

-
目前,全球正面临能源危机和环境污染的双重挑战。光解水制氢将太阳能转化成化学能,既能提供清洁能源又可避免传统能源的环境污染。因此,光解水制氢引起了人们的广泛关注。由于可见光占太阳光的绝大部分,拓宽可见光的利用是提高太阳光利用率的有效手段。基于此,开发可见光驱动的光催化剂对于光解水制氢具有十分重要的意义。
近年来石墨相氮化碳(g-C3N4)在可见光催化领域受到人们的青睐[1-3]。一方面是因石墨相氮化碳的中等宽度带隙(2.7 eV)的能带结构具有与石墨相似的二维结构和优异的光催化性能;另一方面是因为其合成原料易得、制备条件简便,具有推广应用的潜力。然而,单纯的g-C3N4还存在一些不足,如比表面积低、光生载流子复合快、导电性较差等限制了它的推广应用。因此,人们采取多种手段对其进行改性以提升g-C3N4的光催化性能。为了增大其表面积,由块体结构转向超薄片层结构。苏跃涵等[4]将g-C3N4制备成超薄的片层材料,可提升其比表面积,同时提升其光降解抗生素的催化性能。Cao等[5]在超薄片层的g-C3N4表面引入了-NH3官能团,其光催化固氮的反应活性得到增强。为了提高g-C3N4电荷空穴分离效率,采用金属离子或非金属离子改性[6-7]。Liu等[7]发现g-C3N4中引入氯,氯原子以插入层间的方式存在,可有效提高电荷空穴分离能力。
贵金属改性不仅可以提升材料的光催化性能,还可以省略氯铂酸等助催化剂的添加[8]。Huang等[9]发现,在g-C3N4表面沉积Pd,其光解水产氢的性能提升了数百倍。Liu等[10]在超薄TiO2纳米片上负载高度分散Pt纳米颗粒,其光催化还原CO2的性能也显著增强。由于贵金属价格昂贵,在贵金属纳米粒子中引入二元金属,既可减少贵金属的用量降低成本,还可以通过合成方法、改变比例等手段来调控二元金属粒子的形貌及界面作用,从而优化其催化性能。Naulani-Garcia等[11]发现,PdCo/g-C3N4相较于Pd/g-C3N4对甲酸催化降解的活性得到提升;Ye等[12]合成了PdCu/g-C3N4并将其用于硝酸根离子的催化还原反应发现,Cu的引入使催化剂的活性和选择性均有显著提升。本课题组前期的研究发现PdAg粒子改性的g-C3N4比单独Pd粒子改性g-C3N4具有更高的光解水产氢性能,主要是由于PdAg间的界面效应有利于电子的富集,促进光生电荷空穴的分离[13]。在此基础上,进一步合成了PdCu、PdCo、PdIr的3种双金属粒子,考察了引入元素离子半径和电负性变化对二元金属性质及光催化性能的影响规律。
本文通过乙二醇还原法合成了PdM(M=Cu、Co、Ir)纳米粒子,将其负载到g-C3N4表面,评价了PdM/g-C3N4催化剂可见光驱动下光解水产氢的性能,并结合XRD、TEM、XPS及光电化学参数等表征手段对样品性质进行表征,进而讨论其中的作用机理。
-
氯钯酸钠(Na2PdCl4,99.95%,Alfa Aesar),硝酸铜(Cu(NO3)2,99.5%,上海振欣试剂公司),硝酸钴(Co(NO3)2,99%,aladdin),三氯化铱(IrCl3,99.9%,aladdin),聚乙烯吡咯烷酮(Poly vinylpyrrolidone,PVP, MW=58000,Fluka),尿素,抗坏血酸,丙酮,无水乙醇均购自国药集团化学试剂有限公司,溴化钾(KBr)和三乙醇胺购自上海凌峰化学试剂有限公司,乙二醇购自南京化学试剂有限公司。所有试剂都是分析纯,直接使用。
-
金属纳米粒子合成 采用乙二醇还原法制备PdM(PdCo、PdCu、PdIr)二元和单组份Pd金属纳米粒子。样品合成所用试剂的量是按理论产量10 mg,Pd和M的原子比1:1进行投加。以PdCu为例,首先称取Na2PdCl4 17.3 mg和Cu(NO3)2 14.2 mg分别溶解于4 mL乙二醇样品管,超声1 min促进溶解。然后称取50 mg PVP置于100 mL圆底烧瓶中,加入7 mL乙二醇(单组份Pd粒子则加入11 mL,保证每次反应乙二醇总量为15 mL),搅拌、冷凝回流条件下,油浴加热至150 ℃。缓慢同时加入两种金属前体物溶液,反应30 min。反应结束后,冰水浴骤冷。将产物溶液转移至50 mL塑料离心管中并以7倍体积加入丙酮,振荡洗涤,6000 r·min−1转速下离心30 min分离出纳米金属粒子。分离产物在60 ℃烘干72 h,再研磨得到粉末。样品标记 PdCu、PdCo、PdIr和Pd。
g-C3N4制备 高温灼烧法制备g-C3N4。在100 mL瓷坩埚中加入一半体积的尿素,放入马弗炉中,在空气氛中以5 ℃·min−1的速度升温至550 ℃,并在550 ℃下保持4 h。
PdM/g-C3N4催化剂制备 称取前述制备得到的金属纳米粒子60% wt(相当于6 mg PdM)溶解7.5 mL水和无水乙醇(体积比1∶1)的溶剂,记为溶液1;称取600 mg·g−1-C3N4并加入12.5 mL水和无水乙醇(体积比1∶1)的溶剂,记为溶液2。将两个溶液超声10 min。在搅拌状态下,用胶头滴管逐滴将溶液1加入到溶液2。将混合后的溶液再超声10 min。搅拌1 h,60 ℃烘干过夜。烘干后,研磨,称量。PdM的理论负载量1 %wt。样品标记为PdM(M=Cu、Co、Ir)/g-C3N4。
-
用X射线衍射(XRD)的方法测定PdCu、PdCo、PdIr和Pd及负载到g-C3N4表面后样品的晶体结构,测试是在瑞士ARL公司的XRD-6000型仪器上进行,2θ扫描范围是15°—75°,扫描速度是3°·min−1,Cu Kα辐射λ=0.15418 nm。金属粒子的形貌是在日本JEOL公司的JEM-200cx透射电子显微镜上测试,加速电压200 kV。样品中元素结合能在日本UIVAC-PHI公司的PHI 5000 VersaProbe光电子能谱仪上测得,Al Kα光源(1486.6 eV),结合能以C1s = 284.6 eV作为标准进行校正。岛津UV-2401分光光度计(以BaSO4为参比)在200—800 nm范围内记录了UV-漫反射光谱(UV-DRS)。在Fluoromax-4荧光分光光度计上测定光致发光(PL)光谱,实验以固态进行,激发光波长为320 nm,所有测量中的缝隙为1.5 nm,在360 nm至800 nm范围内测量PL光谱。光电流测量是使用标准三电极电池在CHI660E电化学工作站上进行,其中沉积光催化剂的电极用作工作电极,参比电极和对电极分别为Hg/Hg2Cl2和铂丝(平时保存于饱和KCl中)。电解质为0.2 mol·L−1 Na2SO4溶液,并使用Xe灯照射。光照强度为467 mW,光谱范围320—2500 nm。水接触角的测定是利用Kruss公司的DSA-30S液滴形状分析仪进行测量,使用方法为座滴法,角度取液滴完全浸没入样品的前一帧进行计算机测量。
-
光催化水分解实验是在密闭玻璃系统(CEL-SPH2N-D,中国北京中教金源)中进行的。用300 W Xe灯作为光源(420 nm滤光片),光照强度为467 mW。将50 mg催化剂分散在100 mL含三乙醇胺(10% vol)作为牺牲电子给体的水溶液中,水溶液的温度通过循环泵水浴保持在6 ℃,将反应系统密封并抽空30 min后开启光源。氢气的产生量通过配备有热导检测器(TCD)、氩气作为载气的气相色谱仪在线测定。为了确定催化剂的重复利用性能,对催化剂进行重复试验,步骤同上,并且每次重复都补充牺牲剂。其中,0.05 g 催化剂4 h Pd 原子平均产氢量计算方式如下:
-
文中所有的DFT计算都是用VASP软件包完成的,计算中使用PAW平面波的方法来处理核-电相互作用,平面波截断能为520 eV。使用GGA-PBE交换关联泛函描述该体系,使用M-P方法展开电子波函数,展宽为0.2 eV,布里渊区撒点密度为5×5×1,结构优化的力收敛标准为小于0.2 eV·nm−1。
吸附能的定义为:
其中,E (吸附物 + 底物)是吸附物与底物相互作用体系的总能,E (吸附物)是孤立吸附物在气相中的能量,E (底物)是未吸附之前的表面的能量(Pd(111))。吸附能为负值代表吸附过程中体系放热,吸附能为正值代表吸附过程中体系吸热。
-
首先对合成的PdM金属粒子进行了晶体结构和形貌表征,结果如图1所示。从图1a可见,PdM和Pd金属粒子均出现3个衍射峰分别位于2θ = 40°、46°、68°处,对应于面心立方相的金属Pd的(111)、(200)和(220)晶面衍射[14]。此外,每个样品在22°处还有个宽峰,是样品底座玻璃的信号。相对于单组份Pd,PdM样品的(111)晶面的衍射峰均发生了一定程度位移,见表1。再依据布拉格方程,计算出PdM和Pd样品的(111)晶面间距,Pd、PdCu、PdCo和PdIr的d(111)分别为0.225、0.224、0.225、0.229 nm,即Cu和Co的引入对其晶面间距影响不大,而Ir的引入d(111)略有增加。这主要是由于引入元素M和Pd的原子半径不同所致。Cu和Co的原子半径小于Pd,Ir的原子半径大于Pd,导致其晶格分别发生一定程度的收缩和膨胀。晶格收缩和膨胀也说明PdM可能形成了合金[14]。图1b是金属粒子的TEM图。从图1b可见,所合成的金属粒子都是高度分散,尺寸均匀的纳米颗粒。经过对样品进行粒径统计,PdCu、PdCo、PdIr和Pd的平均粒径分别为7.0、6.0、8.6、4.7 nm。
将上述金属粒子负载到g-C3N4表面制得PdM/g-C3N4催化剂,并进行了XRD和TEM表征如图2。从图2a可见,样品和载体g-C3N4均在2θ=13.1°和27.7°处出现特征衍射峰,对应于g-C3N4的构成平面的3-s-三嗪单元结构(100)晶面和π共轭的石墨层状结构(002)晶面衍射[3-4, 15]。未检测到对应于Pd或者PdM的晶相衍射峰,说明PdM金属粒子高度分散在g-C3N4表面。从图2b也可以看出g-C3N4呈薄膜结构,PdCo金属粒子高度分散在其表面。
-
在可将光照射下评价了催化剂光解水产氢的性能,结果如图3所示。经过4 h光催化产氢反应,每个催化剂的产氢量随时间线性增加,载金属粒子的样品PdM/g-C3N4相对于单纯g-C3N4,均大幅度提升。至4 h反应结束,g-C3N4、PdCu/g-C3N4、Pd/g-C3N4、PdCo/g-C3N4和PdIr/g-C3N4的产氢量分别为3.3、117.19、135.6、184.6、214.7 μmol,除了PdCu/g-C3N4外,双金属催化剂产氢活性均较纯Pd单金属催化剂有明显提升。可见,贵金属粒子负载可显著提升催化剂的光解水产氢性能。对于4种金属粒子,其产氢性能顺序:PdCu/g-C3N4 < Pd/g-C3N4 < PdCo/g-C3N4 < PdIr/g-C3N4。即Cu的引入使Pd的催化性能略有降低,Co和Ir的引入则有所提升。由于二元金属引入Pd的用量减少,提升了催化剂的经济性。此外,Co和Cu的引入Pd原子的4 h平均产氢量也提升至原来的2.1倍和1.4倍。这也说明,二元金属的引入,可提升贵金属的利用率,减少贵金属用量。最后,测试了PdIr/g-C3N4催化剂的重复利用性,结果如图3c。第2次和第3次的产氢能力较前一次均略有下降,而第4次与第3次几乎不变,说明稳定性较好。
-
为了分析各样品催化性能产生差异的原因,对催化剂进行了光谱和电化学性质表征。图4a是样品的固体紫外可见吸收光谱,从图上可以看出,所有样品在250—400 nm间均产生了强的光吸收,对应于半导体g-C3N4光生电子由价带向导带的跃迁[7, 9]。相较于单纯的g-C3N4,负载金属粒子后的样品其吸收峰的强度除了紫外光区间外,在450—800 nm可见光区间的吸收也显著增强。Chen等[16]在Pd/TiO2体系中也观察到类似结果,这主要是由于光生电子在Pd金属的d-d轨道间跃迁增强了样品对光的吸收。因此,紫外可见吸收谱图说明金属粒子负载后,催化剂对光的吸收能力显著增强。图4b是样品的光电流结果。所有样品在可见光激发下均产生了不同强度的光电流,主要是由于半导体材料的光电效应所致。从光电流强度来看,金属粒子负载其强度均较单纯的g-C3N4有所增加。尤其是PdCo粒子负载后,显示了最强的光电流。Chen[16]和Chava[17]分别在Pt/TiO2和Au/g-C3N4体系中报道了相同的结果。金属粒子负载后,样品的光电流增强主要是有两个原因,第一是样品对光的吸收能力增强;第二是由于金属粒子在半导体表面沉积后形成了肖特基势垒,增强了光生电荷迁移能力,促进了光生电荷-空穴的分离,前人研究说明双金属对比单金属具有更低的费米能级,从而使得光激发电子能够更容易地从g-C3N4导带转移到合金上,也就是更容易翻越肖特基势垒,从而提升分离效率[18]。而更高的电荷空穴分离能力,意味着更多的光生电子可用于H+还原,以产生更多的氢气。另外从光致发光光谱(PL)可以看出负载金属的催化剂比纯g-C3N4信号弱,说明金属的引入降低了电子-空穴复合效率。其中双金属催化剂活性顺序与PL光谱相对应,而单金属催化剂则有所不同,但文献中有表明,第二金属的引入会改变单金属的结构、电子密度、表面缺陷等性质[18],而颗粒的尺寸、样品表面粗糙度、发光体的浓度、杂志缺陷等性质均会影响PL的强度[19-20]。
-
电子传递是光解水产氢中的关键步骤。为了明确光生电子在催化剂表面的迁移路径,对样品进行了X射线光电子能谱表征,结果如图5所示。首先来看PdM金属粒子中Pd 3d的结合能。从图5可见,4个样品均出现了4组峰,分别位于334.7、337.6、339.9、343.0 eV附近。其中334.7 eV和339.9 eV处的峰归属为Pd0的Pd3d5/2和Pd3d3/2分裂,337.6 eV和343.3 eV则归属为Pd2+的Pd 3d5/2和Pd3d3/2分裂[9, 21-24]。说明4组样品中的Pd均以Pd0和Pd2+的混合形态存在。本研究中,金属粒子采用的是乙二醇还原法,应该主要获得金属态粒子。XPS结果显示样品中有部分氧化态钯,可能是金属粒子表面部分被氧化所致。二元金属的引入,对Pd的结合能和峰面积比例都产生了一些影响。从结合能来看,Cu、Co和Ir的引入,Pd0和Pd2+的结合能都有向低结合能偏移的趋势。结合元素电负性分析,Pd、Cu、Co、Ir的电负性分别为2.2、1.88、1.7和2.2,当合成二元金属粒子时,电负性大的元素有更强的得电子能力,在XPS谱图上就显示结合能向低结合能方向偏移。从文献报道的结果也可以看到,二元金属粒子中由于两种金属相互作用发生了电子转移使其结合能发生了一定的位移[21-23]。这也印证了XRD的结果,PdM形成了合金。此时Pd形成了Pdδ−,而电负性较小的M则形成了Mδ+,Pdδ−···Mδ+的金属界面更有利于电荷传递,当g-C3N4被光激发形成光生电子,光生电子则迁移至费米能级较低的PdM表面,形成捕获光电子的肖特基势垒,从而抑制了电子-空穴对复合[13]。
另外,单组份Pd/g-C3N4样品中Pd0和Pd2+的峰面积较接近说明两种物种比例相近,各占一半。而M的引入,两个峰的面积比例发生了变化。大体上是Pd2+物种的面积减少,说明M的引入稳定了金属态的Pd,这也是跟前述的M元素的电负性相关,M和Pd的电负性差异使得Pd有得电子趋势,从而形成了更多Pd0物种。而Ir的电负性和Pd相同,所以Ir的引入对Pd2+物种的量影响不大。接下来对样品中的氧元素进行分析如图5b。可以看出4个样品都在531.4 eV处出现一个主峰,在532.9 eV处出现一个肩峰。531.4 eV处的峰对应于PdM表面吸附氧物种,532.9 eV的峰对应于吸附的水分子中羟基氧物种[25]。相较于单组份样品,PdM/g-C3N4中吸附氧的峰位置均有向高结合能方向偏移的趋势。这也可能是PdM间的相互作用使表面吸附氧物种的化学环境发生了变化。
图5c、d分别对样品中C、N元素进行了XPS分析。C元素都在287.8 eV和284.6 eV处出现了特征峰,对应于g-C3N4中N—C=N的碳和石墨结构的C—C物种[9, 13, 15, 17]。金属粒子负载后N—C=N峰的位置略向低结合能方向偏移约0.2 eV。N1s谱图在398.4 eV出现主峰,在400.0 eV出现肩峰,分别对应C—N=C中的氮和N-(C)3中的氮[9, 13, 15, 17]。金属粒子负载后主峰也略向低结合能方向偏移约0.2 eV。C1s和N1s的结合能偏移,说明金属粒子和载体间具有一定的相互作用,形成了电子传递作用。
-
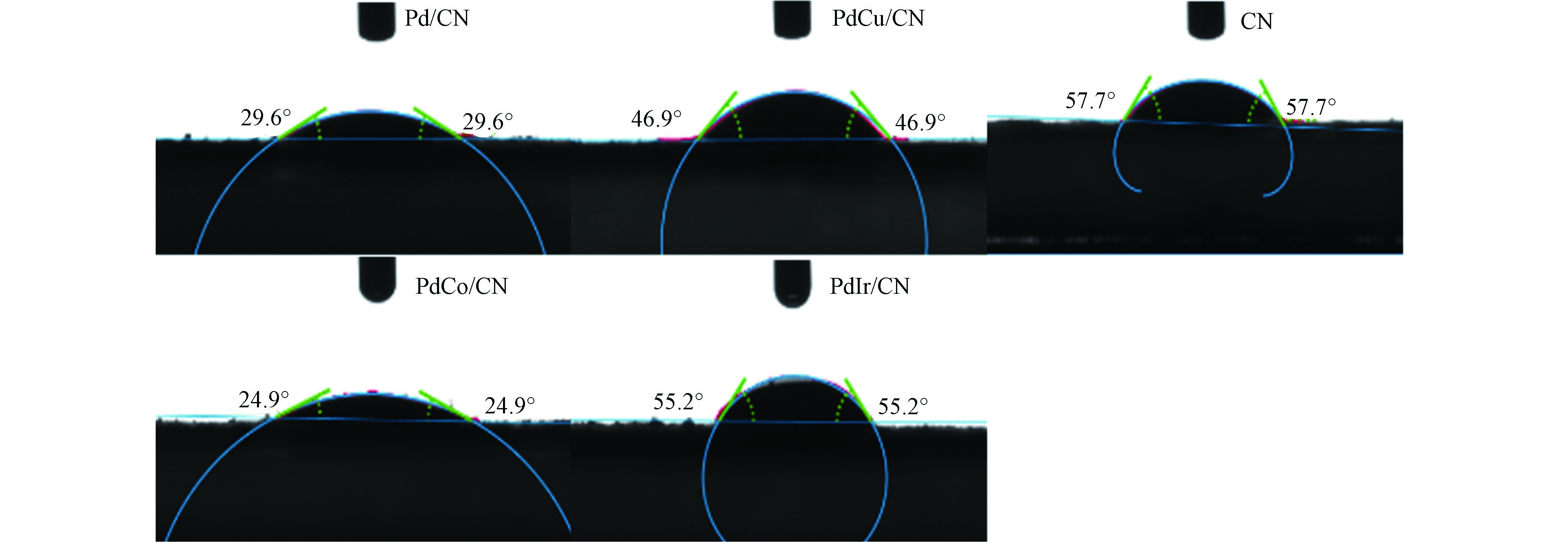
光解水产氢反应中,水是非常重要的反应物分子。水分子与催化剂间的接触能力将直接影响其催化性能。因此,本文测定了样品的水接触角判断其亲疏水性强弱。结果如图6所示。从图上可以看出,单纯g-C3N4的水接触角是57.7°,而金属粒子负载后其水接触角均有不同程度减小。一般来说,水接触角越小其亲水性越好[15]。所以,金属粒子负载后样品的亲水性都有所增加,也说明反应物水分子和样品间的接触机会增加。PdM粒子负载后其亲水性增加,也说明合成的PdM粒子具有良好的亲水性,可与反应物水分子进行充分接触。其中PdCo/g-C3N4的接触角最小,这可能与其具有较高催化活性相关[26-29]。另外,PdIr/g-C3N4的水接触角相对较大,但是活性却是最好的一个,说明亲疏水性并不是影响催化剂产氢活性的关键因素。Pd和Ir同属Pt族元素,均为贵金属,因此PdIr显示优于PdCo和PdCu的催化活性是由Ir的贵金属特性决定的。
此外,对PdCo、PdCu、PdIr表面吸附水分子的吸附能进行了DFT计算,结果如图7所示。水分子在PdCu、PdIr和PdCo粒子表面的吸附能分别是−5.53、−8.99、−10.15 kcal·mol−1。吸附能越负表明水分子在金属粒子表面吸附越稳定。可见,3种双金属粒子对水分子的吸附顺序是PdCo > PdIr >PdCu。在3种双金属粒子中,PdIr活性最高,是由于Pd和Ir均是贵金属。此外,值得一提的是PdCo的性能与PdIr接近,远高于PdCu。由水分子在PdCu和PdCo表面的吸附能及亲水性可推测,PdCo的活性较高与其更易吸附反应物水分子相关。
-
本文采用乙二醇还原法合成了PdCu、PdCo、PdIr和Pd金属粒子,并将其负载于g-C3N4表面制备出光催化剂用于可见光驱动光解水产氢反应。结果发现,二元金属Co, Ir的引入相较于单金属负载的催化剂0.05 g 4 h产氢量都明显提升,而Cu则稍微有所降低,并且使得Pd原子的平均产氢量增加了1.4—4.4倍。并且,PdIr/g-C3N4经过4次循环测试显示了较高的稳定性。催化性能提升主要是由于:(1) 贵金属负载后对光的吸收能力增加,同时由于金属粒子对光生电子具有强的捕获能力,形成更强的光电流;(2) M原子进入Pd晶格形成了纳米合金,降低了费米能级,使g-C3N4的光生电子在PdM合金粒子上更易传输,更利于电子捕获促进光生电子-空穴的分离;(3) 贵金属粒子负载到g-C3N4表面后,增强了样品的亲水性,降低了水分子吸附能,提升了反应速率。
PdM/g-C3N4 (M=Cu、Co、Ir) 催化剂制备及其可见光驱动光解水制氢反应性能
Study of the visible-light photocatalytic water splitting for hydrogen evolution on PdM/g-C3N4(M=Cu,Co,Ir)
-
摘要: 贵金属基光催化剂在可见光驱动分解水产氢反应中具有十分优异的性能,却面临价格贵、利用率较低的问题。本文采用乙二醇还原法,将金属Cu、Co、Ir分别引入到贵金属Pd粒子中形成二元PdM金属粒子,负载到g-C3N4表面制得催化剂。在可见光驱动下分解水产氢性能测试结果表明,二元金属Co、Ir的引入相较于0.05 g单金属负载催化剂的4 h产氢量都有明显提升,而Cu则稍微有所降低,最高产氢量是215 μmol。并且使得Pd原子的平均产氢量增至1.4—4.4倍。光催化性能提升的主要原因是由于PdM粒子负载增强了样品对光的吸收,促进了光生电子与空穴的分离,增加了样品的亲水性降低了水分子吸附能。另外,二元金属的引入可显著提升Pd原子的平均产氢量。尤其是与过渡金属Co形成PdCo粒子,其4 h产氢性能与PdIr接近,但其价格远低于Ir。因而PdCo/g-C3N4催化剂具有很好的经济性。Abstract: Noble metal based photocatalysts have excellent performance toward the hydrogen evolution by water splitting under visible light irradiation, but some disadvantages are present, such as expensive, low utilization, etc. In this study, by the polyol reduction method, the elements of copper (Cu), cobalt (Co) and iridium (Ir) were introduced into Pd nanoparticles to prepare PdM bimetallic nanoparticles, which were loaded on the surface of g-C3N4. The results of hydrogen evolution indicated that compared to pristine Pd/g-C3N4, the hydrogen amounts of 0.05 g Pd(Co, Ir)/g-C3N4 catalysts have significant hydrogen evolution increase in 4 h, while Cu has a slight decrease, the highest one achieved 215 μmol. And the introduction of Cu, Co, Ir increases the hydrogen evolution of average Pd atoms by 1.4—4.4 times. The improvement of catalytic performance is attributed to the increased light absorption, the separation of electron and hole, the hydrophilic property, and the decreased adsorption energy with water reactant of the PdM/g-C3N4 catalysts. Furthermore, the average produced hydrogen amount of Pd atom was increased by introducing of M, PdCo/g-C3N4 displayed a similar catalytic performance with PdIr/g-C3N4. On the basis of that cobalt is cheaper than iridium, PdCo/g-C3N4 is very economical.
-
目前,全球正面临能源危机和环境污染的双重挑战。光解水制氢将太阳能转化成化学能,既能提供清洁能源又可避免传统能源的环境污染。因此,光解水制氢引起了人们的广泛关注。由于可见光占太阳光的绝大部分,拓宽可见光的利用是提高太阳光利用率的有效手段。基于此,开发可见光驱动的光催化剂对于光解水制氢具有十分重要的意义。
近年来石墨相氮化碳(g-C3N4)在可见光催化领域受到人们的青睐[1-3]。一方面是因石墨相氮化碳的中等宽度带隙(2.7 eV)的能带结构具有与石墨相似的二维结构和优异的光催化性能;另一方面是因为其合成原料易得、制备条件简便,具有推广应用的潜力。然而,单纯的g-C3N4还存在一些不足,如比表面积低、光生载流子复合快、导电性较差等限制了它的推广应用。因此,人们采取多种手段对其进行改性以提升g-C3N4的光催化性能。为了增大其表面积,由块体结构转向超薄片层结构。苏跃涵等[4]将g-C3N4制备成超薄的片层材料,可提升其比表面积,同时提升其光降解抗生素的催化性能。Cao等[5]在超薄片层的g-C3N4表面引入了-NH3官能团,其光催化固氮的反应活性得到增强。为了提高g-C3N4电荷空穴分离效率,采用金属离子或非金属离子改性[6-7]。Liu等[7]发现g-C3N4中引入氯,氯原子以插入层间的方式存在,可有效提高电荷空穴分离能力。
贵金属改性不仅可以提升材料的光催化性能,还可以省略氯铂酸等助催化剂的添加[8]。Huang等[9]发现,在g-C3N4表面沉积Pd,其光解水产氢的性能提升了数百倍。Liu等[10]在超薄TiO2纳米片上负载高度分散Pt纳米颗粒,其光催化还原CO2的性能也显著增强。由于贵金属价格昂贵,在贵金属纳米粒子中引入二元金属,既可减少贵金属的用量降低成本,还可以通过合成方法、改变比例等手段来调控二元金属粒子的形貌及界面作用,从而优化其催化性能。Naulani-Garcia等[11]发现,PdCo/g-C3N4相较于Pd/g-C3N4对甲酸催化降解的活性得到提升;Ye等[12]合成了PdCu/g-C3N4并将其用于硝酸根离子的催化还原反应发现,Cu的引入使催化剂的活性和选择性均有显著提升。本课题组前期的研究发现PdAg粒子改性的g-C3N4比单独Pd粒子改性g-C3N4具有更高的光解水产氢性能,主要是由于PdAg间的界面效应有利于电子的富集,促进光生电荷空穴的分离[13]。在此基础上,进一步合成了PdCu、PdCo、PdIr的3种双金属粒子,考察了引入元素离子半径和电负性变化对二元金属性质及光催化性能的影响规律。
本文通过乙二醇还原法合成了PdM(M=Cu、Co、Ir)纳米粒子,将其负载到g-C3N4表面,评价了PdM/g-C3N4催化剂可见光驱动下光解水产氢的性能,并结合XRD、TEM、XPS及光电化学参数等表征手段对样品性质进行表征,进而讨论其中的作用机理。
1. 材料与方法(Materials and Methods)
1.1 药品与试剂
氯钯酸钠(Na2PdCl4,99.95%,Alfa Aesar),硝酸铜(Cu(NO3)2,99.5%,上海振欣试剂公司),硝酸钴(Co(NO3)2,99%,aladdin),三氯化铱(IrCl3,99.9%,aladdin),聚乙烯吡咯烷酮(Poly vinylpyrrolidone,PVP, MW=58000,Fluka),尿素,抗坏血酸,丙酮,无水乙醇均购自国药集团化学试剂有限公司,溴化钾(KBr)和三乙醇胺购自上海凌峰化学试剂有限公司,乙二醇购自南京化学试剂有限公司。所有试剂都是分析纯,直接使用。
1.2 样品合成
金属纳米粒子合成 采用乙二醇还原法制备PdM(PdCo、PdCu、PdIr)二元和单组份Pd金属纳米粒子。样品合成所用试剂的量是按理论产量10 mg,Pd和M的原子比1:1进行投加。以PdCu为例,首先称取Na2PdCl4 17.3 mg和Cu(NO3)2 14.2 mg分别溶解于4 mL乙二醇样品管,超声1 min促进溶解。然后称取50 mg PVP置于100 mL圆底烧瓶中,加入7 mL乙二醇(单组份Pd粒子则加入11 mL,保证每次反应乙二醇总量为15 mL),搅拌、冷凝回流条件下,油浴加热至150 ℃。缓慢同时加入两种金属前体物溶液,反应30 min。反应结束后,冰水浴骤冷。将产物溶液转移至50 mL塑料离心管中并以7倍体积加入丙酮,振荡洗涤,6000 r·min−1转速下离心30 min分离出纳米金属粒子。分离产物在60 ℃烘干72 h,再研磨得到粉末。样品标记 PdCu、PdCo、PdIr和Pd。
g-C3N4制备 高温灼烧法制备g-C3N4。在100 mL瓷坩埚中加入一半体积的尿素,放入马弗炉中,在空气氛中以5 ℃·min−1的速度升温至550 ℃,并在550 ℃下保持4 h。
PdM/g-C3N4催化剂制备 称取前述制备得到的金属纳米粒子60% wt(相当于6 mg PdM)溶解7.5 mL水和无水乙醇(体积比1∶1)的溶剂,记为溶液1;称取600 mg·g−1-C3N4并加入12.5 mL水和无水乙醇(体积比1∶1)的溶剂,记为溶液2。将两个溶液超声10 min。在搅拌状态下,用胶头滴管逐滴将溶液1加入到溶液2。将混合后的溶液再超声10 min。搅拌1 h,60 ℃烘干过夜。烘干后,研磨,称量。PdM的理论负载量1 %wt。样品标记为PdM(M=Cu、Co、Ir)/g-C3N4。
1.3 催化剂表征
用X射线衍射(XRD)的方法测定PdCu、PdCo、PdIr和Pd及负载到g-C3N4表面后样品的晶体结构,测试是在瑞士ARL公司的XRD-6000型仪器上进行,2θ扫描范围是15°—75°,扫描速度是3°·min−1,Cu Kα辐射λ=0.15418 nm。金属粒子的形貌是在日本JEOL公司的JEM-200cx透射电子显微镜上测试,加速电压200 kV。样品中元素结合能在日本UIVAC-PHI公司的PHI 5000 VersaProbe光电子能谱仪上测得,Al Kα光源(1486.6 eV),结合能以C1s = 284.6 eV作为标准进行校正。岛津UV-2401分光光度计(以BaSO4为参比)在200—800 nm范围内记录了UV-漫反射光谱(UV-DRS)。在Fluoromax-4荧光分光光度计上测定光致发光(PL)光谱,实验以固态进行,激发光波长为320 nm,所有测量中的缝隙为1.5 nm,在360 nm至800 nm范围内测量PL光谱。光电流测量是使用标准三电极电池在CHI660E电化学工作站上进行,其中沉积光催化剂的电极用作工作电极,参比电极和对电极分别为Hg/Hg2Cl2和铂丝(平时保存于饱和KCl中)。电解质为0.2 mol·L−1 Na2SO4溶液,并使用Xe灯照射。光照强度为467 mW,光谱范围320—2500 nm。水接触角的测定是利用Kruss公司的DSA-30S液滴形状分析仪进行测量,使用方法为座滴法,角度取液滴完全浸没入样品的前一帧进行计算机测量。
1.4 光催化分解水产氢性能测试
光催化水分解实验是在密闭玻璃系统(CEL-SPH2N-D,中国北京中教金源)中进行的。用300 W Xe灯作为光源(420 nm滤光片),光照强度为467 mW。将50 mg催化剂分散在100 mL含三乙醇胺(10% vol)作为牺牲电子给体的水溶液中,水溶液的温度通过循环泵水浴保持在6 ℃,将反应系统密封并抽空30 min后开启光源。氢气的产生量通过配备有热导检测器(TCD)、氩气作为载气的气相色谱仪在线测定。为了确定催化剂的重复利用性能,对催化剂进行重复试验,步骤同上,并且每次重复都补充牺牲剂。其中,0.05 g 催化剂4 h Pd 原子平均产氢量计算方式如下:
Pd原子平均产氢量=4h产氢总量催化剂中Pd的物质的量 1.5 DFT理论计算
文中所有的DFT计算都是用VASP软件包完成的,计算中使用PAW平面波的方法来处理核-电相互作用,平面波截断能为520 eV。使用GGA-PBE交换关联泛函描述该体系,使用M-P方法展开电子波函数,展宽为0.2 eV,布里渊区撒点密度为5×5×1,结构优化的力收敛标准为小于0.2 eV·nm−1。
吸附能的定义为:
E吸附=E(吸附物+底物)−E(吸附物)−E(底物) 其中,E (吸附物 + 底物)是吸附物与底物相互作用体系的总能,E (吸附物)是孤立吸附物在气相中的能量,E (底物)是未吸附之前的表面的能量(Pd(111))。吸附能为负值代表吸附过程中体系放热,吸附能为正值代表吸附过程中体系吸热。
2. 结果与讨论(Result and Discussion)
2.1 催化剂结构分析
首先对合成的PdM金属粒子进行了晶体结构和形貌表征,结果如图1所示。从图1a可见,PdM和Pd金属粒子均出现3个衍射峰分别位于2θ = 40°、46°、68°处,对应于面心立方相的金属Pd的(111)、(200)和(220)晶面衍射[14]。此外,每个样品在22°处还有个宽峰,是样品底座玻璃的信号。相对于单组份Pd,PdM样品的(111)晶面的衍射峰均发生了一定程度位移,见表1。再依据布拉格方程,计算出PdM和Pd样品的(111)晶面间距,Pd、PdCu、PdCo和PdIr的d(111)分别为0.225、0.224、0.225、0.229 nm,即Cu和Co的引入对其晶面间距影响不大,而Ir的引入d(111)略有增加。这主要是由于引入元素M和Pd的原子半径不同所致。Cu和Co的原子半径小于Pd,Ir的原子半径大于Pd,导致其晶格分别发生一定程度的收缩和膨胀。晶格收缩和膨胀也说明PdM可能形成了合金[14]。图1b是金属粒子的TEM图。从图1b可见,所合成的金属粒子都是高度分散,尺寸均匀的纳米颗粒。经过对样品进行粒径统计,PdCu、PdCo、PdIr和Pd的平均粒径分别为7.0、6.0、8.6、4.7 nm。
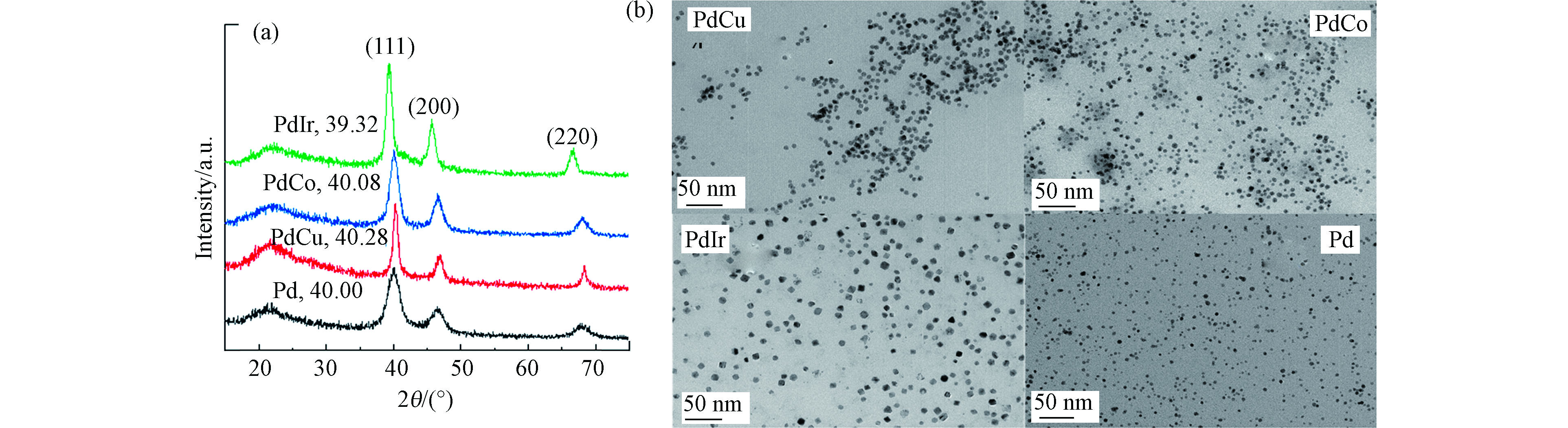 图 1 金属粒子的XRD(a)和TEM(b)Figure 1. the XRD spectra(a) and TEM micrograph(b) of bimetal nanoparticle表 1 PdM粒子物理参数Table 1. Physical parameter of PdM particles
图 1 金属粒子的XRD(a)和TEM(b)Figure 1. the XRD spectra(a) and TEM micrograph(b) of bimetal nanoparticle表 1 PdM粒子物理参数Table 1. Physical parameter of PdM particles样品Samples (111)衍射峰位置 2θ/(°)(111)Difraction peak 2θ (111) 晶面间距d/nm(111)Interplanar spacing d M原子半径/nmMetal atomic radius Pd 40 0.225 0.169 PdCu 40.28 0.224 0.145 PdCo 40.08 0.225 0.152 PdIr 39.32 0.229 0.180 | Show Table DownLoad:
CSV
DownLoad:
CSV
将上述金属粒子负载到g-C3N4表面制得PdM/g-C3N4催化剂,并进行了XRD和TEM表征如图2。从图2a可见,样品和载体g-C3N4均在2θ=13.1°和27.7°处出现特征衍射峰,对应于g-C3N4的构成平面的3-s-三嗪单元结构(100)晶面和π共轭的石墨层状结构(002)晶面衍射[3-4, 15]。未检测到对应于Pd或者PdM的晶相衍射峰,说明PdM金属粒子高度分散在g-C3N4表面。从图2b也可以看出g-C3N4呈薄膜结构,PdCo金属粒子高度分散在其表面。
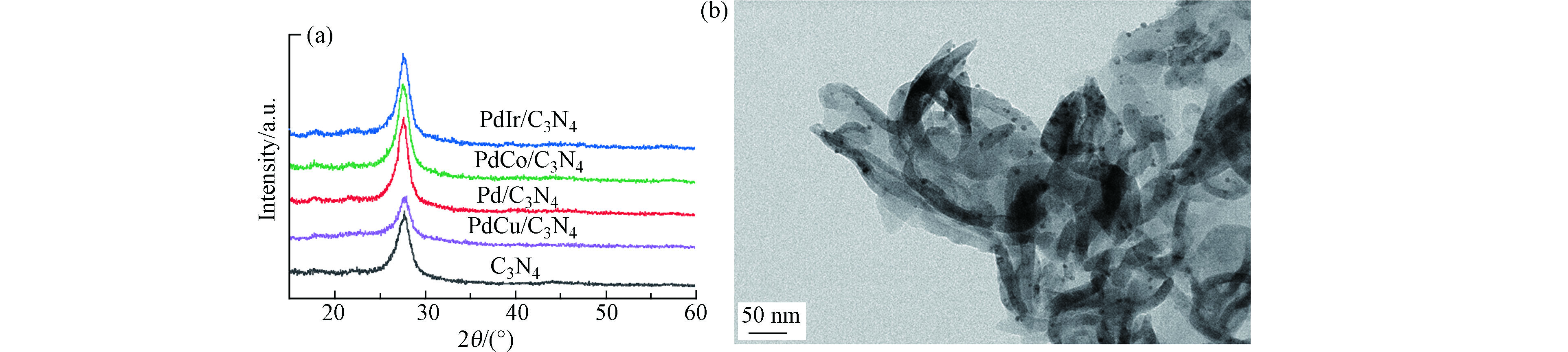 图 2 PdM/g-C3N4的XRD结果(a)和PdCo/g-C3N4 TEM图(b)Figure 2. XRD spectra of(a) PdM/g-C3N4 and (b)TEM micrograph of PdCo/g-C3N4
图 2 PdM/g-C3N4的XRD结果(a)和PdCo/g-C3N4 TEM图(b)Figure 2. XRD spectra of(a) PdM/g-C3N4 and (b)TEM micrograph of PdCo/g-C3N42.2 光催化性能
在可将光照射下评价了催化剂光解水产氢的性能,结果如图3所示。经过4 h光催化产氢反应,每个催化剂的产氢量随时间线性增加,载金属粒子的样品PdM/g-C3N4相对于单纯g-C3N4,均大幅度提升。至4 h反应结束,g-C3N4、PdCu/g-C3N4、Pd/g-C3N4、PdCo/g-C3N4和PdIr/g-C3N4的产氢量分别为3.3、117.19、135.6、184.6、214.7 μmol,除了PdCu/g-C3N4外,双金属催化剂产氢活性均较纯Pd单金属催化剂有明显提升。可见,贵金属粒子负载可显著提升催化剂的光解水产氢性能。对于4种金属粒子,其产氢性能顺序:PdCu/g-C3N4 < Pd/g-C3N4 < PdCo/g-C3N4 < PdIr/g-C3N4。即Cu的引入使Pd的催化性能略有降低,Co和Ir的引入则有所提升。由于二元金属引入Pd的用量减少,提升了催化剂的经济性。此外,Co和Cu的引入Pd原子的4 h平均产氢量也提升至原来的2.1倍和1.4倍。这也说明,二元金属的引入,可提升贵金属的利用率,减少贵金属用量。最后,测试了PdIr/g-C3N4催化剂的重复利用性,结果如图3c。第2次和第3次的产氢能力较前一次均略有下降,而第4次与第3次几乎不变,说明稳定性较好。
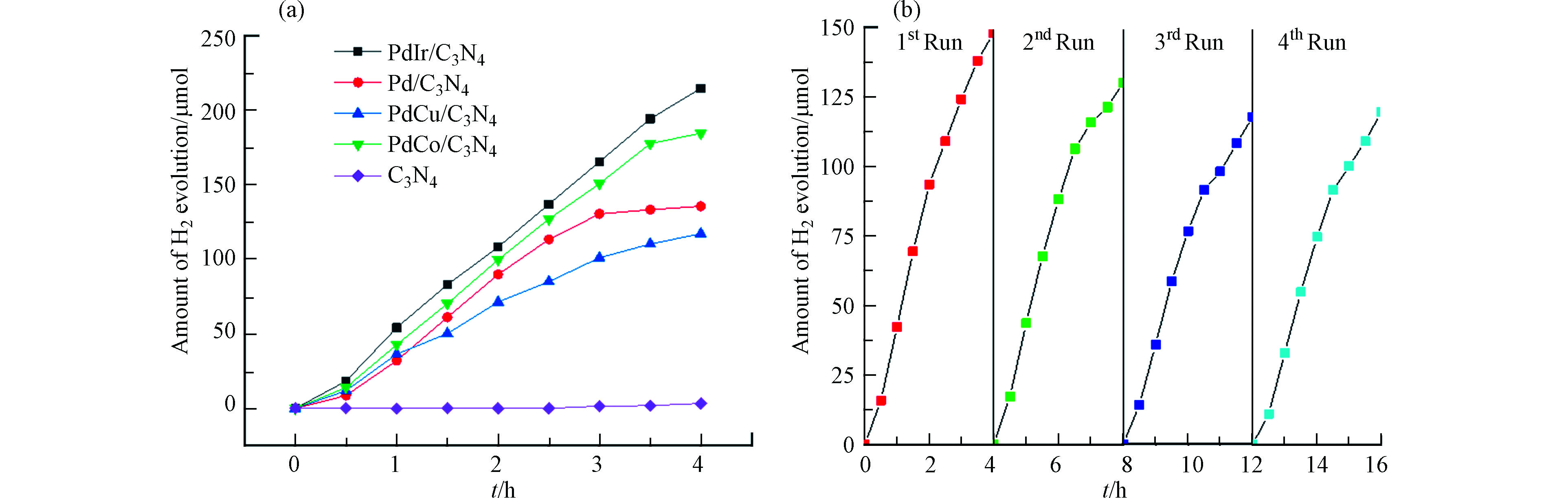 图 3 各催化剂光解水产氢性能(a) 总量; (b) PdIr/g-C3N4的循环产氢性能Figure 3. Performance H2 evolution performance of catalysis (a) Total; (b) The cyclic stability tests for PdIr/g-C3N4
图 3 各催化剂光解水产氢性能(a) 总量; (b) PdIr/g-C3N4的循环产氢性能Figure 3. Performance H2 evolution performance of catalysis (a) Total; (b) The cyclic stability tests for PdIr/g-C3N42.3 光电性质
为了分析各样品催化性能产生差异的原因,对催化剂进行了光谱和电化学性质表征。图4a是样品的固体紫外可见吸收光谱,从图上可以看出,所有样品在250—400 nm间均产生了强的光吸收,对应于半导体g-C3N4光生电子由价带向导带的跃迁[7, 9]。相较于单纯的g-C3N4,负载金属粒子后的样品其吸收峰的强度除了紫外光区间外,在450—800 nm可见光区间的吸收也显著增强。Chen等[16]在Pd/TiO2体系中也观察到类似结果,这主要是由于光生电子在Pd金属的d-d轨道间跃迁增强了样品对光的吸收。因此,紫外可见吸收谱图说明金属粒子负载后,催化剂对光的吸收能力显著增强。图4b是样品的光电流结果。所有样品在可见光激发下均产生了不同强度的光电流,主要是由于半导体材料的光电效应所致。从光电流强度来看,金属粒子负载其强度均较单纯的g-C3N4有所增加。尤其是PdCo粒子负载后,显示了最强的光电流。Chen[16]和Chava[17]分别在Pt/TiO2和Au/g-C3N4体系中报道了相同的结果。金属粒子负载后,样品的光电流增强主要是有两个原因,第一是样品对光的吸收能力增强;第二是由于金属粒子在半导体表面沉积后形成了肖特基势垒,增强了光生电荷迁移能力,促进了光生电荷-空穴的分离,前人研究说明双金属对比单金属具有更低的费米能级,从而使得光激发电子能够更容易地从g-C3N4导带转移到合金上,也就是更容易翻越肖特基势垒,从而提升分离效率[18]。而更高的电荷空穴分离能力,意味着更多的光生电子可用于H+还原,以产生更多的氢气。另外从光致发光光谱(PL)可以看出负载金属的催化剂比纯g-C3N4信号弱,说明金属的引入降低了电子-空穴复合效率。其中双金属催化剂活性顺序与PL光谱相对应,而单金属催化剂则有所不同,但文献中有表明,第二金属的引入会改变单金属的结构、电子密度、表面缺陷等性质[18],而颗粒的尺寸、样品表面粗糙度、发光体的浓度、杂志缺陷等性质均会影响PL的强度[19-20]。
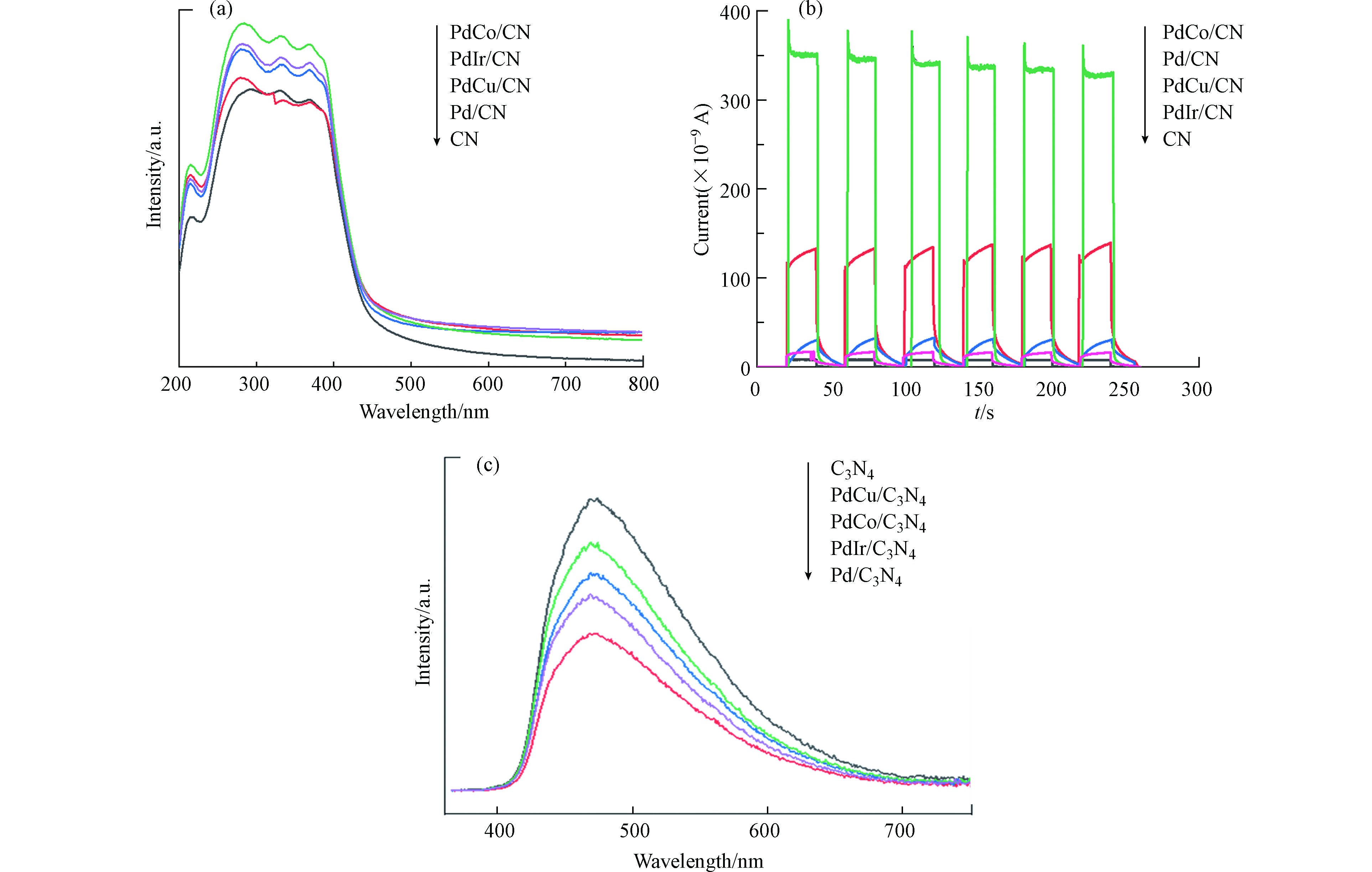 图 4 各催化剂的(a)固体紫外吸收光谱(b)光电流和(c)光致发光光谱Figure 4. (a) UV-Vis DRS spectra, (b) photocurrent and (c) PL spectra of catalysts
图 4 各催化剂的(a)固体紫外吸收光谱(b)光电流和(c)光致发光光谱Figure 4. (a) UV-Vis DRS spectra, (b) photocurrent and (c) PL spectra of catalysts2.4 金属载体间作用
电子传递是光解水产氢中的关键步骤。为了明确光生电子在催化剂表面的迁移路径,对样品进行了X射线光电子能谱表征,结果如图5所示。首先来看PdM金属粒子中Pd 3d的结合能。从图5可见,4个样品均出现了4组峰,分别位于334.7、337.6、339.9、343.0 eV附近。其中334.7 eV和339.9 eV处的峰归属为Pd0的Pd3d5/2和Pd3d3/2分裂,337.6 eV和343.3 eV则归属为Pd2+的Pd 3d5/2和Pd3d3/2分裂[9, 21-24]。说明4组样品中的Pd均以Pd0和Pd2+的混合形态存在。本研究中,金属粒子采用的是乙二醇还原法,应该主要获得金属态粒子。XPS结果显示样品中有部分氧化态钯,可能是金属粒子表面部分被氧化所致。二元金属的引入,对Pd的结合能和峰面积比例都产生了一些影响。从结合能来看,Cu、Co和Ir的引入,Pd0和Pd2+的结合能都有向低结合能偏移的趋势。结合元素电负性分析,Pd、Cu、Co、Ir的电负性分别为2.2、1.88、1.7和2.2,当合成二元金属粒子时,电负性大的元素有更强的得电子能力,在XPS谱图上就显示结合能向低结合能方向偏移。从文献报道的结果也可以看到,二元金属粒子中由于两种金属相互作用发生了电子转移使其结合能发生了一定的位移[21-23]。这也印证了XRD的结果,PdM形成了合金。此时Pd形成了Pdδ−,而电负性较小的M则形成了Mδ+,Pdδ−···Mδ+的金属界面更有利于电荷传递,当g-C3N4被光激发形成光生电子,光生电子则迁移至费米能级较低的PdM表面,形成捕获光电子的肖特基势垒,从而抑制了电子-空穴对复合[13]。
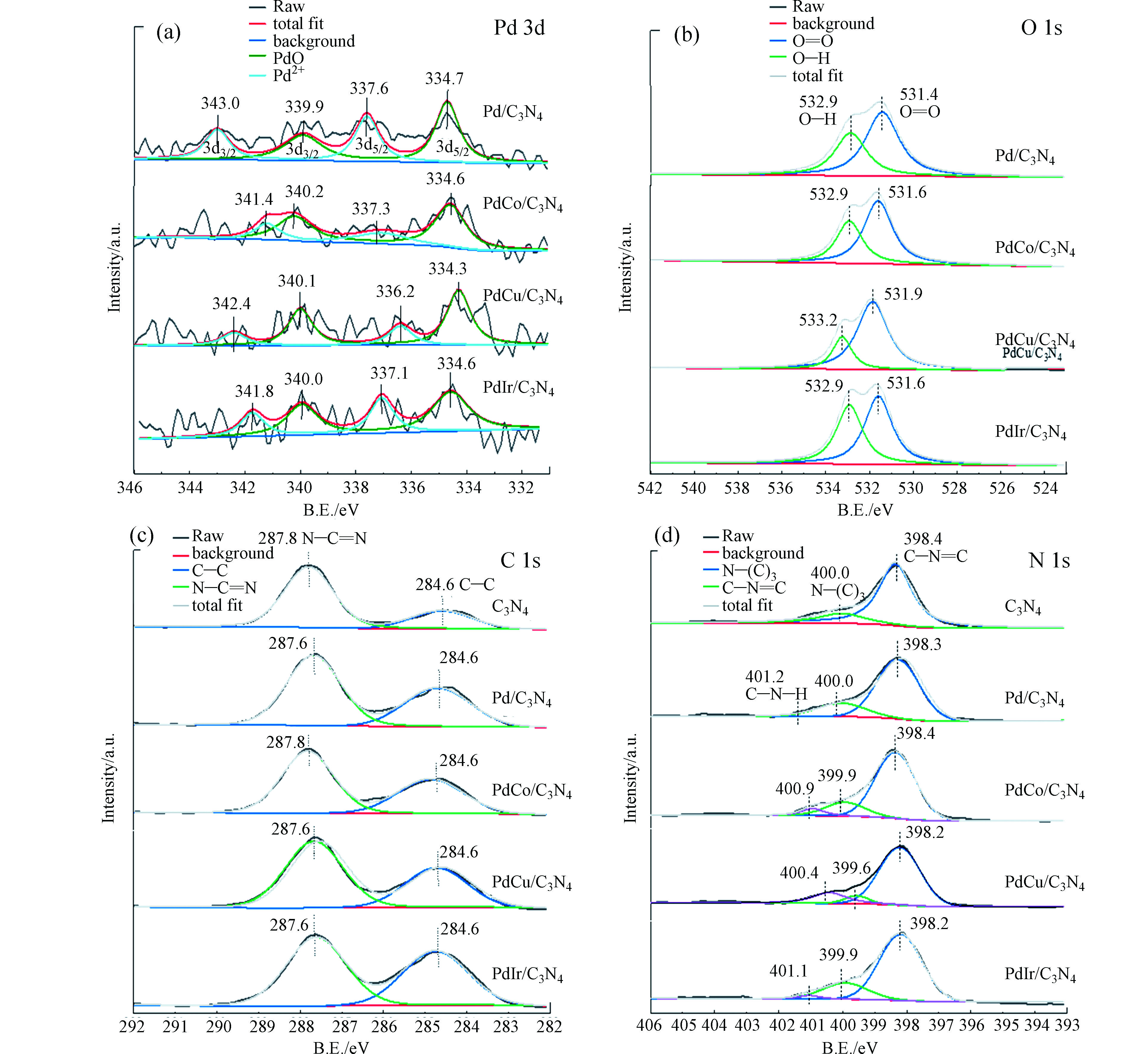 图 5 样品的XPS结果(a) Pd3d, (b) O1s,(c) C1s,(d) N1sFigure 5. XPS spectrum of the (a) Pd3d, (b) O1s,(c) C1s,(d) N1s
图 5 样品的XPS结果(a) Pd3d, (b) O1s,(c) C1s,(d) N1sFigure 5. XPS spectrum of the (a) Pd3d, (b) O1s,(c) C1s,(d) N1s另外,单组份Pd/g-C3N4样品中Pd0和Pd2+的峰面积较接近说明两种物种比例相近,各占一半。而M的引入,两个峰的面积比例发生了变化。大体上是Pd2+物种的面积减少,说明M的引入稳定了金属态的Pd,这也是跟前述的M元素的电负性相关,M和Pd的电负性差异使得Pd有得电子趋势,从而形成了更多Pd0物种。而Ir的电负性和Pd相同,所以Ir的引入对Pd2+物种的量影响不大。接下来对样品中的氧元素进行分析如图5b。可以看出4个样品都在531.4 eV处出现一个主峰,在532.9 eV处出现一个肩峰。531.4 eV处的峰对应于PdM表面吸附氧物种,532.9 eV的峰对应于吸附的水分子中羟基氧物种[25]。相较于单组份样品,PdM/g-C3N4中吸附氧的峰位置均有向高结合能方向偏移的趋势。这也可能是PdM间的相互作用使表面吸附氧物种的化学环境发生了变化。
图5c、d分别对样品中C、N元素进行了XPS分析。C元素都在287.8 eV和284.6 eV处出现了特征峰,对应于g-C3N4中N—C=N的碳和石墨结构的C—C物种[9, 13, 15, 17]。金属粒子负载后N—C=N峰的位置略向低结合能方向偏移约0.2 eV。N1s谱图在398.4 eV出现主峰,在400.0 eV出现肩峰,分别对应C—N=C中的氮和N-(C)3中的氮[9, 13, 15, 17]。金属粒子负载后主峰也略向低结合能方向偏移约0.2 eV。C1s和N1s的结合能偏移,说明金属粒子和载体间具有一定的相互作用,形成了电子传递作用。
2.5 亲疏水性及水分子的吸附能
光解水产氢反应中,水是非常重要的反应物分子。水分子与催化剂间的接触能力将直接影响其催化性能。因此,本文测定了样品的水接触角判断其亲疏水性强弱。结果如图6所示。从图上可以看出,单纯g-C3N4的水接触角是57.7°,而金属粒子负载后其水接触角均有不同程度减小。一般来说,水接触角越小其亲水性越好[15]。所以,金属粒子负载后样品的亲水性都有所增加,也说明反应物水分子和样品间的接触机会增加。PdM粒子负载后其亲水性增加,也说明合成的PdM粒子具有良好的亲水性,可与反应物水分子进行充分接触。其中PdCo/g-C3N4的接触角最小,这可能与其具有较高催化活性相关[26-29]。另外,PdIr/g-C3N4的水接触角相对较大,但是活性却是最好的一个,说明亲疏水性并不是影响催化剂产氢活性的关键因素。Pd和Ir同属Pt族元素,均为贵金属,因此PdIr显示优于PdCo和PdCu的催化活性是由Ir的贵金属特性决定的。
此外,对PdCo、PdCu、PdIr表面吸附水分子的吸附能进行了DFT计算,结果如图7所示。水分子在PdCu、PdIr和PdCo粒子表面的吸附能分别是−5.53、−8.99、−10.15 kcal·mol−1。吸附能越负表明水分子在金属粒子表面吸附越稳定。可见,3种双金属粒子对水分子的吸附顺序是PdCo > PdIr >PdCu。在3种双金属粒子中,PdIr活性最高,是由于Pd和Ir均是贵金属。此外,值得一提的是PdCo的性能与PdIr接近,远高于PdCu。由水分子在PdCu和PdCo表面的吸附能及亲水性可推测,PdCo的活性较高与其更易吸附反应物水分子相关。
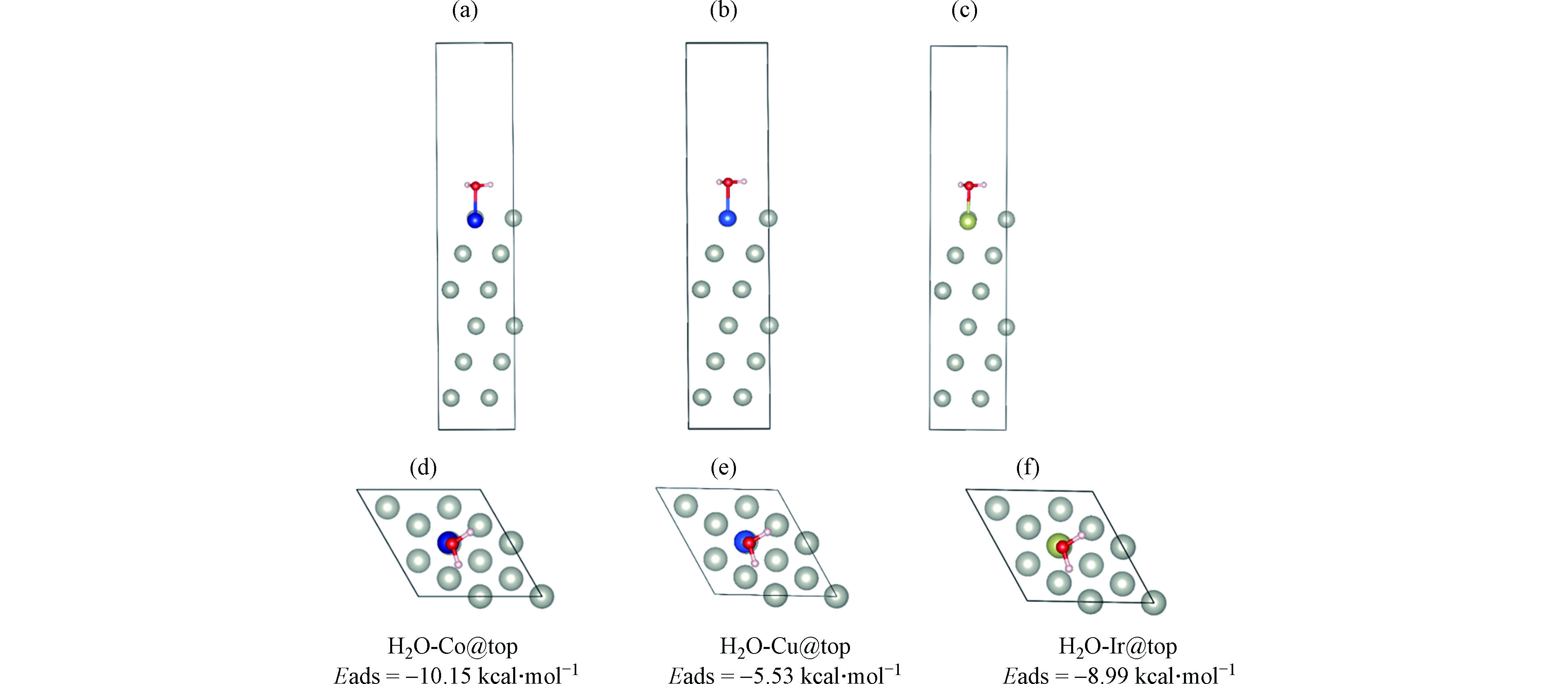 图 7 水分子在PdCo(a、d)、PdCu(b、e)、PdIr(c、f)表面构型及吸附能(a、b、c:侧面图,d、e、f:俯视图)Figure 7. calculated the relaxed geometry of (a) the side view (d) the top view of H2O-Co@top; (b) the side view (e) the top view of H2O-Cu@top; (c) the side view (f) the top view of H2O-Ir@top
图 7 水分子在PdCo(a、d)、PdCu(b、e)、PdIr(c、f)表面构型及吸附能(a、b、c:侧面图,d、e、f:俯视图)Figure 7. calculated the relaxed geometry of (a) the side view (d) the top view of H2O-Co@top; (b) the side view (e) the top view of H2O-Cu@top; (c) the side view (f) the top view of H2O-Ir@top3. 结论(Conclusion)
本文采用乙二醇还原法合成了PdCu、PdCo、PdIr和Pd金属粒子,并将其负载于g-C3N4表面制备出光催化剂用于可见光驱动光解水产氢反应。结果发现,二元金属Co, Ir的引入相较于单金属负载的催化剂0.05 g 4 h产氢量都明显提升,而Cu则稍微有所降低,并且使得Pd原子的平均产氢量增加了1.4—4.4倍。并且,PdIr/g-C3N4经过4次循环测试显示了较高的稳定性。催化性能提升主要是由于:(1) 贵金属负载后对光的吸收能力增加,同时由于金属粒子对光生电子具有强的捕获能力,形成更强的光电流;(2) M原子进入Pd晶格形成了纳米合金,降低了费米能级,使g-C3N4的光生电子在PdM合金粒子上更易传输,更利于电子捕获促进光生电子-空穴的分离;(3) 贵金属粒子负载到g-C3N4表面后,增强了样品的亲水性,降低了水分子吸附能,提升了反应速率。
-
图 1 金属粒子的XRD(a)和TEM(b)
Figure 1. the XRD spectra(a) and TEM micrograph(b) of bimetal nanoparticle
图 2 PdM/g-C3N4的XRD结果(a)和PdCo/g-C3N4 TEM图(b)
Figure 2. XRD spectra of(a) PdM/g-C3N4 and (b)TEM micrograph of PdCo/g-C3N4
图 3 各催化剂光解水产氢性能(a) 总量; (b) PdIr/g-C3N4的循环产氢性能
Figure 3. Performance H2 evolution performance of catalysis (a) Total; (b) The cyclic stability tests for PdIr/g-C3N4
图 4 各催化剂的(a)固体紫外吸收光谱(b)光电流和(c)光致发光光谱
Figure 4. (a) UV-Vis DRS spectra, (b) photocurrent and (c) PL spectra of catalysts
图 5 样品的XPS结果(a) Pd3d, (b) O1s,(c) C1s,(d) N1s
Figure 5. XPS spectrum of the (a) Pd3d, (b) O1s,(c) C1s,(d) N1s
图 7 水分子在PdCo(a、d)、PdCu(b、e)、PdIr(c、f)表面构型及吸附能(a、b、c:侧面图,d、e、f:俯视图)
Figure 7. calculated the relaxed geometry of (a) the side view (d) the top view of H2O-Co@top; (b) the side view (e) the top view of H2O-Cu@top; (c) the side view (f) the top view of H2O-Ir@top
表 1 PdM粒子物理参数
Table 1. Physical parameter of PdM particles
样品Samples (111)衍射峰位置 2θ/(°)(111)Difraction peak 2θ (111) 晶面间距d/nm(111)Interplanar spacing d M原子半径/nmMetal atomic radius Pd 40 0.225 0.169 PdCu 40.28 0.224 0.145 PdCo 40.08 0.225 0.152 PdIr 39.32 0.229 0.180
下载: 导出CSV
-
[1] 邓细宇, 邝鑫雅, 字包叶, 等. 石墨相氮化碳(g-C3N4)用于光催化产氢的研究进展 [J]. 功能材料与器件学报, 2020, 26(1): 7-15. DENG X Y, KAUNG X Y, ZI B Y, et al. Research progress of graphite phase carbon nitride (g-C3N4) for photocatalytic hydrogen production [J]. Journal of Functional Materials and Devices, 2020, 26(1): 7-15(in Chinese).
[2] 柳璐, 张文, 王宇新, 等. 石墨相氮化碳的可控制备及其在能源催化中的应用 [J]. 化工学报, 2018, 69(11): 4577-4591. LIU L, ZHANG W, WANG X Y, et al. Graphitic carbon nitride materials: Controllable preparations and applications in energy catalysis [J]. CIESC Journal, 2018, 69(11): 4577-4591(in Chinese).
[3] 陈克龙, 黄建花, 等. g-C3N4-CdS-NiS2复合纳米管的制备及可见光催化分解水制氢 [J]. 化工学报, 2020, 71(1): 397-408. CHEN K L, HUANG J H, et al. g-C3N4-CdS-NiS2 composite nanotube: Synthesis and its photocatalytic activity for H2 generation under visible light [J]. CIESC Journal, 2020, 71(1): 397-408(in Chinese).
[4] 苏跃涵, 王盈霏, 张钱新, 等. 二维超薄g-C3N4的制备及其光催化性能研究 [J]. 中国环境科学, 2017, 37(10): 3748-3757. doi: 10.3969/j.issn.1000-6923.2017.10.017 SU Y H, WANG Y F, ZHANG Q X, et al. The preparation of two-dimensional ultrathin g-C3N4 and the research of the photo-catalysis properties [J]. China Environment Science, 2017, 37(10): 3748-3757(in Chinese). doi: 10.3969/j.issn.1000-6923.2017.10.017
[5] CAO S, CHEN H, JIANG F, et al. Nitrogen photofixation by ultrathin amine-functionalized graphitic carbon nitride nanosheets as a gaseous product from thermal polymerization of urea [J]. Applied Catalysis B-Environmental, 2018, 224: 222-229. doi: 10.1016/j.apcatb.2017.10.028 [6] 李文博, 郭桂全, 马龙, 等. 金属掺杂改性g-C3N4材料可见光催化性能研究进展 [J]. 冶金管理, 2020(1): 23, 187. LI W B, GUO G Q, MA L, et al. Research progress on visible light catalytic performance of metal-doped modified g-C3N4 materials [J]. China Steel Focus, 2020(1): 23, 187(in Chinese).
[7] LIU C, ZHANG Y, DONG F, et al. Chlorine intercalation in graphitic carbon nitride for efficient photocatalysis [J]. Applied Catalysis B-Environmental, 2017, 203: 465-474. doi: 10.1016/j.apcatb.2016.10.002 [8] KURNARAVEL V, MATHEW S, BARTLETT J, et al. Photocatalytic hydrogen production using metal doped TiO2: A review of recent advances [J]. Applied Catalysis B-Environmental, 2019, 244: 1021-1064. [9] HUANG Z, ZHANG Y, DAI H, et al. Highly dispersed Pd nanoparticles hybridizing with 3D hollow-sphere g-C3N4 to construct 0D/3D composites for efficient photocatalytic hydrogen evolution [J]. Journal of Catalysis, 2019, 378: 331-340. doi: 10.1016/j.jcat.2019.09.007 [10] LIU Y, MIAO C, YANG P, et al. Synergetic promotional effect of oxygen vacancy-rich ultrathin TiO2 and photochemical induced highly dispersed Pt for photoreduction of CO2 with H2O [J]. Applied Catalysis B-Environmental, 2019, 244: 919-930. doi: 10.1016/j.apcatb.2018.12.028 [11] NAULANI-GARCIA M, SALINAS-TORRES D, MORI K, et al. Enhanced formic acid dehydrogenation by the synergistic alloying effect of PdCo catalysts supported on graphitic carbon nitride [J]. International Journal of Hydrogen Energy, 2019, 44(53): 28483-28493. doi: 10.1016/j.ijhydene.2018.11.057 [12] YE T, DURKIN D P, BANEK N A, et al. Graphitic carbon nitride supported ultrafine Pd and Pd-Cu catalysts: Enhanced reactivity, selectivity, and longevity for nitrite and nitrate hydrogenation [J]. Acs Applied Materials & Interfaces, 2017, 9(33): 27421-27426. [13] ZOU W, XU L, PU Y, CAI H, et al. Advantageous interfacial effects of AgPd/g-C3N4 for photocatalytic hydrogen evolution: Electronic structure and H2O dissociation [J]. Chemistry-a European Journal, 2019, 25(19): 5058-5064. doi: 10.1002/chem.201806074 [14] SERAJ S, KUNAL P, LI H, et al. PdAu alloy nanoparticle catalysts: Effective candidates for nitrite reduction in water [J]. Acs Catalysis, 2017, 7(5): 3268-3276. [15] ZHU C, WANG Y, JIANG Z, et al. CeO2 nanocrystal-modified layered MoS2/g-C3N4 as 0D/2D ternary composite for visible-light photocatalytic hydrogen evolution: Interfacial consecutive multi-step electron transfer and enhanced H2O reactant adsorption [J]. Applied Catalysis B-Environmental, 2019, 259: 118072. doi: 10.1016/j.apcatb.2019.118072 [16] CHEN Y, WANG Y, LI W, et al. Enhancement of photocatalytic performance with the use of noble-metal-decorated TiO2 nanocrystals as highly active catalysts for aerobic oxidation under visible-light irradiation [J]. Applied Catalysis B-Environmental, 2017, 210: 352-367. doi: 10.1016/j.apcatb.2017.03.077 [17] CHAVE R K, DO J, KANG M, et al. Strategy for improving the visible photocatalytic H2 evolution activity of 2D graphitic carbon nitride nanosheets through the modification with metal and metal oxide nanocomponents [J]. Applied Catalysis B-Environmental, 2019, 248: 538-551. doi: 10.1016/j.apcatb.2019.01.075 [18] HAN C, LU Y, ZHANG J, et al. Novel PtCo alloy nanoparticle decorated 2D g-C3N4 nanosheets with enhanced photocatalytic activity for H2 evolution under visible light irradiation [J]. Journal of Materials Chemistry A, 2015, 3(46): 23274-23282. [19] ZHONG X H, HAN M Y, DONG Z L, et al. Composition-tunable ZnxCd1-xSe nanocrystals with high luminescence and stability [J]. Journal of the American Chemical Society, 2003, 125(28): 8589-8594. doi: 10.1021/ja035096m [20] ZHONG X H, FENG Y Y, KNOLL W, et al. Alloyed ZnxCd1-xS nanocrystals with highly narrow luminescence spectral width [J]. Journal of the American Chemical Society, 2003, 125(44): 13559-13563. doi: 10.1021/ja036683a [21] LIU H, WANG M, ZHANG X, et al. High efficient photocatalytic hydrogen evolution from formaldehyde over sensitized Ag@Ag-Pd alloy catalyst under visible light irradiation [J]. Applied Catalysis B-Environmental, 2018, 237: 563-573. [22] VERMA P, KUWAHARA Y, MORI K, et al. Pd/Ag and Pd/Au bimetallic nanocatalysts on mesoporous silica for plasmon-mediated enhanced catalytic activity under visible light irradiation [J]. Journal of Materials Chemistry A, 2016, 4(26): 10142-10150. doi: 10.1039/C6TA01664B [23] CAUDILLO-FLORES U, MUNOZ-BATISTA M J, FERNANDEZ-GARCIA M, et al. Bimetallic Pt-Pd co-catalyst Nb-doped TiO2 materials for H2 photo-production under UV and Visible light illumination [J]. Applied Catalysis B-Environmental, 2018, 238: 533-545. doi: 10.1016/j.apcatb.2018.07.047 [24] LIU X, SU P, CHEN, Y, et al. g-C3N4 supported metal (Pd, Ag, Pt) catalysts for hydrogen-production from formic acid [J]. New Journal of Chemistry, 2018, 42(12): 9449-9454. [25] DIAK M, KLEIN M, KLIMCZUK T, et al. Photoactivity of decahedral TiO2 loaded with bimetallic nanoparticles: Degradation pathway of phenol-1-C-13 and hydroxyl radical formation [J]. Applied Catalysis B-Environmental, 2017, 200: 56-71. doi: 10.1016/j.apcatb.2016.06.067 [26] LIN B, YANG G, WANG L, et al. Stacking-layer-number dependence of water adsorption in 3D ordered close-packed g-C3N4 nanosphere arrays for photocatalytic hydrogen evolution [J]. Angewandte Chemie-International Edition, 2019, 58(14): 4587-4591. [27] RAN J, GAO G, LI F.-T, et al Ti3C2 MXene co-catalyst on metal sulfide photo-absorbers for enhanced visible-light photocatalytic hydrogen production [J]. Nature Communications, 2017, 8: 13907. [28] TIAN D, CHEN Q, NIE F Q, et al. Patterned wettability transition by photoelectric cooperative and anisotropic wetting for liquid reprography [J]. Advanced Materials, 2009, 21(37): 3744-3749. doi: 10.1002/adma.200900022 [29] WANG L, GAO Z, LI Y, et al. Photosensitization of CdS by acid red-94 modified alginate: Dual ameliorative effect upon photocatalytic hydrogen evolution [J]. Applied Surface Science, 2019, 492: 598-606. doi: 10.1016/j.apsusc.2019.06.222 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3169
- HTML全文浏览数: 3169
- PDF下载数: 118
- 施引文献: 0
