














 下载:
下载:










-
由于自然过程(风化作用和生化作用)以及在世界范围内许多工业和制造过程中的使用不断增加,水生生态系统中的铬污染已成为一个突出问题,对生态环境和人类健康构成严重威胁。铬以Cr(Ⅲ)和Cr(Ⅵ)的形式存在于环境中,虽然Cr(Ⅲ)是人体必需的微量营养素,但Cr(Ⅵ)在水介质中可高度溶解,具有急性毒性、致突变性和致癌性,Cr(Ⅵ)的毒性是Cr(Ⅲ)的500倍[1]。
铬在制造和工业使用时,由于贮存和泄漏处理不当,导致多次被释放到环境中,造成地下水和土壤的污染。自然过程也会导致地下水中铬的浓度升高,例如,超镁铁质岩石可能具有较高的铬含量,其主要存在于母体矿物中的Cr(Ⅲ)中,但在风化过程中被四价锰氧化物氧化为Cr(Ⅵ)。尽管Cr(Ⅵ)的释放可能来自许多环节,但对环境危害最大的是铬铁矿加工残渣的处置不当。与铬铁矿加工残渣接触的水pH >12,并且水中的Cr(Ⅵ)浓度可超过1mmol·L−1[2-3]。当这种水不可避免地从废物堆中逸出到地球圈,会产生Cr(Ⅵ)羽流,其中pH值从高碱性值向天然土壤pH值变化。在水体环境中,Cr(Ⅵ)通常以可溶阴离子基团形式存在,如
CrO2−4 、HCrO−4 和Cr2O2−7 ,最终通过生物富集作用进入人体,严重影响人类健康[4]。当pH <2 时,溶液中的Cr(Ⅵ)主要是以Cr2O2−7 形式存在,当pH值在5—6之间时,Cr(Ⅵ)存在形式是Cr2O2−7 、HCrO−4 ,当pH >7 时,Cr(Ⅵ)主要以CrO2−4 形式存在[5]。由于其毒性和潜在的迁移性,Cr(Ⅵ)释放到生态系统引起普遍关注。在氧化环境中,Cr(Ⅵ)与铁和氧化铝矿物的表面发生络合反应进而去除酸性溶液中的Cr(Ⅵ) [6];但是,在中性和碱性环境下,由于在矿物表面存在净负表面电荷,因此土壤矿物对Cr(Ⅵ)的吸附通常较弱。铬在还原性土壤环境中的流动性比较低,因为水中的Fe(Ⅱ)和含Fe(Ⅱ)的矿物质以及具有还原性的硫化合物可以将Cr(Ⅵ)迅速还原为Cr(Ⅲ)。一旦还原,Cr(Ⅲ)在周围环境条件下将以Cr(OH)3的形式沉淀,或者当被Fe(Ⅱ)还原时以(CrxFe1−x)(OH)3的形式沉淀(式1,2)[7-8]。当前对环境中铬污染的处理,通常包括将Cr(Ⅵ)还原为Cr(Ⅲ),如传统的化学还原工艺[9],生物学方法以及其它一些先进的工艺,例如,吸附[10]、生物修复[11]、化学沉淀[12]和光催化还原[13]等。上述技术方法尽管在Cr(Ⅵ)还原方面具有很高的效率,但仍具有各种缺点,例如对化学药品和能量的大量需求,大量污泥的产生以及高昂的运行成本。HA是一种通过微生物的分解和转化植物残留物而累积起来的高分子有机物质[14],具有生物化学稳定性高、表面积大、结构复杂、带有多种活性官能团等特点。它在全球总计达数万亿吨,通常存在于河流,湖泊,海洋,土壤和煤矿中。HA广泛应用于农业,林业,畜牧业和化学工业等领域[15-16],多项研究已将HA用作污染物的吸附剂[17-18]。尽管HA的存在会污染地下水和地表水,但其优点胜于其用于处理工业废水的缺点,并且可以在后续深加工中将其完全清洗。此外,HA表面富含官能团,包括羧基、羟基和羰基[19]。这些基团可用于与阳离子物质进行离子交换、吸附、络合和螯合[20-22]。目前国内外对利用HA去除水体和土壤中铬离子的研究尤为重视。
本文旨在系统地阐述HA的结构特征和与Cr(Ⅵ)的作用机理,分析了矿物质和纳米复合材料去除Cr(Ⅵ)的过程中HA的影响,总结了仍然存在的局限性和尚待解决的问题,以便为HA处理含Cr(Ⅵ)废水的应用提供未来展望。
-
HA是包含于腐殖质(HS)的大分子,HS是由细菌和真菌对植物和动物的残留物进行腐烂和转化而产生的深色、异质的有机化合物。主要的HS前体分子是通过植物产生的生物聚合物和蛋白质的解聚和氧化形成的,这些分子含有未分解的聚合物片段和酚类、羟基、羧基和氨基残基[23]。传统的“聚合物模型”假设前体是由微生物合成的随机卷曲的聚合物大分子,而有研究表明HS是超分子缔合的[24-25]。两亲性前体分子聚集在一起形成胶束状粒子,而其它植物降解的生物分子则与疏水或亲水结构域有关[26-27]。虽然目前大多数证据支持这种“胶束模型”,但是在HS数百万年的老化和降解过程中并不排除聚合物大分子的形成 [28-29]。
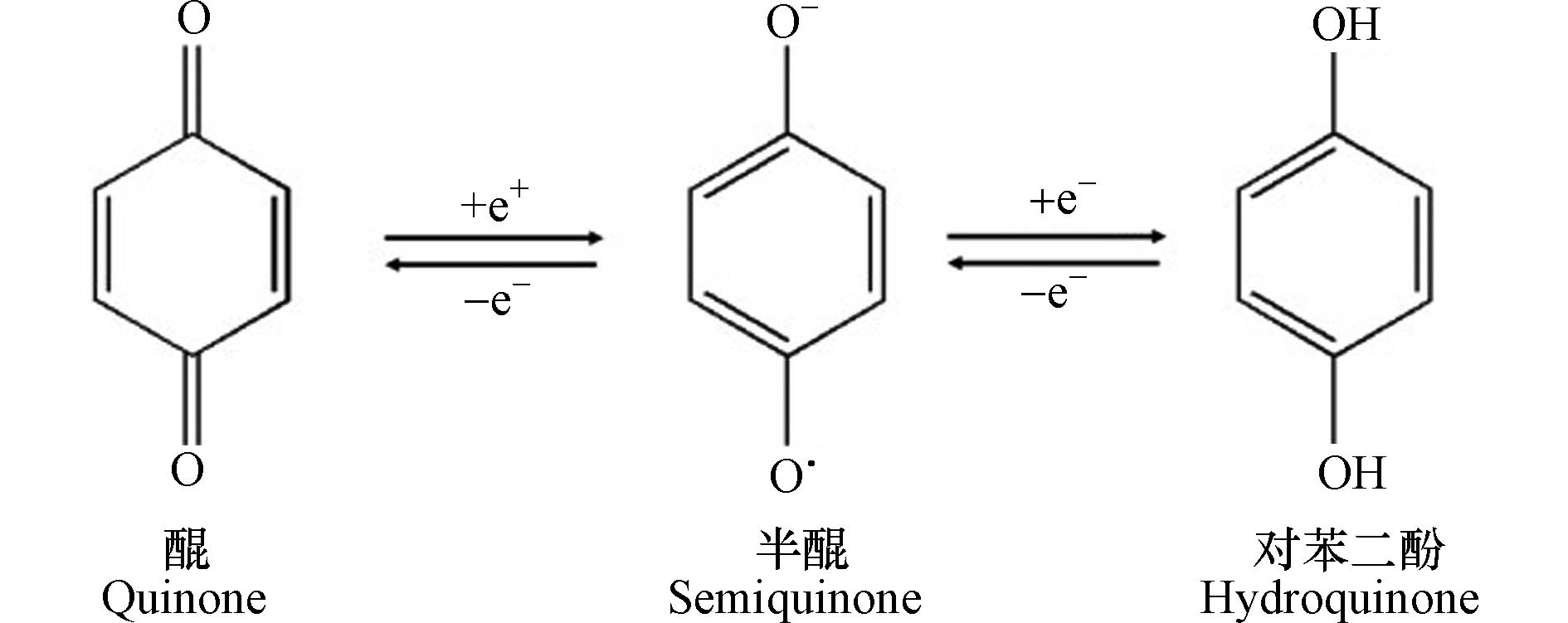
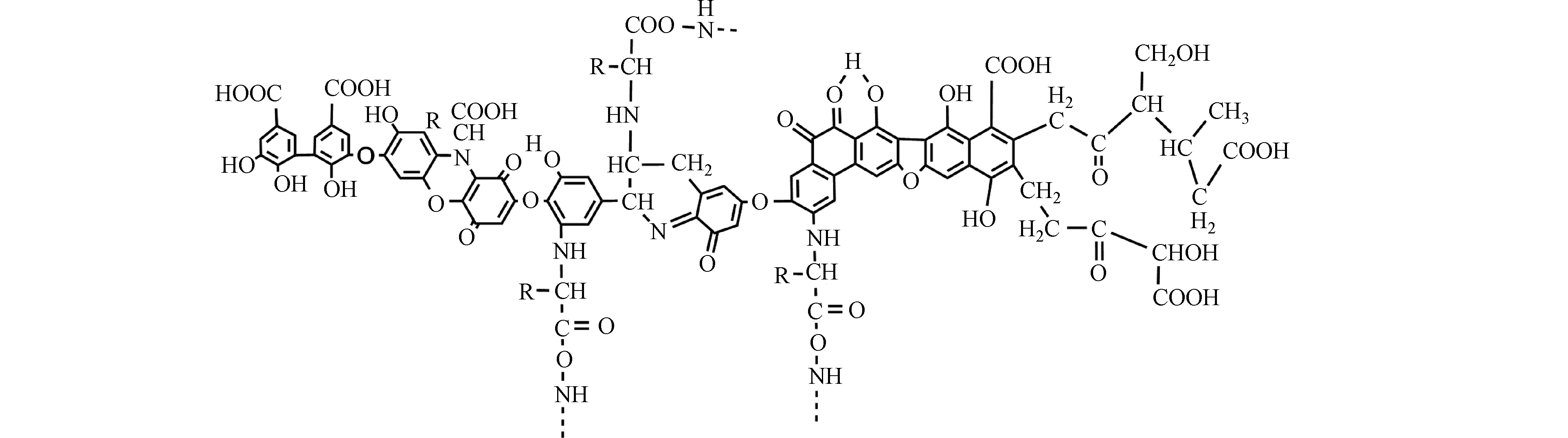
HA是可溶于碱性介质,部分可溶于水而不溶于酸性介质的HS组分[30]。由于具有两亲性,HA在中性至酸性条件下会形成胶束状结构,称为假胶束[31]。已经探索出该性质可用于污染修复[32],并增加疏水性药物的水溶性[33-34]。HA含有不同的官能团,其数量取决于HA的来源、年龄、气候和提取的环境条件。HA的各种功能主要归因于酚和羧酸官能团,它们允许—OH/—OOH的脱质子化。这使得HA拥有了许多功能,如具有改善植物生长和增强营养[35],与重金属络合[36],抗病毒和抗炎活性[37]。此外,HA结构中酚类、羧酸和醌的存在与其抗氧化、抗突变/去突变和杀菌/杀菌活性有关[38]。HA的化学组成可能会因来源,年龄,气候和生物学条件而异,因此很难对这些物质进行精确表征。它们的分子量在2.0 kDa到1300 kDa之间[39],并且它们包含许多官能团,如图1所示[33],HA主要由酚、羧酸、烯醇、醌和醚官能团组成,但也可能包括糖和肽。但是,酚羟基和羧基在HA结构中更为普遍。HA分子的巨大结构由亲水部分(由—OH基团组成)和疏水部分(由脂族链和芳环组成)组成。醌是电子接受基团,负责产生活性氧(ROS)。它们被还原为半醌,由其芳环稳定,并进一步还原为更加稳定的对苯二酚(图2)[40]。Aeschbacher等[41]研究了从不同来源中获得的HA中的电子接受部分(醌)和供电子部分(酚类),发现这些物质的来源和年龄直接影响其氧化还原特性。13C NMR分析表明,与陆生HA相比,水生HA具有更高的给电子数和更低的电子接受基团[40]。Aeschbacher等[41]做出以下假设:HA中的酚羟基会减慢醌的氧化转化,从而提高其在含氧环境中的持久性。
-
水生环境中金属阳离子在溶液中的作用与H+相同,即电荷中和,电荷越高,阳离子在HA假胶束的形成中越有效。此外,多价阳离子与相邻链上的酚羟基和羧基基团相互作用,增强了假胶束结构域和去污作用。von Wandruszka[42]提出这种相互作用的机制如下:HA分子与金属阳离子之间的相互作用最初完全是静电作用,并且阳离子会移动到HA中热力学上更稳定的位置。此过程形成球状HA-金属络合物。对HA与金属离子结合的研究表明,这种相互作用随金属不同而变化,并受金属离子浓度以及HA的来源、分子量和浓度影响[43]。Christl等[43]证明HA与金属离子之间的结合能力与HA的分子大小有关。用尺寸排阻色谱-聚丙烯酰胺凝胶电泳分离HA得到的3个级分的13C NMR分析证实,分子量较低的HA组分具有最多的酚羟基和羧基,因此是可以最有效地结合金属离子的组分。此外,Christl和Kretzschmar[44]发现在较高金属离子浓度下,可观察到两个HA大小级分之间的差异,因为较小的级分由于官能团数较多而具有较高的结合能力。HA通过与金属离子结合形成络合物,从而去除水和土壤中的重金属,是一个很好的重金属污染补救措施。重金属污染的水(包括铬污染)一般是呈酸性,使得HA的溶解度会降低,从而干扰HA-金属配合物的形成。但是铬污染的水中铬以阴离子形式存在,酸性环境下反而有利于HA对铬的还原。
Cr(Ⅵ)能与含有酚羟基、醇羟基的有机物反应从而被还原为Cr(Ⅲ)。与这些基团的反应被认为涉及到一种铬酸盐中间体,该中间体可由这些基团与单体H2CrO4和
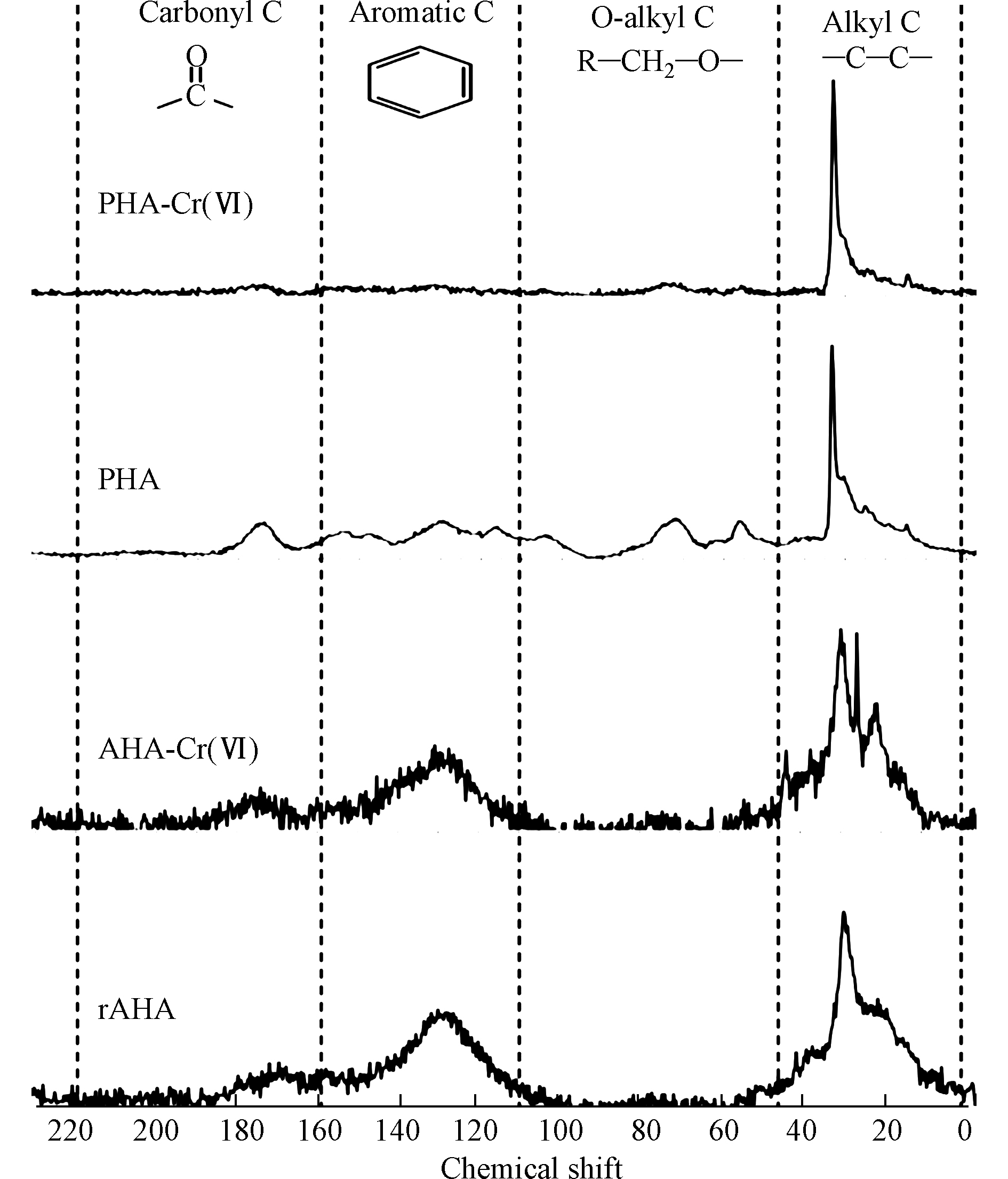
HCrO−4 发生氧化还原相互作用形成[45-46]。在酸性体系中,这种机制对铬的还原速度很快,但随着pH值的增加,还原速度明显降低。通常认为,当pH≥6时,用乙醇、酚类和醛类化合物还原Cr(Ⅵ)是可以忽略不计的,因为铬酸盐的形成对CrO2−4 离子不利,CrO2−4 离子是高pH下Cr(Ⅵ)的主要组成[46]。HA官能团与Cr(Ⅵ)之间的反应受到了广泛关注,研究表明脂肪族极性官能团(主要是羧基)是HA还原Cr(Ⅵ)的主要位点[47-48]。其它研究表明,酚羟基是将Cr(Ⅵ)还原为Cr(Ⅲ)的主要官能团,导致羰基/羧基的形成[49],而羧基可能会为Cr(Ⅲ)提供结合位点[50]。引起争议的原因可能是忽视了HA官能团的变化与Cr(Ⅵ)的反应途径之间的关联,其中Cr(Ⅵ)和HA之间的相互作用可能包括Cr(Ⅵ)还原和Cr(Ⅲ)络合或沉淀[51]。此外,还可能涉及Cr(Ⅵ)的络合,这可能是HA还原Cr(Ⅵ)所必需的。另外,在反应过程的不同阶段,参与相互作用的官能团的类型可能不同。在此基础上,Zhang等[52]基于分子尺度上的2DCOS分析结果,建立了酸性条件(pH为1—3.5范围内)下HA去除Cr(Ⅵ)的机理。HA去除Cr(Ⅵ)的途径遵循络合偶联的还原机制。羧基和酚羟基被证实参与了去除过程,并分别参与了Cr(Ⅵ)的络合和还原,这两个基团可能位于HA 分子的芳香族结构中。此外,用2DCOS表征了HA官能团的变化顺序,揭示了在酸性条件下官能团还原Cr(Ⅵ)的过程:(1)静电吸附的Cr(Ⅵ)被游离羧基和酯络合;(2)酚羟基和多糖将络合的Cr(Ⅵ)还原为Cr(Ⅲ);(3)还原态的Cr(Ⅲ)与羧基络合。HA还原Cr(Ⅵ)降低了铬的毒性,产生的Cr(Ⅲ)与HA中的两个碳原子形成一个内层吸附复合物,大大减少了Cr(Ⅲ)扩散或重新氧化成可移动Cr(Ⅵ)的机会。Cr(Ⅵ)的还原速率和还原Cr(Ⅵ)的总容量与所使用的HA的反应活性呈正比,特别是HA中酚羟基和醇羟基的密度。Zhang等[53]研究了从东北典型黑土中提取的HA在pH=1.0时对Cr(Ⅵ)的还原,结果表明,HA中的酚羟基和醇羟基官能团是还原Cr(Ⅵ)的主要电子给体,而不是羧基和羰基,它们更可能参与铬的络合反应,而且HA颗粒表面的酚羟基和醇羟基对Cr(Ⅵ)还原的活性远高于其内部。Aldmour等[54]研究泥炭腐植酸(PHA)和褐煤腐植酸(AHA)对Cr(Ⅵ)的还原,发现HA将Cr(Ⅵ)还原为Cr(Ⅲ)通常涉及与芳香族的反应,特别是与酚羟基的反应。PHA的变化表明醇羟基也可能发生了反应。HA样品与过量的Cr(Ⅵ)反应,使pH值从3升到7,表明在酸性体系中Cr(Ⅵ)与HA反应消耗了H+。Cr(Ⅵ)的还原必然导致HA的氧化,但13C-NMR谱没有鉴定出产生的官能团(图3)。研究表明,Cr(Ⅲ)吸附在HA上导致羰基Cr(Ⅲ)络合物的形成和与电子再分配相关的屏蔽导致羰基的13C-NMR谱信号减少。因此,用PHA和AHA还原Cr(Ⅵ)很可能会生成新的羰基[52]。
-
铬的界面行为是了解铬在污染环境中的环境效应的基础,然而,复杂的表面会导致铬表现出各种各样的行为。铬的迁移和转化主要受固-液界面的相互作用控制,尤其是沿铁氧化物界面的相互作用。HS与矿物相互作用时,带负电的HS可能会保留在带正电的矿物表面上。因此,HS和矿物之间的相互作用可能会改变任何一种物质的原始特性,并产生复杂的HS矿物[55]。因此,有必要研究铬与具有不同特性的各种HS矿物界面的相互作用。HS是一种富含官能团的有机分子,包含HA和黄腐酸(FA),HA可以吸附在矿物表面,改变矿物的的反应性和表面特性[56]。HA对Cr(Ⅵ)离子具有很强的络合能力[57],并且对矿物表面具有很高的亲和力[58],已被认为是控制水中各种元素行为的重要因素。Kantar等[59]在pH为3—8体系中,研究HA对含Ca2+、Mg2+等阳离子体系中黄铁矿去除Cr(Ⅵ)的影响,发现HA对黄铁矿吸收Cr(Ⅵ)有抑制作用。HA吸附在黄铁矿表面,与Cr(Ⅵ)竞争黄铁矿表面位置;HA通过与黄铁矿反应位点结合改变了黄铁矿表面的静电特性,导致反应位点与
CrO2−4 之间的静电排斥增加。在含有HA和Ca2+/Mg2+的体系中,由于HA与Ca2+/Mg2+的聚集作用,黄铁矿对Cr(Ⅵ)的吸收相对于单独的HA急剧下降,这种HA聚集/沉淀的形成阻止了Cr(Ⅵ)到达其结合位点,从而大量减少Cr(Ⅵ)的吸收.HA作为一种自然水生环境中普遍存在的天然有机质,已被用于磁铁矿的表面改性来修复受Cr(Ⅵ)污染的水。由于HA官能团和磁铁矿的表面羟基之间的络合-配位交换反应,HA对磁铁矿具有高亲和力。完全被HA覆盖的磁铁矿颗粒具有HA的特性和磁铁矿的磁性,与金属氧化物相关的特性相反[55],溶液pH > 3时,零电荷点和负电荷点的低pH值抑制了它们的团聚。磁铁矿中的Fe(Ⅱ)可以将Cr(Ⅵ)还原为Cr(Ⅲ),随后Cr(Ⅲ)被氧化铁表面的羟基螯合,形成内层表面配合物[60]。但是,磁铁矿中的Fe(II)极易自氧化形成磁性降低的Fe(Ⅲ),不利于磁铁矿在磁场条件下的分离。Jiang等[61]用HA包覆磁铁矿纳米颗粒在pH = 4条件下还原Cr(Ⅵ)发现,HA使Fe(Ⅱ)不受自氧化和Cr(Ⅵ)电子转移的影响,并通过HA将Cr(Ⅵ)还原为无毒的Cr(Ⅲ)来保留材料的磁性,使得在磁场条件下HA-Fe3O4颗粒在处理后易于分离。
Zheng等[62]研究了HA在铁矿(磁铁矿和赤铁矿)吸附Cr(Ⅵ)过程中的作用以及Cr(Ⅵ)吸附到铁矿HS络合物表面上的界面行为。发现化学吸附起了重要作用,且在较高温度下,磁铁矿HA复合物、磁铁矿FA复合物和赤铁矿FA复合物对Cr(Ⅵ)的吸附较好;而在较低的温度下,磁铁矿、赤铁矿和赤铁矿HA复合物对Cr(Ⅵ)吸附能力较好。此外,铁矿物上的HA分子对Cr(Ⅵ)的亲和力比FA分子弱。化学吸附和物理吸附同时存在于Cr(Ⅵ)在铁矿物质和铁矿HS络合物上的吸附。此外,除赤铁矿HA复合物外,所有铁矿物质和铁矿HS络合物的化学吸附作用最为明显。
Yu等[63]研究Fe(Ⅱ)诱导水铁矿(Fh)-HA共沉淀转化过程中吸附Cr(Ⅵ)的行为,发现在Fh-HA的老化过程中,在无Fe(Ⅱ)存在下,共沉淀HA通过吸附和络合作用将Cr(Ⅵ)固定在赤铁矿中;在Fe(Ⅱ)存在下,Fe(Ⅱ)能将吸附的Cr(Ⅵ)还原为Cr(Ⅲ)(式3、4)。
此外,吸附的Cr(Ⅵ)在Fh和Fh-HA的相变过程中,可以在水相Fe(Ⅱ)的促进下,通过结构掺入的方式结合到次生矿中。在中性条件下,当Fe(Ⅱ)初始浓度较低(0.2 mmol·L−1)时,水溶性残余Cr(Ⅵ)可并入次生矿物中的Cr(Ⅲ)- Fe(Ⅱ)共沉淀物中。最后,通过共沉淀HA还原和络合次生矿物中的Cr(Ⅵ),进一步促进转化过程中Cr(Ⅵ)的固定化。
-
纳米材料由于大比表面积和高反应活性广泛用于去除废水中重金属。HA普遍存在于水生环境中,并具有多种官能团,可使其与金属离子络合并与纳米材料相互作用。这些相互作用不仅可以改变纳米材料的环境行为,而且可以影响纳米材料对重金属的去除和迁移。因此,所涉及的相互作用和潜在机制值得进行深入研究。
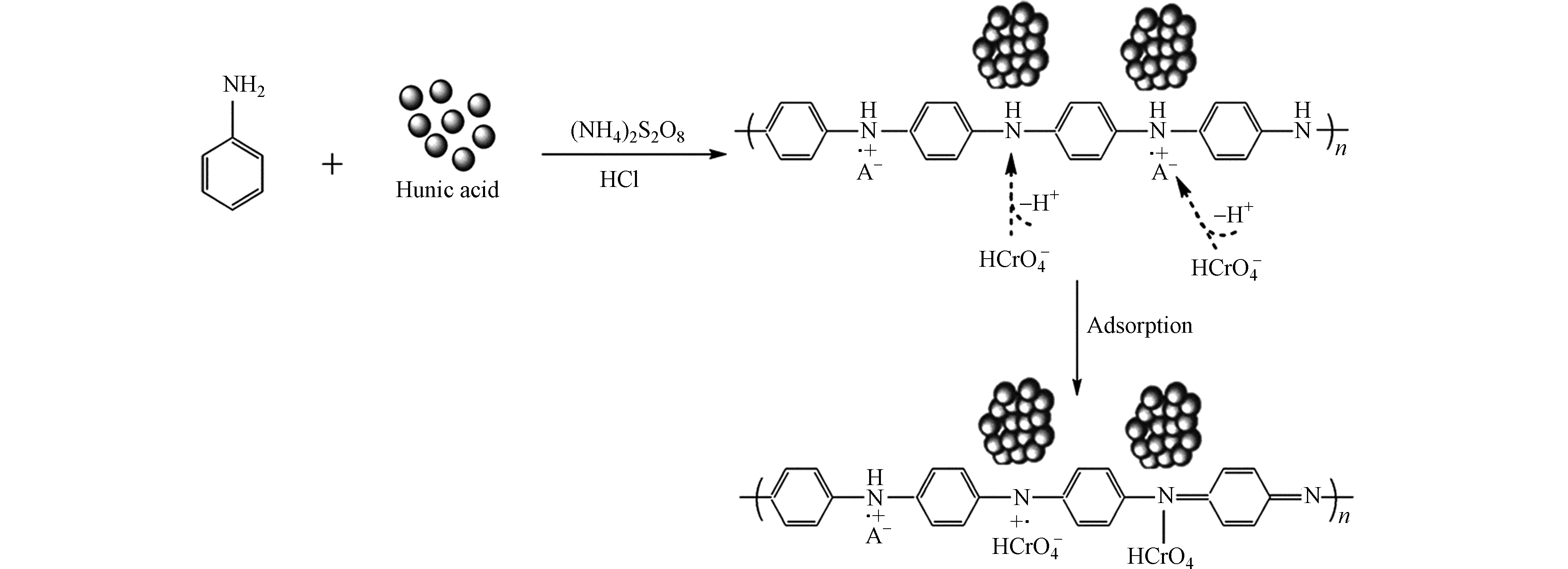
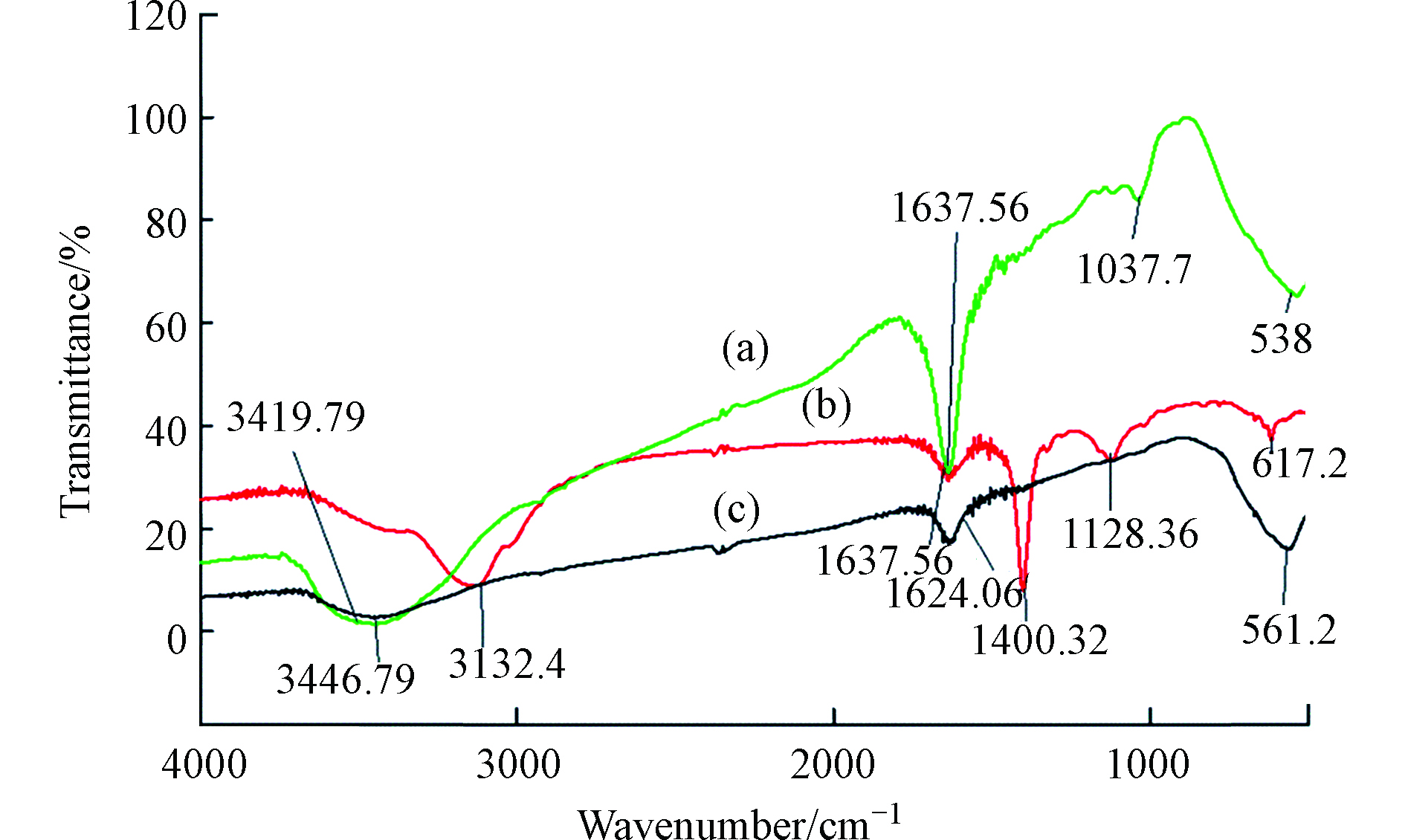
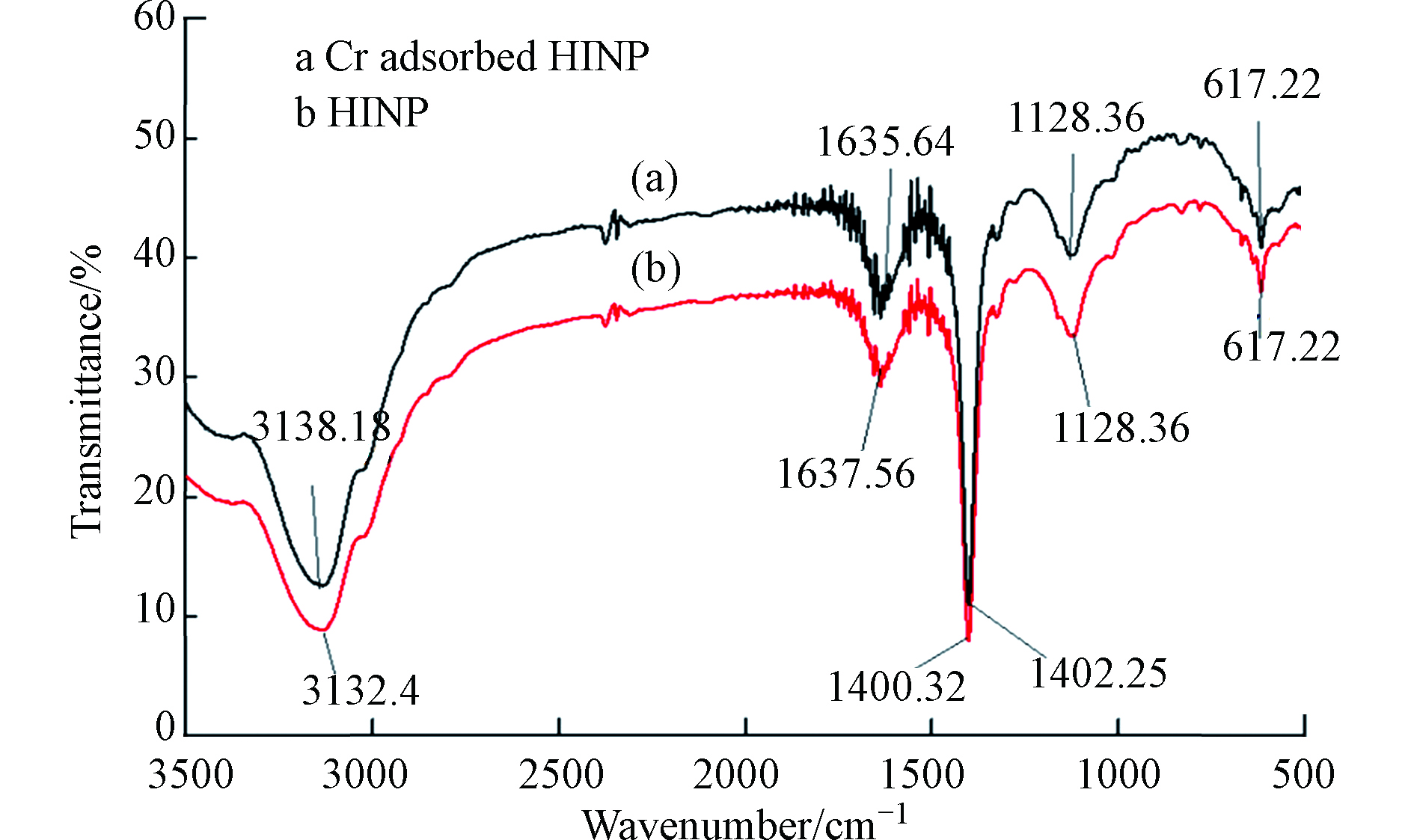
将HA通过各种改性方法制备出不溶性HA基吸附材料,已成为HA吸附铬离子的一种新的研究方向,Lu等[64]通过水热法制备了蒙脱石/ Fe3O4 /HA(MFH)纳米复合材料,将HA涂覆在Fe3O4纳米粒子上以及将HA涂覆的Fe3O4纳米粒子分散在蒙脱石上,可以保护Fe3O4纳米粒子免受氧化并抑制其聚集。研究表明,蒙脱石、Fe3O4纳米粒子和HA之间的协同作用可有效增强Fe3O4纳米粒子从废水中去除Cr(Ⅵ)的活性和效率。对HA、HA包覆(HINP)和未包覆氧化铁纳米粒子(INP)进行FTIR分析[65],如图4所示,在INP和HINP的561 cm−1和617 cm−1处出现了强吸收带,这是磁铁矿为主要相的Fe─O键拉伸振动导致[66]。如预期[67]那样,HINP光谱中在1400 cm−1处由于─CH2基团的振动出现了强吸收峰,而在INP中却没有观察到。在3419.79 cm−1和3421.72 cm−1处存在O─H伸缩振动,表明HA和INP之间的相互作用导致氢键的形成。HINP在1128 cm−1和1637 cm−1的吸收峰分别是由于C─O和芳香族C=C拉伸所致[68]。表明HA与INP表面之间的键合主要是通过配位交换反应。
Cr(Ⅵ)吸附后关键FTIR谱带的移动(图5)表明,金属结合过程发生在吸附剂表面。从3132.4 cm−1(HINP)到3138.18 cm−1(铬负载的HINP)的明显偏移表明表面羟基参与吸附。在铬吸附之后,HINP中烯烃基团的C = C拉伸也从1637.56 cm−1变为1635.64 cm−1[69]。
Zhang等[70]将黑液中的木质素转化为一种HA包覆的氮掺杂磁性多孔碳吸附剂(HA-N-MPC),用于去除水中的Cr(Ⅵ)。研究发现,在pH = 2时,HA-N-MPC对Cr(Ⅵ)的吸附主要是化学吸附且吸附能力非常高,为130.5 mg·g−1。HA-N-MPC对Cr(Ⅵ)去除主要包括3个过程:(1)由于表面积大且介孔结构发达,HA-N-MPC可以轻松快速地捕获Cr(Ⅵ)。(2)在pH值较低的水溶液中,通过静电作用和化学作用将带负电荷的Cr(Ⅵ)物质吸附到HA-N-MPC表面;(3)吸附的Cr(Ⅵ)通过HA-N-MPC吸附剂上某些电子给体的电子转移而还原为Cr(Ⅲ)。Zhang等[71]研究聚苯胺/HA纳米复合材料(PANI/HA)对水中Cr(Ⅵ)的吸附发现PANI/HA对材料稳定性和Cr(Ⅵ)去除效率有显著的提高,HA的存在可以通过阻止初始PANI的聚集来增强其稳定性。
PANI/HA-1吸附前后的形貌(图6)显示,PANI/HA-1粒子表现出短棒状粒子的形态,与其相比,铬负载的PANI/HA-1显示嵌入短棒状颗粒的球形小颗粒消失,这可能是由于负载铬与PANI/HA基质之间的相互作用。在pH值为3—7之间和较低的初始Cr(Ⅵ)浓度下,PANI/HA-1(HA为0.12 g)对Cr(Ⅵ)去除率较高;制备的PANI/HA-1大部分处于完全氧化状态,Cr(Ⅵ)在pH 5.0时被PANI / HA-1吸收,而PANI / HA-1从初始的完全氧化状态变为部分还原状态(图7)。PANI/HA-1对Cr(Ⅵ)的脱附研究表明,1.0 mol·L−1 NaOH洗脱液比HCl洗脱液具有更高的脱附效率,并且通过对电化学解吸,可以将PANI/HA中加载的Cr(Ⅵ)从PANI/HA基体中去除(图8),为重金属解吸开辟了新的途径。
-
HA是一种对Cr(Ⅵ)有强烈的络合和吸附能力的清洁的物质,能有效降低废水中Cr(Ⅵ)的毒性,减少铬的迁移,是废水中铬污染的优良去除剂。现有研究多集中于加强HA对含铬废水的去除效果,对HA与铬离子的微观作用机理研究较少。同时,HA与矿物质相互作用改变了矿物的反应性和表面特性,使得与铬相互作用的途径和动力学发生改变。因此,有必要对铬在不同性质HA矿物界面上的结合行为进行系统的研究,加深对铬离子的去除和迁移的理解。HA与铬离子络合并与纳米材料相互作用,不仅改变了纳米材料的环境行为,而且影响着纳米材料对铬离子的去除和迁移,需要进一步研究纳米材料、HA和铬离子之间的相互作用机理。HA在土壤中广泛存在强Cr(Ⅵ)截留效应,进一步研究HA与Cr(Ⅵ)的作用机制将有助于提高HA对环境中Cr(Ⅵ)的去除能力。
腐植酸对环境中Cr(Ⅵ)去除机理探究及展望
Research and perspect on the removal mechanism of humic acid to Cr(Ⅵ) in the environment
-
摘要: 铬是地下水中的主要污染物之一,尤其是六价
CrO2−4 ,HCrO−4 和Cr2O2−7 。由于其高迁移率、生物毒性以及潜在的对人类的致癌性,对人体健康和生态系统构成威胁。腐植酸(HA)具有特殊的物理化学性质,能与Cr(Ⅵ)发生吸附、离子交换和络合等作用,能有效迁移和转化环境中的Cr(Ⅵ)。HA与环境中Cr(Ⅵ)的结合机制已经有相关研究,但对于HA微观结构特点尚不清晰。因此,本文系统地阐述了HA的结构特征和与Cr(Ⅵ)的作用机理,分析了矿物质和纳米复合材料去除Cr(Ⅵ)的过程中HA的影响。总结了仍然存在的局限性和尚待解决的问题,并对HA处理含Cr(Ⅵ)废水的未来应用进行了展望。Abstract: Chromium is one of the main pollutants in groundwater, especiallyCrO2−4 ,HCrO−4 andCr2O2−7 . Due to its high mobility, biological toxicity and potential carcinogenicity to human beings, it poses a threat to human health and ecosystem. Humic acid (HA) has special physical and chemical properties, which can adsorb, exchange and complexate with Cr(Ⅵ), and and can be used to effectively transport and transform Cr(Ⅵ) in the environment. The binding mechanism of HA with Cr(Ⅵ) in the environment has been studied, but the microstructure of HA is still unclear. Therefore, the structural characteristics of HA and its interaction mechanism with Cr(Ⅵ) were systematically described, the effects of HA on Cr(Ⅵ) removal by minerals and nanocomposites were analyzed. The limitations and problems to be solved were summarized, and the future application of HA in Cr(Ⅵ) - containing wastewater treatment was prospected.-
Key words:
- humic acid /
- Cr(Ⅵ) /
- minerals /
- nanocomposites
-
由于自然过程(风化作用和生化作用)以及在世界范围内许多工业和制造过程中的使用不断增加,水生生态系统中的铬污染已成为一个突出问题,对生态环境和人类健康构成严重威胁。铬以Cr(Ⅲ)和Cr(Ⅵ)的形式存在于环境中,虽然Cr(Ⅲ)是人体必需的微量营养素,但Cr(Ⅵ)在水介质中可高度溶解,具有急性毒性、致突变性和致癌性,Cr(Ⅵ)的毒性是Cr(Ⅲ)的500倍[1]。
铬在制造和工业使用时,由于贮存和泄漏处理不当,导致多次被释放到环境中,造成地下水和土壤的污染。自然过程也会导致地下水中铬的浓度升高,例如,超镁铁质岩石可能具有较高的铬含量,其主要存在于母体矿物中的Cr(Ⅲ)中,但在风化过程中被四价锰氧化物氧化为Cr(Ⅵ)。尽管Cr(Ⅵ)的释放可能来自许多环节,但对环境危害最大的是铬铁矿加工残渣的处置不当。与铬铁矿加工残渣接触的水pH >12,并且水中的Cr(Ⅵ)浓度可超过1mmol·L−1[2-3]。当这种水不可避免地从废物堆中逸出到地球圈,会产生Cr(Ⅵ)羽流,其中pH值从高碱性值向天然土壤pH值变化。在水体环境中,Cr(Ⅵ)通常以可溶阴离子基团形式存在,如
CrO2−4 HCrO−4 Cr2O2−7 Cr2O2−7 Cr2O2−7 HCrO−4 CrO2−4 xCr3++(1−x)Fe3++3H2O⟶(CrxFe1−x)(OH)3(s)+3H+ (1) xCr3++(1−x)Fe3++2H2O⟶CrxFe1−xOOH(s)+3H+ (2) HA是一种通过微生物的分解和转化植物残留物而累积起来的高分子有机物质[14],具有生物化学稳定性高、表面积大、结构复杂、带有多种活性官能团等特点。它在全球总计达数万亿吨,通常存在于河流,湖泊,海洋,土壤和煤矿中。HA广泛应用于农业,林业,畜牧业和化学工业等领域[15-16],多项研究已将HA用作污染物的吸附剂[17-18]。尽管HA的存在会污染地下水和地表水,但其优点胜于其用于处理工业废水的缺点,并且可以在后续深加工中将其完全清洗。此外,HA表面富含官能团,包括羧基、羟基和羰基[19]。这些基团可用于与阳离子物质进行离子交换、吸附、络合和螯合[20-22]。目前国内外对利用HA去除水体和土壤中铬离子的研究尤为重视。
本文旨在系统地阐述HA的结构特征和与Cr(Ⅵ)的作用机理,分析了矿物质和纳米复合材料去除Cr(Ⅵ)的过程中HA的影响,总结了仍然存在的局限性和尚待解决的问题,以便为HA处理含Cr(Ⅵ)废水的应用提供未来展望。
1. 腐植酸的结构特征及去除Cr(Ⅵ)机理(Structure characteristics and Cr(Ⅵ) removal mechanism of humic acid)
1.1 腐植酸的结构特征
HA是包含于腐殖质(HS)的大分子,HS是由细菌和真菌对植物和动物的残留物进行腐烂和转化而产生的深色、异质的有机化合物。主要的HS前体分子是通过植物产生的生物聚合物和蛋白质的解聚和氧化形成的,这些分子含有未分解的聚合物片段和酚类、羟基、羧基和氨基残基[23]。传统的“聚合物模型”假设前体是由微生物合成的随机卷曲的聚合物大分子,而有研究表明HS是超分子缔合的[24-25]。两亲性前体分子聚集在一起形成胶束状粒子,而其它植物降解的生物分子则与疏水或亲水结构域有关[26-27]。虽然目前大多数证据支持这种“胶束模型”,但是在HS数百万年的老化和降解过程中并不排除聚合物大分子的形成 [28-29]。
HA是可溶于碱性介质,部分可溶于水而不溶于酸性介质的HS组分[30]。由于具有两亲性,HA在中性至酸性条件下会形成胶束状结构,称为假胶束[31]。已经探索出该性质可用于污染修复[32],并增加疏水性药物的水溶性[33-34]。HA含有不同的官能团,其数量取决于HA的来源、年龄、气候和提取的环境条件。HA的各种功能主要归因于酚和羧酸官能团,它们允许—OH/—OOH的脱质子化。这使得HA拥有了许多功能,如具有改善植物生长和增强营养[35],与重金属络合[36],抗病毒和抗炎活性[37]。此外,HA结构中酚类、羧酸和醌的存在与其抗氧化、抗突变/去突变和杀菌/杀菌活性有关[38]。HA的化学组成可能会因来源,年龄,气候和生物学条件而异,因此很难对这些物质进行精确表征。它们的分子量在2.0 kDa到1300 kDa之间[39],并且它们包含许多官能团,如图1所示[33],HA主要由酚、羧酸、烯醇、醌和醚官能团组成,但也可能包括糖和肽。但是,酚羟基和羧基在HA结构中更为普遍。HA分子的巨大结构由亲水部分(由—OH基团组成)和疏水部分(由脂族链和芳环组成)组成。醌是电子接受基团,负责产生活性氧(ROS)。它们被还原为半醌,由其芳环稳定,并进一步还原为更加稳定的对苯二酚(图2)[40]。Aeschbacher等[41]研究了从不同来源中获得的HA中的电子接受部分(醌)和供电子部分(酚类),发现这些物质的来源和年龄直接影响其氧化还原特性。13C NMR分析表明,与陆生HA相比,水生HA具有更高的给电子数和更低的电子接受基团[40]。Aeschbacher等[41]做出以下假设:HA中的酚羟基会减慢醌的氧化转化,从而提高其在含氧环境中的持久性。
1.2 腐植酸去除铬离子机理
水生环境中金属阳离子在溶液中的作用与H+相同,即电荷中和,电荷越高,阳离子在HA假胶束的形成中越有效。此外,多价阳离子与相邻链上的酚羟基和羧基基团相互作用,增强了假胶束结构域和去污作用。von Wandruszka[42]提出这种相互作用的机制如下:HA分子与金属阳离子之间的相互作用最初完全是静电作用,并且阳离子会移动到HA中热力学上更稳定的位置。此过程形成球状HA-金属络合物。对HA与金属离子结合的研究表明,这种相互作用随金属不同而变化,并受金属离子浓度以及HA的来源、分子量和浓度影响[43]。Christl等[43]证明HA与金属离子之间的结合能力与HA的分子大小有关。用尺寸排阻色谱-聚丙烯酰胺凝胶电泳分离HA得到的3个级分的13C NMR分析证实,分子量较低的HA组分具有最多的酚羟基和羧基,因此是可以最有效地结合金属离子的组分。此外,Christl和Kretzschmar[44]发现在较高金属离子浓度下,可观察到两个HA大小级分之间的差异,因为较小的级分由于官能团数较多而具有较高的结合能力。HA通过与金属离子结合形成络合物,从而去除水和土壤中的重金属,是一个很好的重金属污染补救措施。重金属污染的水(包括铬污染)一般是呈酸性,使得HA的溶解度会降低,从而干扰HA-金属配合物的形成。但是铬污染的水中铬以阴离子形式存在,酸性环境下反而有利于HA对铬的还原。
Cr(Ⅵ)能与含有酚羟基、醇羟基的有机物反应从而被还原为Cr(Ⅲ)。与这些基团的反应被认为涉及到一种铬酸盐中间体,该中间体可由这些基团与单体H2CrO4和
HCrO−4 CrO2−4 CrO2−4 HA还原Cr(Ⅵ)降低了铬的毒性,产生的Cr(Ⅲ)与HA中的两个碳原子形成一个内层吸附复合物,大大减少了Cr(Ⅲ)扩散或重新氧化成可移动Cr(Ⅵ)的机会。Cr(Ⅵ)的还原速率和还原Cr(Ⅵ)的总容量与所使用的HA的反应活性呈正比,特别是HA中酚羟基和醇羟基的密度。Zhang等[53]研究了从东北典型黑土中提取的HA在pH=1.0时对Cr(Ⅵ)的还原,结果表明,HA中的酚羟基和醇羟基官能团是还原Cr(Ⅵ)的主要电子给体,而不是羧基和羰基,它们更可能参与铬的络合反应,而且HA颗粒表面的酚羟基和醇羟基对Cr(Ⅵ)还原的活性远高于其内部。Aldmour等[54]研究泥炭腐植酸(PHA)和褐煤腐植酸(AHA)对Cr(Ⅵ)的还原,发现HA将Cr(Ⅵ)还原为Cr(Ⅲ)通常涉及与芳香族的反应,特别是与酚羟基的反应。PHA的变化表明醇羟基也可能发生了反应。HA样品与过量的Cr(Ⅵ)反应,使pH值从3升到7,表明在酸性体系中Cr(Ⅵ)与HA反应消耗了H+。Cr(Ⅵ)的还原必然导致HA的氧化,但13C-NMR谱没有鉴定出产生的官能团(图3)。研究表明,Cr(Ⅲ)吸附在HA上导致羰基Cr(Ⅲ)络合物的形成和与电子再分配相关的屏蔽导致羰基的13C-NMR谱信号减少。因此,用PHA和AHA还原Cr(Ⅵ)很可能会生成新的羰基[52]。
2. 腐植酸对矿物质和纳米复合材料去除Cr(Ⅵ)的影响(Effect of humic acid on Cr(Ⅵ) removal by minerals and nanocomposites)
2.1 腐植酸对矿物质去除Cr(Ⅵ)的影响
铬的界面行为是了解铬在污染环境中的环境效应的基础,然而,复杂的表面会导致铬表现出各种各样的行为。铬的迁移和转化主要受固-液界面的相互作用控制,尤其是沿铁氧化物界面的相互作用。HS与矿物相互作用时,带负电的HS可能会保留在带正电的矿物表面上。因此,HS和矿物之间的相互作用可能会改变任何一种物质的原始特性,并产生复杂的HS矿物[55]。因此,有必要研究铬与具有不同特性的各种HS矿物界面的相互作用。HS是一种富含官能团的有机分子,包含HA和黄腐酸(FA),HA可以吸附在矿物表面,改变矿物的的反应性和表面特性[56]。HA对Cr(Ⅵ)离子具有很强的络合能力[57],并且对矿物表面具有很高的亲和力[58],已被认为是控制水中各种元素行为的重要因素。Kantar等[59]在pH为3—8体系中,研究HA对含Ca2+、Mg2+等阳离子体系中黄铁矿去除Cr(Ⅵ)的影响,发现HA对黄铁矿吸收Cr(Ⅵ)有抑制作用。HA吸附在黄铁矿表面,与Cr(Ⅵ)竞争黄铁矿表面位置;HA通过与黄铁矿反应位点结合改变了黄铁矿表面的静电特性,导致反应位点与
CrO2−4 HA作为一种自然水生环境中普遍存在的天然有机质,已被用于磁铁矿的表面改性来修复受Cr(Ⅵ)污染的水。由于HA官能团和磁铁矿的表面羟基之间的络合-配位交换反应,HA对磁铁矿具有高亲和力。完全被HA覆盖的磁铁矿颗粒具有HA的特性和磁铁矿的磁性,与金属氧化物相关的特性相反[55],溶液pH > 3时,零电荷点和负电荷点的低pH值抑制了它们的团聚。磁铁矿中的Fe(Ⅱ)可以将Cr(Ⅵ)还原为Cr(Ⅲ),随后Cr(Ⅲ)被氧化铁表面的羟基螯合,形成内层表面配合物[60]。但是,磁铁矿中的Fe(II)极易自氧化形成磁性降低的Fe(Ⅲ),不利于磁铁矿在磁场条件下的分离。Jiang等[61]用HA包覆磁铁矿纳米颗粒在pH = 4条件下还原Cr(Ⅵ)发现,HA使Fe(Ⅱ)不受自氧化和Cr(Ⅵ)电子转移的影响,并通过HA将Cr(Ⅵ)还原为无毒的Cr(Ⅲ)来保留材料的磁性,使得在磁场条件下HA-Fe3O4颗粒在处理后易于分离。
Zheng等[62]研究了HA在铁矿(磁铁矿和赤铁矿)吸附Cr(Ⅵ)过程中的作用以及Cr(Ⅵ)吸附到铁矿HS络合物表面上的界面行为。发现化学吸附起了重要作用,且在较高温度下,磁铁矿HA复合物、磁铁矿FA复合物和赤铁矿FA复合物对Cr(Ⅵ)的吸附较好;而在较低的温度下,磁铁矿、赤铁矿和赤铁矿HA复合物对Cr(Ⅵ)吸附能力较好。此外,铁矿物上的HA分子对Cr(Ⅵ)的亲和力比FA分子弱。化学吸附和物理吸附同时存在于Cr(Ⅵ)在铁矿物质和铁矿HS络合物上的吸附。此外,除赤铁矿HA复合物外,所有铁矿物质和铁矿HS络合物的化学吸附作用最为明显。
Yu等[63]研究Fe(Ⅱ)诱导水铁矿(Fh)-HA共沉淀转化过程中吸附Cr(Ⅵ)的行为,发现在Fh-HA的老化过程中,在无Fe(Ⅱ)存在下,共沉淀HA通过吸附和络合作用将Cr(Ⅵ)固定在赤铁矿中;在Fe(Ⅱ)存在下,Fe(Ⅱ)能将吸附的Cr(Ⅵ)还原为Cr(Ⅲ)(式3、4)。
3Fe2++HCrO−4+7H+⟶3Fe3++Cr3++4H2O (3) 3Fe2++CrO2−4+8H+⟶3Fe3++Cr3++4H2O (4) 此外,吸附的Cr(Ⅵ)在Fh和Fh-HA的相变过程中,可以在水相Fe(Ⅱ)的促进下,通过结构掺入的方式结合到次生矿中。在中性条件下,当Fe(Ⅱ)初始浓度较低(0.2 mmol·L−1)时,水溶性残余Cr(Ⅵ)可并入次生矿物中的Cr(Ⅲ)- Fe(Ⅱ)共沉淀物中。最后,通过共沉淀HA还原和络合次生矿物中的Cr(Ⅵ),进一步促进转化过程中Cr(Ⅵ)的固定化。
2.2 腐植酸对纳米复合材料去除Cr(Ⅵ)的影响
纳米材料由于大比表面积和高反应活性广泛用于去除废水中重金属。HA普遍存在于水生环境中,并具有多种官能团,可使其与金属离子络合并与纳米材料相互作用。这些相互作用不仅可以改变纳米材料的环境行为,而且可以影响纳米材料对重金属的去除和迁移。因此,所涉及的相互作用和潜在机制值得进行深入研究。
将HA通过各种改性方法制备出不溶性HA基吸附材料,已成为HA吸附铬离子的一种新的研究方向,Lu等[64]通过水热法制备了蒙脱石/ Fe3O4 /HA(MFH)纳米复合材料,将HA涂覆在Fe3O4纳米粒子上以及将HA涂覆的Fe3O4纳米粒子分散在蒙脱石上,可以保护Fe3O4纳米粒子免受氧化并抑制其聚集。研究表明,蒙脱石、Fe3O4纳米粒子和HA之间的协同作用可有效增强Fe3O4纳米粒子从废水中去除Cr(Ⅵ)的活性和效率。对HA、HA包覆(HINP)和未包覆氧化铁纳米粒子(INP)进行FTIR分析[65],如图4所示,在INP和HINP的561 cm−1和617 cm−1处出现了强吸收带,这是磁铁矿为主要相的Fe─O键拉伸振动导致[66]。如预期[67]那样,HINP光谱中在1400 cm−1处由于─CH2基团的振动出现了强吸收峰,而在INP中却没有观察到。在3419.79 cm−1和3421.72 cm−1处存在O─H伸缩振动,表明HA和INP之间的相互作用导致氢键的形成。HINP在1128 cm−1和1637 cm−1的吸收峰分别是由于C─O和芳香族C=C拉伸所致[68]。表明HA与INP表面之间的键合主要是通过配位交换反应。
Cr(Ⅵ)吸附后关键FTIR谱带的移动(图5)表明,金属结合过程发生在吸附剂表面。从3132.4 cm−1(HINP)到3138.18 cm−1(铬负载的HINP)的明显偏移表明表面羟基参与吸附。在铬吸附之后,HINP中烯烃基团的C = C拉伸也从1637.56 cm−1变为1635.64 cm−1[69]。
Zhang等[70]将黑液中的木质素转化为一种HA包覆的氮掺杂磁性多孔碳吸附剂(HA-N-MPC),用于去除水中的Cr(Ⅵ)。研究发现,在pH = 2时,HA-N-MPC对Cr(Ⅵ)的吸附主要是化学吸附且吸附能力非常高,为130.5 mg·g−1。HA-N-MPC对Cr(Ⅵ)去除主要包括3个过程:(1)由于表面积大且介孔结构发达,HA-N-MPC可以轻松快速地捕获Cr(Ⅵ)。(2)在pH值较低的水溶液中,通过静电作用和化学作用将带负电荷的Cr(Ⅵ)物质吸附到HA-N-MPC表面;(3)吸附的Cr(Ⅵ)通过HA-N-MPC吸附剂上某些电子给体的电子转移而还原为Cr(Ⅲ)。Zhang等[71]研究聚苯胺/HA纳米复合材料(PANI/HA)对水中Cr(Ⅵ)的吸附发现PANI/HA对材料稳定性和Cr(Ⅵ)去除效率有显著的提高,HA的存在可以通过阻止初始PANI的聚集来增强其稳定性。
PANI/HA-1吸附前后的形貌(图6)显示,PANI/HA-1粒子表现出短棒状粒子的形态,与其相比,铬负载的PANI/HA-1显示嵌入短棒状颗粒的球形小颗粒消失,这可能是由于负载铬与PANI/HA基质之间的相互作用。在pH值为3—7之间和较低的初始Cr(Ⅵ)浓度下,PANI/HA-1(HA为0.12 g)对Cr(Ⅵ)去除率较高;制备的PANI/HA-1大部分处于完全氧化状态,Cr(Ⅵ)在pH 5.0时被PANI / HA-1吸收,而PANI / HA-1从初始的完全氧化状态变为部分还原状态(图7)。PANI/HA-1对Cr(Ⅵ)的脱附研究表明,1.0 mol·L−1 NaOH洗脱液比HCl洗脱液具有更高的脱附效率,并且通过对电化学解吸,可以将PANI/HA中加载的Cr(Ⅵ)从PANI/HA基体中去除(图8),为重金属解吸开辟了新的途径。


3. 结语(Conclusion)
HA是一种对Cr(Ⅵ)有强烈的络合和吸附能力的清洁的物质,能有效降低废水中Cr(Ⅵ)的毒性,减少铬的迁移,是废水中铬污染的优良去除剂。现有研究多集中于加强HA对含铬废水的去除效果,对HA与铬离子的微观作用机理研究较少。同时,HA与矿物质相互作用改变了矿物的反应性和表面特性,使得与铬相互作用的途径和动力学发生改变。因此,有必要对铬在不同性质HA矿物界面上的结合行为进行系统的研究,加深对铬离子的去除和迁移的理解。HA与铬离子络合并与纳米材料相互作用,不仅改变了纳米材料的环境行为,而且影响着纳米材料对铬离子的去除和迁移,需要进一步研究纳米材料、HA和铬离子之间的相互作用机理。HA在土壤中广泛存在强Cr(Ⅵ)截留效应,进一步研究HA与Cr(Ⅵ)的作用机制将有助于提高HA对环境中Cr(Ⅵ)的去除能力。
-
-
[1] KUMAR P A, RAY M, CHAKRABORTY S. Hexavalent chromium removal from wastewater using aniline formaldehyde condensate coated silica gel [J]. J Hazard Mater, 2007, 143(1/2): 24-32. [2] STEWART D I, BURKE I T, HUGHES-BERRY D V, et al. Microbially mediated chromate reduction in soil contaminated by highly alkaline leachate from chromium containing waste [J]. Ecol Eng, 2010, 36: 211-221. doi: 10.1016/j.ecoleng.2008.12.028 [3] MATERN K, WEIGAND H, SINGH A, et al. Environmental status of groundwater affected by chromite ore processing residue (COPR) dumpsites during pre-monsoon and monsoon seasons [J]. Environ Sci Pollut Res, 2017, 24: 3582-3592. doi: 10.1007/s11356-016-8110-2 [4] 代启虎, 李冉, 葛俊苗, 等. 短乳杆菌对Cr3+的吸附及动力学和热力学拟合 [J]. 环境化学, 2019, 38(3): 626-634. doi: 10.7524/j.issn.0254-6108.2018042602 DAI Q H, LI R, GE J M, et al. Adsorption and kinetic and thermodynamic fitting of lactobacillus brevis to Cr3+ [J]. Environmental Chemistry, 2019, 38(3): 626-634(in Chinese). doi: 10.7524/j.issn.0254-6108.2018042602
[5] ZHANG J H, ZHANG L L, ZHOU S Y. Magnetically separable attapulgite -TiO2-FexOy composites with superior activity towards photodegradation of methyl orange under visible light radiation [J]. Journal of Industrial and Engineering Chemistry, 2014, 20: 3884-3889. doi: 10.1016/j.jiec.2013.12.094 [6] RAI D, EARY L, ZACHARA J. Environmental chemistry of chromium [J]. Sci Total Environ, 1989, 86: 15-23. doi: 10.1016/0048-9697(89)90189-7 [7] EARY L, RAI D. Chromate removal from aqueous wastes by reduction with ferrous ion [J]. Environmental Science & Technology, 1988, 22: 972-977. [8] SASS B M, RAI D. Solubility of amorphous chromium (Ⅲ)-iron (Ⅲ) hydroxide solid solutions [J]. Inorg Chem, 1987, 26: 2228-2232. doi: 10.1021/ic00261a013 [9] 高卫国, 钱林波, 韩璐, 等. 锰铁氧体吸附及催化柠檬酸还原六价铬的过程及机理 [J]. 环境化学, 2018, 37(7): 1525-1533. doi: 10.7524/j.issn.0254-6108.2017101302 GAO W G, QIN L B, HAN L, et al. Iron manganese minerals catalyzed Cr(Ⅵ) reduction by citric acid and its mechanism [J]. Environmental Chemistry, 2018, 37(7): 1525-1533(in Chinese). doi: 10.7524/j.issn.0254-6108.2017101302
[10] WU B, LIU C, FU C, et al. Selective separation of Cr(Ⅵ) and V(Ⅴ) from solution by simple pH controlled two-step adsorption/desorption process with ZrO2 [J]. Chem Eng J, 2019, 373: 1030-1041. doi: 10.1016/j.cej.2019.05.131 [11] SINGH R, KUMAR A, KIRROLIA A, et al. Removal of sulphate, COD and Cr(Ⅵ) in simulated and real wastewater by sulphate reducing bacteria enrichment in small bioreactor and FTIR study [J]. Bioresour Technol, 2011, 102: 677-682. doi: 10.1016/j.biortech.2010.08.041 [12] XIE B, SHAN C, XU Z, et al. One-step removal of Cr(Ⅵ) at alkaline pH by UV/sulfite process: Reduction to Cr(Ⅲ) and in situ Cr(Ⅲ) precipitation [J]. Chem Eng J, 2017, 308: 791-797. doi: 10.1016/j.cej.2016.09.123 [13] ZHANG J, GAO N, CHEN F, et al. Improvement of Cr (Ⅵ) photoreduction under visible-light by g-C3N4 modified by nano-network structured palygorskite [J]. Chem Eng J, 2019, 358: 398-407. doi: 10.1016/j.cej.2018.10.083 [14] GOLI E, HIEMSTRA T, RAHNEMAIE R. Interaction of boron with humic acid and natural organic matter: Experiments and modeling [J]. Chem. Geol, 2019, 515: 1-8. doi: 10.1016/j.chemgeo.2019.03.021 [15] BIJANZADEH E, NADERI R, EGAN T P. Exogenous application of humic acid and salicylic acid to alleviate seedling drought stress in two corn (Zea mays L. ) hybrids [J]. J. Plant Nutr, 2019, 42(13): 1483-1495. doi: 10.1080/01904167.2019.1617312 [16] GHANBARPOUR E, REZAEI M, LAWSON S. Reduction of cracking in pomegranate fruit after foliar application of humic acid, calcium-boron and kaolin during water stress [J]. Erwerbs-obstbau, 2019, 61: 29-37. doi: 10.1007/s10341-018-0386-6 [17] CHEN Q, YIN D, ZHU S, et al. Adsorption of cadmium (II) on humic acid coated titanium dioxide [J]. J. Colloid Interface Sci, 2012, 367: 241-248. doi: 10.1016/j.jcis.2011.10.005 [18] ZHANG X, ZHANG P, WU Z, et al. Adsorption of methylene blue onto humic acid-coated Fe3O4 nanoparticles [J]. Colloids Surf A, 2013, 435: 85-90. doi: 10.1016/j.colsurfa.2012.12.056 [19] MACHIANI M A, REZAEI-CHIYANEH E, JAVANMARD A, et al. Evaluation of common bean (Phaseolus vulgaris L. ) seed yield and quali-quantitative production of the essential oils from fennel (Foeniculum vulgare) and dragonhead (Dracocephalum moldavica) in intercropping system under humic acid application [J]. J Cleaner Prod, 2019, 235: 112-122. doi: 10.1016/j.jclepro.2019.06.241 [20] LUO H, HE D, ZHU W, et al. Humic acid induced for mation of tobermorite upon hydrothermal treatment with municipal solid waste in cineration bottom ash and its application for efficient removal of Cu (Ⅱ) ions [J]. Waste Manage, 2019, 84: 83-90. doi: 10.1016/j.wasman.2018.11.037 [21] ZHOU L, MONREAL C M, XU S, et al. Effect of bentonite-humic acid application on the improvement of soil structure and maize yield in a sandy soil of a semi-arid region [J]. Geoderma, 2019, 338: 269-280. doi: 10.1016/j.geoderma.2018.12.014 [22] LALONDE K, MUCCI A, OUELLET A, et al. Preservation of organic matter in sediments promoted by iron[J]. Nature 2012, 483 (7388): 198−200. [23] ARO N, PAKULA T, PENTTILÄ M. Transcriptional regulation of plant cell wall degradation by filamentous fungi [J]. FEMS Microbiol Rev, 2005, 29: 719-739. doi: 10.1016/j.femsre.2004.11.006 [24] BROOKES P C, CAYUELA M L, CONTIN M, et al. The mineralisation of fresh and humified soil organic matter by the soil microbial biomass [J]. Waste Manag, 2008, 28: 716-722. doi: 10.1016/j.wasman.2007.09.015 [25] SUTTON R, SPOSITO G. Molecular structure in soil humic substances: the new view [J]. Environ Sci Technol, 2005, 39: 9009-9015. doi: 10.1021/es050778q [26] KÖGEL-KNABNER I. Analytical approaches for characterizing soil organic matter [J]. Org Geochem, 2000, 31: 609-625. doi: 10.1016/S0146-6380(00)00042-5 [27] SIMPSON A J, KINGERY W L, HAYES M H, et al. Molecular structures and associations of humic substances in the terrestrial environment [J]. Naturwissenschaften, 2002, 89: 84-88. doi: 10.1007/s00114-001-0293-8 [28] FAN T M, LANE A, CHEKMENEV E, et al. Synthesis and physico-chemical properties of peptides in soil humic substances [J]. J Pept Res, 2004, 63: 253-264. doi: 10.1111/j.1399-3011.2004.00142.x [29] BURDON J. Are the traditional concepts of the structures of humic substances realistic? [J]. Soil Sci, 2001, 166: 752-769. doi: 10.1097/00010694-200111000-00004 [30] KLUČÁKOVÁ M, PEKAŘ M. Solubility and dissociation of lignitic humic acids in water suspension [J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2005, 252(2/3): 157-163. [31] VON WANDRUSZKA R, RAGLE C, ENGEBRETSON R. The role of selected cations in the formation of pseudomicelles in aqueous humic acid [J]. Talanta, 1997, 44(5): 805-809. doi: 10.1016/S0039-9140(96)02116-9 [32] TEJEDA-AGREDANO M C, MAYER P, ORTEGA-CALVO J J. The effect of humic acids on biodegradation of polycyclic aromatic hydrocarbons depends on the exposure regime [J]. Environ Pollut, 2014, 184: 435-442. doi: 10.1016/j.envpol.2013.09.031 [33] MIRZA M A, AGARWAL S P, RAHMAN M A, et al. Role of humic acid on oral drug delivery of an antiepileptic drug[J], Drug Dev Ind Pharm, 2011, 37(3): 310–319. [34] MARTINI S, D'ADDARIO C, BONECHI C, et al. Increasing photostability and water-solubility of carotenoids: Synthesis and characterization of β-carotene-humic acid complexes [J]. J Photochem Photobiol B Biol, 2010, 101(3): 355-361. doi: 10.1016/j.jphotobiol.2010.08.008 [35] TAHIR M M, KHURSHID M, KHAN M Z, et al. Lignite-derived humic acid effect on growth of wheat plants in different soils [J]. Pedosphere, 2011, 21(1): 124-131. doi: 10.1016/S1002-0160(10)60087-2 [36] YATES L M, VON WANDRUSZKA R. Decontamination of polluted water by treatment with a crude humic acid blend [J]. Environ Sci Technol, 1999, 33(12): 2076-2080. doi: 10.1021/es980408k [37] JUNEK R, MORROW R, SCHOENHERR J I, et al. Bimodal effect of humic acids on the LPS-induced TNF-α release from differentiated U937 cells[J], Phytomedicine, 2009, 16(5): 470-476. [38] KHIL'KO S L, EFIMOVA I V, SMIRNOVA O V. Antioxidant properties of humic acids from brown coal[J], Solid Fuel Chem, 2011, 45(6): 367-371. [39] DE MELO B A G, MOTTA F L, SANTANA M H A. Humic acids: Structural properties and multiple functionalities for novel technological developments [J]. Materials Science and Engineering C, 2016, 62: 967-974. doi: 10.1016/j.msec.2015.12.001 [40] SCOTT D T, MCKNIGHT D M, BLUNT-HARRIS E L, et al. Quinone moieties act as electron acceptors in the reduction of humic substances by humics-reducing microorganisms [J]. Environ Sci Technol, 1998, 32(19): 2984-2989. doi: 10.1021/es980272q [41] AESCHBACHER M, GRAF C, SCWARZENBACH R P, et al. Antioxidant properties of humic substances [J]. Environ Sci Technol, 2012, 46: 4916-4925. doi: 10.1021/es300039h [42] VON WANDRUSZKA R. Humic acids: their detergent qualities and potential uses in pollution remediation [J]. Geochem Trans, 2000, 1: 10-15. . doi: 10.1186/1467-4866-1-10 [43] CHRISTL I, MILNE C J, KINNIBURGH D G, et al. Relating ion binding by fulvic and humic acids to chemical composition and molecular size. 2. Metal binding [J]. Environ Sci Technol, 2001, 35(12): 2512-2517. doi: 10.1021/es0002520 [44] CHRISTL I, KRETZSCHMAR R. Relating ion binding by fulvic and humic acids to chemical composition and molecular size. 1. Proton binding [J]. Environ Sci Technol, 2001, 35(12): 2505-2511. doi: 10.1021/es0002518 [45] LEE D G, STEWART R. Oxidation of aliphatic secondary alcohols by chromium (Ⅵ) in concentrated sulfuric acid solutions [J]. J Org Chem, 1967, 32: 2868-2871. doi: 10.1021/jo01284a047 [46] ELOVITZ M S, FISH W. Redox interactions of Cr (Ⅵ) and substituted phenols: products and mechanism [J]. Environ Sci Technol, 1995, 29: 1933-1943. doi: 10.1021/es00008a010 [47] CHEN S Y, HUANG S W, CHIANG P N, et al. Influence of chemical compositions and molecular weights of humic acids on Cr(Ⅵ) photo-reduction [J]. J Hazard Mater, 2011, 197: 337-344. doi: 10.1016/j.jhazmat.2011.09.091 [48] HUANG S W, CHIANG P N, LIU J C, et al. Chromate reduction on humic acid derived from a peat soil e exploration of the activated sites on HAs for chromate removal [J]. Chemosphere, 2012, 87: 587-594. doi: 10.1016/j.chemosphere.2012.01.010 [49] ZHAO T T, GE W Z, NIE Y X, et al. Highly efficient detoxification of Cr(Ⅵ) by brown coal and kerogen: process and structure studies [J]. Fuel Process Technol, 2016, 150: 71-77. doi: 10.1016/j.fuproc.2016.05.001 [50] OHTA A, KAGI H, TSUNO H, et al. Speciation study of Cr(VI/III) reacting with humic substances and determination of local structure of Cr binding humic substances using XAFS spectroscopy [J]. Geochem J, 2012, 46: 409-420. doi: 10.2343/geochemj.2.0222 [51] OHTA A. Speciation study of Cr in a geochemical reference material sediment series using sequential extraction and XANES spectroscopy [J]. Geostand Geoanal Res, 2015, 39: 87-103. doi: 10.1111/j.1751-908X.2014.00292.x [52] ZHANG J, CHEN L, YIN H, et al. Mechanism study of humic acid functional groups for Cr(Ⅵ) retention: two-dimensional FTIR and 13C CP/MAS NMR correlation spectroscopic analysis [J]. Environ Pollut, 2017, 225: 86-92. doi: 10.1016/j.envpol.2017.03.047 [53] ZHANG J, YIN H L, WANG H, et al. Reduction mechanism of hexavalent chromium by functional groups of undissolved humic acid and humin fractions of typical black soil from Northeast China [J]. Environmental Science and Pollution Research, 2018, 25(17): 1-9. [54] ALDMOUR S T, BURKE I T, BRAY A W, et al. Abiotic reduction of Cr(Ⅵ) by humic acids derived from peat and lignite: kinetics and removal mechanism [J]. Environmental Science and Pollution Research, 2019, 26: 4717-4729. doi: 10.1007/s11356-018-3902-1 [55] HU J D, ZEVI Y, KOU X M, et al. Effect of dissolved organic matter on the stability of magnetite nanoparticles under different pH and ionic strength conditions [J]. Sci Total Environ, 2010, 408(16): 3477-3489. doi: 10.1016/j.scitotenv.2010.03.033 [56] AVENA M J, VERMEER A W P, KOOPAL L K. Volume and structure of humic acids studied by viscometry pH and electrolyte concentration effects [J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 1999, 151: 213-224. [57] LI Y, YUE Q Y, GAO B Y, et al. Adsorption thermodynamic and kinetic studies of dissolved chromium onto humic acids [J]. Colloids and Surfaces B:Biointerfaces, 2008, 65: 25-29. doi: 10.1016/j.colsurfb.2008.02.014 [58] SALMAN M, EL-ESWED B, KHALILI F. Adsorption of humic acid on bentonite [J]. Applied Clay Science, 2007, 38: 51-56. doi: 10.1016/j.clay.2007.02.011 [59] KANTAR C, BULBUL M S, KESKIN S. Role of Humic Substances on Cr(Ⅵ) Removal from Groundwater with Pyrite [J]. Water Air Soil Pollut, 2017, 228: 48-58. doi: 10.1007/s11270-016-3233-0 [60] KENDELEWICZ T, LIU P, DOYLE C S, et al. Spectroscopic study of the reaction of aqueous Cr(Ⅵ) with Fe3O4 (Ⅲ) surfaces [J]. Surf Sci, 2000, 469(2-3): 144-163. doi: 10.1016/S0039-6028(00)00808-6 [61] JIANG W J, CAI Q, XU W, et al. Cr(Ⅵ) adsorption and reduction by humic acid coated on magnetite [J]. Environ Sci Technol, 2014, 48: 8078-8085. doi: 10.1021/es405804m [62] ZHENG Z Y, ZHENG Y, TIAN X C, et al. Interactions between iron mineral-humic complexes and hexavalent chromium and the corresponding bio-effects [J]. Environmental Pollution, 2018, 241: 265-271. doi: 10.1016/j.envpol.2018.05.060 [63] YU G D, FU F L, YE C J, et al. Behaviors and fate of adsorbed Cr(Ⅵ) during Fe(Ⅱ)-induced transformation of ferrihydrite-humic acid coprecipitates [J]. Journal of Hazardous Materials, 2020: doi. [64] LU H J, WANG J K, LI F, et al. Highly efficient and reusable montmorillonite/Fe3O4/humic acid nanocomposites for simultaneous removal of Cr(Ⅵ) and aniline [J]. Nanomaterials, 2018, 8: 537-551. doi: 10.3390/nano8070537 [65] SINGARAJ S G, MAHANTY B, BALACHANDRAN D, et al. Adsorption and desorption of chromium with humic acid coated iron oxide nanoparticles [J]. Environmental Science and Pollution Research, 2019, 26: 30044-30054. doi: 10.1007/s11356-019-06164-0 [66] NIU H, ZHANG D, ZHANG S, et al. Humic acid coated Fe3O4 magnetic nanoparticles as highly efficient Fenton-like catalyst for complete mineralization of sulfathiazole [J]. J Hazard Mater, 2011, 190: 559-565. doi: 10.1016/j.jhazmat.2011.03.086 [67] HONG-BO F, XIE Q, SHUO C, et al. Interaction of humic substances and hematite: FTIR study [J]. J Environ Sci, 2005, 17: 43-47. [68] KOESNARPADI S, SANTOSA S J, SISWANTA D, et al. Synthesis and characterization of magnetite nanoparticle coated humic acid (Fe3O4/HA) [J]. Procedia Environ Sci, 2015, 30: 103-108. doi: 10.1016/j.proenv.2015.10.018 [69] NAIYA T K, SINGHA B, DAS S K. FTIR study for the Cr(Ⅵ) removal from aqueous solution using rice waste [J]. International Conference on Chemistry and Chemical Process-IPCBEE, 2011, 10: 114-119. [70] ZHANG T, WEI S, WATERHOUSE G I N, et al. Chromium (Ⅵ) adsorption and reduction by humic acid coated nitrogen-doped magnetic porous carbon [J]. Powder Technology, 2020, 360: 55-64. doi: 10.1016/j.powtec.2019.09.091 [71] ZHANG Y, LI Q, SUN L, et al. Batch adsorption and mechanism of Cr(Ⅵ) removal from aqueous solution by polyaniline/humic acid [J]. J Environ Eng, 2011, 137: 1158-1164. doi: 10.1061/(ASCE)EE.1943-7870.0000453 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4971
- HTML全文浏览数: 4971
- PDF下载数: 133
- 施引文献: 0











