









-
沙尘颗粒物是全球大气颗粒物的主要贡献来源之一,最新研究表明,全球的沙尘排放量随着土地荒漠化和气候变化的加剧而增加[1 − 3],其中全球沙尘年排放总量预估达
1000 —8000 Tg[4]. 沙尘颗粒物能够通过吸收和反射太阳光影响区域热辐射和气候变化[5]. 沙尘天气不仅对起沙源及附近区域造成空气质量影响,沙尘细颗粒物也可通过远距离传输并造成半球甚至全球范围的影响. 例如,2021年3月蒙古国和我国西北地区发生大规模沙尘天气,受沙尘影响部分地区PM10峰值浓度超过5000 µg·m−3[6],同时沙尘颗粒物通过高层气流远距离传输并与我国华南地区的城市大气污染物耦合形成了复合型大气污染事件[7]. 2023年春季(3—4月),我国共爆发数十次高强度、大范围的沙尘天气。在季风和蒙古气旋的影响下,沙尘的发生和传输对下风向城市的空气质量和居民生活产生了显著的影响[8]. 沙尘天气的产生是非常复杂且难以预测的,我国2021年和2023年相继发生的大范围沙尘也引发了人们对于沙尘暴发生的频率和强度增加的担忧. 研究沙尘暴对城市环境的影响以及其与城市大气污染物的复合过程也变得越加重要,其中污染物的耦合、分布、迁移、转化和沉降仍然具有高度的不确定性,阻碍了对沙尘复合污染事件的溯源、归因和环境风险等分析.本文讨论了沙尘颗粒物的化学组成、传输和大气化学过程,重点梳理沙尘颗粒物与气态化合物非均相反应机理的研究现状,对气态分子在沙尘颗粒物表面的吸附规律、氧化机制、非均相过程和间接环境影响展开讨论,并对未来研究进行了展望.
-
沙尘颗粒物主要指由强风吹过裸露的土壤和荒漠地表而形成的大气悬浮颗粒物. 其颗粒物的粒径通常小于100 µm,且富含各类矿物质,主要包括二氧化硅、硅酸盐、碳酸盐以及金属氧化物等. 大气中沙尘颗粒物的矿物质组分可以通过X射线衍射(XRD)、X射线荧光光谱(XRF)、电感耦合等离子体质谱和离子色谱等进行分析. 沙尘颗粒物的元素组成与地壳基本一致,其中含量最高的元素是O和Si,含量最高的金属元素是Al[9]. 沙尘通常同时含有Fe、Ca、K、Mg、Na、Ti等常见的金属元素,以及Mn、P等微量元素. 沙尘中的矿物质元素主要以金属氧化物、碱性氢氧化物和硅酸盐等形式存在[10]. 沙尘颗粒物的矿物质组分差异通常受到母岩种类、岩石风化和土壤发生过程、气候条件、植被种类和人类活动等多种因素影响[11],因此不同地区的沙尘颗粒物通常具有不同的元素类型及比例(图1)[12 − 23]. 例如,由于岩石类型和风化过程的区别,亚洲地区的沙尘颗粒物(黄土高原和戈壁沙漠等)中的碱性金属元素(Ca和Mg)含量较其他地区的沙尘更高,而撒哈拉沙漠南部区域的沙尘颗粒物中则含有较多的Al元素. 不同区域的沙尘颗粒物因风化程度和传输过程等差异,颗粒物比表面积也不一样. 例如,亚利桑那测试沙尘(Arizona Test Dust, ATD)、腾格里沙漠沙尘和北京沙尘天气期间采集的东亚沙尘的比表面积(Brunauer–Emmett–Teller,BET)分别为4.0、16.5 、 6.1 m2·g−1 [24]. 此外,前期研究发现沙尘颗粒物经过大气复合过程后,其理化性质有所改变. 例如,有报道采用X射线扫描电镜分析黄河三角洲区域的沙尘与霾混合颗粒并发现沙尘暴后颗粒物被硝酸钙包裹,从而对颗粒物的吸湿性造成一定的影响 [25].
沙尘颗粒物中粒径较小的细颗粒物通常能够在表层紊流和对流空气的作用下悬浮,并进一步在季风等高空大气环流的作用下实现中、远距离的迁移和传输. 沙尘颗粒物的形成、传输和沉降与地表的裸露程度、区域气候的条件以及植被覆盖的程度都有一定的关联. 在冬春季节,我国西北部地区干燥、多风,同时植被覆盖率低,而土壤和岩石风化频率的增加也导致了沙尘释放量的升高[26]. 近期有研究指出,在我国西北部的起沙区域内,由于受到上风向大范围起尘的影响,位于下风向的城市整体沙尘的传输和沉降量呈现自西向东和自北向南逐渐增加的趋势[27]. 沙尘颗粒物的矿物质组分对传输过程中的大气化学过程具有重要的影响,而矿物质类型则主要由起沙源决定. 目前常用的沙尘颗粒物溯源技术包括同位素示踪法[28 − 31]和基于拉格朗日轨迹模型的理论计算. 例如,Chen等[32]结合区域气象模式和轨迹模型分析了2023年3月至4月我国北部强沙尘暴事件,发现蒙古和塔克拉玛干沙漠是主要的沙尘源,其中蒙古对我国北部沙尘浓度有超过42%的贡献. 在全球范围内,沙尘颗粒物的主要影响范围涉及非洲撒哈拉沙漠区域、中东、亚洲、澳大利亚和北美洲西海岸部分区域. 其中,撒哈拉沙漠的沙尘通过跨大西洋传输到加勒比海地区和美洲东部,并维持了亚马逊区域的矿物质输入[33];中东地区的沙尘则主要源自伊拉克、叙利亚和沙特阿拉伯等地区,并可通过中远距离传输到东亚地区[34];而亚洲的沙尘主要来自中国西北和蒙古地区,通过季风和气旋传输到朝鲜半岛、日本,甚至北美洲和北极地区[35].
-
沙尘颗粒物的平均寿命可达数周,从起沙源头至远距离迁移的过程中,沙尘通过其表面的自然孔隙吸附、摄取各类气态污染物分子. 与大气中其他有机或无机盐颗粒物相比,沙尘颗粒物的表面形状多变且比表面积更大. 沙尘颗粒物对气体分子的吸附主要通过气体分子扩散与颗粒物表面碰撞并发生摄取,其中吸附速率常数或摄取系数(
γ )可以由公式(1)进行计算[36]:其中,[X]为气体化合物的空气质量浓度,
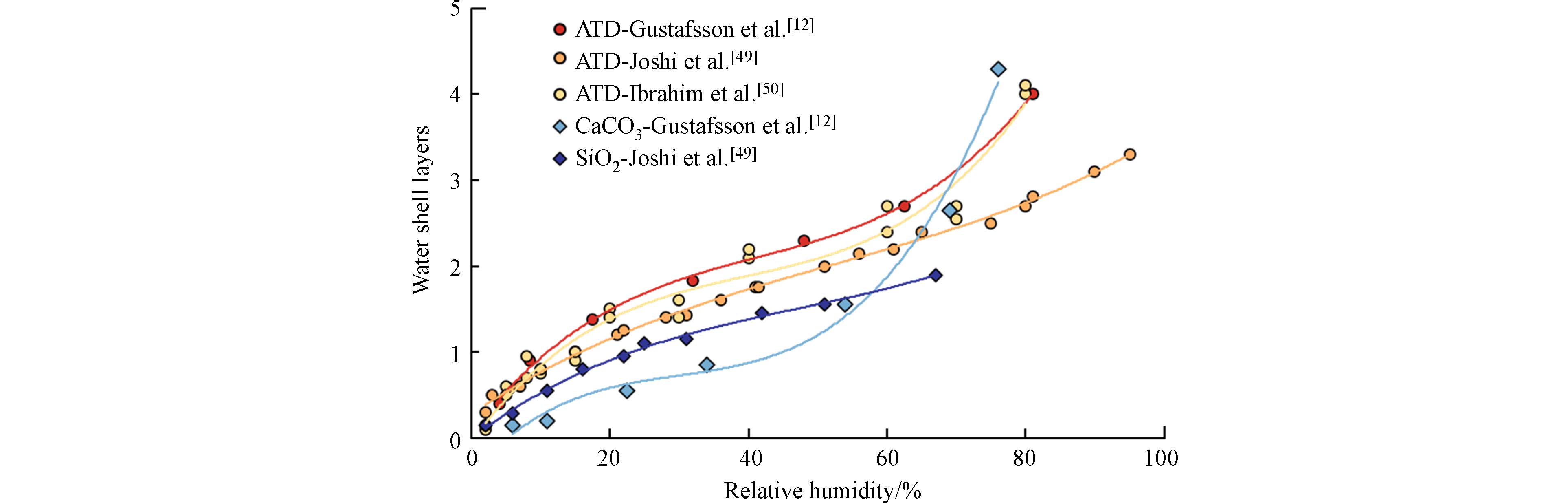
ˉc 是分子X的平均热力学速度(cm·s−1),A为沙尘颗粒物的比表面积(cm2·cm−3). 颗粒物的比表面积通常采用几何比表面积或通过BET(Brunauer–Emmett–Teller)方法测定表征. 在稳定状态下由BET比表面积计算所得的吸附速率常数则可表示为γBETss . 国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry, IUPAC)于2009年起陆续汇总了各类气体分子在沙尘颗粒物表面的非均相反应速率、计算方法和单位等参数,其中气态化合物主要包括O3、H2O2、OH自由基、NOx、HNOx和SO2等[36]. 气体分子在沙尘颗粒物表面的吸附速率受众多因素的影响. 例如颗粒物表面的活性位点、气体分子的极性以及相应的空气质量浓度等[36 − 38]. Underwood等[39]通过对NO2和气态HNO3在沙尘颗粒物表面的吸附速率进行精确测量,发现沙尘颗粒物中的矿物质含量和比表面积差异对污染物的吸附速率有较大的影响,同等条件下氮氧化物在中国黄土高原的沙尘颗粒物表面的吸附速率比在源自撒哈拉沙漠的沙尘颗粒物表面的吸附速率高约2—10倍.沙尘作为主要的大气颗粒物之一,既参与了大气污染物的多相反应过程,也是对流层凝结核重要来源之一,因此沙尘颗粒物对水分子的吸附特性即吸湿性对大气成云、降水、热辐射和区域气候都有一定的影响[40]. 研究表明,沙尘颗粒物的吸湿性受物理化学因素和环境条件的影响,包括沙尘颗粒物表面的亲水基团、化学组成、颗粒物尺寸和几何形状等[38, 40]. 另一方面,颗粒物吸湿性的提高显著增加了水分子在其表面的吸附速率、覆盖厚度和覆盖范围,也对气体分子的吸附和非均相反应造成了一定的影响. 在低湿条件下,颗粒物表面尚未形成单层水分子壳,水分子和其他气体分子可能发生竞争机制,争夺颗粒表面的吸附位点. Mogili等[41]通过吸附实验发现O3在Fe2O3和Al2O3颗粒表面的初始吸附速率随湿度增加(RH<30%)而降低. 随着环境湿度的不断升高,颗粒物吸附的水分子将逐渐超过并覆盖其表面积并最终形成多层水合壳(图2)[12, 42 − 43]. 同时,颗粒物表面的部分空隙被水分子填充并导致比表面积的降低,从而使气体分子的吸附从气-固交换逐渐转变为气-液交换,颗粒表面水层则作为反应媒介进一步加速推动液相反应的进行[40]. 目前采用较多的沙尘吸湿性分析方法包括水分子气压变化测定[44]、质量动态测定[45]、FTIR测定含水量[46]、扫描电镜和扫描隧道显微镜等手段观测颗粒物的吸湿变化[47]、基于双差分电迁移分析仪(Differential Mobility Analyzer, DMA)的H-TMDA颗粒物吸湿增长测定法等[48]. 研究表明,沙尘颗粒物的吸湿性随颗粒物的迁移、老化等一系列环境化学过程而改变. 例如,硝酸盐具有较强的亲水性,沙尘颗粒物在吸附或表面形成硝酸盐后其吸湿性有明显的增强[49, 50]. 外场观测证实,沙尘过程后约有32%的沙尘颗粒由于硝酸盐的包裹而发生了吸湿增强[20].
-
吸附于沙尘颗粒物表面的化合物分子往往会与颗粒物表面的氧化物或自由基发生氧化还原反应,并生成二次产物. 黑暗条件下,颗粒物表面的活性位点上的氧原子能够与表面吸附的化合物分子发生缓慢的氧化还原反应,即表面自发性氧化反应[51]. 其反应速率主要受到化合物分子的吸附速率、氧分子的吸附速率以及表面反应速率的影响. 在光照条件下,沙尘颗粒物中具有一定光催化活性的金属氧化物,如Fe2O3、Al2O3和TiO2等,能够激发形成电子-空穴对(e−cb-h+vb),并与颗粒物表面的氧气和水等反应生成强氧化自由基(如羟基自由基和超氧自由基)[48, 52 − 54]. 具体反应可由方程(2—5)表示:
其中,“d”表示沙尘表面的反应物或产物. 反应产生的羟基自由基和超氧自由基会进一步氧化吸附于颗粒物表面的化合物分子(如SO2和NO2等). 大部分电子-空穴对会发生泯灭反应并以能量的形式发散[55, 56],具体反应如式(6):
由于电子空穴对的不稳定性,反应式(2)中的光激发速率较难定量,而通过假定电子对的泯灭速率(10−2)再结合紫外-可见光谱对沙尘颗粒物吸收光谱的测定,可以近似推算沙尘颗粒物的光吸收截面和量子产率并计算光激发速率[48, 51]. 进一步采用光激发参数化方程可以对反应速率(J·s−1)进行计算,如式(7)[57-58]:
其中l、m和n为经验参数,
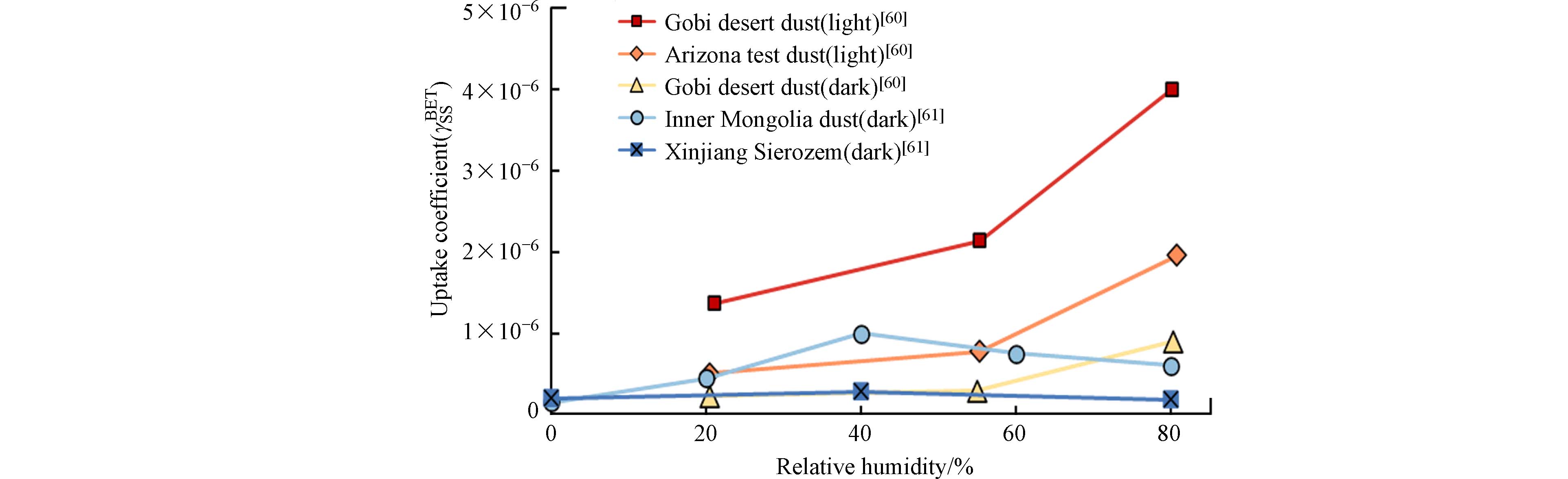
χ 是太阳天顶角. 表1中列举了蒙古国戈壁沙漠沙尘颗粒物和亚利桑那商业测试沙尘颗粒物的光激发近似参数.沙尘表面氧化物的生成速率受到多个方面因素的影响,包括光照强度、温湿度、具有光激发活性的金属氧化物含量以及电子空穴对泯灭速率等[51]. 例如,Dupart等[59]通过分析NO2在亚利桑那试验沙尘表面的吸附速率发现,光照下NO2的吸附速率是无光下的4倍以上. 此外,研究发现湿度对SO2在沙尘表面的光催化氧化反应速率也有较大的影响,在特定的光照下SO2在蒙古国戈壁沙尘表面的吸附反应速率增加了约4—7倍(图3)[60,-61].
目前,沙尘颗粒物对气体分子的吸附摄取系数主要由反应物浓度变化或二次产物的生成速率计算得到. 然而,沙尘表面的光催化反应过程既包括光激发、电子-空穴对和表面自由基的生成,也涉及到气体分子的氧化还原反应,因此并不能完全由单步反应进行简化计算得到. 受限于氧化自由基和多相反应中间产物的测量难度,目前沙尘颗粒物光催化反应的研究还十分匮乏,相关反应过程参数的定量分析十分有限. 此外,由于化学机理和反应参数的缺失,目前区域空气质量模式对受沙尘影响下的二次产物的模拟预测精度整体较无沙尘条件的结果更低[62].
-
沙尘颗粒物通过提供吸附位点和反应界面,与各类气态分子耦合并发生非均相反应,同时加快了大气污染物的演化速度并增加了大气化学过程的复杂度[63]. 本段落将进一步讨论沙尘颗粒物与臭氧、二氧化硫和氮氧化物、有机物以及二氧化碳等化合物的非均相反应过程.
-
臭氧作为城市大气环境中主要的二次污染物之一,其与沙尘颗粒物之间的相互作用和耦合机制受到广泛的关注. 黑暗条件下,臭氧在沙尘颗粒物表面的吸附和反应过程可以由方程(8—9)表示[64]:
光照条件下,吸附于沙尘颗粒物表面的臭氧分子会参与光催化反应,通过与电子-空穴对反应,快速转化并生成羟基自由基[65- 66],如式(10—12):
目前,较多研究测定并报道了臭氧在各类矿质颗粒物和沙尘颗粒物表面的吸附过程及反应参数[16, 41, 66 − 70]. 图4结合了文献数据并基于公式(1)中的吸附模型,分析了不同光照条件和臭氧初始浓度下,臭氧在沙尘颗粒物表面的稳定态吸附摄取常数(
γBETss ). 结果表明,光照或者增加活性金属氧化物的含量都能显著提高臭氧在颗粒物表面的吸附速率. 臭氧作为一种氧化剂,在沙尘颗粒物表面的吸附和反应可以促进颗粒物表面的氧化作用,加速沙尘颗粒物的大气老化过程和二次产物的形成. 例如,在早期的模式中,Zhang等[71]模拟发现臭氧在沙尘颗粒物表面的吸附作用解释了其在对流层中约10%—20%的减少现象. -
城市大气中的二氧化硫(SO2)主要来自煤炭和石化燃料中含硫元素的燃烧和释放[72]. SO2在排放进入大气后会参与大气氧化还原反应生成SO3,最终形成硫酸盐并由干、湿沉降进入地表[73]. 其中,在颗粒物表面发生的氧化还原反应是SO2的关键转化路径之一. 吸附于沙尘颗粒物表面的SO2能够自发地与颗粒物表面的氧分子或氧化自由基反应,形成硫酸根离子和硫酸盐,相关反应可由以下方程(13—14)简化表示[74]:
SO2在自然沙尘表面的吸附摄取系数
γBETss 在10−8 —10−6之间[74]. SO2在沙尘颗粒物表面的非均相反应过程受到环境温湿度、表面氧化物浓度、二级反应速率等影响. 如2.1节所述,环境湿度对颗粒物表面的水分子和水合壳厚度起到决定性的作用. 实验发现高湿环境下,水分子会与SO2分子竞争沙尘颗粒物表面的吸附位点,从而抑制了SO2的吸附速率[75-76]. 另一方面,沙尘表面吸附的水分子能够参与SO2的水解和氧化反应,促进其在沙尘颗粒物表面的转化过程[19, 37]. 不同来源和地区的沙尘颗粒物中的矿物质组分有较大的差异,其中富含碳酸盐、氧化钙等矿物质的沙尘颗粒物能够与非均相反应形成的硫酸盐等酸性产物发生反应,促进了硫酸盐的形成和固定[77]. Ruan等[78]通过模拟颗粒物中的无机盐离子平衡发现,沙尘天气期间受钙离子浓度增加的影响,大气颗粒物的pH值较平均值(pH = 3.6)更高并呈弱酸性(pH = 4.2—5.7). Wang等[79 − 80]通过原位漫反射傅立叶红外光谱(DRIFTS)和扫描电迁移光谱研究了SO2在沙尘颗粒的非均相反应,发现沙尘表面的游离铁离子可能对二氧化硫的吸附具有潜在的促进作用. -
氮氧化物(NOx)是交通运输过程中产生的主要大气污染物之一,能够参与一系列大气光化学反应,对臭氧(O3)和颗粒物的形成都有重要的影响. 沙尘期间, NO2能够通过在沙尘颗粒物表面吸附并进行自发性氧化反应和光催化氧化反应,最终形成氧化产物包括HONO[59, 81]、HNO3[59, 82-83]、N2O4[81, 84- 85]和N2O5[86]等. 在傍晚和夜间,大气中NO3自由基的光解速率降低而N2O5的浓度逐渐升高,因此NO3和N2O5在沙尘颗粒物表面的吸附和非均相反应也是夜间硝酸盐生成的关键演化路径[38, 87 − 89]. 在光照条件下,沙尘表面的NO2分子会与电子空穴对发生电子转移,并进一步氧化产生自由基和硝酸根离子,如式(15—16) [48, 82-83]:
氮氧化物同时也能够作为弱氧化剂,氧化吸附于沙尘颗粒物表面的其他化合物. 例如,研究发现在黑暗条件下,吸附于沙尘颗粒物表面的NO2也能够生成N2O4,并通过异构化和电离水解反应促进SO2向硫酸盐的转化,如式(17—22) [84-85]:
其中,M是沙尘颗粒表面金属位点,MO则是表面活性氧位点. 反应形成的HONO会通过快速吸附平衡过程,与空气中的HONO交换,并在沙尘颗粒物表面达到稳定[90]. He等[91]通过对中国京津冀地区2013年大气观测,并证实NO2和SO2在沙尘颗粒物表面的协同反应促进了SO2向硫酸盐的快速转变,且由沙尘、氮氧化物和硫酸盐等复合污染产生的新粒子可能是引发城市重雾霾天气的关键因素之一.
颗粒物中的钙、镁氧化物和碳酸钙等碱性碳酸盐能够通过酸碱反应捕捉和固定非均相反应过程中形成的硝酸根离子. 前期研究发现硝酸根和硫酸根离子在沙尘颗粒物表面与钙、镁离子的反应存在一定的竞争机制,随着硫酸盐的形成和气溶胶酸性的降低,硝酸根离子更易从颗粒物表面逃逸[48]. 由于硝酸盐具有较强的吸湿性,由硝酸盐形成主导的非均相化学过程会使沙尘颗粒通过吸湿增长而逐渐改变其形状和表面积,从而影响颗粒物的光学特性和区域沉降特性[92, 93]. 随着我国SO2排放的逐年降低,氮氧化物和颗粒态硝酸盐也将逐渐成为我国城市大气污染的主导因素之一,与此相关的大气反应过程亟需进一步评估. 另一方面,光照条件下的颗粒态硝酸盐存在部分光解现象[94],虽然反应本身与气溶胶酸度的相关性较小,但是反应产物的分配则受酸度变化的影响(主要产物为NO2−(pH < 3)和 HONO(pH > 3))[95 − 96]. 研究表明,酸性条件下颗粒态硝酸盐的光解也会间接生成氧化自由基,并可能激活氯离子的转化和日间大气Cl2的形成[97].
此外,NOx在大气化学过程中也同时与臭氧、挥发性有机物(VOC)和有机自由基反应,并与其他氧化物或自由基存在一定的反应竞争,由此形成复杂的NOx-VOC-O3大气污染体系. 目前,沙尘对NOx-VOC-O3复合污染体系的影响仍然不太清楚,亟需结合实验数据和外场观测结果来进一步厘清其中的关键大气化学机理.
-
沙尘颗粒物不仅能与气态无机化合物进行耦合,还能与有机化合物发生非均相反应. 众多沙尘天气期间的观测研究发现,沙尘颗粒物对大气中的有机化合物有一定的吸附和非均相转化作用. Falkovich等[98]分析了2000—2001年沙尘暴期间的大气颗粒物样品,发现沙尘能够作为载体吸附并携带传输多环芳烃、杀虫剂和短链有机酸等半挥发和低挥发性的有机物. 最近研究也发现在沙尘天气下,大气颗粒物样品中有机酸和可溶性有机氮的质量浓度都有一定的提高[99 − 100]. Romanias等[15]使用漫反射傅里叶变换红外光谱仪(DRIFTS)等仪器研究了撒哈拉沙漠沙尘颗粒物样品对柠檬烯和甲苯的吸附作用. Wang等[17]分析了甲酸和乙酸在沙尘颗粒物中的富集量,发现富含碳酸钙的新疆灰钙土沙尘对有机酸的吸附速率远高于亚利桑那试验沙尘. Al-Hosney等[101]在实验室环境中采用Kundsen反应器测定了甲酸在碳酸钙颗粒物表面的吸附摄取常数(3×10−3). Lederer等[102]通过气相色谱质谱联用仪分析了d-柠檬烯在亚利桑那测试沙尘表面的吸附和转化过程,生成的主要产物包括香芹醇、葛缕酮、1,2-柠檬烯醇以及α-松油醇. 沙尘颗粒物的光催化特性对吸附于其表面的有机污染物也有一定的催化转化效应,研究发现光照条件下的沙尘颗粒物能够显著促进甲苯和三甲苯的大气氧化过程[103]. Ponczek等[104]采用质子转移反应飞行时间质谱(Proton-Transfer-Reaction Time-of-flight Mass Spectrometer,PTR-ToF-MS)和超高分辨液相质谱(Ultra-High-Performance Liquid Chromatograph Mass Spectrometer, UHPLC-MS)分别分析了气态和沙尘颗粒物表面吸附的有机酸浓度,发现颗粒物对有机酸的催化反应具有显著的促进作用.
除了实验室的模拟研究外,外场观测研究也证实了沙尘颗粒物对挥发性有机物的转化具有显著影响. 例如,Xue等[105]通过西安地区的外场观测实验发现,沙尘天气期间沙尘颗粒物加速了挥发性有机物的转化过程,导致其浓度迅速降低. 沙尘颗粒物在促进大气中碳氢化合物的转化的同时也影响了有机化合物的大气生命周期和沉降规律,然而目前大部分的研究主要集中在外场观测和特定污染物的靶向分析,尚缺少与有机污染物和沙尘颗粒物非均相反应过程相关的化学参数和机理研究.
-
TiO2等过渡金属氧化物,由于其易产生光激发并进行电子传递,因此也对CO2也有一定的非均相反应催化作用,如式(23) [106 − 107]:
光照和常温条件下的TiO2能通过光激发反应和电子传递推动颗粒物表面吸附的CO2和H2O之间的反应,生成甲烷和甲醇等产物[108 − 110]. 自然沙尘颗粒物也具有类似的效应,其表面活性位点能够吸附CO2,且颗粒物中的过渡金属氧化物也对CO2的非均相转化有一定的催化作用. 例如,Deng等[111]采用13C同位素标记的方法,研究了CO2在TiO2、亚利桑那测试沙尘、伊利石、蒙脱石和高岭土颗粒物表面的光催化降解机理,通过测定CO的生成证实自然沙尘对CO2具有催化降解作用. 相关反应及产物如下(24—26):
实验测得CO2在亚利桑那测试沙尘表面的吸附摄取常数(
γBETss )约为1.9×10−8. 氧分子作为主要的电子受体,也同时能够与CO2竞争沙尘表面激发的电子,因此CO2在自然沙尘表面的吸附转化速率较其他气体污染物慢1—2个数量级(如SO2等). Liu等[112 − 113]发现CO2在沙尘颗粒物表面的非均相反应同时可能对SO2的氧化过程造成潜在的影响,其中CO2的非均相反应生成的碳酸根自由基(CO3•−)可能是SO2转化为硫酸盐的潜在氧化物之一. 随着全球温度的不断升高,研究以CO2等温室气体的大气过程对厘清碳排放和碳循环具有重要意义,而金属氧化物和沙尘颗粒物表面的吸附和非均相反应过程也CO2在大气环境中潜在的转化路径之一. 虽然CO2在沙尘颗粒物表面的吸附速率较SO2和NOx等气体慢数个数量级,但是沙尘颗粒物是全球大气中质量浓度最高且排放量最大的颗粒物,因此其与CO2的大气化学过程有待进一步评估. -
随着近年来中亚和东亚地区沙尘天气的频繁发生,沙尘传输带来的城市空气污染和影响受到政府和居民的广泛关注. 大量实验室研究和外场观测结果表明,沙尘颗粒物能够以载体的形式,对各类大气污染物进行吸附、催化、转化和携带传输. 沙尘天气期间,沙尘颗粒物表面发生的非均相反应则可能在一定程度上改变传统初次排放污染物的沉降通路,通过影响二次产物的生成加速初次产物的分解和转化. 众多实验结果证实,沙尘颗粒物对以SO2和NO2等为代表的无机氧化物具有一定的吸附和催化转化作用,然而仍有部分机理和观测结果需要进一步解释. 目前,大气污染物在沙尘颗粒物表面的非均相反应研究主要集中在单一污染物系统,少数研究讨论了二元无机污染物在颗粒物表面的复合效应. 然而城市环境下的大气污染通常呈现多源、多类型的污染特征,因此对多元系统中的复合过程和反应机理研究提出了新的挑战. 此外,沙尘颗粒物的大气化学过程参数化和模式模拟仍然停留在一次反应机理(吸附反应机理)的假设阶段,难以对复杂的污染过程进行描述,同时缺少关键的反应速率常数和参数化方案. 随着中国大气污染治理工作的不断推进,城市大气中二氧化硫的浓度逐年降低,而氮氧化物和挥发性有机物的复合污染将成为目前以及未来一段时间内的主要污染类型,因此氮氧化物和有机物在沙尘颗粒物中的多元复合污染过程和模拟预测方法的研究有待深入开展. 此外,沙尘颗粒物表面非均相反应的机理尚未完全阐明,沙尘颗粒物与有机气态污染物的非均相反应机制、与人为源大气颗粒物的相互作用机制都有待进一步研究.
沙尘作为主要的大气颗粒物成分,不仅对城市空气质量产生显著影响,同时也影响着区域辐射平衡. 然而,目前对沙尘颗粒物的矿物类型、形貌特征和粒径分布与其光学辐射的影响研究较为有限. 此外,沙尘与城市大气污染的相互作用,特别是在沙尘颗粒物与其他污染物的复合过程中,不仅改变了沙尘颗粒物在传输过程中的形态,还影响了其化学组成. 这些变化如何影响沙尘颗粒物的光学特性、可溶性离子的分布,以及如何进一步影响区域气候和矿质元素的循环,都是值得进一步探讨. 沙尘颗粒物在大气老化过程中发生改变的特性还可能影响凝结核及云的形成,进而影响局部甚至全球的降水模式. 另一方面,最新研究提出沙尘颗粒物和TiO2等金属氧化物是CO2的潜在固碳路径,而相关反应机理和对大气碳循环的贡献则需要进一步的系统性分析.
沙尘颗粒物大气化学过程的全面研究不仅对区域空气质量的改善至关重要,对于分析气候变化和理解大气污染过程都具有重要意义. 通过深入研究其中的化学机理和反应参数,可以更好地理解沙尘颗粒物对气溶胶二次产物的形成以区域气候变化的影响,将有助于更好地应对环境变化带来的挑战.
沙尘颗粒物大气化学过程研究进展
Research progress of atmospheric chemical process of airborne dust particles
-
摘要: 沙尘颗粒物与各类气态大气污染物的非均相反应,对沙尘天气下的区域空气质量和污染物演化有重要影响. 研究气态污染物在沙尘表面的吸附和非均相反应机理,有助于加深理解城市环境下污染物大气化学过程和多元污染物的复合过程,为沙尘颗粒物的大气化学影响和气候效应评估提供科学依据. 本文简要介绍了大气沙尘颗粒物的组分和传输,对污染物在沙尘颗粒物表面的吸附、自发性氧化反应和光激发催化氧化反应机理进行了总结,并讨论了城市大气中常见的气态化合物(包括臭氧、二氧化硫、二氧化氮、有机化合物和二氧化碳)与沙尘颗粒物的非均相反应过程,最后提出了沙尘颗粒物大气化学过程研究中亟待解决的问题.Abstract: Heterogeneous reaction of urban gaseous pollutants on the surface of mineral dust particles during sandstorm are an important pathway for the evolution and deposition of atmospheric pollutants. Research on the mechanisms of adsorption and heterogeneous reaction of various pollutants on dust promotes our understanding of atmospheric process and fate of these pollutants, as well as the analysis and simulation of combined air pollution scenarios. This study introduces the composition, sources, and transport of airborne dust particles; summarizes the mechanisms of the typical adsorption process, autooxidation, and photocatalytic oxidation of gaseous pollutants on the dust surface; and discusses the heterogeneous reactions of ozone, sulfur dioxide, nitrogen dioxide, hydrocarbon, and carbon dioxide on dust particles. In addition, the urgent issues that need to be addressed in studying atmospheric chemical process of airborne dust particles are identified.
-
Key words:
- airborne dust /
- heterogeneous reaction /
- atmospheric chemistry /
- photocatalytic reaction /
- air quality
-
沙尘颗粒物是全球大气颗粒物的主要贡献来源之一,最新研究表明,全球的沙尘排放量随着土地荒漠化和气候变化的加剧而增加[1 − 3],其中全球沙尘年排放总量预估达
1000 —8000 Tg[4]. 沙尘颗粒物能够通过吸收和反射太阳光影响区域热辐射和气候变化[5]. 沙尘天气不仅对起沙源及附近区域造成空气质量影响,沙尘细颗粒物也可通过远距离传输并造成半球甚至全球范围的影响. 例如,2021年3月蒙古国和我国西北地区发生大规模沙尘天气,受沙尘影响部分地区PM10峰值浓度超过5000 µg·m−3[6],同时沙尘颗粒物通过高层气流远距离传输并与我国华南地区的城市大气污染物耦合形成了复合型大气污染事件[7]. 2023年春季(3—4月),我国共爆发数十次高强度、大范围的沙尘天气。在季风和蒙古气旋的影响下,沙尘的发生和传输对下风向城市的空气质量和居民生活产生了显著的影响[8]. 沙尘天气的产生是非常复杂且难以预测的,我国2021年和2023年相继发生的大范围沙尘也引发了人们对于沙尘暴发生的频率和强度增加的担忧. 研究沙尘暴对城市环境的影响以及其与城市大气污染物的复合过程也变得越加重要,其中污染物的耦合、分布、迁移、转化和沉降仍然具有高度的不确定性,阻碍了对沙尘复合污染事件的溯源、归因和环境风险等分析.本文讨论了沙尘颗粒物的化学组成、传输和大气化学过程,重点梳理沙尘颗粒物与气态化合物非均相反应机理的研究现状,对气态分子在沙尘颗粒物表面的吸附规律、氧化机制、非均相过程和间接环境影响展开讨论,并对未来研究进行了展望.
1. 沙尘颗粒物的化学组分及大气传输(Chemical composition and atmospheric transport of mineral dust particles)
沙尘颗粒物主要指由强风吹过裸露的土壤和荒漠地表而形成的大气悬浮颗粒物. 其颗粒物的粒径通常小于100 µm,且富含各类矿物质,主要包括二氧化硅、硅酸盐、碳酸盐以及金属氧化物等. 大气中沙尘颗粒物的矿物质组分可以通过X射线衍射(XRD)、X射线荧光光谱(XRF)、电感耦合等离子体质谱和离子色谱等进行分析. 沙尘颗粒物的元素组成与地壳基本一致,其中含量最高的元素是O和Si,含量最高的金属元素是Al[9]. 沙尘通常同时含有Fe、Ca、K、Mg、Na、Ti等常见的金属元素,以及Mn、P等微量元素. 沙尘中的矿物质元素主要以金属氧化物、碱性氢氧化物和硅酸盐等形式存在[10]. 沙尘颗粒物的矿物质组分差异通常受到母岩种类、岩石风化和土壤发生过程、气候条件、植被种类和人类活动等多种因素影响[11],因此不同地区的沙尘颗粒物通常具有不同的元素类型及比例(图1)[12 − 23]. 例如,由于岩石类型和风化过程的区别,亚洲地区的沙尘颗粒物(黄土高原和戈壁沙漠等)中的碱性金属元素(Ca和Mg)含量较其他地区的沙尘更高,而撒哈拉沙漠南部区域的沙尘颗粒物中则含有较多的Al元素. 不同区域的沙尘颗粒物因风化程度和传输过程等差异,颗粒物比表面积也不一样. 例如,亚利桑那测试沙尘(Arizona Test Dust, ATD)、腾格里沙漠沙尘和北京沙尘天气期间采集的东亚沙尘的比表面积(Brunauer–Emmett–Teller,BET)分别为4.0、16.5 、 6.1 m2·g−1 [24]. 此外,前期研究发现沙尘颗粒物经过大气复合过程后,其理化性质有所改变. 例如,有报道采用X射线扫描电镜分析黄河三角洲区域的沙尘与霾混合颗粒并发现沙尘暴后颗粒物被硝酸钙包裹,从而对颗粒物的吸湿性造成一定的影响 [25].
 Figure 1. Elemental compositions of major mineral dust particles around the world(注:该图基于国家测绘地理信息局标准地图服务网站下载的审图号GS(2016)1611号的标准地图制作)(Note: The figure is generated based on the standard map with the review number GS(2016)1611 from the State Bureau of Surveying and Mapping of China).
Figure 1. Elemental compositions of major mineral dust particles around the world(注:该图基于国家测绘地理信息局标准地图服务网站下载的审图号GS(2016)1611号的标准地图制作)(Note: The figure is generated based on the standard map with the review number GS(2016)1611 from the State Bureau of Surveying and Mapping of China).沙尘颗粒物中粒径较小的细颗粒物通常能够在表层紊流和对流空气的作用下悬浮,并进一步在季风等高空大气环流的作用下实现中、远距离的迁移和传输. 沙尘颗粒物的形成、传输和沉降与地表的裸露程度、区域气候的条件以及植被覆盖的程度都有一定的关联. 在冬春季节,我国西北部地区干燥、多风,同时植被覆盖率低,而土壤和岩石风化频率的增加也导致了沙尘释放量的升高[26]. 近期有研究指出,在我国西北部的起沙区域内,由于受到上风向大范围起尘的影响,位于下风向的城市整体沙尘的传输和沉降量呈现自西向东和自北向南逐渐增加的趋势[27]. 沙尘颗粒物的矿物质组分对传输过程中的大气化学过程具有重要的影响,而矿物质类型则主要由起沙源决定. 目前常用的沙尘颗粒物溯源技术包括同位素示踪法[28 − 31]和基于拉格朗日轨迹模型的理论计算. 例如,Chen等[32]结合区域气象模式和轨迹模型分析了2023年3月至4月我国北部强沙尘暴事件,发现蒙古和塔克拉玛干沙漠是主要的沙尘源,其中蒙古对我国北部沙尘浓度有超过42%的贡献. 在全球范围内,沙尘颗粒物的主要影响范围涉及非洲撒哈拉沙漠区域、中东、亚洲、澳大利亚和北美洲西海岸部分区域. 其中,撒哈拉沙漠的沙尘通过跨大西洋传输到加勒比海地区和美洲东部,并维持了亚马逊区域的矿物质输入[33];中东地区的沙尘则主要源自伊拉克、叙利亚和沙特阿拉伯等地区,并可通过中远距离传输到东亚地区[34];而亚洲的沙尘主要来自中国西北和蒙古地区,通过季风和气旋传输到朝鲜半岛、日本,甚至北美洲和北极地区[35].
2. 沙尘颗粒物的大气化学过程(Atmospheric chemical process of dust particles)
2.1 沙尘颗粒物对气体分子的吸附摄取
沙尘颗粒物的平均寿命可达数周,从起沙源头至远距离迁移的过程中,沙尘通过其表面的自然孔隙吸附、摄取各类气态污染物分子. 与大气中其他有机或无机盐颗粒物相比,沙尘颗粒物的表面形状多变且比表面积更大. 沙尘颗粒物对气体分子的吸附主要通过气体分子扩散与颗粒物表面碰撞并发生摄取,其中吸附速率常数或摄取系数(
γ stringUtils.convertMath(!{formula.content}) (1) 其中,[X]为气体化合物的空气质量浓度,
ˉc γBETss 沙尘作为主要的大气颗粒物之一,既参与了大气污染物的多相反应过程,也是对流层凝结核重要来源之一,因此沙尘颗粒物对水分子的吸附特性即吸湿性对大气成云、降水、热辐射和区域气候都有一定的影响[40]. 研究表明,沙尘颗粒物的吸湿性受物理化学因素和环境条件的影响,包括沙尘颗粒物表面的亲水基团、化学组成、颗粒物尺寸和几何形状等[38, 40]. 另一方面,颗粒物吸湿性的提高显著增加了水分子在其表面的吸附速率、覆盖厚度和覆盖范围,也对气体分子的吸附和非均相反应造成了一定的影响. 在低湿条件下,颗粒物表面尚未形成单层水分子壳,水分子和其他气体分子可能发生竞争机制,争夺颗粒表面的吸附位点. Mogili等[41]通过吸附实验发现O3在Fe2O3和Al2O3颗粒表面的初始吸附速率随湿度增加(RH<30%)而降低. 随着环境湿度的不断升高,颗粒物吸附的水分子将逐渐超过并覆盖其表面积并最终形成多层水合壳(图2)[12, 42 − 43]. 同时,颗粒物表面的部分空隙被水分子填充并导致比表面积的降低,从而使气体分子的吸附从气-固交换逐渐转变为气-液交换,颗粒表面水层则作为反应媒介进一步加速推动液相反应的进行[40]. 目前采用较多的沙尘吸湿性分析方法包括水分子气压变化测定[44]、质量动态测定[45]、FTIR测定含水量[46]、扫描电镜和扫描隧道显微镜等手段观测颗粒物的吸湿变化[47]、基于双差分电迁移分析仪(Differential Mobility Analyzer, DMA)的H-TMDA颗粒物吸湿增长测定法等[48]. 研究表明,沙尘颗粒物的吸湿性随颗粒物的迁移、老化等一系列环境化学过程而改变. 例如,硝酸盐具有较强的亲水性,沙尘颗粒物在吸附或表面形成硝酸盐后其吸湿性有明显的增强[49, 50]. 外场观测证实,沙尘过程后约有32%的沙尘颗粒由于硝酸盐的包裹而发生了吸湿增强[20].
2.2 沙尘颗粒物表面的氧化物
吸附于沙尘颗粒物表面的化合物分子往往会与颗粒物表面的氧化物或自由基发生氧化还原反应,并生成二次产物. 黑暗条件下,颗粒物表面的活性位点上的氧原子能够与表面吸附的化合物分子发生缓慢的氧化还原反应,即表面自发性氧化反应[51]. 其反应速率主要受到化合物分子的吸附速率、氧分子的吸附速率以及表面反应速率的影响. 在光照条件下,沙尘颗粒物中具有一定光催化活性的金属氧化物,如Fe2O3、Al2O3和TiO2等,能够激发形成电子-空穴对(e−cb-h+vb),并与颗粒物表面的氧气和水等反应生成强氧化自由基(如羟基自由基和超氧自由基)[48, 52 − 54]. 具体反应可由方程(2—5)表示:
stringUtils.convertMath(!{formula.content}) (2) stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) stringUtils.convertMath(!{formula.content}) (5) 其中,“d”表示沙尘表面的反应物或产物. 反应产生的羟基自由基和超氧自由基会进一步氧化吸附于颗粒物表面的化合物分子(如SO2和NO2等). 大部分电子-空穴对会发生泯灭反应并以能量的形式发散[55, 56],具体反应如式(6):
stringUtils.convertMath(!{formula.content}) (6) 由于电子空穴对的不稳定性,反应式(2)中的光激发速率较难定量,而通过假定电子对的泯灭速率(10−2)再结合紫外-可见光谱对沙尘颗粒物吸收光谱的测定,可以近似推算沙尘颗粒物的光吸收截面和量子产率并计算光激发速率[48, 51]. 进一步采用光激发参数化方程可以对反应速率(J·s−1)进行计算,如式(7)[57-58]:
stringUtils.convertMath(!{formula.content}) (7) 其中l、m和n为经验参数,
χ 表 1 自然沙尘颗粒物的光激发近似参数Table 1. Photoactivation coefficients of mineral dust particles样品名称Sample Name 光激发参数Photoactivation parameters 参考文献Reference l m n 蒙古国戈壁沙尘(Gobi Desert Dust) 1.8×10−4 1.7 0.5 [48] 亚利桑那测试沙尘 Arizona Test Dust) 7.0×10−5 1.7 0.5 [51] | Show Table DownLoad:
CSV
DownLoad:
CSV
沙尘表面氧化物的生成速率受到多个方面因素的影响,包括光照强度、温湿度、具有光激发活性的金属氧化物含量以及电子空穴对泯灭速率等[51]. 例如,Dupart等[59]通过分析NO2在亚利桑那试验沙尘表面的吸附速率发现,光照下NO2的吸附速率是无光下的4倍以上. 此外,研究发现湿度对SO2在沙尘表面的光催化氧化反应速率也有较大的影响,在特定的光照下SO2在蒙古国戈壁沙尘表面的吸附反应速率增加了约4—7倍(图3)[60,-61].
目前,沙尘颗粒物对气体分子的吸附摄取系数主要由反应物浓度变化或二次产物的生成速率计算得到. 然而,沙尘表面的光催化反应过程既包括光激发、电子-空穴对和表面自由基的生成,也涉及到气体分子的氧化还原反应,因此并不能完全由单步反应进行简化计算得到. 受限于氧化自由基和多相反应中间产物的测量难度,目前沙尘颗粒物光催化反应的研究还十分匮乏,相关反应过程参数的定量分析十分有限. 此外,由于化学机理和反应参数的缺失,目前区域空气质量模式对受沙尘影响下的二次产物的模拟预测精度整体较无沙尘条件的结果更低[62].
2.3 沙尘颗粒物的非均相反应
沙尘颗粒物通过提供吸附位点和反应界面,与各类气态分子耦合并发生非均相反应,同时加快了大气污染物的演化速度并增加了大气化学过程的复杂度[63]. 本段落将进一步讨论沙尘颗粒物与臭氧、二氧化硫和氮氧化物、有机物以及二氧化碳等化合物的非均相反应过程.
2.3.1 臭氧的非均相反应
臭氧作为城市大气环境中主要的二次污染物之一,其与沙尘颗粒物之间的相互作用和耦合机制受到广泛的关注. 黑暗条件下,臭氧在沙尘颗粒物表面的吸附和反应过程可以由方程(8—9)表示[64]:
stringUtils.convertMath(!{formula.content}) (8) stringUtils.convertMath(!{formula.content}) (9) 光照条件下,吸附于沙尘颗粒物表面的臭氧分子会参与光催化反应,通过与电子-空穴对反应,快速转化并生成羟基自由基[65- 66],如式(10—12):
stringUtils.convertMath(!{formula.content}) (10) stringUtils.convertMath(!{formula.content}) (11) stringUtils.convertMath(!{formula.content}) (12) 目前,较多研究测定并报道了臭氧在各类矿质颗粒物和沙尘颗粒物表面的吸附过程及反应参数[16, 41, 66 − 70]. 图4结合了文献数据并基于公式(1)中的吸附模型,分析了不同光照条件和臭氧初始浓度下,臭氧在沙尘颗粒物表面的稳定态吸附摄取常数(
γBETss 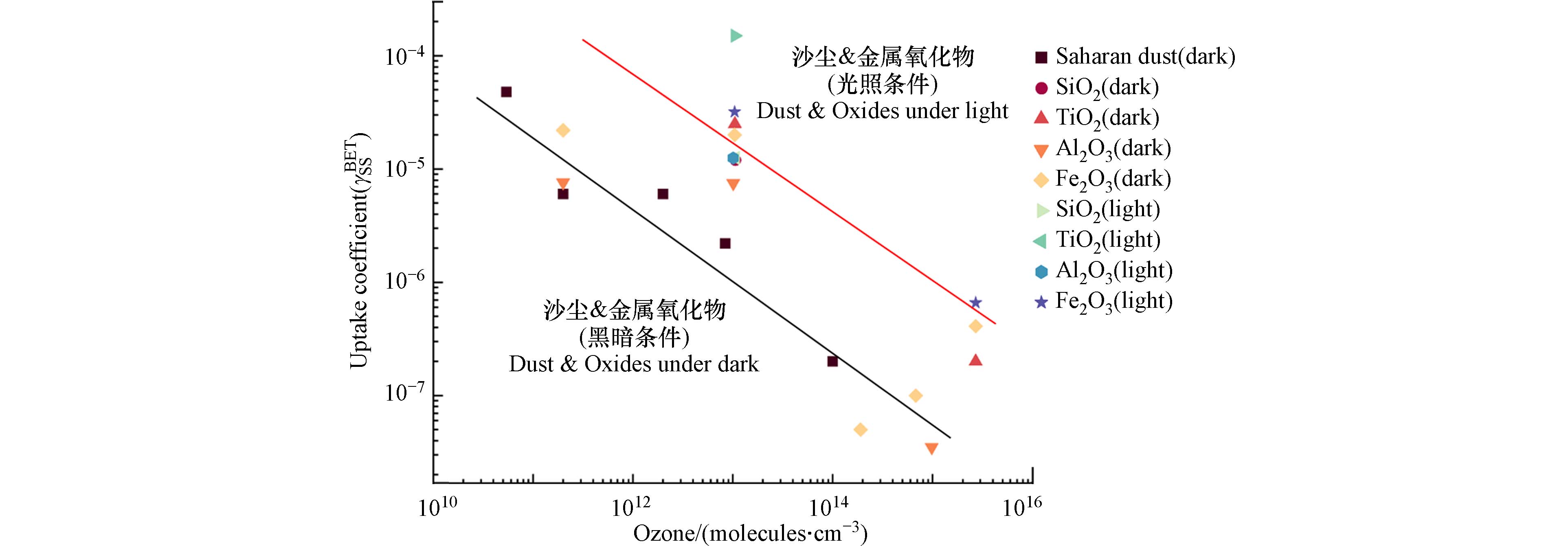
2.3.2 二氧化硫的非均相反应
城市大气中的二氧化硫(SO2)主要来自煤炭和石化燃料中含硫元素的燃烧和释放[72]. SO2在排放进入大气后会参与大气氧化还原反应生成SO3,最终形成硫酸盐并由干、湿沉降进入地表[73]. 其中,在颗粒物表面发生的氧化还原反应是SO2的关键转化路径之一. 吸附于沙尘颗粒物表面的SO2能够自发地与颗粒物表面的氧分子或氧化自由基反应,形成硫酸根离子和硫酸盐,相关反应可由以下方程(13—14)简化表示[74]:
stringUtils.convertMath(!{formula.content}) (13) stringUtils.convertMath(!{formula.content}) (14) SO2在自然沙尘表面的吸附摄取系数
γBETss 2.3.3 氮氧化物与多元污染物的非均相反应过程
氮氧化物(NOx)是交通运输过程中产生的主要大气污染物之一,能够参与一系列大气光化学反应,对臭氧(O3)和颗粒物的形成都有重要的影响. 沙尘期间, NO2能够通过在沙尘颗粒物表面吸附并进行自发性氧化反应和光催化氧化反应,最终形成氧化产物包括HONO[59, 81]、HNO3[59, 82-83]、N2O4[81, 84- 85]和N2O5[86]等. 在傍晚和夜间,大气中NO3自由基的光解速率降低而N2O5的浓度逐渐升高,因此NO3和N2O5在沙尘颗粒物表面的吸附和非均相反应也是夜间硝酸盐生成的关键演化路径[38, 87 − 89]. 在光照条件下,沙尘表面的NO2分子会与电子空穴对发生电子转移,并进一步氧化产生自由基和硝酸根离子,如式(15—16) [48, 82-83]:
stringUtils.convertMath(!{formula.content}) (15) stringUtils.convertMath(!{formula.content}) (16) 氮氧化物同时也能够作为弱氧化剂,氧化吸附于沙尘颗粒物表面的其他化合物. 例如,研究发现在黑暗条件下,吸附于沙尘颗粒物表面的NO2也能够生成N2O4,并通过异构化和电离水解反应促进SO2向硫酸盐的转化,如式(17—22) [84-85]:
stringUtils.convertMath(!{formula.content}) (17) stringUtils.convertMath(!{formula.content}) (18) stringUtils.convertMath(!{formula.content}) (19) stringUtils.convertMath(!{formula.content}) (20) stringUtils.convertMath(!{formula.content}) (21) stringUtils.convertMath(!{formula.content}) (22) 其中,M是沙尘颗粒表面金属位点,MO则是表面活性氧位点. 反应形成的HONO会通过快速吸附平衡过程,与空气中的HONO交换,并在沙尘颗粒物表面达到稳定[90]. He等[91]通过对中国京津冀地区2013年大气观测,并证实NO2和SO2在沙尘颗粒物表面的协同反应促进了SO2向硫酸盐的快速转变,且由沙尘、氮氧化物和硫酸盐等复合污染产生的新粒子可能是引发城市重雾霾天气的关键因素之一.
颗粒物中的钙、镁氧化物和碳酸钙等碱性碳酸盐能够通过酸碱反应捕捉和固定非均相反应过程中形成的硝酸根离子. 前期研究发现硝酸根和硫酸根离子在沙尘颗粒物表面与钙、镁离子的反应存在一定的竞争机制,随着硫酸盐的形成和气溶胶酸性的降低,硝酸根离子更易从颗粒物表面逃逸[48]. 由于硝酸盐具有较强的吸湿性,由硝酸盐形成主导的非均相化学过程会使沙尘颗粒通过吸湿增长而逐渐改变其形状和表面积,从而影响颗粒物的光学特性和区域沉降特性[92, 93]. 随着我国SO2排放的逐年降低,氮氧化物和颗粒态硝酸盐也将逐渐成为我国城市大气污染的主导因素之一,与此相关的大气反应过程亟需进一步评估. 另一方面,光照条件下的颗粒态硝酸盐存在部分光解现象[94],虽然反应本身与气溶胶酸度的相关性较小,但是反应产物的分配则受酸度变化的影响(主要产物为NO2−(pH < 3)和 HONO(pH > 3))[95 − 96]. 研究表明,酸性条件下颗粒态硝酸盐的光解也会间接生成氧化自由基,并可能激活氯离子的转化和日间大气Cl2的形成[97].
此外,NOx在大气化学过程中也同时与臭氧、挥发性有机物(VOC)和有机自由基反应,并与其他氧化物或自由基存在一定的反应竞争,由此形成复杂的NOx-VOC-O3大气污染体系. 目前,沙尘对NOx-VOC-O3复合污染体系的影响仍然不太清楚,亟需结合实验数据和外场观测结果来进一步厘清其中的关键大气化学机理.
2.3.4 有机化合物的非均相反应
沙尘颗粒物不仅能与气态无机化合物进行耦合,还能与有机化合物发生非均相反应. 众多沙尘天气期间的观测研究发现,沙尘颗粒物对大气中的有机化合物有一定的吸附和非均相转化作用. Falkovich等[98]分析了2000—2001年沙尘暴期间的大气颗粒物样品,发现沙尘能够作为载体吸附并携带传输多环芳烃、杀虫剂和短链有机酸等半挥发和低挥发性的有机物. 最近研究也发现在沙尘天气下,大气颗粒物样品中有机酸和可溶性有机氮的质量浓度都有一定的提高[99 − 100]. Romanias等[15]使用漫反射傅里叶变换红外光谱仪(DRIFTS)等仪器研究了撒哈拉沙漠沙尘颗粒物样品对柠檬烯和甲苯的吸附作用. Wang等[17]分析了甲酸和乙酸在沙尘颗粒物中的富集量,发现富含碳酸钙的新疆灰钙土沙尘对有机酸的吸附速率远高于亚利桑那试验沙尘. Al-Hosney等[101]在实验室环境中采用Kundsen反应器测定了甲酸在碳酸钙颗粒物表面的吸附摄取常数(3×10−3). Lederer等[102]通过气相色谱质谱联用仪分析了d-柠檬烯在亚利桑那测试沙尘表面的吸附和转化过程,生成的主要产物包括香芹醇、葛缕酮、1,2-柠檬烯醇以及α-松油醇. 沙尘颗粒物的光催化特性对吸附于其表面的有机污染物也有一定的催化转化效应,研究发现光照条件下的沙尘颗粒物能够显著促进甲苯和三甲苯的大气氧化过程[103]. Ponczek等[104]采用质子转移反应飞行时间质谱(Proton-Transfer-Reaction Time-of-flight Mass Spectrometer,PTR-ToF-MS)和超高分辨液相质谱(Ultra-High-Performance Liquid Chromatograph Mass Spectrometer, UHPLC-MS)分别分析了气态和沙尘颗粒物表面吸附的有机酸浓度,发现颗粒物对有机酸的催化反应具有显著的促进作用.
除了实验室的模拟研究外,外场观测研究也证实了沙尘颗粒物对挥发性有机物的转化具有显著影响. 例如,Xue等[105]通过西安地区的外场观测实验发现,沙尘天气期间沙尘颗粒物加速了挥发性有机物的转化过程,导致其浓度迅速降低. 沙尘颗粒物在促进大气中碳氢化合物的转化的同时也影响了有机化合物的大气生命周期和沉降规律,然而目前大部分的研究主要集中在外场观测和特定污染物的靶向分析,尚缺少与有机污染物和沙尘颗粒物非均相反应过程相关的化学参数和机理研究.
2.3.5 二氧化碳的非均相反应
TiO2等过渡金属氧化物,由于其易产生光激发并进行电子传递,因此也对CO2也有一定的非均相反应催化作用,如式(23) [106 − 107]:
stringUtils.convertMath(!{formula.content}) (23) 光照和常温条件下的TiO2能通过光激发反应和电子传递推动颗粒物表面吸附的CO2和H2O之间的反应,生成甲烷和甲醇等产物[108 − 110]. 自然沙尘颗粒物也具有类似的效应,其表面活性位点能够吸附CO2,且颗粒物中的过渡金属氧化物也对CO2的非均相转化有一定的催化作用. 例如,Deng等[111]采用13C同位素标记的方法,研究了CO2在TiO2、亚利桑那测试沙尘、伊利石、蒙脱石和高岭土颗粒物表面的光催化降解机理,通过测定CO的生成证实自然沙尘对CO2具有催化降解作用. 相关反应及产物如下(24—26):
stringUtils.convertMath(!{formula.content}) (24) stringUtils.convertMath(!{formula.content}) (25) stringUtils.convertMath(!{formula.content}) (26) 实验测得CO2在亚利桑那测试沙尘表面的吸附摄取常数(
γBETss 3. 总结和展望(Conclusions and perspectives)
随着近年来中亚和东亚地区沙尘天气的频繁发生,沙尘传输带来的城市空气污染和影响受到政府和居民的广泛关注. 大量实验室研究和外场观测结果表明,沙尘颗粒物能够以载体的形式,对各类大气污染物进行吸附、催化、转化和携带传输. 沙尘天气期间,沙尘颗粒物表面发生的非均相反应则可能在一定程度上改变传统初次排放污染物的沉降通路,通过影响二次产物的生成加速初次产物的分解和转化. 众多实验结果证实,沙尘颗粒物对以SO2和NO2等为代表的无机氧化物具有一定的吸附和催化转化作用,然而仍有部分机理和观测结果需要进一步解释. 目前,大气污染物在沙尘颗粒物表面的非均相反应研究主要集中在单一污染物系统,少数研究讨论了二元无机污染物在颗粒物表面的复合效应. 然而城市环境下的大气污染通常呈现多源、多类型的污染特征,因此对多元系统中的复合过程和反应机理研究提出了新的挑战. 此外,沙尘颗粒物的大气化学过程参数化和模式模拟仍然停留在一次反应机理(吸附反应机理)的假设阶段,难以对复杂的污染过程进行描述,同时缺少关键的反应速率常数和参数化方案. 随着中国大气污染治理工作的不断推进,城市大气中二氧化硫的浓度逐年降低,而氮氧化物和挥发性有机物的复合污染将成为目前以及未来一段时间内的主要污染类型,因此氮氧化物和有机物在沙尘颗粒物中的多元复合污染过程和模拟预测方法的研究有待深入开展. 此外,沙尘颗粒物表面非均相反应的机理尚未完全阐明,沙尘颗粒物与有机气态污染物的非均相反应机制、与人为源大气颗粒物的相互作用机制都有待进一步研究.
沙尘作为主要的大气颗粒物成分,不仅对城市空气质量产生显著影响,同时也影响着区域辐射平衡. 然而,目前对沙尘颗粒物的矿物类型、形貌特征和粒径分布与其光学辐射的影响研究较为有限. 此外,沙尘与城市大气污染的相互作用,特别是在沙尘颗粒物与其他污染物的复合过程中,不仅改变了沙尘颗粒物在传输过程中的形态,还影响了其化学组成. 这些变化如何影响沙尘颗粒物的光学特性、可溶性离子的分布,以及如何进一步影响区域气候和矿质元素的循环,都是值得进一步探讨. 沙尘颗粒物在大气老化过程中发生改变的特性还可能影响凝结核及云的形成,进而影响局部甚至全球的降水模式. 另一方面,最新研究提出沙尘颗粒物和TiO2等金属氧化物是CO2的潜在固碳路径,而相关反应机理和对大气碳循环的贡献则需要进一步的系统性分析.
沙尘颗粒物大气化学过程的全面研究不仅对区域空气质量的改善至关重要,对于分析气候变化和理解大气污染过程都具有重要意义. 通过深入研究其中的化学机理和反应参数,可以更好地理解沙尘颗粒物对气溶胶二次产物的形成以区域气候变化的影响,将有助于更好地应对环境变化带来的挑战.
-
-
[1] URBAN F E, GOLDSTEIN H L, FULTON R, et al. Unseen dust emission and global dust abundance: Documenting dust emission from the mojave desert (USA) by daily remote camera imagery and wind-erosion measurements[J]. Journal of Geophysical Research: Atmospheres, 2018, 123(16): 8735-8753. doi: 10.1029/2018JD028466 [2] WU C L, LIN Z H, LIU X H. The global dust cycle and uncertainty in CMIP5 (Coupled Model Intercomparison Project phase 5) models[J]. Atmospheric Chemistry and Physics, 2020, 20(17): 10401-10425. doi: 10.5194/acp-20-10401-2020 [3] YU Y, GINOUX P. Enhanced dust emission following large wildfires due to vegetation disturbance[J]. Nature Geoscience, 2022, 15: 878-884. doi: 10.1038/s41561-022-01046-6 [4] CHEN W X, MENG H, SONG H Q, et al. Progress in dust modelling, global dust budgets, and soil organic carbon dynamics[J]. Land, 2022, 11(2): 176. doi: 10.3390/land11020176 [5] 田雨, 潘小乐, 姚维杰, 等. 基于颗粒物光学检测技术的大气沙尘气溶胶形貌、混合态研究进展[J]. 大气与环境光学学报, 2022, 17(1): 65-91. doi: 10.3969/j.issn.1673-6141.2022.01.005 TIAN Y, PAN X L, YAO W J, et al. Research progress on atmospheric aerosol morphology and mixing state properties based on particle optical detection technology[J]. Journal of Atmospheric and Environmental Optics, 2022, 17(1): 65-91 (in Chinese). doi: 10.3969/j.issn.1673-6141.2022.01.005
[6] ZHANG T L, ZHENG M, SUN X G, et al. Environmental impacts of three Asian dust events in the Northern China and the northwestern Pacific in spring 2021[J]. The Science of the Total Environment, 2023, 859(Pt 1): 160230. [7] 雷蕾, 张金谱, 裴成磊, 等. 2021年一次春季北方沙尘过程对广州空气质量的影响[J]. 环境科学学报, 2023, 43(1): 247-254. LEI L, ZHANG J P, PEI C L, et al. Influence of a northern dust weather process on air quality of Guangzhou in spring 2021[J]. Acta Scientiae Circumstantiae, 2023, 43(1): 247-254 (in Chinese).
[8] 尹志聪, 霍芊伊, 麻晓晴, 等. 触发2023年春季中国北方沙尘暴的沙源累积和天气扰动机制[J]. 大气科学学报, 2023, 46(3): 321-331. YIN Z C, HUO Q Y, MA X Q, et al. Mechanisms of dust source accumulation and synoptic disturbance triggering the 2023 spring sandstorm in Northern China[J]. Transactions of Atmospheric Sciences, 2023, 46(3): 321-331 (in Chinese).
[9] GOUDIE A, MIDDLETON N. Desert dust in the global system[M]. Berlin: Springer, 2006. [10] SCHEUVENS D, SCHÜTZ L, KANDLER K, et al. Bulk composition of northern African dust and its source sediments—a compilation[J]. Earth-Science Reviews, 2013, 116: 170-194. doi: 10.1016/j.earscirev.2012.08.005 [11] DREWNIK M, SKIBA M, SZYMAŃSKI W, et al. Mineral composition vs. soil forming processes in loess soils—a case study from Kraków (Southern Poland)[J]. CATENA, 2014, 119: 166-173. doi: 10.1016/j.catena.2014.02.012 [12] JOSHI N, ROMANIAS M N, RIFFAULT V, et al. Investigating water adsorption onto natural mineral dust particles: Linking DRIFTS experiments and BET theory[J]. Aeolian Research, 2017, 27: 35-45. doi: 10.1016/j.aeolia.2017.06.001 [13] DONG F Q, CHEN W, DAI Q W, et al. Characterization of mineralogy and surface zeta potential of atmospheric dust fall in Northwest China[J]. Mineralogy and Petrology, 2015, 109(3): 387-395. doi: 10.1007/s00710-014-0347-1 [14] FORMENTI P, RAJOT J L, DESBOEUFS K, et al. Regional variability of the composition of mineral dust from western Africa: Results from the AMMA SOP0/DABEX and DODO field campaigns[J]. Journal of Geophysical Research: Atmospheres, 2008, 113(D23): D00C13. [15] ROMANÍAS M N, OURRAD H, THÉVENET F, et al. Investigating the heterogeneous interaction of VOCs with natural atmospheric particles: Adsorption of limonene and toluene on Saharan mineral dusts[J]. The Journal of Physical Chemistry. A, 2016, 120(8): 1197-1212. doi: 10.1021/acs.jpca.5b10323 [16] HANISCH F, CROWLEY J N. Ozone decomposition on Saharan dust: An experimental investigation[J]. Atmospheric Chemistry and Physics, 2003, 3(1): 119-130. doi: 10.5194/acp-3-119-2003 [17] WANG Y D, ZHOU L, WANG W G, et al. Heterogeneous uptake of formic acid and acetic acid on mineral dust and coal fly ash[J]. ACS Earth and Space Chemistry, 2020, 4(2): 202-210. doi: 10.1021/acsearthspacechem.9b00263 [18] ARYAL R, KANDEL D, ACHARYA D, et al. Unusual Sydney dust storm and its mineralogical and organic characteristics[J]. Environmental Chemistry, 2012, 9(6): 537. doi: 10.1071/EN12131 [19] NOWAK S, LAFON S, CAQUINEAU S, et al. Quantitative study of the mineralogical composition of mineral dust aerosols by X-ray diffraction[J]. Talanta, 2018, 186: 133-139. doi: 10.1016/j.talanta.2018.03.059 [20] SADRIAN M R, CALVIN W M, McCORMACK J. Contrasting mineral dust abundances from X-ray diffraction and reflectance spectroscopy[J]. Atmospheric Measurement Techniques, 2022, 15(9): 3053-3074. doi: 10.5194/amt-15-3053-2022 [21] KANDLER K, SCHÜTZ L, JÄCKEL S, et al. Ground-based off-line aerosol measurements at Praia, Cape Verde, during the Saharan Mineral Dust Experiment: Microphysical properties and mineralogy[J]. Tellus B: Chemical and Physical Meteorology, 2011, 63(4): 459. doi: 10.1111/j.1600-0889.2011.00546.x [22] SENTHIL KUMAR R, RAJKUMAR P. Characterization of minerals in air dust particles in the state of Tamilnadu, India through FTIR, XRD and SEM analyses[J]. Infrared Physics & Technology, 2014, 67: 30-41. [23] SCHLEICHER N J, DONG S F, PACKMAN H, et al. A global assessment of copper, zinc, and lead isotopes in mineral dust sources and aerosols[J]. Frontiers in Earth Science, 2020, 8: 167. doi: 10.3389/feart.2020.00167 [24] HUANG L B, ZHAO Y, LI H, et al. Kinetics of heterogeneous reaction of sulfur dioxide on authentic mineral dust: Effects of relative humidity and hydrogen peroxide[J]. Environmental Science & Technology, 2015, 49(18): 10797-10805. [25] LI W J, SHAO L Y, SHI Z B, et al. Mixing state and hygroscopicity of dust and haze particles before leaving Asian continent[J]. Journal of Geophysical Research: Atmospheres, 2014, 119(2): 1044-1059. doi: 10.1002/2013JD021003 [26] 黄晓霞, 程宏, 蒋宁, 等. 京津风沙源治理对生态系统服务的影响及其效益核算[J]. 科学通报, 2023, 68(11): 1367-1380. doi: 10.1360/TB-2022-0295 HUANG X X, CHENG H, JIANG N, et al. Impact of the Beijing-Tianjin sandstorm source control project on ecosystem services and an evaluation of economic benefits[J]. Chinese Science Bulletin, 2023, 68(11): 1367-1380 (in Chinese). doi: 10.1360/TB-2022-0295
[27] 程宏, 张恺笛, 蒋宁, 等. 京津风沙源地表释尘到达典型城市沙尘量及其源解析[J]. 科学通报, 2023, 68(7): 801-816. doi: 10.1360/TB-2022-0477 CHENG H, ZHANG K D, JIANG N, et al. Dust amount reaching typical cities from dust emissions due to soil wind erosion in Beijing-Tianjin sandstorm source regions and its source analysis[J]. Chinese Science Bulletin, 2023, 68(7): 801-816 (in Chinese). doi: 10.1360/TB-2022-0477
[28] YANG J D, LI G J, RAO W B, et al. Isotopic evidences for provenance of East Asian dust[J]. Atmospheric Environment, 2009, 43(29): 4481-4490. doi: 10.1016/j.atmosenv.2009.06.035 [29] LI G, CHEN J, JI J, et al. Natural and anthropogenic sources of East Asian dust[J]. Geology, 2009, 37(8): 727-730. doi: 10.1130/G30031A.1 [30] DEWAN N, MAJESTIC B J, KETTERER M E, et al. Stable isotopes of lead and strontium as tracers of sources of airborne particulate matter in Kyrgyzstan[J]. Atmospheric Environment, 2015, 120: 438-446. doi: 10.1016/j.atmosenv.2015.09.017 [31] ERHARDT A M, DOUGLAS G, JACOBSON A D, et al. Assessing sedimentary detrital Pb isotopes as a dust tracer in the Pacific Ocean[J]. Paleoceanography and Paleoclimatology, 2021, 36(4): e2020PA004144. doi: 10.1029/2020PA004144 [32] CHEN S Y, ZHAO D, HUANG J P, et al. Mongolia contributed more than 42% of the dust concentrations in Northern China in March and April 2023[J]. Advances in Atmospheric Sciences, 2023, 40(9): 1549-1557. doi: 10.1007/s00376-023-3062-1 [33] RIZZOLO J A, BARBOSA C G G, BORILLO G C, et al. Soluble iron nutrients in Saharan dust over the central Amazon rainforest[J]. Atmospheric Chemistry and Physics, 2017, 17(4): 2673-2687. doi: 10.5194/acp-17-2673-2017 [34] SALMABADI H, KHALIDY R, SAEEDI M. Transport routes and potential source regions of the Middle Eastern dust over Ahvaz during 2005–2017[J]. Atmospheric Research, 2020, 241: 104947. doi: 10.1016/j.atmosres.2020.104947 [35] CHEN K Y. The northern path of Asian dust transport from the Gobi Desert to North America[J]. Atmospheric and Oceanic Science Letters, 2010, 3(3): 155-159. doi: 10.1080/16742834.2010.11446858 [36] CROWLEY J N, AMMANN M, COX R A, et al. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume V–heterogeneous reactions on solid substrates[J]. Atmospheric Chemistry and Physics, 2010, 10(18): 9059-9223. doi: 10.5194/acp-10-9059-2010 [37] PARK J Y, JANG M. Heterogeneous photooxidation of sulfur dioxide in the presence of airborne mineral dust particles[J]. RSC Advances, 2016, 6(63): 58617-58627. doi: 10.1039/C6RA09601H [38] TANG M J, HUANG X, LU K D, et al. Heterogeneous reactions of mineral dust aerosol: Implications for tropospheric oxidation capacity[J]. Atmospheric Chemistry and Physics, 2017, 17(19): 11727-11777. doi: 10.5194/acp-17-11727-2017 [39] UNDERWOOD G M, SONG C H, PHADNIS M, et al. Heterogeneous reactions of NO2 and HNO3 on oxides and mineral dust: A combined laboratory and modeling study[J]. Journal of Geophysical Research: Atmospheres, 2001, 106(D16): 18055-18066. doi: 10.1029/2000JD900552 [40] TANG M J, CZICZO D J, GRASSIAN V H. Interactions of water with mineral dust aerosol: Water adsorption, hygroscopicity, cloud condensation, and ice nucleation[J]. Chemical Reviews, 2016, 116(7): 4205-4259. doi: 10.1021/acs.chemrev.5b00529 [41] MOGILI P K, KLEIBER P D, YOUNG M A, et al. Heterogeneous uptake of ozone on reactive components of mineral dust aerosol: An environmental aerosol reaction chamber study[J]. The Journal of Physical Chemistry. A, 2006, 110(51): 13799-13807. doi: 10.1021/jp063620g [42] GUSTAFSSON R J, ORLOV A, BADGER C L, et al. A comprehensive evaluation of water uptake on atmospherically relevant mineral surfaces: DRIFT spectroscopy, thermogravimetric analysis and aerosol growth measurements[J]. Atmospheric Chemistry and Physics, 2005, 5(12): 3415-3421. doi: 10.5194/acp-5-3415-2005 [43] IBRAHIM S, ROMANIAS M N, ALLEMAN L Y, et al. Water interaction with mineral dust aerosol: Particle size and hygroscopic properties of dust[J]. ACS Earth and Space Chemistry, 2018, 2(4): 376-386. doi: 10.1021/acsearthspacechem.7b00152 [44] CHEN L, PENG C, GU W J, et al. On mineral dust aerosol hygroscopicity[J]. Atmospheric Chemistry and Physics, 2020, 20(21): 13611-13626. doi: 10.5194/acp-20-13611-2020 [45] GU W J, LI Y J, ZHU J X, et al. Investigation of water adsorption and hygroscopicity of atmospherically relevant particles using acommercial vapor sorption analyzer[J]. Atmospheric Measurement Techniques, 2017, 10(10): 3821-3832. doi: 10.5194/amt-10-3821-2017 [46] GOODMAN A L, BERNARD E T, GRASSIAN V H. Spectroscopic study of nitric acid and water adsorption on oxide particles: enhanced nitric acid uptake kinetics in the presence of adsorbed water[J]. The Journal of Physical Chemistry A, 2001, 105(26): 6443-6457. doi: 10.1021/jp003722l [47] SONG X W, BOILY J F. Water vapor adsorption on goethite[J]. Environmental Science & Technology, 2013, 47(13): 7171-7177. [48] TANG M J, CHAN C K, LI Y J, et al. A review of experimental techniques for aerosol hygroscopicity studies[J]. Atmospheric Chemistry and Physics, 2019, 19(19): 12631-12686. doi: 10.5194/acp-19-12631-2019 [49] MA Q X, HE H. Synergistic effect in the humidifying process of atmospheric relevant calcium nitrate, calcite and oxalic acid mixtures[J]. Atmospheric Environment, 2012, 50: 97-102. doi: 10.1016/j.atmosenv.2011.12.057 [50] YU Z C, JANG M. Simulation of heterogeneous photooxidation of SO2 and NO x in the presence of Gobi Desert dust particles under ambient sunlight[J]. Atmospheric Chemistry and Physics, 2018, 18(19): 14609-14622. doi: 10.5194/acp-18-14609-2018 [51] YU Z C, JANG M, PARK J. Modeling atmospheric mineral aerosol chemistry to predict heterogeneous photooxidation of SO2[J]. Atmospheric Chemistry and Physics, 2017, 17(16): 10001-10017. doi: 10.5194/acp-17-10001-2017 [52] SCHMIDT M, JANSEN van BEEK S M, ABOU-GHANEM M, et al. Production of atmospheric organosulfates via mineral-mediated photochemistry[J]. ACS Earth and Space Chemistry, 2019, 3(3): 424-431. doi: 10.1021/acsearthspacechem.8b00178 [53] MA Q X, ZHONG C, MA J Z, et al. Comprehensive study about the photolysis of nitrates on mineral oxides[J]. Environmental Science & Technology, 2021, 55(13): 8604-8612. [54] CHEN H H, NANAYAKKARA C E, GRASSIAN V H. Titanium dioxide photocatalysis in atmospheric chemistry[J]. Chemical Reviews, 2012, 112(11): 5919-5948. doi: 10.1021/cr3002092 [55] QIAN R F, ZONG H X, SCHNEIDER J, et al. Charge carrier trapping, recombination and transfer during TiO2 photocatalysis: An overview[J]. Catalysis Today, 2019, 335: 78-90. doi: 10.1016/j.cattod.2018.10.053 [56] THIEBAUD J, THÉVENET F, FITTSCHEN C. OH radicals and H2O2 molecules in the gas phase near to TiO2 surfaces[J]. The Journal of Physical Chemistry C, 2010, 114(7): 3082-3088. doi: 10.1021/jp9102542 [57] JENKIN M E, SAUNDERS S M, PILLING M J. The tropospheric degradation of volatile organic compounds: A protocol for mechanism development[J]. Atmospheric Environment, 1997, 31(1): 81-104. doi: 10.1016/S1352-2310(96)00105-7 [58] SAUNDERS S M, JENKIN M E, DERWENT R G, et al. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part a): Tropospheric degradation of non-aromatic volatile organic compounds[J]. Atmospheric Chemistry and Physics, 2003, 3(1): 161-180. doi: 10.5194/acp-3-161-2003 [59] DUPART Y, FINE L, D’ANNA B, et al. Heterogeneous uptake of NO2 on Arizona Test Dust under UV-a irradiation: An aerosol flow tube study[J]. Aeolian Research, 2014, 15: 45-51. doi: 10.1016/j.aeolia.2013.10.001 [60] PARK J, JANG M, YU Z C. Heterogeneous photo-oxidation of SO2 in the presence of two different mineral dust particles: Gobi and Arizona dust[J]. Environmental Science & Technology, 2017, 51(17): 9605-9613. [61] ZHOU L, WANG W G, GAI Y B, et al. Knudsen cell and smog chamber study of the heterogeneous uptake of sulfur dioxide on Chinese mineral dust[J]. Journal of Environmental Sciences (China), 2014, 26(12): 2423-2433. doi: 10.1016/j.jes.2014.04.005 [62] YU Z C, JANG M, KIM S, et al. Simulating the impact of long-range-transported Asian mineral dust on the formation of sulfate and nitrate during the KORUS-AQ campaign[J]. ACS Earth and Space Chemistry, 2020, 4(7): 1039-1049. doi: 10.1021/acsearthspacechem.0c00074 [63] LI X P, FENG L N, HUANG C C, et al. Chemical characteristics of atmospheric fallout in the south of Xi’an during the dust episodes of 2001–2012 (NW China)[J]. Atmospheric Environment, 2014, 83: 109-118. doi: 10.1016/j.atmosenv.2013.10.004 [64] USHER C. Laboratory studies of ozone uptake on processed mineral dust[J]. Atmospheric Environment, 2003, 37(38): 5337-5347. doi: 10.1016/j.atmosenv.2003.09.014 [65] CHEN H H, NAVEA J G, YOUNG M A, et al. Heterogeneous photochemistry of trace atmospheric gases with components of mineral dust aerosol[J]. The Journal of Physical Chemistry. A, 2011, 115(4): 490-499. doi: 10.1021/jp110164j [66] CHEN H H, STANIER C O, YOUNG M A, et al. A kinetic study of ozone decomposition on illuminated oxide surfaces[J]. The Journal of Physical Chemistry. A, 2011, 115(43): 11979-11987. doi: 10.1021/jp208164v [67] MICHEL A E, USHER C R, GRASSIAN V H. Reactive uptake of ozone on mineral oxides and mineral dusts[J]. Atmospheric Environment, 2003, 37(23): 3201-3211. doi: 10.1016/S1352-2310(03)00319-4 [68] CHANG R Y W, SULLIVAN R C, ABBATT J P D. Initial uptake of ozone on Saharan dust at atmospheric relative humidities[J]. Geophysical Research Letters, 2005, 32(14): L14815. [69] LASNE J, ROMANIAS M N, THEVENET F. Ozone uptake by clay dusts under environmental conditions[J]. ACS Earth and Space Chemistry, 2018, 2(9): 904-914. doi: 10.1021/acsearthspacechem.8b00057 [70] FAN L, SHEN Z Z, WANG Z Y, et al. Effect of photothermal conversion on ozone uptake over deposited mineral dust[J]. The Science of the Total Environment, 2023, 871: 162047. doi: 10.1016/j.scitotenv.2023.162047 [71] ZHANG Y, SUNWOO Y, KOTAMARTHI V, et al. Photochemical oxidant processes in the presence of dust: An evaluation of the impact of dust on particulate nitrate and ozone formation[J]. Journal of Applied Meteorology and Climatology, 1994, 33(7): 813-824. doi: 10.1175/1520-0450(1994)033<0813:POPITP>2.0.CO;2 [72] CRIPPA M, GUIZZARDI D, PISONI E, et al. Global anthropogenic emissions in urban areas: Patterns, trends, and challenges[J]. Environmental Research Letters, 2021, 16(7): 074033. doi: 10.1088/1748-9326/ac00e2 [73] JOHNSTON D T. Multiple sulfur isotopes and the evolution of Earth’s surface sulfur cycle[J]. Earth-Science Reviews, 2011, 106(1/2): 161-183. [74] MA Q X, ZHANG C Y, LIU C, et al. A review on the heterogeneous oxidation of SO2 on solid atmospheric particles: Implications for sulfate formation in haze chemistry[J]. Critical Reviews in Environmental Science and Technology, 2023, 53(21): 1888-1911. doi: 10.1080/10643389.2023.2190315 [75] ADAMS J W, RODRIGUEZ D, COX R A. The uptake of SO2 on Saharan dust: A flow tube study[J]. Atmospheric Chemistry and Physics, 2005, 5(10): 2679-2689. doi: 10.5194/acp-5-2679-2005 [76] MA Q X, WANG L, CHU B W, et al. Contrary role of H2O and O2 in the kinetics of heterogeneous photochemical reactions of SO2 on TiO2[J]. The Journal of Physical Chemistry. A, 2019, 123(7): 1311-1318. doi: 10.1021/acs.jpca.8b11433 [77] AL-HOSNEY H A, GRASSIAN V H. Water, sulfur dioxide and nitric acid adsorption on calcium carbonate: A transmission and ATR-FTIR study[J]. Physical Chemistry Chemical Physics: PCCP, 2005, 7(6): 1266-1276. doi: 10.1039/b417872f [78] RUAN X Y, ZHAO C, ZAVERI R A, et al. Simulations of aerosol pH in China using WRF-Chem (v4.0): Sensitivities of aerosol pH and its temporal variations during haze episodes[J]. Geoscientific Model Development, 2022, 15(15): 6143-6164. doi: 10.5194/gmd-15-6143-2022 [79] WANG Z Z, WANG T, FU H B, et al. Enhanced heterogeneous uptake of sulfur dioxide on mineral particles through modification of iron speciation during simulated cloud processing[J]. Atmospheric Chemistry and Physics, 2019, 19(19): 12569-12585. doi: 10.5194/acp-19-12569-2019 [80] WANG T, LIU Y Y, DENG Y, et al. Emerging investigator series: Heterogeneous reactions of sulfur dioxide on mineral dust nanoparticles: From single component to mixed components[J]. Environmental Science: Nano, 2018, 5(8): 1821-1833. doi: 10.1039/C8EN00376A [81] ANGELINI M M, GARRARD R J, ROSEN S J, et al. Heterogeneous reactions of gaseous HNO3 and NO2 on the clay minerals kaolinite and pyrophyllite[J]. The Journal of Physical Chemistry. A, 2007, 111(17): 3326-3335. doi: 10.1021/jp0672656 [82] NDOUR M, D’ANNA B, GEORGE C, et al. Photoenhanced uptake of NO2 on mineral dust: Laboratory experiments and model simulations[J]. Geophysical Research Letters, 2008, 35(5): L05812. [83] NDOUR M, NICOLAS M, D’ANNA B, et al. Photoreactivity of NO2 on mineral dusts originating from different locations of the Sahara Desert[J]. Physical Chemistry Chemical Physics, 2009, 11(9): 1312-1319. doi: 10.1039/b806441e [84] MA Q X, LIU Y C, HE H. Synergistic effect between NO2 and SO2 in their adsorption and reaction on gamma-alumina[J]. The Journal of Physical Chemistry. A, 2008, 112(29): 6630-6635. doi: 10.1021/jp802025z [85] LIU C, MA Q X, LIU Y C, et al. Synergistic reaction between SO2 and NO2 on mineral oxides: A potential formation pathway of sulfate aerosol[J]. Physical Chemistry Chemical Physics, 2012, 14(5): 1668-1676. doi: 10.1039/C1CP22217A [86] CHU B W, LIU Y, LI H, et al. Photocatalytic oxidation of NO2 on TiO2: Evidence of a new source of N2 O5[J]. Angewandte Chemie (International Ed. in English), 2023, 62(25): e202304017. doi: 10.1002/anie.202304017 [87] WAGNER C, SCHUSTER G, CROWLEY J N. An aerosol flow tube study of the interaction of N2O5 with calcite, Arizona dust and quartz[J]. Atmospheric Environment, 2009, 43(32): 5001-5008. doi: 10.1016/j.atmosenv.2009.06.050 [88] TANG M J, THIESER J, SCHUSTER G, et al. Uptake of NO3 and N2O5 to Saharan dust, ambient urban aerosol and soot: A relative rate study[J]. Atmospheric Chemistry and Physics, 2010, 10(6): 2965-2974. doi: 10.5194/acp-10-2965-2010 [89] TANG M J, THIESER J, SCHUSTER G, et al. Kinetics and mechanism of the heterogeneous reaction of N2O5 with mineral dust particles[J]. Physical Chemistry Chemical Physics, 2012, 14(24): 8551-8561. doi: 10.1039/c2cp40805h [90] MA Q X, WANG T, LIU C, et al. SO2 initiates the efficient conversion of NO2 to HONO on MgO surface[J]. Environmental Science & Technology, 2017, 51(7): 3767-3775. [91] HE H, WANG Y S, MA Q X, et al. Mineral dust and NO x promote the conversion of SO2 to sulfate in heavy pollution days[J]. Scientific Reports, 2014, 4: 4172. doi: 10.1038/srep04172 [92] PAN X L, GE B Z, WANG Z, et al. Synergistic effect of water-soluble species and relative humidity on morphological changes in aerosol particles in the Beijing megacity during severe pollution episodes[J]. Atmospheric Chemistry and Physics, 2019, 19(1): 219-232. doi: 10.5194/acp-19-219-2019 [93] TIAN Y, PAN X L, WANG Z, et al. Transport patterns, size distributions, and depolarization characteristics of dust particles in East Asia in spring 2018[J]. Journal of Geophysical Research: Atmospheres, 2020, 125(16): e2019JD031752. doi: 10.1029/2019JD031752 [94] GEN M S, LIANG Z C, ZHANG R F, et al. Particulate nitrate photolysis in the atmosphere[J]. Environmental Science: Atmospheres, 2022, 2(2): 111-127. doi: 10.1039/D1EA00087J [95] RIORDAN E, MINOGUE N, HEALY D, et al. Spectroscopic and optimization modeling study of nitrous acid in aqueous solution[J]. The Journal of Physical Chemistry. A, 2005, 109(5): 779-786. doi: 10.1021/jp040269v [96] SU H, CHENG Y F, OSWALD R, et al. Soil nitrite as a source of atmospheric HONO and OH radicals[J]. Science, 2011, 333(6049): 1616-1618. doi: 10.1126/science.1207687 [97] PENG X, WANG T, WANG W H, et al. Photodissociation of particulate nitrate as a source of daytime tropospheric Cl2[J]. Nature Communications, 2022, 13: 939. doi: 10.1038/s41467-022-28383-9 [98] FALKOVICH A H, SCHKOLNIK G, GANOR E, et al. Adsorption of organic compounds pertinent to urban environments onto mineral dust particles[J]. Journal of Geophysical Research: Atmospheres, 2004, 109(D2): D02208. [99] WANG G H, CHENG C L, MENG J J, et al. Field observation on secondary organic aerosols during Asian dust storm periods: Formation mechanism of oxalic acid and related compounds on dust surface[J]. Atmospheric Environment, 2015, 113: 169-176. doi: 10.1016/j.atmosenv.2015.05.013 [100] LIU Q Y, LIU Y J, ZHAO Q, et al. Increases in the formation of water soluble organic nitrogen during Asian dust storm episodes[J]. Atmospheric Research, 2021, 253: 105486. doi: 10.1016/j.atmosres.2021.105486 [101] AL-HOSNEY H A, CARLOS-CUELLAR S, BALTRUSAITIS J, et al. Heterogeneous uptake and reactivity of formic acid on calcium carbonate particles: A Knudsen cell reactor, FTIR and SEM study[J]. Physical Chemistry Chemical Physics: PCCP, 2005, 7(20): 3587-3595. doi: 10.1039/b510112c [102] LEDERER M R, STANIEC A R, COATES FUENTES Z L, et al. Heterogeneous reactions of limonene on mineral dust: Impacts of adsorbed water and nitric acid[J]. The Journal of Physical Chemistry. A, 2016, 120(48): 9545-9556. doi: 10.1021/acs.jpca.6b09865 [103] YU Z C, JANG M. Atmospheric processes of aromatic hydrocarbons in the presence of mineral dust particles in an urban environment[J]. ACS Earth and Space Chemistry, 2019, 3(11): 2404-2414. doi: 10.1021/acsearthspacechem.9b00195 [104] PONCZEK M, HAYECK N, EMMELIN C, et al. Heterogeneous photochemistry of dicarboxylic acids on mineral dust[J]. Atmospheric Environment, 2019, 212: 262-271. doi: 10.1016/j.atmosenv.2019.05.032 [105] XUE Y G, HUANG Y, HO S S H, et al. Origin and transformation of ambient volatile organic compounds during a dust-to-haze episode in Northwest China[J]. Atmospheric Chemistry and Physics, 2020, 20(9): 5425-5436. doi: 10.5194/acp-20-5425-2020 [106] BALTRUSAITIS J, SCHUTTLEFIELD J, ZEITLER E, et al. Carbon dioxide adsorption on oxide nanoparticle surfaces[J]. Chemical Engineering Journal, 2011, 170(2/3): 471-481. [107] NANAYAKKARA C E, LARISH W A, GRASSIAN V H. Titanium dioxide nanoparticle surface reactivity with atmospheric gases, CO2, SO2, and NO2: Roles of surface hydroxyl groups and adsorbed water in the formation and stability of adsorbed products[J]. The Journal of Physical Chemistry C, 2014, 118(40): 23011-23021. doi: 10.1021/jp504402z [108] ANPO M, YAMASHITA H, ICHIHASHI Y, et al. Photocatalytic reduction of CO2 with H2O on various titanium oxide catalysts[J]. Journal of Electroanalytical Chemistry, 1995, 396(1/2): 21-26. [109] CHO-CHING L O, HUNG C H, YUAN C S, et al. Parameter effects and reaction pathways of photoreduction of CO2 over TiO2/SO42–photocatalyst[J]. Chinese Journal of Catalysis, 2007, 28(6): 528-534. doi: 10.1016/S1872-2067(07)60046-1 [110] INDRAKANTI V P, KUBICKI J D, SCHOBERT H H. Photoinduced activation of CO2 on TiO2 surfaces: Quantum chemical modeling of CO2 adsorption on oxygen vacancies[J]. Fuel Processing Technology, 2011, 92(4): 805-811. doi: 10.1016/j.fuproc.2010.09.007 [111] DENG Y, LIU Y Y, WANG T, et al. Photochemical reaction of CO2 on atmospheric mineral dusts[J]. Atmospheric Environment, 2020, 223: 117222. doi: 10.1016/j.atmosenv.2019.117222 [112] LIU Y Y, WANG T, FANG X Z, et al. Impact of greenhouse gas CO2 on the heterogeneous reaction of SO2 on alpha-Al2O3[J]. Chinese Chemical Letters, 2020, 31(10): 2712-2716. doi: 10.1016/j.cclet.2020.04.037 [113] LIU Y Y, DENG Y, LIU J R, et al. A novel pathway of atmospheric sulfate formation through carbonate radicals[J]. Atmospheric Chemistry and Physics, 2022, 22(13): 9175-9197. doi: 10.5194/acp-22-9175-2022 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1018
- HTML全文浏览数: 1018
- PDF下载数: 35
- 施引文献: 0







