


-
2022年全球塑料总量达4亿吨,同比增长约600万吨,其中90.6%为化石基塑料,仅有不到9%被机械、化学等回收利用,大量垃圾被最终填埋或因管理不善等各种原因排放到环境中[1]. 自20世纪70年代,首次在水生环境中发现塑料垃圾以来[2],已陆续在世界各地的空气[3]、海洋[4]、土壤[5]和地表水[6],甚至在北极与深海[1,7]等人类活动稀少的偏远地区都检测到NPs的存在. 在风化的长期影响与紫外线辐射(UV)以及其他环境因素[8]的作用下,环境中的塑料垃圾可降解成小于5 mm的微塑料(MPs)和100 nm纳米塑料(NPs)[9]. 并通过吸入、摄入,特别是食物链的方式进入人体,在人类粪便中检测到的NPs也为其提供了进一步的证据[10]. MPs、NPs可通过诱导氧化压力或内质网压力等进入不同生物体内,影响生殖系统、消化系统、神经系统等不同身体系统,造成胃肠道毒性、肝脏毒性、生殖毒性、神经毒性和关节毒性等多方面毒性[11]. 由于NPs尺寸较小,比表面积较大,且细胞亲和性高于MPs,更易穿过生物屏障进入血液、细胞、组织和器官,对生物造成更严重的危害[12].
近年来,已有研究关注NPs在胃肠道当中的毒性. 在细胞水平上,Li等研究发现,暴露于聚苯乙烯纳米塑料会改变肠道微生物群的组成,从而诱发菌群失调,引发肠道炎症反应[13];Mahler等[14]研究则集中探讨了口服聚苯乙烯纳米粒子对肠上皮细胞体外模型和鸡肠环体内模型中的铁吸收和铁运输的影响,发现暴露于高剂量纳米颗粒的肠道细胞显示出铁转运增加,而急性暴露于羧化颗粒(直径50 nm)的鸡比未暴露或长期暴露的鸡具有更低的铁吸收率. 在分子水平上,研究主要关注纳米粒子的毒性机制,包括氧化应激、脂质代谢失调、能量代谢异常、以及由此引发的炎症反应和细胞凋亡[15],Hu等[16]发现,摄入聚苯乙烯微塑料会增加炎症和氧化应激相关基因的表达,从而诱导哺乳动物细胞炎症和氧化应激增加. 塑料毒性效应与纳米粒子的粒径、比表面积和形态等物理化学特性密切相关. 当食物基质参与其中,不同分子与胶体物质会与NPs表面相互作用[17],产生生物电晕,对NPs的结构、电荷、聚集状态与生物毒性等产生潜在影响[18]. Ersöz等[19]研究发现,在消化模拟后,牛奶的存在会改变TiO2 NPs的细胞毒性,对细胞的生存能力造成不利影响,同时,裸露的以及与食物基质相互作用的NPs在经过消化后所获得的不同电晕结构,可能对其肠道吸收和慢性毒性产生巨大影响;Zhang等[20]则发现标准食物模型的存在下,NPs的毒性进一步降低. 因此,确定NPs与消化道食物基质的相互作用与作用机制,对了解NPs的口服毒性,评估人体的健康安全风险具有重要意义.
然而,当前对于NPs与人体健康的研究聚焦于颗粒在消化系统中的最终形态,大多关注纳米颗粒与消化液在接触时间终点的物化性质变化,忽略了食物基质与纳米颗粒的相互作用及其口胃肠消化过程的影响[18]. 本实验以应用范围较广的聚苯乙烯纳米塑料为研究对象,建立人体胃肠道(GIT)三级体外模拟消化系统,探究在蛋白质、碳水化合物和油等多种食物基质作用下,NPs在胃肠道的命运与稳定性.
-
聚苯乙烯纳米塑料购自纽邦生物科技有限公司,酪蛋白酸钠、玉米油、淀粉、果胶、蔗糖、胆盐购自上海麦克林生化科技股份有限公司,α-淀粉酶、胃蛋白酶、胆汁盐、胰液购自Sigma-Aldrich,NaCl来自阿拉丁试剂(上海)有限公司.
-
根据制造商给定的相关参数,新鲜的聚苯乙烯纳米塑料悬浮液的质量浓度为10.06%. 取0.099 mL该悬浮液于容量瓶中,并用超纯水定容至100 mL,摇匀后制成实验所需的100 mg·L−1聚苯乙烯纳米塑料悬浮液[21]. 将制备好的溶液转入棕瓶,于4 ℃冰箱内避光储存,在使用前利用NaOH或HCl调整至合适pH.
-
配制浓度为5 mmol·L−1的磷酸盐缓冲液,调pH为7.0[22].
-
标准食物模型(SFM)依据中国居民膳食指南[23](即2.89%蛋白质,1.44%糖类,1.32%膳食纤维,13.8%淀粉,3.21%脂肪,0.37%矿物)制成,如表1所示.
将酪蛋白酸钠(1%,W/W)在室温下溶解于磷酸盐缓冲溶液(10 mmol·L−1,pH 7)中,连续搅拌2 h,至完全溶解,随后对溶解的酪蛋白酸钠盐溶液进行过滤,去除剩余未溶解的粉末微粒. 将玉米油(3.21%,W/W)加入该酪蛋白酸钠溶液中,连续摇晃10 min后,在高剪切搅拌机
16000 r·min−1转速下搅拌2 min. 产生的粗乳剂再通过高压均质器,在8.27×104 kPa上通过3次,形成细乳剂(蛋白浓度为2.89%,W/W,脂肪浓度为3.21%,W/W). 最后将果胶(1.32%,W/W)、淀粉(13.8%,W/W)、蔗糖(1.44%,W/W)和氯化钠(0.37%,W/W)等其他食品成分依次加入到乳液相中,充分搅拌,确保每一成分完全溶解,达到规定浓度. 通过喷雾干燥法将其从流体形式转化为粉末状,液体形式放入台式喷雾干燥机(进样速率0.45 L·h−1,入口温度170 ℃,喷雾器喷嘴0.7 mm),喷雾干燥后在4 ℃下储存,以蒸馏水进行重组[20]. -
酪蛋白酸钠、淀粉和玉米油组成的标准化食品基质分别代表日常摄入的蛋白质、碳水化合物和油的来源,依次制备其他食物模型[24].
将酪蛋白酸钠在室温条件下溶解于磷酸盐缓冲溶液(10 mmol·L−1,pH 7)中,配制其浓度为2.89% W/W,并利用磁力搅拌器连续搅拌,以确保粉末完全分散. 将淀粉加入磷酸盐缓冲溶液(10 mmol·L−1,pH 7)中,利用磁力搅拌器连续搅拌,最终配制淀粉浓度为13.8% W/W. 以同样的方法,配制玉米油浓度为3.21% W/W.
-
为研究不同消化阶段和不同食物基质对NPs大小和分布变化的影响,建立一个包括口腔、胃和小肠3个阶段的GIT模拟器模拟体内的消化过程. 体外消化方案改编自先前的研究[25],将先前配好的100 mg·L−1 NPs加入到食品模型中进行重悬,37 ℃,100 r·min−1的水浴磁力搅拌中孵育. 在唾液步骤中,取上述制备的悬浮液20 g加入20 g含23.6 mg·L−1α-淀粉酶,2 g·L−1氯化钠的模拟唾液,以1 mol·L−1 HCl调节pH至6.8,持续孵育10 min. 取出20 g唾液消化后的样品进行分析. 在胃消化步骤中,以1 mol·L−1 HCl调节pH至2.5,加入20 mL 2 g·L−1的氯化钠溶液,并加入胃蛋白酶,使消化体系中胃蛋白浓度达到1.6 mg·mL−1. 100 r·min−1孵育2 h后,取出10 g胃期消化的样品. 随后加入1 mol·L−1 NaOH调节pH至7.0,再孵育2 h. 每一步消化后,收集样品并用于表征NPs. 在烧瓶中加入1.5 mL含0.25 mol·L−1 CaCl2·2H2O和3.75 mol·L−1 NaCl的模拟肠液,然后加入3.5 mL(0.05 g·mL−1)的胆汁盐溶液. 将系统调整到pH 7.0. 然后,将加入2.5 mL胰酶溶液(0.036 g·mL−1)加入烧杯中,在37 ℃下,100 r·min−1的转速下孵育2 h.
-
利用Zetasizer Nano ZS90,采用时间动态光散射法(DLS),使用波长为633 nm的4.0 mW氦氖激光器,测定溶液背景下NPs的水动力学直径(Dh)与粒径分布. 需注意,在测量前应用磷酸缓冲溶液对样品进行适当稀释,以避免多重散射效应[26].
根据相分析光散射法(PALS),使用Zetasizer Nano ZS90仪器测量不同溶液化学条件下聚苯乙烯纳米颗粒的电泳迁移率(electrophoretic mobility,EPM). 对于每个溶液条件,对样品连续测定10次取平均值,并重复3次以确保数据质量,然后利用公式(1)换算为zeta(ζ)电位:
式中,ζ为表面电位(mV);η为黏度(Pa·s−1);UE为电泳迁移率(cm2·V−1·s−1);ε为介电常数;f(Ka)为Henry函数.
-
在将NPs悬浮液加入标准化食品基质中并模拟其通过GIT之前,检测其在纯水中的物理化学性质(表2). 可测得NPs的平均粒径为54.47 nm. 随着pH从2.5升高到6.8和7,NPs的ζ电位从−17.47 mV增强至−25.52 mV和−29.19 mV,这可能是由于更多的氢氧根离子吸附到NPs的表面所致[26]. 表明在纯水环境中,随着pH的升高,NPs的胶体稳定性逐渐增强[27].
-
对不同食物基质下,NPs在口、胃、肠的3个阶段消化样品进行了检测,并设置空白消化液、加入食物基质后的消化液等实验组别进行对照,表现出不同食物基质下NPs粒径分布的不同变化趋势. 由于塑料的粒径超出仪器的检测范围,在淀粉和油类处理下,未显示出胃期的粒径分布曲线. 整体上,与空白消化液(图1(a))、加入食物基质后的消化液(图1(b)-(f))相比,加入纳米塑料后,粒径分布曲线大多从单峰态转变为多峰态,且表现出整体向粒径较小方向移动的趋势,有别于空白消化液、加入食物基质后的消化液等实验组别. 这表明实验测定的为纳米塑料,且可能受到食物、蛋白酶等颗粒的干扰,加入纳米塑料后,塑料会与食物、消化液组分发生相互作用,并结合形成新的团聚体.
与空腹食物模型(FFM)相比(图1(b)),加入了SFM、protein、starch、oil不同食物基质后,未消化的NPs粒径分布呈现多峰态,平均粒径增加,体系较为分散. 在标准食物模型(图1(c))下,相较于未消化阶段,在口、胃、肠三级消化阶段中的粒径分布都较为集中,均为单峰态,这表明标准食物模型的聚集状态相对稳定[23],可能与标准食物模型内食物基质丰富均匀,分阶段消化的特性有关. 在蛋白食物基质(图1(d))下,口腔与肠期都与消化前阶段都表现出相似的粒径分布形态,这可能归因于蛋白质在NPs表面形成生物电晕的干扰[28]. 而在胃期,由于胃蛋白酶对蛋白的消化作用,弱酸集团的质子化增强了其静电排斥力与空间电阻,使其进一步稳定下来[29]. 在淀粉处理(图1(e))下,口腔阶段和未处理阶段表现出几乎一致的粒径分布曲线,这可以用:淀粉完全溶解在唾液中,并且与NPs之间不会发生静电相互作用来解释[30]. 最后,在油处理(图1(f))下,呈现出在肠期较为宽敞的粒径分布曲线,这也有油在肠期阶段被分解为脂肪酸的特性有关[31].
-
如图2所示,加入纳米塑料后的Dh区别于空白消化液(图2(a))、加入食物基质后的消化液等实验组别,进一步证实所测定的颗粒为纳米塑料颗粒,且会与食物、消化液等发生相互作用.
未消化阶段,空腹食物模型(图2(b))下的Dh与初始平均粒径(54.47 nm)相差不大,标准食物模型(图2(c))、蛋白质(图2(d))、淀粉(图2(e))、油(图2(f))等食物基质作用下则显著增大,分别增大至666.1、405.17、89.03、618.22 nm. 这可能是由于食物基质会被吸附于NPs表面,增大其团聚体的粒径,并根据各食物基质的不同性质表现出不同的大小(标准食物模型>油>蛋白>淀粉>空腹食物模型). 与未消化的NPs-食物基质模型相比,经口、胃、肠各消化处理后Dh显著增大,进一步佐证了体内消化过程对NPs凝聚动力学的重要作用.
在口腔阶段,除蛋白质下降以外,其他食物基质下的Dh都表现出一定程度的增加. Zhou等[28]之前的研究也观察到类似的现象,这可能是由于蛋白质与NPs的表面快速结合形成了蛋白质电晕,并通过提供空间位阻和静电斥力分散NPs. 一些关于银纳米塑料的研究也表明,口腔阶段食物基质对NPs的粒径几乎没有影响[32],与本次实验结果相一致. 这表明在此阶段,消化酶、pH变化等因素对液体的化学性质影响可能发挥了更大的作用[18].
在胃期,与口腔阶段相比,所有处理下的Dh都增加,这可能是由于pH下降,黏蛋白的羧酸基团被质子化,大大降低了NPs的胶体稳定性,影响NPs与酶的相互作用,最终促进酶对NPs的吸附并增强它们的凝聚[32]. 此外,高离子强度的介质也可能是另一个原因[33]. 在此阶段,蛋白质(图2(d))的变化(Dh从口期的377.73 nm增加至
4942.67 nm)还可归因于被胃蛋白酶消化为氨基酸、肽和氢氧根离子,降低了NPs之间的静电排斥和空间电阻,增强了其的团聚[34]. 对于油的处理,据报道,静电排斥的减少可能是由于液滴电荷的减少和低pH下离子强度的增加[32],这一点可从图3胃期表面ζ电位接近于0的实验数据中得到进一步的证实.在肠期,NPs的Dh相较于胃期都呈现出明显的减少,而油类基质(图2(f))表现最为显著. 这可能是由于pH升高,胰酶的加入则会分解在胃期中形成的团聚体. 而在油类食物基质的处理下,NPs表面的玉米油被脂肪酶(如胰蛋白酶)完全或部分消化为脂肪酸,并与胆盐混合形成混合胶束,以影响NPs的凝聚[31]. DeLoid等[29]也观察到了类似的现象,即由于表面三酰甘油的消化,部分Fe2O3纳米粒子在小肠阶段的粒径减小.
从整体来看,相较于初始状态,淀粉(图2(e))对NPs的流体动力学直径增幅最大,从口期至胃期, Dh由229.7 nm增加至
30036.67 nm,相较于其他食物基质,肠期阶段粒径也最大,Dh为1096.33 nm,对NPs的凝聚与胶体稳定性的影响最显著. 这可能是由于淀粉中的亲水大分子的添加量较大,能更有效地影响胶体的稳定性. 这表明食用淀粉类物质可能意味着增强其生物毒性与人体健康风险. -
如图3所示,测量了NPs在不同食物模型下,口胃肠三级消化阶段的ζ电位的变化,这与生物分子和NPs表面的结合密切相关.
口腔阶段,NPs-食物模型均带有负电荷,范围从−5.77 mV到−34.27 mV,这表明其可能都具有较强的胶体稳定性. 而到达胃期,所有的NPs表现出几乎完全的电荷消除,ζ电位接近于0,甚至在FFM、food matrix、starch参与下,NPs呈现出电荷反转并带有轻微正电荷,这可能是由于胃蛋白酶通过吸附形成蛋白冠,对颗粒表面电荷进行了屏蔽作用[24]. 也有一些研究表明,可能是与低pH值(~1.5)下弱酸基团的质子化[29]以及低于黏蛋白的等电点(pI 3—5)等有关[25]. 肠期阶段,NPs又重新表现为负电荷,且范围在−21.29 mV到−28.02 mV. 在之前的一些研究中就有强调消化酶、胆汁盐与NPs的结合[32],而这次实验中得出了较为一致的结果,在食物基质的参与之下,食物以及消化后形成的肽聚糖或脂肪酸可与NPs相结合,并在他们的周围形成电晕,进一步稳定NPs悬浮液,这一点可从肠期阶段ζ电位的上升中可见得. 吸附的胆汁表面带有的负电荷[35]与具有表面活性剂的性质[36]都可使得NPs更为分散.
而不同食物基质的作用下,胶体的稳定性则表现为口腔阶段:油(−34.27 mV)>标准食物模型(−19.67 mV)>淀粉(−11.97 mV)>空腹食物模型(−9.93 mV)>蛋白(−5.77 mV),胃期:空腹食物模型((9.30 ± 0.58)mV)>标准食物模型(5.31 mV)>油(−1.41 mV)>蛋白质(−1.31 mV)>淀粉(0.33 mV),肠期:油(−28.02 mV)>标准食物模型(−23.44 mV)>蛋白(−21.64 mV)>空腹食物模型(−21.4 mV)>淀粉(−21.29 mV). 其中,淀粉在胃期的极不稳定性表明,淀粉能更有效地通过增加平均粒径,降低ζ电位来降低胶体的稳定性. 对比于淀粉与蛋白相似的不稳定性,油类食物基质在三级消化道中表现出较强的稳定性,特别是从胃期到肠期的转变(ζ电位由−1.41 mV转变为−28.02 mV),这可归因于在肠期中脂肪酶的存在. 肠液中含有的胰液中含有脂肪酶,可进一步催化脂肪的水解,因此油类基质可在肠期被转化为脂肪酸,从而稳定NPs使其ζ电位的绝对值增加[32]. 同样的,在口腔阶段,淀粉相较于蛋白质更稳定的原因也可能归因于口腔阶段淀粉酶的作用. 而在先前一些研究当中,发现的油和淀粉在凝聚作用中更相似,而牛奶起更稳定的作用,这一点的差异也用NPs的类型以及消化液制备的方式不同进行解释. 而从消化的整体过程来看,则表现出标准食物模型比空腹食物模型更强的稳定性的效果.
-
以往研究往往忽略食物基质与消化过程的影响,本实验通过建立GIT模拟体外消化三级模型,探究标准食物基质、蛋白质、碳水化合物、油等日常摄入的食物基质对人体内NPs粒径变化与胶体稳定性的影响. 实验结果表明,食物基质的参与会影响人体内NPs的粒径与表面电位的变化,与未消化阶段的54.47 nm相比,蛋白、淀粉、油类等食物基质加入后,Dh增至405.17、89.03、618.22 nm. 总体呈现口、胃期粒径增大、稳定性降低,肠期粒径减小、稳定性增大,并表现为各食物基质下不同消化阶段的不同特性. 淀粉通过增大粒径,降低ζ电位来达到较好的胶体稳定的效果,在胃、肠期的粒径最大,分别为
30036.67 、1096.33 nm,ζ电位分别为0.326、−21.29 nm. 蛋白则主要通过吸附于NPs表面形成蛋白冠与在胃期被胃蛋白酶消化,进一步降低NPs之间的静电排斥和空间电阻,增强其的团聚,胃期粒径从377.73 nm增加至4942.67 nm. 油由于易被肠期的胰酶分解为脂肪酸,并与胆盐混合形成混合胶束,在肠期表现出最稳定的性质,ζ电位为−28.017 nm. 以上结果显示,NPs由于受到各消化酶对各食物基质的分解、pH的变化、介质电解质等的影响,在模拟体外消化道中与不同的食物基质发生复杂的相互作用,从而改变其胃肠道行为与胶体稳定性. 前人研究大多关注接触时间终点的物化性质变化,即使有食物基质的考量也往往局限于单种物质的研究. 基于此,本研究选择了代表中国居民膳食摄入的标准食物模型与蛋白质、淀粉、油类等多种食物基质,并结合体外消化模型,尽可能真实地模拟纳米塑料在人体胃肠道的行为与胶体稳定性,为进一步评估人类在日常饮食中所面临的纳米塑料危机与健康安全风险提供了依据,也同时为纳米营养品的运输与功能发挥提供了新的思路与研究路径,推动纳米塑料健康影响评估研究的发展.
不同食物基质对纳米塑料的胃肠道行为与胶体稳定性的影响
Effects of different food matrices on gastrointestinal fate and colloidal stability of nanoplastics
-
摘要: 随着塑料产量的持续增加和回收率的低下,大量塑料进入环境,并通过机械磨损、光照和生物降解等途径分解为小于100 nm的纳米塑料(NPs). 它们通过口服摄入、呼吸吸入和皮肤接触等途径进入人体,造成生殖、神经和关节等多方面的毒性,严重威胁人体健康. 目前,NPs的研究主要集中在水环境和胃液中的迁移转化,且大多数忽视了食物基质的作用. 为进一步阐明NPs在人体内的作用机制并评估其安全风险,本研究拟建立人体胃肠道(GIT)体外消化模型,使用时间动态光散射法(DLS)探讨NPs在不同食物基质下的胃肠道行为和胶体稳定性. 结果表明,食物基质会吸附在NPs表面,增大其初始粒径. 在加入蛋白、淀粉、油类等食物基质后,NPs的水动力学直径(Dh)从54.47 nm增加到405.17、89.03、618.22 nm. 该变化受到消化过程中的pH、溶液电解质和消化酶的影响. 在胃期阶段,zeta(ζ)电位接近0;而在肠期阶段,ζ电位表现为负电荷,范围从−21.29 mV到−28.02 mV. 总体而言,NPs在胃期阶段表现为粒径增大和胶体不稳定,而在肠期阶段则表现为粒径减小和胶体稳定. 食物基质在这一过程中发挥了重要作用. 蛋白质会被蛋白酶降解,在NPs表面形成蛋白冠,这使其在口腔阶段表现出独特的粒径增加,并在胃期呈现出更强的胶体不稳定性(ζ电位为−1.308 mV. 淀粉作为中性分子,不影响体系中的静电相互作用,但能够通过增加粒径和降低ζ电位来更有效地降低胶体稳定性. 从口腔到胃期,Dh由229.7 nm增加至
30036.67 nm. 油类物质在胰酶作用下被消化为脂肪酸,并吸附在NPs表面,与胆盐形成胶束,在肠期表现出更强的胶体稳定性,ζ电位为−28.017 mV,高于其它食物基质模型. 本研究结果可为NPs在食物基质与消化道作用下的胃肠道行为提供新思路,对人体健康风险的评估具有重要意义.Abstract: With the continuous increase in plastic production and the low recycling rates, a significant amount of plastic enters the environment and is decomposed into nanoplastics (NPs) smaller than 100 nm through processes including mechanical abrasion, light irradiation, and biodegradation. They enter the human body via various pathways, such as oral ingestion, inhalation, and dermal contact, to cause reproductive, neurological, and joint toxicity, posing a serious threat to human health. Currently, research on NPs mainly focuses on their migration and transformation in aqueous environments and gastric fluids, while most studies overlook the role of food matrices. To further elucidate the interaction mechanisms of NPs in the human body and assess their safety risks, this study aims to establish an in vitro gastrointestinal tract (GIT) digestion model and use time-resolved dynamic light scattering (DLS) to investigate the gastrointestinal behavior and colloidal stability of NPs under different food matrices. The results showed that food matrices adsorbed onto the surface of NPs, increasing their initial particle size. After adding food matrices such as protein, starch, and oil, the hydrodynamic diameter (Dh) of NPs increased from 54.47 nm to 405.17, 89.03 and 618.22 nm. This change was affected by the pH, solution electrolytes, and digestive enzymes during the digestion process. In the gastric phase, the zeta (ζ) potential was close to 0; while in the intestinal phase, the ζ potential exhibited negative charges, ranging from −21.29 mV to −28.02 mV. Overall, NPs exhibited increased particle size and colloidal instability in the gastric phase, while showing reduced particle size and colloidal stability in the intestinal phase. Food matrices played an important role in this process. Proteins were degraded by proteases, forming protein corona on the surface of NPs, leading to unique increases in particle size in the oral phase and stronger colloidal instability in the gastric phase (ζ potential of −1.308 mV). Starch, as a neutral molecule, did not affect the electrostatic interactions in the system but more effectively reduced colloidal stability by increasing particle size and decreasing ζ potential. From the oral to the gastric phase, Dh increased from 229.7 nm to30036.67 nm. Oily substances were digested by pancreatic enzymes into fatty acids, adsorbed onto the surface of NPs, and mixed with bile salts to form micelles. This resulted in stronger colloidal stability in the intestinal phase, with a ζ potential of −28.017 mV, higher than that of other food matrix models. The results of this study provide new insights into the gastrointestinal behavior of NPs under the influence of food matrices and the digestive tract, which is of great significance for the assessment of human health risks. -
作为由空气污染引发的全球性灾难之一,酸雨被认为是当今最严重的一种跨境环境问题[1]。酸雨问题主要源于工业化石燃料的燃烧(产生SO2)和汽车尾气的排放(产生NOx)[1],因此,人们治理酸雨的主要方法就是限制SO2和NOx等酸性污染物的产生。早在工业革命时期,酸雨问题就已经在美国出现,但直到1970年以前都未曾受到应有的关注,只是被用来反映空气质量状况的一项指标。随着1972年在瑞典斯德哥尔摩举行的“联合国人类环境会议”上被首次报道,加之酸雨危机愈演愈烈,美国从联邦政府层面,开始了对酸雨治理模式的探索之路。
1. 2种酸雨治理模式的主要特征
在当今酸雨治理领域中,命令—控制(Command-and-Control,CAC)和“市场调节”(Market-Based Instrument, MBI)是2种最具代表性的环境管制模式,美国完成由前者到后者的转型历经了二十余载。鉴于这2种模式在构成要素及运作机制等方面具有本质性的区别,在此,以酸雨治理实践为例,从实施主体、动力来源和运行机制3个方面,首先对2种模式的主要特征加以阐释。
1.1 酸雨治理模式的实施主体
酸雨治理模式的实施主体,是指对酸雨进行治理的行为主体,它承担着酸雨治理的责任,并被赋予实施治理的相应权力。实施主体对酸雨治理的主观认知和态度,及对酸雨进行治理的能力,都会对酸雨治理的客观效果产生重要影响。
命令—控制模式的实施主体主要是国家机构(包括:行政、立法和司法3个组成部分),它们借助其在公共事务管理中法定的权力和权威,以发布酸雨管制命令、制定酸雨防治法规、设定SO2排放标准、开展对排污单位的监督审查等方式,实施对酸雨的治理。
市场调节模式则通过将酸雨治理的环境成本与企业的经济利益相挂钩的方式,将酸雨治理的行为主体由国家机构转向企业。受经济利益的驱使,企业会自发地依据其自身经营的成本和收益状况,灵活地做出“污染多少,治理多少”的选择,而不必受制于环境监管者设定的酸雨管制模式。
1.2 酸雨治理模式的动力来源
酸雨治理模式的动力来源,是指推动酸雨治理模式得以实施的主要动因和激励因素,它在很大程度上影响到酸雨治理主体的积极性和努力程度。
命令—控制模式的动力来源,是在世界环保运动蓬勃兴起与美国国内环境污染日益严峻的背景下,国家机构对于本国经济、社会发展,特别是环境保护事业所必须担负的责任、义务和职能。
市场调节模式的动力来源,则是市场运行中成本—收益衡量下的经济效益。在市场调节模式中,市场之所以能够发挥作用的关键在于其借助一些经济激励措施,将企业治理酸雨的费用内化到其经济运行的成本当中。为了花费最少的治污成本换取最大的经济效益,企业便会想方设法地减少污染排放。因此,较之命令—控制模式,市场调节模式在酸雨治理的过程中会呈现出更加灵活、自觉的特点。
1.3 酸雨治理模式的运行机制
酸雨治理模式的运行机制,是指推动酸雨治理模式运行并发挥作用的逻辑、程序、方式与路径,它随酸雨治理模式中其他要素所产生的变化而变化,直接决定了某一酸雨治理模式的具体表现形态。
命令—控制模式依靠国家机构的行政和法律手段,以外部约束的方式对致使酸雨产生的排污企业实施最直接的管控。在这一过程中,国家机构首先需要通过立法手段界定法律所允许的SO2等酸性污染物的最大排放量,然后再借助行政手段对那些超过最大排放量的行为施以惩罚。由于该模式的运行建立在各种严格的酸雨管制标准和严厉的行政处罚措施基础之上,因此实施主体对于各企业排污信息的掌握尤为重要。
市场调节模式则主要依靠经济和市场手段,以内在约束的方式对致使酸雨产生的排污企业实施间接性管控。为了充分调动企业治理酸雨的积极性,在制定总体的SO2减排目标后,环境监管者会借助一些经济激励措施,以经济诱导环保的方式将企业治理酸雨的责任与其追求经济效益的动机结合起来,以此来促使其自发地采取一些酸雨治理措施。由于该模式的运行依靠市场经济的自我调节机制,因此合理恰当的经济激励措施是其得以有效运转的重要保障。
2. 美国酸雨治理的“命令—控制”时期(1970—1990年)
20世纪70年代以前是美国酸雨治理的“无政府时代”。彼时,以联邦政府为主的国家机构对于国内酸雨乃至空气污染问题并未施加过多的干预。而后,随着酸雨等跨境污染问题愈发严峻,各州和地方政府的“画地为牢”式治理方式,以及民间环保运动的“分散性”弊端日显。美国最终于1970年伴随《国家环境政策法》的实施确立了以国家管制为主要特征的命令—控制模式,并由此开启了借助行政、立法和司法手段共同管制空气污染的“命令—控制”时期。
2.1 实施主体
“命令—控制”时期美国治理酸雨的主体是以美国联邦政府为主的国家机构——既包括一般意义上以总统为首的行政机构,还包括以国会为代表的立法机构和以联邦法院为代表的司法机构。
在国家机构的三方实施主体中,起主导性作用的是以总统为首的行政机构。1970年,尼克松政府开启了以联邦政府的名义就每年所需要解决的环境问题向国会提交咨文的先例[2],还特别成立了以美国环保局为代表的专门性环境管制机构。自此,“环境保护不再仅仅是各州和地方政府的职能,美国联邦政府开始承担起对环境保护的主要领导责任”[3]。国会作为各方利益集团相互博弈的场所,也在此期间表现出强烈的环保价值取向。一般而言,在参众两院表决时,民主党人支持环保的热情要远高于共和党人。在民主党人掌控着参众两院多数席位的70年代,国会先后于1970和1977年2次修订《清洁空气法》。联邦法院虽然没有像上述主体的态度倾向那样明显,但在总统和国会这两大利益主体的共同影响下,其作为宪政秩序维护者的形象,在不损害宪法效力的情况下也是会考虑到维护联邦政府和国会的权威,从而向环保势力一方倾斜的。
由此可见,在美国治理酸雨的命令—控制模式中,尽管作为共同实施主体的三大部门行使权力的领域各不相同,但其在此时期内支持美国酸雨治理的态度却是基本趋于一致的。
2.2 动力来源
20世纪70年代是美国酸雨问题的全面爆发时期,也是命令—控制模式的高度发展时期,其发展的主要动因在于国家机构承担环保责任的需要。
从1970年1月1日《国家环境政策法》正式生效的那一刻起,美国进入到了“环境保护的十年”。这一时期,无论是国家机构对于环境污染问题的重视程度,还是在出台环保政策的数量和水平上,都较之前有了显著提高。与此同时,美国的酸雨现象在70年代初期愈发严峻,甚至已损害到美国国家的经济利益和人民的生活质量乃至生命安全。在此影响下,特别是在热衷于环保的民主党人占参众两院多数席位的70年代,作为“最具有政治责任感的联邦机构”[4],国会和各州议会首先应承担起作为国家立法机构的重大使命——通过环境立法的方式对酸雨问题实施管控。
鉴于酸雨问题不仅是一个环境问题,更是一个国际关系问题——一方面,它涉及美国与邻国加拿大之间在共同应对跨境空气污染问题时进行的合作与博弈;另一方面,在国内环保运动高涨和经济下行压力增大的背景下,作为国家机构的核心组成部分,联邦政府也需要向公众对其所必须承担的环保责任作出积极回应,以求弥补在经济问题上损伤的政治权威。特别是酸雨本就是一个跨境性质的空气污染问题,污染程度的加深使得各州与地区之间各自治污的状态逐渐被打破,在环境产权界定不清晰的情况下,市场的自发调节机制只会使污染者的“搭便车”行为愈演愈烈。因此,无论是出于环境质量改善的目的,还是出于国家机构所必须担负的环保责任的考量,都迫切需要联邦从国家政治层面对酸雨问题加以协调和管制。
2.3 运行机制
命令—控制模式的运行围绕国家立法领域的措施展开,配合行政和司法手段的干预,三者共同开创了美国以国家强制力方式来解决酸雨问题的先河。
首先,从国家立法层面来看。命令—控制时期是联邦以立法手段应对酸雨问题的开端,这为日后美国“酸雨计划”的正式出台奠定了充实的法理依据。在《1970年清洁空气法修正案》中,法案授权美国环保局对于6种危害公众健康与福利的空气污染物质设置“国家环境空气质量标准(National Ambient Air Quality Standards, NAAQS)”[5],其中就包括致使酸雨产生的SO2和NOx。在《1977年清洁空气法修正案》中,法案对于酸雨问题的管控更加具体。在第123条条款中,法案增加了限制高烟囱的使用,使之不能经常作为一项污染控制技术的决定[6];在第110和第126条条款中,法案首次就各州酸性污染物的跨州际传播及相关申诉问题进行了界定[6];在第115条条款中,法案则对国家解决酸性污染物跨境传播的程序问题作出了说明[6]。
其次,从行政措施层面来看。联邦政府在此期间发布的酸雨管制命令主要表现在以下3个方面:第一,根据1970年法案的要求制定SO2和NOx等酸性污染物的排放标准[7],见表1。
表 1 国家环境空气质量标准Table 1. National Ambient Air Quality Standards污染物 国家一级环境质量标准 国家二级环境质量标准 均值类型 标准浓度水平 均值类型 标准浓度水平 一氧化碳 8小时 10 mg·m−3 未设定 无 1小时 40 mg·m−3 未设定 无 铅 平均每季最大值 1.5 μg·m−3 平均每季最大值 1.5 μg·m−3 氮氧化物 每年算数平均值 100 μg·m−3 每年算数平均值 100 μg·m−3 臭氧 每天1小时平均最大值 235 μg·m−3 每天1小时平均最大值 235 μg·m−3 颗粒物(PM10) 每年算数平均值 50 μg·m−3 每年算数平均值 50 μg·m−3 24小时 150 μg·m−3 24小时 150 μg·m−3 二氧化硫 每年算数平均值 80 μg·m−3 3小时 1300 μg·m−3 24小时 365 μg·m−3 | Show Table DownLoad:
CSV
DownLoad:
CSV
1971年4月,美国环保局分别将SO2和NOx的“国家一级空气质量标准”的年度算数平均值设为80和100 μg/m3[8]。自此,美国的酸雨管控工作有了科学的依据和准则。第二,为了使更多地区尽快达到NAAQS的要求,美国环保局采纳了通过建造高烟囱的方式来将SO2等酸性污染物排放到高空中,以此来稀释和分散地表污染物浓度的高烟囱政策。据悉,1960年,美国烟囱的平均高度约74 m;1970年,已经存在2个高度超过152 m的烟囱;到了1980年,烟囱的平均高度已达到223 m[9]。第三,开始就跨境酸雨防治问题与加拿大政府展开合作。1978年,两国建立了“空气污染物远距离传输双边研究磋商小组”,开始对酸雨问题展开科学研究[10]。到了1979年,两国又共同起草了一份关于酸雨在北美地区的起因、传播和影响等内容的科学报告[11],同时还签署了《跨界空气质量联合声明》,表达了美加双方想要“共同减少或防止损害公众健康和财产安全的跨界空气污染问题的决心”[10]。
最后,从司法诉讼层面来看。美国联邦法院支持为酸雨治理申诉的态度主张也在此期间表现得愈发明显,一个典型的案例是1973年“西宾夕法尼亚电力公司诉特雷恩案”[12]。西宾夕法尼亚电力公司是一家主要排放含硫化合物的电力企业。1973年,它因不满所在州向美国环保局提交的州实施计划(State Implementation Plan, SIP)中对于SO2等酸性污染物设定了高于NAAQS的做法向联邦法院提起诉讼。最终,最高法院以凡是符合1970年法案要求的SIP均可被执行为由驳回了电力公司因过高的经济技术成本而想要执行另外一种“不那么严格”的排污标准的主张[12]。此外,在70年代中期的“美国自然资源委员会诉环保局案”“美国大河电力公司诉环保局案”“美国肯尼科特铜业公司诉特雷恩案”等案件中,联邦法院同样对于工业企业试图通过建造高烟囱以规避NAAQS的做法进行了“抵制”[9]。
3. 美国酸雨治理的“市场调节”时期(1990年至今)
从20世纪70年代末期起,受滞胀危机的持续影响,全国对于不计成本治理污染的命令—控制模式的信心开始动摇。但鉴于酸雨危机的不断恶化乃至国际化,又使得美国无法完全放弃对酸雨的治理,因而从这时开始,酸雨治理模式中逐渐孕育出一些市场因素。这些市场因素在大力倡导“反环保、促发展”的里根政府时期逐渐完善和发展,最终,随着《1990年清洁空气法修正案》第四章“酸雨计划”(Acid Rain Program)的出台,美国正式确立了酸雨治理的市场调节模式。与命令—控制模式不同,在该模式中,市场机制逐渐开始取代联邦管控的统治地位,在酸雨乃至空气污染治理领域中日渐发挥起主导性作用。
3.1 实施主体
作为市场经济的主要参与者,美国在1990年之后进行酸雨治理的主体,主要就是受到国家“酸雨计划”管制的各类排放SO2和NOx等酸性污染物的工业企业。但是,鉴于市场调节机制本身所具有的开放性,与排污企业密切相关的一些诸如美国联邦政府、工商业投机者以及环保主义者等“非污染物排放主体”也积极参与其中。
首先来说市场调节模式中最重要的实施主体——排污企业。通常而言,主动参与到酸雨治理过程中的排污企业有2种:一种是在“酸雨计划”中被明确列出的法定执行者,包括被列入“酸雨计划”第一阶段管控范围的位于美国东部和中西部21个州的110家发电厂的263个污染源,和第二阶段在此基础上增加的2 000多家生产规模大于2.5万KW的SO2排放企业[13],它们是导致美国SO2排放总量增加的主要力量;另一种则是通过“选择—加入计划(Opt-in program)”参与其中的自愿加入者,包括一些生产规模小于2.5万千瓦的发电设备、工业锅炉以及未被列入“酸雨计划”的城市废物焚烧炉等污染源[14]。由于它们自主降低1 t的SO2排放的成本通常会低于排污交易市场中1 t的SO2排放配额的平均售价,因此加入“酸雨计划”的SO2排污交易机制中能够使其“有利可图”。
此外,市场调节模式还有3种“非污染物排放主体”。其一是以美国环保局为代表的政府监管者,他们主要通过监督和审查的方式对市场调节模式辅之以必要的行政管控措施。其二是工商业投机者,他们通过将SO2排放配额视作一种投机产品,以“低买高卖、赚取差价”的方式活跃着SO2排污交易市场。其三是环保主义者,他们购买SO2排放配额的主要目的是将其储蓄不再卖出,以此来减少排污配额在交易市场中的流通总量,从而达到控制社会中SO2总体排放量的目的。当然,除了环保主义者之外,政府有时也会扮演这一角色。
3.2 动力来源
1990年“酸雨计划”的出台是美国市场调节模式取代命令—控制模式的最终胜利,该模式之所以能够发挥作用,其主要驱动力在于各排污企业对经济效益的不断追求。
早在“酸雨计划”出台前的数十年间,国会便已经开始就如何处理SO2排放的问题进行了反复的辩论。截至1989年(即“酸雨计划”正式出台的前一年),国会已然对酸雨问题的解决提出了大大小小共计70多个议案[15],这些议案所反映出的共同主旨在于,在最大限度节省治污成本的基础上寻找到解决酸雨问题的新路径。由于滞胀危机余波的影响,国家机构和排污企业在此期间不约而同地将目光聚焦于注重成本—收益分析的市场手段上。随后,在各方势力就如何通过市场机制的创新来节省酸雨治理成本、提高酸雨治理效率等问题展开了数轮博弈与交锋后,最终得以兼顾企业和环保主义者利益的、既清洁又廉价的“酸雨计划”应运而生。
不同于命令—控制模式中不计成本的强制性污染管控模式,经济发展的诉求在“酸雨计划”的运行过程中始终占据着最重要的位置。一方面,这是出于对20世纪80年代国家经济需要快速恢复和发展的现实需要的考虑;另一方面,唯有将企业对经济效益的追求引入污染治理领域,换言之,唯有通过经济刺激的方式才更能引导企业自觉地在最大程度上、且最持久地对空气污染问题作出改善。而从后续“酸雨计划”的实施过程来看,这的确是传统意义上的命令—控制模式所不能及的。
在市场调节模式的实施下,借助于排污配额的交易过程,美国不仅成功建立了世界上首个SO2排污权交易市场,还通过允许合理买卖SO2排放权利的方式为企业常常会选择“搭便车”的污染治理责任赋予了经济价值。其实,无论是在“酸雨计划”出台之前的讨论设计阶段,还是在市场调节模式的正式运行阶段,以企业经济效益为重的理念贯穿于“酸雨计划”的运行始终。因为从某种意义上来说,它不仅是政策制定者引导排污企业自觉参与到酸雨治理进程中的一座桥梁,更是在市场经济自发调节的机制下保障排污权交易过程得以正常运转的最大动力。
3.3 运行机制
以“酸雨计划”为代表的市场调节模式的运行机制主要包含以下4个方面。
3.3.1 总量上限的控制机制
这一机制具有2层含义:首先,对致使酸雨产生的SO2和NOx等酸性污染物设定明确的总量上限。《1990年清洁空气法修正案》在第四章开篇中明确表示:“为减少酸沉积带来的不利影响,预将国家本土48个州及哥伦比亚特区的SO2年排放总量在1980年的排放水平上减少1 000万t,将NOx的排放量在1980年的排放水平上减少200万t”[16]。随后,国会还将达成总量上限目标的具体过程划分为两个阶段,并分别设定更具体的排污上限。其次,在实现总量上限目标的方式选择上,“酸雨计划”给了各排污单位足够的灵活性。换句话说,在规定的期限内完成总量上限目标的前提下,各排污单位可根据其自身的运营情况选择“最能使其效益最大化”的污染治理方式。此外,“酸雨计划”还鼓励各排污单位通过自主研发或引进治污新技术的方式来实现SO2排放总量的减少。
3.3.2 配额的初始分配机制
排污配额(allowance),即美国环保局授予各排污单位排放定额SO2的权利[16]。在“酸雨计划”中,各排污单位获得SO2排放配额的方式主要有3种:无偿获取、有偿交易和技术奖励。其中,无偿获取方式是三者中最主要且最稳定的一种配额分配来源,大约占初始分配配额总量的97.2%[17]。按照1990年法案规定,美国环保局局长需对所有被管制的排污单位分配为期1年的排污配额,且配额分配的数量应等于法案允许的SO2最高排放上限[16]。除了无偿获取,各排污单位还可通过有偿交易的方式购买到一定数量的排污配额。根据法案要求,美国环保局会从“酸雨计划”第一、二阶段无偿发放的排污配额总量中各扣除2.8%的数额,用于建立一个“特定配额储备”,以供排污配额的有偿出售[16]。通常情况下,美国环保局出售排污配额的方式主要有2种:直接出售和拍卖出售。此外,各排污单位还可通过更新现有排污技术设备的方式来获取额外的“奖励性配额”。
3.3.3 排污交易机制
这是整个“酸雨计划”中最核心的环节。根据“排污配额可在各排污单位和任意配额持有者之间进行交易和转让”[16]的要求,美国建立了世界上首个SO2排污交易市场。在此基础上,各排污单位可对其削减1 t的SO2的成本与市场中一单位排污配额的售价进行比较,以此来自行决定其实现SO2总量上限目标的方式。当然,对于本年度没有抵消完SO2实际排放量的剩余配额,可转入下一年使用;对于“酸雨计划”第一阶段内没有使用完的排污配额,可转入第二阶段使用;但是不可在配额未正式发放前就“预使用”排污配额[16]。
3.3.4 监督审查机制
这一机制包含以下3个子系统:排污跟踪系统、配额跟踪系统和年度调整系统。排污跟踪系统是监督审查机制的核心组成部分,主要依靠在各污染源上安装和运行对于SO2的“连续排放监测系统”来实现[16]。通常情况下,各排污单位至少要以1个季度为期限,定期将其监测到的SO2排放数据向美国环保局如实汇报,以此来确保美国环保局在执行“酸雨计划”其他各运行机制时所需要的可靠数据来源。配额跟踪系统是美国环保局为掌握其自身及各排污单位的配额持有情况而建立的一种计算机控制系统,也是查阅配额分配、交易、年终审查等官方记录的唯一途径。最后,在对于排污单位的年度SO2排放总量和年度排污配额持有量等信息都准确掌握之后,美国环保局便会通过年度调整系统核算出这一年各排污单位应被扣除的排污配额总量。当然,对于最终年度排污配额持有量不足以抵消其年度SO2排放总量的排污单位,美国环保局要对其超额排放SO2的行为施以严厉的行政和经济处罚。
4. 对我国环境污染管制的启示
4.1 树立人与自然和谐共生的可持续发展理念
在经济至上的发展观和功利主义自然观的指引下,以美国为首的发达国家在环境治理领域曾走上了一条“先污染后治理”式的发展道路。在70年代生态危机空前加剧的背景下,尽管美国一度确立了以国家管制为主要特征的命令—控制模式,但随着滞胀危机的爆发及国内经济形势的下滑,很快又出现了将经济发展的重要性置于环境保护之上的倾向。虽然经过各个利益集团的反复博弈,致力于平衡经济与环境效益的“酸雨计划”最终产生,但还是对环境造成了不可逆的损害。作为发展中国家,我们要充分利用好后发优势,吸取资本主义国家经济发展过程中的经验教训,同时要将美国“酸雨计划”中所体现出的这种经济与环境平衡发展的理念,与我国传统文化中的“和谐”思想相融合,秉承人与自然和谐共生的优秀传统文化,树立“绿水青山就是金山银山”的科学发展理念,坚持在发展中保护,在保护中发展。
4.2 坚持行政管制与市场调节有机结合的发展模式
美国酸雨治理模式的转型历经了从“依靠市场力量—市场失灵—政府干预—政府失灵—重新重视市场力量”这一“失灵”与“回归”相交错的探索路径。1990年“酸雨计划”出台后,美国建立了世界上第一个国家管制层面的SO2排污交易市场,较之改革路径上最初依靠的“单一型市场”而言,这个新型市场的建立与其说是再次依靠市场力量,不如说是有政府的干预为其保驾护航。这也是90年代以前美国单纯依靠市场或政府力量为何会频繁出现“失灵”的原因,这种“回归”实则是对命令—控制模式进一步市场化的结果。由此可见,任何一种单一型污染治理模式都并非完美无瑕,在进一步深化我国环境管制模式的市场化改革之际,我们一定要从单纯依靠政府或市场力量的一元论思维中解脱出来,本着“管制为先,市场并重”的原则将二者有机结合起来,以此来制定出更契合新时代中国特色社会主义市场经济体制的环境管制模式。
4.3 积极参与多边化、区域化、全球化的环保合作
酸雨、温室气体排放等跨境环境问题的解决不仅是某一国的个体行为,还是一种国际行为,需要国家与国家之间的团结协作。美加酸雨谈判始于1977年,直至1991年双方才最终签署美加《空气质量协定》(Air Quality Agreement)。在此15年间,即便是在双方政府曾就共同治理酸雨的问题多次达成共识的前提下,还是有协议因美方政府的单方面否认而夭折。这是霸权主义国家以牺牲他国环境为代价维护本国利益的惯用手段,我们不但不应效仿还应对其予以谴责和抵制。作为全球生态文明建设的参与者、贡献者与引领者,我们要摒弃任何国家任何形式的单边主义,加强与邻国及世界其他各国在国际环保事务中的多边化、区域化与全球化合作,努力提升本国对国际环保事业的自主贡献率,为积极构建人类命运共同体贡献出中国力量,在全球环境治理中充分彰显大国担当。
4.4 发挥制度优势,建立健全社会主义环境立法体系
美国酸雨治理模式变迁既有《1970年清洁空气法修正案》授权同意将酸雨治理的主导权收归联邦政府作为法律保障,同时也与美国数百年形成的市场经济制度相匹配。但在此之前,美国曾耗时半个多世纪的时间去确立联邦政府管制环境进而解决酸雨危机的合法性。如此,不仅可以使美国在一定程度上避免环境管制又回到70年代以前的“州权时代”,还能够防止日后这些酸雨管控条例成为一纸空文。对比美国这一环境治理体系,作为社会主义国家,我国在发挥中央政府治理环境的主导权方面具有天然的优势;但鉴于我国当前还处于社会主义发展的初级阶段,社会主义市场经济体制本身发展得并不完善。为此,在社会主义市场经济与治污模式的市场化改革双项并举的时代,我国应从国情出发,在充分发挥中国特色社会主义集中力量办大事制度优势的前提下,不断提升市场力量在污染治理过程中的杠杆性作用,同时进一步健全与我国社会主义市场经济体制相匹配的环境法律体系,以使我国的环境治理真正做到有法可依、有章可循。
-

图 1 100 mg·L−1 PSNPs 在体外消化过程中与不同食物基质作用下的尺寸分布
Figure 1. Size distributions of 100 mg·L−1 PSNPs during each phase in vitro digestion with the different food matrices
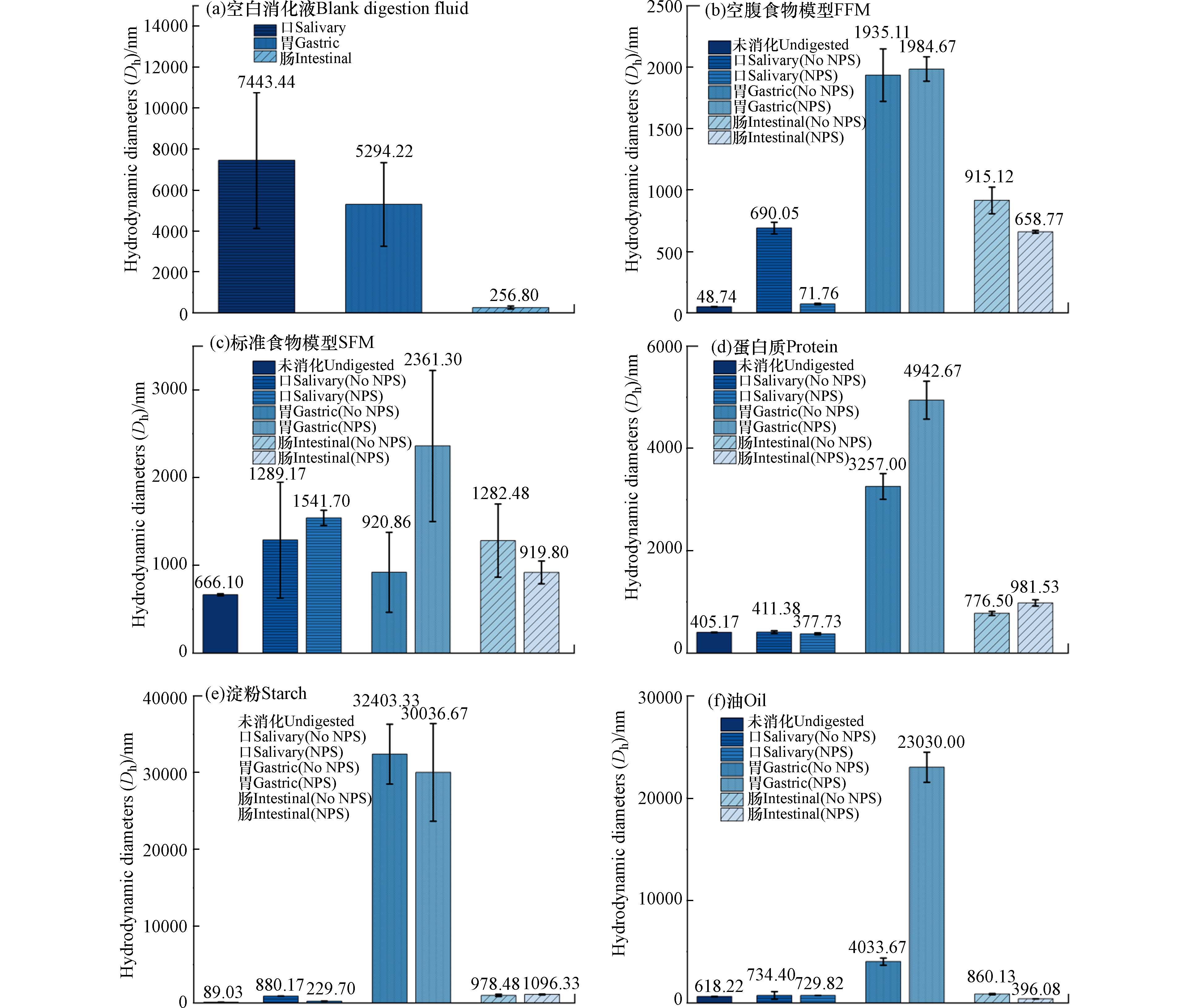
图 2 100 mg·L−1NPs在不同食物基质体外消化各阶段的流体动力学直径(Dh)
Figure 2. Hydrodynamic diameters (Dh) of 100 mg·L−1PSNPs during each phase of in vitro digestion with different food matrices
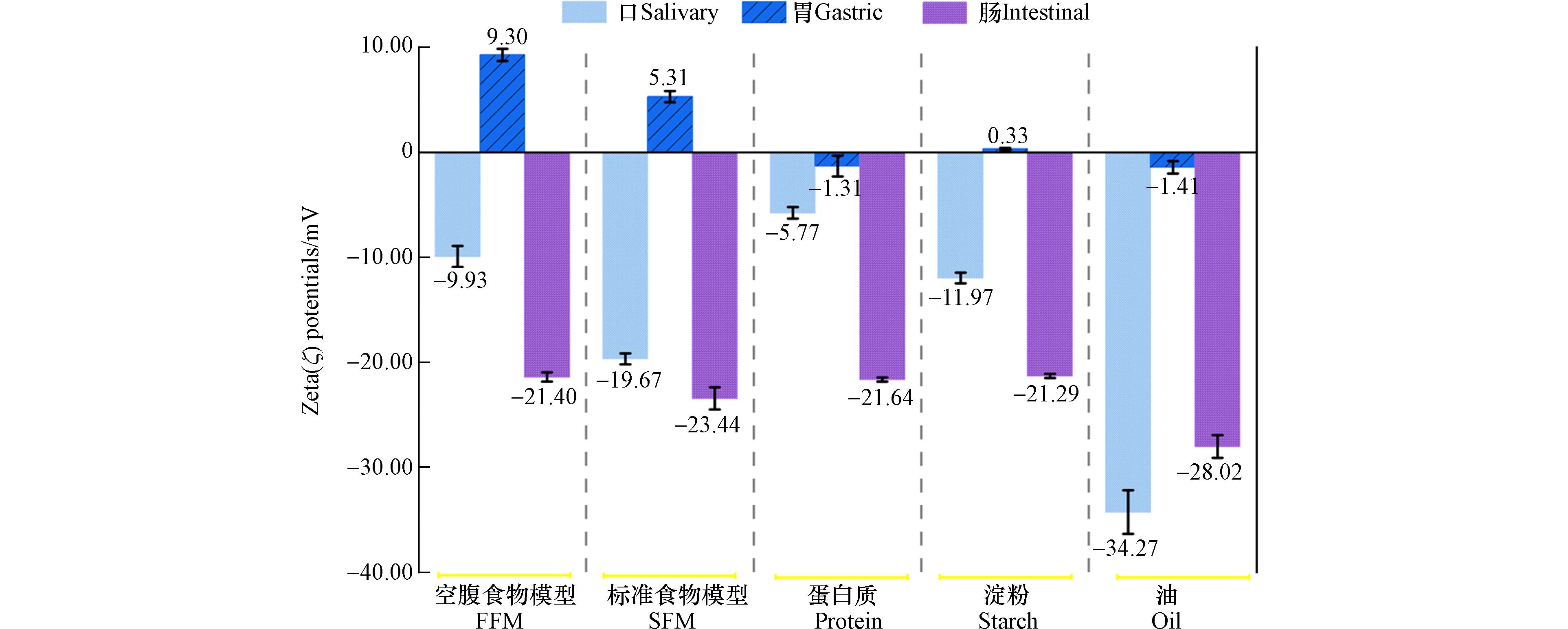
图 3 100 mg·L−1 NPs在不同食物基质体外消化的各个阶段的Zeta (ζ)电位.
Figure 3. Zeta (ζ) potentials of 100 mg·L−1NPs during each phase of in vitro digestion with different food matrices.
组成成分Composition 含量浓度/%Concentration 蛋白质(酪蛋白酸钠) 2.89 糖类(蔗糖) 1.44 膳食纤维(果胶) 1.32 淀粉(玉米淀粉) 13.8 脂肪(玉米油) 3.21 矿物(氯化钠) 0.37
下载: 导出CSV
表 2 NPs的初始性质
Table 2. Initial properties of nanoplastics
平均粒径/nmAverage particle size 表面电位/mVSurface potential pH 2.5 pH 6.8 pH 7 54.47 −17.47 −25.52 −29.19
下载: 导出CSV
-
[1] ALIMI O S, FARNER BUDARZ J, HERNANDEZ L M, et al. Microplastics and nanoplastics in aquatic environments: Aggregation, deposition, and enhanced contaminant transport[J]. Environmental Science & Technology, 2018, 52(4): 1704-1724. [2] CARPENTER E J, JR SMITH K L. Plastics on the Sargasso sea surface[J]. Science, 1972, 175(4027): 1240-1241. doi: 10.1126/science.175.4027.1240 [3] PANKO J M, CHU J, KREIDER M L, et al. Measurement of airborne concentrations of tire and road wear particles in urban and rural areas of France, Japan, and the United States[J]. Atmospheric Environment, 2013, 72: 192-199. doi: 10.1016/j.atmosenv.2013.01.040 [4] Da COSTA J P, SANTOS P S M, DUARTE A C, et al. (Nano)plastics in the environment–Sources, fates and effects[J]. Science of the Total Environment, 2016, 566: 15-26. [5] NIZZETTO L, BUSSI G, FUTTER M N, et al. A theoretical assessment of microplastic transport in river catchments and their retention by soils and river sediments[J]. Environmental Science. Processes & Impacts, 2016, 18(8): 1050-1059. [6] DRIS R, GASPERI J, ROCHER V, et al. Microplastic contamination in an urban area: A case study in greater Paris[J]. Environmental Chemistry, 2015, 12(5): 592. doi: 10.1071/EN14167 [7] CHAE Y, AN Y J. Effects of micro- and nanoplastics on aquatic ecosystems: Current research trends and perspectives[J]. Marine Pollution Bulletin, 2017, 124(2): 624-632. doi: 10.1016/j.marpolbul.2017.01.070 [8] LIU Z Q, LI Y M, PÉREZ E, et al. Polystyrene nanoplastic induces oxidative stress, immune defense, and glycometabolism change in Daphnia pulex: Application of transcriptome profiling in risk assessment of nanoplastics[J]. Journal of Hazardous Materials, 2021, 402: 123778. doi: 10.1016/j.jhazmat.2020.123778 [9] TER HALLE A, LADIRAT L, GENDRE X, et al. Understanding the fragmentation pattern of marine plastic debris[J]. Environmental Science & Technology, 2016, 50(11): 5668-5675. [10] SCHWABL P, KÖPPEL S, KÖNIGSHOFER P, et al. Detection of various microplastics in human stool: A prospective case series[J]. Annals of Internal Medicine, 2019, 171(7): 453-457. doi: 10.7326/M19-0618 [11] CHANG X R, XUE Y Y, LI J Y, et al. Potential health impact of environmental micro- and nanoplastics pollution[J]. Journal of Applied Toxicology, 2020, 40(1): 4-15. doi: 10.1002/jat.3915 [12] YANG S L, LI M Z, KONG R Y C, et al. Reproductive toxicity of micro- and nanoplastics[J]. Environment International, 2023, 177: 108002. doi: 10.1016/j.envint.2023.108002 [13] CHAMAS A, MOON H, ZHENG J J, et al. Degradation rates of plastics in the environment[J]. ACS Sustainable Chemistry & Engineering, 2020, 8(9): 3494-3511. [14] MAHLER G J, ESCH M B, TAKO E, et al. Oral exposure to polystyrene nanoparticles affects iron absorption[J]. Nature Nanotechnology, 2012, 7(4): 264-271. doi: 10.1038/nnano.2012.3 [15] XU X, FENG Y D, HAN C J, et al. Autophagic response of intestinal epithelial cells exposed to polystyrene nanoplastics[J]. Environmental Toxicology, 2023, 38(1): 205-215. doi: 10.1002/tox.23678 [16] HU Q L, WANG H, HE C, et al. Polystyrene nanoparticles trigger the activation of p38 MAPK and apoptosis via inducing oxidative stress in zebrafish and macrophage cells[J]. Environmental Pollution, 2021, 269: 116075. doi: 10.1016/j.envpol.2020.116075 [17] McCLEMENTS D J, DeLOID G, PYRGIOTAKIS G, et al. The role of the food matrix and gastrointestinal tract in the assessment of biological properties of ingested engineered nanomaterials (iENMs): State of the science and knowledge gaps[J]. NanoImpact, 2016, 3: 47-57. [18] YIN C Y, ZHAO W L, LIU R, et al. TiO2 particles in seafood and surimi products: Attention should be paid to their exposure and uptake through foods[J]. Chemosphere, 2017, 188: 541-547. doi: 10.1016/j.chemosphere.2017.08.168 [19] ERSÖZ N, ÇANGA E M, YILDIRIM-ELIKOGLU S, et al. Effect of real food matrix on the behavior and toxicity of TiO2 nanoparticles[J]. J Nanopart Res, 2022, 10: 211. [20] ZHANG Z P, ZHANG R J, XIAO H, et al. Development of a standardized food model for studying the impact of food matrix effects on the gastrointestinal fate and toxicity of ingested nanomaterials[J]. NanoImpact, 2019, 13: 13-25. doi: 10.1016/j.impact.2018.11.002 [21] DeLOID G M, CAO X Q, COREAS R, et al. Incineration-generated polyethylene micro-nanoplastics increase triglyceride lipolysis and absorption in an in vitro small intestinal epithelium model[J]. Environmental Science & Technology, 2022, 56(17): 12288-12297. [22] COREAS R, CAO X Q, DeLOID G M, et al. Lipid and protein Corona of food-grade TiO2 nanoparticles in simulated gastrointestinal digestion[J]. NanoImpact, 2020, 20: 100272. doi: 10.1016/j.impact.2020.100272 [23] LI Y, JIANG K, CAO H, et al. Establishment of a standardized dietary model for nanoparticles oral exposure studies[J]. Food Science & Nutrition, 2021, 9(3): 1441-1451. [24] LALOUX L, KASTRATI D, CAMBIER S, et al. The food matrix and the gastrointestinal fluids alter the features of silver nanoparticles[J]. Small, 2020, 16(21): 1907687. doi: 10.1002/smll.201907687 [25] LI Y, McCLEMENTS D J. Influence of non-ionic surfactant on electrostatic complexation of protein-coated oil droplets and ionic biopolymers (alginate and chitosan)[J]. Food Hydrocolloids, 2013, 33(2): 368-375. doi: 10.1016/j.foodhyd.2013.04.016 [26] LIU Y J, HU Y B, YANG C, et al. Aggregation kinetics of UV irradiated nanoplastics in aquatic environments[J]. Water Research, 2019, 163: 114870. doi: 10.1016/j.watres.2019.114870 [27] LU S H, ZHU K R, SONG W C, et al. Impact of water chemistry on surface charge and aggregation of polystyrene microspheres suspensions[J]. Science of the Total Environment, 2018, 630: 951-959. doi: 10.1016/j.scitotenv.2018.02.296 [28] ZHOU P F, GUO M F, CUI X Y. Effect of food on orally-ingested titanium dioxide and zinc oxide nanoparticle behaviors in simulated digestive tract[J]. Chemosphere, 2021, 268: 128843. doi: 10.1016/j.chemosphere.2020.128843 [29] DeLOID G M, WANG Y L, KAPRONEZAI K, et al. An integrated methodology for assessing the impact of food matrix and gastrointestinal effects on the biokinetics and cellular toxicity of ingested engineered nanomaterials[J]. Particle and Fibre Toxicology, 2017, 14(1): 40. doi: 10.1186/s12989-017-0221-5 [30] GO M R, BAE S H, KIM H J, et al. Interactions between food additive silica nanoparticles and food matrices[J]. Frontiers in Microbiology, 2017, 8: 1013. doi: 10.3389/fmicb.2017.01013 [31] ZHANG Y Y, PIGNATELLO J J, TAO S, et al. Bioaccessibility of PAHs in fuel soot assessed by an in vitro digestive model with absorptive sink: Effect of food ingestion[J]. Environmental Science & Technology, 2015, 49(24): 14641-14648. [32] KÄSTNER C, LICHTENSTEIN D, LAMPEN A, et al. Monitoring the fate of small silver nanoparticles during artificial digestion[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2017, 526: 76-81. [33] WALCZAK A P, FOKKINK R, PETERS R, et al. Behaviour of silver nanoparticles and silver ions in an in vitro human gastrointestinal digestion model[J]. Nanotoxicology, 2013, 7(7): 1198-1210. doi: 10.3109/17435390.2012.726382 [34] CAO X Q, HAN Y H, LI F, et al. Impact of protein-nanoparticle interactions on gastrointestinal fate of ingested nanoparticles: Not just simple protein Corona effects[J]. NanoImpact, 2019, 13: 37-43. doi: 10.1016/j.impact.2018.12.002 [35] van AKEN G A. Relating food emulsion structure and composition to the way it is processed in the gastrointestinal tract and physiological responses: What are the opportunities?[J]. Food Biophysics, 2010, 5(4): 258-283. doi: 10.1007/s11483-010-9160-5 [36] DEGEN L P, PHILLIPS S F. Variability of gastrointestinal transit in healthy women and men[J]. Gut, 1996, 39(2): 299-305. doi: 10.1136/gut.39.2.299 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 318
- HTML全文浏览数: 318
- PDF下载数: 21
- 施引文献: 0
