



-
砷(As)广泛分布于环境中,世界上许多国家的地下水受到砷的污染[1-2].地下水的砷污染已成为一个世界性的重大问题.流行病学研究表明,人类长期接触含砷化合物会导致皮肤损害、血管疾病、高血压和癌症等疾病[3-4].砷在环境中主要以无机砷和有机胂两种形态存在[5],无机砷主要有亚砷酸盐和砷酸盐,有机胂主要以一甲基胂酸盐、二甲基胂酸盐以及胂胆碱、胂甜菜碱、胂糖、胂脂类化合物等形态存在[6].在人类体内摄取和代谢过程中,有机态胂化合物与无机砷会产生含有二甲基胂酸尿代谢物[7].在一定条件下,水环境中的二甲基胂酸会转化为毒性较高的无机砷.因此,研究二甲基胂酸在地下水污染研究中具有相当重大的意义.
砷从土壤、地表水和沉积物中向地下水中释放或迁移的过程受诸多因素控制[8].腐殖酸(HA)是水体中广泛存在的天然有机物,腐殖酸可以通过络合、竞争吸附位点和电子穿梭等化学反应强烈的影响地下水中砷的迁移[9-10].腐殖酸抑制砷的吸附主要是通过腐殖酸和砷在铁氧化物表面的竞争吸附作用[11].As(Ⅴ)/As(Ⅲ)/二甲基胂酸与HA络合的影响因素有离子强度和pH等,As(Ⅴ)/As(Ⅲ)和二甲基胂酸与 HA 络合程度随着离子强度的增加而减少,离子强度对砷吸附的影响可能归因于腐殖酸分子表面化学性质的变化.在弱酸条件下,As(Ⅴ)和二甲基胂酸的去除率较高,随着pH增大,As(Ⅴ)和二甲基胂酸的去除率急剧下降,可能是由于 HA 与 As(Ⅴ)/二甲基胂酸竞争含铁矿物表面的吸附位点,因为随着 pH增加,As(Ⅴ)和二甲基胂酸带负电荷,这会导致 As(Ⅴ) 和二甲基胂酸与含铁矿物的静电斥力增加[12].在pH为中性时,As(Ⅲ)的去除率最高,低pH下As(Ⅲ)的去除率低,是因为在低pH条件下As(Ⅲ)主要以As(OH)3形式存在,其在低pH下与氧化物的相互作用最小,然而,随着pH值的升高,腐殖酸变得更易溶,在地下水中以腐殖酸盐或络合物的形式存在,这些腐殖酸盐中的离子可能在高pH下解离产生含水氧化物 (例如Fe,Al)吸附剂[13],使砷和腐殖酸的络合减弱.腐殖酸对无机砷酸中As(Ⅴ)和As(Ⅲ)的影响机制有所不同,腐殖酸中的亲核基团能与As(Ⅴ)中的亲电原子(砷原子)发生亲电反应,腐殖酸中的羧酸基团与As(Ⅲ)通过酯基相结合[14].当腐殖酸和有机胂共存时,由于腐殖酸带负电荷,它可以被带正电荷的吸附剂吸引,腐殖酸的羧基和酚基可以与质子化的胺基相互作用形成有机络合物,这些有机络合物对结合位点的直接竞争可能导致有机胂的去除率下降[15].
水体中的铁、锰氧化物或氢氧化物是砷的主要载体[16],砷可以吸附在含铁矿物表面,形成络合物,降低了砷在水中的迁移能力[17-18].磁铁矿是水体中常见的含铁矿物,其不仅能够选择性的吸附砷,在低磁场下还可以从水相中迅速分离,在环境工程领域已被用于去除水体中的砷.在铁(Fe) 存在的情况下,溶解性有机质(DOM)可以通过Fe 桥联结合大量的砷而形成 As-Fe-DOM 络合物[19].在As-Fe-DOM 络合物中,砷和DOM的络合会使DOM的荧光基团发生荧光猝灭,并被三维荧光技术所检测.本论文以腐殖酸-二甲基胂酸体系的荧光特性、分子表面电荷特性以及红外特征为抓手,探讨了不同吸附时间、吸附剂用量和pH下,腐殖酸对二甲基胂酸在磁铁矿上吸附的影响.通过红外光谱、Zeta电位、X 射线光电子能谱和三维荧光光谱的数据分析,解析磁铁矿-腐殖酸-二甲基胂酸三者之间的相互作用机制,为砷污染环境风险评估与治理提供了科学依据.
-
FeCl3·6H2O、结晶乙酸钠、乙二醇、二甲基胂酸、腐殖酸(Aladdin-阿拉丁试剂(上海)有限公司)、优级纯盐酸(天津市富宇精细化工有限公司);氢氧化钠(天津市红岩化学试剂厂);硝酸(天津永晟精细化工有限公司).所有试剂均为分析纯.
-
在常温下将4.0 gFeCl3·6H2O和8.0 g结晶乙酸钠溶解在40 mL乙二醇溶液中,搅拌约30 min后,将溶液转移到50 mL的聚四氟乙烯内衬不锈钢高压釜中,在200 ℃保存8 h,自然冷却至室温后将黑色沉淀物离心,用乙醇洗涤至中性后在真空烘箱中60 ℃条件下烘干,得到磁铁矿样品.
-
取适量磁铁矿样品,测定条件为:扫描电压40 kV,电流200 mA,步长0.02(°)·min−1,扫描速度17.7s·步−1,θ范围10°—90°,Cu靶Kα射线(λ=0.15406 nm).
-
取适量磁铁矿样品,使用LakeShore公司的7404仪器,测试条件为:极头直径:5 cm;室温测量灵敏度:5×10−7 emu;矩测量范围:5×10−7 emu到103 emu;最大磁场:2.17T @ 16.2 mm极头间距.
-
用0.01 mol·L−1盐酸和0.01 mol·L−1NaOH调节其pH值,在pH=7时,称取1.5 g腐殖酸定容到100 mL,得到15000 g·L−1的腐殖酸溶液,二甲基胂酸初始浓度为1 mg·L−1,腐殖酸与二甲基胂酸的摩尔比为1.84:1,加入0.064 g磁铁矿后将离心管置于振荡器上开始吸附并计时.分别在0、0.17、0.5、1、2、10、24 h时取样2 mL, 用0.22 µm过滤器过滤后测定滤液中的二甲基胂酸浓度.每组数据测定三次,取平均值.
式中,Qe、Qt分别为吸附平衡和t时刻二甲基胂酸在磁铁矿上的吸附量(mg·g−1);K1、K2分别为准一级和准二级动力学吸附速率常数(单位分别为h−1、mg·g−1·h−1).
-
用0.01 mol·L−1盐酸和0.01 mol·L−1NaOH调节其pH值.当pH=7、9、11时,在50 mL离心管中,加入0.4 mL 50 mg·L−1的二甲基胂酸溶液.二甲基胂酸初始浓度为1 mg·L−1,根据腐殖酸与二甲基胂酸的摩尔比为1.84:1,计算腐殖酸的用量.再分别加入0.008、0.016、0.032、0.064、0.128 g磁铁矿,将离心管置于振荡器上开始吸附计时.吸附24 h后,取样2 mL,经0.22 µm过滤器过滤后测定滤液中的二甲基胂酸浓度.每组数据测定3次,取平均值.
Langmuir 吸附等温方程:
式中,Qe为吸附平衡浓度(mg·g−1);Ce为溶液中剩余二甲基胂酸浓度(mg·L−1);KL为Langmuir吸附常数(L·mg−1);Qm最大吸附量(mg·g−1).
Freundlich吸附等温方程:
式中,Qe为吸附平衡浓度(mg·g−1);Ce为溶液中剩余二甲基胂酸浓度(mg·L−1);KF为Freundlich吸附常数(mg(1-n)Ln·g−1);n为吸附强度.
-
用安捷伦5900型等离子发射光谱仪(ICP)测定溶液中总砷浓度.每组数据测定3次,取平均值.用下式计算二甲基胂酸的吸附量:
式中Qe为吸附量,单位为mg·g−1;V为溶液体积,单位升(L);C0为初始浓度,单位是mg·L−1;Ce为平衡后的浓度,单位为mg·L−1;m是吸附材料的质量,在实验中即为磁铁矿的质量,单位为g.
-
pH=7时,二甲基胂酸初始浓度为1 mg·L−1,腐殖酸浓度为150 mg·L−1,加入0.064 g磁铁矿,单独或混合吸附,吸附时间为24 h,得到磁铁矿-二甲基胂酸、磁铁矿-腐殖酸和磁铁矿-二甲基胂酸-腐殖酸的样品,使用Zetasizer2000型马尔文Zeta电位仪测定样品的Zeta电位.与纯磁铁矿样品的Zeta电位进行比较.
-
pH=7时,二甲基胂酸初始浓度为1 mg·L−1,腐殖酸浓度为150 mg·L−1,加入0.064 g磁铁矿,单独或混合吸附吸附24 h,得到磁铁矿、磁铁矿-二甲基胂酸、和磁铁矿-二甲基胂酸-腐殖酸体系的样品,经离心和烘干后,用KBr压片法在红外仪测定样品的红外谱图.
-
pH=7时,二甲基胂酸初始浓度为1 mg·L−1,腐殖酸浓度为150 mg·L−1,加入0.064 g磁铁矿,单独或混合吸附24 h,得到磁铁矿、磁铁矿-二甲基胂酸、和磁铁矿-二甲基胂酸-腐殖酸样品压片后,贴于样品盘上,将样品放进Thermo Scientific K-Alpha XPS仪器样品室中,在样品室的压力小于2.0×10−7 mbar时,将样品送入分析室,光斑大小为400 μm,工作电压12 kV,灯丝电流6 Ma;全谱扫描通能为150 eV,步长1 eV;窄谱扫描通能为50 eV,步长0.1 eV.
-
用0.01 mol·L−1盐酸和0.01 mol·L−1NaOH调节pH为7、9、11,二甲基胂酸初始浓度为1 mg·L−1,腐殖酸浓度为150 mg·L−1,加入0.064 g磁铁矿吸附24 h,得到不同pH时的磁铁矿-腐殖酸和磁铁矿-腐殖酸-二甲基胂酸体系的样品,过滤后进行三维荧光光谱测定.测定使用AQUALOG型三维荧光光谱仪获得EEM光谱,设定激发波长(Ex)的范围为200—800 nm,发射波长(Em)的范围为200—600 nm,激发和发射狭缝宽度为5.0 nm;激发波长扫描间隔为5.0 nm;扫描速度为1200 nm·min−1.通过减去去离子水的背景荧光信号来调节原始的EEM荧光光谱.测定过程中去除了瑞利和拉曼散射.
-
对合成的磁铁矿进行了X射线衍射分析,结果如图1所示.合成的磁铁矿和磁铁矿的标准卡(PDF#:79- 417)非常吻合,为尖晶石型结构且有良好的结晶度.合成的磁铁矿在18.20º(111)、30.0º(220)、35.3º(311)、42.9º(400)、53.4º(422)、56.9º(511)、62.5º(440)、74.20º(533)等衍射峰与标准图谱数据一致[20].衍射峰尖锐,强度强,这说明磁铁矿的物相单一,结晶度很好[21].
-
为了考察磁铁矿的磁化强度,对其进行了磁滞回线分析,结果如图1所示.磁铁矿的磁滞回线接近“S”型闭合曲线,磁铁矿的磁化强度随着磁场强度的增大而增大,在磁场强度为20 kA·m−1时,磁化强度基本达到饱和,磁铁矿的饱和磁化强度为83.23 emu·g−1.这说明通过水热合成法制备的磁铁矿磁化强度较强.
-
在pH=7时,二甲基胂酸初始浓度为1 mg·L−1,腐殖酸浓度为150 mg·L−1,加入0.064 g磁铁矿,分别在0、0.17、0.5、1、2、10、24 h时取样测定二甲基胂酸的浓度. 结果如图2所示.在pH为7时,添加腐殖酸前后,二甲基胂酸在磁铁矿上吸附24 h后均达到了吸附平衡,在2 h内二甲基胂酸在磁铁矿上的吸附比较快,2 h后吸附速率逐渐减慢.与磁铁矿-二甲基胂酸体系对比,加入腐殖酸后,磁铁矿对二甲基胂酸的吸附量明显下降.说明腐殖酸抑制了磁铁矿对二甲基胂酸的吸附.
分别用行准一级动力学和准二级动力学方程对二甲基胂酸在磁铁矿上的吸附过程进行拟合,考察了共存腐殖酸对二甲基胂酸吸附动力学的影响,结果如图3所示,拟合参数如表1所示.由图3和表1可知,二甲基胂酸在磁铁矿上的吸附符合准二级动力学模型(R2=0.99).加入腐殖酸后,二甲基胂酸在磁铁矿上的吸附仍然符合准二级动力学模型(R2=0.99).
由于准二级动力学方程被用来描述化学吸附过程,因此,二甲基胂酸在磁铁矿上的吸附属于化学吸附[22].当未添加腐殖酸时,二甲基胂酸的吸附速率常数K2为0.18 mg·g−1·h−1,加入腐殖酸后,二甲基胂酸的吸附速率常数K2为0.00018 mg·g−1·h−1.这说明腐殖酸的加入明显降低了二甲基胂酸的吸附速率,这可能是腐殖酸和二甲基胂酸竞争磁铁矿表面的吸附位点,降低了磁铁矿对二甲基胂酸的吸附[23-24].
-
当pH为7、9、11时,在二甲基胂酸-磁铁矿和二甲基胂酸-磁铁矿-腐殖酸吸附体系中,吸附24 h后取样测定二甲基胂酸的平衡浓度,计算二甲基胂酸的平衡吸附量.结果如图4所示.在pH=7、9、11时,在磁铁矿-二甲基胂酸体系中,二甲基胂酸的最大吸附量分别是0.47、0.17、0.18 mg·g−1;在磁铁矿-二甲基胂酸-腐殖酸体系中,二甲基胂酸的吸附量分别是0.08、0.06、0.04 mg·g−1. 与未添加腐殖酸的吸附体系相比,加入腐殖酸后,二甲基胂酸的吸附量均明显减小.说明在不同pH时,腐殖酸均抑制了二甲基胂酸在磁铁矿上的吸附[25].在二甲基胂酸-磁铁矿和二甲基胂酸-磁铁矿-腐殖酸吸附体系中,pH=7时,二甲基胂酸的吸附量最大,pH=11时,二甲基胂酸的吸附量最小.表明随着pH值升高,磁铁矿对二甲基胂酸的吸附能力也逐渐下降[26].
用Langmuir吸附等温模型和Freundlich吸附等温模型对磁铁矿-二甲基胂酸和磁铁矿-二甲基胂酸-腐殖酸体系吸附等温线进行非线性拟合,拟合曲线如图4所示,拟合参数如表2所示.拟合结果表明,在Langmuir吸附等温模型中,Qm和KL均为负值.说明二甲基胂酸在磁铁矿上的吸附不符合Langmuir吸附等温模型.pH=7、9、11时, 在磁铁矿-二甲基胂酸体系中, Freundlich吸附等温模型的R2分别为0.90、0.97、0.90,二甲基胂酸-磁铁矿-腐殖酸吸附体系中,Freundlich吸附等温模型的R2分别为0.63、0.97、0.82,Qm和KL均为正值.表明二甲基胂酸在磁铁矿上的吸附更符合Freundlich吸附等温模型,属于多层吸附[27].n值均<1,为弱吸附.
磁铁矿与腐殖酸之间的配位交换机理与其表面羟基离子化产生的可变电荷有关.腐殖酸在天然水体中呈现负电性,二甲基胂酸也以砷酸根阴离子存在,腐殖酸可以有效地与二甲基胂酸竞争磁铁矿表面的吸附位点.导致磁铁矿对二甲基胂酸的吸附减弱[28].磁铁矿可以通过表面配位交换、酚羟基相互作用、阴离子交换、氢键以及阳离子键桥等方式与腐殖酸发生作用,腐殖酸的羧基官能团可通过形成带负电荷的化合物与二甲基胂酸结合[29],并通过H+桥形成稳定的络合物.腐殖酸对磁铁矿有很强的亲和力,可以与磁铁矿形成表面内层络合物.而这些金属络合物又可以与溶解的砷酸根阴离子强烈结合,通过金属桥联机制,减弱了砷酸根阴离子与磁铁矿形成表面络合物的趋势[30].因此,腐殖酸抑制了磁铁矿对二甲基胂酸的去除.
-
在水环境中,磁铁矿表面通常带有—OH,这些羟基随着pH的改变从而结合或者释放H+,形成了表面电荷.为了探究二甲基胂酸和腐殖酸对磁铁矿表面Zeta电位的影响,测定了纯磁铁矿、磁铁矿-二甲基胂酸、磁铁矿-腐殖酸和二甲基胂酸-磁铁矿-腐殖酸体系在pH值为3—11范围内的Zeta电位,结果如图5所示.由图5可知,纯磁铁矿Zeta电位在3.6— −41.5之间,等电点为3.95,加入二甲基胂酸后,磁铁矿-二甲基胂酸的Zeta电位在44.6— −45.8之间,等电点为6.54,加入腐殖酸后,二甲基胂酸-磁铁矿-腐殖酸体系的Zeta电位在−24.5— −57.8之间.
纯磁铁矿等电点的与文献中磁铁矿的等电点相差较大的原因可能是合成的磁铁矿粒径不同[31].磁铁矿-二甲基胂酸等电点比纯磁铁矿体系等电点高.等电点向右移动,说明磁铁矿和二甲基胂酸发生了静电作用[32].在磁铁矿-二甲基胂酸体系中,二甲基胂酸以砷酸根的形式存在,吸附在磁铁矿表面并与磁铁矿表面的羟基发生交换,使砷酸根离子在磁铁矿表面富集,从而使磁铁矿表面的负电荷增加,测定的Zeta电位降低[33].加入二甲基胂酸后,二甲基胂酸-磁铁矿-腐殖酸体系的Zeta电位整体偏左移,这说明二甲基胂酸与磁铁矿形成了内层络合物,这与其他有机物在不同铁氧化物上的吸附一致[34].当pH=3时,磁铁矿表面呈现正电性,所以磁铁矿会吸附大量二甲基胂酸,加入腐殖酸后,呈负电性的腐殖酸会阻碍二甲基胂酸与磁铁矿作用.随着pH的增加,磁铁矿Zeta电位由正转为负,而且负电性越来越大[35].腐殖酸与磁铁矿发生静电吸附,占据了磁铁矿表面的吸附位点,降低了二甲基胂酸在磁铁矿表面的吸附.
-
通过KBr压片法测定了磁铁矿和二甲基胂酸加入腐殖酸前后吸附平衡时的红外谱图,探究了腐殖酸对磁铁矿吸附二甲基胂酸的影响机制,结果如图6所示.由图6可知,在586 cm−1处是磁铁矿的特征吸收峰[36].在850 cm−1处出现了二甲基胂酸中As—O键的伸缩振动峰[37].在1686 cm−1处是H—O—H的弯曲振动, Fe—OH的特征峰在882 cm−1处.并且400 —800 cm−1区间的红外峰发生了较大的变化,这个范围的峰主要为Fe—O的伸缩振动,表明磁铁矿与腐殖酸形成了络合物.2921 cm−1处是二甲基胂酸分子中甲基的C—H键对称伸缩振动[38].
腐殖酸中含有羧基、羟基、胺基和酚羟基等多种基团,其中羧基和酚羟基常与某些金属元素(如Fe、Mn和Al)形成各种络合物[5],以促进砷在络合物中的结合并增强砷的迁移.3450 cm−1处的特征峰属于腐殖酸中酚羟基或醇羟基(—OH)的伸缩振动吸收峰[38],在1390 cm−1处是腐殖酸中羧基对称振动峰[39].加入腐殖酸后,1390 cm−1处特征峰的强度发生了明显的增强,表明羧基参与了反应,通过配体交换形成内层络合物[40],综上所述,腐殖酸中的羧基和酚羟基与磁铁矿发生反应,形成了Fe—O键,而二甲基胂酸也能与Fe—O发生作用,形成了As—O—Fe键.1637 cm−1处的峰是酰胺中的C=O和芳环中的C—C的伸缩振动峰.加入腐殖酸后,1637 cm−1处的特征吸收峰的强度增强也说明了腐殖酸与磁铁矿和二甲基胂酸发生了络合作用,形成了二甲基胂酸-磁铁矿-腐殖酸络合物[41-42].Wang等[43]也发现腐殖酸的羧基(—COOH)和酚类(—OH)可以通过配体交换机制与As(Ⅲ)/(Ⅴ)和Fe3+结合,形成As(Ⅲ)/(Ⅴ)—Fe—HA络合物.
-
X射线光电子能谱揭示吸附剂表面各元素化学形态和结构信息.在pH=7时,二甲基胂酸初始浓度为1 mg·L−1,腐殖酸浓度为150 mg·L−1,测定和比较了添加腐殖酸前后,磁铁矿-二甲基胂酸样品的X射线光电子能谱.结果如图7所示.图7a给出了添加腐殖酸前后,磁铁矿-二甲基胂酸样品Fe2p、O1s、C1s、As3d全谱图.图7b-e分别是添加腐殖酸前后磁铁矿-二甲基胂酸样品Fe2p、O1s、C1s、As3d分峰拟合谱.
从图7b中,Fe2p分峰拟合结果可知,结合能在710.19 eV和723.65 eV左右属于Fe(II)—O, 结合能在713.74 eV和727.38 eV左右属于Fe(Ⅲ)—O.磁铁矿-二甲基胂酸的结合能在710.18 eV,加入腐殖酸作用后,Fe2p变为709.81 eV.说明Fe2p的峰位置发生了偏移,Fe2p的结合能发生了变化可能是二甲基胂酸和磁铁矿发生了络合[44].
从图7c 中,O1s的拟合结果可知,结合能在531—532 eV的O1s 主要属于金属氢氧化物或有机物中的C=O,结合能在532—533.5 eV之间的O1s属于C—O、C=O和C—O的含量为26.95%和11.22%,加入腐殖酸后,C=O和C—O为35.31%和14.02%,这说明腐殖酸参与了反应,Fe—O的含量由61.83%变为50.66%,也说明磁铁矿和二甲基胂酸与腐殖酸发生了络合.Shimizu等[45-46]发现,一甲基胂酸和二甲基胂酸都可以在 Fe/Al 氧化物上形成双齿双核配合物.
从图7d中,C1s的拟合结果可知,结合能在284.8 eV附近的C1s 主要属于为吸附碳和有机碳(C—C/C—H),C—O的结合能通常在286—287 eV之间,O—C=O的结合能通常在288—289 eV之间,说明磁铁矿表面碳的官能团主要由C—H、C—C、C—O、O—C=O组成,其中,C—H/C—C 结构最为丰富(约 84.56%).C—O和O—C=O含量分别为9.03%和6.41%.加入腐殖酸后,C—C/C—H、C—O和O—C=O含量分别为10.63%和7.94%,O—C=O和C—O结构的含量升高,C—O、C—C、O—C—O和—COO—是具有很强得失电子能力的含氧碳化物,有利于砷在地下水中的迁移和转化[47]. 这也说明了腐殖酸中含有O—C=O和C—O基团,与二甲基胂酸发生了络合,形成了磁铁矿-二甲基胂酸-腐殖酸络合物.
从图7e中,As3d的拟合结果可知,结合能在45 eV左右的As3d归属于五价态的氧化砷,加入腐殖酸前后五价态的砷含量均为100%,说明加入腐殖酸前后,二甲基胂酸的价态没有发生变化[48].有研究表明,二甲基胂酸只有在一定的条件下有可能会被还原为二甲基亚胂酸或一甲基亚胂酸,例如光催化降解,土壤中微生物的氧化还原等[13-49].
-
三维荧光光谱可以定量(或半定量)地分析DOM组分与砷的络合作用[9],为了探究pH值对磁铁矿上腐殖酸与二甲基胂酸络合作用的影响.在pH=7、9、11条件下,二甲基胂酸初始浓度为1 mg·L−1,腐殖酸浓度为150 mg·L−1条件下,测定和比较了添加二甲基胂酸前后,腐殖酸在磁铁矿上吸附平衡样品的三维荧光.结果如图8所示.当pH=7、9、11时,添加二甲基胂酸前后,在(Ex/Em=250—300/442—530)处均出现了较强的荧光峰,但是荧光强度各不相同.未添加二甲基胂酸时,在pH=7、9、11时,该荧光峰强度分别为5900、6100、6900.加入二甲基胂酸后,在pH=7、9、11时,该荧光峰强度分别为5700、5800、6000.
在(Ex/Em=250—300/442—530)处的荧光峰可归属于类腐殖酸荧光峰[50].加入二甲基胂酸后, 在pH=7、9、11时,类腐殖酸荧光峰的强度均明显降低,这说明在3种pH条件下,类腐殖酸组分均发生了荧光猝灭[51].这可能是由于二甲基胂酸中的As=O与腐殖酸中的荧光基团相作用后,形成了络合物,改变了腐殖酸荧光基团的性质,导致其发光基团被部分猝灭.
另外,随着pH增大,类腐殖酸荧光峰的荧光强度均变强.这可能是在碱性条件下腐殖酸的官能团带负电荷,而二甲基胂酸也带负电荷,腐殖酸和二甲基胂酸之间相互排斥,腐殖酸的静电斥力和空间位阻减弱,导致大量的腐殖酸以自由形式存在[52],在碱性条件下会暴露更多的荧光基团,导致荧光强度增强[53].加入二甲基胂酸后,随着pH增大, 二甲基胂酸对类腐殖酸荧光峰的猝灭作用越强.在pH=7时,猝灭程度较小,说明在中性条件下,腐殖酸的荧光强度受二甲基胂酸的影响较小.在pH=9时,二甲基胂酸在溶液中以砷酸根阴离子存在,由于静电斥力,与羧基发生相互作用几率较小,因此对类腐殖酸荧光峰猝灭程度较小[54].在pH=11时,猝灭较明显,这可能是在较高pH时,腐殖酸和二甲基胂酸更容易在磁铁矿表面形成络合物[55].
不同pH时的类腐殖酸荧光峰被二甲基胂酸不同程度的猝灭,类腐殖酸荧光峰没有被完全猝灭的原因可能是加二甲基胂酸前后均有磁铁矿的存在.腐殖酸和磁铁矿发生络合后,依然会保留腐殖酸的部分荧光强度,即使是形成阳离子桥,对腐殖酸的荧光强度也有一定的增强作用.还有可能是磁铁矿与二甲基胂酸中的As=O发生作用,导致直接与腐殖酸发生作用的二甲基胂酸减少,二甲基胂酸对腐殖酸构象的改变减少[56].
-
本实验合成的磁铁矿粒子的物相单一,结晶度好,磁化强度较强.考察了腐殖酸对二甲基胂酸在磁铁矿上吸附的影响机制.
(1)吸附动力学结果表明,二甲基胂酸的吸附速率常数为0.18 mg·g−1·h−1,加入腐殖酸后,吸附速率常数为0.00018 mg·g−1·h−1.说明腐殖酸抑制了二甲基胂酸在磁铁矿上的吸附.R2均为0.99,符合准二级动力学方程.
(2)吸附等温线结果表明,pH=7、9、11时,二甲基胂酸的最大吸附量分别是0.47、0.17、0.18 mg·g−1;加入腐殖酸后,二甲基胂酸的最大吸附量分别是0.08、0.06、0.04 mg·g−1.表明在pH=7、9、11的条件下腐殖酸均抑制了磁铁矿上二甲基胂酸的吸附,且符合Freundlich吸附等温模型.随着pH增大,磁铁矿对二甲基胂酸的吸附速率降低.
(3)Zeta电位和红外光谱表明腐殖酸和二甲基胂酸在磁铁矿表面存在竞争吸附,可以通过表面配位交换、酚羟基等相互作用形成了二甲基胂酸-磁铁矿-腐殖酸络合物.XPS分析结果也表明腐殖酸和磁铁矿及二甲基胂酸形成了络合物.三维荧光光谱表明随着pH增大,磁铁矿-腐殖酸体系和磁铁矿-腐殖酸-二甲基胂酸体系的荧光强度均增强.同一pH时,加入二甲基胂酸后,腐殖酸的荧光强度降低,表明二甲基胂酸和腐殖酸的荧光基团发生了猝灭作用.
腐殖酸对二甲基胂酸在磁铁矿上吸附过程的影响
Effect of humic acid on the adsorption of dimethylarsinic acid by magnetite
-
摘要: 二甲基胂酸作为一种有毒物质,对水环境造成了严重的危害.水中的溶解有机质可以抑制或促进二甲基胂酸在水中的迁移,本研究探究了腐殖酸对二甲基胂酸在磁铁矿上吸附过程的影响.结果表明,二甲基胂酸在磁铁矿表面的吸附动力学符合准二级吸附动力学模型,吸附等温线符合Freundlich等温吸附模型,从而证实了二甲基胂酸在磁铁矿上的吸附是多层化学吸附过程.在pH为7、9、11时,腐殖酸对二甲基胂酸在磁铁矿上的吸附均有抑制作用.Zeta电位分析说明腐殖酸通过静电斥力作用抑制磁铁矿吸附二甲基胂酸.红外光谱分析表明形成了磁铁矿-腐殖酸-二甲基胂酸三元络合物.X 射线光电子能谱表明吸附后含氧官能团含量的增多也进一步验证了磁铁矿-腐殖酸-二甲基胂酸络合物的形成.不同pH时,腐殖酸-磁铁矿的三维荧光光谱证实发生了荧光猝灭.Abstract: As a kind of toxic substance, dimethylarsinic acid has caused serious harm to water environment. Dissolved organic matter in water can inhibit or promote the migration of dimethylarsinic acid in water. This study investigated the effect of humic acid on the adsorption process of dimethylarsinic acid by magnetite. The results show that the adsorption kinetics of dimethylarsinic acid on magnetite surface conformed to the quasi-second-order adsorption kinetics model, and the adsorption isotherm conformed to the Friedrich isotherm model. Therefore, it is confirmed that the adsorption of dimethylarsinic acid on magnetite is a multi-layer chemical adsorption process. The adsorption of dimethylarsinic acid on magnetite was inhibited by humic acid at pH 7, 9 and 11. Zeta potential analysis suggested that humic acid inhibited the adsorption of dimethylarsinic acid on magnetite by electrostatic interaction. Infrared spectrum analysis showed that the ternary complex of magnetite-humic acid-dimethylarsinic acid was formed. X-ray photoelectron spectroscopy showed that the content of oxygen-containing functional groups increased after adsorption, further verifying the formation of magnetite-humic acid-dimethylarsinic acid complex. The appearance of fluorescence quenching was confirmed by three-dimensional fluorescence spectra of humic acid-magnetite at different pH.
-
Key words:
- dimethylarsinic acid /
- magnetite /
- humic acid /
- complexation.
-
近年来,药物和个人护理品(pharmaceutical and personal care products,PPCPs)在地下水、地表水和饮用水中被广泛检出,引起研究者们关注。双氯芬酸钠(diclofenac sodium,DCF)是一种典型的PPCPs,作为一种消炎止痛类药物已被广泛使用,因其具有难生物降解和生物积累性的特点,故给生态环境和人类健康带来极大的威胁。常规的处理技术无法有效地去除DCF,传统的生物处理技术对DCF的去除率只能达到30%左右[1]。因此,亟需寻找一种新型有效的处理方法去除水中的DCF。
近年来,基于硫酸根自由基(
SO⋅−4 )的高级氧化技术受到研究者们的广泛关注[2-3]。与·OH相比,SO⋅−4 具有氧化还原电位高、pH适用范围广及半衰期长等优点,有利于污染物的降解。过硫酸盐(persulfate,PS)可在紫外光、热、碱、过渡金属离子(Mn2+)和零价铁等活化下产生SO⋅−4 。然而,不同的方法具有各自的优点和缺点,如:热活化不产生二次污染,但在使用过程中会消耗很多能量;过渡金属离子可以在室温下活化PS,但是易受溶液pH影响而产生沉淀。零价铁可以在室温下活化PS产生SO⋅−4 ,其主要反应如式(1)和式(2)所示。Fe0+S2O2−8→Fe2++2SO⋅−4 (1) Fe2++S2O2−8→Fe3++SO⋅−4+SO2−4 (2) 零价铁的化学性质活泼,在制备和存储的过程中与氧气接触,会形成氧化膜覆盖在零价铁表面,从而影响其在反应过程中的活性。为了解决零价铁钝化问题,研究者们提出了一些改进方法,如利用纳米零价铁[4]、酸洗[5]、制备零价铁双金属[6]和氢气还原等[7]。这些方法可以在一定程度上改善零价铁去除污染物的活性,但是在实际应用方便仍然会存在一定的问题,如增加使用的成本、操作复杂等。有关磁场效应影响零价铁去除污染物的研究是近几年来新兴的研究方向,受到了研究者的广泛关注。KIM等[8]研究发现,在零价铁降解4-氯酚的过程中加入磁场时,可以提高4-氯酚的去除效率。研究者认为磁场可以加速零价铁的腐蚀作用,并促进氧气扩散到零价铁的表面,使其相互作用生成·OH降解污染物。一些研究对磁场强化零价铁降解污染物进行了一系列的探讨,证明了在不同的反应体系中,磁场均可以促进零价铁的腐蚀和Fe2+的溶出,可以不同程度地提高污染物的降解速率[9-13]。综上所述,磁场可以明显改善零价铁的反应活性,且操作简单、成本低、无二次污染。因此,将磁场与其他污染物处理技术相结合具有非常广泛的应用前景。由于零价铁是铁磁性物质,在磁场中磁化后离开磁场仍能保持剩磁,具有“磁记忆性”。因此,本研究利用零价铁的磁记效应来提高其反应活性,以DCF为模型污染物,采用预磁化零价铁活化PS体系对DCF进行降解,考察了零价铁投加量、PS投加量、pH等因素对DCF降解的影响,并探讨了DCF的降解机理,为DCF实际废水的降解提供了科学依据。
1. 材料与方法
1.1 实验试剂和仪器
过硫酸盐(K2S2O8,PS)、氢氧化钠(NaOH)、硫酸(H2SO4)购于天津科密欧试剂有限公司;盐酸羟胺(NH2OH·HCl)、邻菲啰啉(C12H8N2.H2O)购于天津博迪化工有限公司;双氯芬酸钠(C14H10Cl2NNaO2)购于北京百灵威科技有限公司;甲醇(CH3OH)、5,5-二甲基-1-吡咯啉-氮-氧化物(DMPO)均为色谱纯并购于上海阿拉丁生化科技股份有限公司,实验用水为超纯水。
BS124S电子天平(赛多利斯科学仪器有限公司);PHS-3C型pH计(上海仪电科学仪器股份有限公司);VI-1501可见分光光度计(天津港东科技发展有限公司);D2004W搅拌器(上海司乐仪器有限公司);DH101-3BS型电热鼓风干燥箱(天津中环实验电炉有限公司);EMX-6/1电子自旋共振波谱仪(德国Bruker公司);FL2200液相色谱仪(浙江福立分析仪器有限公司)。
1.2 实验方法
实验采用1 000 mL的烧杯为反应器,以2片圆形钕-铁-硼永久磁铁提供磁场,用特斯拉计测定并调整所需磁场强度。将零价铁置于磁场中磁化2 min,磁化过程中以机械搅拌器搅拌使零价铁均匀悬浮于烧杯中。磁化后,将调节好pH的DCF溶液加入反应器中,然后加入一定量的PS开始计时,每隔一定的时间取样,最后加入叔丁醇终止反应,过0.22 μm滤膜后待测。同时,在其他条件相同的情况下,以非磁化零价铁作对照组。实验在常温常压下进行。
1.3 分析测试方法
DCF采用液相色谱法测定,流动相为甲醇∶水=75∶25(体积比),流速为1 mL·min−1,检测波长为278 nm,柱温为40 ℃,进样量为10 μL;铁离子浓度采用邻菲啰啉分光光度法测定;pH采用玻璃电极法测定;利用电子自选共振波谱法(electron spin resonance,ESR)测定体系中自由基产生情况。
1.4 数据分析方法
DCF的去除率计算方法如式(3)所示。
R=(C0−Ct)/C0×100% (3) DCF氧化分解的反应符合动力学一级反应的特征,拟一级动力学方程如式(4)所示。
ln(Ct/C0)=−kt (4) 式中:R为DCF的去除率;C0为DCF的初始浓度,mg·L–1;Ct为t时间的DCF浓度,mg·L–1;k为DCF降解的一级动力学速率常数,min−1。
2. 结果与讨论
2.1 不同体系对DCF降解效果的对比
实验对比了DCF在PS、零价铁、预磁化零价铁、Fe0/PS和Pre-Fe0/PS几种体系中的去除效果,结果如图1所示。在零价铁体系中,反应60 min仅有6.8%的DCF被去除;Pre-Fe0体系在60 min内可以去除9.8%的DCF;在PS体系中,反应60 min,DCF的去除率为29.3%。这3种体系对DCF去除率较低的原因是零价铁和预磁化零价铁在溶液中无法参与反应,对DCF有较低的去除可能是由于吸附作用,预磁化零价铁比表面积有所增加[14],对DCF的吸附作用比零价铁稍强。而PS体系中尽管其氧化还原电位较高[15],但仍无法有效降解DCF。当向零价铁体系中加入PS时,DCF的降解率可在60 min达到99%,说明零价铁可以有效活化PS氧化降解DCF。值得注意的是,Pre-Fe0/PS体系中,反应5 min时,DCF的降解率可达99.7%。前期研究[14]表明,预磁化可以加速体系中Fe2+溶出,因此,预磁化零价铁可以更快的催化PS产生更多的
SO⋅−4 ,使污染物更快降解。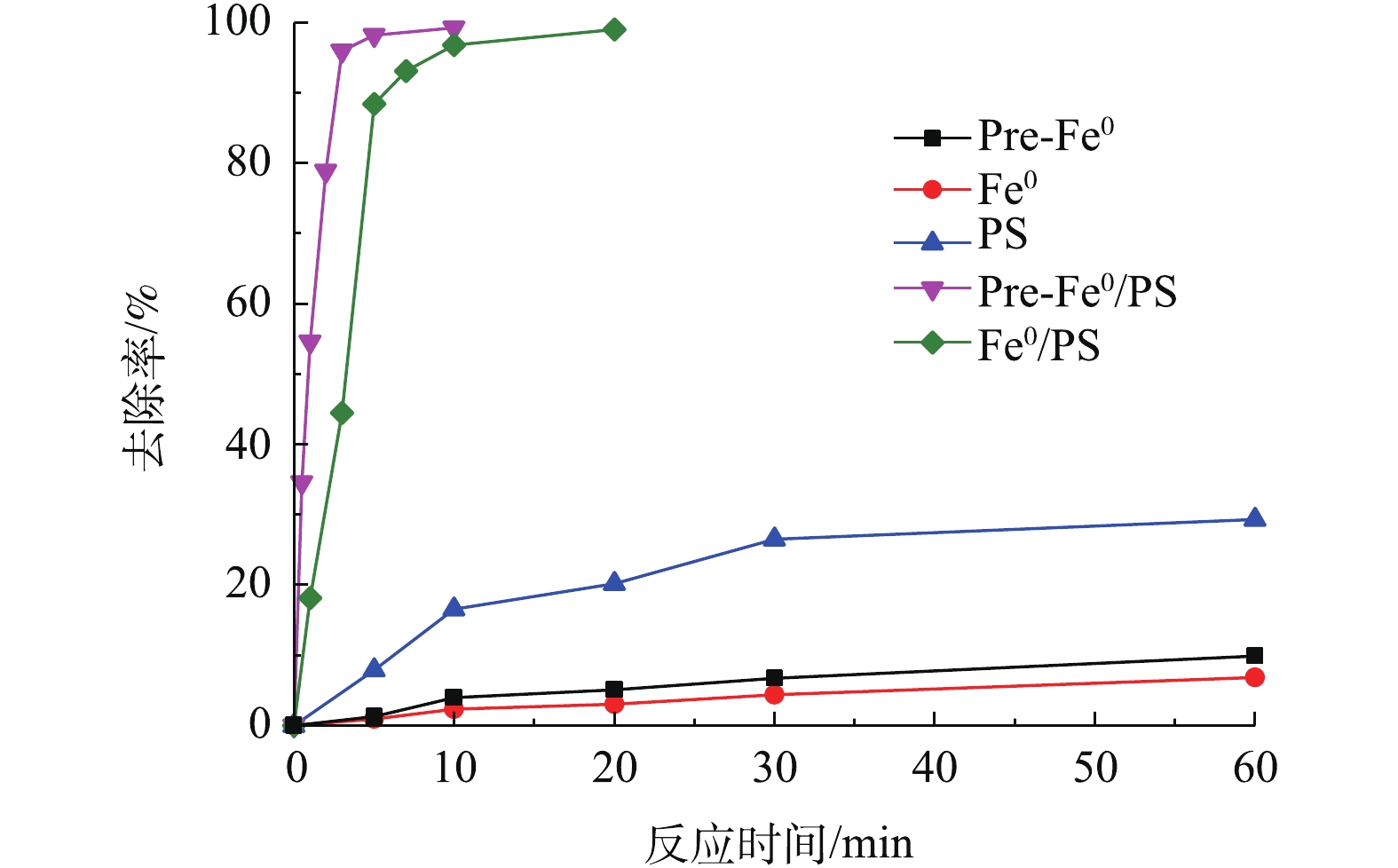 图 1 不同体系对DCF去除率的影响Figure 1. Removal efficiency of diclofenac sodium by different system
图 1 不同体系对DCF去除率的影响Figure 1. Removal efficiency of diclofenac sodium by different system2.2 PS初始投加量对DCF降解效果的影响
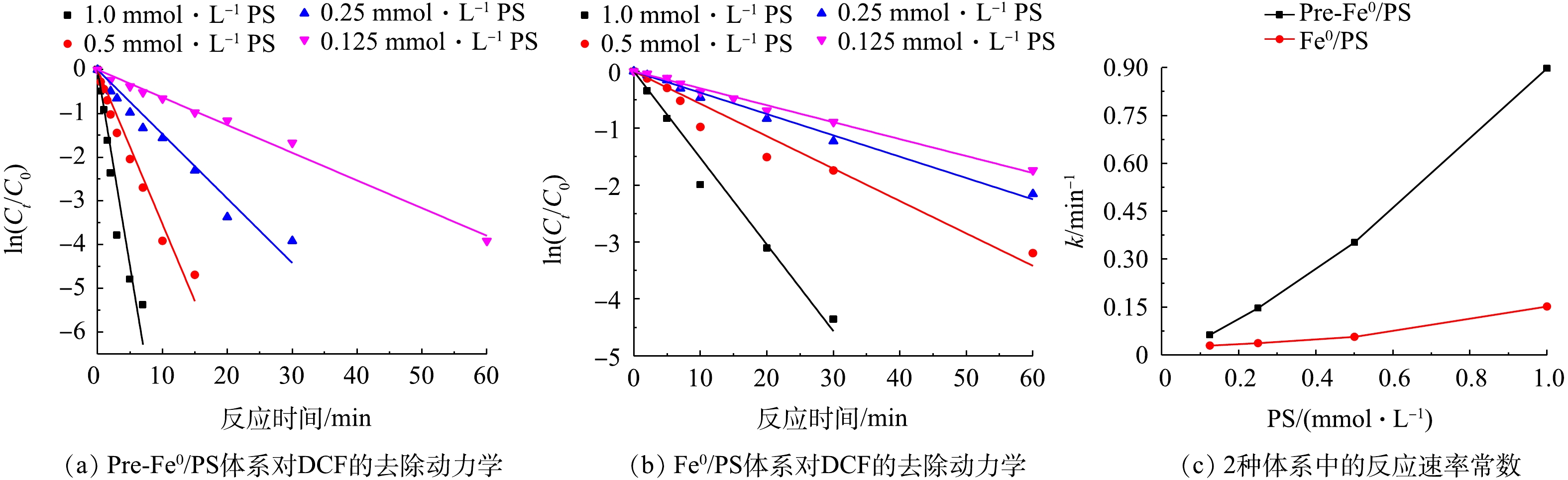
体系中PS的浓度决定了产生
SO⋅−4 的量,进而影响DCF的降解率。为了研究不同PS投加量对DCF的降解,实验考察了PS浓度分别为0.125、0.25、0.5、1.0 mmol·L−1时,Pre-Fe0/PS和Fe0/PS体系对DCF的降解情况,结果如图2所示。由图2可知,随着PS浓度的增加,2种体系对DCF的去除率都呈升高趋势,Pre-Fe0/PS体系对DCF的去除率和去除速率均大于Fe0/PS体系。当PS投加量为0.125、0.25、0.5和1.0 mmol·L−1时,在反应30 min后,Fe0/PS体系对DCF的去除率分别为58.5%、70.2%、75.8%和96.8%;在Pre-Fe0/PS体系中,PS投加量为0.125 mmol·L−1和0.25 mmol·L−1时,反应30 min后,DCF的去除率为81.9%和98.1%;当PS的投加量为0.5 mmol·L−1时,反应进行10 min时,DCF的去除率可达98.2%,继续增加PS的量为1.0 mmol·L−1时,Pre-Fe0/PS体系对DCF的去除率在5 min达99.3%。这是因为随着PS的量增加,会有更多的SO⋅−4 产生,故2种体系中DCF的去除速率均会升高。在Pre-Fe0/PS体系中,由于零价铁的腐蚀速率加快,会促进PS的分解加速产生SO⋅−4 ,进而使DCF的降解速率更快。利用拟一级动力学反应方程对2种体系在不同PS投加量时的实验结果进行拟合。当PS投加量由0.125 mmol·L−1增加到1.0 mmol·L−1时,Fe0/PS体系降解DCF的反应速率常数由0.029 min−1增加到0.152 min−1,在实验条件范围内,任一浓度过硫酸钾条件下,Pre-Fe0/PS体系降解DCF的反应速率常数均高于Fe0/PS体系,可从0.063 min−1提高到0.898 min−1。2.3 零价铁初始投加量对DCF降解效果的影响
零价铁在反应过程中释放的铁离子对
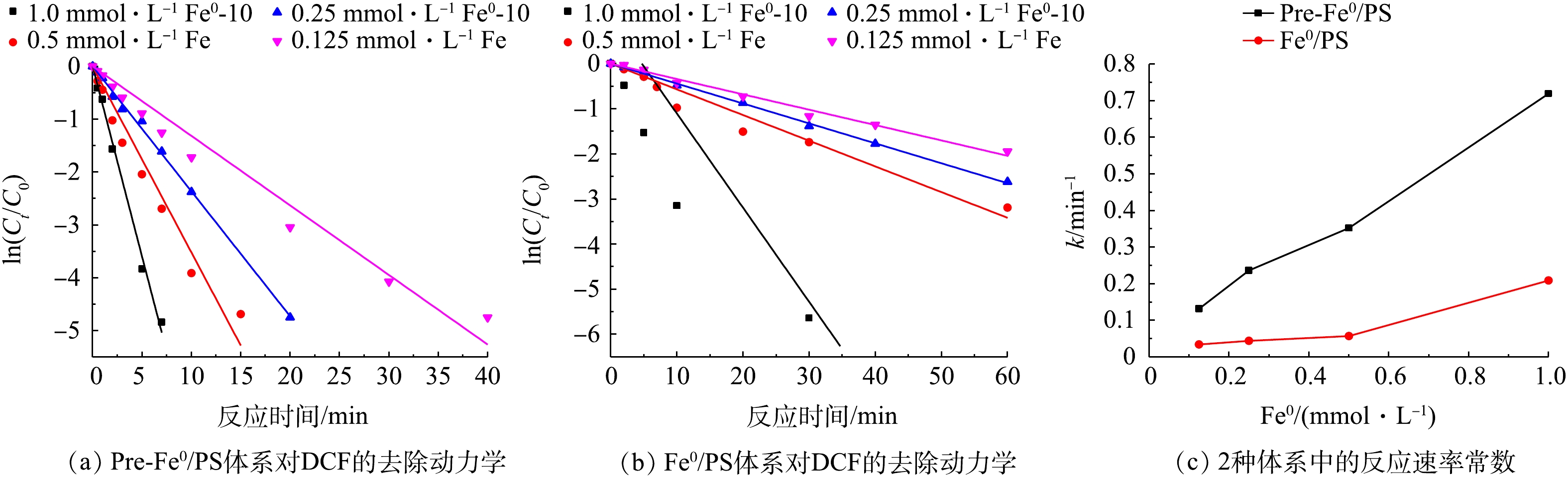
SO⋅−4 的产生起着非常重要的作用[16],因此,零价铁的浓度对污染物的降解有较大影响。为了研究不同零价铁投加量对DCF去除过程的影响,实验选取了0.125、0.25、0.5、1.0 mol·L−1零价铁投加量,在DCF初始浓度为20 mg·L−1,初始pH为7,PS投加量为0.5 mol·L−1时,同浓度的零价铁体系中DCF的反应速率如图3所示。可以看出,随着零价铁投加量的增加,2种体系对DCF的去除率有很大提升,且在相同零价铁投加量时,Pre-Fe0/PS体系的去除速率远大于化Fe0/PS体系。当零价铁投加量为0.125 mmol·L−1时,反应60 min后,Fe0/PS体系对DCF的去除率分别为80%;当零价铁投加量为1.0 mmol·L−1时,DCF在30 min的去除率可以达到99%。其原因是因为随着零价铁投加量的增加,体系中能产生更多的铁离子,进而活化PS产生SO⋅−4 ,最终加速DCF的去除。而Pre-Fe0/PS体系中零价铁投加量为0.125 mmol·L−1时,反应20 min时,对DCF的去除率为83.3%;随着零价铁投加量的增加,Pre-Fe0/PS体系对DCF的去除速率迅速增加,当零价铁投加量为0.25 mmol·L−1时,反应20 min后DCF的降解率接近100%。原因可能是因为在Pre-Fe0/PS体系中零价铁腐蚀速率较快,当零价铁为0.25 mmol·L−1时,溶出的铁离子能够在短时间内将体系中DCF完全去除,当零价铁的投加量继续增大时,对Pre-Fe0/PS体系的影响较小。2种体系的反应速率常数如图3(c)所示。2种体系对DCF降解的表观速率常数随零价铁投加量的增加而升高,在Pre-Fe0/PS体系中,反应速率常数由0.132 min−1增大到0.719 min−1;Fe0/PS体系由0.034 min−1增加到0.209 min−1。为了更好地表明零价铁投加量在Pre-Fe0/PS体系中对DCF去除率的影响,本研究增加了DCF浓度(40 mg·L−1),结果如图4所示。由图4可知,当零价铁浓度由0.125 mmol·L−1增加到0.5 mmol·L−1时,DCF的去除率随着零价铁投加量的增加而升高;当零价铁浓度继续增加,DCF去除率基本不变,这是因为当零价铁投加量过大时,体系中产生过多的铁离子会与
SO⋅−4 发生反应[17]。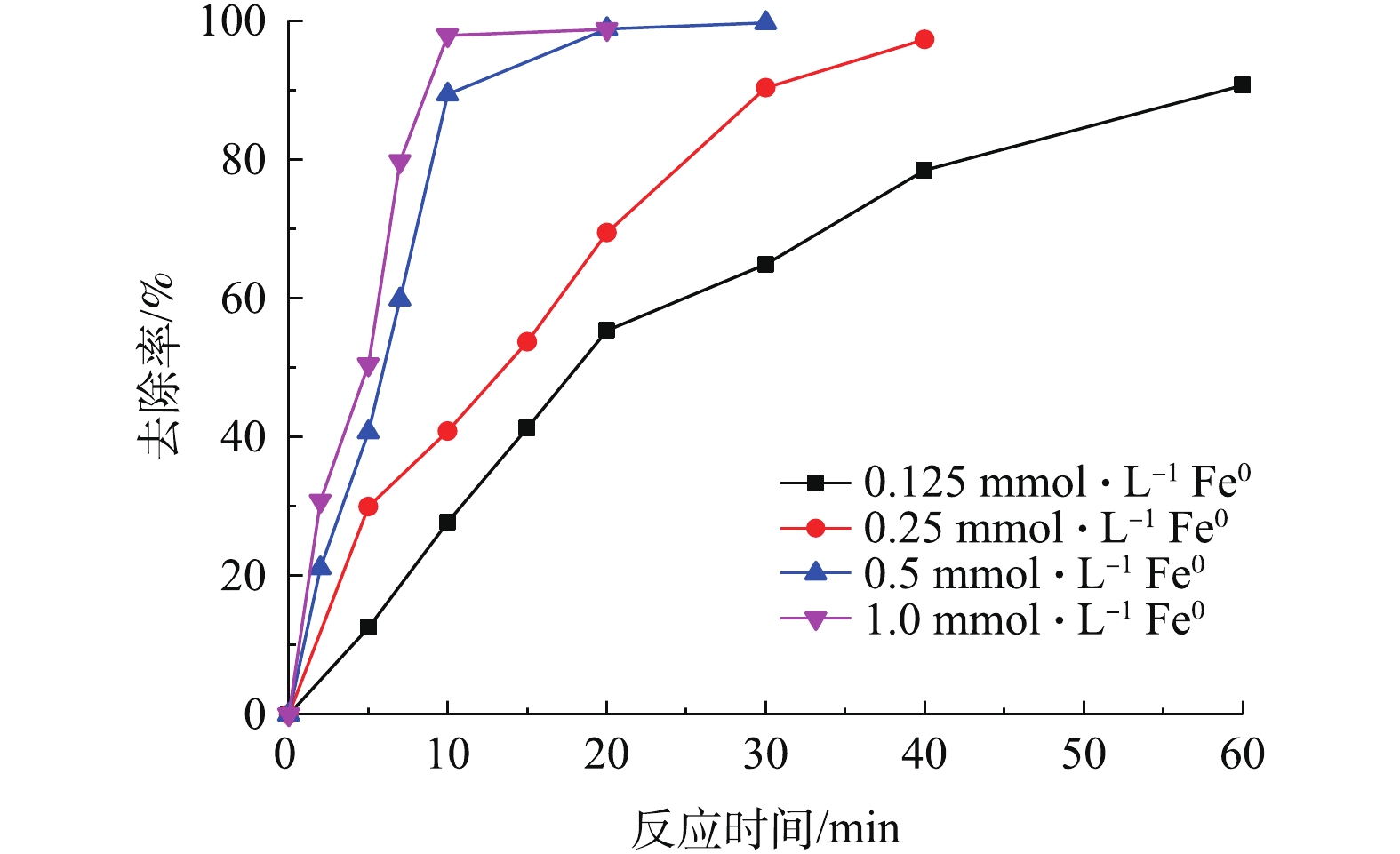 图 4 零价铁投加量对Pre-Fe0/PS体系降解DCF的影响Figure 4. Influence of dosage of Fe0 on DCF degradation by Pre-Fe0/PS process
图 4 零价铁投加量对Pre-Fe0/PS体系降解DCF的影响Figure 4. Influence of dosage of Fe0 on DCF degradation by Pre-Fe0/PS process2.4 初始pH对DCF降解效果的影响
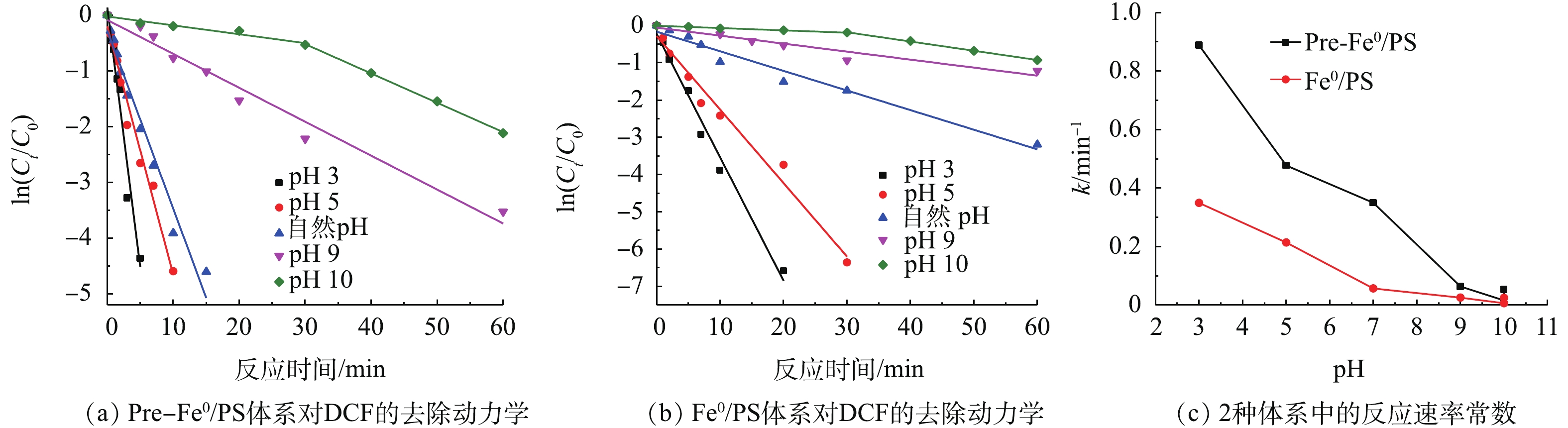
实验考察了当DCF初始浓度为20 mg·L−1,零价铁投加量为0.5 mmol·L−1,PS投加量为0.5 mmol·L−1,初始pH分别为3,5,7,9和10时DCF的降解效果,结果如图5所示。由图5可知,在Fe0/PS体系中,在初始pH为3~10时,DCF的去除率随初始pH值的升高而下降,特别是初始pH为10时下降尤为明显。当初始pH为3,反应20 min时,DCF的去除率为99%;初始pH为10时,DCF在60 min的去除率为50%左右。在Pre-Fe0/PS体系中,当初始pH为3、反应15 min时,对DCF的去除率可达100%;初始pH为10时,DCF在60 min时的降解率可达90.4%。
与Fe0/PS体系相比,在相同的pH下,Pre-Fe0/PS体系中DCF的去除率均有较大的提升,特别是在pH较低时,2种体系中DCF的降解较快。分析其原因可能是:零价铁在储存和运输过程中被氧化形成一层钝化膜覆盖其表面,当反应体系pH较低时,零价铁表面的氧化膜更容易被溶解[18]。因此,零价铁在体系pH较低时的腐蚀速率和反应活性较高,在反应过程中会产生更多的氢参与加成反应[19]。由图5(b)可知,随着体系初始pH的升高,Fe0/PS体系对DCF的去除率急剧下降;而Pre-Fe0/PS体系对DCF的去除率仍能保持在较高的水平,当pH为10时,对DCF的降解率在60 min时仍可达到90.4%,是Fe0/PS体系的2倍左右。
由图5(c)可知,2种体系的反应速率常数随初始pH的升高而迅速减小,Pre-Fe0/PS体系的反应速率常数是Fe0/PS体系的2.1~6.2倍,Pre-Fe0/PS体系对反应速率常数提升的倍数并没有因初始pH的升高而下降,其原因为当体系的初始pH较高时,零价铁在参与反应时会形成铁氧化物或铁氢氧化物钝化膜覆盖其表面阻止反应的进行。然而,目前有研究显示,预磁化可以加速零价铁的腐蚀,阻止钝化膜的形成[20],从而提高DCF的降解。因此,Pre-Fe0可以在一定程度上使该体系pH适用范围增大,减少其在应用过程中pH调节剂的使用,降低污染物的降解成本。
2.5 降解机理
1)铁离子的产生。在Fe0/PS体系中,零价铁可以与体系中的氧气、水和H+反应生成Fe2+,活化PS生成
SO⋅−4 ,而本身被氧化为Fe3+,为了研究体系中零价铁、Fe2+和Fe3+的作用,实验测定了在近中性条件下,体系中亚铁离子和铁离子的变化。2种体系的反应过程中都没有测出Fe2+(测定方法的最低检测限为0.03 mg·L−1),这一现象与XIONG等[21]的研究结果吻合,即Fe2+的溶出是反应活化PS的限速步骤。实验研究了当DCF初始浓度为20 mg·L−1,零价铁投加量为0.5 mmol·L−1,PS投加量为0.5 mmol·L−1,自然初始pH下,体系中铁离子浓度和pH变化情况。图6为2种反应体系中总铁离子浓度的变化。由图6可见,Pre-Fe0/PS体系在反应过程中铁离子浓度高于Fe0/PS体系,说明在Pre-Fe0/PS体系中铁离子的快速溶出导致了DCF的降解效率的升高。此外,我们还测定了反应过程中体系pH的变化,随着反应的进行,2种体系的pH都呈降低的趋势。其原因可能是在反应过程中生成的Fe3+会发生水解作用产生H+(式(5)),另外,在部分
SO⋅−4 转化为·OH(式(6))的过程中也会产生H+,从而使体系pH下降。由于Pre-Fe0/PS体系中能产生更多的的Fe3+和SO⋅−4 ,因此,Pre-Fe0/PS体系中的pH下降较Fe0/PS体系更为明显。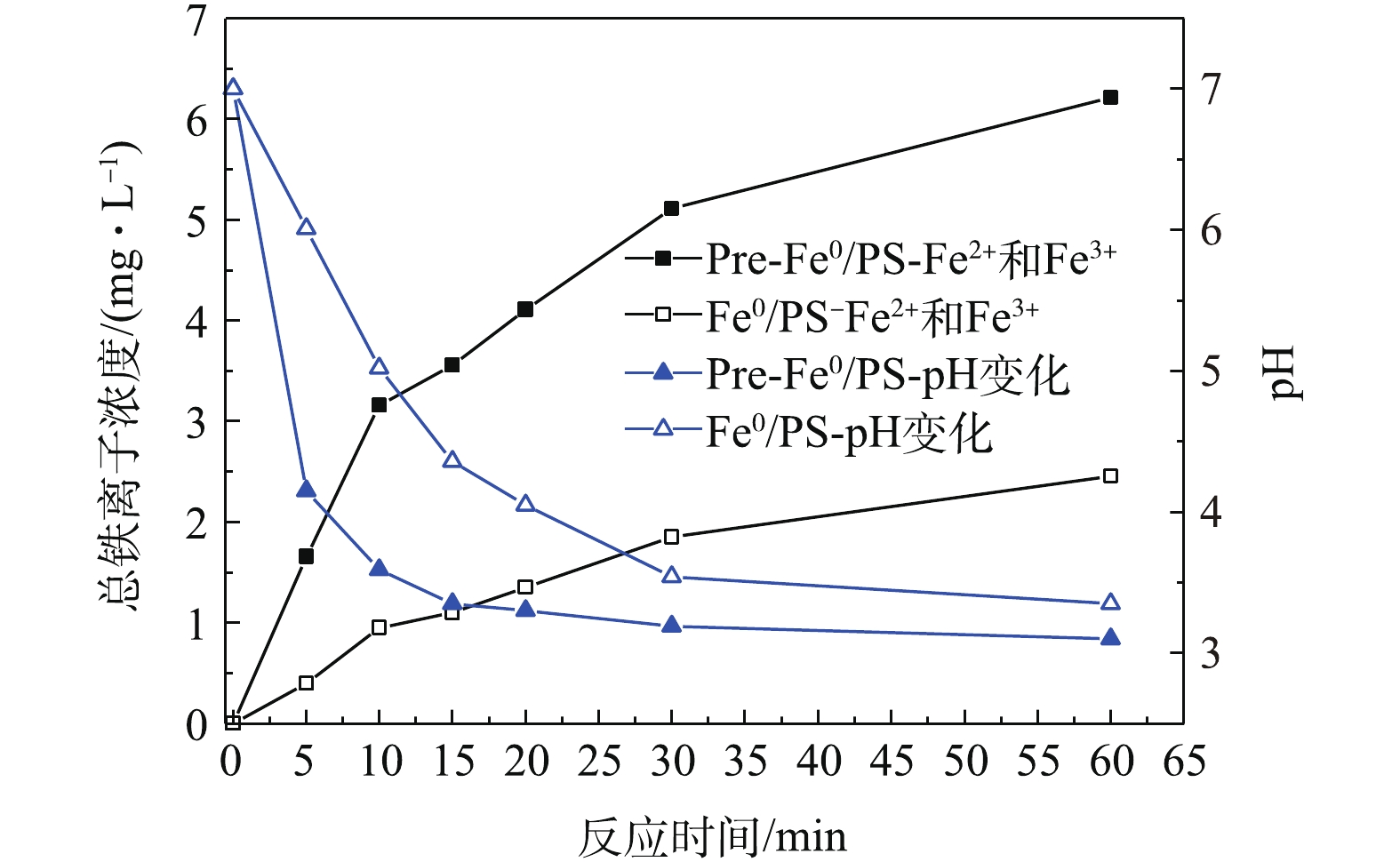 图 6 反应过程中2种体系中铁离子浓度和pH变化Figure 6. Changes of total iron concentration and pH in Fe0/PS and pre-Fe0/PS process.
图 6 反应过程中2种体系中铁离子浓度和pH变化Figure 6. Changes of total iron concentration and pH in Fe0/PS and pre-Fe0/PS process.Fe3++xH2O→Fe(OH)3−xx+xH+ (5) SO⋅−4+xH2O→⋅OH+H++SO2−4 (6) 2)自由基的产生。自由基是降解污染物重要的活性物质,其在体系中的产生量决定了污染物的降解率。电子自旋共振波谱法(ESR)是测定短寿命自由基非常有效的手段,其信号可以半定量地反映自由基的产生量。由于自由基的寿命非常短暂,在水溶液中存在的时间小于10−4 s[22],实验过程中以5,5-二甲基-1-吡咯啉-氮-氧化物(DMPO)为捕获剂,生成寿命较长的自旋加合物进行测定。由图7可知,2种体系中均出现了DMPO-
SO−4 和DMPO-OH加合物的典型特征峰[23]。对比图7(a)和图7(b)可以看出,当反应条件相同时,Pre-Fe0/PS体系在任一取样时间点的加合物对应的峰高均大于Fe0/PS体系,即产生的SO⋅−4 和·OH量比Fe0/PS体系中多。由图7还可以看出,Pre-Fe0/PS体系在2 min时产生的SO⋅−4 和·OH比Fe0/PS体系在5 min时产生的量还要多,而且能在相当长的时间内保持较高的浓度水平,当取样时间为5 min时,DMPO-SO−4 和DMPO-OH加合物的信号峰仍然很强。然而,Fe0/PS体系中SO⋅−4 和·OH产生速度相对较慢,DMPO-SO−4 和DMPO-OH加合物的信号峰衰减较快。这一结果解释了Pre-Fe0/PS体系对DCF的去除率大于Fe0/PS体系的原因。3. 结论
1)预磁化后的零价铁能够显著提升其对PS活化作用,进而提高其降解DCF的能力。
2) PS浓度、零价铁投加量及初始pH对Pre-Fe0/PS和Fe0/PS体系降解DCF均有较大影响。其中,在零价铁投加量为0.125~1.0 mmol·L−1、PS浓度为0.125~1.0 mmol·L−1条件中,反应速率常数均呈升高趋势,而DCF可在Fe0为 0.5 mmol·L−1,PS为0.5 mmol·L−1条件下几乎被完全去除;2种体系的反应速率常数随初始pH的升高而迅速减小,Pre-Fe0/PS体系的反应速率常数是Fe0/PS体系的2.1~6.2倍,在pH为6~8的条件下有利于反应进行。
3) Pre-Fe0/PS体系中铁离子溶出和pH下降趋势均比Fe0/PS体系快。
4) ESR结果表明,2种体系中都会产生
SO⋅−4 和·OH,且其对污染物的降解起主要作用,预磁化可以加速SO⋅−4 和·OH的产生,并能使其在较长的时间保持较高的浓度水平。 -
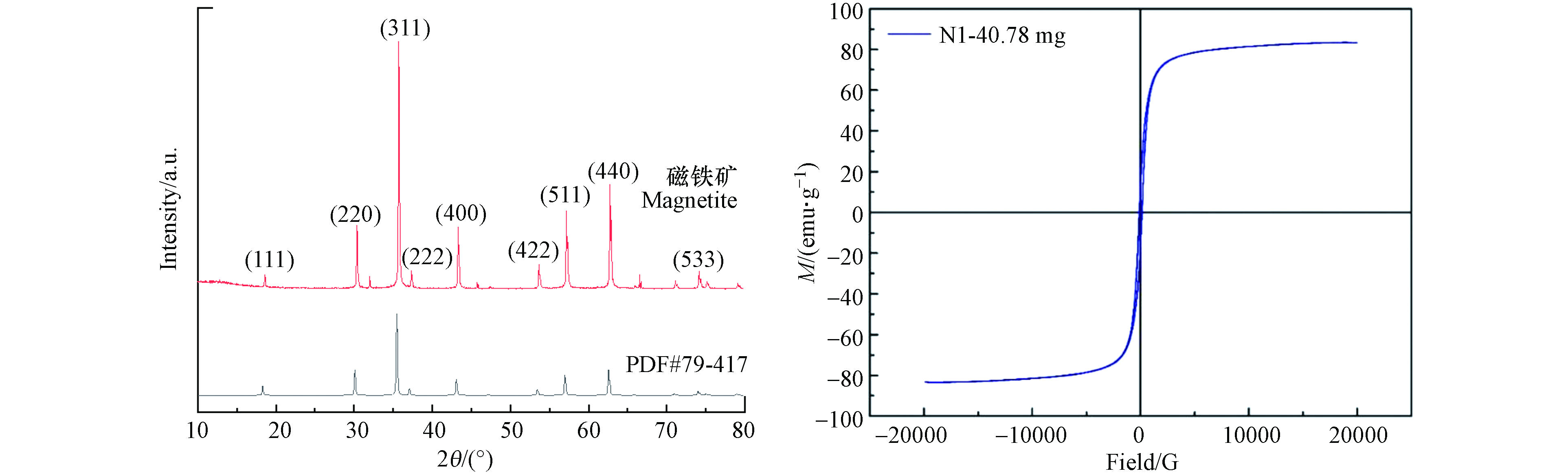
图 1 磁铁矿X-射线衍射和磁滞回线分析
Figure 1. Analysis of magnetite by X-ray diffraction and hysteresis loop
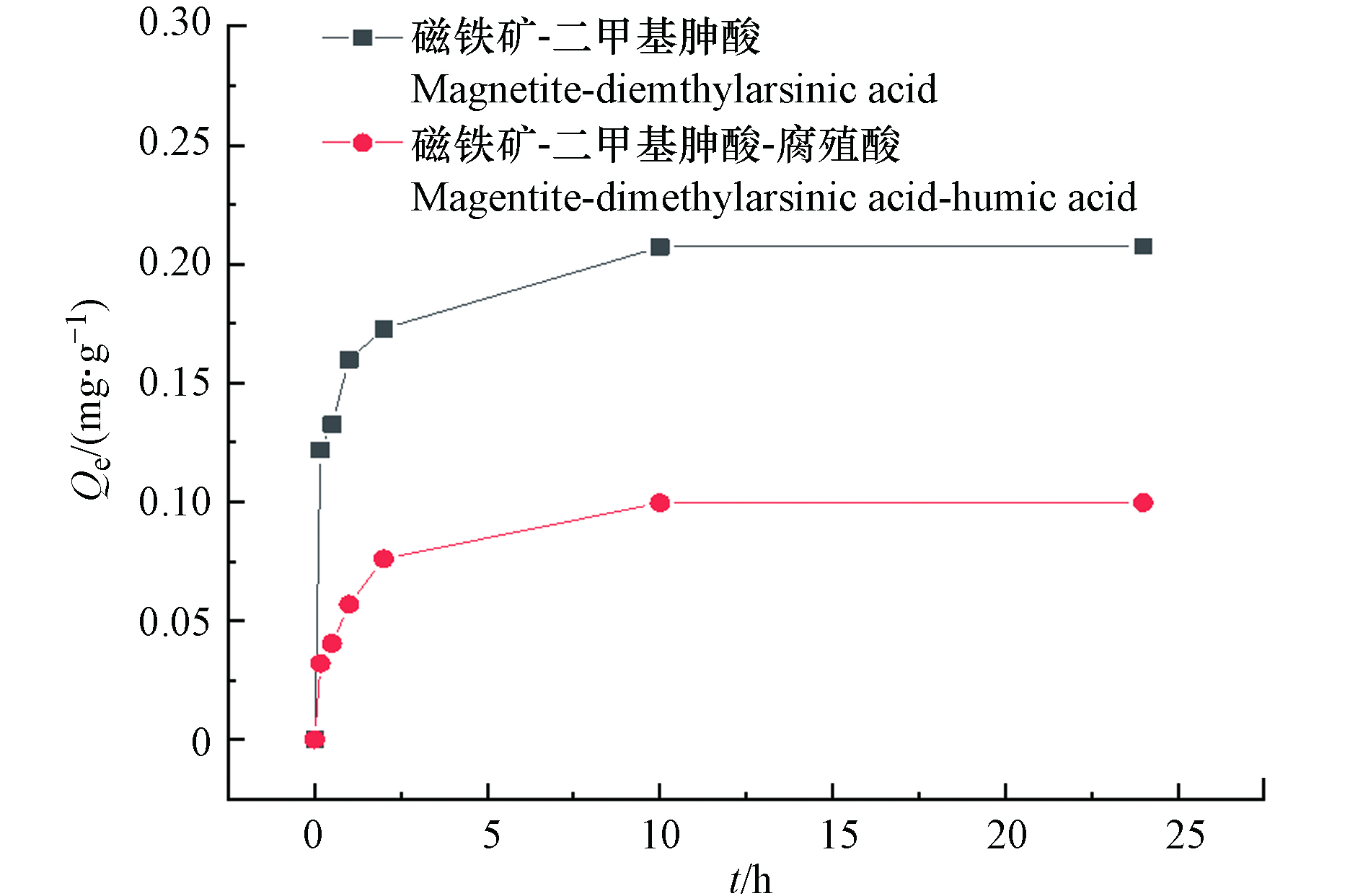
图 2 pH=7时,二甲基胂酸在磁铁矿上的吸附动力学C二甲基胂酸= 1 mg·L−1,C腐殖酸= 150 mg·L−1
Figure 2. Adsorption kinetics of dimethylarsinic acid on magnetite at pH=7
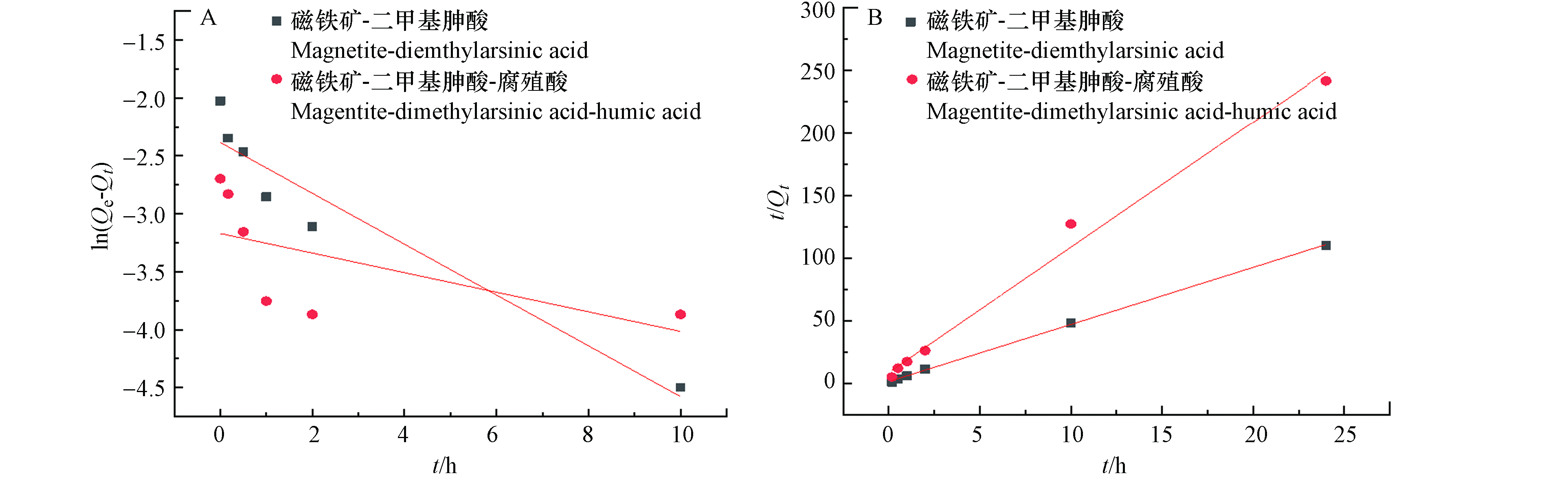
图 3 pH=7时,二甲基胂酸在磁铁矿表面吸附准一级动力学拟合(A)和准二级动力学拟合(B)C二甲基胂酸= 1 mg·L−1,C腐殖酸= 150 mg·L−1
Figure 3. Quasi-first-order kinetic fitting (A) and quasi-second-order kinetic fitting (B) for adsorption of dimethylarsinic acid on magnetite surface at pH=7
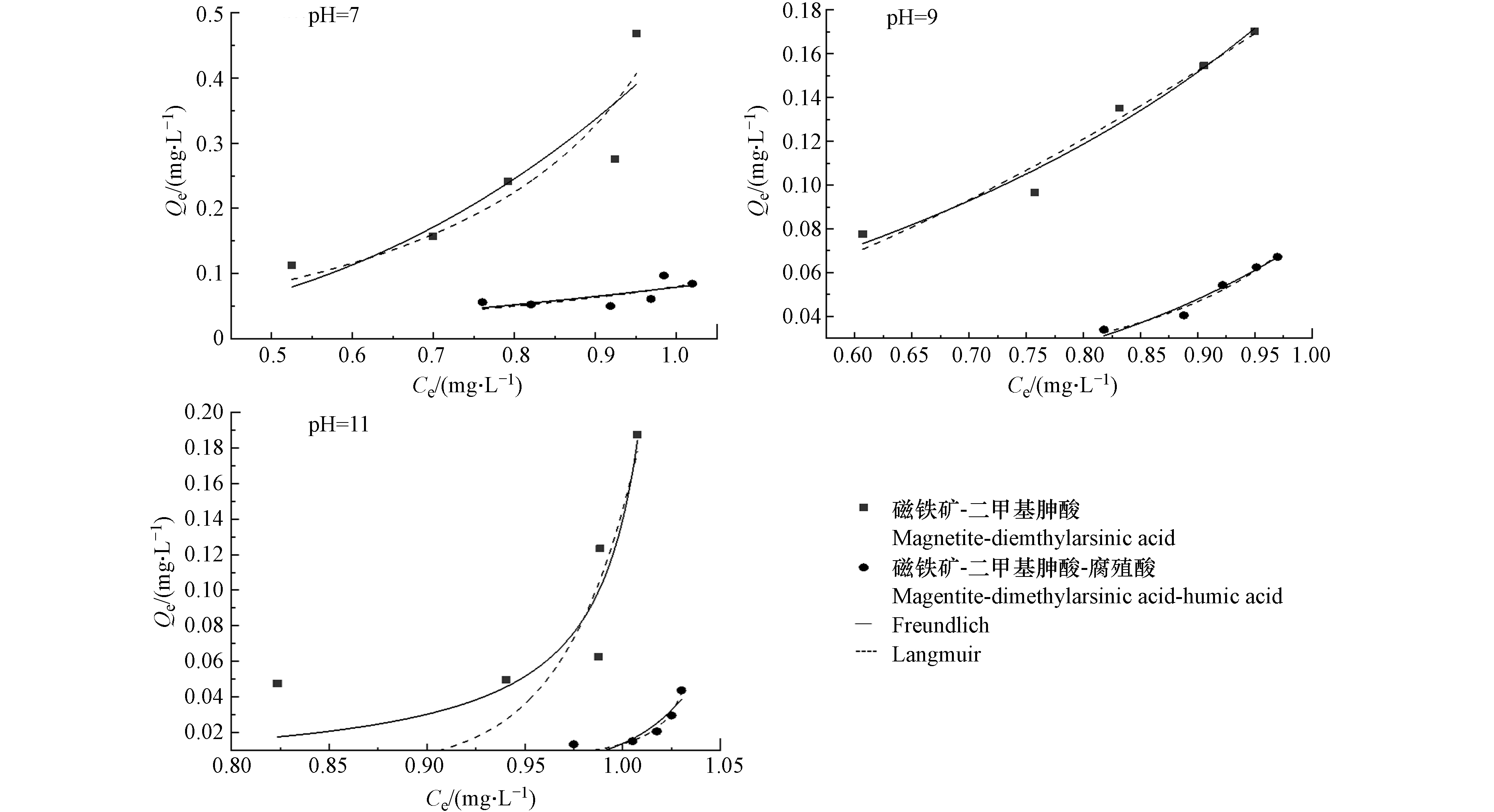
图 4 pH=7、9、11时,腐殖酸存在时二甲基胂酸在磁铁矿上吸附等温线拟合曲线C二甲基胂酸= 1 mg·L−1,C腐殖酸= 150 mg·L−1
Figure 4. When pH=7、9、11, the adsorption isotherm curve of dimethylarsinic acid on magnetite in the presence of humic acid was fitted
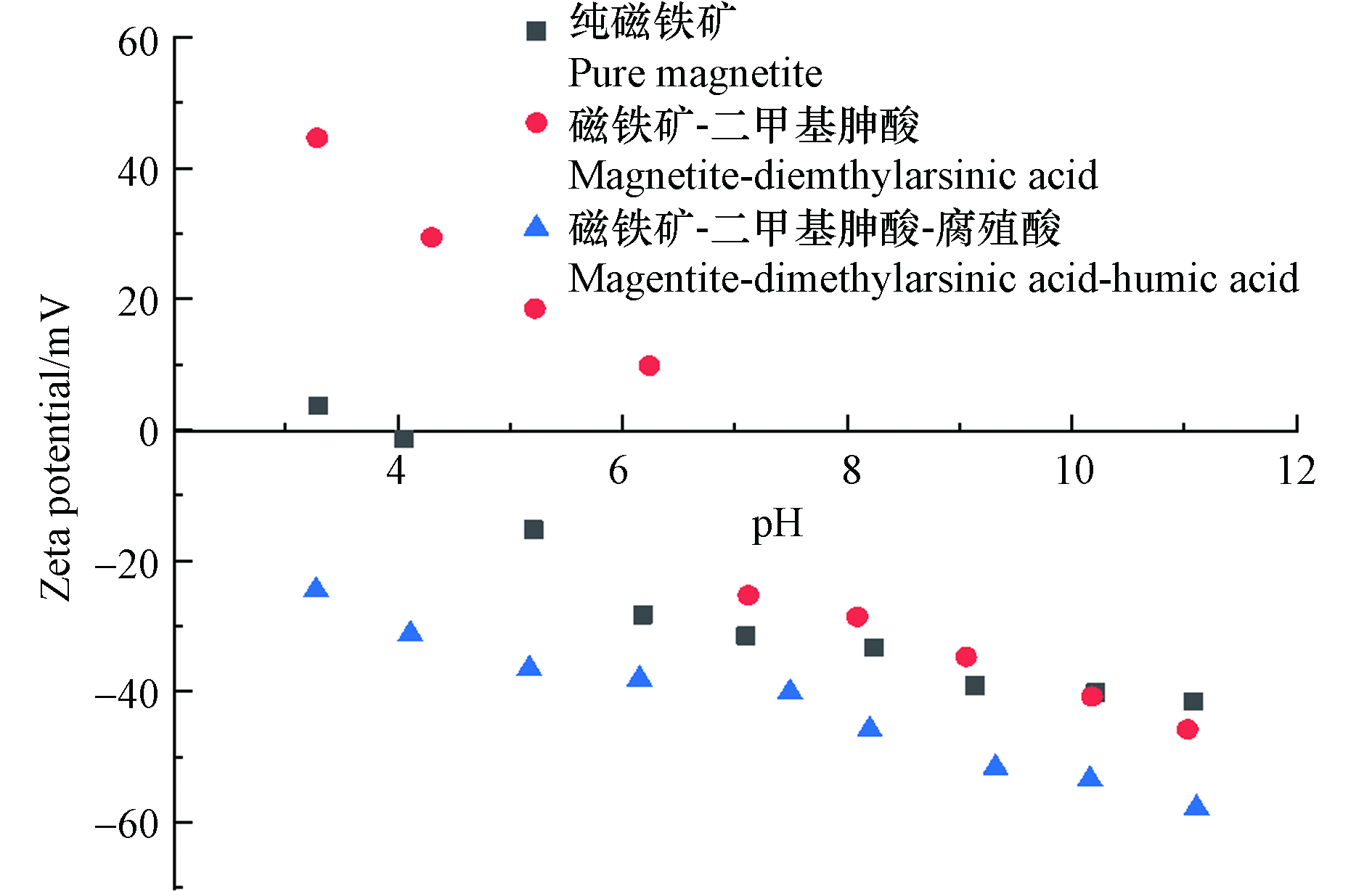
图 5 二甲基胂酸和腐殖酸对磁铁矿表面Zeta电位的影响
Figure 5. Effect of dimethylarsinic acid and humic acid on Zeta potential of magnetite surface

图 6 二甲基胂酸吸附体系红外光谱
Figure 6. Infrared spectra of dimethylarsinic acid adsorption system
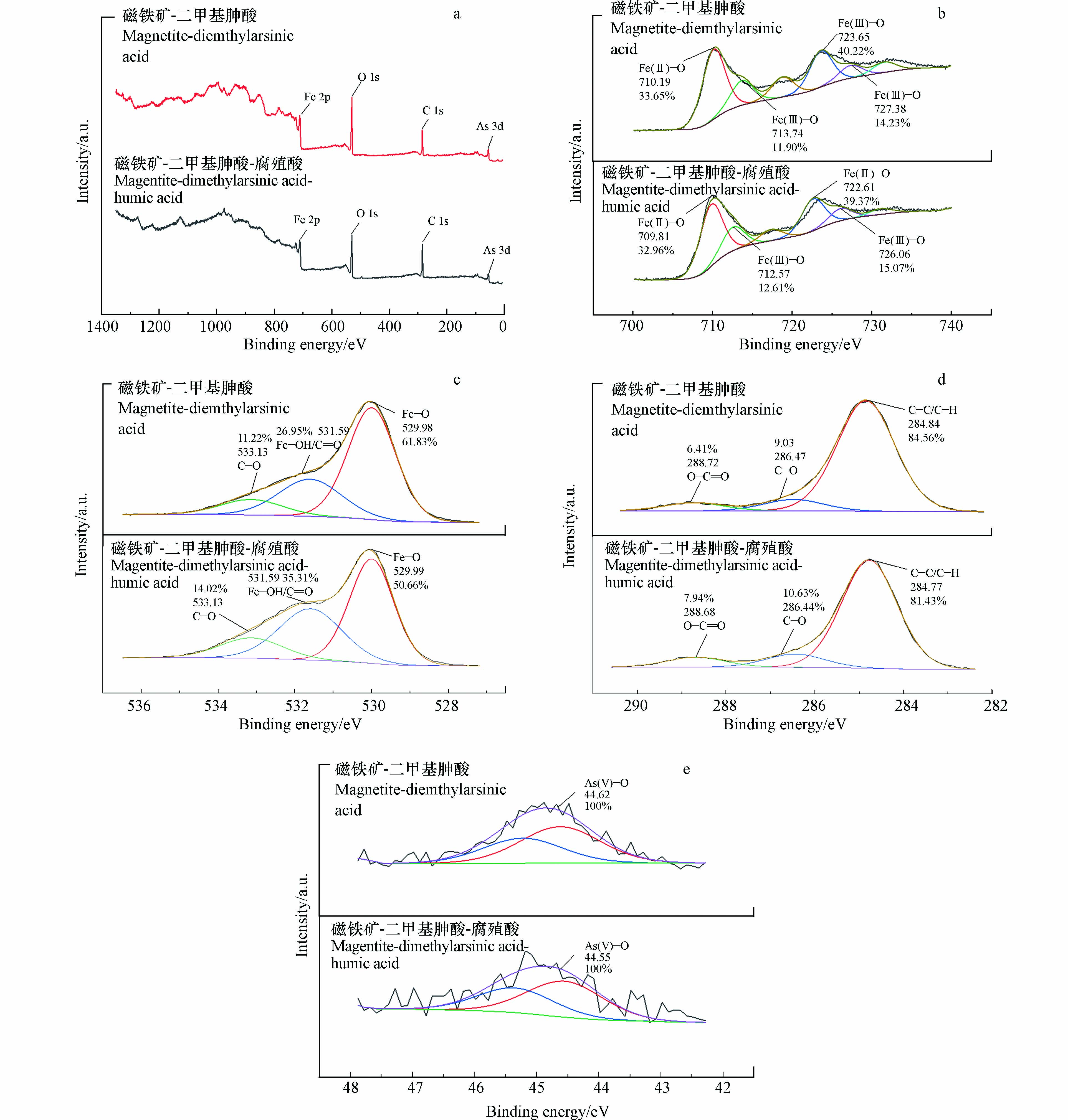
图 7 二甲基胂酸吸附体系X射线光电子能谱
Figure 7. X ray photoelectron spectroscopy of dimethylarsinic acid adsorption system
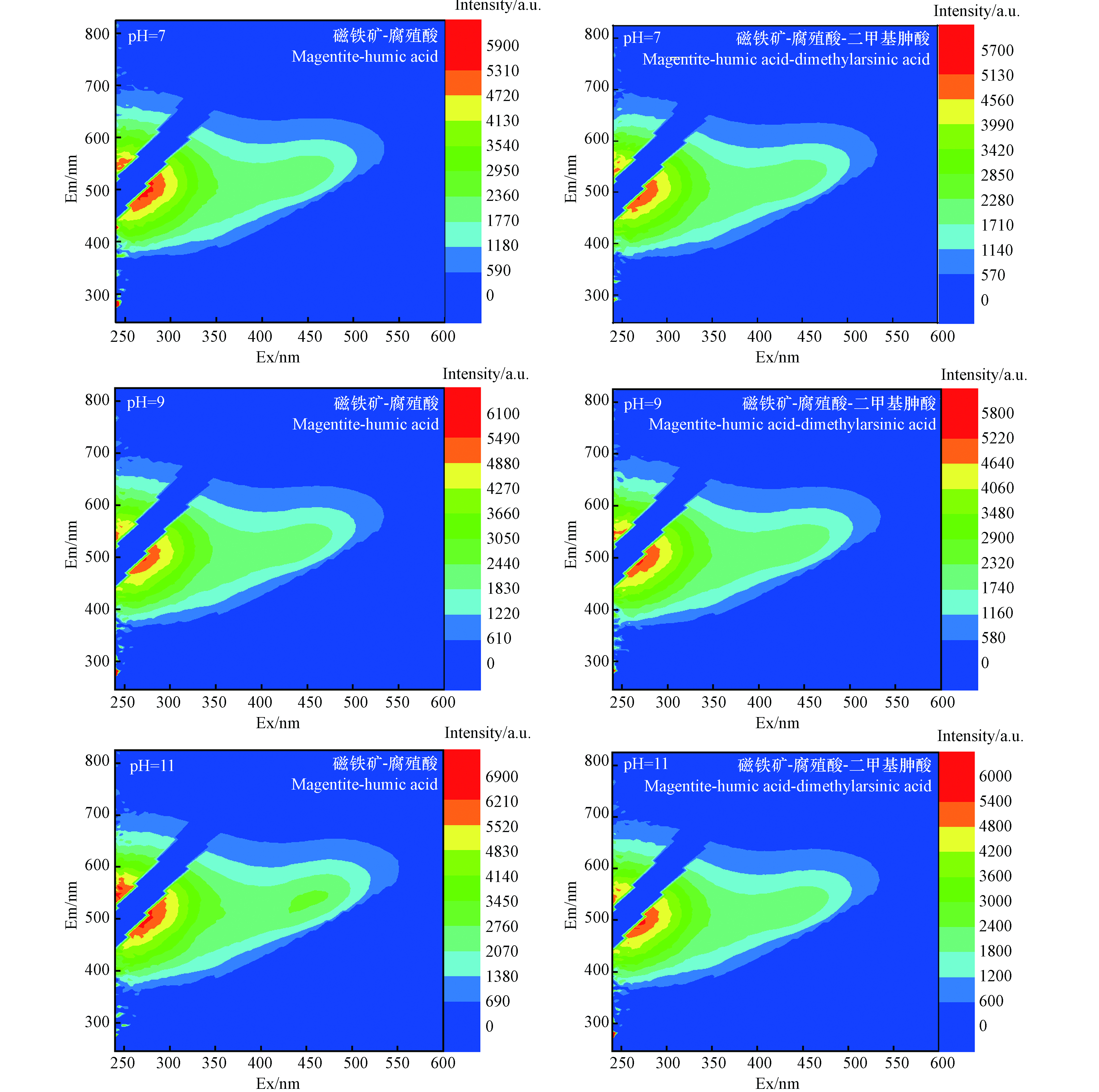
图 8 不同pH加二甲基胂酸前后的腐殖酸三维荧光光谱图
Figure 8. Three-dimensional fluorescence spectra of humic acid before and after adding dimethylarsinic acid
表 1 二甲基胂酸吸附动力学拟合结果
Table 1. Fitting results of adsorption kinetics of dimethylarsinic acid
实验体系Experimental system pH Qe,exp/ (mg·g−1) 准一级动力学Pseudo-first order dynamics 准二级动力学Pseudo-second order dynamics Qe,cal/(mg·g−1) K1/h−1 R2 Qe,cal/(mg·g−1) K2/(mg·g−1·h−1) R2 磁铁矿-二甲基胂酸 7 0.22 0.10 0.004 0.91 0.23 0.18 0.99 磁铁矿-二甲基胂酸-腐殖酸 7 0.10 0.04 0.0014 0.61 0.10 0.00018 0.99  下载: 导出CSV
下载: 导出CSV
表 2 腐殖酸存在时二甲基胂酸在磁铁矿上吸附等温线拟合参数
Table 2. Adsorption isotherm fitting parameters of dimethylarsinic acid on magnetite in the presence of humic acid
pH 实验体系Experimental system Langmuir吸附等温模型Langmuir adsorption isothermal model Freundlich吸附等温模型Freundlich adsorption isothermal model Qm/(mg·g−1) KL/(mg·L−1) R2 n KF/(mg(1-n)·Ln·g−1) R2 7 磁铁矿-二甲基胂酸 −0.12 −0.81 0.91 0.37 0.45 0.90 磁铁矿-二甲基胂酸-腐殖酸 −0.06 −5.8 0.63 0.53 0.08 0.63 9 磁铁矿-二甲基胂酸 −0.13 −0.61 0.98 0.51 0.20 0.97 磁铁矿-二甲基胂酸-腐殖酸 −1.20 −0.86 0.98 0.22 0.08 0.97 11 磁铁矿-二甲基胂酸 −6.14 −0.95 0.97 0.03 0.02 0.90 磁铁矿-二甲基胂酸-腐殖酸 4.00×10−3 −0.97 0.87 0.04 0.15 0.82
下载: 导出CSV
-
[1] AGUSA T, TRANG P T K, LAN V M, et al. Human exposure to arsenic from drinking water in Vietnam [J]. Science of the Total Environment, 2014, 488/489: 562-569. doi: 10.1016/j.scitotenv.2013.10.039 [2] HE J, CHARLET L. A review of arsenic presence in China drinking water [J]. Journal of Hydrology, 2013, 492: 79-88. doi: 10.1016/j.jhydrol.2013.04.007 [3] DAS N, PAUL S, CHATTERJEE D, et al. Arsenic exposure through drinking water increases the risk of liver and cardiovascular diseases in the population of West Bengal, India [J]. BMC Public Health, 2012, 12: 639. doi: 10.1186/1471-2458-12-639 [4] PAN W C, SEOW W J, KILE M L, et al. Association of low to moderate levels of arsenic exposure with risk of type 2 diabetes in Bangladesh [J]. American Journal of Epidemiology, 2013, 178(10): 1563-1570. doi: 10.1093/aje/kwt195 [5] LIU L, YANG Y P, DUAN G L, et al. The chemical-microbial release and transformation of arsenic induced by citric acid in paddy soil [J]. Journal of Hazardous Materials, 2022, 421: 126731. doi: 10.1016/j.jhazmat.2021.126731 [6] ZOU H M, ZHOU C, LI Y X, et al. Speciation analysis of arsenic in edible mushrooms by high-performance liquid chromatography hyphenated to inductively coupled plasma mass spectrometry [J]. Food Chemistry, 2020, 327: 127033. doi: 10.1016/j.foodchem.2020.127033 [7] TAYLOR V, GOODALE B, RAAB A, et al. Human exposure to organic arsenic species from seafood [J]. Science of the Total Environment, 2017, 580: 266-282. doi: 10.1016/j.scitotenv.2016.12.113 [8] LIU C W, LAI C C, CHEN Y, et al. Hydrogeochemical and mineralogical investigations of arsenic- and humic substance-enriched aquifers [J]. Journal of Hydrology, 2013, 498: 59-75. doi: 10.1016/j.jhydrol.2013.06.017 [9] MIADENOV N, ZHENG Y, SIMONE B, et al. Dissolved organic matter quality in a shallow aquifer of Bangladesh: Implications for arsenic mobility [J]. Environmental Science & Technology, 2015, 49(18): 10815-10824. [10] WANG S L, MULLIGAN C N. Effect of natural organic matter on arsenic release from soils and sediments into groundwater [J]. Environmental Geochemistry and Health, 2006, 28(3): 197-214. doi: 10.1007/s10653-005-9032-y [11] JIN J, ZIMMERMAN A R, NORTON S B, et al. Arsenic release from Floridan Aquifer rock during incubations simulating aquifer storage and recovery operations [J]. Science of the Total Environment, 2016, 551/552: 238-245. doi: 10.1016/j.scitotenv.2016.02.028 [12] KONG Y L, KANG J, SHEN J M, et al. Influence of humic acid on the removal of arsenate and arsenic by ferric chloride: Effects of pH, As/Fe ratio, initial As concentration, and co-existing solutes [J]. Environmental Science and Pollution Research, 2017, 24(3): 2381-2393. doi: 10.1007/s11356-016-7994-1 [13] WARWICK P, INAM E, EVANS N. Arsenic's interaction with humic acid [J]. Environmental Chemistry, 2005, 2(2): 119. doi: 10.1071/EN05025 [14] RASHID M, STERBINSKY G E, PINILLA M Á G, et al. Kinetic and mechanistic evaluation of inorganic arsenic species adsorption onto humic acid grafted magnetite nanoparticles [J]. The Journal of Physical Chemistry C, 2018, 122(25): 13540-13547. doi: 10.1021/acs.jpcc.7b12438 [15] WEI Y T, ZHENG Y M, CHEN J P. Uptake of methylated arsenic by a polymeric adsorbent: Process performance and adsorption chemistry [J]. Water Research, 2011, 45(6): 2290-2296. doi: 10.1016/j.watres.2011.01.002 [16] JAIN N, MAITI A. Arsenic adsorbent derived from the ferromanganese slag [J]. Environmental Science and Pollution Research, 2021, 28(3): 3230-3242. doi: 10.1007/s11356-020-10745-9 [17] 柳凤娟, 张国平, 罗绪强, 等. Fe(Ⅱ)浓度对硫酸盐还原菌去除水体中砷和锑的影响 [J]. 环境化学, 2021, 40(10): 3171-3179. doi: 10.7524/j.issn.0254-6108.2020060401 LIU F J, ZHANG G P, LUO X Q, et al. Effect of different contents of Fe(Ⅱ) on removal of arsenic and antimony from water by sulfate reducing bacteria [J]. Environmental Chemistry, 2021, 40(10): 3171-3179(in Chinese). doi: 10.7524/j.issn.0254-6108.2020060401
[18] 杨厅, 王强, 李二平, 等. 铁盐类复合稳定剂对砷钙渣中As的稳定化作用及机理 [J]. 环境化学, 2020, 39(11): 2999-3008. doi: 10.7524/j.issn.0254-6108.2020042202 YANG T, WANG Q, LI E P, et al. Stabilization treatment of arsenic calcium residue using Fe-containing compound stabilizers [J]. Environmental Chemistry, 2020, 39(11): 2999-3008(in Chinese). doi: 10.7524/j.issn.0254-6108.2020042202
[19] LIU G L, FERNANDEZ A, CAI Y. Complexation of arsenite with humic acid in the presence of ferric iron [J]. Environmental Science & Technology, 2011, 45(8): 3210-3216. [20] 王晓丹, 梁康, 杨佘维. 高度分散的球状纳米磁铁矿的水热合成及其结构表征[C]//中国环境科学学会2019年科学技术年会——环境工程技术创新与应用分论坛论文集(二). 西安, 2019: 46-48. [21] 韩建军, 廖党, 席壮民, 等. 磁铁矿防辐射超高性能混凝土制备及性能研究 [J]. 硅酸盐通报, 2021, 40(9): 2930-2938. doi: 10.16552/j.cnki.issn1001-1625.20210629.001 HAN J J, LIAO D, XI Z M, et al. Preparation and properties of ultra-high performance concrete for radiation protection of magnetite [J]. Bulletin of the Chinese Ceramic Society, 2021, 40(9): 2930-2938(in Chinese). doi: 10.16552/j.cnki.issn1001-1625.20210629.001
[22] AYAZ I, RIZWAN M, ULLMAN J L, et al. Lignocellulosic based biochar adsorbents for the removal of fluoride and arsenic from aqueous solution: Isotherm and kinetic modeling [J]. Polymers, 2022, 14(4): 715. doi: 10.3390/polym14040715 [23] MIKUTTA R, LORENZ D, GUGGENBERGER G, et al. Properties and reactivity of Fe-organic matter associations formed by coprecipitation versus adsorption: Clues from arsenate batch adsorption [J]. Geochimica et Cosmochimica Acta, 2014, 144: 258-276. doi: 10.1016/j.gca.2014.08.026 [24] 王喆, 赵志西. 砷迁移释放过程中吸附-脱附作用机制的研究进展[J]. 化学通报, 2020, 83(1): 23-29. WANG Z, ZHAO Z X. Research advances of the adsorption-desorption mechanism in arsenic mobilization and retention[J]. Chemistry, 2020, 83(1): 23-29(in Chinese). Chemistry, 2020, 83(1): 23-29(in Chinese).
[25] ZSOLNAY Á. Dissolved organic matter: Artefacts, definitions, and functions [J]. Geoderma, 2003, 113(3/4): 187-209. [26] 吴少雄, 邢志, 陈红兵, 等. 磁性纳米四氧化三铁选择性富集-电感耦合等离子体原子发射光谱测定砷 [J]. 分析化学, 2009, 37(5): 711-714. doi: 10.3321/j.issn:0253-3820.2009.05.017 WU S X, XING Z, CHEN H B, et al. Nanomagnetic material ferriferrous oxide separation/enrichment and inductively coupled plasma-atomic emission spectrometry for determination of arsenic [J]. Chinese Journal of Analytical Chemistry, 2009, 37(5): 711-714(in Chinese). doi: 10.3321/j.issn:0253-3820.2009.05.017
[27] SARMAH S, SAIKIA J, PHUKAN A, et al. Adsorption of As(V) from water over a hydroxyl-alumina modified paddy husk ash surface and its sludge immobilization [J]. Water, Air, & Soil Pollution, 2019, 230(2): 32. [28] REDMAN A D, MACALADY D L, AHMANN D. Natural organic matter affects arsenic speciation and sorption onto hematite [J]. Environmental Science & Technology, 2002, 36(13): 2889-2896. [29] BUSCHMANN J, KAPPELER A, LINDAUER U, et al. Arsenite and arsenate binding to dissolved humic acids: Influence of pH, type of humic acid, and aluminum [J]. Environmental Science & Technology, 2006, 40(19): 6015-6020. [30] LIN H T, WANG M C, LI G C. Complexation of arsenate with humic substance in water extract of compost [J]. Chemosphere, 2004, 56(11): 1105-1112. doi: 10.1016/j.chemosphere.2004.05.018 [31] 罗梦, 何绪文, 林宏宇, 等. 混凝—气浮工艺预处理ABS树脂生产废水 [J]. 化工环保, 2017, 37(1): 55-61. doi: 10.3969/j.issn.1006-1878.2017.01.010 LUO M, HE X W, LIN H Y, et al. Pretreatment of ABS resin production wastewater by coagulation-air flotation process [J]. Environmental Protection of Chemical Industry, 2017, 37(1): 55-61(in Chinese). doi: 10.3969/j.issn.1006-1878.2017.01.010
[32] ZHANG X, DENG J S, HUANGFU M Z, et al. Novel insights into the influence of ferric ion as a surface modifier to enhance the floatability of specularite [J]. Powder Technology, 2022, 398: 117141. doi: 10.1016/j.powtec.2022.117141 [33] 王喆, 赵志西, 刘杨秋凡, 等. 砷在新疆奎屯河沉积物上的吸附及有机酸对吸附的影响 [J]. 生态学杂志, 2021, 40(6): 1766-1774. doi: 10.13292/j.1000-4890.202106.008 WANG Z, ZHAO Z X, LIU Y, et al. Adsorption of arsenic on the sediments of Kuitun River in Xinjiang and the effects of organic acids on adsorption [J]. Chinese Journal of Ecology, 2021, 40(6): 1766-1774(in Chinese). doi: 10.13292/j.1000-4890.202106.008
[34] ZUBAIR Y O, FUCHIDA S, TOKORO C. Insight into the mechanism of arsenic(III/V) uptake on mesoporous zerovalent iron-magnetite nanocomposites: Adsorption and microscopic studies [J]. ACS Applied Materials & Interfaces, 2020, 12(44): 49755-49767. [35] 温全宝, 滕青, 杨志超, 等. 苛化淀粉对磁铁矿和金云母浮选分离的影响及机理研究 [J]. 矿产保护与利用, 2020, 40(2): 62-69. doi: 10.13779/j.cnki.issn1001-0076.2020.02.009 WEN Q B, TENG Q, YANG Z C, et al. Effect and mechanism of causticized starch on flotation separation of magnetite and phlogopite [J]. Conservation and Utilization of Mineral Resources, 2020, 40(2): 62-69(in Chinese). doi: 10.13779/j.cnki.issn1001-0076.2020.02.009
[36] 张帆. 改性材料在石油污染水体中的应用 [J]. 北方环境, 2013, 25(12): 142-148. ZHANG F. Application of inodifiled material in oilpoclution in water bodies [J]. Northern Environment, 2013, 25(12): 142-148(in Chinese).
[37] ZHANG G S, QU J H, LIU H J, et al. Removal mechanism of As(III) by a novel Fe-Mn binary oxide adsorbent: Oxidation and sorption [J]. Environmental Science & Technology, 2007, 41(13): 4613-4619. [38] ZHENG X, KHAN M T, CROUÉ J P. Contribution of effluent organic matter (EfOM) to ultrafiltration (UF) membrane fouling: Isolation, characterization, and fouling effect of EfOM fractions [J]. Water Research, 2014, 65: 414-424. doi: 10.1016/j.watres.2014.07.039 [39] RAO P H, MAK M S H, LIU T Z, et al. Effects of humic acid on arsenic(V) removal by zero-valent iron from groundwater with special references to corrosion products analyses [J]. Chemosphere, 2009, 75(2): 156-162. doi: 10.1016/j.chemosphere.2008.12.019 [40] XIE L, SHANG C. Role of humic acid and ouinone model compounds in bromate reduction by zerovalent iron [J]. Environmental Science & Technology, 2005, 39(4): 1092-1100. [41] SHARMA P, OFNER J, KAPPLER A. Formation of binary and ternary colloids and dissolved complexes of organic matter, Fe and As [J]. Environmental Science & Technology, 2010, 44(12): 4479-4485. [42] WANG Y, ZHANG D, SHEN Z Y, et al. Investigation of the interaction between As and Sb species and dissolved organic matter in the Yangtze Estuary, China, using excitation-emission matrices with parallel factor analysis [J]. Environmental Science and Pollution Research, 2015, 22(3): 1819-1830. doi: 10.1007/s11356-014-3380-z [43] WANG S, ZHENG K, LI H P, et al. Arsenopyrite weathering in acidic water: Humic acid affection and arsenic transformation [J]. Water Research, 2021, 194: 116917. doi: 10.1016/j.watres.2021.116917 [44] HU C Z, CHEN Q X, LIU H J, et al. Coagulation of methylated arsenic from drinking water: Influence of methyl substitution [J]. Journal of Hazardous Materials, 2015, 293: 97-104. doi: 10.1016/j.jhazmat.2015.03.055 [45] SHIMIZU M, GINDER-VOGEL M, PARIKH S J, et al. Molecular scale assessment of methylarsenic sorption on aluminum oxide [J]. Environmental Science & Technology, 2010, 44(2): 612-617. [46] SHIMIZU M, ARAI Y, SPARKS D L. Multiscale assessment of methylarsenic reactivity in soil. 2. Distribution and speciation in soil [J]. Environmental Science & Technology, 2011, 45(10): 4300-4306. [47] SUN Y, LAN J R, CHEN X H, et al. High arsenic levels in sediments, Jianghan Plain, central China: Vertical distribution and characteristics of arsenic species, dissolved organic matter, and microbial community [J]. Journal of Geochemical Exploration, 2021, 228: 106822. doi: 10.1016/j.gexplo.2021.106822 [48] HUANG S B, WANG Y X, CAO L, et al. Multidimensional spectrofluorometry characterization of dissolved organic matter in arsenic-contaminated shallow groundwater [J]. Journal of Environmental Science and Health. Part A, Toxic/Hazardous Substances & Environmental Engineering, 2012, 47(10): 1446-1454. [49] CHEN C, LI L Y, HUANG K, et al. Sulfate-reducing bacteria and methanogens are involved in arsenic methylation and demethylation in paddy soils [J]. The ISME Journal, 2019, 13(10): 2523-2535. doi: 10.1038/s41396-019-0451-7 [50] LÜ W, YAO X, REN H Y, et al. Characterizing the interactions between sediment dissolved organic matter and zinc using multispectroscopic techniques [J]. Environmental Pollution, 2020, 261: 113644. doi: 10.1016/j.envpol.2019.113644 [51] 朱江鹏, 梅婷, 彭云, 等. 荧光猝灭法研究洛克沙胂与腐殖酸的相互作用 [J]. 环境科学, 2014, 35(7): 2620-2626. doi: 10.13227/j.hjkx.2014.07.026 ZHU J P, MEI T, PENG Y, et al. Characterizing the interaction between roxarsone and humic acid by fluorescence quenching experiment [J]. Environmental Science, 2014, 35(7): 2620-2626(in Chinese). doi: 10.13227/j.hjkx.2014.07.026
[52] PALMER N E, von WANDRUSZKA R. Humic acids as reducing agents: The involvement of quinoid moieties in arsenate reduction [J]. Environmental Science and Pollution Research, 2010, 17(7): 1362-1370. doi: 10.1007/s11356-010-0322-2 [53] KONG Y L, SHEN J M, CHEN Z L, et al. Influence of potassium permanganate pre-oxidation on the interaction of humic acid with cadmium/arsenic [J]. RSC Advances, 2016, 6(4): 3048-3057. doi: 10.1039/C5RA22043B [54] 董丽娴. 溶解性有机质对砷形态及藻类有效性的影响[D]. 上海: 同济大学, 2008. DONG L X. Influence of dissolved organic matter on the arsenic speciation and availability to algae[D]. Shanghai: Tongji University, 2008(in Chinese).
[55] KIM E J, HWANG B R, BAEK K. Effects of natural organic matter on the coprecipitation of arsenic with iron [J]. Environmental Geochemistry and Health, 2015, 37(6): 1029-1039. doi: 10.1007/s10653-015-9692-1 [56] 葛思怡. 金属离子对溶解性有机质与砷类化合物之间相互作用的影响研究[D]. 南京: 南京师范大学, 2017. GE S Y. Effect of metal ions on the interaction between dissolved organic matter and arsenic compounds[D]. Nanjing: Nanjing Normal University, 2017(in Chinese).
期刊类型引用(2)
1. 裴曼一,李梅,裴建川,杨金艳,单骁,郑砚石,赖阳阳,宋博涵,梁佳雨. 铁铜改性稻壳炭对双氯芬酸钠吸附性能. 环境工程学报. 2024(03): 737-746 .  百度学术
百度学术
2. 华菁,贾莹刚,吕媛,杨潇,高羽,苏艳超,商建英,阎秀兰. 酸性条件下饱和多孔介质中赤铁矿胶体和Cr(Ⅵ)的协同运移及影响机制. 环境化学. 2024(11): 3845-3853 . 本站查看
其他类型引用(2)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 3214
- HTML全文浏览数: 3214
- PDF下载数: 61
- 施引文献: 4
