







-
近几年,臭氧(O3)已成为影响我国环境空气质量的重要因素,其中京津冀及周边地区、长三角地区以O3为首要污染物的超标天数占比已经超过PM2.5[1-3]。研究表明,挥发性有机物(VOCs)可在紫外线照射下与氮氧化物(NOx)发生光化学反应,产生光化学烟雾,光化学烟雾的主要成分为O3[4-5]。因此,作为O3重要前体物的VOCs到研究学者的广泛关注。
VOCs种类繁多,不同种类的VOCs化学反应活性也不相同,研究VOCs的组成和来源特征对控制O3污染和揭示复合型大气污染的形成都具有重要意义[6]。目前国内关于VOCs的监测和研究主要集中在长江三角洲[7-8]、珠江三角洲[9-10]和京津冀[11-12]等地区。山东半岛相关的研究较少,刘泽常等[13]研究表明,济南市区VOCs的优势组分为C3—C5的烷烃、丙烯、顺-2-丁烯、间/对二甲苯和甲苯等,主要来源为汽车尾气、工业源和燃烧源。薛莲等[14]发现青岛市大气VOCs中烯烃对臭氧的生成贡献远高于烷烃和芳香烃。张桢超[15]发现威海市大气中,C2—C4烯烃类、烷烃类和苯系物对臭氧的生成贡献率较高,VOCs主要来源于机动车排放、工艺过程和溶剂使用。
泰安市地处山东省中部的泰山南麓,三面环山,属于内陆中小型城市。2016—2017年,泰安市O3最大8 h平均浓度分别为197 μg·m−3和210 μg·m−3,在全省分别排名第二位和第一位。臭氧已成为泰安市夏、秋季节环境空气的首要污染物[16]。了解臭氧前体物VOCs的污染现状及来源对泰安市采取适当措施改善空气质量具有重要意义。
本研究在泰安市城区建立一个观测站点,采用在线观测法,连续对站点大气中的VOCs进行监测,分析其浓度特征,并利用特征比值和模型分析对VOCs进行来源解析,同时评估其臭氧生成潜势,以期为泰安市大气环境VOCs和O3污染管控提供科学支撑。
-
本次观测时间为2018年6月1日—7月11日,可以反应泰安市夏季大气中VOCs的污染特点。监测地点位于泰安市泰山区的山东电力高等专科学校校院内(36.18°N,117.11°E),该观测点是泰安市的国控监测点,周边紧邻交通干线,同时分布着农贸市场、工业区、商业区和居民区,是典型的城市中心站点,观测点位置如图1所示。
NMHCs的观测采用由中国科学院生态环境研究中心自主研发的GC-FID-VOCs在线监测仪24 h连续监测,采样时间分辨率为1 h,毛细管色谱柱型号为OV-1(30 m 柱长× 0.32 mm直径 × 1.0 μm厚度);采样时,通过采样泵以50 mL·min−1的流速将环境气体浓缩至温度为−80 ℃的吸附管中,然后升温加热至100 ℃进行热脱附,保持6 min;同时以5 mL·min−1的N2流速将解吸的样品吹入GC毛细管色谱柱中进行分离,此时将吸附管的温度升高至220 ℃,以60 mL·min−1反吹10 min以清除残留;色谱柱的程序升温如下:初始温度为−60 ℃,保持3 min;以12 ℃·min−1升温至−20 ℃;以6 ℃·min−1升温至30 ℃;以10 ℃·min−1升温至170 ℃,保持2 min;FID检测器的温度为250 ℃,仪器共检测到51种物质,其中包含27种烷烃、9种烯烃和15种芳香烃[16]。醛酮类化合物(OVOCs)的观测采用涂有2,4-二硝基苯肼(DNPH)衍生化试剂的硅胶小柱采集,每2 h采集1个样品,并采用高效液相色谱(HPLC)方法检测,共检测出15种OVOCs。
为了保证观测数据的有效性和可靠性,GC-FID-VOCs在线监测仪每2 d采用美国环保署认可的Linde SPECTRA Environmental Gases标准气体进行5点校准,校准时相关系数均在0.992—0.995;同时,为了避免一些高反应性的VOCs物种的氧化损失,在采样吸附管的前端连接填充亚硫酸钠的捕集器,用于去除空气中的氧化剂,每2 d更换1次亚硫酸钠捕集器;高效液相色谱每2 d进行曲线校准,每20个样品分析一次校准曲线中间浓度点,每个目标化合物的测定结果与初始浓度值相对偏差≤30%[16-17]。除VOCs的观测外,同时观测环境空气中的CO、SO2和NOx等参数,监测仪器均采用赛默飞世尔科技公司i系列的自动连续检测仪。
-
不同城市中大气VOCs的来源各异,VOCs中各组分的浓度水平和化学活性也不同,对大气O3生成的贡献也有差异。臭氧生成潜势(OFP)是用最大增量反应活性方法评估挥发性有机化合物的光化学反应性,并估算臭氧形成过程中单个有机化合物的贡献率[18],计算公式为:
式中,OFPi为第i个VOCs物种的臭氧生成潜势,μg·m−3;[VOCsi]表示物种i的环境质量浓度,μg·m−3;MIRi为VOCs第i个物种最大增量反应中臭氧生成系数,可在文献[19]中查出。
-
正交矩阵因子分析模型(positive matrix factorization,PMF)作为受体模型,根据长时间序列的受体化学组分数据集进行VOCs来源解析[20]。PMF计算过程中的基本公式为:
式中,Xij为样本i中污染物j的浓度,×10−9;p表示污染源的数量;gik为第k个来源对第i个因子的贡献量,%;fkj为第k个源中第j个组分的分布占比,%;eij为样本残差。PMF模型主要是将目标函数Q最小化[21-22],目标函数Q定义为:
式中,n为样本个数,m为物种个数;uij表示样本中物种的不确定性。根据PMF5.0指导方法要求,不确定度的计算公式为:
式中,unc. 表示样本中物种的不确定度;C表示样本中物种的浓度;RSD表示相对标准偏差;MDL表示检出限。
-
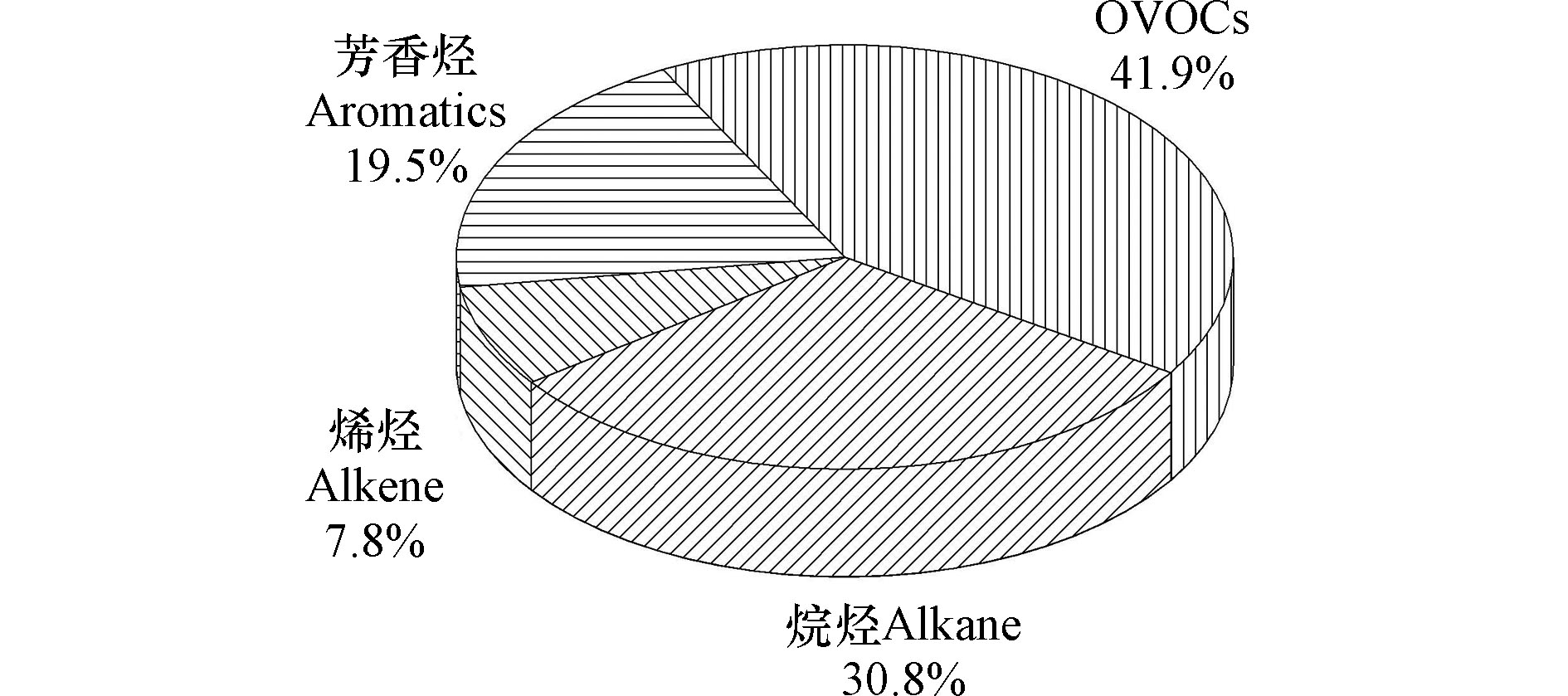
观测期间采样频率为1 h,VOCs浓度平均值为(16.57±7.99)×10−9(体积分数)。由表1可知,观测期间VOCs浓度水平最高的物种是甲醛(3.18±2.09)×10−9和丙酮(2.02±1.27)×10−9,其次为丙烷(1.71±1.41)×10−9、乙醛(1.39±0.61)×10−9和丁烷(0.92±0.88)×10−9。由图2可以看出,整个监测期间VOCs四大组分浓度顺序依次为:OVOCs(41.9%)> 烷烃(30.8%)> 芳香烃(19.5%) > 烯烃(7.8%)。
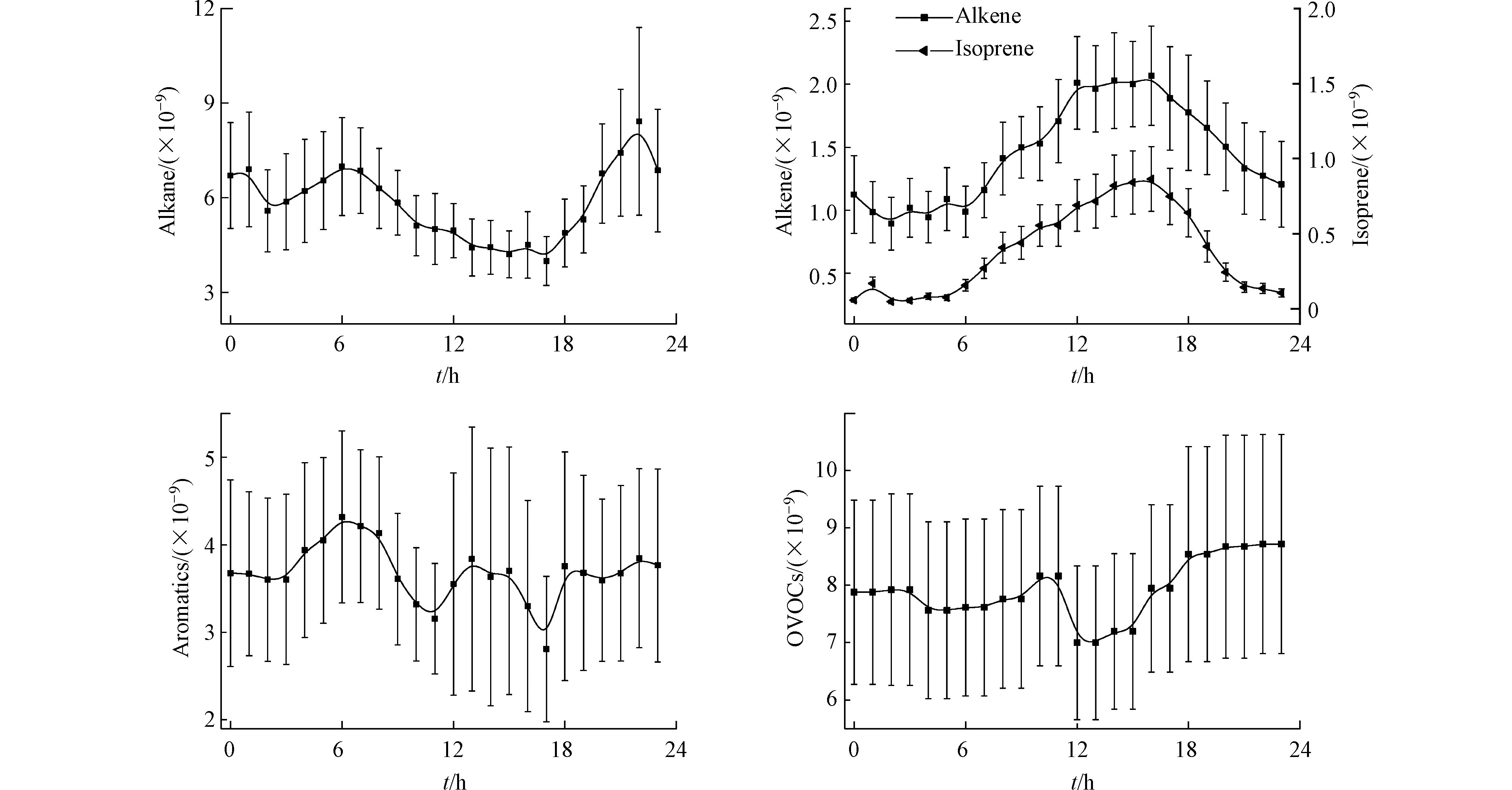
由于VOCs来源的不同和化学活性的差异,导致VOCs组分浓度的日变化特征也不同,分析VOCs浓度日变化特征是探讨其来源的重要手段之一。图3给出了观测站点大气中烷烃、烯烃、芳香烃和OVOCs的日变化趋势。烷烃、芳香烃和OVOCs日变化趋势较为一致,整体呈现夜间高白天低的变化特征;因为早晚存在较强的人类活动,如城市地区机动车尾气排放等,同时早晚大气较稳定,不利于VOCs的扩散;中午及下午对流强,边界层抬升,有利于污染物的扩散,同时中午和下午太阳辐射强,大气光化学反应活性剧烈,也会消耗一定量的VOCs,造成大气中VOCs的浓度下降。然而,烯烃在白天出现了显著抬升的变化特征,这主要是来自于植物活动排放异戊二烯的重要贡献[23-24],在白天随着太阳辐射的增强,异戊二烯排放量明显增加,在午后达到峰值,16:00以后由于植物活性等影响,其浓度快速下降。
对于烷烃,由于观测站点靠近蔬菜批发市场,运输车辆工作较早,受交通早高峰影响,在06:00出现峰值,随后太阳辐射增强,光化学反应消耗增加,浓度逐渐降低,在17:00以后,光化学反应消耗停止并随着市内交通晚高峰到来,污染物的浓度逐渐积累。芳香烃和OVOCs相对于烷烃和烯烃峰型规律没那么明显,芳香烃在中午出现了浓度升高的趋势,说明芳香烃类除来源于机动车排放外,还受溶剂挥发和化工企业排放的影响;OVOCs夜晚浓度较高,日出后出现微弱下降,随后缓慢抬升,在正午12:00左右发生快速下降达到最低,而后又快速抬升一直维持到深夜,说明除来自一次排放外,二次贡献生成和区域气象因素对其也有重要影响。
-
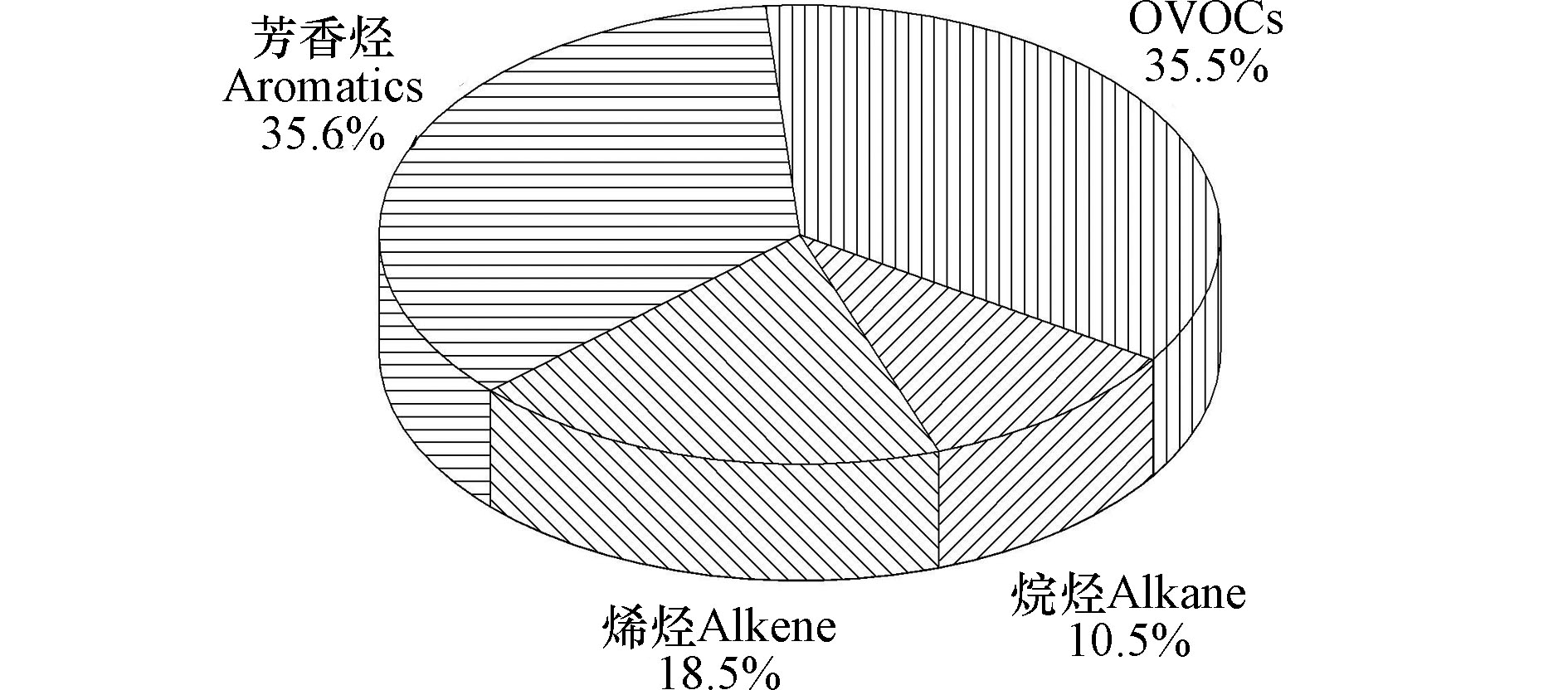
环境空气中VOCs组分的化学反应活性不同,对O3的形成影响也不同,识别对大气O3贡献较大VOCs物种,对于制定有效的减排控制措施意义重大[25]。由表1可知,观测点观测期间甲醛(40.34±26.52) μg·m−3、间/对-二甲苯(20.51±14.96) μg·m−3、乙醛(17.87±7.90) μg·m−3、异戊二烯(15.44±14.10) μg·m−3和间二乙基苯(14.59±11.71) μg·m−3是OFP值最高的5种VOCs物种。由图4可知,观测期间大气VOCs四大类别对OFP贡献率顺序为:芳香烃(35.6%) > OVOCs(35.5%) > 烯烃(18.5%) > 烷烃(10.5%)。烷烃化合物VOCs浓度占比最高,但化学反应活性低,故对OFP的贡献较小[26-27];烯烃中异戊二烯的浓度水平较高,且所含的碳碳双键化学反应活性强,对OFP的贡献较大,异戊二烯主要来源于植物活动排放,说明植物源对大气中O3的生成有重要贡献[20];芳香烃和OVOCs浓度水平和臭氧生成潜势都较高,因此,控制芳香烃和OVOCs的排放是未来控制泰安市臭氧污染的关键。
-
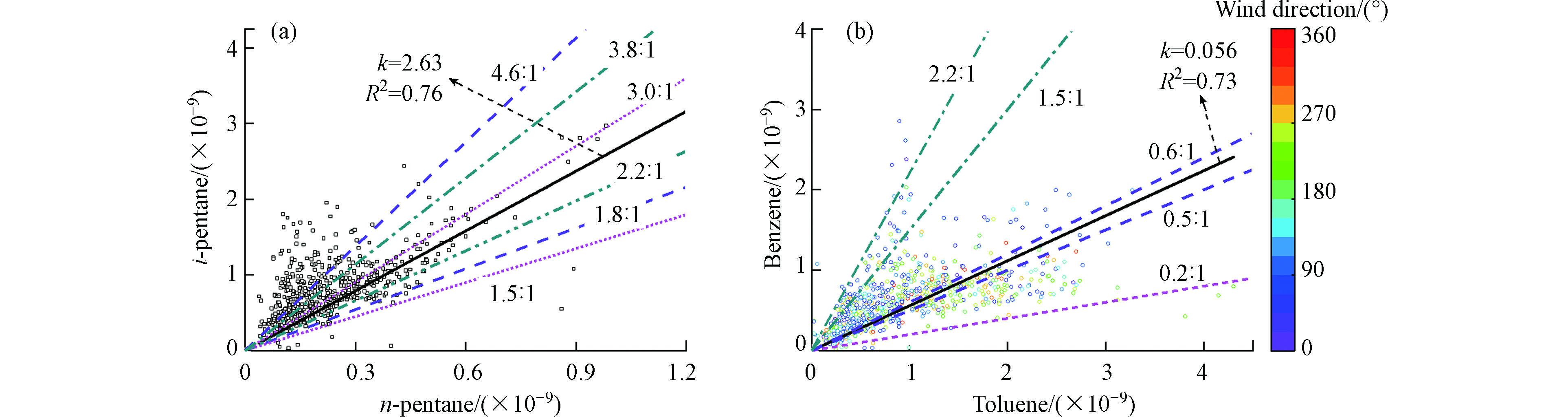
VOCs组分中,与·OH具有相似反应速率的的特征污染物之间的比值,可以反映其来源特征。戊烷在环境中主要来源于机动车尾气、天然气排放、燃料和液体汽油挥发,正戊烷(n-pentane)和异戊烷(i-pentane)具有相似的物理和化学性质,异戊烷/正戊烷的值可以初步判断其来源,当比值范围为0.82—0.89时,来源为天然气排放,比值范围为2.20—3.80时,来源为机动车尾气,比值范围在1.50—3.00时为液态汽油排放,比值范围在1.80—4.60时为燃料挥发,比值范围大于4.60时为其他源[28]。图5(a)分别给出了异戊烷/正戊烷不同的比值线,可知观测点异戊烷和正戊烷的比值大部分分布在1.80—4.60之间,表明机动车尾气、液态汽油排放和燃料挥发都对其有贡献作用,而天然气排放对戊烷的来源贡献较少,其它源对其来源也有贡献。利用特征比值法分析可以看出异戊烷和正戊烷的比值较为分散,异戊烷和正戊烷的来源复杂,并不是单一来源,特征比值法无法很好的解析其来源。
芳香烃是泰安市大气VOCs中对O3生成贡献最大的物种,苯(benzene)与甲苯(toluene)的比值(B/T)常用来判断芳香烃在环境中的来源,当苯/甲苯值不大于0.20时,判断其来源为工业溶剂,机动车尾气源为0.50—0.60,燃煤源为1.50—2.20,生物质燃烧源比值约为2.50[29]。由图5(b)分别给出了苯/甲苯的不同比值线,可以看出苯和甲苯比值分布在0.20—2.20之间,表明机动车尾气和燃煤源对其有贡献作用,经线性拟合的比值为0.56,与机动车尾气排放比值相近,进一步表明机动车尾气排放对观测点大气VOCs和O3有重要贡献。B/T的值不只受到其来源的影响,还受到大气氧化性的影响,甲苯的光化学反应活性要大于苯,B/T的值相较于异戊烷/正戊烷也更为分散;另一方面B/T的值受风向影响较大,西南风向上有较高的甲苯浓度,表明污染物传输对其比值有影响。
综上,观测点位于城市中心点,VOCs的来源更为复杂,特征污染物的比值范围较为分散,简单的特征比值法无法准确的解析出污染物的来源,需要与PMF模型解析的结果进行相互印证。
-
针对观测点的VOCs数据,选取了来源指示性强和监测数据相对完整VOCs物种输入到PMF模型中,同时将观测的CO、NOx和SO2数据纳入计算,帮助识别排放源,最终共识别出6类因子,各类因子的源成分谱特征如图6所示。
第一类因子中甲醛、乙醛和丙酮等OVOCs所占的百分比较高,因此将此类因子归为OVOCs源。第二类因子中,C2—C4烯烃和烷烃为优势组分,C2—C4烯烃和烷烃是LPG和溶剂挥发的关键物种[12,30],因此,将此类因子归为LPG和溶剂挥发源。第三类因子具有高组分的CO和苯,工业燃烧过程中可排放大量的CO,苯也是工业燃烧过程中重要的特征指示物[31],故将此类因子归为工业排放。第四类因子具有高组分的SO2,电厂可排放大量的SO2,因此,将此类因子归为电厂排放。第五类因子的优势组分为苯系物和烷烃,根据之前特征比值分析出异戊烷、正戊烷、苯和甲苯的主要来源都有机动车尾气排放,且C8—C10烷烃是柴油发动机排放尾气的标志[32],因此,将此类因子归为包括汽油车与柴油车的机动车尾气排放。第六类因子的优势组分为异戊二烯,城市中的异戊二烯大部分来源于植物活动排放,另外机动车尾气也会排放一部分排放,但该因子中与机动车尾气相关的其它VOCs物种贡献率都不高,因此将此类因子定义为植物排放源。
图7为PMF解析出6类VOCs排放源在观测期间对VOCs的相对贡献结果,可以看出,观测期间VOCs最大的排放源为LPG和溶剂挥发源(40.2%),其次分别为OVOCs源(17.8%)、机动车排放(17.4%)、工业排放(11.8%)、植物源(10.5%)和电厂排放(2.3%)。因此,控制燃烧源和工业溶剂排放是控制泰安市夏季VOCs污染的重要途径。
-
(1)观测期间VOCs浓度平均值为(16.57±7.99)×10−9,其中OVOCs占比最高为41.9%,烷烃占比为30.8%,芳香烃为19.5%,烯烃为7.8%。烷烃、芳香烃和OVOCs日变化趋势较为一致,整体呈现夜间高白天低的变化特征,而烯烃受到植物源排放异戊二烯的影响,在白天出现了显著抬升的变化特征。VOCs物种中平均浓度水平最高的前5种是甲醛、丙酮、丙烷、乙醛和丁烷。观测期间大气VOCs四大类别对OFP贡献率顺序为:芳香烃 > OVOCs > 烯烃 > 烷烃,其中甲醛、间/对-二甲苯、乙醛、异戊二烯和间二乙基苯是OFP水平最高的5种VOCs物种。
(2)观测点VOCs来源解析结果显示,观测期间泰安市VOCs最大的排放源为LPG和溶剂挥发(40.2%),其次分别为OVOCs源(17.8%)、机动车排放(17.4%)、工业排放(11.8%)、植物源(10.5%)和电厂排放(2.3%)。因此,控制LPG和溶剂挥发是控制泰安市夏季VOCs和O3污染的重要途径。
泰安市大气挥发性有机物污染特征及来源解析
Characteristics and source apportionment of ambient volatile organic compounds in Taian
-
摘要: 2018年夏季对泰安市城区站点的挥发性有机物(VOCs)进行监测,研究了其污染特征、臭氧生成潜势(OFP)和特征污染物比值,利用PMF源解析模型对VOCs的来源进行了解析。结果表明,观测期间泰安市VOCs体积分数平均值为(16.57±7.99)×10−9,VOCs中浓度占比最高的为OVOCs(41.9%),其次为烷烃(30.8%)、芳香烃(19.5%)和烯烃(7.8%),对OFP的贡献率最高的为芳香烃(35.6%),其次为OVOCs(35.5%)、烯烃(18.5%)和烷烃(10.5%);PMF来源解析结果显示,观测点VOCs最大的排放源为LPG和溶剂挥发(40.2%),其次分别为OVOCs源(17.8%)、机动车排放(17.4%)、工业排放(11.8%)、植物源(10.5%)和电厂排放(2.3%)。控制LPG和溶剂挥发是控制泰安市夏季VOCs污染的重要途径。
-
关键词:
- 挥发性有机物 /
- 污染特征 /
- 臭氧生成潜势(OFP) /
- 来源解析 /
- 泰安
Abstract: Ambient volatile organic compounds (VOCs) were monitored in Taian, Shandong Province, China in 2018. The VOC pollution characteristics, ozone generation potential (OFP), characteristic pollutants ratios and source apportionment conducted based on PMF were comprehensively analyzed. The results showed that the average concentration of VOCs was (16.57±7.99) ×10−9 during the observation and VOCs was dominated by OVOCs (41.9%), alkane (30.8%), aromatics (19.5%), alkene (7.8%). Aromatics, occupying 41.9%, made the greatest contribution to OFP, which were followed by OVOCs (35.5%), alkene (18.5%), alkane (10.5%). The source apportionment results of the PMF model showed six main sources of VOCs, namely LPG and solvent volatilization sources (40.2%), OVOCs sources (17.8%), motor vehicle emissions (17.4%), industrial emissions (11.8%), plant emissions (10.5%) and power plant emissions (2.3%). Overall, the concentration of ambient VOCs in Taian was strongly influenced by LPG and solvent volatilization, and the control of VOCs emitted from LPG and solvent volatilization should be strengthened to reduce the concentration of VOCs in Taian, further reducing the generation of ozone.-
Key words:
- volatile organic compounds(VOCs) /
- pollution characteristics /
- OFP /
- source apportionment /
- Taian
-
全氟和多氟类化合物(per-and polyfluoroalkyl substances, PFAS)是一类分子结构中烷基上的氢原子全部(全氟)或部分(多氟)被氟原子取代的脂肪族化合物[1],具有独特的理化性质。自20世纪50年代以来,PFAS被制造并广泛应用于许多军事、工业、民用领域[2]。全氟烷基酸(perfluoroalkyl acids, PFAAs),如全氟烷基羧酸(perfluoroalkyl carboxylic acids, PFCAs)和全氟烷基磺酸(perfluoroalkane sulfonic acids, PFSAs),是应用和研究最为广泛的一类PFAS,其中又以全氟辛基羧酸(perfluorooctanoic acid, PFOA)和全氟辛基磺酸(perfluorooctane sulfonic acid, PFOS)为典型。越来越多的研究表明了PFOA和PFOS具有持久性、生物累积性、毒性和长距离迁移的特性,因此这两种物质均已列入《关于持久性有机污染物斯德哥尔摩公约》的管制范围[3-4]。许多国家和国际组织已停止生产PFOA和PFOS,并不断寻求更安全、更环保的替代品[5-8]。六氟环氧丙烷二聚体(hexafluoropropylene oxide dimer acid, HFPO-DA,其铵盐的商品名为GenX),最初作为PFOA替代品而开发,但目前由于其可能对生态和人体健康产生严重影响而正在接受审查,如荷兰通过了将HFPO-DA,其盐和酰卤作为高度关注物质的提案,并得到了欧盟的支持。含8个碳和10个碳的氯代多氟烷基醚磺酸盐(potassium 9-chlorohexadecafluoro-3-oxanonane-1-sulfonate, 9Cl-PF3ONS和potassium 11-chloroeicosafluoro-3-oxaundecane-1-sulfonate, 11Cl-PF3OUdS,又称 6∶2 Cl-PFESA和8∶2 Cl-PFESA),是PFOS替代商品F53-B的主要和次要组分,用以替代PFOS作为铬雾抑制剂,其潜在的生态和人体健康影响也受到越来越多的关注。
环境固体介质对PFAS有较强的吸附性[9],使得土壤/大气颗粒物/沉积物/污泥等成为PFAS在环境中重要的汇,并可能成为其它环境介质中该类污染物的潜在来源。极限萃取(Exhaustive extraction)是环境样品中有机污染物分析的常用技术,即利用极性有机试剂一次或多次萃取样品中的目标化合物,萃取过程中通常会存在大量的基质干扰物质与目标化合物共萃取的现象。为了减少基质干扰,需要对萃取液进行净化。在土壤/大气颗粒物/沉积物/污泥等环境固体样品PFAS的萃取过程中,净化萃取液的常用手段是固相萃取(Solid phase extraction, SPE),即利用固体吸附剂将萃取液中的目标化合物吸附使其与干扰化合物分离,然后再用洗脱液洗脱,达到分离和富集目标化合物的目的。然而,繁琐的超声、振荡、离心、转移、浓缩、复溶、净化、干燥等过程非常耗时,使得基于SPE的极限萃取法样品制备所用时间占到整个污染物分析过程所用时间的75%以上[10],这往往构成分析技术的瓶颈[11]。繁琐的萃取、净化过程同时增大了目标污染物损失的可能性,使得方法的回收率不稳定。由于步骤繁琐,消耗了较多的有机试剂、超纯水、离心管、固相萃取柱、高纯氮气等,成本较高。为了提高萃取效率,快速分析环境中PFAS的赋存水平,进而探讨其风险和管控措施,亟需完善固体环境介质中PFAS的萃取方法,发展样品制备效率和成本效益较高的处理技术。
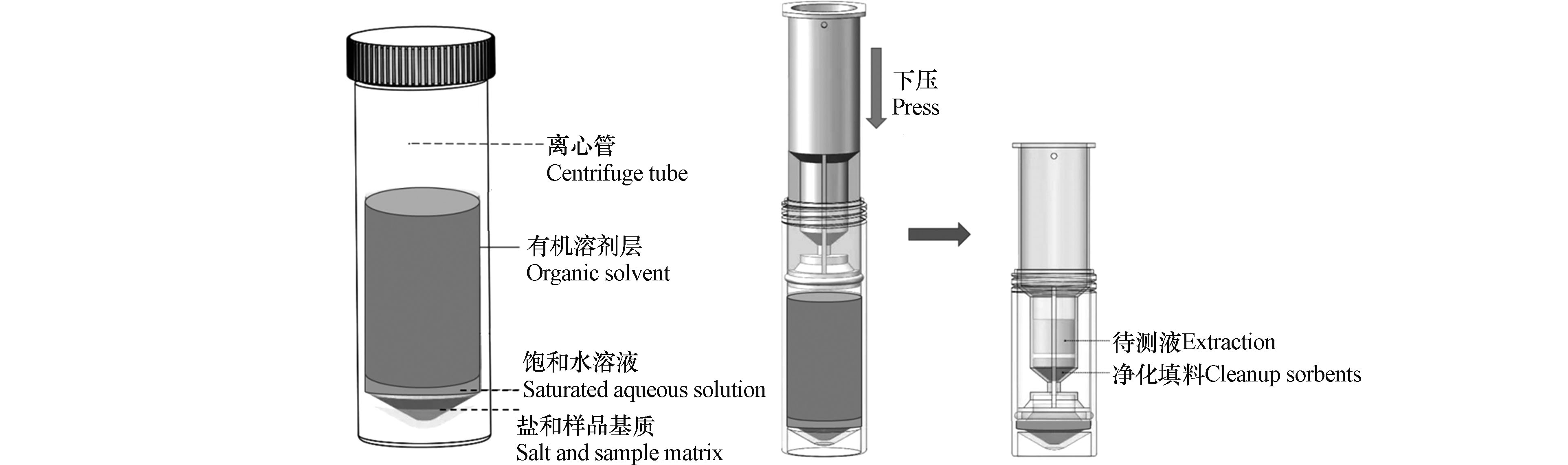
QuEChERS技术是由美国农业部在本世纪初提出的一种提取和净化农作物中农残的样品前处理方法[12],该技术利用乙腈提取目标化合物,之后采用硫酸镁等盐析分层,再加入吸附剂进行分散固相萃取,达到净化样品的目的。该技术具有快速(Quick)、简单(Easy)、便宜(Cheap)、有效(Effective)、可靠(Rugged)和安全(Safe)的特点,但存在稀释倍数大,重复性较差等问题[13]。SinCHERS技术是在SPE、QuEChERS等技术基础上开发出的一种新型快速样品制备技术,具有一步(Single–step)、经济(Cheap)、有效(Effective)、可靠(Rugged)、安全(Safe)的特点。基于SinCHERS技术的样品制备流程如图1所示。首先在碱性条件下利用有机试剂充分浸提固体样品中的目标化合物,之后向体系中加入酸调节体系pH值,振荡、离心,使得目标化合物在该体系内分配平衡,使得有机萃取液与其他组分之间分层良好。将SinCHERS前处理净化小柱(简称SinCHERS柱)置入离心管,缓慢下压,有机萃取液通过净化填料净化,并进入储液槽,取出槽内液体,浓缩、过滤后完成样品制备。相较于传统的固相萃取净化技术,SinCHERS技术节省了样品制备时间,减少了溶剂和各类器皿的使用量,并可以减少样品转移过程中目标化合物的损失,通常具有更好的稳定性和回收率。目前,SinCHERS技术已经成功应用于植物性样品中农药残留的测定[13-17]、动物性样品中兽药残留的测定以及纺织品中偶氮染料的测定[18]。
本文主要研究内容为SinCHERS技术在土壤/大气颗粒物/沉积物/污泥等固体样品PFAS检测中的应用,包括SinCHERS柱净化填料的优选和样品萃取净化流程的确定等,其目的在于提高土壤/大气颗粒物/沉积物/污泥等固体样品PFAS的萃取效率,缩短萃取时间,降低萃取成本,从而提高这些化合物的检测效率。
1. 材料与方法(Materials and methods)
1.1 试剂耗材
土壤/大气颗粒物/沉积物/污泥等固体样品;标准样品,含目标化合物PFAC-MXB (含全氟丁酸(perfluorobutanoic acid, PFBA)、全氟戊酸(perfluoropentanoic acid, PFPeA)、全氟己酸(perfluorohexanoic acid, PFHxA)、全氟庚酸(perfluoroheptanoic acid, PFHpA)、PFOA、全氟壬酸(perfluorononanoic acid, PFNA)、全氟癸酸(perfluorodecanoic acid, PFDA)、全氟十一酸(perfluoroundecanoic acid, PFUnDA)、全氟十二酸(perfluorododecanoic acid, PFDoDA)、全氟十三酸(perfluorotridecanoic acid, PFTrDA)、全氟十四酸(perfluorotetradecanoic acid, PFTeDA)、全氟十六酸(perfluorohexadecanoic acid, PFHxDA)、全氟十八酸(perfluorooctadecanoic acid, PFODA)、全氟丁烷磺酸(perfluorobutane sulfonic acid, PFBS)、全氟己烷磺酸(perfluorohexane sulfonic acid, PFHxS)、PFOS、全氟癸烷磺酸(perfluorodecane sulfonic acid, PFDS)), HFPO-DA, 9Cl-PF3ONS, 11Cl-PF3OUdS, 以及内标物质MPFAC-MXA (含MPFBA, MPFHxA, MPFOA, MPFNA, MPFDA, MPFUnDA, MPFDoDA, MPFHxS, MPFOS), M3HFPO-DA (Wellington 实验室, 加拿大);甲醇、乙腈(HPLC级,Fisher,美国);氢氧化钠(AR,麦克林,上海);盐酸(AR);乙酸铵(纯度≥98%, Sigma,美国);0.2 µm GHP针式滤器(PALL,美国);高纯氮(海科,北京);Milli-Q 超纯水(Millipore,美国);SinCHERS柱(Anybond,天津);ACQUITY BEH C18色谱柱(1.7 µm, 2.1 mm×100 mm, Waters, 美国)。
SinCHERS柱是天津安邦键合(Anybond)科技有限公司开发出的产品[19],其结构如图2所示。其中净化填料能够起到吸附有机萃取液内溶解的杂质,净化萃取液的目的,是SinCHERS柱的核心结构。填料内容需要根据目标污染物和样品所含杂质情况进行选择和配比。SinCHERS柱柱体采用医疗级聚丙烯材质,密封圈采用医疗级高级纯硅胶材质,可有效避免柱体应用带来的背景污染。
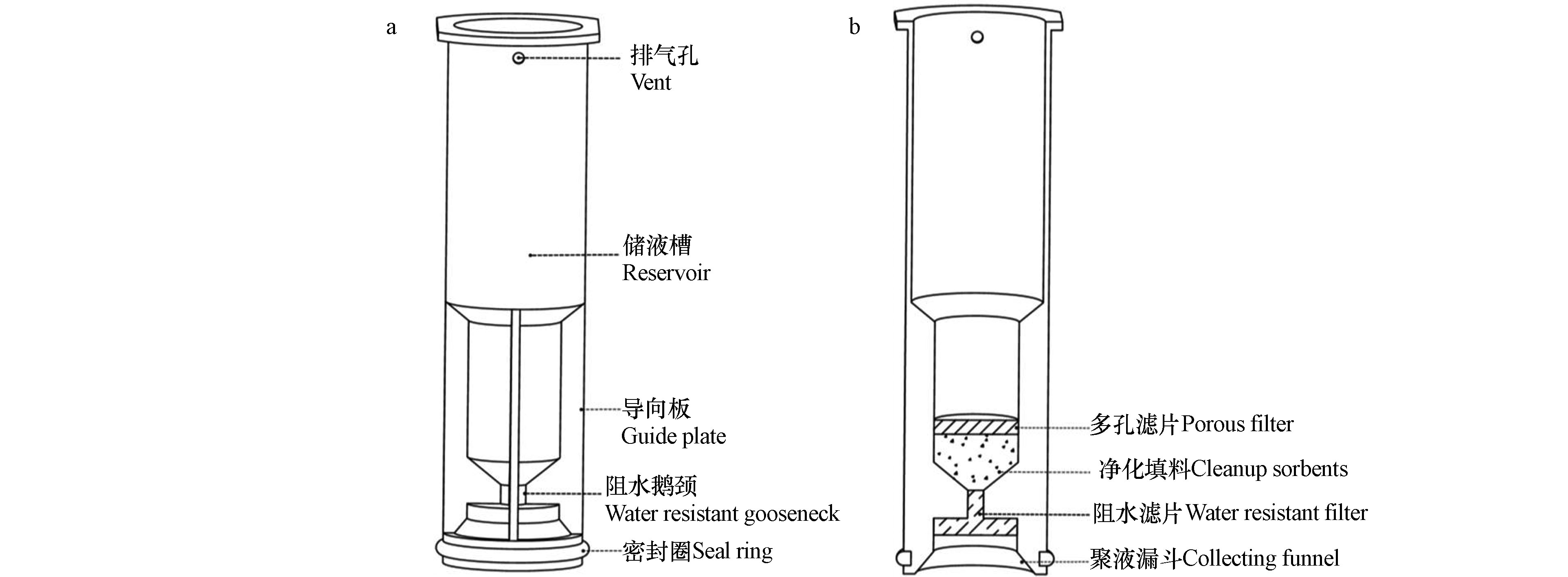 图 2 SinCHERS柱结构图(a:外观;b:内部结构)Figure 2. The structure of SinCHERS cartridge (a: external shape;b: internal structure).
图 2 SinCHERS柱结构图(a:外观;b:内部结构)Figure 2. The structure of SinCHERS cartridge (a: external shape;b: internal structure).1.2 仪器设备
Agilent 1290高效液相色谱/6460三重四极杆质谱系统(HPLC-MS/MS, Agilent,美国);电动辅助工具(Anybond,天津);分析天平;超声波清洗器;振荡器;离心机;氮吹仪。
1.3 样品制备与检测
土壤/大气颗粒物/沉积物/污泥等环境固体样品中PFAS萃取净化步骤如下:称取1—2 g干燥研磨后固体样品至50 mL离心管中,加入2—10 ng同位素标记的化合物,用于内标法定量。然后加入1 mL 0.5 mol·L−1氢氧化钠溶液和4 mL乙腈溶液,涡旋混匀后水浴条件下超声30 min。再加入16 mL乙腈,振荡30 min,使样品中的待测组分充分转移至有机萃取液中。之后加入250 μL 2 mol·L−1盐酸调节pH值,涡旋混匀,然后使用离心机5000r·min−1离心5 min。将SinCHERS柱置入离心管,缓慢下压,使上清液净化后进入储液槽,下压至SinCHERS柱体无法下行,净化过程完成。将净化液转移至另一离心管中,氮吹浓缩至近干,用甲醇或初始流动相比例的混合溶液定容至0.5—1 mL,将定容后的溶液过0.2 µm GHP针式滤器,上机分析检测。
色谱条件:流动相A组分为10 mmol·L−1乙酸铵水溶液,B组分为乙腈,采用梯度洗脱:0 min A组分80%,14 min A组分10%,16 min A组分10%,20 min A组分80%;流速0.3 mL·min−1;柱温为40 ℃;进样量为5 µL。
质谱条件:离子化模式为电喷雾离子化(ESI)负离子模式;数据采集模式为多反应检测扫描模式(Multi reaction monitoring, MRM);离子源温度350 ℃;载气流量9 L·min−1;雾化器压力40 psi;毛细管电压3500 V;ΔEMV电压400 V。所分析20种PFAS及10种内标物的质谱相关参数如表1所示。
表 1 20种PFAS目标化合物及10种内标物的质谱相关参数Table 1. MS conditions for 20 PFASs and 10 Internal standards目标化合物Compound 母离子(m/z)→子离子(m/z)Precursor ion (m/z)→Production (m/z) 裂解电压/VFragmentor 碰撞能量/eVCollision energy 内标Internal standard 目标化合物 PFBA 213.0 → 169.1 57 1 MPFBA PFPeA 263.0 → 218.9 68 2 MPFBA PFHxA 313.0 → 269.0 68 3 MPFHxA PFHpA 363.0 → 318.9 68 4 MPFOA PFOA 413.0 → 368.9 82 4 MPFOA PFNA 463.0 → 419.0 82 3 MPFNA PFDA 513.0 → 468.9 86 3 MPFDA PFUnDA 563.0 → 519.0 90 5 MPFUnDA 目标化合物 PFDoDA 613.0 → 569.0 90 5 MPFDoDA PFTrDA 662.9 → 619.0 80 5 MPFDoDA PFTeDA 713.1 → 669.0 100 7 MPFDoDA PFHxDA 813.0 → 769.0 100 9 MPFDoDA PFODA 913.0 → 869.0 118 10 MPFDA PFBS 299.0 → 80.0 135 32 MPFHxS PFHxS 399.0 → 80.0 150 40 MPFHxS PFOS 498.9 → 80.0 154 47 MPFOS PFDS 599.0 → 79.9 160 65 MPFOS HFPO-DA 329.0 → 169.0 49 5 M3HFPO-DA 9Cl-PF3ONS 530.8 → 350.9 120 22 MPFOS 11Cl-PF3OUdS 630.7 → 450.9 123 25 MPFOS 内标 MPFBA 217.0 → 172.0 57 1 MPFHxA 315.0 → 270.0 68 3 MPFOA 417.0 → 372.0 82 4 MPFNA 468.0 → 423.0 82 3 MPFDA 515.0 → 470.0 86 3 MPFUnDA 565.0 → 520.0 90 5 MPFDoDA 615.0 → 570.0 90 5 MPFHxS 403.0 → 103.0 150 40 MPFOS 503.0 → 80.0 154 47 M3HFPO-DA 331.2 → 169.0 44 5 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.4 质量保证与质量控制
将标准物质用甲醇配制混合标准溶液,标准化合物的质量浓度依次为0.01、0.05、0.1、0.5、1、5、10、50、100 ng·mL−1,内标物质的质量浓度均为5 ng·mL−1。以标准化合物浓度为横坐标,以标准化合物和内标的相对浓度为纵坐标,得到标准曲线。
PFAS的方法检出限(Method detection limit, MDL)可基于加标样品的检测结果,根据美国环保署提供的方法计算[20]:
MDL=t(n−1,1−α=0.99)SS 其中,
MDL t(n−1,1−α=0.99) SS 不添加土壤样品,用本方法全流程进行定量分析,得到该方法的程序空白,能够反应SinCHERS柱体、所使用试剂、器皿、管路等的背景污染情况。
取青海省海南藏族自治州共和县廿地乡草场土壤,将其烘干、均质化、研磨至100目后作为土壤基质,添加10 ng各目标化合物,按照本文“1.3”部分所述步骤进行PFAS的分析检测,扣除本底值后计算加标回收率。由于所分析样品的理化性质受到样品种类、采样时间、采样地点等多种因素的影响,不同样品间可能存在回收率的差异,将本文所描述方法应用于实际样品时,需进行预实验,并在必要时做出适度调整。
2. 结果与讨论(Results and discussion)
2.1 HPLC-MS/MS条件优化
本研究采用Waters ACQUITY BEH C18色谱柱(1.7 µm, 2.1 mm×100 mm)进行目标化合物的分离。对比了流动相A组分中不同浓度乙酸铵含量和不同流速对20种目标化合物的分离效果的影响。基于以2 mmol·L−1与10 mmol·L−1乙酸铵水溶液作为流动相A相时得到的色谱图,发现乙酸铵含量主要影响目标化合物的保留时间和响应强度,当乙酸铵含量为10 mmol·L−1时,物质间的分离效果更好,峰形对称性更好,而乙酸铵含量为2 mmol·L−1时,各物质的响应强度更高。基于流动相速度分别为0.25、0.3 mL·min−1时得到的色谱图,发现流速主要影响目标化合物的保留时间和峰形,较高流速时目标化合物的出峰时间更早,整体比低流速时出峰时间提前约0.5 min。较高流速时,峰形更好,表现在峰宽较窄,峰形较尖,峰高较高,因此改善了相邻物质的分离度,如PFTrDA和11Cl-PF3OUdS之间,PFUnDA和9Cl-PF3ONS之间的分离度。综合考虑不同条件下的分离效果,优选10 mmol·L−1乙酸铵水溶液作为流动相A相,优选0.3 mL·min−1为流动相流速,使目标化合物达到一个更好的分离检测效果。
许多研究已经对产品PFAC-MXB中17种PFAAs,以及产品MPFAC-MXA中9种同位素内标化合物的质谱条件进行了优化[21-22],本研究重点优化了HFPO-DA及其同位素内标M3HFPO-DA,9Cl-PF3ONS,以及11Cl-PF3OUdS的质谱参数。负离子模式下,首先通过全扫确定目标化合物的母离子,之后进行离子扫描优化裂解电压,再进行子离子扫描找到子离子并优化碰撞能,最后在多反应监测模式下选择响应更高的子离子作为定量子离子。优化后的参数见表1。
2.2 吸附填料优选
为有效净化有机萃取液中所含的杂质,使用无水硫酸镁、石墨化碳黑(Graphitized carbon black, GCB)、离子交换材料的混合物作为SinCHERS柱中的净化填料,三类填料的作用分别为:吸收萃取液中可能含有的微量水分、去除色素等杂质、去除其他干扰性离子化合物。为考察具有不同层间距的GCB和不同离子交换材料对SinCHERS柱净化效果的影响,在两个污染水平下(基质样品中额外添加目标化合物10 ng、50 ng)对比了使用不同层间距的GCB和不同离子交换填料时PFAS的回收率,每种处理设置了6个重复。
层间距不同,GCB的净化能力也不同。为了选择合适的层间距,将层间距分别为10、20、30 nm的GCB与无水硫酸镁和离子交换材料HLB混合后作为SinCHERS柱内的净化填料,在两种污染水平下对比了各PFAS的回收率(图3a, b)。结果表明,当加标水平为10 ng时,若使用层间距为10 nm或30 nm的GCB,PFODA的回收率下降到57%以下。在50 ng的加标水平下,所有20种PFAS的回收率均在可接受范围内(70%—130%)[23-24],但层间距为30 nm时,SinCHERS柱去除色素的能力降低,净化后基质的颜色更深。根据回收率和净化效果对比,优选层间距为20 nm的GCB作为净化填料。GCB能吸附非极性/弱极性化合物,层间距越小,净化效果越好。但过小的层间距条件下不可逆吸附更容易发生,特别是对大分子物质。这可能是使用层间距为10 nm的GCB 时PFODA回收率较低的原因。相反,层间距为30 nm的GCB,其净化色素和其他干扰物质的能力也会降低。
 图 3 不同组合净化填料条件下SinCHERS技术萃取PFAS回收率对比(n=6)Figure 3. Recoveries of PFAS by Sin-CHERS extraction approach with different sorbent combination at two spiked levels, n=6.
图 3 不同组合净化填料条件下SinCHERS技术萃取PFAS回收率对比(n=6)Figure 3. Recoveries of PFAS by Sin-CHERS extraction approach with different sorbent combination at two spiked levels, n=6.为了选择合适的离子交换材料,对比了HLB、SAX、WCX 3种离子交换填料,分别将其与层间距为20 nm的GCB、无水硫酸镁混合后作为SinCHERS柱内的净化填料,在两种污染水平下对比PFAS的回收率(图3c, d)。结果表明,当使用HLB时,20种PFAS在两种加标水平下的平均回收率分别为71.0%—110.6%和74.0%—127%。而使用SAX和WCX时,无论在什么加标水平下,PFODA的回收率都下降到63%以下。在10 ng的加标水平下,使用HLB时,有12种PFAS的回收率更稳定,即回收率的标准方差低于另外两种填料。当使用SAX和WCX时,分别有4种PFAS的回收率的标准方差低于另外两种填料。HLB作为一种弱阴离子交换材料,在乙腈溶液中能吸收比PFAS酸性更强的阴离子化合物,降低了负离子模式下杂质阴离子对质谱的干扰。SAX作为一种强阴离子交换材料,对PFAS的吸附能力比HLB强。这可能是使用SAX时,PFODA回收率较低的原因。而阳离子交换材料WCX对干扰性阴离子化合物没有净化作用。
综合考虑不同填料组合条件下目标化合物的回收率范围和稳定性,优选层间距为200 Å的GCB、HLB与无水硫酸镁组合作为SinCHERS柱中的净化填料,使目标化合物同时具有较好的回收率和检测稳定性。
2.3 方法验证
用甲醇配制不同浓度梯度的标准物质混合标准溶液,拟合得到标准曲线的R2值均在0.99以上,线性范围在0.01—100 ng·mL−1,对应环境固体样品的可检测范围为0.005—50 ng·g−1 dw。
浓度为100 ng·mL−1的20种目标化合物的MRM色谱图如图4所示。可以发现,在优化的条件下,HPLC-MS/MS可以实现20种PFAS的分离。
 图 4 浓度为100 ng·mL−1时20种PFAS MRM色谱图Figure 4. Chromatogram of 20 PFAS by MRM with the concentration of 100 ng·mL−1设PFDoDA的响应强度为100%(the response of PFDoDA was set to 100%)
图 4 浓度为100 ng·mL−1时20种PFAS MRM色谱图Figure 4. Chromatogram of 20 PFAS by MRM with the concentration of 100 ng·mL−1设PFDoDA的响应强度为100%(the response of PFDoDA was set to 100%)基于该方法得到20种目标化合物的方法检出限见表2。PFAS的检出限范围0.46—2.31 ng·g-1 dw。当不添加土壤样品时,得到该方法程序空白,各目标化合物含量均低于检出限。
表 2 20种PFAS目标化合物的方法检出限(MDL, n=10)Table 2. Method detection limit (MDL) of 20 PFASs目标化合物Compound 方法检出限/(ng·g−1 dw)MDL 目标化合物Compound 方法检出限/ (ng·g−1 dw)MDL PFBA 0.95 PFTeDA 1.03 PFPeA 0.75 PFHxDA 1.44 PFHxA 0.97 PFODA 1.07 PFHpA 0.70 PFBS 0.80 PFOA 0.48 PFHxS 1.27 PFNA 0.64 PFOS 0.58 PFDA 0.75 PFDS 0.46 PFUnDA 0.62 HFPO-DA 2.31 PFDoDA 1.06 9Cl-PF3ONS 0.55 PFTrDA 0.95 11Cl-PF3OUdS 0.68 | Show TableDownLoad:
CSV
基于该方法得到20种PFAS的方法回收率如表3所示。其中,PFODA和PFBS的方法回收率为71.0%和78.4%,其余PFAS的回收率范围为84.1%—110.6%。该方法PFOA、PFODA、PFDS的回收率的标准偏差最小,为3.1%、3%、2.7%。其余PFAS的标准偏差在3.8%—13.4%之间。
表 3 20种PFAS目标化合物的方法回收率(平均值±标准偏差), n=6Table 3. Method recoveries of 20 PFASs (average±sd), n=6.目标化合物Compound 方法回收率/%Recovery rate 目标化合物Compound 方法回收率/%Recovery rate PFBA 110.2±6.9 PFTeDA 88±6.6 PFPeA 103.2±5.5 PFHxDA 95.3±10.1 PFHxA 98±6.6 PFODA 71.0±3 PFHpA 84.1±6.2 PFBS 78.4±7.7 PFOA 107.7±3.1 PFHxS 100.3±10.8 PFNA 110.6±3.8 PFOS 108.3±6.1 PFDA 99.2±4.7 PFDS 96.1±2.7 PFUnDA 103.8±5.5 HFPO-DA 91.3±13.4 PFDoDA 104.1±7.6 9Cl-PF3ONS 89.7±3.8 PFTrDA 101.5±5.9 11Cl-PF3OUdS 93.8±5.3 | Show TableDownLoad:
CSV
2.4 方法对比
再次利用青海省土壤基质样品(本文“1.4”部分),同时应用本方法和SPE样品前处理过程[22, 25-28]对样品进行处理。当在基质样品中额外添加10 ng目标化合物时,两种技术在回收率上的差异如图5所示,对于大部分PFAS,即碳链长度为4—14的全氟羧酸化合物,碳链长度为6和8的全氟磺酸化合物,HFPO-DA,以及11Cl-PF3OUdS,两种技术的回收率均在80%—120%之间。对于碳链长度为16和18的全氟羧酸化合物,即PFHxDA和PFODA,传统SPE技术的回收率仅为20%和1%,而SinCHERS技术的回收率为95%和71%,增大了可检测污染物的种类。对于PFDS和9Cl-PF3ONS,传统SPE技术的回收率为77%和79%,SinCHERS技术的回收率为96%和90%,回收效率显著提高。对于全氟丁烷磺酸来说,SinCHERS技术的回收率78%低于SPE技术102%,但仍可用于环境样品中该物质的检测定量[29]。
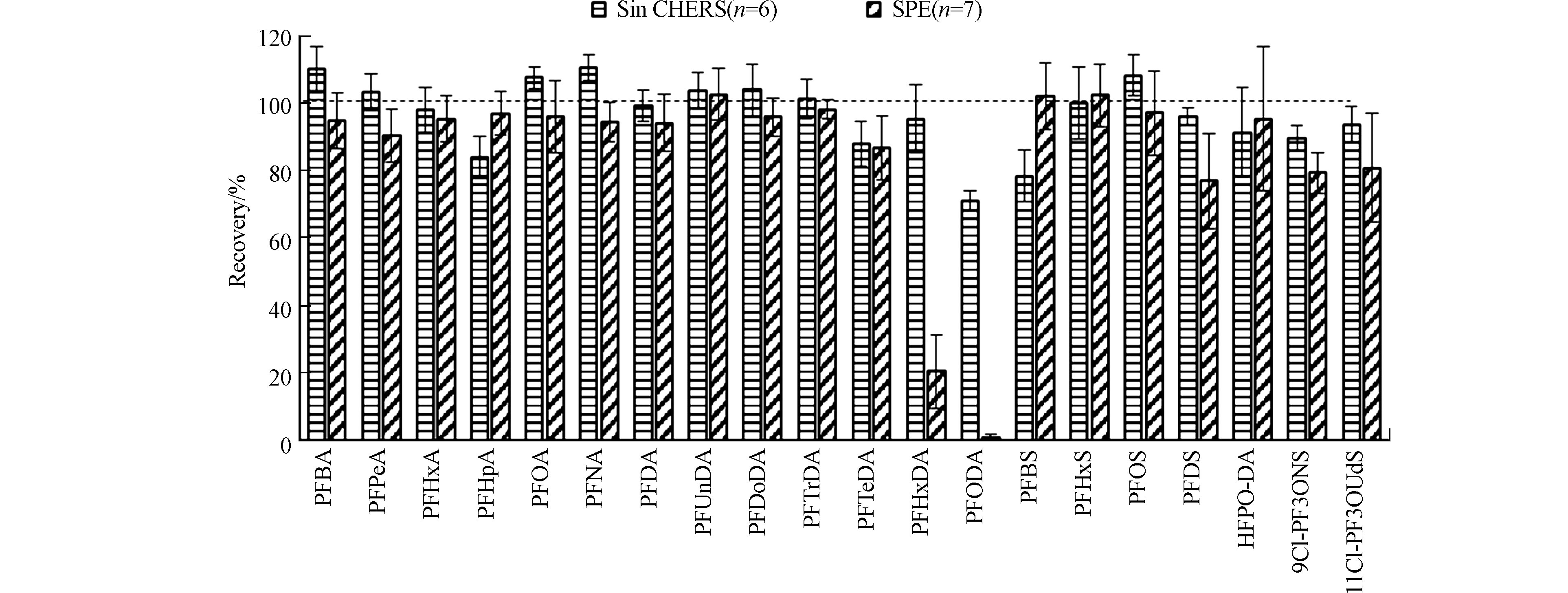 图 5 SinCHERS技术与SPE技术目标化合物回收率的对比Figure 5. Comparison of SinCHERS technology and SPE technology based on method recoveries of target compounds
图 5 SinCHERS技术与SPE技术目标化合物回收率的对比Figure 5. Comparison of SinCHERS technology and SPE technology based on method recoveries of target compounds对于大部分PFAS,即碳链长度为4—11,14,16的全氟羧酸化合物,碳链长度为4、8、10的全氟磺酸化合物,HFPO-DA,9Cl-PF3ONS,以及11Cl-PF3OUdS,SinCHERS技术回收率的方差低于传统的SPE技术,说明对于大部分PFAS,该方法具有更好的稳定性。同时,SinCHERS技术在步骤上更为简洁,可节约大量的试剂耗材,且具有更高的时效性。
2.5 方法应用
为验证本方法对PFAS提取效果的稳定性和适用性,2020年7月,采集了黄河流域河套灌区大棚和露天条件下共67个农田土壤样品,应用本方法进行了PFAS样品加标回收率和实际样品浓度的测定,分析结果如表4所示。其中,PFODA的方法回收率为71.9%,其余PFAS的回收率范围为87.0%—110.5%。PFPeA、PFOA、PFODA的标准偏差最小,为2.9%、2.4%、2.1%,其余PFAS的标准偏差在3.5%—12.1%之间。67份土壤样品中,20种PFAS的总浓度
∑ ∑ 表 4 河套灌区土壤20种PFAS目标化合物的样品加标回收率(平均值±标准偏差, n=6)和浓度(范围)Table 4. Spiked recoveries (average ± sd, n=6) and concentrations (range) of 20 PFASs in soils from Hetao Irrigation District目标化合物Compound 浓度/(ng·g−1 dw)Concentration 加标回收率/%Spiked recovery 目标化合物Compound 浓度/(ng·g−1 dw)Concentration 加标回收率/%Spiked recovery PFBA <MDL—5.21 104.6±5.2 PFTeDA <MDL 107.7±6.9 PFPeA <MDL—3.09 87.0±2.9 PFHxDA <MDL 98.4±7.6 PFHxA <MDL 98.4±3.9 PFODA <MDL 71.9±2.1 PFHpA <MDL 88.1±3.5 PFBS <MDL 90.0±9.7 PFOA <MDL—2.62 99.6±2.4 PFHxS <MDL 92.5±10.9 PFNA <MDL—0.92 91.4±3.7 PFOS <MDL 110.5±7.3 PFDA <MDL—5.63 94.0±5.5 PFDS <MDL 100.7±4.5 PFUnDA <MDL 103.7±5.2 HFPO-DA <MDL 93.1±12.1 PFDoDA <MDL—1.23 99.4±4.3 9Cl-PF3ONS <MDL 102.5±5.3 PFTrDA <MDL 99.6±5.0 11Cl-PF3OUdS <MDL 106.7±5.7 | Show TableDownLoad:
CSV
通过对比大棚和露天条件下的污染特征可以发现,多数样点的
∑ 3. 结论(Conclusion)
该方法建立了基于SinCHERS-HPLC-MS/MS技术的环境固体样品中20种PFAS的萃取、净化和检测方法。优选出了无水硫酸镁、层间距为20 nm的GCB、HLB离子交换材料的组合作为SinCHERS柱内的净化填料,并优化了HPLC-MS/MS条件以分离并定量检测20种目标化合物。目标化合物的方法回收率在71.0%—110.6%之间。相较于SPE样品前处理流程,该方法大大缩短了样品中污染物萃取净化的时间,降低了经济成本,优化了某些长链PFAS的萃取效率。
-
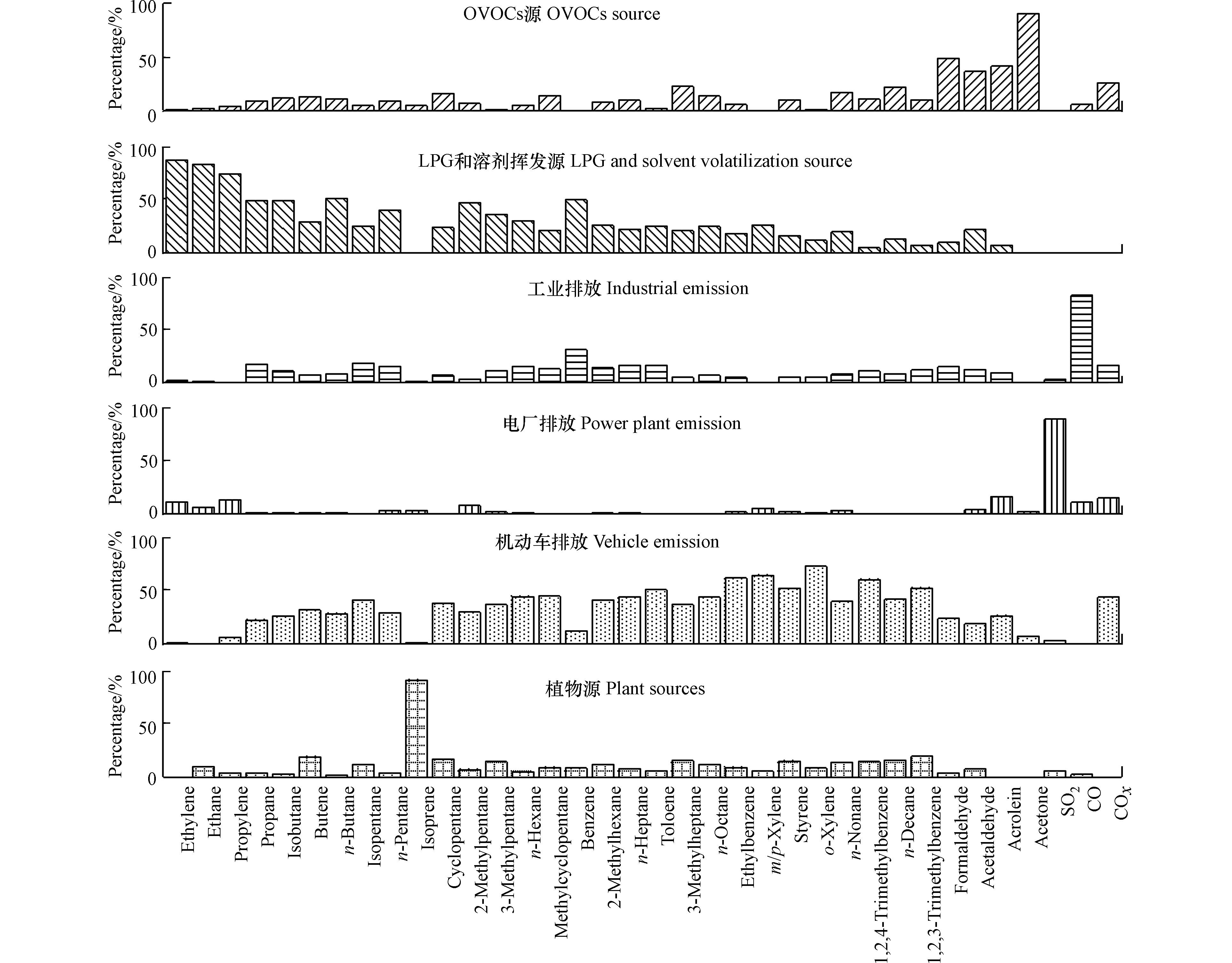
图 6 观测期间大气VOCs源成分谱图
Figure 6. Source composition spectrum of VOCs species during observation period
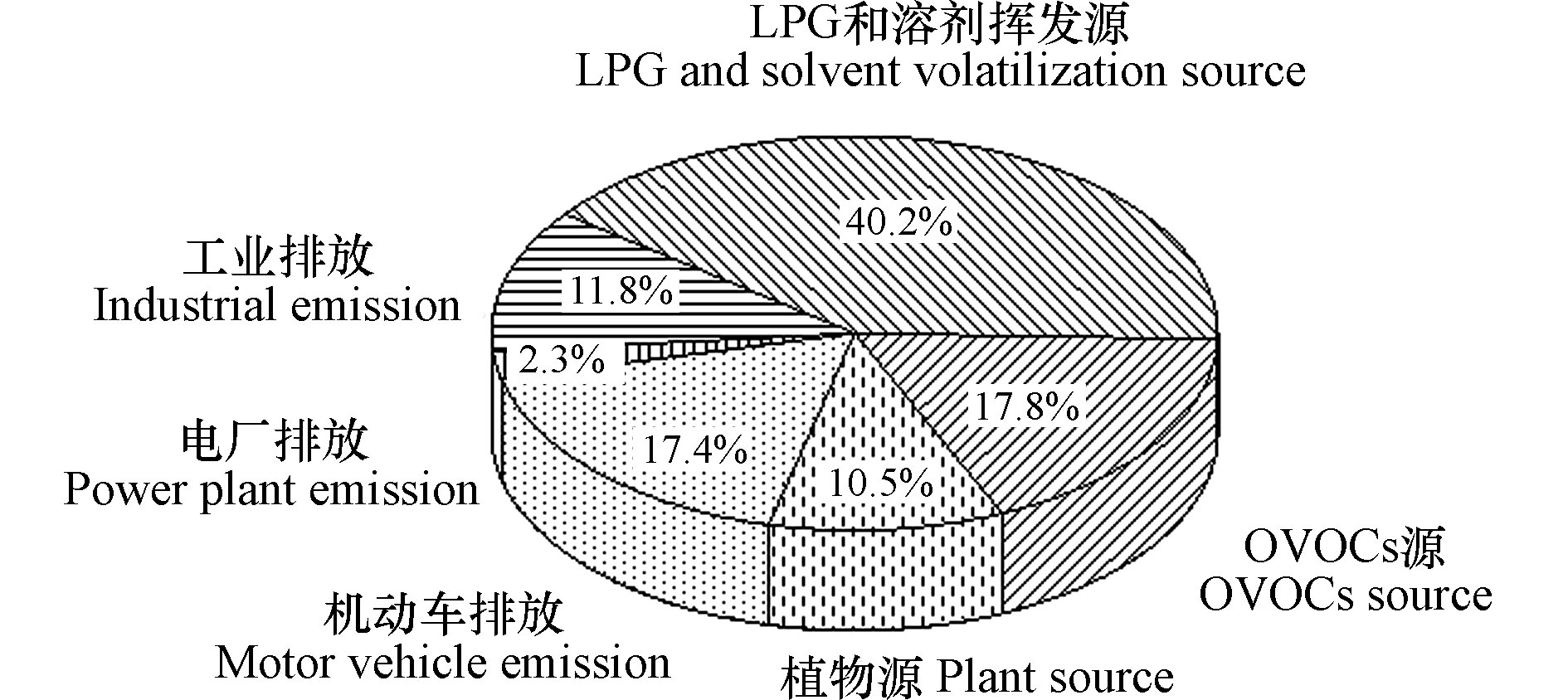
图 7 观测期间不同源排放对VOCs的贡献率
Figure 7. Contribution rate of emissions from different sources to VOCs during the observation period
表 1 观测期间主要VOCs物种的浓度和OFP值
Table 1. Concentrations and OFP of main VOCs species during the observation period
序号Serial number 平均浓度Average concentration 臭氧生成潜势OFP 组分Species 数值(×10−9)Value 组分Species 数值/(μg·m−3)Value 1 甲醛 3.18±2.09 甲醛 40.34±26.52 2 丙酮 2.02±1.27 间/对-二甲苯 20.51±14.96 3 丙烷 1.71±1.41 乙醛 17.87±7.90 4 乙醛 1.39±0.61 异戊二烯 15.44±14.10 5 丁烷 0.92±0.88 间二乙基苯 14.59±11.71 6 甲苯 0.88±0.67 甲苯 14.52±10.96 7 异戊烷 0.81±0.43 戊醛 12.10±8.01 8 苯乙烯 0.72±1.13 正戊烯 11.90±7.36 9 戊醛 0.62±0.41 丁醛 10.74±2.46 10 苯 0.60±0.39 间-甲基苯甲醛 10.01±7.20 11 丁醛 0.56±0.13 邻-二甲苯 8.54±8.36 12 正戊烯 0.53±0.33 1,2,4-三甲基苯 7.86±4.77 13 异丁烷 0.50±0.44 1,3,5-三甲基苯 7.77±6.62 14 异戊二烯 0.48±0.44 对二乙基苯 7.76±5.27 15 丁烯醛 0.47±0.33 反-2-戊烯 6.15±3.47 16 乙苯 0.33±0.30 丙烯 5.83±5.59 17 十二烷 0.32±0.13 苯乙烯 5.77±9.12 18 戊烷 0.31±0.27 顺-2-丁烯 5.62±3.80 19 间/对-二甲苯 0.28±0.20 苯甲醛 5.28±2.75 20 丙醛 0.27±0.14 丙醛 5.02±2.57
下载: 导出CSV
-
[1] WANG T, WEI X L, DING A J, et al. Increasing surface ozone concentrations in the background atmosphere of Southern China, 1994—2007 [J]. Atmospheric Chemistry and Physics, 2009, 9(16): 6217-6227. doi: 10.5194/acp-9-6217-2009 [2] SUN L, XUE L K, WANG T, et al. Significant increase of summertime ozone at Mount Tai in Central Eastern China [J]. Atmospheric Chemistry and Physics, 2016, 16(16): 10637-10650. doi: 10.5194/acp-16-10637-2016 [3] ZHANG Q, YUAN B, SHAO M, et al. Variations of ground-level O3 and its precursors in Beijing in summertime between 2005 and 2011 [J]. Atmospheric Chemistry and Physics, 2014, 14(12): 6089-6101. doi: 10.5194/acp-14-6089-2014 [4] ATKINSON R, AREY J. Atmospheric degradation of volatile organic compounds [J]. Chemical Reviews, 2003, 103(12): 4605-4638. doi: 10.1021/cr0206420 [5] ZHU Y H, YANG L X, CHEN J M, et al. Characteristics of ambient volatile organic compounds and the influence of biomass burning at a rural site in Northern China during summer 2013 [J]. Atmospheric Environment, 2016, 124: 156-165. doi: 10.1016/j.atmosenv.2015.08.097 [6] DENG Y Y, LI J, LI Y Q, et al. Characteristics of volatile organic compounds, NO2, and effects on ozone formation at a site with high ozone level in Chengdu [J]. Journal of Environmental Sciences, 2019, 75: 334-345. doi: 10.1016/j.jes.2018.05.004 [7] XU Z N, HUANG X, NIE W, et al. Influence of synoptic condition and holiday effects on VOCs and ozone production in the Yangtze River Delta region, China [J]. Atmospheric Environment, 2017, 168: 112-124. doi: 10.1016/j.atmosenv.2017.08.035 [8] CAI C J, GENG F H, TIE X X, et al. Characteristics and source apportionment of VOCs measured in Shanghai, China [J]. Atmospheric Environment, 2010, 44(38): 5005-5014. doi: 10.1016/j.atmosenv.2010.07.059 [9] 罗玮, 王伯光, 刘舒乐, 等. 广州大气挥发性有机物的臭氧生成潜势及来源研究 [J]. 环境科学与技术, 2011, 34(5): 80-86. doi: 10.3969/j.issn.1003-6504.2011.05.019 LUO W, WANG B G, LIU S L, et al. VOC ozone formation potential and emission sources in the atmosphere of Guangzhou [J]. Environmental Science & Technology, 2011, 34(5): 80-86(in Chinese). doi: 10.3969/j.issn.1003-6504.2011.05.019
[10] 邹宇, 邓雪娇, 王伯光, 等. 广州番禺大气成分站挥发性有机物的污染特征 [J]. 中国环境科学, 2013, 33(5): 808-813. doi: 10.3969/j.issn.1000-6923.2013.05.006 ZOU Y, DENG X J, WANG B G, et al. Pollution characteristics of volatile organic compounds in Panyu composition station [J]. China Environmental Science, 2013, 33(5): 808-813(in Chinese). doi: 10.3969/j.issn.1000-6923.2013.05.006
[11] LI L Y, XIE S D, ZENG L M, et al. Characteristics of volatile organic compounds and their role in ground-level ozone formation in the Beijing-Tianjin-Hebei region, China [J]. Atmospheric Environment, 2015, 113: 247-254. doi: 10.1016/j.atmosenv.2015.05.021 [12] WU R R, LI J, HAO Y F, et al. Evolution process and sources of ambient volatile organic compounds during a severe haze event in Beijing, China [J]. Science of the Total Environment, 2016, 560/561: 62-72. doi: 10.1016/j.scitotenv.2016.04.030 [13] 刘泽常, 李娜, 侯鲁健, 等. 济南市环境空气VOCs污染特征及来源识别 [J]. 中国环境监测, 2014, 30(6): 83-88. doi: 10.3969/j.issn.1002-6002.2014.06.014 LIU Z C, LI N, HOU L J, et al. Pollution characteristics and source identification of VOCs in ambient air of Ji'nan [J]. Environmental Monitoring in China, 2014, 30(6): 83-88(in Chinese). doi: 10.3969/j.issn.1002-6002.2014.06.014
[14] 薛莲, 王静, 冯静, 等. 青岛市环境空气中VOCs的污染特征及化学反应活性 [J]. 环境监测管理与技术, 2015, 27(2): 26-30. doi: 10.3969/j.issn.1006-2009.2015.02.007 XUE L, WANG J, FENG J, et al. Pollution characteristics and chemical reactivity of ambient VOCs in Qingdao [J]. The Administration and Technique of Environmental Monitoring, 2015, 27(2): 26-30(in Chinese). doi: 10.3969/j.issn.1006-2009.2015.02.007
[15] 张桢超. 某典型沿海城市VOCs源清单的建立及其来源解析研究[D]. 济南: 山东师范大学, 2019. ZHANG Z C. Establishment and source analysis of VOCs source list in A typical coastal city[D]. Jinan: Shandong Normal University, 2019(in Chinese).
[16] 李凯, 刘敏, 梅如波. 泰安市大气臭氧污染特征及敏感性分析 [J]. 环境科学, 2020, 41(8): 3539-3546. LI K, LIU M, MEI R B. Pollution characteristics and sensitivity analysis of atmospheric ozone in Taian city [J]. Environmental Science, 2020, 41(8): 3539-3546(in Chinese).
[17] LIU C T, MU Y J, ZHANG C L, et al. Development of gas chromatography-flame ionization detection system with a single column and liquid nitrogen-free for measuring atmospheric C2—C12 hydrocarbons [J]. Journal of Chromatography A, 2016, 1427: 134-141. doi: 10.1016/j.chroma.2015.11.060 [18] 杨帆. 长治市环境空气中挥发性有机物特征及来源研究[D]. 北京: 华北电力大学(北京), 2019. YANG F. Characteristics and sources apportionment of ambient volatile organic compounds in Changzhi[D]. Beijing: North China Electric Power University, 2019(in Chinese).
[19] CARTER W P L. Development of ozone reactivity scales for volatile organic compounds [J]. Air & Waste, 1994, 44(7): 881-899. [20] 任义君, 马双良, 王思维, 等. 郑州市春季大气污染过程VOCs特征、臭氧生成潜势及源解析 [J]. 环境科学, 2020, 41(6): 2577-2585. REN Y J, MA S L, WANG S W, et al. Ambient VOCs characteristics, ozone formation potential, and source apportionment of air pollution in spring in Zhengzhou [J]. Environmental Science, 2020, 41(6): 2577-2585(in Chinese).
[21] BROWN S G, FRANKEL A, HAFNER H R. Source apportionment of VOCs in the Los Angeles area using positive matrix factorization [J]. Atmospheric Environment, 2007, 41(2): 227-237. doi: 10.1016/j.atmosenv.2006.08.021 [22] 杨健. 安阳市城区臭氧污染特征及影响因素研究[D]. 郑州: 郑州大学, 2020. YANG J. Study on the characteristics and influencing factors of ozone pollution in Anyang city[D]. Zhengzhou: Zhengzhou University, 2020(in Chinese).
[23] GAO J, ZHANG J, LI H, et al. Comparative study of volatile organic compounds in ambient air using observed mixing ratios and initial mixing ratios taking chemical loss into account - A case study in a typical urban area in Beijing [J]. Science of the Total Environment, 2018, 628/629: 791-804. doi: 10.1016/j.scitotenv.2018.01.175 [24] 刘成堂. 大气中挥发性有机物的检测技术及其应用[D]. 北京: 中国科学院大学, 2016. LIU C T. Detection technology and application of volatile organic compounds in the atmosphere[D]. Beijing: University of Chinese Academy of Sciences, 2016(in Chinese).
[25] 张敬巧, 吴亚君, 李慧, 等. 廊坊开发区秋季VOCs污染特征及来源解析 [J]. 中国环境科学, 2019, 39(8): 3186-3192. doi: 10.3969/j.issn.1000-6923.2019.08.007 ZHANG J Q, WU Y J, LI H, et al. Characteristics and source apportionment of ambient volatile organic compounds in autumn in Langfang development zones [J]. China Environmental Science, 2019, 39(8): 3186-3192(in Chinese). doi: 10.3969/j.issn.1000-6923.2019.08.007
[26] 贾双庆, 周速, 程远. 新乡市环境空气中挥发性有机物(VOCs)污染特征及来源解析 [J]. 中国环境管理干部学院学报, 2019, 29(3): 68-71,76. JIA S Q, ZHOU S, CHENG Y. Characteristics and sources apportionment of volatile organic compounds(VOCs) in Xinxiang [J]. Journal of Environmental Management College of China, 2019, 29(3): 68-71,76(in Chinese).
[27] 赵乐, 刘新军, 范莉茹, 等. 石家庄夏季典型时段臭氧污染特征及来源解析 [J]. 中国环境监测, 2019, 35(4): 78-84. ZHAO L, LIU X J, FAN L R, et al. Pollution characteristic and source apportionment of VOCs during summer typical periods in Shijiazhuang [J]. Environmental Monitoring in China, 2019, 35(4): 78-84(in Chinese).
[28] ZHENG H, KONG S F, XING X L, et al. Monitoring of volatile organic compounds (VOCs) from an oil and gas station in northwest China for 1 year [J]. Atmospheric Chemistry and Physics, 2018, 18(7): 4567-4595. doi: 10.5194/acp-18-4567-2018 [29] HSIEH L T, YANG H H, CHEN H W. Ambient BTEX and MTBE in the neighborhoods of different industrial Parks in Southern Taiwan [J]. Journal of Hazardous Materials, 2006, 128(2/3): 106-115. [30] YAN Y L, PENG L, LI R M, et al. Concentration, ozone formation potential and source analysis of volatile organic compounds (VOCs) in a thermal power station centralized area: A study in Shuozhou, China [J]. Environmental Pollution, 2017, 223: 295-304. doi: 10.1016/j.envpol.2017.01.026 [31] 张玉欣, 安俊琳, 林旭, 等. 南京北郊冬季挥发性有机物来源解析及苯系物健康评估 [J]. 环境科学, 2017, 38(1): 1-12. doi: 10.21608/jes.2017.19095 ZHANG Y X, AN J L, LIN X, et al. Source apportionment of volatile organic compounds and health assessment of benzene series in northern suburb of Nanjing in winter [J]. Environmental Science, 2017, 38(1): 1-12(in Chinese). doi: 10.21608/jes.2017.19095
[32] FUJITA E M. Hydrocarbon source apportionment for the 1996 Paso del Norte Ozone Study [J]. Science of the Total Environment, 2001, 276(1/2/3): 171-184. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4327
- HTML全文浏览数: 4327
- PDF下载数: 172
- 施引文献: 0
