

-
土壤有机质(SOM)作为土壤的一个重要组成部分,它控制着进入土壤环境中有机污染物的去向及其迁移转化[1-2],而不同来源及形成年代的土壤有机质常表现出化学组成和物理结构的差异[3],因而对土壤中有机污染物的影响各异[4-6]。活性污泥是源于市政污水处理的一种絮凝物,我国每年产生的污泥量可达6000万吨左右,其有机质含量通常介于6.5%—48%,且生物源组分占到其总量的70%以上[7];泥炭土广泛分布于我国云贵高原及东北部地区,其有机质含量可达10%—60%,且具有含水率高、密度小和孔隙比大等特点[8]。活性污泥主要靠微生物活动降解污水中的有机质,而泥炭土则是依靠大量的生物质堆积腐熟,两者的有机质构成就有所差异。
近年来的研究表明,土壤有机质组分一直处在动态更替中,分子组成的异质性成为土壤有机质的普遍特征[4]。13C核磁共振、红外荧光等传统技术手段往往不能精确地区分土壤有机质组分差异。而生物标志物分子鉴别技术为探索土壤有机质异质性提供了新的途径[9]。分子生物标志物在环境中能够很好的保留其母源有机质的碳骨架分子信息,因此,采用分子生物标志物表征土壤有机质组成时,可利用一些特征参数指示有机组分的来源与降解程度[10]。而去除与土壤有机质结合的活性矿物时,一部分生物标志物被释放,可能使这些特征参数更准确地用于指示其来源[11-12]。
本研究以主要是微生物来源的活性污泥和植物性来源的滇池泥炭土两种材料为研究对象,分别进行水处理和酸处理,进而实验观察对憎水性有机污染物菲(PHE)和离子型有机污染物双酚A(BPA)的吸附特征,采用分子生物标志物技术结合元素分析和比表面积测定,探讨不同来源有机质分子组成的差异和活性矿物对于游离态脂和木质素酚类的保护机制,期望更好地理解有机污染物与SOM组分的非均质相互作用。
-
本实验采用不同性质的两种土壤,分别是来自滇池周边的泥炭土和昆明市政活性污泥,采取土壤后经自然风干,研磨并过100目筛,人工除去肉眼可见植物残体。为去除活性矿物,用10%氢氟酸,以1∶4(W∶V)比例混合,在摇床内振荡2 h,随后以2500 r·min−1离心30 min,去除上清液,重复6次后,用UP水洗至中性,收集固体并冷干研磨过100目筛备用,此过程收集到活性污泥去矿(HA)及泥炭土去矿(PA)两种样品。原始土样用UP水洗作为对照得到活性污泥水洗(HW)及泥炭土水洗(PW)。
-
采用元素分析(MicroCube, Elementar)和BET分析仪(Autosorbe1C, Quantachrome)的表征手段对两种土壤及其酸处理前后的元素组成以及比表面积进行表征。
-
500 mg土样设置2个平行,依次用40 mL的甲醇、甲醇:二氯甲烷(1:1, V/V)、二氯甲烷3种有机溶剂,在水浴式超声机中以520 W功率超声15 min,每萃取1次,在2500 r·min−1下离心20 min,上清液过玻璃纤维膜(Whatman GF/A 1.6 μm)保存,残余物在相同条件下用下一种溶剂进行萃取,收集3次过膜滤液,旋转蒸发浓缩后转移至2 mL玻璃样品瓶内,氮气吹干等待下一步衍生化。3次萃取后的土壤残余物风干后备用。
-
游离态脂萃取过的残余物风干后,称取400 mg土壤样品、1 g的CuO、100 mg的Fe(NH4)2(SO4)2·6H2O加入到20 mL高压反应釜内,加入15 mL事先经过氮吹除氧的2 mol·L−1 NaOH,在盖上反应釜盖子之前先用氮气吹除掉顶部空气,后在170 ℃下反应2 h,反应釜冷却后加100 μL乙基香兰素作为内标,上清液倒入离心管内,残余固体加入10 mL的UP水用磁力转子搅拌10 min导入离心管混合,重复两次后以2500 r·min−1离心20 min,转移上清液并用6 mol·L−1的HCl调制pH值为0.8—1,暗反应1 h,离心后取上清液用50 mL乙酸乙酯进行液液萃取,萃取液经旋转蒸发浓缩后转移至2 mL玻璃样品瓶中,氮气吹干后备用,每个样品设置2个平行。
-
在上述游离态脂及木质素样品瓶中加入10 μL的无水吡啶和90 μL的BSTFA,70 ℃下反应3 h进行硅烷化取代。两种生物标志物均采用GC-MS(Agilent, 7890A)进行测定,采用DB-5MS色谱柱分离,其参数为: 65 ℃保持2 min,随后以 6 ℃·min−1升至300 ℃并保持20 min,2∶1的分流比,氦气载流,280 ℃进样。其中,正十七烷酸、麦角固醇、正二十五烷和葡萄糖作为游离态脂的外标,木质素则以其单体作为外标进行定量分析。
-
本实验选取了两种憎水性有机污染物PHE和BPA(pH=7条件下主导形态为显正电分子[13]),根据预备实验,设置了11个浓度梯度,分别是PHE: 0.01—1 mg·L−1及BPA∶1—16 mg·L−1,污染物溶于含有100 mg·L−1的叠氮化钠和0.02 mol·L−1的氯化钠背景溶液中,并稀释至不同浓度。在8 mL带有聚四氟乙烯隔垫的棕色液相瓶中进行实验,固液比分别为8000∶0.5—8000∶4、8000∶25—8000∶40。每个浓度设置2个平行,相同条件下设置空白对照,在摇床内进行振荡((25±0.5)℃,90 r·min−1),并在pH值为(7±0.2)的条件下吸附7 d,吸附平衡后液相浓度变化为20%—80%。7 d后取出,2500 r·min−1离心,取1 mL上清液转移至2 mL棕色玻璃样品瓶内,用高效液相色谱仪(Agilent Technologies, 1200)对上清液中的污染物浓度进行测定。两种污染物均用SB-C18反向柱(5 μm,4.6 mm × 150 mm)进行测定,PHE在荧光检测器下检测,流动相为95∶5(V∶V)的甲醇∶水,1 mL·L−1流速,262/365(激发波长/发射波长)条件下检测;BPA则是在紫外检测器下检测,流动相为40∶60(V∶V)的乙腈∶水,1 mL·min−1流速,280 nm波长下检测。
-
烷酸碳优势指数CPIAF=(C20+C22+C24+C26+C28+C30)/(C21+C23+C25+C27+C29);
烷烃碳优势指数CPIALK=(C21+C23+C25+C27+C29)/(C20+C22+C24+C26+C28);
长链烷酸含量与短链烷酸含量比值RLS=(C20+C21+…+C30)/(C12+C13+…+C18);
烷酸平均碳链长度ACL=∑(
x×Cx )/∑Cx ,式中,x 为烷酸碳原子数量,Cx为碳原子数为x的烷酸的浓度,单位为 mg·g−1 土壤.对吸附数据进行吸附等温线绘制,并用SigmaPlot12.5对吸附数据进行拟合,选用Freundlich模型:lgQe = lgKF + nlgCe。式中,Qe 为固相浓度(mg·kg−1),Ce 为液相浓度(mg·L−1),KF 为 Freundlich 模型吸附系数,n 为非线性指数。
数据拟合后,对单点吸附系数Kd(L·kg−1)进行计算并对比不同等温线之间的吸附差异:Kd = Qe/Ce = KF.Cen−1.
-
元素分析结果表明(表1),泥炭土中有机碳含量远高于活性污泥(表1),这主要是因为泥炭土中具有大量丰富的植物源有机碳存在,更多天然有机质的沉降腐熟也使得泥炭土的有机质积累约是活性污泥的3倍。另外,泥炭土的C/H比值明显高于活性污泥,而(N+O)/C比值低于活性污泥。这一结果也表明,泥炭土相对于活性污泥而言,具有更高的芳香性和更低的极性。然而,值得注意的是,泥炭土和活性污泥的比表面积是比较相近的。经酸处理后,得益于活性矿物的大量溶解,使得有机质富集,导致泥炭土和活性污泥的有机碳含量均有一定增加。从两种土壤样品的质量损失比例(WL,%)和有机碳损失比例(CL,%)可以得知(表1),活性污泥的质量损失比例较泥炭土高,而与之不同的是,两种土壤样品的有机碳损失比例却较为相近,考虑到活性污泥相较于泥炭土含有更多的活性矿物,因此,两种土壤样品活性矿物的差异可能是造成这一现象的原因。酸处理后,可以观察到活性污泥和泥炭土的芳香性均有一定程度的增加,而极性却呈现出减少的趋势,可以认为,酸处理后部分极性有机质如蛋白质等有一定程度的损失[14]。而在表1中也可以看出,活性污泥的极性相比泥炭土的极性降低更多,这可能是因为活性污泥含有更多的极性物质。此外,在活性矿物去除后,活性污泥和泥炭土的比表面积分别减少了33%和31%。
-
为了深入研究两种不同源有机质的分子水平信息,本研究利用游离态脂(FL: 主要包括烷酸、烷烃和烷醇)和木质素(VSC: 愈创木基单体V;紫丁香基单体S;对羟基肉桂基单体C)生物标志物方法探究其在分子组成上的差异。从游离态脂的分布和含量可知,活性污泥和泥炭土游离态脂具有显著的分布差异,活性污泥中的正构烷酸、烷醇和烷烃主要以短链碳为主,而在泥炭土中显示以长链碳为主,其中,烷烃具有明显的奇偶优势差异(图1b),烷酸和烷醇呈现显著的偶奇优势(图1c,d),过去的研究表明,高等植物的游离态脂生物标志物分布主要呈现出明显的长链、烷酸的偶数分布特征,可以以此表征土壤有机质的积累源[15],因此,上述研究结果也进一步验证了泥炭土的有机质主要来自于高等植物的积累。
此外,相对于烷烃和烷醇,烷酸在游离态脂中占比更高(表2),在泥炭土和活性污泥的游离态脂含量中分别占到91.15%和49.80%,这与大多数文献报道是一致的[16-17]。从游离态脂的降解参数来看,活性污泥中的烷酸和烷烃的碳优势指数(CPI)比值均低于泥炭土,表明活性污泥中游离态脂的降解程度较泥炭土更高,这可能是由于活性污泥中更多更强的微生物活性所导致的。使用有机溶剂萃取样品时,除游离态脂外,一些糖类也有被检测到(图1a)。在酸处理后,对于泥炭土而言,游离态脂中烷酸有一定的增加,其他组分基本上变化不大。然而,糖类在酸处理后的大量检出被明显观察到,且增加程度要高于游离态脂,表明活性矿物对糖类保护作用可能比游离态脂强。这也验证了,易分解有机碳组分在土壤中被更稳定地保存,可能是这些组分与矿物形成稳定的有机无机复合体导致[18]。此外,活性污泥的可提取游离态脂含量并没有增加,推测是因为活性污泥中游离态脂主要来源于微生物作用下的短链脂肪碳,在酸处理过程中,短链脂肪碳更易流失。这一点也可以从酸处理后的降解参数CPI反映,相对于酸处理前,两种土壤的CPI均有所增加(表3),表明长链脂肪酸的相对富集。
木质素是SOM中重要的芳香族组分贡献者[19],木质素酚类产物可以识别其在土壤有机质中的来源以及降解程度[20]。木质素酚类单体主要包含:愈创木基(V)、紫丁香基(S)及对羟基肉桂基(C)[21]。被子植物木本组织中主要以V和S为主;裸子植物主要以V为主不含S;草本或非木质组织(non-woody)还包含丰富的C[22]。从S/V和C/V的比值可以看出(表3),S/V和C/V均接近1,表明活性污泥和泥炭土中木质素的来源主要来自于被子植物的非木质组织或草本植物。泥炭土中木质素含量(VSC)高于活性污泥(表2),这也反映出泥炭土比活性污泥具有更多的植物来源。3,5-DHBA/V比值常被用于指示的有机质的降解程度[23],活性污泥中具有更高的3,5-DHBA/V比值,表明活性污泥中的木质素比泥炭土具有更高的降解程度,这一结果与P/(V+S) 比值显示结果一致。活性污泥由于含有丰富的微生物,其活性比泥炭土要高,因此,活性污泥中有机质发生了更强的降解。然而,酸处理后,VSC含量显著减少,其降解程度显著增加,这一结果与以前报道是一致的[2],这也说明,矿物更易与氧化程度高的木质素酚类发生相互作用。
-
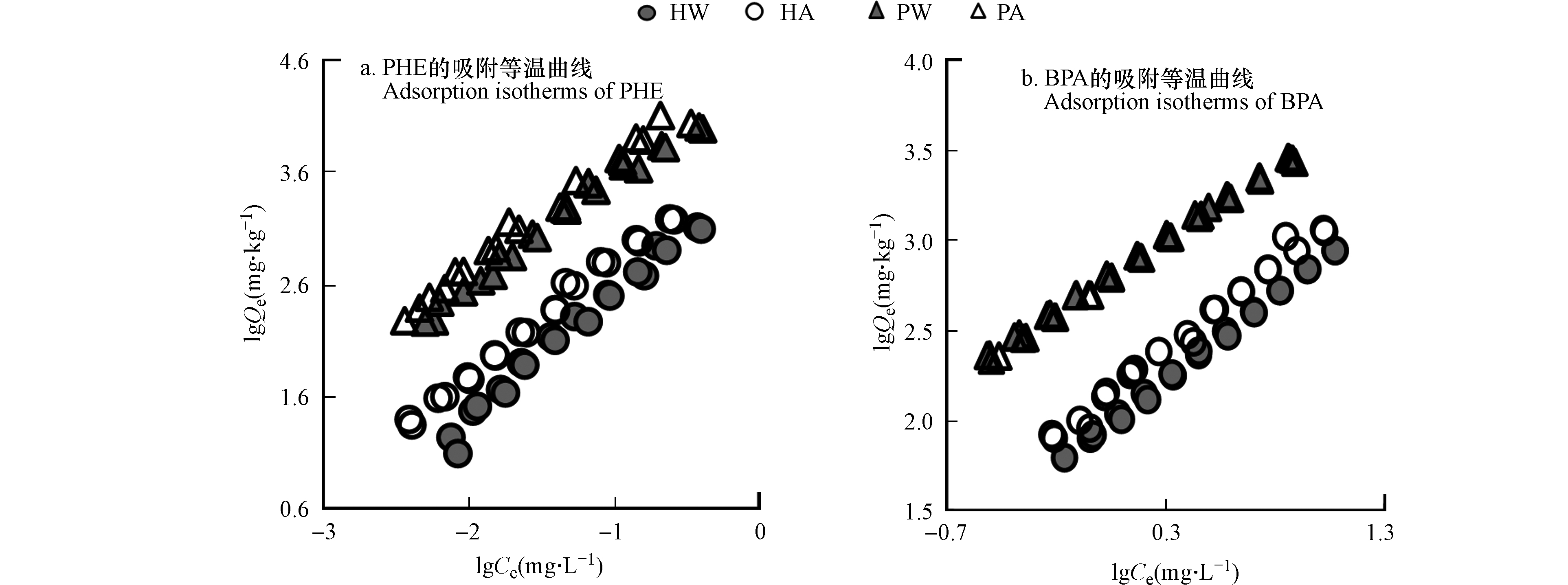
为了进一步探究不同源有机质组成对不同类型的污染物的吸附特性,本研究进行了等温批量吸附实验。由于有更好的拟合效果,因此本实验采用了Freundlich模型拟合吸附结果,由可调可决系数R2adj可以看出该模型对于两种污染物的吸附均有非常好的拟合度(表4)。模型中的n(无量纲常数)用于表征吸附质位点的异质性和吸附线性,可以看到,两种吸附质在酸处理前后,对于PHE的n值影响不大,均有很强的线性吸附。相对于活性污泥,泥炭土有更低的n值,说明其吸附点位的异质性更强,另一方面,PHE的吸附因为其较BPA更为单一的构型和不可电离的性质,因而主要是靠憎水性作用和π-π电子供受体作用,所以,泥炭土上更多的芳香族碳和丰富的脂质都能为其提供充足的吸附位点(表2)。而对比BPA,PHE的吸附有更高的n值,这可能与BPA的双蝴蝶立体结构适应更多的吸附位点有关[24]。两种吸附质在去除活性矿物后n值均有所增加,说明针对BPA而言,活性矿物的去除,减少了其吸附位点的异质性,可能与极性物质在这一过程中的减少有关。
活性污泥和泥炭土两种吸附质在酸处理前后对两种污染物吸附时,更高有机碳的吸附质表现出更强的吸附能力,这种SOM对HOCs吸附能力的控制与文献报道相一致[25]。具体来说,针对于两种不同的污染物,两种吸附剂在吸附能力上也呈现出了较大差异。PHE因为憎水性更强,在两种吸附质上有着比BPA更高的吸附能力,而BPA则因为其本身是介于强疏水和离子型的污染物,因此在土壤上表现出较低的吸附能力。
对于PHE的吸附,泥炭土的吸附能力均比活性污泥要强(表4),在活性矿物去除后,尽管泥炭土的吸附仍高于活性污泥,但这一差距有所减小。可以看到,酸处理后,活性污泥较泥炭土对PHE吸附增加更显著,这可能与活性污泥酸处理后流失了更高的极性物质有关,这一解释可以从活性污泥中测出了更高降解程度的木质素酚类产物以及酸洗后减少了更多的VSC反映,说明酸处理后,活性污泥暴露出了更多的吸附位点。此外,在酸处理后,对于BPA,活性污泥的吸附有一定增加,但泥炭土基本上没有变化,这说明对于有机质更为丰富的泥炭土而言,酸处理过程中流失的部分有机碳对于BPA的吸附位点贡献较小,且能够观察到,在有机碳标准化后,两种土壤的Koc值在活性矿物去除前后基本保持一致,这也表明BPA的吸附可能主要受有机碳的控制。
在拟合绘制出两种污染物的吸附等温曲线之后,可以看出,无论是PHE还是BPA,泥炭土的吸附能力都更强(图2),与元素分析有机碳含量的趋势一致。在酸处理后,泥炭土对于两种污染物的吸附能力没有太大的提升,而活性污泥则都有一定的增强,这可能是由于活性污泥在酸处理过程中流失了更多的游离态脂和木质素酚类,从而暴露出更多的吸附位点,而泥炭土总体来说变化不大。
-
从组成上来看,活性污泥的游离态脂分布因为其有机质来源与微生物相关,呈现出比泥炭土更低的碳数分布。从降解参数来看,活性污泥拥有更高的降解程度,这与木质素酚类生物标志物结果所一致。在活性矿物去除后,因为有机无机复合体和团聚体的保护机制和保护能力存在差异,生物标志物含量发生了不同的变化,具体表现为活性污泥流失了更多的游离态脂有机质,而泥炭土则得到了更高的萃取量,说明并不是所有的土壤在酸处理后都会增加萃取效率,而两种土壤的木质素酚类的提取量均有所降低,可能由于与矿物结合的木质素酚类因其较高的氧化程度而拥有更高的溶解度。从降解参数来看,两种土壤在酸处理后所萃取的游离态脂降解程度均有所降低,这说明矿物倾向于保护降解程度较低的游离态脂。而在酸处理后,两种土壤木质素酚类的降解程度均有所增加,表明矿物倾向于保护降解程度更高的木质素。
PHE和BPA是两种不同性质的HOCs,活性污泥和泥炭土在吸附PHE时,仍然以极性和芳香性变化为主导,酸处理后因为极性组分的流失导致两种吸附质的吸附能力均有提升,但是对于BPA的吸附,活性污泥因其游离态脂和木质素酚类在酸处理后的有所流失而暴露出更多的吸附位点,呈现出了吸附能力升高的趋势,泥炭土则因其被丰富的有机质包裹,在酸处理后吸附位点分布变化不大,结果也表明,两种吸附质对其的吸附主要还是以憎水性作用为主。
活性污泥和泥炭土有机质分子组成差异及其对有机污染物的吸附影响
The difference in molecular composition of organic matter between biosolids and peat and their effects on the adsorption of organic contaminants
-
摘要: 土壤有机质(SOM)是控制土壤环境中有机污染物迁移转化的关键组分,不同来源土壤有机质的组成、矿物保护机制及其对有机污染物的吸附差异还不够明确。本研究采用分子生物标志物技术结合元素组成及比表面积分析表征酸处理前后的活性污泥和滇池底泥的有机质变化,进一步考察其对菲(PHE)和双酚A(BPA)的吸附特征。结果表明,活性污泥中可提取游离态脂组分和木质素酚类组分要低于泥炭土,但降解程度要远高于泥炭土。活性矿物去除后,泥炭土中游离态脂含量明显增加,而活性污泥的游离态脂含量稍有减少,这可能是因为活性污泥游离态脂主要以短链脂肪碳为主,在酸处理过程中容易导致流失。而糖类的显著增加证明了易分解有机碳组分在土壤中被稳定保存。活性污泥和泥炭土中的木质素酚类的含量随着酸处理显著减少,这表明与活性矿物结合的木质素酚类具有更高的氧化程度,导致其溶解性增强。此外,游离态脂降解程度在去除活性矿物后呈降低趋势,而木质素酚类的降解程度却呈增大趋势,这说明活性矿物选择性保护降解程度低的游离态脂和降解程度高的木质素酚类。对于菲的吸附,两种土壤在去除活性矿物后因为极性组分的流失,吸附能力均有所提升,但对于双酚A的吸附,活性污泥由于在酸处理后木质素酚类和游离态脂的减少暴露出更多的吸附位点,而呈现出了吸附能力升高的趋势,泥炭土则因其被丰富的有机质包裹,在去除活性矿物后没太大变化。本研究为揭示不同源SOM与有机污染物之间的相互作用具有重要意义。Abstract: Soil organic matter (SOM) is a key component for controlling the migration and transformation of organic contaminants in the soil environment. The molecular composition of SOM from different sources, mineral protection mechanisms and their differences in the adsorption of organic contaminants remain unclear. In this study, molecular biomarker technology combined with elemental analysis and specific surface area was used to characterize the organic matter composition in biosolids and Dianchi peat before and after acid treatment, and further investigated its adsorption characteristics for phenanthrene (PHE) and bisphenol A (BPA). The results showed that the amounts of extractable free lipids and lignin derived phenols are lower in the biosolids than in the peat, but the degradation degree of biosolids were much higher than that of peat. After reactive minerals were removed, the free lipids content in the peat increased significantly, while the free lipids in the biosolids decreased slightly. This may be attributed to the fact that the free lipids in the biosolids, mainly including short-chain fatty carbons, were dissolved during the acid treatment. In addition, a large number of sugars were released and detected. This result proved that the easily degraded organic matters are well-preserved in the soil. The contents of lignin derived phenols in biosolids and peat decreased significantly after acid treatment, which indicated that the lignin derived phenols associated with reactive minerals have a higher oxidation degree. In addition, the degradation of free lipids showed a decreasing trend after the removal of reactive minerals, while the degradation of lignin phenols showed an increasing trend. This result suggested that reactive minerals selectively protected free lipids with a low degree of degradation and lignin phenols with a high degree of degradation. For the adsorption of PHE, the adsorption capacity of the two soils were improved due to the loss of polar components after the removal of reactive minerals. For the adsorption of BPA, the biosolids showed an increasing trend of adsorption capacity because of the reduction of lignin derived phenols and free lipids after acid treatment to expose more adsorption sites. But the adsorption of peat soil for BPA had no changed after the removal of reactive minerals, probably because adsorption sites were wrapped in rich organic matter. This work is of great significance for revealing the interaction mechanism between SOM and organic contaminants.
-
Key words:
- soil organic matter /
- biomarkers /
- mineral removal /
- organic contaminant
-

图 1 酸处理前后土壤中游离态脂的单体分布
Figure 1. The distribution of individual free lipids in soils before and after acid treatment
表 1 元素分析及土壤基础表征
Table 1. Elemental analysis and soil basic characterization
元素分析Elemental analysis SA /(m2·g−1) WL/% CL/% N/% C/% H/% O/% C/H (N+O)/C HW 2.07 14.5 3.96 24.2 0.31 1.37 21.8 — — HA 3.34 21.2 4.70 22.1 0.38 0.91 14.5 55.8 34.9 PW 2.66 42.8 6.35 32.2 0.56 0.62 23.5 — — PA 2.90 47.4 6.38 29.7 0.62 0.52 16.3 36.6 30.7 注:SA为比表面积;质量损失比WL=酸处理损失的土壤质量/酸处理前的土壤质量;碳损失比CL=酸处理损失的碳含量/酸处理前碳的含量. Note: SA is the surface area; weight loss ratio WL=soil weight lost after acid treatment/soil weight before acid treatment; carbon loss ratio CL=carbon content lost after acid treatment/carbon content before acid treatment.  下载: 导出CSV
下载: 导出CSV
表 2 不同处理活性污泥和泥炭土有机质的生物标志物丰度(mg·g−1土壤)
Table 2. Relative abundance of molecule biomarkers in organic matter of the treated biosolids and peat (mg·g−1 soil)
n-alkanes n-alkanoic acids n-alkanols Carbohydrates FL V S C VSC HW 0.04 2.06 0.16 0.05 2.26 0.20 0.19 0.18 0.58 HA 0.03 1.21 0.07 0.95 1.31 0.02 0.02 0.01 0.06 PW 0.12 1.26 1.15 0.03 2.53 0.64 0.54 0.55 1.72 PA 0.13 2.01 0.98 3.98 3.12 0.28 0.27 0.34 0.88 注:FL,游离态脂;V,愈创木基;S,紫丁香基;C,对羟基肉桂基;VSC,愈创木基+紫丁香基+对羟基肉桂基的总和. Note: FL, free lipids; V, vanilly units; S, syringyl units; C, cinnamyl units; VSC, the sum of vanilly units + syringyl units + cinnamyl units.
下载: 导出CSV
表 3 生物标志物的降解参数
Table 3. The degradation parameters of molecular biomarkers
CPIAF CPIALK RLS ACL S/V C/V P/(V+S) 3,5-DHBA/V HW — 0.68 0.04 16.18 0.93 0.90 0.79 0.36 HA 12.09 1.76 0.11 17.08 1.00 0.43 3.11 0.51 PW 17.76 3.55 7.10 25.30 0.85 0.86 0.53 0.16 PA 19.42 5.16 11.30 25.85 0.95 1.21 0.62 0.34 注:CPIAF,脂肪酸的碳优势指数;CPIALK,烷烃的碳优势指数;RLS,长链烷酸(>C20)含量与短链烷酸含量的比值;ACL,烷酸的平均碳链长度;S/V,紫丁香基/愈创木基;C/V,对羟基肉桂基/愈创木基;P/(V+S) ,对羟基苯酚/(愈创木基+紫丁香基);3,5-DHBA/V,3,5-二羟基苯甲酸/愈创木基;HW的CPIAF没有数值是因为HW中长链烷酸含量低于检出限. Note: CPIAF, carbon preference index of n-alkanoic acids; CPIALK, carbon preference index of n-alkanes; RLS, ratio of long chain carbon alkanoic acids (C>20) to short chain carbon alkanoic acids; ACL, averaged carbon length of alkanoic acids; S/V, ratio of syringyl to vanillyl; C/V, ratio of cinnamyl to vanillyl; P/(V+S), ratio of p-hydroxyphenols (P) to (vanilly + syringyl); 3,5-DHBA/V, ratio of 3,5-Dihydroxybenzoic acid to vanilly; CPIAF of HW has no value because the content of long chain carbon alkanoic acids in HW is below the detection limit.
下载: 导出CSV
表 4 两种污染物吸附实验的Freundlich参数
Table 4. Freundlich parameters of two contaminants adsorption experiments
土壤 Soil n SE lgKF R2adj Kd/(L·kg−1) Koc/(L·kg−1 C) Ce=0.01Cs Ce=0.04Cs Ce=0.01Cs Ce=0.04Cs PHE HW 1.117 0.08 3.63 0.982 2576 3028 17878 21015 HA 1.036 0.05 3.85 0.993 6100 6408 28747 30198 PW 0.936 0.07 4.45 0.985 37320 34161 87283 79895 PA 0.936 0.08 4.61 0.982 53526 49009 112810 103290 BPA HW 0.942 0.01 1.94 0.999 80 74 556 513 HA 0.960 0.04 2.10 0.988 119 112 560 530 PW 0.793 0.01 2.77 0.998 447 335 1045 784 PA 0.828 0.01 2.74 0.999 435 343 916 722
下载: 导出CSV
-
[1] KANG S, XING B S. Phenanthrene sorption to sequentially extracted soil humic acids and humins [J]. Environmental Science & Technology, 2005, 39(1): 134-140. [2] LI F F, PAN B, LIANG N, et al. Reactive mineral removal relative to soil organic matter heterogeneity and implications for organic contaminant sorption [J]. Environmental Pollution, 2017, 227: 49-56. doi: 10.1016/j.envpol.2017.04.047 [3] LEINWEBER P, BLUMENSTEIN O, SCHULTEN H R. Organic matter composition in sewage farm soils: Investigations by 13C-NMR and pyrolysis-field ionization mass spectrometry [J]. European Journal of Soil Science, 2005, 47(1): 71-80. [4] XING B. Sorption of naphthalene and phenanthrene by soil humic acids [J]. Environmental Pollution, 2001, 111(2): 303-309. doi: 10.1016/S0269-7491(00)00065-8 [5] KILE D E, WERSHAW R L, CHIOU C T. Correlation of soil and sediment organic matter polarity to aqueous sorption of nonionic compounds [J]. Environmental Science & Technology, 1999, 33(12): 2053-2056. [6] HU W G, MAO J D, XING B S, et al. Poly(methylene) crystallites in humic substances detected by nuclear magnetic resonance [J]. Environmental Science & Technology, 2000, 34(3): 530-534. [7] 王彦, 左宁, 姜媛媛, 等. 污泥生物炭中氮硫行为及环境效应研究进展 [J]. 化工进展, 2020, 39(4): 1539-1549. WANG Y, ZUO N, JIANG Y Y, et al. Behavior and environmental effects of nitrogen and sulfur in sludge biochar [J]. Chemical Industry and Engineering Progress, 2020, 39(4): 1539-1549(in Chinese).
[8] 张帆舸, 杨敏, 刘侃. 昆明滇池泥炭土物理指标相关性研究[J]. 建筑科学, 2020, 36(增刊1): 1-7. ZHANG F G, YANG M, LIU K. Study on the relevance between the physical parameters of peaty soils in Dianchi district of Kunming[J]. Building Science, 2020, 36(Sup 1): 1-7(in Chinese). Science, 2020, v. 36(S1): 8-14.
[9] OTTO A, SIMPSON M J. Analysis of soil organic matter biomarkers by sequential chemical degradation and gas chromatography-mass spectrometry [J]. Journal of Separation Science, 2007, 30(2): 272-282. doi: 10.1002/jssc.200600243 [10] SIMONEIT B R. A review of current applications of mass spectrometry for biomarker/molecular tracer elucidation [J]. Mass Spectrometry Reviews, 2005, 24(5): 719-765. doi: 10.1002/mas.20036 [11] LI F F, LIANG N, ZHANG P C, et al. Protection of extractable lipid and lignin: Differences in undisturbed and cultivated soils detected by molecular markers [J]. Chemosphere, 2018, 213: 314-322. doi: 10.1016/j.chemosphere.2018.09.043 [12] LIN L H, SIMPSON M J. Enhanced extractability of cutin- and suberin-derived organic matter with demineralization implies physical protection over chemical recalcitrance in soil [J]. Organic Geochemistry, 2016, 97: 111-121. doi: 10.1016/j.orggeochem.2016.04.012 [13] ZHANG D, PAN B, ZHANG H, et al. Contribution of different sulfamethoxazole species to their overall adsorption on functionalized carbon nanotubes [J]. Environmental Science & Technology, 2010, 44(10): 3806-3811. [14] ZHANG J, TIAN Y, CUI Y N, et al. Key intermediates in nitrogen transformation during microwave pyrolysis of sewage sludge: A protein model compound study [J]. Bioresource Technology, 2013, 132: 57-63. doi: 10.1016/j.biortech.2013.01.008 [15] KOLATTUKUDY P E, CROTEAU R, BUCKNER J S. Biochemistry of plant waxes [J]. Chemistry & Biochemistry of Natural Waxes, 1976: 290-349. [16] OTTO A, SHUNTHIRASINGHAM C, SIMPSON M J. A comparison of plant and microbial biomarkers in grassland soils from the Prairie Ecozone of Canada [J]. Organic Geochemistry, 2005, 36(3): 425-448. doi: 10.1016/j.orggeochem.2004.09.008 [17] WIESENBERG G L B, DORODNIKOV M, KUZYAKOV Y. Source determination of lipids in bulk soil and soil density fractions after four years of wheat cropping [J]. Geoderma, 2010, 156(3/4): 267-277. [18] TAMURA M, THARAYIL N. Plant litter chemistry and microbial priming regulate the accrual, composition and stability of soil carbon in invaded ecosystems [J]. New Phytologist, 2014, 203(1): 110-124. doi: 10.1111/nph.12795 [19] GUL S, YANNI S, WHALEN J K. Lignin controls on soil ecosystem services: Implications for biotechnological advances in biofuel crops[M]. Lignin: Structural analysis, applications in biomaterials and ecological significance, New York: Biochemistry Research Trends, Nova Science Publishers, 2014, 375-416. ISBN 978-1-56973-641-8. [20] DING X D, BAO H Y, ZHENG L W, et al. Lacustrine lignin biomarker record reveals a severe drought during the late Younger Dryas in southern Taiwan [J]. Journal of Asian Earth Sciences, 2017, 135: 281-290. doi: 10.1016/j.jseaes.2017.01.003 [21] TAREQ S M, TANAKA N, OHTA K. Biomarker signature in tropical wetland: Lignin phenol vegetation index (LPVI) and its implications for reconstructing the paleoenvironment [J]. Science of the Total Environment, 2004, 324(1/2/3): 91-103. [22] HEDGES J I, MANN D C. The characterization of plant tissues by their lignin oxidation products [J]. Geochimica et Cosmochimica Acta, 1979, 43(11): 1803-1807. doi: 10.1016/0016-7037(79)90028-0 [23] MOINGT M, LUCOTTE M, PAQUET S. Lignin biomarkers signatures of common plants and soils of Eastern Canada [J]. Biogeochemistry, 2016, 129(1/2): 133-148. [24] PAN B, LIN D, MASHAYEKHI H, et al. Adsorption and hysteresis of bisphenol A and 17 alpha-ethinyl estradiol on carbon nanomaterials [J]. Environmental Science & Technology, 2008, 42(15): 5480-5485. [25] HAN L, SUN K, JIN J, et al. Role of structure and microporosity in phenanthrene sorption by natural and engineered organic matter [J]. Environmental Science & Technology, 2014, 48(19): 11227-11234. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3667
- HTML全文浏览数: 3667
- PDF下载数: 108
- 施引文献: 0



