibility of the catalytic wet air oxidation of thiosulfate at atmospheric pressure with MnOx@TiO2 as the catalyst. This process provided a possibility for improving productivity and economic efficiency of pre-treatment technology for the industrial sulfur-containing spent caustic solution.-
Key words:
- MnOx@TiO2 /
- sodium thiosulfate /
- atmospheric pressure /
- catalytic wet air oxidation
-
油田落地油泥是在油田生产过程中,井口溢流、作业落地以及集输过程中跑冒滴漏等造成的含油物质[1-2]。落地油泥的成分比较复杂,一般含有石油烃、水、矿物质和泥砂等物质。目前,落地油泥主要采用焚烧、热解和热水洗等工艺处置,但这些处置方法均存在处理成本高、工艺复杂或处理不彻底等问题[3-5]。因此,开发出经济、高效、环保的落地油泥无害化处置技术有利于油田企业的绿色和可持续发展。
落地油泥的生物降解处置对环境影响小、处理后不会改变泥砂的性质,是油泥无害处置中较为环保的技术之一[6-7]。在处置过程中,利用生物材料,就可以实现油泥的无害化处理,与其他技术相比经济、生态优势明显。然而,生物降解处置适用于含油率低的油泥[8],含油率一般需低于50 mg·g−1。这是因为,在含油率高于50 mg·g−1的条件下,一方面需要更长的生物处理周期;另一方面含油率过高会影响微生物的生长代谢。为解决这一问题,国外有学者提出采用掺土的办法降低含油率,但这种方法又会大幅度增加处理总量,存在一定局限性[1, 9]。从减量化的角度来看,热水洗法与生物降解处置具有良好的兼容性,在有效控制处理成本的前提下,可实现高含油落地油泥的经济、快速处置。
本研究以油田高含油落地油泥为供试材料,拟建立一种热水洗辅助微生物修复高含油落地油泥组合工艺。通过清洗剂类型及清洗温度的优化,以去除大部分的石油烃,使水洗后的含油残砂适用于进一步生物处理。在生物降解过程,探究了填料和烃降解菌对生物降解速率的影响,以期为油田石油污染土壤的治理提供参考。
1. 材料与方法
1.1 供试原料及检测方法
落地油泥样品取自某采油厂油泥缓存站,参照《污染场地环境监测技术导则》(HJ 25.2-2014)[10]和《工业固体废物采样制样技术规范》(HJ/T 20-1998)[11]设计了6个采样点取样。采用红外光谱法测定油泥含油率,方法依据《危险废物鉴别标准-毒性物质含量鉴别》(GB 5085.6-2007)[12]中附录O的要求,结果见表1。本实验将6个不同取样点的样品混合均匀后使用,混合后的油泥含油率为107 mg·g−1。利用索氏抽提器,以氯仿为溶剂,提取落地油泥中的石油烃,用于嗜烃微生物的筛选及界面张力测试。清洗剂分别由鼠李糖脂或脂肽发酵液与OP10(辛基酚聚氧乙烯醚)配制而成。填料秸秆和沼渣均取自东营某农产品公司,使用前经过粉碎处理,长度小于3 cm。客土取自东营市黄河口文化园,填料的化学成分见表2。实验用水为落地油泥缓存站临近联合站注入水,其离子组成及性质见表3。石油醚、尿素、磷酸氢二钾(K2HPO4)均为分析纯。
表 1 现场不同取样点油泥组分组成Table 1. Composition of oil sludge from different sampling points取样点 含油率/(mg·g−1) 含水率/% 固含率/% 干物质含油率/(mg·g−1) a 58.4 29.3 53.6 98.3 b 82.8 18.0 73.7 101 c 131 22.6 64.3 169 d 43.4 16.7 78.9 52.1 e 64.9 14.2 79.5 75.6 f 95.5 19.0 71.4 118 | Show Table DownLoad:
CSV
表 2 不同填料的化学成分Table 2. Chemical properties of various bulking agents
DownLoad:
CSV
表 2 不同填料的化学成分Table 2. Chemical properties of various bulking agents填料名称 含油率/(mg·g−1) 总N/% 总P/% pH 有机质/% 客土 0.62 0.04 0.10 7.9 1.28 秸秆 — 0.69 0.12 7.4 75.1 沼渣 — 1.69 1.03 8.6 81.3 | Show TableDownLoad:
CSV
表 3 油田注入水基本理化性质Table 3. Properties of the oil filed injected water主要离子质量浓度/ (mg·L−1) 总矿化度/(mg·L−1) pH 温度/℃ 含油率/(mg·g−1) Ca2+ Mg2+ HCO−3 Cl− SO2−4 Na++K+ 162.3 88.4 1032 5487 201.7 6421 13392 6.90 53.0 0.007 5 | Show TableDownLoad:
CSV
1.2 实验仪器
实验中使用的主要仪器包括:红外测油仪(OIL450,北京华夏科创仪器股份有限公司);恒温振荡器(Climo-shaker 1SF1-X,瑞士阿道夫科耐公司);机械搅拌器(Eurostar 20,德国伊卡公司);高速冷冻离心机(J26SXPI,美国贝克曼公司);荧光显微镜(BX53,日本奥林巴斯公司);高通量测序仪(X5/XL,美国赛默飞世尔科技公司);界面张力仪(TX500C,美国CNG公司);总N/P分析仪(Multi C/N 2100,德国耶拿仪器股份有限公司);pH计(FE28,梅特勒-托利多公司);电热恒温鼓风干燥箱(DHG-9023A,上海林频仪器股份有限公司)。
1.3 油泥水洗预处理
由于落地油泥含油量高(含油率107 mg·g−1),不适于直接生物修复,因此有必要采用水洗预处理工艺降低含油率[13]。使用配注水配制质量浓度为0.5%的鼠李糖脂/OP10溶液和脂肽/OP10溶液,调节溶液pH为7。利用界面张力仪测试上述清洗剂与从落地油泥中提取的原油间的界面张力,测试条件为50 ℃、6 000 r·min−1。在250 mL锥形瓶内分别加入25 g落地油泥和75 mL清洗剂,在控制转速800 r·min−1和50 ℃水浴条件下,考察清洗剂的洗油效率;同时,考察水洗温度对洗油效率的影响;随后,将水洗后的残砂加入无菌水,质量体积比(m/V)为1∶5,分析水洗后残砂浸出液pH、总N、总P、微生物菌浓、细微粒组分(粒径小于50 μm)含量。
1.4 油泥中微生物菌群分析与烃降解菌分离
将100 g落地油泥、200 mL无菌生理盐水和50 mL石油醚混合,经机械振荡清洗、离心(12 000 r·min−1、15 min)后收集水相中的微生物菌体。提取油泥中土著菌群的DNA及进行微生物群落高通量测序分析,具体步骤参考胡婧等[14]报道的方法。利用涂布平板法从油泥中分离出烃降解菌,将适量油泥用无菌生理盐水分散后,移取菌悬液涂布在原油平板上,在37 ℃下恒温培养2 d,观察以原油为碳源的菌落生长及产生排油圈情况。将平板上形成排油圈的菌落再次培养后,挑取单菌落保存于LB斜面培养基中;将单菌落接种于原油平板上恒温培养2 d,挑取出排油圈与菌落直径差大的菌株接种于原油液体培养基中作进一步驯化处理,从而获得油泥中的土著烃降解菌;随后,使用普通培养基培养上述分离出的烃降解菌。该培养基组成为葡萄糖3 g·L−1、蛋白胨3 g·L−1、酵母粉3 g·L−1、磷酸氢二钾2.7 g·L−1、氯化钠5 g·L−1。培养条件,37 ℃、180 r·min−1。
1.5 生物降解用填料优选
有效提升生物处置过程的好氧降解是加速生物修复速率的关键[15],而通过添加填料提升土壤溶氧量是其中一种有效的方式[16-17]。在本研究中,选择秸秆、客土、沼渣等填料作为研究对象,将上述物质与油泥混合拌匀,使混合物中填料质量分数分别为3%、5%、8%和10%,并转移至1 L烧杯后在室内环境下静置7 d。随后,利用环刀法测定混合物最大持水量和土壤容重指标[18]。
1.6 生物降解性能评价
对水洗处理后的残砂进行生物降解性能评价,考察投加烃降解菌及填料对生物降解效率的影响。实验设置空白组、生物强化组和生物投加组。前述3个处理组的区别在于,空白实验过程不投加任何营养元素,生物强化组加入填料和无机盐,生物投加组加入嗜烃菌、填料和无机盐。实验中,将残砂、填料及适宜比例营养元素(尿素和K2HPO4,用量按C∶N∶P=100∶10∶1加入)拌匀后装入塑料盆内。对于生物投加组,按其质量3%比例平均喷洒上述2株烃降解菌发酵液。对上述所有实验对象补充自来水,使含水保持在35%~50%。将塑料盆置于室外(实验在5—11月之间进行),定期检测含水率、含油率、总N/P含量,并根据需要补充水或总N/P。
以上每组实验均设置3个平行实验,实验结果取平均值。
2. 结果与讨论
2.1 清洗剂与水洗温度的确定
在生物降解前,对落地油泥进行水洗处理,来降低其含油率,以有利于提升后续的生物修复效率。实验考察了鼠李糖脂清洗剂(配方1)和脂肽清洗剂(配方2)的水洗效果,测试结果见图1。从图1中可见,相比于脂肽清洗剂,鼠李糖脂清洗剂具有更好的清洗效率,水洗后残砂的含油率最低可降至35.4 mg·g−1。这是因为,配方1与原油(从落地油泥中提取的原油)的油水界面张力为0.21 mN·m−1,低于配方2与原油间的界面张力(0.89 mN·m−1),因此能更有效地降低原油从固体颗粒表面剥离所要克服的粘附功。此外,从图1中可见,水洗效率随水洗时间呈逐渐下降趋势,30 min后基本趋于平缓。因此,从能耗和日处理量考虑,水洗时间以30 min为宜,此时水洗后残砂的含油率为39.3 mg·g−1。
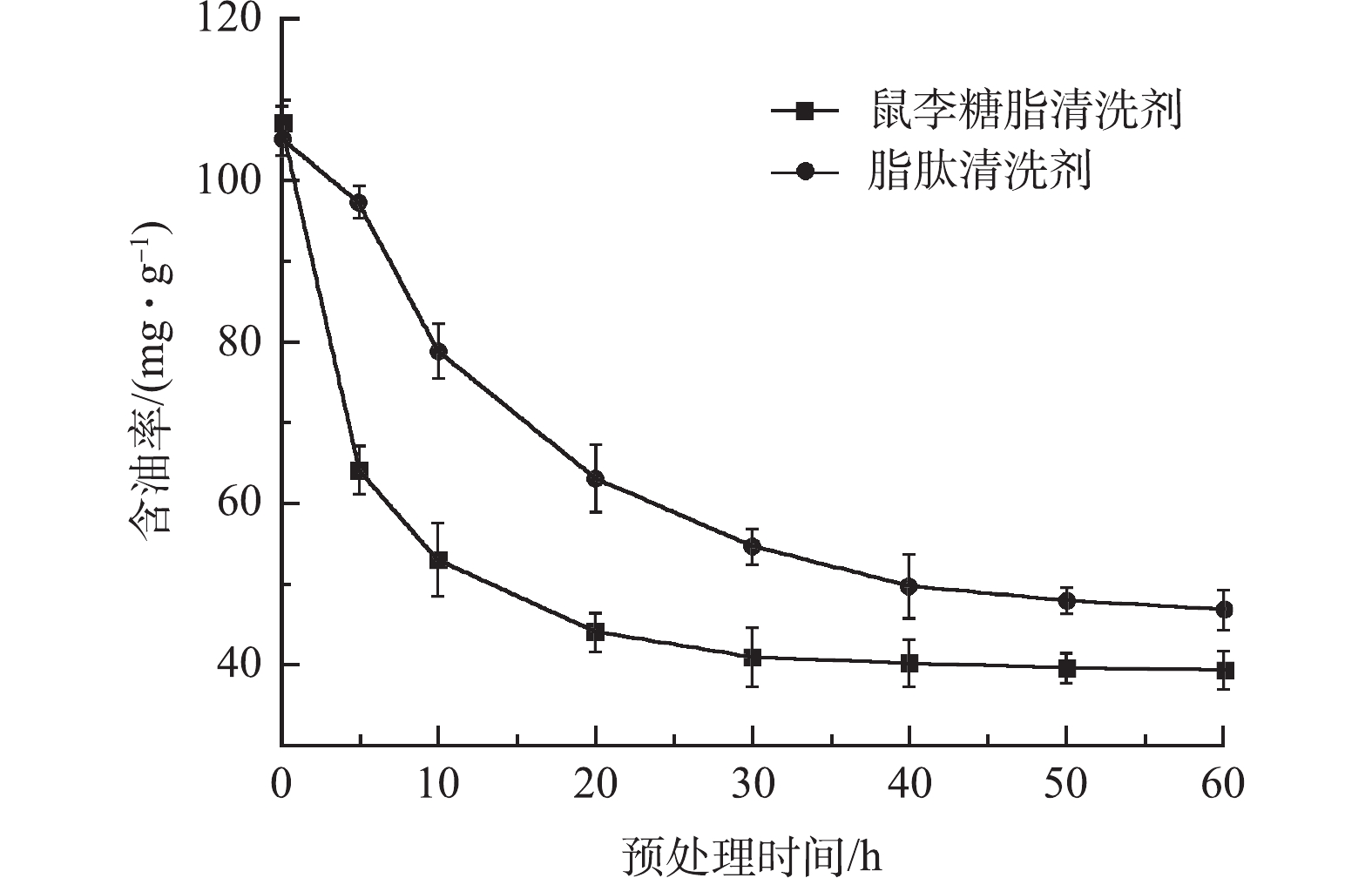 图 1 不同生物型清洗剂配方水洗后含油率变化Figure 1. Effect of various washing agents on oil content of the oil sludge-contaminated soil
图 1 不同生物型清洗剂配方水洗后含油率变化Figure 1. Effect of various washing agents on oil content of the oil sludge-contaminated soil考察了配方1条件下水温对落地油泥洗油效率的影响,结果见图2。从测试结果可知,随着水洗温度的升高,洗油效率亦大幅提升,在水洗温度为30 ℃时,残砂中含油率为89.2 mg·g−1;当水洗温度升至50 ℃时,含油率降至39.3 mg·g−1;随后,洗油效率提升缓慢,到80 ℃时含油率降至34.5 mg·g−1。水洗过程影响洗油效率的关键因素之一是石油烃的黏度,在水温由30 ℃升至50 ℃时,石油烃黏度大幅下降,流动性增强,其在搅拌剪切力作用下从固体颗粒表面剥离,此时洗油效率大幅提升。当温度升至50 ℃上后,黏度对洗油效率的影响作用减弱,因此洗油效率提升速率减慢[19]。综合考虑水洗的能耗和水洗效率,采用水温50 ℃即可以达到预期热水洗目标。
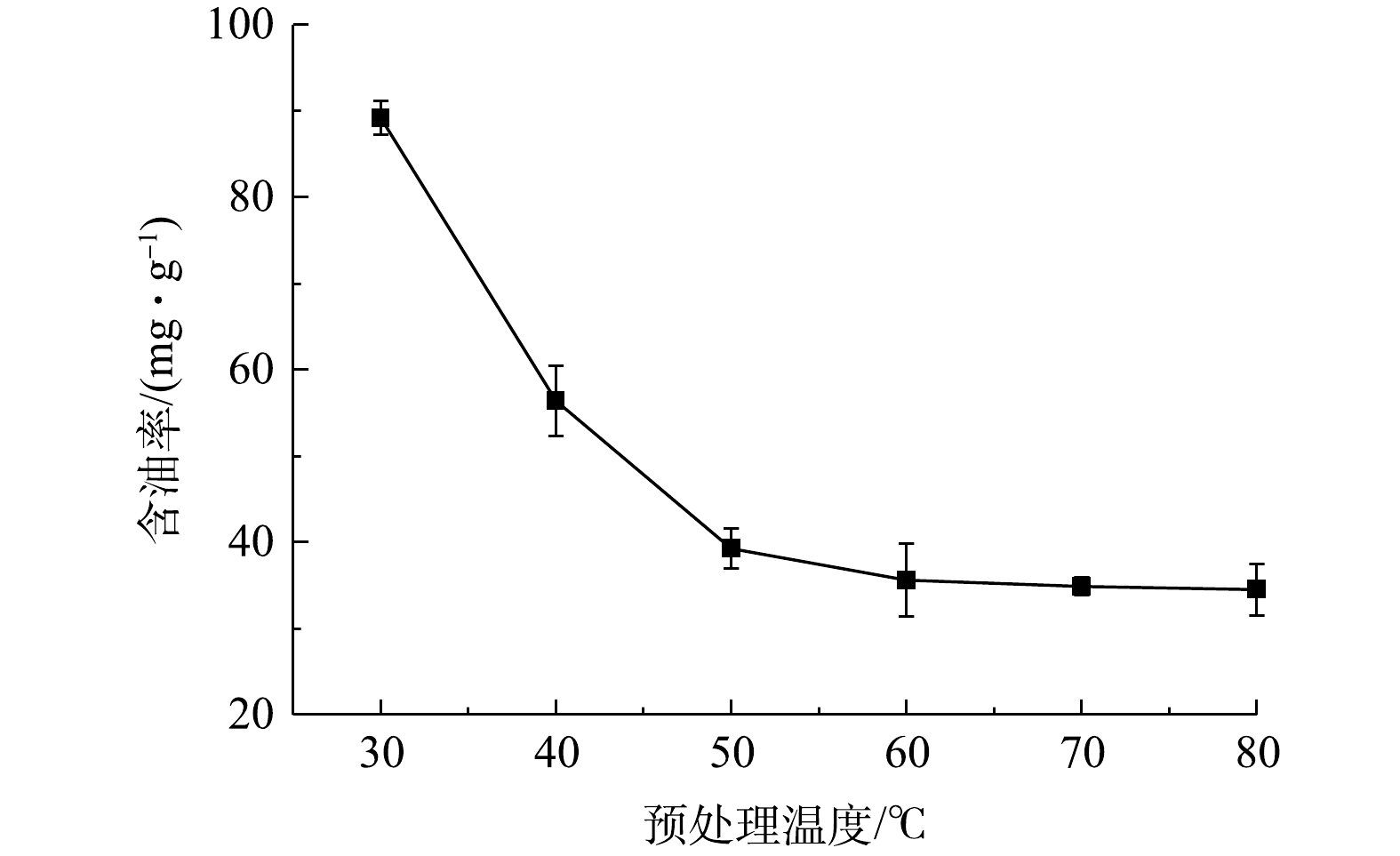 图 2 水洗温度对油泥含油率的影响Figure 2. Effect of washing temperature on the oil content of the oil sludge-contaminated soil
图 2 水洗温度对油泥含油率的影响Figure 2. Effect of washing temperature on the oil content of the oil sludge-contaminated soil分析了水洗后残砂的理化性质,包括浸出液总N、总P、pH、细微粒组分含量及微生物菌含量,结果见表4。从表中可以看出,相比于水洗前的落地油泥,水洗后残砂中的总N、总P、细微粒组分含量及微生物菌含量均出现了不同程度的下降。这是因为,在水洗过程,加热和搅拌作用会致使大部分的水溶物质(包括无机盐、矿物质和微生物等)进入水相,并随固液分离而流失。而经过水洗后的残砂,从含油率、pH、细微粒组分含量等多个参数来看,能够满足生物修复的基本要求[13]。但由于残砂中总N、总P含量偏低,无法满足微生物的正常生长代谢。因此,需要在后续的生物处理过程补充外源菌剂、足够量的营养物质,以保障微生物的嗜烃代谢活动。
表 4 水洗前后落地油泥的理化性质变化Table 4. Changes in properties of the water-washed oil-contaminated soil after and before washing供试材料 总N/(mg·L−1) 总P/(mg·L−1) pH 细微粒组分含量/% 微生物菌含量/(个·mL−1) 落地油泥 21.6 3.55 7.3 24.6 8×106 水洗后残砂 7.8 0.66 6.9 22.7 1×105 | Show TableDownLoad:
CSV
2.2 落地油泥的群落结构分析及烃降解菌分离
利用高通量测序分析了落地油泥中土著微生物的群落结构,结果见图3。从图中可以看到,土著微生物菌群以食烷菌属(Alcanivorax)、海杆菌属(Marinobacter)、假海源菌属(Pseudidiomarina)及假交替单胞菌属(Pseudoalteromonas)等为主。有报道称,食烷菌属能降解烷烃类有机物[20-21]。另外,有研究[22-24]结果表明,海杆菌属、假海源菌属和假交替单胞菌属均能降解多环芳烃类有机物。以上菌群出现在落地油泥中,说明落地油泥在其产生、转移及场地堆放过程能逐渐形成以烃降解微生物为主的群落结构。这是因为,油泥中的石油烃能为这些微生物的代谢生长提供必要的碳源和营养,只是由于油泥中氮、磷元素含量太低(见表4),难以满足微生物的正常代谢活动,故微生物的数量维持在较低水平,对石油烃的降解作用弱。同时,前人报道的烃降解菌多数来源于海洋环境,而本实验选取的落地油泥的产生地点位于黄河三角洲沿海滩涂地区,故其中的群落结构受海洋环境影响较大。因此,在其长期存放过程中,以石油烃为碳源的微生物逐渐形成优势菌群[25]。
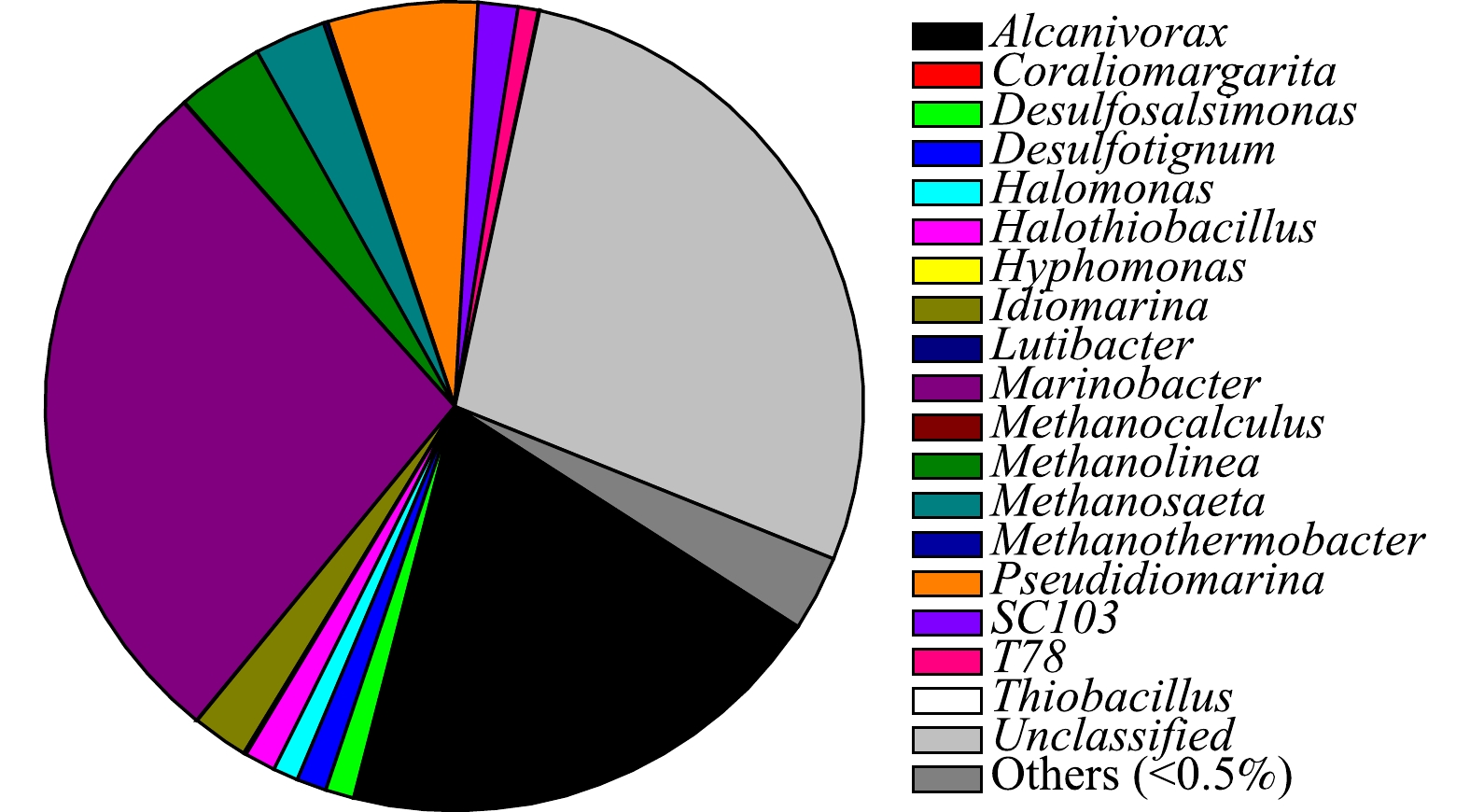 图 3 落地油泥中土著微生物群落结构Figure 3. Indigenous bacterial community structure from the oil sludge-contaminated soil
图 3 落地油泥中土著微生物群落结构Figure 3. Indigenous bacterial community structure from the oil sludge-contaminated soil采用涂布平板法分离烃降解菌,原油平板上出现3个扩油圈,如图4所示。从图中可见,标记为“a”、“b”和“c”的菌株能够利用原油为碳源代谢生长,并产生表面活性物质,形成排油圈。出现排油圈则表明上述菌株能够利用平板内的原油为唯一碳源进行代谢生长,并代谢合成表面活性剂。而表面活性剂的产生,则能够进一步乳化原油,提升微生物利用效率[26-28]。进一步分离后,挑取出排油圈与菌落直径差大的2株嗜烃菌,命名为G-3和G-6。随后,使用培养基培养G-3和G-6,获得烃降解菌发酵液,经显微镜检测,发酵液中菌含量均能够达到2×108个·mL−1,表明这2株烃降解微生物具备现场应用的基本条件。
 图 4 落地油泥土著烃降解菌在原油平板上生长情况Figure 4. Surface morphologies of indigenous microorganisms grew in the crude oil culture plate
图 4 落地油泥土著烃降解菌在原油平板上生长情况Figure 4. Surface morphologies of indigenous microorganisms grew in the crude oil culture plate2.3 填料的优选
考察加入填料对提高油泥溶氧量的影响,评价的指标包括油泥密度和最大持水量,结果见图5和图6。从图5可以看出,残砂中加入秸秆、客土和沼渣后均能有效降低油泥密度;而且随着填料加入比例的增加,密度呈线性下降。与沼渣和客土相比,加入质量分数为10%的秸秆后,则密度下降13.5%,而加入相同质量分数的客土后,密度下降了7.97%。这是因为,在加入相同质量分数的填料中,秸秆体积最大,掺入残砂后最有利于提高油泥的蓬松度。而在土壤修复领域,特别是采用地耕法处置污染土壤过程,秸秆等廉价、疏松的填料常用于提高土壤通透性[29-30],以达到增加溶氧的目的。
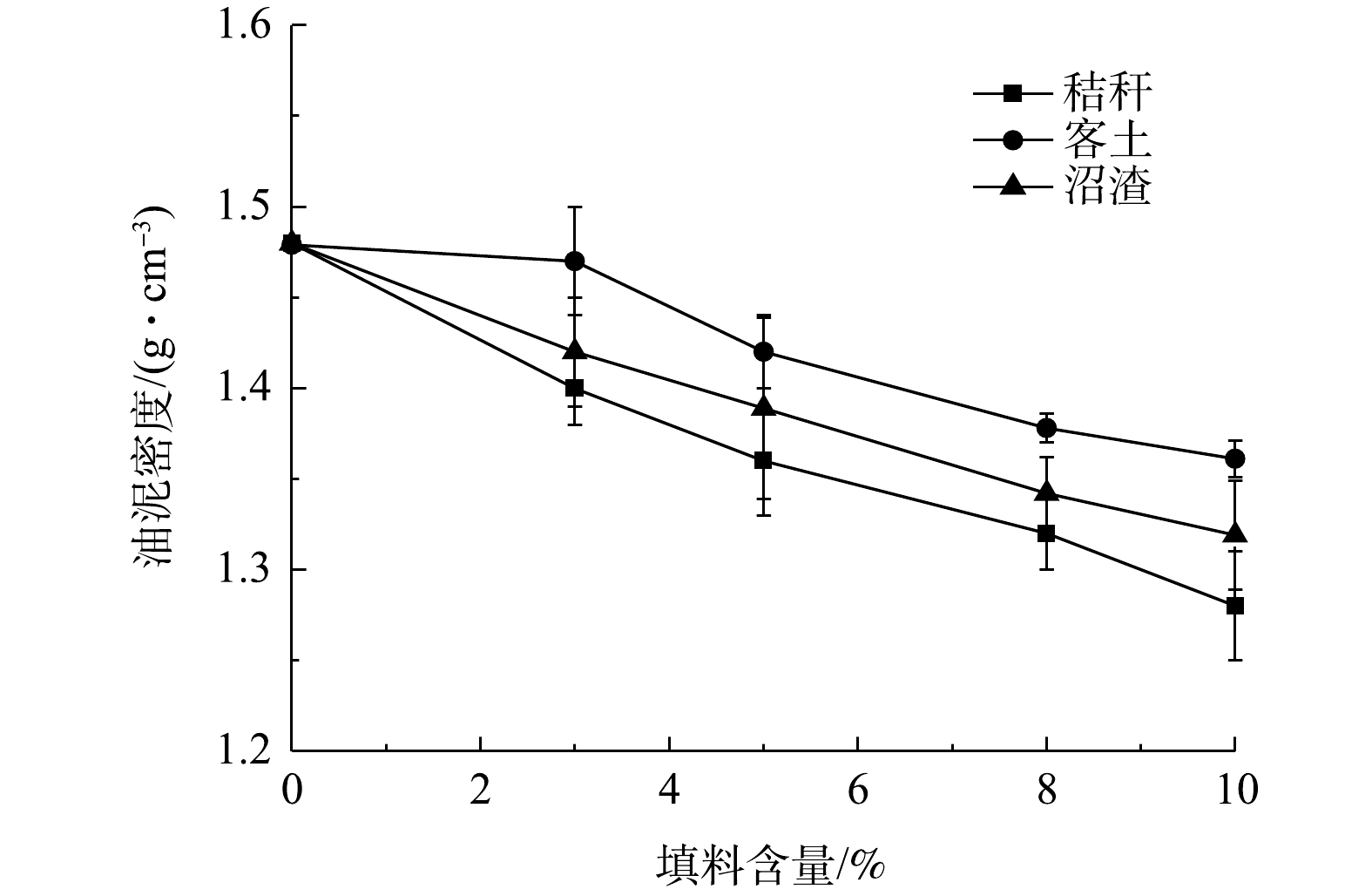 图 5 不同填料加入对油泥密度的影响Figure 5. Effect of different bulking agent content on the soil bulk density
图 5 不同填料加入对油泥密度的影响Figure 5. Effect of different bulking agent content on the soil bulk density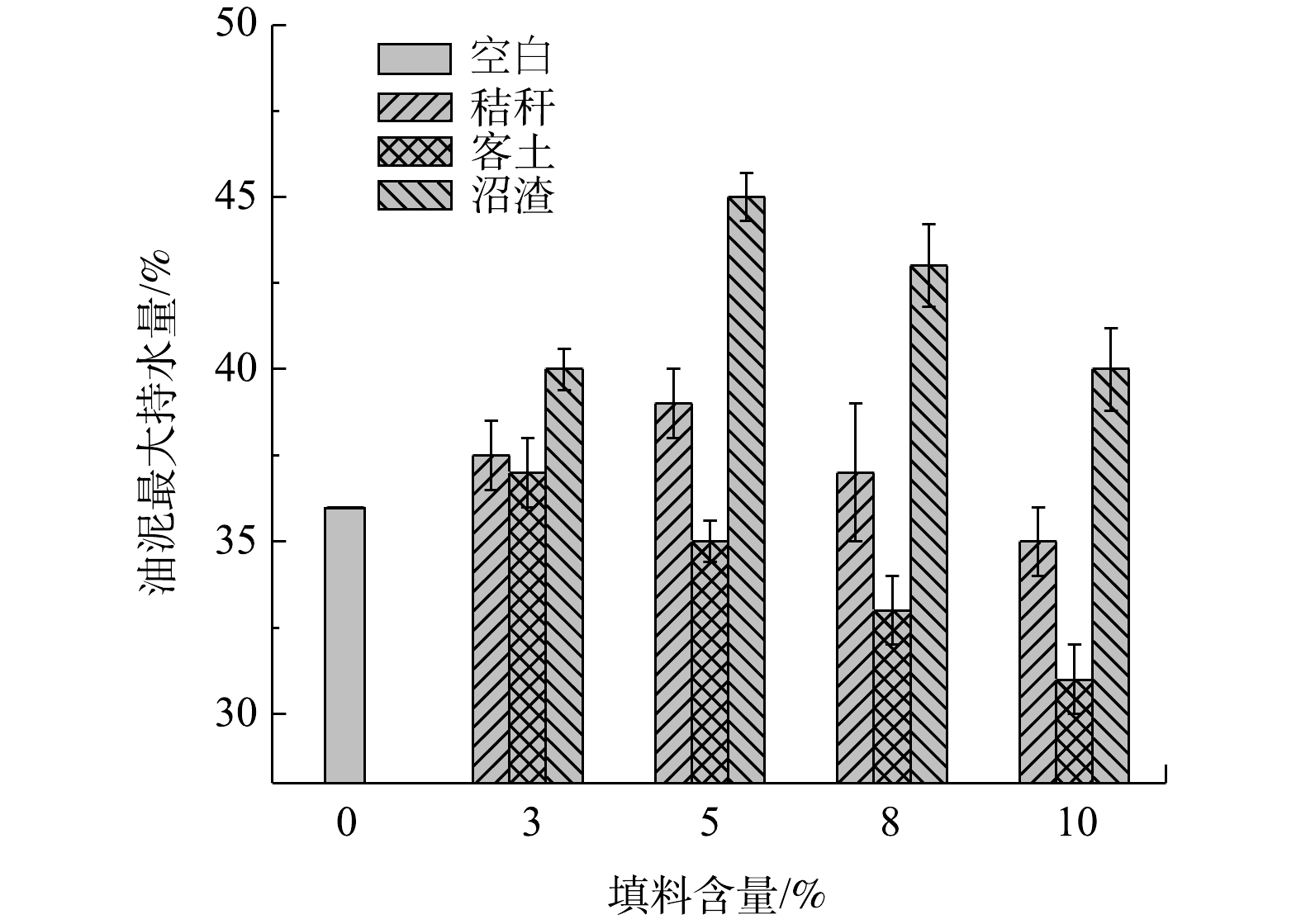 图 6 不同填料加入对油泥最大持水量的影响Figure 6. Effect of different bulking agent content on maximum moisture capacity of the soil
图 6 不同填料加入对油泥最大持水量的影响Figure 6. Effect of different bulking agent content on maximum moisture capacity of the soil图6给出了添加不同填料后对油泥最大持水量的影响。从测试结果来看,添加秸秆和沼渣对提高最大持水量有一定的提高作用,而添加客土后最大持水量出现下降趋势。相比于秸秆,沼渣具有更好的持水能力,添加量为5%时,最大持水量由36%提升到45%;此后,继续增加沼渣含量时,油泥的最大持水量开始下降。对于秸秆,增加量在3%以内,有利于保水,随后出现下降趋势。这是因为,管状空心结构的秸秆能够增加油泥表层的水分流动通道,使水分蒸发加剧。而对于处理后的沼渣,能够有效改良油泥结构,提高其含水饱和度和持水能力,从而降低油泥脱水速度,最终提高油泥保水、抗旱性能。
此外,通过比较客土、秸秆和沼渣的化学性质来看(表2),沼渣具有丰富的N、P元素和有机物,能够为微生物的代谢生长提供营养物质。此外,沼渣中还含富含有腐殖质,而这类物质能作为电子受体参与有机物的厌氧生物降解过程,提升微生物对有机物的代谢速率[31]。而这一推论在大量的研究报道中已被证实,即在厌氧环境下,微生物以腐殖质为电子受体氧化石油类有机物[32-33]。因此,沼渣作为一种新型的填充物不仅能够有效提高油泥的通透性,同时其富含的腐殖质能够使厌氧区域发生更高速率的生物降解。因此,通过以上实验筛选出沼渣用量为5%并用于其后的生物降解实验。
2.4 生物降解实验
在生物降解性能评价过程中,考察了添加G-3和G-6外源烃降解菌对处理效果的影响,结果见图7。从图中看到,相比于空白组,单纯通过激活油泥中土著微生物进行生物修复时,效果较差,处理至200 d时残砂样品的含油率仍然达到26.3 mg·g−1。相反,投加烃降解菌组对石油烃有着较强的降解效果,处理87 d时残砂样品的含油率降至10.0 mg·g−1以下,处理200 d时则降至4.62 mg·g−1。由此可认为,在预处理水洗过程中,在水溶液环境下的搅拌剪切作用破坏了油泥的微生态环境,高温水洗、投加药剂造成大量的土著烃降解菌死亡或随水体流失(见表4),从而导致残砂中土著烃降解菌的数量和种类大幅降低,最终造成生物修复效果较差。而通过添加筛选的烃降解菌,补充和提升了残砂样品中烃降解菌的种类和数量,在营养元素富余的条件下,嗜烃微生物的生物量大幅提升,并能够有效降解残砂中的石油烃,因此取得了理想的处理效果。由此可得出,在选择水洗技术为预处理工艺时,在生物修复过程中,需要投加适量的烃降解外源菌,从而提高处置的效果。
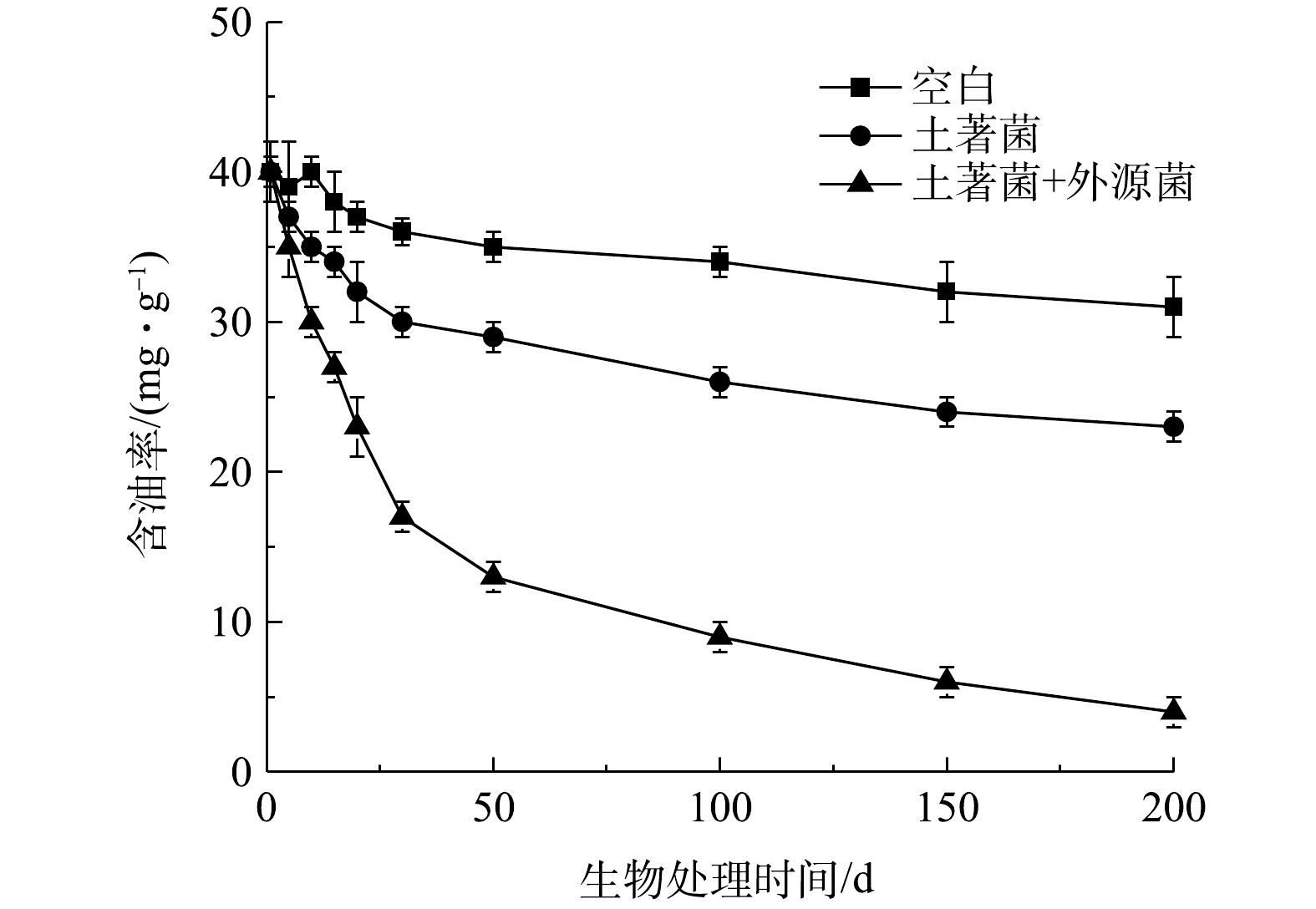 图 7 添加烃降解外源菌对残砂中含油率的影响Figure 7. Effect of the added isolated bacterias on degradation of the residual sands
图 7 添加烃降解外源菌对残砂中含油率的影响Figure 7. Effect of the added isolated bacterias on degradation of the residual sands测试了未处理时、处理第200 d时的对照组及投加菌剂组样品中含油率及各组分含量的变化,结果如表5所示。由表可知,在实验前,空白组和投加菌剂组含油率分别为39.3 mg·g−1和39.1 mg·g−1;经过200 d的生物处理后,空白组含油率下降至31.2 mg·g−1,而投加菌剂组含油率下降至4.62 mg·g−1。其中,不同石油烃组分经过生物处理后含量均下降,其中以饱和烃和芳香烃降解率最为显著,而胶质和沥青质则较难被降解。
表 5 生物处理前后残砂中石油烃组分Table 5. Component analysis of the hydrocarbons from various residual sands检测项目或组分 未处理的残砂中石油烃组分质量分数/(mg·g−1) 处理200 d后的残砂中石油烃组分质量分数/(mg·g−1) 对照组 投加菌剂组 对照组 投加菌剂组 含油率 39.3±3.4 39.1±2.9 31.2±1.2 4.62±0.6 饱和烃 16.2±1.5 15.9±1.0 10.2±0.3 0.33±0.03 芳香烃 12.4±0.9 11.8±0.5 11.1±0.2 1.02±0.1 胶质 8.62±1.3 9.52±0.8 7.09±0.3 1.24±0.2 沥青质 2.10±0.5 1.88±0.4 2.86±0.4 2.03±0.1 | Show TableDownLoad:
CSV
3. 结论
1)从高含油落地油泥中分离获得2株烃降解菌,用于微生物修复实验。添加5%的沼渣能能够提升油泥的溶氧能力,最大持水量增加到45%。
2)在鼠李糖脂清洗剂总用量为0.5%、水洗时间30 min和50 ℃条件下,油泥含油率由107 mg·g−1降至39.3 mg·g−1。水洗后残砂中总N/P、细微粒组分(粒径≤50 μm)及土著微生物菌浓均不同程度下降,在随后的生物修复过程需要补充外源菌和营养物质。
3)生物修复过程投加沼渣和2株烃降解菌,生物降解速率得到提升;经过200 d的生物修复后,残砂含油率降至4.62 mg·g−1。在此过程中,微生物优先降解饱和烃和芳香烃,对胶质和沥青质的降解作用弱。
-
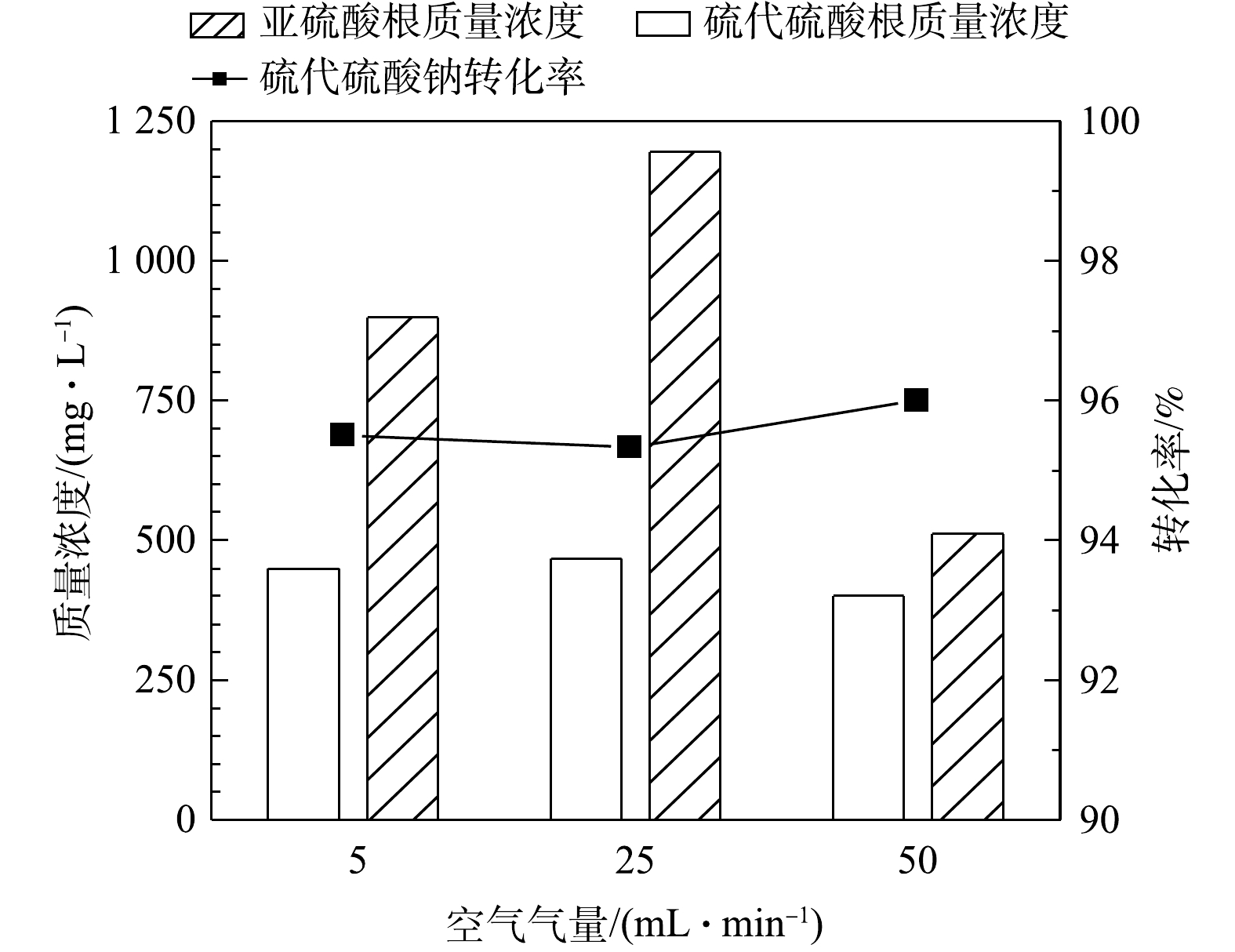
图 2 空气气量对硫代硫酸钠转化的影响
Figure 2. Effect of air flow on the conversion of sodium thiosulfate
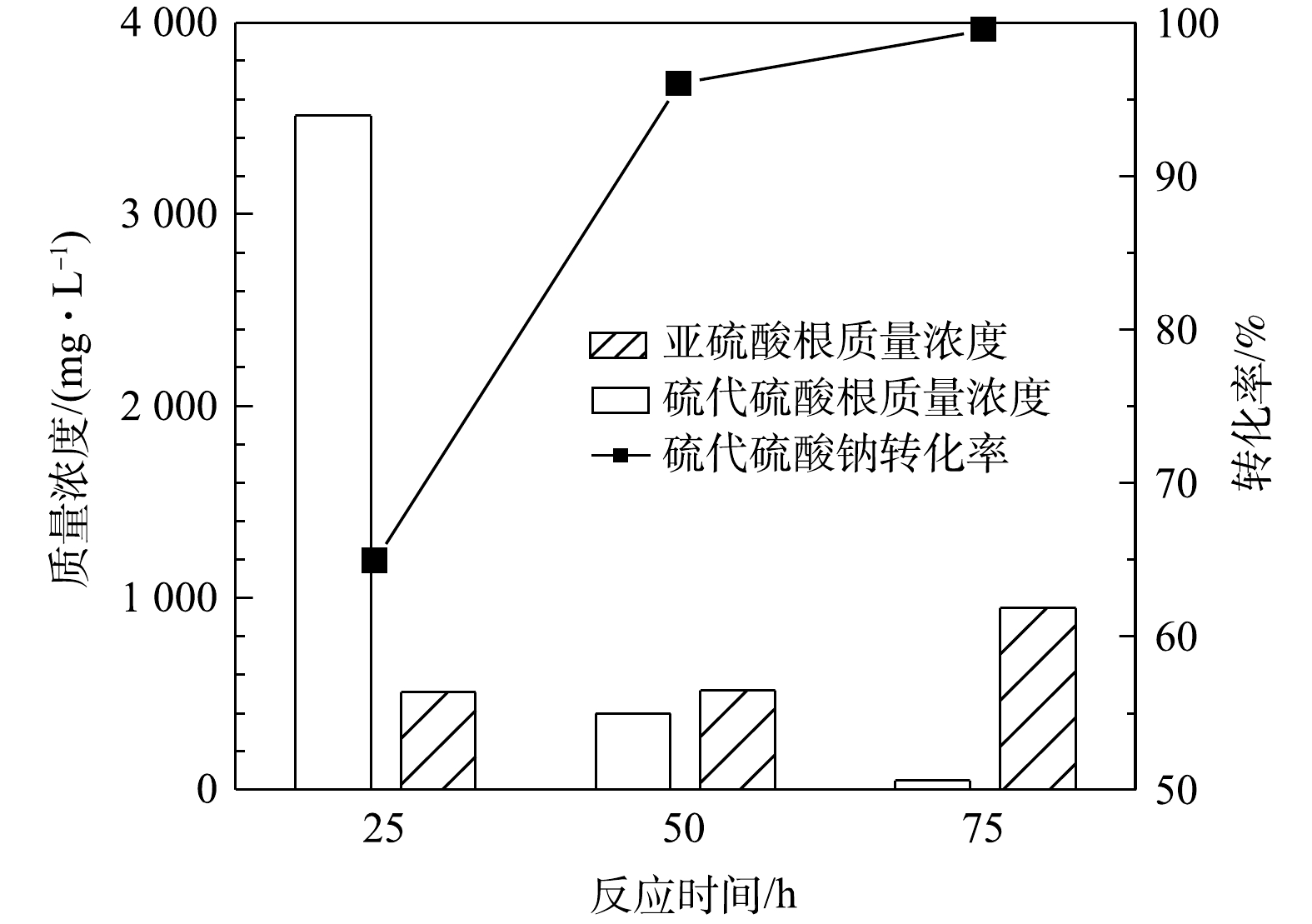
图 3 反应时间对硫代硫酸钠转化的影响
Figure 3. Effect of reaction time on the conversion of sodium thiosulfate
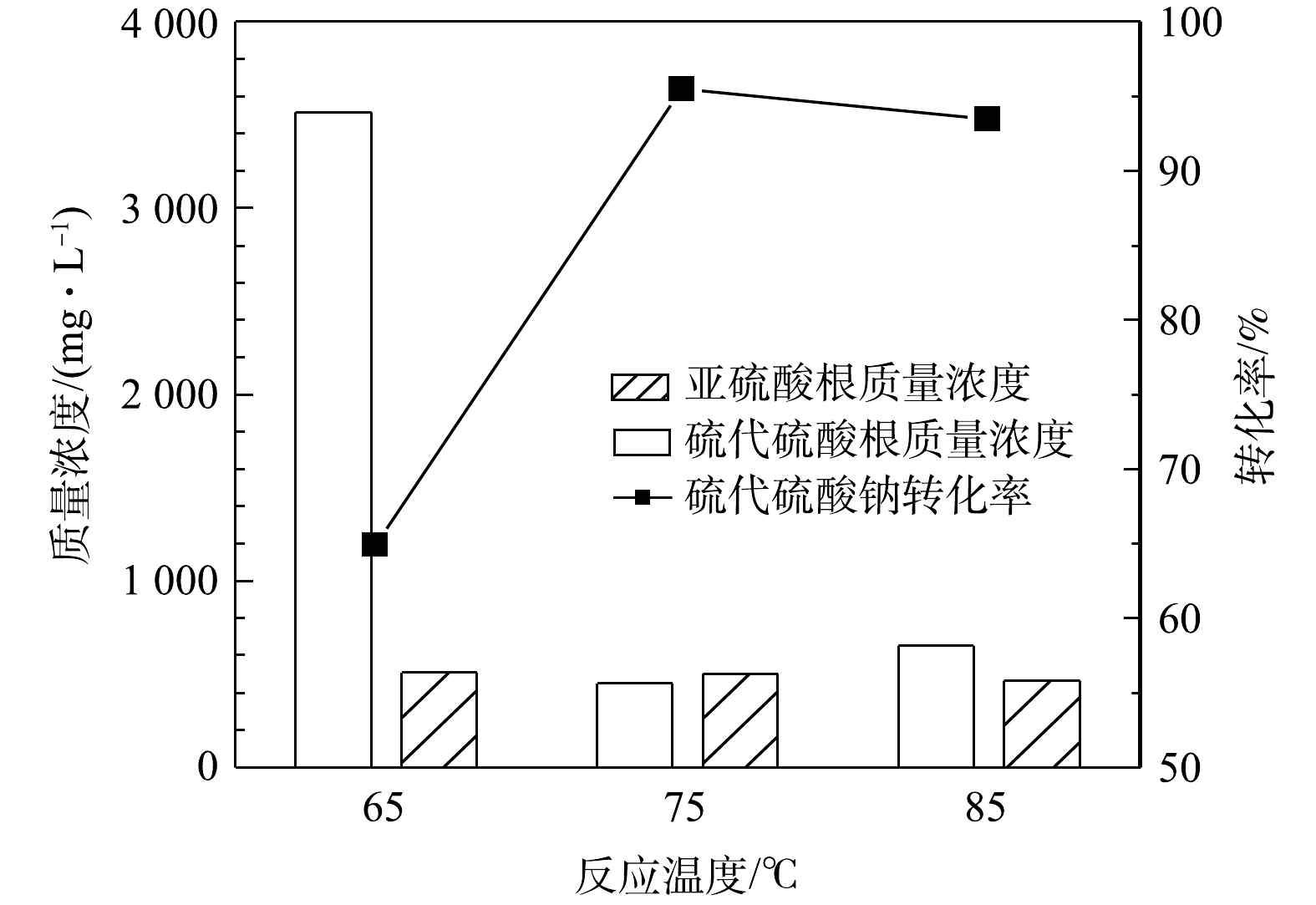
图 4 反应温度对硫代硫酸钠转化的影响
Figure 4. Effect of temperature on the conversion of sodium thiosulfate
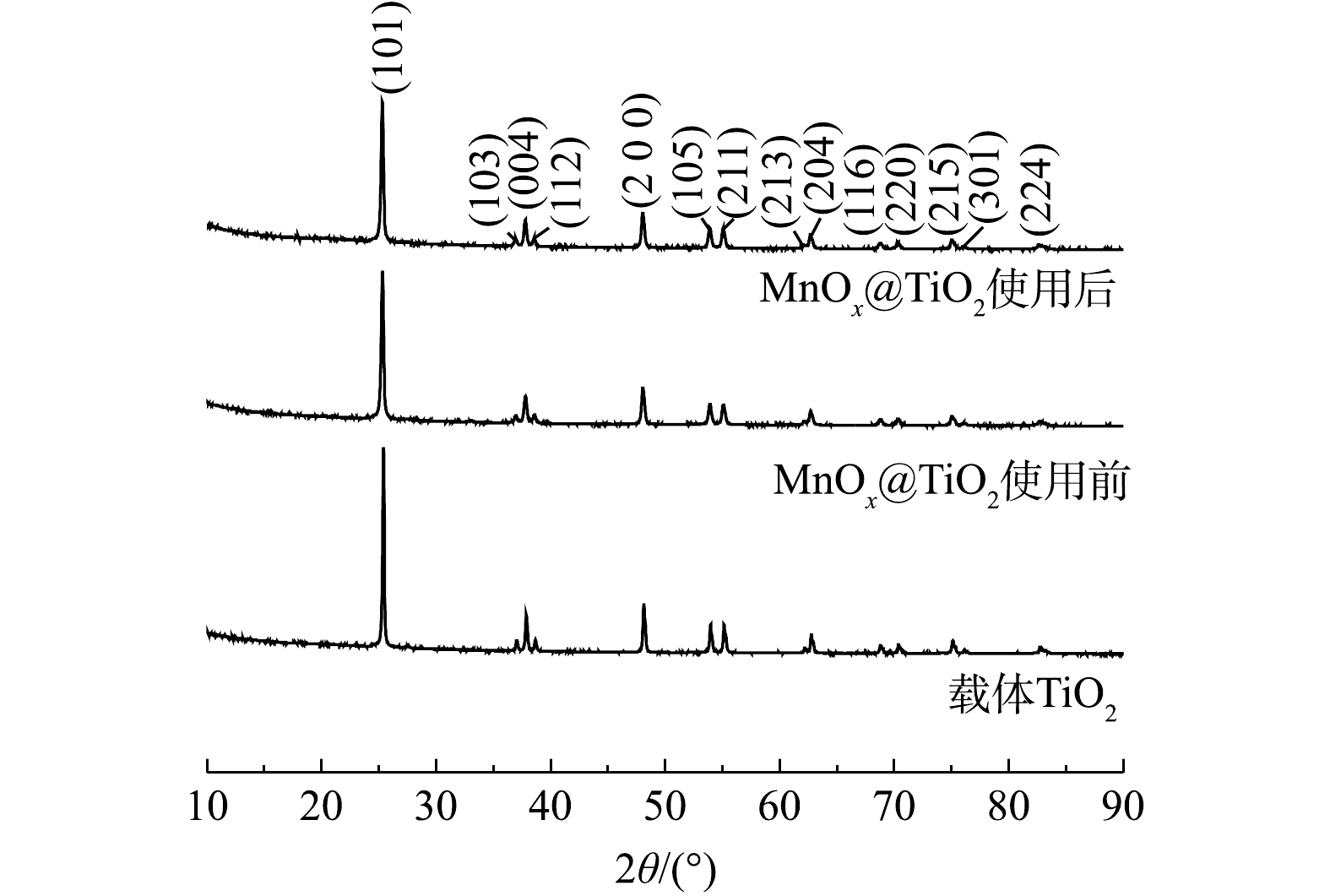
图 6 TiO2和MnOx@TiO2使用前后的XRD衍射图谱
Figure 6. XRD patterns of TiO2 and MnOx@TiO2 before and after the reaction
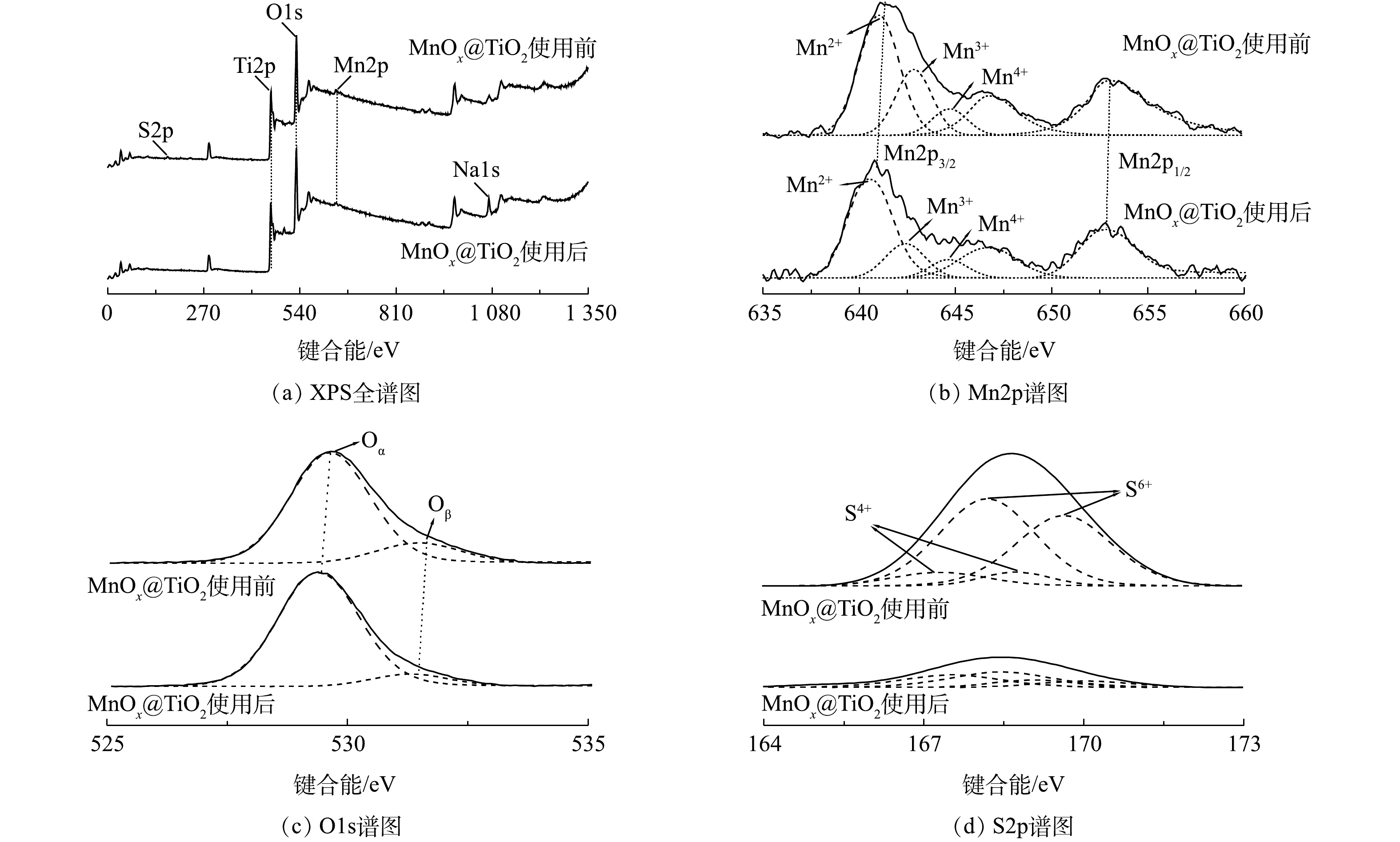
图 7 MnOx@TiO2使用前后的XPS谱图
Figure 7. XPS spectra of MnOx@TiO2 before and after the reaction
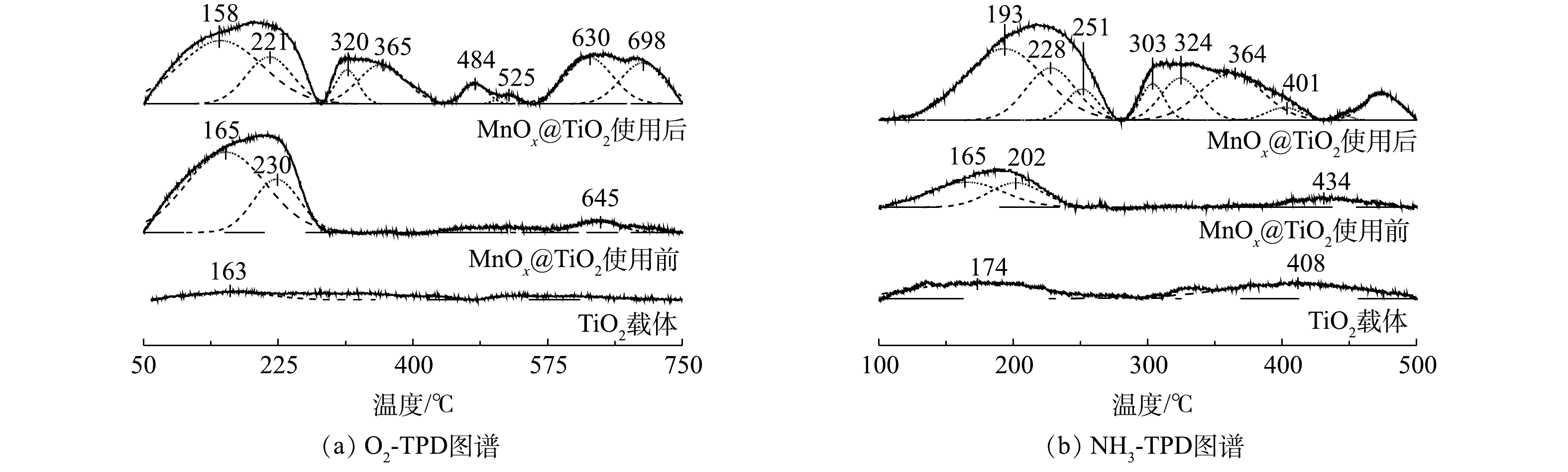
图 8 TiO2和MnOx@TiO2使用前后O2-/NH3-TPD图谱
Figure 8. TPD-Profiles of TiO2 and MnOx@TiO2 before and after the reaction
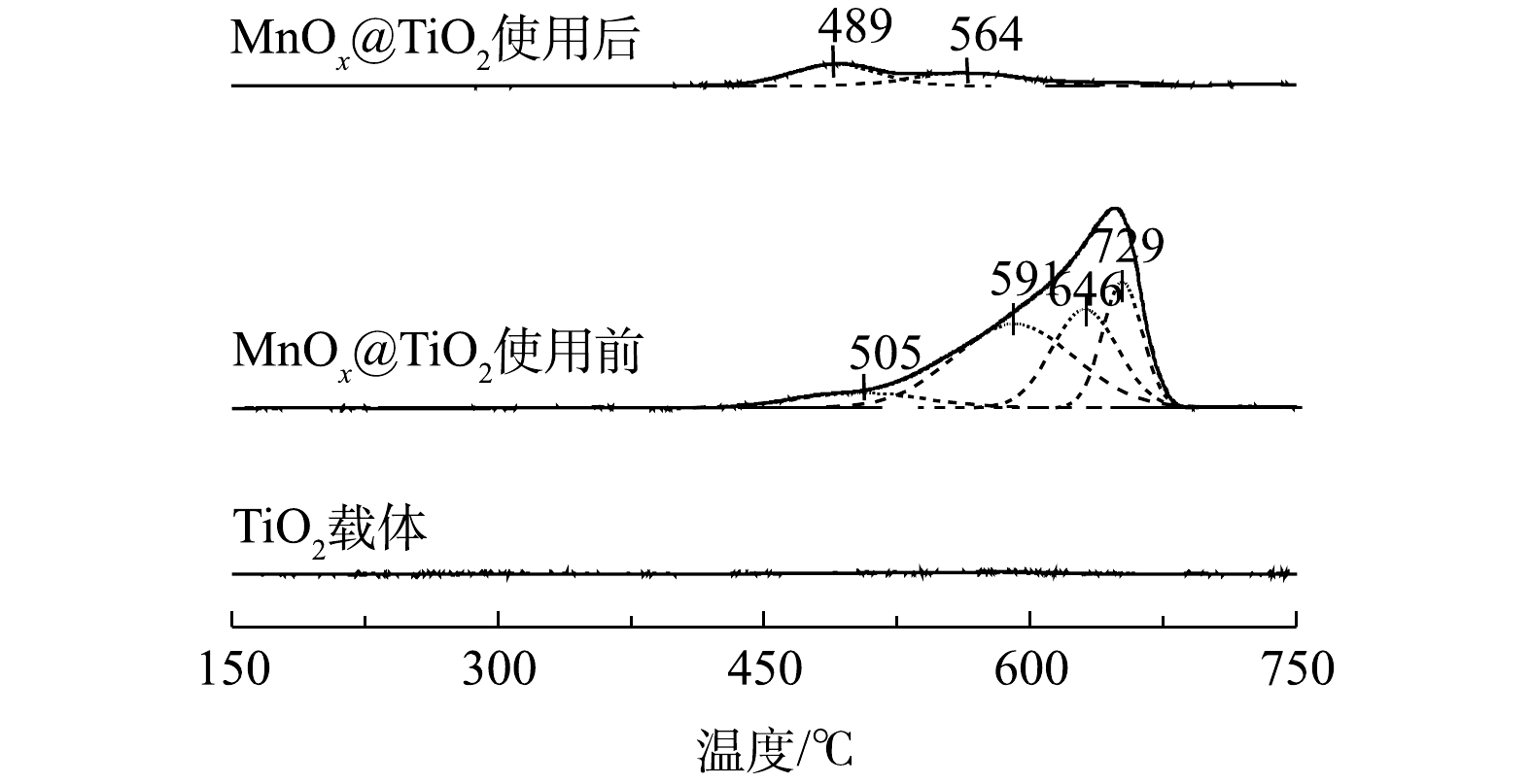
图 9 TiO2和MnOx@TiO2使用前后H2-TPR图谱
Figure 9. H2-TPR-Profiles of TiO2 and MnOx@TiO2 before and after the reaction
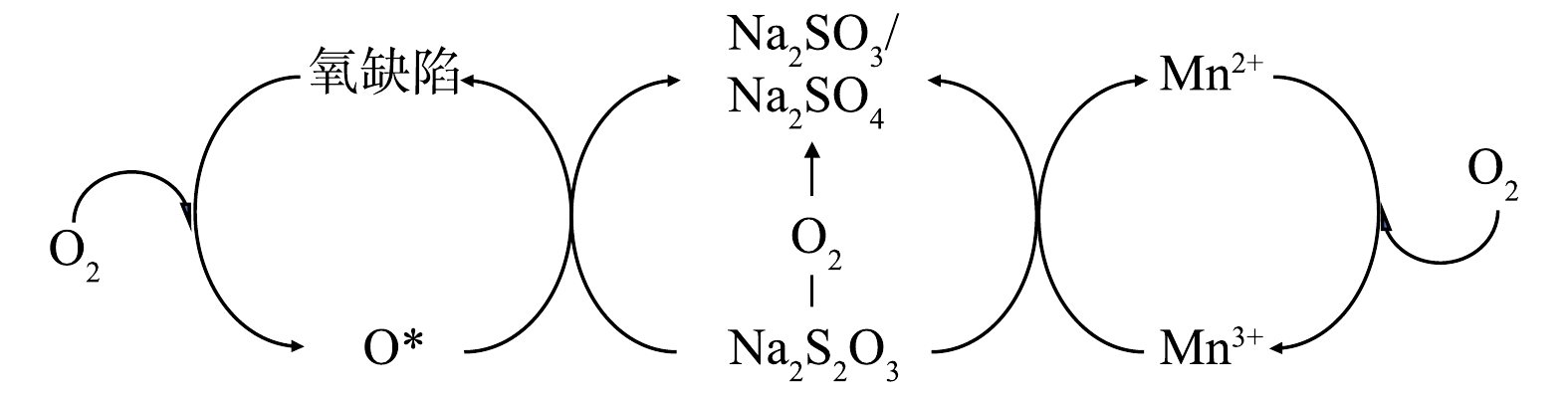
图 10 MnOx@TiO2催化氧化硫代硫酸钠可能的反应机制
Figure 10. Scheme illustrating the possible mechanism for catalytic oxidation process with MnOx@TiO2
表 1 湿式空气氧化脱硫工艺汇总
Table 1. A summary of desulfurization process with wet air oxidation process
工艺类型 代表公司 反应温度/℃ 反应压力/MPa 低温湿式氧化 美国斯通韦伯 115 0.7 中温湿式氧化 西门子 200 3.2 高温湿式氧化 西门子 260 8.6 催化湿式氧化 德国拜尔 220 8.6
下载: 导出CSV
表 2 催化空气氧化脱硫效果汇总
Table 2. A summary of desulfurization effect with catalytic air oxidation process
下载: 导出CSV
表 3 TiO2和MnOx@TiO2使用前后的XRF元素分析结果
Table 3. Elemental analyses of TiO2 and MnOx@TiO2 before and after the reaction
催化剂 质量分数/% Ti O Mn 其他 TiO2 59.3 40.0 0 0.6 MnOx@TiO2使用前 56.9 39.9 1.9 1.4 MnOx@TiO2使用后 58.6 39.9 0.2 1.3
下载: 导出CSV
表 4 MnOx@TiO2使用前后XPS结果
Table 4. XPS results of MnOx@TiO2 before and after the reaction
催化剂 键合能/eV (Mn3+/Mnx+)/% (Oβ/O)/% Mn2+ Mn3+ Mn4+ Oβ MnOx@TiO2 使用前 640.4 642.7 644.6 531.5 23 15 MnOx@TiO2 使用后 641.0 642.4 644.1 531.3 16 10
下载: 导出CSV
-
[1] 赵兴龙, 刘丽军, 张克利. 湿式氧化处理含硫废碱液的技术进展[J]. 石油化工安全环保技术, 2008, 24(1): 57-60. doi: 10.3969/j.issn.1673-8659.2008.01.019 [2] 高英. 处理废碱液中硫化物和有机物的方法研究[J]. 化学工程师, 2001(6): 79-50. [3] 何志祥, 宋远清, 戴友芝. 湿式空气氧化法处理废碱液运行调试[J]. 石油化工环境保护, 2004, 27(2): 48-51. [4] 王婷婷. 硫化钠废碱液催化氧化研究[D]. 北京: 北京化工大学, 2011. [5] FU F L, XIONG Y, XIE B P, et al. Adsorption of acid red 73 on copper dithiocarbamate precipitate-type solid wastes[J]. Chemosphere, 2007, 66(1): 1-7. doi: 10.1016/j.chemosphere.2006.05.054 [6] 刘崇华, 周皓, 刘晓群等. 催化氧化法处理含硫废碱液新技术的开发与应用[J]. 油气田环境保护, 2006, 16(3): 14-17. doi: 10.3969/j.issn.1005-3158.2006.03.005 [7] 马艺璇, 马冬晨, 蒋齐光. 湿式催化氧化处理塔河油田脱硫废碱液实验研究[J]. 仪器仪表与分析检测, 2015(4): 42-45. [8] 何长明, 李俊, 刘晓晶. 催化氧化法处理废碱液中硫化物的工程应用研究[J]. 应用化工, 2018, 47(1): 123-125. doi: 10.3969/j.issn.1671-3206.2018.01.031 [9] BAI B, LI J, HAO J. 1D-MnO2, 2D-MnO2 and 3D-MnO2 for low-temperature oxidation of ethanol[J]. Applied Catalysis B:Environmental, 2015, 164: 241-250. doi: 10.1016/j.apcatb.2014.08.044 [10] TANG X F, CHEN J L, HUANG X M, et al. Pt/MnOx-CeO2 catalysts for the complete oxidation of formaldehyde at ambient temperature[J]. Applied Catalysis B:Environmental, 2008, 81(1/2): 115-121. [11] ZHAO B G, RUI R, GUO X G, et al. Nb-modified Mn/Ce/Ti catalyst for the selective catalytic reduction of NO with NH3 at low temperature[J]. Applied Catalysis B:Environmental, 2017, 545: 64-71. doi: 10.1016/j.apcata.2017.07.024 [12] SUN C Z, LIU H, CHEN W, et al. Insights into the Sn/Zr co-doping effects on N2 selectivity and SO2 resistance of a MnOx-TiO2 catalyst for the NH3-SCR reaction[J]. Chemical Engineering Journal, 2018, 347: 27-40. doi: 10.1016/j.cej.2018.04.029 [13] 马兵兵, 苏中华, 弥海鹏, 等. 间接碘量法测定氧化铝生产流程样品铝酸钠溶液中硫离子、硫代硫酸钠和亚硫酸根[J]. 冶金分析, 2016, 36(11): 41-45. [14] KWON D W, NAM K B, HONG S C. Influence of tungsten on the activity of a Mn/Ce/W/Ti catalyst for the selective catalytic reduction of NO with NH3 at low temperatures[J]. Applied Catalysis A:General, 2015, 497: 160-166. doi: 10.1016/j.apcata.2015.01.013 [15] CHEN H H, ZHANG H P, YAN Y. Fabrication of porous copper/manganese binary oxides modified ZSM-5 membrane catalyst and potential application in the removal of VOCs[J]. Chemical Engineering Journal, 2014, 254: 133-142. doi: 10.1016/j.cej.2014.05.083 [16] WANG X M, LI X Y, ZHAO Q D, et al. Improved activity of W-modified MnOx-TiO2 catalysts for the selective catalytic reduction of NO with NH3[J]. Chemical Engineering Journal, 2016, 288: 216-222. [17] DUPIN J C, GONBEAU D, VINATIER P, et al. Systematic XPS studies of metal oxides, hydroxides and peroxides[J]. Physical Chemistry Chemical Physics, 2000, 2(6): 1319-1324. doi: 10.1039/a908800h [18] LIU S X, CHEN X Y. A visible light response TiO2 photocatalyst realized by cationic S-doping and its application for phenol degradation[J]. Journal of Hazardous Materials, 2008, 152: 48-55. doi: 10.1016/j.jhazmat.2007.06.062 [19] PUTLURU S S R, SCHILL L, JENSEN A D, et al. Mn/TiO2 and Mn-Fe/TiO2 catalysts synthesized by deposition precipitation-promising for selective catalytic reduction of NO with NH3 at low temperature[J]. Applied Catalysis B:Environmental, 2015, 165: 628-635. doi: 10.1016/j.apcatb.2014.10.060 [20] GUO R T, WANG Q S, PAN W G, et al. The poisoning effect of heavy metals doping on Mn/TiO2 catalyst for selective catalytic reduction of NO with NH3[J]. Journal of Molecular Catalysis A:Chemical, 2015, 407: 1-7. doi: 10.1016/j.molcata.2015.06.017 [21] SUN P, HUANG S X, GUO R T, et al. The enhanced SCR performance and SO2 resistance of MnOx/TiO2 catalyst by the modification with Nb: A mechanistic study[J]. Applied Surface Science, 2018, 447: 479-488. doi: 10.1016/j.apsusc.2018.03.245 [22] CHEN Q L, GUO R T, WANG Q S, et al. The catalytic performance of Mn/TiWOx catalyst for selective catalytic reduction of NOx with NH3[J]. Fuel, 2016, 181: 852-858. doi: 10.1016/j.fuel.2016.05.045 [23] QU R Y, PENG Y, SUN X X, et al. Identification of the reaction pathway and reactive species for the selective catalytic reduction of NO with NH3 over cerium-niobium oxide catalysts[J]. Catalysis Science & Technology, 2016, 6: 2136-2142. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4656
- HTML全文浏览数: 4656
- PDF下载数: 60
- 施引文献: 0




















