

























-
抗生素广泛用于人类医疗和畜禽水产养殖中,以治疗疾病和促进动物生长等,其被服用后,大部分会以原形或代谢产物的形式进入到污水处理厂中[1-2]。由于传统污水处理厂对此类生物活性物质的去除不完全,大量抗生素在污水处理厂出水中检出,其中,克拉霉素、红霉素、脱水红霉素、阿奇霉素、罗红霉素、磺胺甲恶唑、甲氧苄胺嘧啶、氧氟沙星、环丙沙星、诺氟沙星和四环素是最常检出的抗生素[3]。这些抗生素最终通过污水处理厂出水排放进入到地表水环境中,并对非靶生物表现出不同程度的生态风险[4]。更令人担忧的是,抗生素的普遍存在可能导致抗性细菌的产生和抗性基因的扩散传播,严重威胁人类健康[5]。因此,必须发展有效的降解技术以削减污水处理厂出水中的抗生素。
近年来,基于UV、热、过渡金属、碳材料等活化过硫酸盐(PS)的高级氧化技术在抗生素降解方面表现出广阔的应用前景[6]。在各种活化方式中,Fe(Ⅱ)因具有无毒、成本低和环境友好的特点,是最常用的PS活化方式之一[7]。但是,Fe(Ⅱ)/PS体系在应用上还具有明显的缺点:Fe(Ⅱ)可与PS迅速反应生成Fe(Ⅲ)(式(1)),而Fe(Ⅲ)还原为Fe(Ⅱ)的过程则十分缓慢,这使得Fe(Ⅱ)被迅速消耗,导致PS的活化持续效果较差;而且,过量Fe(Ⅱ)还会淬灭反应体系中的
SO⋅−4 和·OH(式(2)和(3)),降低对有机污染物的去除效果[8-10]。针对以上不足,研究者提出添加Fe的螯合剂,使Fe(Ⅱ)缓慢释放并在更宽的pH范围内保持可溶状态,同时引入UV光,促进Fe(Ⅲ)向Fe(Ⅱ)的还原,充分提高Fe(Ⅱ)的利用率[11-13]。但是,UV光的引入往往增加了处理成本,限制其推广使用。而太阳光作为一种清洁的可再生能源,也可促进Fe(Ⅲ)向Fe(Ⅱ)的光解还原,提高反应体系对污染物的降解效率[8]。但目前有关同时引入太阳光和螯合剂强化Fe(Ⅱ)/PS对抗生素的降解的研究尚未见报道。因此,本研究以典型抗生素罗红霉素为目标污染物,以柠檬酸作为Fe(Ⅱ)的螯合剂,研究太阳光/Fe(Ⅱ)/柠檬酸/PS高级氧化体系对污水二级出水中罗红霉素的降解效能、影响因素和降解机制,为污水中抗生素的深度处理技术的发展提供科学依据。
-
罗红霉素(纯度>98%)购于百灵威科技有限公司(中国);色谱纯的乙腈和甲醇购于美国Tedia试剂公司;色谱纯乙酸铵和叔丁醇购于Aladdin试剂公司;一水合柠檬酸、冰乙酸、乙醇、七水合硫酸亚铁、过硫酸钾购于国药集团;5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)购于TCL化成工业有限公司。所有试剂均至少为分析纯。实验用水为Millipore超纯水。
污水二级出水取自南京某污水处理厂二沉池出水,运送至实验室后过0.45 μm滤膜,放置于4 ℃冰箱保存。其水质特征为:化学需氧量(COD)为16.7 mg·L−1,总有机碳(TOC)为8.04 mg·L−1,pH 7.8,
NO−3 浓度为28.48 mg·L−1,Cl−浓度为50.09 mg·L−1。 -
降解实验在50 mL石英试管和XPA-7型光化学反应仪中进行。模拟太阳光通过500 W氙灯和截止290 nm的滤光片来获得,其光照强度为97.17 mW·cm−2(CEL-FZ-A辐照计,北京中教金源科技有限公司)。在污水二级出水样品中加入一定量的罗红霉素母液,使其初始浓度为10 mg·L−1;加入一定量的Fe(Ⅱ)母液和柠檬酸,使Fe(Ⅱ)的浓度为0.1、0.25、0.5 mmol·L−1,Fe与柠檬酸的物质的量比为2∶1、1∶1、1∶1.5、1∶2、1∶4,置于黑暗中搅拌30 min,使Fe(Ⅱ)和柠檬酸充分络合;再加入一定量的PS母液,使PS的浓度分别为0.5、1、2、4、8 mmol·L−1。然后将反应溶液置于光化学反应仪中,反应一段时间后(0、1、2、5、10、20、30、45、60 min),取1.5 mL的样品放入含有50 μL乙醇(终止由任何残留氧化剂或自由基引起的氧化)液相色谱小瓶中,测定罗红霉素的浓度。同时做黑暗/Fe(Ⅱ)/PS、太阳光/Fe(Ⅱ)/PS、太阳光/Fe(Ⅱ)/柠檬酸、黑暗/Fe(Ⅱ)/柠檬酸/PS对照组,每个处理组至少重复2次。
-
在上述的反应体系中,除加入罗红霉素外,再加入100 mmol·L−1的乙醇或叔丁醇分别作为
SO⋅−4 和/或·OH的淬灭剂,以考察活性物种对罗红霉素的贡献。为验证·OH和
SO⋅−4 的存在,在合适的反应条件下,在反应0、15、30、45 min时加入50 mmol·L−1的DMPO作为·OH和SO⋅−4 的特异性捕获剂,捕获15 min后取样,采用电子自旋共振波谱仪(EMX-10/12,德国Bruker公司)鉴定活性物种。 -
罗红霉素的浓度采用Ultimate 3000液相色谱仪(Dionex, 美国)测定[14]。
罗红霉素的降解产物采用固相萃取-LTQ-Orbitrap-XL高分辨液质联用仪(Thermo Scientific,美国)来测定[15]。反应60 min后的样品用于罗红霉素降解产物的测定,反应0、5、15、30、45、60 min的样品用于了解罗红霉素降解产物随时间的变化情况。
-
在Fe(Ⅱ)浓度为0.25 mmol·L−1,PS为4 mmol·L−1,Fe(Ⅱ)与柠檬酸物质的量比为1∶1.5和1∶2的条件下,研究了黑暗/Fe(Ⅱ)/PS,太阳光/Fe(Ⅱ)/PS、太阳光/Fe(Ⅱ)/柠檬酸、黑暗/Fe(Ⅱ)/柠檬酸/PS和太阳光/Fe(Ⅱ)/柠檬酸/PS对罗红霉素的降解作用(图1)。
由图1可见,反应60 min后,黑暗/Fe(Ⅱ)/PS和太阳光/Fe(Ⅱ)/PS对污水二级出水中罗红霉素的去除率分别仅为5.9%和9.5%,这可能是由于污水的pH(7.8)较高造成的,在较高的pH下,Fe主要以沉淀或胶体状态存在[16],无法有效的激活PS,导致罗红霉素的去除率较低。在Fe/柠檬酸物质的量比为1∶1.5和1∶2时,太阳光/Fe(Ⅱ)/柠檬酸对罗红霉素的去除率分别为13.5%和23.0%。以往研究发现,在紫外光或太阳光条件下,Fe(Ⅲ)/柠檬酸络合物可光解产生·OH,促进磺胺甲恶唑等有机物的降解[17-18]。但是,较高的pH不利于·OH的产生[17],因此,太阳光/Fe(Ⅱ)/柠檬酸对污水二级出水中罗红霉素的去除效果并不理想。
黑暗/Fe(Ⅱ)/柠檬酸/PS体系中罗红霉素的去除率也较低,仅为10%左右。相较而言,太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的去除率显著加强。在Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5时和1∶2时,反应60 min后,罗红霉素的去除率分别为46.9%和91.7%。柠檬酸中有3个羧基配位基,在Fe(Ⅱ)与柠檬酸的物质的量比为1∶2时,Fe(Ⅱ)可以与柠檬酸完全络合形成六配位络合物[11]。Fe(Ⅱ)/柠檬酸络合物与PS反应可转化成Fe(Ⅲ)/柠檬酸络合物。在太阳光照射下,Fe(Ⅲ)/柠檬酸络合物可通过配位到金属的电荷转移过程生成Fe(Ⅱ)和柠檬酸自由基(公式(4)),柠檬酸自由基通过与O2的反应生成O2·-和H2O2等活性物种(式(5)—(8)),Fe(Ⅱ)可进一步与PS和H2O2反应生成
SO⋅−4 和·OH(式(1)和(9)),促进罗红霉素的降解[19, 20]。式中,R表示CH2COOH。
污水二级出水的初始pH 7.8,当加入的物质的量比分别为1∶1.5和1∶2的Fe(Ⅱ)/柠檬酸后,反应体系的pH值降低至6.4和6.1。随着反应的进行,反应体系的pH逐渐减低,反应60 min后,Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5和1∶2体系的pH值可降低至5.6和2.4。以往研究也发现Fe(Ⅱ)/PS体系反应后的pH降低至3左右[21-22]。这主要是由PS分解和
SO⋅−4 自由基与H2O反应生成H+,以及SO⋅−4 自由基对OH−的消耗造成的(方程式(10)—(12))[11, 23-24]。因此,Fe(Ⅱ)/柠檬酸摩尔比为1∶2的反应体系最终pH较低也表明反应体系中产生了更多的SO⋅−4 和·OH。采用一级动力学和二级动力学对不同反应体系中罗红霉素的降解数据进行拟合,结果见表1。
由表1可见,黑暗/Fe(Ⅱ)/PS体系和太阳光/Fe(Ⅱ)/PS体系中罗红霉素的降解符合一级反应动力学和二级反应动力学;太阳光/Fe(Ⅱ)/柠檬酸体系和黑暗/Fe(Ⅱ)/柠檬酸/PS体系中,可能由于柠檬酸的存在,一级动力学和二级动力学的拟合效果均不佳。而太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的降解更符合一级动力学模型。在Fe浓度为0.25 mmol·L−1,PS浓度为4 mmol·L−1,Fe:柠檬酸的物质的量比为1∶1.5和1∶2时,罗红霉素降解的一级反应动力学常数分别为0.0103 min−1和0.0409 min−1(表1)。这说明螯合剂柠檬酸的添加实现了Fe(Ⅱ)的缓慢释放,使Fe(Ⅱ)能够平稳的活化PS,保持反应体系的持续氧化效果。
-
图2为Fe(Ⅱ)的浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响。在PS为4 mmol·L−1,Fe/柠檬酸的物质的量比为1:1时,反应体系对罗红霉素的去除速率随Fe(Ⅱ)浓度增加而增加。在Fe(Ⅱ)浓度为0.1、0.25 mmol·L−1时,反应60 min后,罗红霉素的去除率分别为12.0%和21.7%;当Fe(Ⅱ)升高至0.5 mmol·L−1后,罗红霉素的去除率显著增加,反应20 min后,罗红霉素的去除率可达到90.7%,但是当反应时间继续延长时,罗红霉素的去除率增加缓慢。这可能是由于后期反应体系中Fe(Ⅱ)浓度降低,产生的活性物种浓度降低以及罗红霉素降解产物对活性自由基的竞争造成的。而且,在Fe(Ⅱ)浓度为0.5 mmol·L−1时,反应20 min后,反应体系的pH值降至2.9。在此pH条件下,罗红霉素以质子化的形式存在,其与·OH等自由基的反应速率也较慢[25]。
不同Fe(Ⅱ)浓度体系中罗红霉素的快速降解阶段符合一级反应动力学。当Fe(Ⅱ)浓度为0.1、0.25、0.5 mmol·L−1时,罗红霉素的一级反应动力学常数分别为0.0022、0.0043、0.118 min−1(前20 min)。这说明较高浓度的Fe(Ⅱ)可以激活PS产生更多的·OH和
SO⋅−4 ,促进罗红霉素的降解[26]。但是Fe(Ⅱ)含量过高时也可能淬灭自由基[22],并产生较多的污泥,不利于后续的处理,因此Fe(Ⅱ)的投加量不宜过高。再者,本实验中所选用的Fe(Ⅱ)/柠檬酸的物质的量比为1∶1,当Fe与柠檬酸摩尔的物质的量比提高时,也可能促进活性自由基的产生和罗红霉素的降解。因此,从节约资源和减少Fe污泥的角度出发,选择Fe(Ⅱ)浓度为0.25、0.5 mmol·L−1进行后续实验。 -
图3为PS浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响。在Fe(Ⅱ)浓度为0.25 mmol·L−1时,太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的去除率和反应速率常数随PS增加而略有增加,这可能是因为反应体系中Fe(Ⅱ)浓度较低,不足以活化过量的PS,因此,Fe(Ⅱ)浓度低时,PS并不是限制该反应体系对罗红霉素降解效能的主要因素。当Fe(Ⅱ)浓度为0.5 mmol·L−1时,太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的去除率和反应速率常数随PS增加而显著增加,在PS浓度为4 mmol·L−1时,罗红霉素的去除率即可达到95%,罗红霉素的降解的一级反应速率常数(0.1034 min−1)可达到PS浓度为0.5 mmol·L−1条件下的8.4倍。这说明在保证Fe(Ⅱ)浓度充足的情况下,高浓度的PS生成了更多的活性自由基,促进了罗红霉素的降解[10]。
-
图4为Fe(Ⅱ)与柠檬酸的摩尔比对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响。在Fe(Ⅱ)浓度为0.25 mmol·L−1时,太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的去除率和反应速率常数随Fe(Ⅱ)/柠檬酸的物质的量比增加而增大。当Fe(Ⅱ)/柠檬酸的物质的量比从2∶1增加到1∶4时,罗红霉素的去除率从19.6%增加到94.3%,罗红霉素降解的一级反应速率常数从0.0045 min−1增加至0.0778 min−1(图4(A))。值得注意的是,尽管Fe(Ⅱ)/柠檬酸的物质的量比为1∶4的反应速率比Fe(Ⅱ)/柠檬酸的物质的量比为1∶2的大,但反应60 min后,两者对罗红霉素的去除率非常接近。
在Fe(Ⅱ)浓度为0.5 mmol·L−1时,当Fe(Ⅱ)/柠檬酸的物质的量比从2:1增加到1∶1时,罗红霉素的去除率从35.5%增加到了95.4%,一级反应速率常数从0.0094 min−1显著增加到了0.1034 min−1,这说明柠檬酸的用量对太阳光/Fe(Ⅱ)/柠檬酸/PS体系的降解效果影响很大。但是当Fe(Ⅱ)/柠檬酸的摩尔比继续增加到1∶2时,罗红霉素的去除率和一级反应速率常数均降低。Tan等[27]研究发现Fe(Ⅱ)/柠檬酸的物质的量比从1∶1增加到1∶5时,Fe(Ⅱ)/柠檬酸/PS对敌草隆的去除率从80%降低到了57%。过量柠檬酸对反应体系中污染物降解的抑制作用可能是由两方面的原因造成的,一是柠檬酸与污染物竞争消耗反应体系中的活性自由基;二是柠檬酸与Fe(Ⅱ)的过分螯合阻碍了Fe与PS的反应,导致反应体系中产生的活性物种量减少[28]。
综上,结合试剂用量和对罗红霉素的去除效果,可确定太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素降解的反应条件为Fe(Ⅱ)浓度为0.25 mmol·L−1,Fe(Ⅱ)/柠檬酸的物质的量比为1:2,PS为4 mmol·L−1,反应时间60 min,在此条件下,罗红霉素的去除率可达到92%以上。
-
在Fe(Ⅱ)浓度为0.25 mmol·L−1,PS为4 mmol·L−1,Fe/柠檬酸的物质的量比为1∶1.5和1∶2的太阳光/Fe(Ⅱ)/柠檬酸/PS体系中分别加入乙醇和叔丁醇,以考察反应体系中的活性物种及其贡献(表2)。乙醇可同时淬灭·OH和
SO⋅−4 ,而叔丁醇仅可淬灭·OH[29]。加入叔丁醇后,Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5和1∶2体系中罗红霉素降解的一级反应速率常数分别从0.0120 min−1和0.0397 min−1降低为0.0026 min−1和0.0054 min−1,根据公式(13)计算得到·OH的贡献率分别为78.3%和86.4%。加入乙醇后,Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5和1∶2体系中罗红霉素降解的一级反应速率常数分别为0.0011 min−1和0.0025 min−1 ,根据公式(14)计算得到SO⋅−4 的贡献分别为12.5%和7.3%。这说明反应体系中·OH是起主要作用的活性物种,与以往研究报道的Fe(Ⅱ)/柠檬酸/PS体系中·OH是主要的活性物种相一致[11, 23]。此外,反应体系也可能通过Fe(Ⅱ)/柠檬酸络合物的光解(方程(4)—(8))和方程(15)—(18)产生O2·-和HO2·等活性物种[19, 30],这些活性物种对两个体系中罗红霉素的去除的贡献分别为9.2%和6.3%。式中,C·OH和
CSO4⋅− 分别表示·OH和SO⋅−4 对罗红霉素降解的贡献;kobs表示未添加淬灭剂时反应体系中罗红霉素降解的表观反应速率常数;kTBA和kEtOH表示加入叔丁醇和乙醇时罗红霉素降解的反应速率常数.为了更直观地验证反应体系中活性物种的存在,分别在反应0、15、30、45 min的时候,添加DMPO作为·OH和
SO⋅−4 的捕获剂,测定反应体系的电子自旋共振波谱,结果见图5。Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5和1∶2体系中均可明显观察到·OH的1∶2∶2∶1的信号,但并未观察到SO⋅−4 的信号,这可能是由于SO⋅−4 的浓度较低和DMPO的捕获时间较短(15 min)造成的。同时这也证明了·OH是太阳光/Fe(Ⅱ)/柠檬酸/PS体系的主要活性物种。图5也表明了不同反应时间段内反应体系中·OH的相对含量。在Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5的体系中,在不同时间段内,·OH的信号强度基本一致,说明Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5时,反应体系的·OH浓度保持稳定。而在Fe(Ⅱ)/柠檬酸的物质的量为1∶2的反应体系中,·OH的信号强度在30—60 min要比0—30 min时弱,这可能是因为柠檬酸含量高时,Fe(Ⅱ)/柠檬酸络合物与PS的反应速率较快,而Fe(Ⅲ)/柠檬酸络合物光解还原为Fe(Ⅱ)的速率相对较慢,导致后期反应体系中Fe(Ⅱ)浓度降低,降低了·OH的产率。这与反应体系中罗红霉素的降解情况是一致的。
-
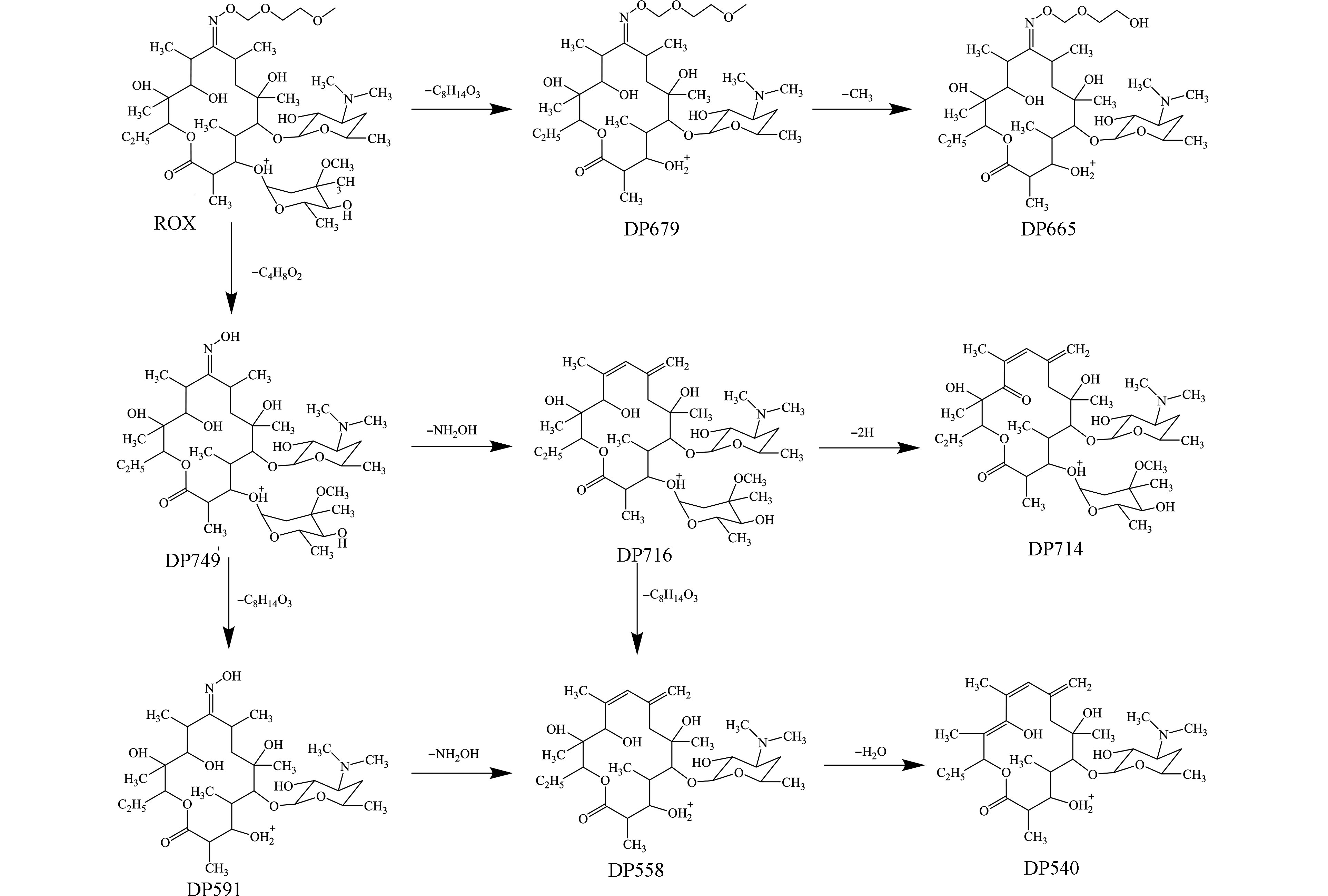
在Fe(Ⅱ)浓度为0.25 mmol·L−1,PS为4 mmol·L−1,Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5和1∶2的条件下,研究了太阳光/Fe(Ⅱ)/柠檬酸/PS体系中罗红霉素的降解产物。根据罗红霉素降解前后的总离子流图、降解产物的精确分子量、二级碎裂质谱图以及Xcalibur的分子式计算功能,共识别出8种罗红霉素的降解产物。罗红霉素及其降解产物(以DP+m/z命名)的精确分子量和元素组成等信息见表3,分子结构见图6。
在8种降解产物中,DP749在UV/H2O2降解罗红霉素的体系中报道过[14],DP679、DP665和DP591在罗红霉素的光降解过程中报道过[15]。而DP716、DP714、DP558和DP540尚未见报道。这4种降解产物的二级质谱中都有碎片离子m/z158.1172,说明红霉脱氧糖胺保持完整。DP716的分子量比DP749的少33 Da,说明DP716是由DP749上脱去羟胺(NH2OH)形成的。相似的,DP558是由DP591上脱去羟胺形成的。DP714和DP540分别是DP716脱去2H和DP558脱去H2O形成的。此外,和文献中报道的罗红霉素的降解产物相比[14-15, 31-32],太阳光/Fe(Ⅱ)/柠檬酸/PS体系中产生的降解产物的分子量普遍要小,这说明太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的氧化降解性能可能更强。
图7为罗红霉素的降解产物的量(以降解产物的峰面积与罗红霉素的初始峰面积比值A/A0表示)随反应时间的变化情况。
由图7可见,反应15 min后,DP749即可达到最大的A/A0值,这说明罗红霉素首先生成DP749。在Fe(Ⅱ)/柠檬酸物质的量比为1∶1.5时,DP679、DP716、DP558和DP714在反应30 min时达到最大值,DP591、DP665和DP540在反应45 min时达到最大值,而在Fe(Ⅱ)/柠檬酸为1∶2时,DP679和DP716在反应30 min后达到最大,其它5种降解产物均在反应45 min时达到最大值。除DP714外,Fe(Ⅱ)/柠檬酸物质的量比为1∶2体系中的降解产物的A/A0值均高于Fe(Ⅱ)/柠檬酸为1∶1.5条件下。这进一步说明了Fe(Ⅱ)/柠檬酸为1∶2的反应体系中产生了更多的·OH,对罗红霉素的降解程度要高于Fe(Ⅱ)/柠檬酸物质的量比为1∶1.5体系.
-
(1)太阳光/Fe(Ⅱ)/柠檬酸/PS高级氧化体系能有效去除污水处理厂二级出水中的罗红霉素,在Fe(Ⅱ)浓度为0.25 mmol·L−1,Fe(Ⅱ)/柠檬酸物质的量比为1∶2,PS为4 mmol·L−1,反应时间60 min时,罗红霉素的去除率可达到92%以上。
(2)太阳光/Fe(Ⅱ)/柠檬酸/PS高级氧化技术可同时产生
SO⋅−4 和·OH,其中·OH是最主要的活性物质,其对罗红霉素降解的贡献可达到78.3%—86.4%,SO⋅−4 的贡献仅为7.3%—12.5%。(3)基于高效液相色谱-高分辨质谱鉴定出8种罗红霉素的降解产物,这些降解产物主要通过脱去克拉定糖(—C8H14O3)、肟侧链的断裂(—C4H8O2)、脱羟胺(—NH2OH)等过程产生,其中,罗红霉素通过肟侧链断裂和脱去克拉定糖生成的DP749和DP679是最主要的降解产物。
太阳光/Fe(Ⅱ)/柠檬酸/激活过硫酸盐对污水二级出水中罗红霉素的降解效能
Degradation of roxithromycin in secondary effluent by persulfate activated by the combination of sunlight, Fe(Ⅱ) and citric acid
-
摘要: 本文以柠檬酸作为Fe(Ⅱ)的螯合剂,引入模拟太阳光强化Fe(Ⅱ)/柠檬酸/螯合物活化过硫酸盐(PS)氧化降解污水处理厂二级出水中的罗红霉素,探讨了该体系对罗红霉素的降解效果、影响因素、活性物种的贡献和降解路径。结果表明,太阳光/Fe(Ⅱ)/柠檬酸/PS体系能有效降解罗红霉素,在Fe(Ⅱ)浓度为0.25 mmol·L−1,PS为4 mmol·L−1,Fe(Ⅱ)/柠檬酸物质的量比为1∶2,反应60 min时,罗红霉素的去除率可达到92%以上。淬灭实验表明,·OH是反应体系的主要活性物种,其对罗红霉素降解的贡献为78.3%—86.4%,而
SO⋅−4 的作用相对较弱,其贡献仅为7.3%—12.5%。基于高效液相色谱高分辨质谱鉴别出8种罗红霉素的降解产物,它们主要是由罗红霉素的肟侧链断裂(—C4H8O2)、脱克拉定糖(—C8H14O3)和脱羟胺(—NH2OH)形成的,其中DP679(C33H63O12N2)和DP749(C37H69O13N2)是最主要的降解产物。-
关键词:
- 罗红霉素 /
- 过硫酸盐 /
- Fe(Ⅱ)-柠檬酸螯合物 /
- 活性物种 /
- 降解产物
Abstract: In this study, citric acid was used as the chelating agent of Fe(Ⅱ), and simulated sunlight was introduced to enhance the degradation of roxithromycin in secondary wastewater by persulfate (PS) activated by Fe(Ⅱ)-citrate chelate complex. The removal efficiency, impact factors, contribution of active species and degradation pathway of roxithromycin were examined. The results showed that sunlight / Fe(Ⅱ) / citrate / PS system can degrade roxithromycin effectively. When Fe(Ⅱ) concentration was 0.25 mmol·L−1, PS concentration was 4 mmol·L−1, molar ratio of Fe(Ⅱ) / citrate was 1∶2, and reaction time was 60 min, the removal efficiency of roxithromycin reached higher than 92%. Quenching experiments showed that ·OH was the principal active species in the reaction system, and its contribution to the degradation of roxithromycin was 78.3%—86.4%; In contrast,SO⋅−4 played a minor role in the degradation of roxithromycin, with the contribution of only 7.3%—12.5%. Eight degradation products (DPs) of roxithromycin were identified using high performance liquid chromatography and high resolution mass spectrometry. They were mainly formed by the cleavage of C4H8O2 from the oxime side chain, cleavage of cladinose (C8H14O3) and hydroxylamine (NH2OH). DP679 (C33H63O12N2) and DP749 (C37H69O13N2) were the primary degradation products.-
Key words:
- roxithromycin /
- persulfate /
- Fe(Ⅱ)-citrate chelate /
- reactive species /
- degradation products
-
抗生素广泛用于人类医疗和畜禽水产养殖中,以治疗疾病和促进动物生长等,其被服用后,大部分会以原形或代谢产物的形式进入到污水处理厂中[1-2]。由于传统污水处理厂对此类生物活性物质的去除不完全,大量抗生素在污水处理厂出水中检出,其中,克拉霉素、红霉素、脱水红霉素、阿奇霉素、罗红霉素、磺胺甲恶唑、甲氧苄胺嘧啶、氧氟沙星、环丙沙星、诺氟沙星和四环素是最常检出的抗生素[3]。这些抗生素最终通过污水处理厂出水排放进入到地表水环境中,并对非靶生物表现出不同程度的生态风险[4]。更令人担忧的是,抗生素的普遍存在可能导致抗性细菌的产生和抗性基因的扩散传播,严重威胁人类健康[5]。因此,必须发展有效的降解技术以削减污水处理厂出水中的抗生素。
近年来,基于UV、热、过渡金属、碳材料等活化过硫酸盐(PS)的高级氧化技术在抗生素降解方面表现出广阔的应用前景[6]。在各种活化方式中,Fe(Ⅱ)因具有无毒、成本低和环境友好的特点,是最常用的PS活化方式之一[7]。但是,Fe(Ⅱ)/PS体系在应用上还具有明显的缺点:Fe(Ⅱ)可与PS迅速反应生成Fe(Ⅲ)(式(1)),而Fe(Ⅲ)还原为Fe(Ⅱ)的过程则十分缓慢,这使得Fe(Ⅱ)被迅速消耗,导致PS的活化持续效果较差;而且,过量Fe(Ⅱ)还会淬灭反应体系中的
SO⋅−4 Fe(Ⅱ)+S2O2−8⟶Fe(Ⅲ)+SO2−4+SO⋅−4 (1) Fe(Ⅱ)+SO⋅−4⟶Fe(Ⅲ)+SO2−4 (2) Fe(Ⅱ)+⋅OH⟶Fe(Ⅲ)+OH− (3) 因此,本研究以典型抗生素罗红霉素为目标污染物,以柠檬酸作为Fe(Ⅱ)的螯合剂,研究太阳光/Fe(Ⅱ)/柠檬酸/PS高级氧化体系对污水二级出水中罗红霉素的降解效能、影响因素和降解机制,为污水中抗生素的深度处理技术的发展提供科学依据。
1. 材料与方法(Materials and methods)
1.1 实验试剂和水样
罗红霉素(纯度>98%)购于百灵威科技有限公司(中国);色谱纯的乙腈和甲醇购于美国Tedia试剂公司;色谱纯乙酸铵和叔丁醇购于Aladdin试剂公司;一水合柠檬酸、冰乙酸、乙醇、七水合硫酸亚铁、过硫酸钾购于国药集团;5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)购于TCL化成工业有限公司。所有试剂均至少为分析纯。实验用水为Millipore超纯水。
污水二级出水取自南京某污水处理厂二沉池出水,运送至实验室后过0.45 μm滤膜,放置于4 ℃冰箱保存。其水质特征为:化学需氧量(COD)为16.7 mg·L−1,总有机碳(TOC)为8.04 mg·L−1,pH 7.8,
NO−3 1.2 降解实验
降解实验在50 mL石英试管和XPA-7型光化学反应仪中进行。模拟太阳光通过500 W氙灯和截止290 nm的滤光片来获得,其光照强度为97.17 mW·cm−2(CEL-FZ-A辐照计,北京中教金源科技有限公司)。在污水二级出水样品中加入一定量的罗红霉素母液,使其初始浓度为10 mg·L−1;加入一定量的Fe(Ⅱ)母液和柠檬酸,使Fe(Ⅱ)的浓度为0.1、0.25、0.5 mmol·L−1,Fe与柠檬酸的物质的量比为2∶1、1∶1、1∶1.5、1∶2、1∶4,置于黑暗中搅拌30 min,使Fe(Ⅱ)和柠檬酸充分络合;再加入一定量的PS母液,使PS的浓度分别为0.5、1、2、4、8 mmol·L−1。然后将反应溶液置于光化学反应仪中,反应一段时间后(0、1、2、5、10、20、30、45、60 min),取1.5 mL的样品放入含有50 μL乙醇(终止由任何残留氧化剂或自由基引起的氧化)液相色谱小瓶中,测定罗红霉素的浓度。同时做黑暗/Fe(Ⅱ)/PS、太阳光/Fe(Ⅱ)/PS、太阳光/Fe(Ⅱ)/柠檬酸、黑暗/Fe(Ⅱ)/柠檬酸/PS对照组,每个处理组至少重复2次。
1.3 活性物种的贡献
在上述的反应体系中,除加入罗红霉素外,再加入100 mmol·L−1的乙醇或叔丁醇分别作为
SO⋅−4 为验证·OH和
SO⋅−4 SO⋅−4 1.4 罗红霉素及其降解产物的分析测定
罗红霉素的浓度采用Ultimate 3000液相色谱仪(Dionex, 美国)测定[14]。
罗红霉素的降解产物采用固相萃取-LTQ-Orbitrap-XL高分辨液质联用仪(Thermo Scientific,美国)来测定[15]。反应60 min后的样品用于罗红霉素降解产物的测定,反应0、5、15、30、45、60 min的样品用于了解罗红霉素降解产物随时间的变化情况。
2. 结果与讨论(Results and discussion)
2.1 不同过硫酸盐体系对罗红霉素的降解作用
在Fe(Ⅱ)浓度为0.25 mmol·L−1,PS为4 mmol·L−1,Fe(Ⅱ)与柠檬酸物质的量比为1∶1.5和1∶2的条件下,研究了黑暗/Fe(Ⅱ)/PS,太阳光/Fe(Ⅱ)/PS、太阳光/Fe(Ⅱ)/柠檬酸、黑暗/Fe(Ⅱ)/柠檬酸/PS和太阳光/Fe(Ⅱ)/柠檬酸/PS对罗红霉素的降解作用(图1)。
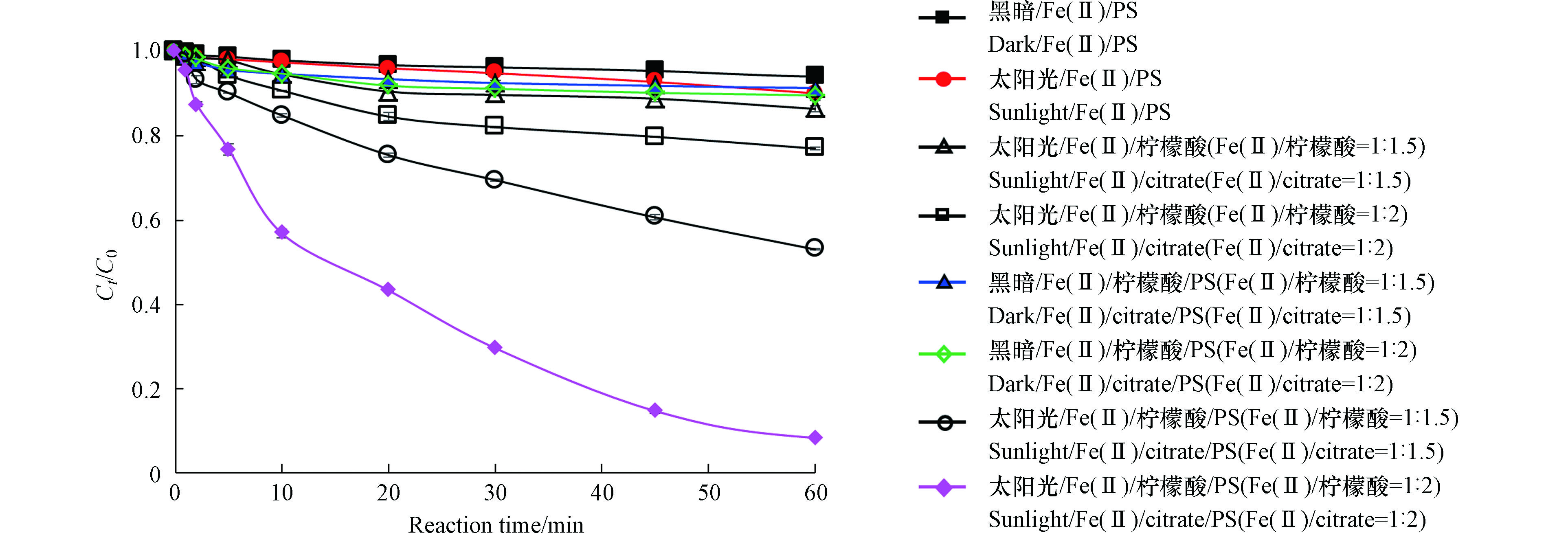 图 1 不同处理系统中罗红霉素的去除率[Fe(Ⅱ)]0=0.25 mmol·L−1,[PS]0=4 mmol·L−1Figure 1. Removal efficiency of roxithromycin in different treatment system
图 1 不同处理系统中罗红霉素的去除率[Fe(Ⅱ)]0=0.25 mmol·L−1,[PS]0=4 mmol·L−1Figure 1. Removal efficiency of roxithromycin in different treatment system由图1可见,反应60 min后,黑暗/Fe(Ⅱ)/PS和太阳光/Fe(Ⅱ)/PS对污水二级出水中罗红霉素的去除率分别仅为5.9%和9.5%,这可能是由于污水的pH(7.8)较高造成的,在较高的pH下,Fe主要以沉淀或胶体状态存在[16],无法有效的激活PS,导致罗红霉素的去除率较低。在Fe/柠檬酸物质的量比为1∶1.5和1∶2时,太阳光/Fe(Ⅱ)/柠檬酸对罗红霉素的去除率分别为13.5%和23.0%。以往研究发现,在紫外光或太阳光条件下,Fe(Ⅲ)/柠檬酸络合物可光解产生·OH,促进磺胺甲恶唑等有机物的降解[17-18]。但是,较高的pH不利于·OH的产生[17],因此,太阳光/Fe(Ⅱ)/柠檬酸对污水二级出水中罗红霉素的去除效果并不理想。
黑暗/Fe(Ⅱ)/柠檬酸/PS体系中罗红霉素的去除率也较低,仅为10%左右。相较而言,太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的去除率显著加强。在Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5时和1∶2时,反应60 min后,罗红霉素的去除率分别为46.9%和91.7%。柠檬酸中有3个羧基配位基,在Fe(Ⅱ)与柠檬酸的物质的量比为1∶2时,Fe(Ⅱ)可以与柠檬酸完全络合形成六配位络合物[11]。Fe(Ⅱ)/柠檬酸络合物与PS反应可转化成Fe(Ⅲ)/柠檬酸络合物。在太阳光照射下,Fe(Ⅲ)/柠檬酸络合物可通过配位到金属的电荷转移过程生成Fe(Ⅱ)和柠檬酸自由基(公式(4)),柠檬酸自由基通过与O2的反应生成O2·-和H2O2等活性物种(式(5)—(8)),Fe(Ⅱ)可进一步与PS和H2O2反应生成
SO⋅−4 Fe(Ⅲ)−cithν→Fe(Ⅱ)+Cit⋅ (4) Cit⋅⟶HO−CR⋅2+CO2 (5) HO−CR⋅2+Fe(Ⅲ)⟶R2C=O+H++Fe(Ⅱ) (6) HO−CR⋅2+O2⟶R2C=O+H++O⋅−2 (7) {\rm{2}}{{\rm{H}}^{\rm{ + }}}{\rm{ + 2O}}_2^{ \cdot - }\rightleftharpoons}_{\rm{2}}}{{\rm{O}}_{\rm{2}}}{\rm{ + }}{{\rm{O}}_{\rm{2}}} (8) H2O2+Fe(Ⅱ)⟶Fe(Ⅲ)+⋅OH+OH− (9) 式中,R表示CH2COOH。
污水二级出水的初始pH 7.8,当加入的物质的量比分别为1∶1.5和1∶2的Fe(Ⅱ)/柠檬酸后,反应体系的pH值降低至6.4和6.1。随着反应的进行,反应体系的pH逐渐减低,反应60 min后,Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5和1∶2体系的pH值可降低至5.6和2.4。以往研究也发现Fe(Ⅱ)/PS体系反应后的pH降低至3左右[21-22]。这主要是由PS分解和
SO⋅−4 SO⋅−4 SO⋅−4 S2O82−+H2O⟶2SO2−4+HO2−+3H+ (10) SO⋅−4+H2O⟶⋅OH+SO2−4+H+ (11) SO⋅−4+OH−⟶SO2−4+⋅OH (12) 采用一级动力学和二级动力学对不同反应体系中罗红霉素的降解数据进行拟合,结果见表1。
表 1 不同反应体系中,罗红霉素的降解反应动力学拟合常数Table 1. The degradation kinetics fitting constants of roxithromycin in different reaction systems反应体系(物质的量比)Reaction system 一级动力学Pseudo-first order kinetic 二级动力学Second order kinetics k1/min−1 R2 k2/(L·mol−1·s−1) R2 黑暗/Fe(Ⅱ)/PS 0.001 0.9564 1.6143 0.9593 太阳光/Fe(Ⅱ)/PS 0.0016 0.9886 2.8060 0.9887 太阳光/Fe(Ⅱ)/柠檬酸(1∶1.5)Sunlight/Fe(Ⅱ)/citrate(1∶1.5) 0.0024 0.8957 4.0228 0.9042 太阳光/Fe(Ⅱ)/柠檬酸1∶2)Sunlight/Fe(Ⅱ)/citrate(1∶2) 0.0045 0.9082 8.5515 0.9247 黑暗/Fe(Ⅱ)/柠檬酸/PS(1∶1.5)Dark/Fe(Ⅱ)/citrate/PS(1∶1.5) 0.0013 0.7795 2.5793 0.7887 黑暗/Fe(Ⅱ)/柠檬酸/PS(1∶2)Dark/Fe(Ⅱ)/citrate/PS(1∶2) 0.0018 0.8343 3.1015 0.8428 太阳光/Fe(Ⅱ)/柠檬酸/PS(1∶1.5)Sunlight/Fe(Ⅱ)/citrate/PS(1∶1.5) 0.0103 0.9848 22.866 0.9962 太阳光/Fe(Ⅱ)/柠檬酸/PS(1∶2)Sunlight/Fe(Ⅱ)/citrate/PS(1∶2) 0.0409 0.9966 290.33 0.9056 | Show Table DownLoad:
CSV
DownLoad:
CSV
由表1可见,黑暗/Fe(Ⅱ)/PS体系和太阳光/Fe(Ⅱ)/PS体系中罗红霉素的降解符合一级反应动力学和二级反应动力学;太阳光/Fe(Ⅱ)/柠檬酸体系和黑暗/Fe(Ⅱ)/柠檬酸/PS体系中,可能由于柠檬酸的存在,一级动力学和二级动力学的拟合效果均不佳。而太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的降解更符合一级动力学模型。在Fe浓度为0.25 mmol·L−1,PS浓度为4 mmol·L−1,Fe:柠檬酸的物质的量比为1∶1.5和1∶2时,罗红霉素降解的一级反应动力学常数分别为0.0103 min−1和0.0409 min−1(表1)。这说明螯合剂柠檬酸的添加实现了Fe(Ⅱ)的缓慢释放,使Fe(Ⅱ)能够平稳的活化PS,保持反应体系的持续氧化效果。
2.2 太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响因素
2.2.1 Fe(Ⅱ)的浓度
图2为Fe(Ⅱ)的浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响。在PS为4 mmol·L−1,Fe/柠檬酸的物质的量比为1:1时,反应体系对罗红霉素的去除速率随Fe(Ⅱ)浓度增加而增加。在Fe(Ⅱ)浓度为0.1、0.25 mmol·L−1时,反应60 min后,罗红霉素的去除率分别为12.0%和21.7%;当Fe(Ⅱ)升高至0.5 mmol·L−1后,罗红霉素的去除率显著增加,反应20 min后,罗红霉素的去除率可达到90.7%,但是当反应时间继续延长时,罗红霉素的去除率增加缓慢。这可能是由于后期反应体系中Fe(Ⅱ)浓度降低,产生的活性物种浓度降低以及罗红霉素降解产物对活性自由基的竞争造成的。而且,在Fe(Ⅱ)浓度为0.5 mmol·L−1时,反应20 min后,反应体系的pH值降至2.9。在此pH条件下,罗红霉素以质子化的形式存在,其与·OH等自由基的反应速率也较慢[25]。
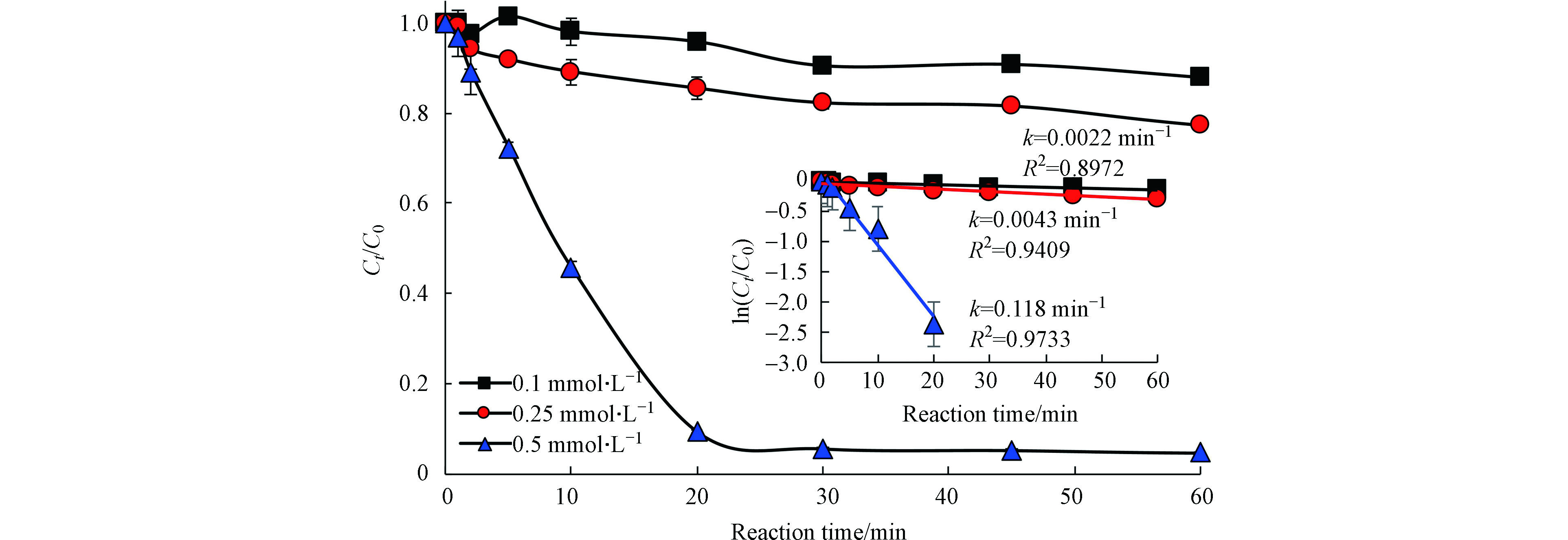 图 2 Fe(Ⅱ)浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响[PS]0=4 mmol·L−1,[Fe(Ⅱ)]/[柠檬酸]=1∶1Figure 2. Effect of Fe(Ⅱ) concentration on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system [PS]0=4 mmol·L−1, [Fe(Ⅱ)]/[citrate]=1∶1
图 2 Fe(Ⅱ)浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响[PS]0=4 mmol·L−1,[Fe(Ⅱ)]/[柠檬酸]=1∶1Figure 2. Effect of Fe(Ⅱ) concentration on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system [PS]0=4 mmol·L−1, [Fe(Ⅱ)]/[citrate]=1∶1不同Fe(Ⅱ)浓度体系中罗红霉素的快速降解阶段符合一级反应动力学。当Fe(Ⅱ)浓度为0.1、0.25、0.5 mmol·L−1时,罗红霉素的一级反应动力学常数分别为0.0022、0.0043、0.118 min−1(前20 min)。这说明较高浓度的Fe(Ⅱ)可以激活PS产生更多的·OH和
SO⋅−4 2.2.2 PS的浓度
图3为PS浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响。在Fe(Ⅱ)浓度为0.25 mmol·L−1时,太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的去除率和反应速率常数随PS增加而略有增加,这可能是因为反应体系中Fe(Ⅱ)浓度较低,不足以活化过量的PS,因此,Fe(Ⅱ)浓度低时,PS并不是限制该反应体系对罗红霉素降解效能的主要因素。当Fe(Ⅱ)浓度为0.5 mmol·L−1时,太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的去除率和反应速率常数随PS增加而显著增加,在PS浓度为4 mmol·L−1时,罗红霉素的去除率即可达到95%,罗红霉素的降解的一级反应速率常数(0.1034 min−1)可达到PS浓度为0.5 mmol·L−1条件下的8.4倍。这说明在保证Fe(Ⅱ)浓度充足的情况下,高浓度的PS生成了更多的活性自由基,促进了罗红霉素的降解[10]。
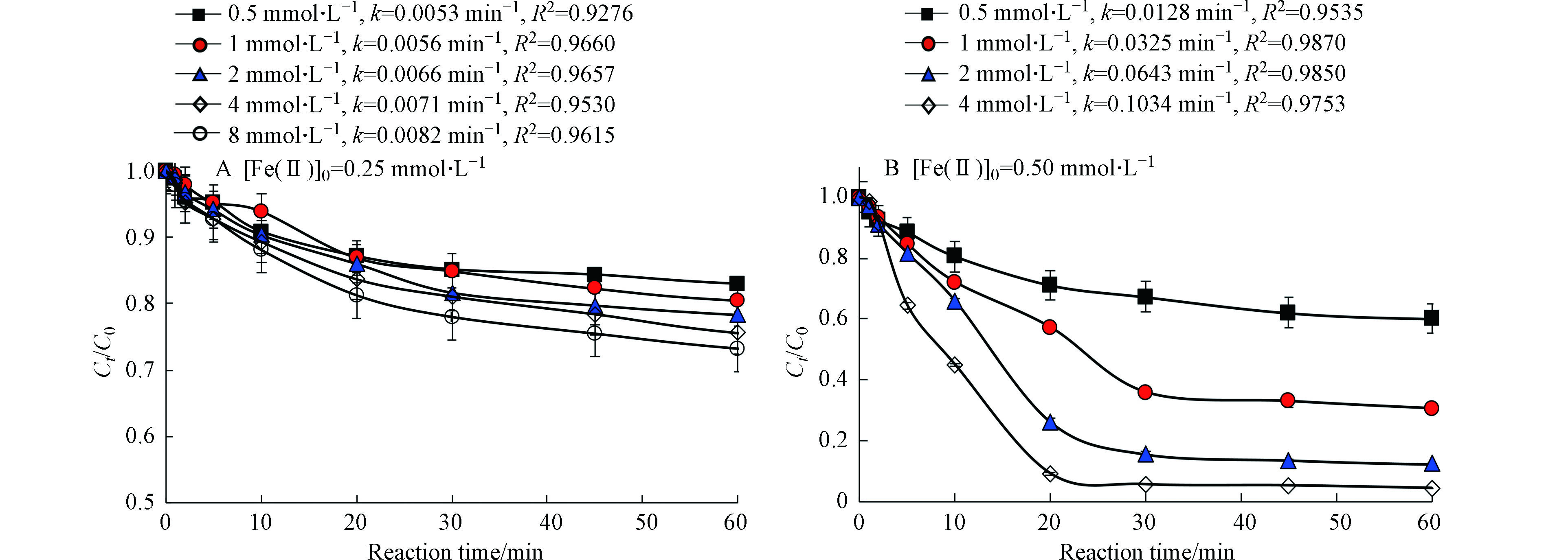 图 3 PS浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响Fe(Ⅱ)]/[柠檬酸]=1∶1Figure 3. Effect of PS concentration on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system[Fe(Ⅱ)]/[citrate]=1∶1
图 3 PS浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响Fe(Ⅱ)]/[柠檬酸]=1∶1Figure 3. Effect of PS concentration on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system[Fe(Ⅱ)]/[citrate]=1∶12.2.3 Fe(Ⅱ)与柠檬酸的比例
图4为Fe(Ⅱ)与柠檬酸的摩尔比对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响。在Fe(Ⅱ)浓度为0.25 mmol·L−1时,太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的去除率和反应速率常数随Fe(Ⅱ)/柠檬酸的物质的量比增加而增大。当Fe(Ⅱ)/柠檬酸的物质的量比从2∶1增加到1∶4时,罗红霉素的去除率从19.6%增加到94.3%,罗红霉素降解的一级反应速率常数从0.0045 min−1增加至0.0778 min−1(图4(A))。值得注意的是,尽管Fe(Ⅱ)/柠檬酸的物质的量比为1∶4的反应速率比Fe(Ⅱ)/柠檬酸的物质的量比为1∶2的大,但反应60 min后,两者对罗红霉素的去除率非常接近。
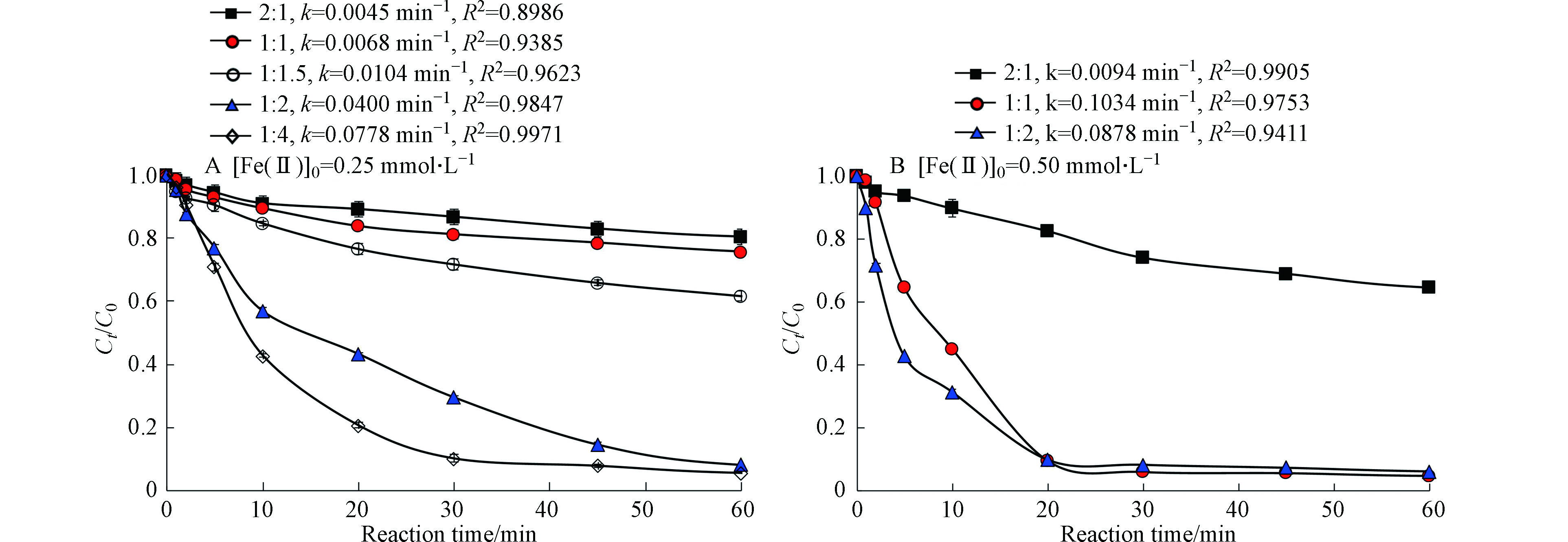 图 4 Fe/柠檬酸物质的量比对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响Figure 4. Effect of molar ratio of Fe/citrate on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system[PS]0=4 mmol·L−1,k为30 min时,罗红霉素降解的一级反应速率常数[PS]0=4 mmol·L−1, k represents the degradation rate constant of roxithromycin in 30 min
图 4 Fe/柠檬酸物质的量比对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响Figure 4. Effect of molar ratio of Fe/citrate on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system[PS]0=4 mmol·L−1,k为30 min时,罗红霉素降解的一级反应速率常数[PS]0=4 mmol·L−1, k represents the degradation rate constant of roxithromycin in 30 min在Fe(Ⅱ)浓度为0.5 mmol·L−1时,当Fe(Ⅱ)/柠檬酸的物质的量比从2:1增加到1∶1时,罗红霉素的去除率从35.5%增加到了95.4%,一级反应速率常数从0.0094 min−1显著增加到了0.1034 min−1,这说明柠檬酸的用量对太阳光/Fe(Ⅱ)/柠檬酸/PS体系的降解效果影响很大。但是当Fe(Ⅱ)/柠檬酸的摩尔比继续增加到1∶2时,罗红霉素的去除率和一级反应速率常数均降低。Tan等[27]研究发现Fe(Ⅱ)/柠檬酸的物质的量比从1∶1增加到1∶5时,Fe(Ⅱ)/柠檬酸/PS对敌草隆的去除率从80%降低到了57%。过量柠檬酸对反应体系中污染物降解的抑制作用可能是由两方面的原因造成的,一是柠檬酸与污染物竞争消耗反应体系中的活性自由基;二是柠檬酸与Fe(Ⅱ)的过分螯合阻碍了Fe与PS的反应,导致反应体系中产生的活性物种量减少[28]。
综上,结合试剂用量和对罗红霉素的去除效果,可确定太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素降解的反应条件为Fe(Ⅱ)浓度为0.25 mmol·L−1,Fe(Ⅱ)/柠檬酸的物质的量比为1:2,PS为4 mmol·L−1,反应时间60 min,在此条件下,罗红霉素的去除率可达到92%以上。
2.3 不同活性物种的贡献
在Fe(Ⅱ)浓度为0.25 mmol·L−1,PS为4 mmol·L−1,Fe/柠檬酸的物质的量比为1∶1.5和1∶2的太阳光/Fe(Ⅱ)/柠檬酸/PS体系中分别加入乙醇和叔丁醇,以考察反应体系中的活性物种及其贡献(表2)。乙醇可同时淬灭·OH和
SO⋅−4 SO⋅−4 表 2 太阳光/Fe(Ⅱ)/柠檬酸/PS体系中的活性物种贡献Table 2. The contribution of reactive species in sunlight/Fe(Ⅱ)/citrate/PS systemFe/柠檬酸的物质的量比Fe(Ⅱ)/citrate kobs/ min−1 kEtOH/min−1 kTBA/min−1 ·OH的贡献/%The contribution of ·OH SO4·-贡献/%The contribution of SO4·- 其他贡献/%The contribution of others 1:1.5 0.0120 0.0011 0.0026 78.3 12.5 9.2 1:2 0.0397 0.0025 0.0054 86.4 7.3 6.3 注 [Fe(Ⅱ)]0=0.25 mmol·L−1,[PS]0=4 mmol·L−1. | Show TableDownLoad:
CSV
C⋅OH=kobs−kTBAkobs×100% (13) CSO⋅−4=kobs−kEtOHkobs×100%−C⋅OH (14) 式中,C·OH和
CSO4⋅− SO⋅−4 S2O82−+HO2−⟶SO2−4+SO⋅−4+O2⋅−+H+ (15) ⋅OH+⋅OH⟶H2O2 (16) Fe(III)+H2O2⟶Fe(II)+HO2⋅+H2O (17) HO2⋅⟶H++O2⋅− (18) 为了更直观地验证反应体系中活性物种的存在,分别在反应0、15、30、45 min的时候,添加DMPO作为·OH和
SO⋅−4 SO⋅−4 SO⋅−4 图5也表明了不同反应时间段内反应体系中·OH的相对含量。在Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5的体系中,在不同时间段内,·OH的信号强度基本一致,说明Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5时,反应体系的·OH浓度保持稳定。而在Fe(Ⅱ)/柠檬酸的物质的量为1∶2的反应体系中,·OH的信号强度在30—60 min要比0—30 min时弱,这可能是因为柠檬酸含量高时,Fe(Ⅱ)/柠檬酸络合物与PS的反应速率较快,而Fe(Ⅲ)/柠檬酸络合物光解还原为Fe(Ⅱ)的速率相对较慢,导致后期反应体系中Fe(Ⅱ)浓度降低,降低了·OH的产率。这与反应体系中罗红霉素的降解情况是一致的。
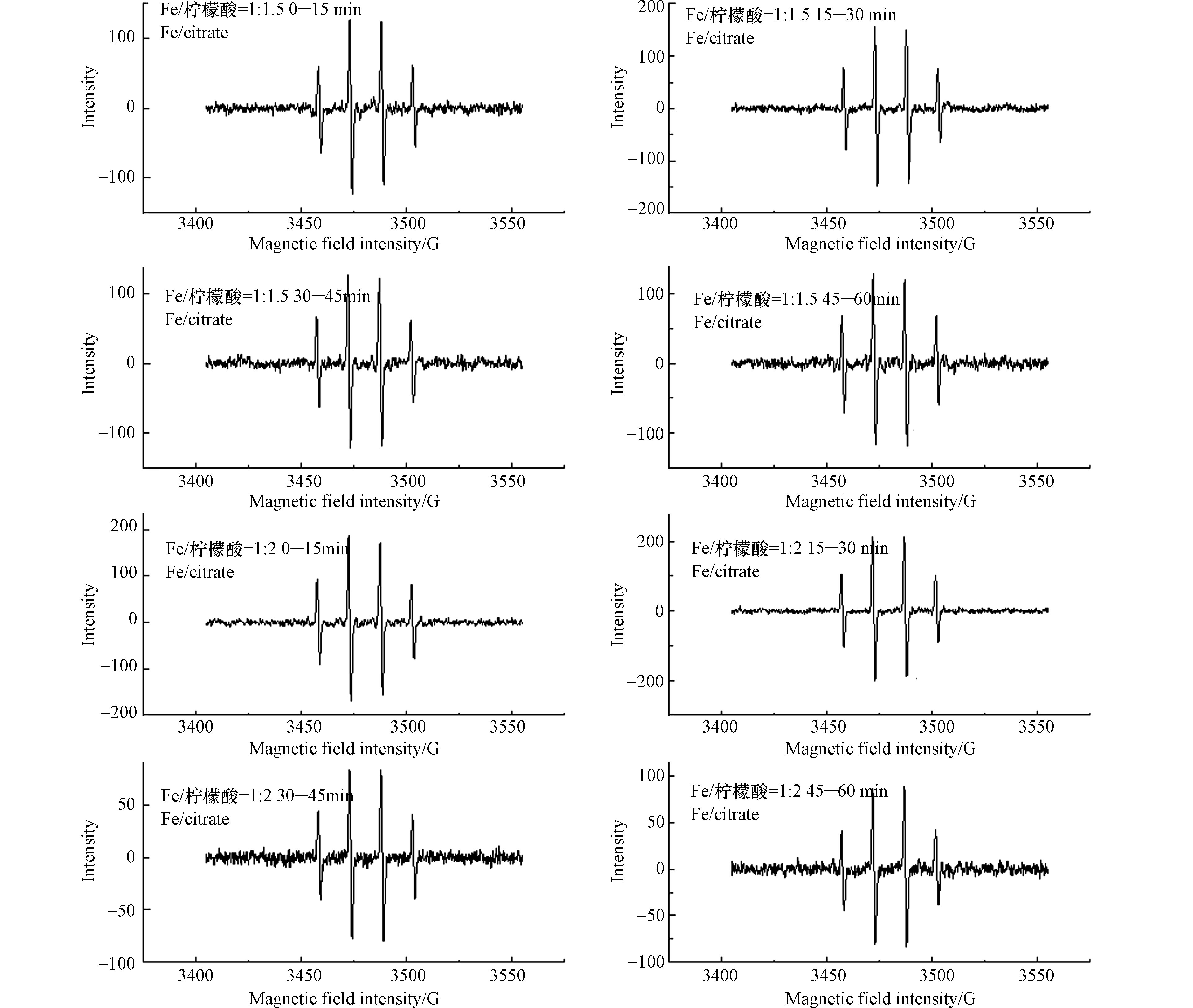 图 5 太阳光/Fe(Ⅱ)/柠檬酸/PS体系的电子自旋共振波谱Figure 5. ESR spectra of the slight/Fe(Ⅱ)/citrate/PS system
图 5 太阳光/Fe(Ⅱ)/柠檬酸/PS体系的电子自旋共振波谱Figure 5. ESR spectra of the slight/Fe(Ⅱ)/citrate/PS system2.4 罗红霉素的降解产物
在Fe(Ⅱ)浓度为0.25 mmol·L−1,PS为4 mmol·L−1,Fe(Ⅱ)/柠檬酸的物质的量比为1∶1.5和1∶2的条件下,研究了太阳光/Fe(Ⅱ)/柠檬酸/PS体系中罗红霉素的降解产物。根据罗红霉素降解前后的总离子流图、降解产物的精确分子量、二级碎裂质谱图以及Xcalibur的分子式计算功能,共识别出8种罗红霉素的降解产物。罗红霉素及其降解产物(以DP+m/z命名)的精确分子量和元素组成等信息见表3,分子结构见图6。
表 3 罗红霉素及其降解产物的精确分子量、元素组成及碎片离子Table 3. Accurate mass, elemental composition and fragmentation ion of roxithromycin and its degradation products化合物Compounds 保留时间Retention time 元素组成Element composition 精确分子量Accurate molecular weight/[m+H]+ 碎片离子fragmentations 误差Error(×10−6) 环和双键值 Ring double bond equivalent(RDB) 罗红霉素Roxithromycin 14.94 C41H77O15N2 837.5315 158.1173, 679.4366 −0.425 4.5 DP591 5.00 C29H55O10N2 591.3834 522.3422, 158.1174 −2.878 3.5 DP665 6.22 C32H61O12N2 665.4210 158.1172 −1.415 3.5 DP558 7.38 C29H52O9N 558.3624 158.1172 −2.308 4.5 DP679 9.49 C33H63O12N2 679.4371 158.1172 −0.709 3.5 DP749 10.21 C37H69O13N2 749.4780 591.3837, 158.1171 −1.850 4.5 DP540 11.14 C29H50O8N 540.3521 158.1171 −1.821 5.5 DP716 12.18 C37H66O12N 716.4565 158.1171 −1.972 5.5 DP714 16.23 C37H64O12N 714.4417 556.3465, 158.1171 −0.914 6.5 | Show TableDownLoad:
CSV
在8种降解产物中,DP749在UV/H2O2降解罗红霉素的体系中报道过[14],DP679、DP665和DP591在罗红霉素的光降解过程中报道过[15]。而DP716、DP714、DP558和DP540尚未见报道。这4种降解产物的二级质谱中都有碎片离子m/z158.1172,说明红霉脱氧糖胺保持完整。DP716的分子量比DP749的少33 Da,说明DP716是由DP749上脱去羟胺(NH2OH)形成的。相似的,DP558是由DP591上脱去羟胺形成的。DP714和DP540分别是DP716脱去2H和DP558脱去H2O形成的。此外,和文献中报道的罗红霉素的降解产物相比[14-15, 31-32],太阳光/Fe(Ⅱ)/柠檬酸/PS体系中产生的降解产物的分子量普遍要小,这说明太阳光/Fe(Ⅱ)/柠檬酸/PS体系对罗红霉素的氧化降解性能可能更强。
图7为罗红霉素的降解产物的量(以降解产物的峰面积与罗红霉素的初始峰面积比值A/A0表示)随反应时间的变化情况。
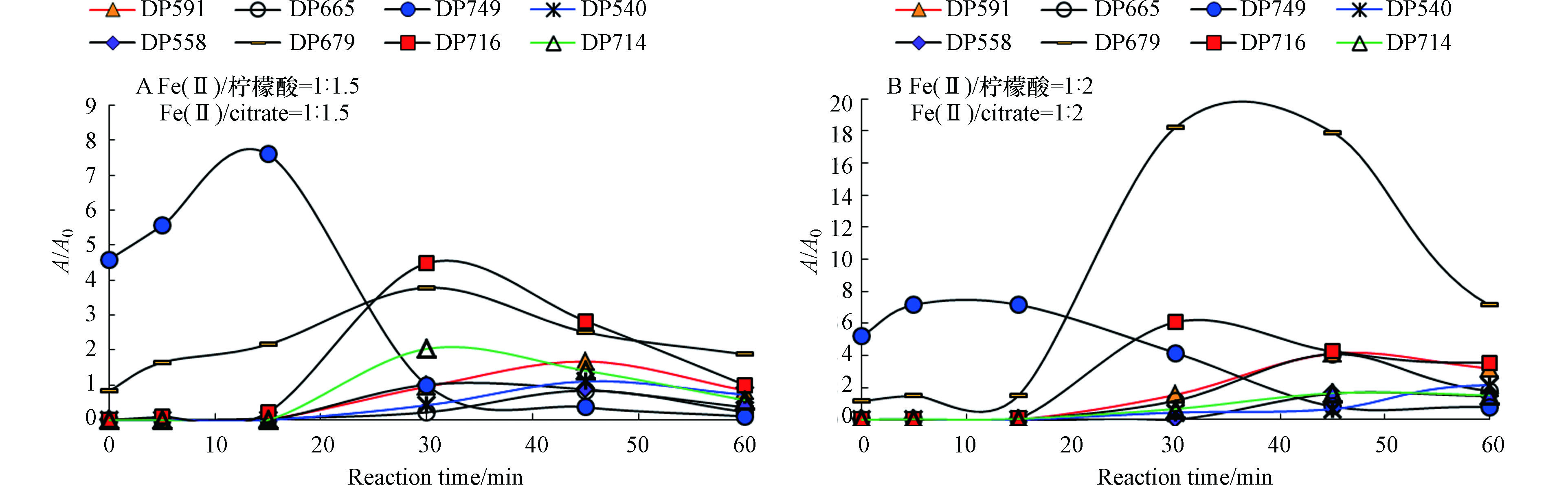 图 7 罗红霉素的降解产物随反应时间的变化[Fe(Ⅱ)]0=0.25 mmol·L−1,[PS]0=4 mmol·L−1Figure 7. The evolution of degradation products of roxithromycin with varying reaction time
图 7 罗红霉素的降解产物随反应时间的变化[Fe(Ⅱ)]0=0.25 mmol·L−1,[PS]0=4 mmol·L−1Figure 7. The evolution of degradation products of roxithromycin with varying reaction time由图7可见,反应15 min后,DP749即可达到最大的A/A0值,这说明罗红霉素首先生成DP749。在Fe(Ⅱ)/柠檬酸物质的量比为1∶1.5时,DP679、DP716、DP558和DP714在反应30 min时达到最大值,DP591、DP665和DP540在反应45 min时达到最大值,而在Fe(Ⅱ)/柠檬酸为1∶2时,DP679和DP716在反应30 min后达到最大,其它5种降解产物均在反应45 min时达到最大值。除DP714外,Fe(Ⅱ)/柠檬酸物质的量比为1∶2体系中的降解产物的A/A0值均高于Fe(Ⅱ)/柠檬酸为1∶1.5条件下。这进一步说明了Fe(Ⅱ)/柠檬酸为1∶2的反应体系中产生了更多的·OH,对罗红霉素的降解程度要高于Fe(Ⅱ)/柠檬酸物质的量比为1∶1.5体系.
3. 结论(Conclusions)
(1)太阳光/Fe(Ⅱ)/柠檬酸/PS高级氧化体系能有效去除污水处理厂二级出水中的罗红霉素,在Fe(Ⅱ)浓度为0.25 mmol·L−1,Fe(Ⅱ)/柠檬酸物质的量比为1∶2,PS为4 mmol·L−1,反应时间60 min时,罗红霉素的去除率可达到92%以上。
(2)太阳光/Fe(Ⅱ)/柠檬酸/PS高级氧化技术可同时产生
SO⋅−4 SO⋅−4 (3)基于高效液相色谱-高分辨质谱鉴定出8种罗红霉素的降解产物,这些降解产物主要通过脱去克拉定糖(—C8H14O3)、肟侧链的断裂(—C4H8O2)、脱羟胺(—NH2OH)等过程产生,其中,罗红霉素通过肟侧链断裂和脱去克拉定糖生成的DP749和DP679是最主要的降解产物。
-
图 1 不同处理系统中罗红霉素的去除率[Fe(Ⅱ)]0=0.25 mmol·L−1,[PS]0=4 mmol·L−1
Figure 1. Removal efficiency of roxithromycin in different treatment system
图 2 Fe(Ⅱ)浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响[PS]0=4 mmol·L−1,[Fe(Ⅱ)]/[柠檬酸]=1∶1
Figure 2. Effect of Fe(Ⅱ) concentration on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system [PS]0=4 mmol·L−1, [Fe(Ⅱ)]/[citrate]=1∶1
图 3 PS浓度对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响Fe(Ⅱ)]/[柠檬酸]=1∶1
Figure 3. Effect of PS concentration on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system[Fe(Ⅱ)]/[citrate]=1∶1
图 4 Fe/柠檬酸物质的量比对太阳光/Fe(Ⅱ)/柠檬酸/PS体系降解罗红霉素的影响
Figure 4. Effect of molar ratio of Fe/citrate on the degradation of roxithromycin by slight/Fe(Ⅱ)/citrate/PS system
图 5 太阳光/Fe(Ⅱ)/柠檬酸/PS体系的电子自旋共振波谱
Figure 5. ESR spectra of the slight/Fe(Ⅱ)/citrate/PS system
图 7 罗红霉素的降解产物随反应时间的变化[Fe(Ⅱ)]0=0.25 mmol·L−1,[PS]0=4 mmol·L−1
Figure 7. The evolution of degradation products of roxithromycin with varying reaction time
表 1 不同反应体系中,罗红霉素的降解反应动力学拟合常数
Table 1. The degradation kinetics fitting constants of roxithromycin in different reaction systems
反应体系(物质的量比)Reaction system 一级动力学Pseudo-first order kinetic 二级动力学Second order kinetics k1/min−1 R2 k2/(L·mol−1·s−1) R2 黑暗/Fe(Ⅱ)/PS 0.001 0.9564 1.6143 0.9593 太阳光/Fe(Ⅱ)/PS 0.0016 0.9886 2.8060 0.9887 太阳光/Fe(Ⅱ)/柠檬酸(1∶1.5)Sunlight/Fe(Ⅱ)/citrate(1∶1.5) 0.0024 0.8957 4.0228 0.9042 太阳光/Fe(Ⅱ)/柠檬酸1∶2)Sunlight/Fe(Ⅱ)/citrate(1∶2) 0.0045 0.9082 8.5515 0.9247 黑暗/Fe(Ⅱ)/柠檬酸/PS(1∶1.5)Dark/Fe(Ⅱ)/citrate/PS(1∶1.5) 0.0013 0.7795 2.5793 0.7887 黑暗/Fe(Ⅱ)/柠檬酸/PS(1∶2)Dark/Fe(Ⅱ)/citrate/PS(1∶2) 0.0018 0.8343 3.1015 0.8428 太阳光/Fe(Ⅱ)/柠檬酸/PS(1∶1.5)Sunlight/Fe(Ⅱ)/citrate/PS(1∶1.5) 0.0103 0.9848 22.866 0.9962 太阳光/Fe(Ⅱ)/柠檬酸/PS(1∶2)Sunlight/Fe(Ⅱ)/citrate/PS(1∶2) 0.0409 0.9966 290.33 0.9056
下载: 导出CSV
表 2 太阳光/Fe(Ⅱ)/柠檬酸/PS体系中的活性物种贡献
Table 2. The contribution of reactive species in sunlight/Fe(Ⅱ)/citrate/PS system
Fe/柠檬酸的物质的量比Fe(Ⅱ)/citrate kobs/ min−1 kEtOH/min−1 kTBA/min−1 ·OH的贡献/%The contribution of ·OH SO4·-贡献/%The contribution of SO4·- 其他贡献/%The contribution of others 1:1.5 0.0120 0.0011 0.0026 78.3 12.5 9.2 1:2 0.0397 0.0025 0.0054 86.4 7.3 6.3 注 [Fe(Ⅱ)]0=0.25 mmol·L−1,[PS]0=4 mmol·L−1.
下载: 导出CSV
表 3 罗红霉素及其降解产物的精确分子量、元素组成及碎片离子
Table 3. Accurate mass, elemental composition and fragmentation ion of roxithromycin and its degradation products
化合物Compounds 保留时间Retention time 元素组成Element composition 精确分子量Accurate molecular weight/[m+H]+ 碎片离子fragmentations 误差Error(×10−6) 环和双键值 Ring double bond equivalent(RDB) 罗红霉素Roxithromycin 14.94 C41H77O15N2 837.5315 158.1173, 679.4366 −0.425 4.5 DP591 5.00 C29H55O10N2 591.3834 522.3422, 158.1174 −2.878 3.5 DP665 6.22 C32H61O12N2 665.4210 158.1172 −1.415 3.5 DP558 7.38 C29H52O9N 558.3624 158.1172 −2.308 4.5 DP679 9.49 C33H63O12N2 679.4371 158.1172 −0.709 3.5 DP749 10.21 C37H69O13N2 749.4780 591.3837, 158.1171 −1.850 4.5 DP540 11.14 C29H50O8N 540.3521 158.1171 −1.821 5.5 DP716 12.18 C37H66O12N 716.4565 158.1171 −1.972 5.5 DP714 16.23 C37H64O12N 714.4417 556.3465, 158.1171 −0.914 6.5
下载: 导出CSV
-
[1] KÜMMERER K. Antibiotics in the aquatic environment - A review - Part I [J]. Chemosphere, 2009, 75(4): 417-434. doi: 10.1016/j.chemosphere.2008.11.086 [2] ZHANG Q Q, YING G G, PAN C G, et al. Comprehensive evaluation of antibiotics emission and fate in the river basins of China: Source analysis, multimedia modeling, and linkage to bacterial resistance [J]. Environmental Science & Technology, 2015, 49(11): 6772-6782. [3] 张国栋, 董文平, 刘晓晖, 等. 我国水环境中抗生素赋存、归趋及风险评估研究进展 [J]. 环境化学, 2018, 37(7): 1491-1500. doi: 10.7524/j.issn.0254-6108.2017112003 ZHANG G D, DONG W P, LIU X H, et al. Occurrence, fate and risk assessment of antibiotics in water environment of China [J]. Environmental Chemistry, 2018, 37(7): 1491-1500(in Chinese). doi: 10.7524/j.issn.0254-6108.2017112003
[4] KOVALAKOVA P, CIZMAS L, MCDONALD T J, et al. Occurrence and toxicity of antibiotics in the aquatic environment: A review [J]. Chemosphere, 2020, 251: 126351. doi: 10.1016/j.chemosphere.2020.126351 [5] EPOLD I, DULOVA N. Oxidative degradation of levofloxacin in aqueous solution by S2O82-/Fe2+, S2O82-/H2O2 and S2O82-/OH- processes: A comparative study [J]. Journal of Environmental Chemical Engineering, 2015, 3(2): 1207-1214. doi: 10.1016/j.jece.2015.04.019 [6] 张凌星, 肖鹏飞. 活化过硫酸盐氧化处理抗生素废水的研究进展[J]. 工业水处理, 2021, 40(5): 29-35. ZHANG L X, XIAO P F. Research progress on treatment of antibiotic wastewater by activated persulfate oxidation[J]. Industrial Water Treatment, 2021, 40(5): 29-35 (in Chinese).
[7] JI Y F, FERRONATO C, SALVADOR A, et al. Degradation of ciprofloxacin and sulfamethoxazole by ferrous-activated persulfate: Implications for remediation of groundwater contaminated by antibiotics [J]. Science of the Total Environment, 2014, 472: 800-808. doi: 10.1016/j.scitotenv.2013.11.008 [8] NIE M H, YAN C X, XIONG X Y, et al. Degradation of chloramphenicol using a combination system of simulated solar light, Fe2+ and persulfate [J]. Chemical Engineering Journal, 2018, 348: 455-463. doi: 10.1016/j.cej.2018.04.124 [9] MAJUMDER A, GUPTA B, GUPTA A K. Pharmaceutically active compounds in aqueous environment: A status, toxicity and insights of remediation [J]. Environmental Research, 2019, 176: 108542. doi: 10.1016/j.envres.2019.108542 [10] WANG S L, WU J F, LU X Q, et al. Removal of acetaminophen in the Fe2+/persulfate system: Kinetic model and degradation pathways [J]. Chemical Engineering Journal, 2019, 358: 1091-1100. doi: 10.1016/j.cej.2018.09.145 [11] HAN D H, WAN J Q, MA Y W, et al. New insights into the role of organic chelating agents in Fe(II) activated persulfate processes [J]. Chemical Engineering Journal, 2015, 269: 425-433. doi: 10.1016/j.cej.2015.01.106 [12] 尹汉雄, 唐玉朝, 黄显怀, 等. 紫外光强化Fe(Ⅱ)-EDTA活化过硫酸盐降解直接耐酸大红4BS [J]. 环境科学研究, 2017, 30(7): 1105-1111. YIN H X, TANG Y C, HUANG X H, et al. Decolorization effect of direct fast scarlet 4BS by Fe (Ⅱ)-EDTA activated peroxodisulfate under ultraviolet light [J]. Research of Environmental Sciences, 2017, 30(7): 1105-1111(in Chinese).
[13] 韩东晖, 李瑛, 李开明, 等. UV强化草酸络合Fe2+活化过硫酸盐氧化苯胺研究 [J]. 环境科学学报, 2018, 38(7): 2659-2666. HAN D H, LI Y, LI K M, et al. Enhanced degradation of aniline by PS oxidation in the presence of UV and ferrous oxalate [J]. Acta Scientiae Circumstantiae, 2018, 38(7): 2659-2666(in Chinese).
[14] LI W, XU X J, LYU B L, et al. Degradation of typical macrolide antibiotic roxithromycin by hydroxyl radical: Kinetics, products, and toxicity assessment [J]. Environmental Science and Pollution Research, 2019, 26(14): 14570-14582. doi: 10.1007/s11356-019-04713-1 [15] LI W, LYU B L, LI J P, et al. Phototransformation of roxithromycin in the presence of dissolved organic matter: Characteriazation of the degradation products and toxicity evaluation [J]. Science of the Total Environment, 2020, 733: 139348. doi: 10.1016/j.scitotenv.2020.139348 [16] XU X R, LI X Z. Degradation of azo dye Orange G in aqueous solutions by persulfate with ferrous ion [J]. Separation and Purification Technology, 2010, 72(1): 105-111. doi: 10.1016/j.seppur.2010.01.012 [17] CHEN Y, LIU Z Z, WANG Z P, et al. Photodegradation of propranolol by Fe(III)-citrate complexes: Kinetics, mechanism and effect of environmental media [J]. Journal of Hazardous Materials, 2011, 194: 202-208. doi: 10.1016/j.jhazmat.2011.07.081 [18] OUYANG Z Z, YANG C, HE J H, et al. Homogeneous photocatalytic degradation of sulfamethazine induced by Fe(III)-carboxylate complexes: Kinetics, mechanism and products [J]. Chemical Engineering Journal, 2020, 402: 126122. doi: 10.1016/j.cej.2020.126122 [19] OU X X, QUAN X, CHEN S, et al. Photocatalytic reaction by Fe(III)-citrate complex and its effect on the photodegradation of atrazine in aqueous solution [J]. Journal of Photochemistry and Photobiology A:Chemistry, 2008, 197(2/3): 382-388. [20] AHILE U J, WUANA R A, ITODO A U, et al. A review on the use of chelating agents as an alternative to promote photo-Fenton at neutral pH: Current trends, knowledge gap and future studies [J]. Science of the Total Environment, 2020, 710: 134872. doi: 10.1016/j.scitotenv.2019.134872 [21] NIE M H, YAN C X, LI M, et al. Degradation of chloramphenicol by persulfate activated by Fe2+ and zerovalent iron [J]. Chemical Engineering Journal, 2015, 279: 507-515. doi: 10.1016/j.cej.2015.05.055 [22] LUO T, WAN J, MA Y, et al. Sulfamethoxazole degradation by an Fe (Ⅱ)-activated persulfate process: insight into the reactive sites, product identification and degradation pathways [J]. Environmental Science: Processes & Impacts, 2019, 21(9): 1560-1569. [23] WU X L, GU X G, LU S G, et al. Degradation of trichloroethylene in aqueous solution by persulfate activated with citric acid chelated ferrous ion [J]. Chemical Engineering Journal, 2014, 255: 585-592. doi: 10.1016/j.cej.2014.06.085 [24] NIE M H, YANG Y, ZHANG Z J, et al. Degradation of chloramphenicol by thermally activated persulfate in aqueous solution [J]. Chemical Engineering Journal, 2014, 246: 373-382. doi: 10.1016/j.cej.2014.02.047 [25] LIU P X, ZHANG H M, FENG Y J, et al. Removal of trace antibiotics from wastewater: A systematic study of nanofiltration combined with ozone-based advanced oxidation processes [J]. Chemical Engineering Journal, 2014, 240: 211-220. doi: 10.1016/j.cej.2013.11.057 [26] RAO Y, QU L, YANG H, et al. Degradation of carbamazepine by Fe (Ⅱ)-activated persulfate process [J]. Journal of Hazardous Materials, 2014, 268: 23-32. doi: 10.1016/j.jhazmat.2014.01.010 [27] TAN C Q, GAO N Y, CHU W H, et al. Degradation of diuron by persulfate activated with ferrous ion [J]. Separation and Purification Technology, 2012, 95: 44-48. doi: 10.1016/j.seppur.2012.04.012 [28] HOU K J, PI Z J, YAO F B, et al. A critical review on the mechanisms of persulfate activation by iron-based materials: Clarifying some ambiguity and controversies [J]. Chemical Engineering Journal, 2021, 407: 127078. doi: 10.1016/j.cej.2020.127078 [29] LIANG C J, SU H W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate [J]. Industrial & Engineering Chemistry Research, 2009, 48(11): 5558-5562. [30] ZHANG Y, ZHOU M H. A critical review of the application of chelating agents to enable Fenton and Fenton-like reactions at high pH values [J]. Journal of Hazardous Materials, 2019, 362: 436-450. doi: 10.1016/j.jhazmat.2018.09.035 [31] RADJENOVIĆ J, GODEHARDT M, et al. Evidencing generation of persistent ozonation products of antibiotics roxithromycin and trimethoprim [J]. Environmental Science & Technology, 2009, 43(17): 6808-6815. [32] KWIECIEŃ A, KRZEK J, ŻMUDZKI P, et al. Roxithromycin degradation by acidic hydrolysis and photocatalysis [J]. Analytical Methods, 2014, 6(16): 6414-6423. doi: 10.1039/C4AY00708E -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3587
- HTML全文浏览数: 3587
- PDF下载数: 112
- 施引文献: 0



















