



-
工业废水的水处理问题是我国面临的主要环境问题之一。电催化氧化净水技术具有高效、适应性强、反应条件温和,在工业废水处理上具有极大的优势,电催化氧化过程通常分为直接氧化过程和间接氧化过程[1]。在阳极发生的直接氧化作用是指污染物颗粒吸附在阳极材料表面,通过阳极上电子的转移实现的污染物降解[2],但是通常对污染物的降解作用较小。间接氧化过程是通过阳极表面产生的活性中间物质(如·OH、OCl、O3)或具有高氧化性的高价态金属氧化物来氧化降解水中有机污染物,是污染物降解的最主要形式[3]。值得注意的是,工业废水中通常含有大量的氯离子,氯离子可以在(光)电化学催化氧化下形成强氧化性的氯自由基和活性氯物种(·Cl、·ClO−、Cl2)[4-5],这些含氯氧化剂可与羟基自由基(·OH)共同氧化降解很多有机污染物[6]。然而,对于含盐废水中有机污染物的降解,间接氧化产生的氯活性物种的利用率受限于传质效率,尤其是针对于大分子难降解有机物,较难实现污染物的完全矿化,因此提升氯活性物种的产生和利用效率则有望极大促进含盐废水中有机污染物的降解[7]。
在光电协同降解污染物的体系中,传质效率和提升光的利用率是提升含盐废水中有机污染物的降解效果的重要途径。已有大量研究表明,通过外力作用使含污染物的废水强制性通过多孔极板的穿透式电极构型可以数量级提升传质效率和电子转移效率,从而提升净水性能[8-9]。ISRAEL等[10]通过穿透式和传统的浸没式电极反应器对水中阿莫西林电化学氧化实验对比发现,穿透式构型可以带来更高的电化学氧化效率和更低的能耗,相比于浸没式电极,穿透式反应器运行下对阿莫西林的去除效率提升了70%,降解速率和传质速率分别提升了3.46和10.74倍,强氧化性的中间活性物质·OH的生成量高了5.64倍,每单元能耗降低了19.89倍。WANG等[11]通过TiO2/Ti多孔膜对甲基蓝(MB)降解中发现,在相同的水力停留时间下,穿透式构型中MB去除率可达99.5%,浸没式构型仅为21.0%,强制对流促进的高传质效率是提升污染物降解的关键因素。合理搭建穿透式电极反应器,减少传质距离,多极板串联,可以有效提升传质效率和净水性能。相比于单独的电化学催化,光的引入可以进一步提升氯活性物种的产生量,研究发现,光催化产氯的主要波段在254 nm[12]。然而常规的将光源引入会存在两个问题,一是光源的能量不足以高效将氯离子向氯活性物质激发转化,二是利用光催化处理废水时,光在水中穿透性有限,光能的有效利用率不足。基于此,我们考虑到在实验中引入具有瞬间高强度光能释放的脉冲灯管作为光源,脉冲光是将相同的能量以高峰值形式瞬间释放,在水中具有很好的穿透性,同时在反应过程中可以保持低温,无需像汞灯一样依赖一定的温度对汞进行汽化,两个脉冲之间也有足够的冷却时间,因而在实际应用过程中,比常规的紫外灯有更广的适用范围。现有的研究主要集中于电化学氧化作用提高污染物的降解效率,对于光电协同直接将废水中的氯离子利用并转化为高氧化性的活性氯物质的研究尚不充足,而提高传质并协同提升光利用效率是促进含盐废水中有机物被高效降解的关键。此外,穿透式电极常受限于电极材料的尺寸,以往研究中基本是采用较小体积的过滤器构型反应器,单次穿透时水停留时间很短[13]。因此构建处理水量达到数十升级的较大规模单个反应器,延长水力停留时间,使单次处理出水即可达标,实现连续运行具有更好的实际应用价值。
本研究选用了4-硝基酚(4-np)作为目标污染物,4-硝基酚作为重要的工业原料,广泛应用于医药、农药、染料和防腐等行业。在实际生产加工过程中,这些行业常常会有大量的4-硝基酚排放[14]。构建了具有大体积的光电协同的穿透式电极反应器,将3层多孔电极进行套管式排布,中心放置具有高强度的脉冲光源,通过脉冲光电协同作用,将废水中的氯离子转化为具有氧化活性的氯活性物质(reactive chlorine species,RCS),实现水中有机污染物的快速降解和高效矿化。实验比较了脉冲光和常规紫外光源在污染物降解上的差异性,并考察了流量、施加电压、含盐量、pH值等因素对污染物降解速率的影响,分析了污染物降解的主导因素,确定反应器长期稳定运行的最佳工作条件。本研究为脉冲光电协同反应器在实际工程中的应用奠定了基础并构建了一种高效的含盐废水中有机污染物去除的水处理技术。
-
实验中所用化学试剂4-硝基酚、氯化钠、硫酸钠、硫酸、氢氧化钠、硫代硫酸钠、磷酸、硝酸等均为国药沪试分析纯,DPD粉包为美国哈希(型号
2105569 )。 -
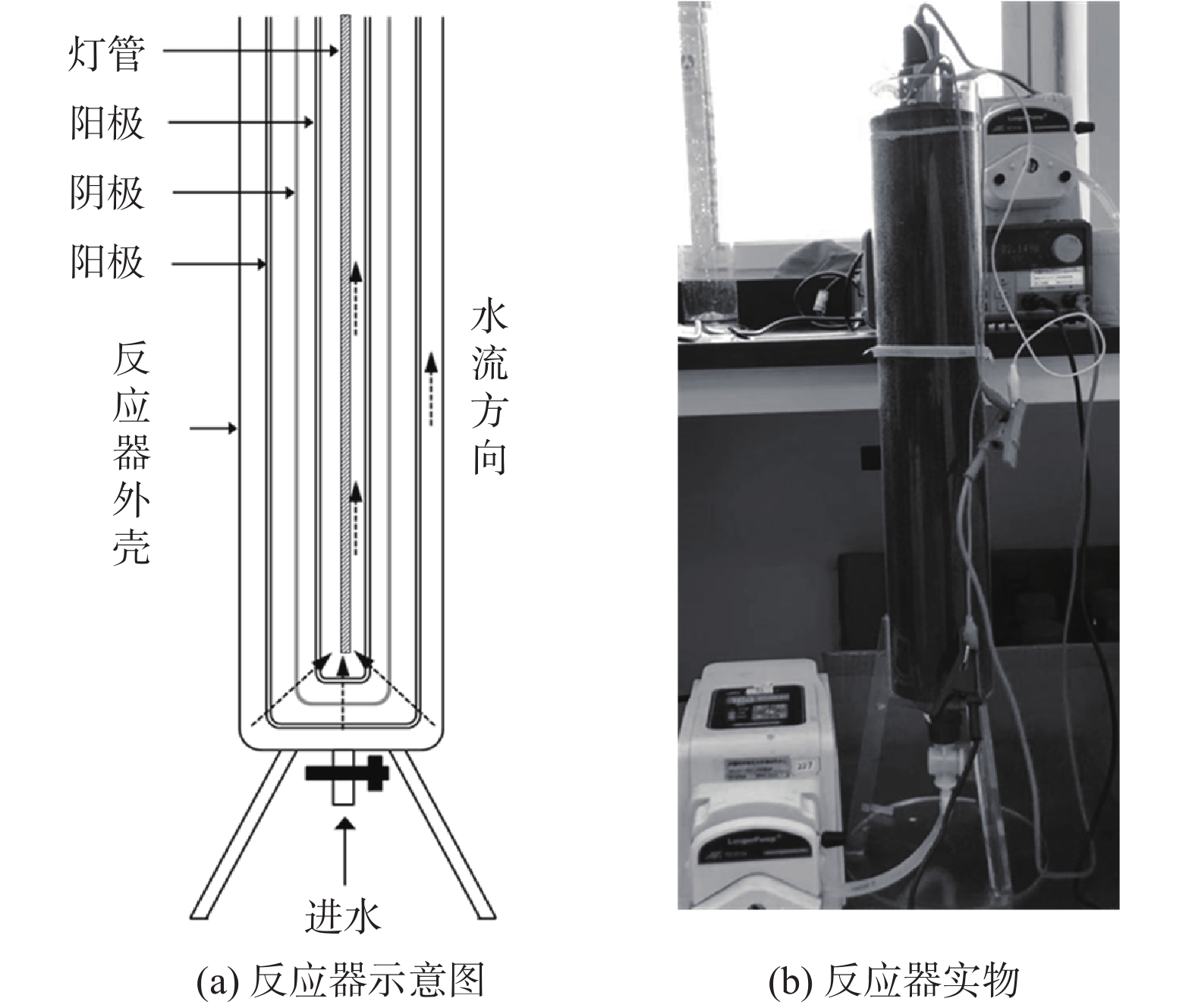
实验采用升流式光电协同穿透式电极反应器,反应器设计如图1(a)所示。反应器主体部分由3层套管电极构成,由外向内为阳极-阴极-阳极;阴极为泡沫钛,平均孔径为50 μm,厚度为2 mm;阳极选用的为相同材质的泡沫钛上喷涂金属钌。3个套管电极的尺寸分别为 Φ65 mm×500 mm、Φ50 mm×490 mm、Φ35 mm×480 mm,电极管上部边缘焊接1 cm×2 cm的钛片以连接电源,该孔径下的3个电极,水流均可无外加压力渗透透过。3层套管中间用硅胶垫片隔离开,以避免短路,极板间距为2 m。反应器外壳为聚四氟乙烯(PTFE)材质作为支撑,腔室下部中间为进水口;反应器上部中心开孔为抽水孔,以便于溢流抽水,反应器顶盖侧面开槽,以便于连接电源线。反应器中心放置灯管光源,光源分别选用定制的脉冲强光灯(IPL)和相等功率的紫外光源(入射波长λ=254 nm,15 W)。脉冲灯管外包裹保护层石英管,光谱透过率大于 90%以上,灯管有效寿命为 5 000 h,脉冲强光的光电能转换率为45%~50%,紫外线能量占约38%,脉冲灯管的能量10 J,功率为15 W。实验中,水流为升流式,水流从底部进水,穿透3层电极后,从反应器顶部抽出,反应器的有效容积为1.9 L。反应器的实物图如图1(b)所示。
-
实验选取4-硝基酚(4-np)为目标污染物进行光电协同降解实验。实验中用直流电源(AMERLLDS302A,大华,中国)进行供电,在未加光照的对照中,灯管仍然放置于电极套管内,以保证平行实验中反应器有效容积的一致性,降解实验分为循环流实验和单次穿透两种模式,实验中,在蠕动泵的作用下从底部进水时刻连接电源,并开启光源,当水流充满反应器,溢流出水时刻开始计时,此时开启蠕动泵在顶端进行溢流排水。循环流实验中间隔一定时长取5 mL水样进行后续分析测试;单次穿透实验中,待反应器稳定运行30 min后每隔10 min取5 mL出水进行分析。水样经过0.45 μm滤膜过滤后,转移到离心管中,加入硫代硫酸钠进行淬灭反应和保存,以测试4-硝基酚的浓度,并测试水样中的活性氯和总有机碳(TOC)。污染物降解实验中,初始4-硝基酚浓度设置为1 mmol·L−1(139.1 mg·L−1);不同流量的实验中,分别设置了200、100、80、50 mL·min−1,计算获得的水力停留时间如表1所示。
不同盐浓度实验中,氯化钠浓度设置为25、50、171、256、512 mmol·L−1;用0.1 mol·L−1硫酸和1 mol·L−1氢氧化钠调节溶液pH,pH值设置为2.0、4.0、6.0、8.00和10.00。不同电压对污染物去除的影响中,施加电压设置为0、1、2、3 V;实验中使用两种光源时均在上述各种条件下进行。污染物降解效果η根据式(1)进行计算。
式中:C0和C分别为4-硝基酚初始浓度(初始总有机碳值)(mg·L−1)和t时刻(min)溶液中的4-硝基酚浓度(t时刻总有机碳值)(mg·L−1)。
-
通过高效液相色谱(Agilent
1260 ,美国)测量样品中 4-硝基酚的浓度,Agilent C18柱(250 mm×4.6 mm, 5 μm)和紫外检测器,柱温35 ℃,进样量50 μL,流动相为10 mM 磷酸和超纯水(50∶50,v/v),流速1 mL·min−1,紫外检测器的检测波长为318 nm;通过N, N-二乙基对苯二胺(DPD)分光光度法(Hitachi,美国)测试样品中活性氯的浓度,每次测试时取1 mL水样,将样品稀释定容到10 mL,加入DPD粉包试剂,晃动20 s,显色3 min后进行读数(Hitachi UV-3100 ,美国);4-硝基酚的矿化程度通过总有机碳(TOC)(Shimadz TOC-L,日本)测定,通过电感协同等离子体发射光谱仪(ICP-OES,Agilent 710,美国)分析测定水中金属钛和钌的浓度,表征是否有钌从电极上剥落,以衡量电极的稳定使用情况实验使用蠕动泵将水从反应器底部泵入,上端溢流出水并收集,以确保液位恒定。测试样品预先用2%硝酸酸化,过0.45 μm滤膜后进行测试。矿化效率一般通过总有机碳(TOC)或化学需氧量(CODCr)衡量,本研究中以TOC为指标,衡量4-硝基酚的矿化率,见式(2)。式中:TOC0和TOCt分别代表初始时刻和t时刻的TOC(mg·L-1)。
本光电反应器的单位能耗的计算根据式(3)。
式中:Ec为单位能耗(kWh·m−3);U为反应时电压值(V);I为反应时电流值(A);t为反应时长(h);V为处理溶液体积(m3)。
-
为了考察相同能耗的光源对污染物的降解能力和本装置的可行性,实验通过循环流模式分别用紫外光(UV)和脉冲光作为光源,对低浓度的4-硝基酚(10 mg·L−1)进行了降解实验。实验采用恒电流模式,施加电流为0.1 A,电解质为50 mmol·L−1氯化钠,实验结果如图2所示。在仅有光照的条件下,污染物降解效率极有限,120 min内4-硝基酚的降解效率分别为15.9%(紫外光源)和14.9%(脉冲光源);电催化性能优于光催化性能,在仅有电的作用下,30 min内4-硝基酚去除效率可达40.0%,60 min时,去除效率达到了98.7%。在同时施加电压和光照的情况下,45 min后4-硝基酚降解效果分别达到了96.6%和91.8%,60 min后溶液中几乎检测不到4-硝基酚。因此,该反应器可用于运行降解4-硝基酚,光电协同效果远优于单独的光/电催化氧化过程。
-
相比于循环流模式,单次穿透更适合于工程化应用,满足实际处理废水的情况,实验在不同的光电条件下进行了单次穿透实验,并考察了流量对于4-硝基酚降解性能的影响。在穿透式电极反应中,流量会直接影响水流和电极的接触时间,不同的水力停留时间会影响传质过程,从而导致降解性能的不同。在反应器稳定运行的条件下,研究了不同流量下不同光电处理中出水4-硝基酚的降解性能,外加电压为3 V,结果如图3所示。实验发现,在仅加电、加电协同紫外光以及加电协同脉冲光3种处理下,随着流量的减少,停留时间的增加,单次穿透后的出水浓度均有明显的下降。更长的停留时间可以使得污染物与电极充分反应,在3种处理模式下,50 mL·min−1流量都足以使污染物完全被降解,在200 mL·min−1的处理中,由于流量较大,停留时间很短,使得出水浓度并不稳定。更大的流量被认为可以促进离子的对流传质和扩散传质,促进反应速率[15],但是更长的停留时间更利于污染物的降解。
实验横向对比了相同流量下,不同光电处理的降解效果,如图4所示。在低流量50 mL·min−1的处理中,3种光电协同处理下的4-硝基酚降解没有明显的区别,出水的降解效果均在99%以上,说明在足够长的停留时间中,电催化直接氧化占了主导地位,可以直接将污染物降解。而在较高流量的处理中(80 mL·min−1 和100 mL·min−1),以脉冲灯为光源的光电协同处理下,污染物降解效率仍能保持在90%以上,而紫外灯为光源的光电协同单元,降解效率下降到80%~85%,这表明在相同的能耗下,强脉冲灯可以带来更好的光电协同效率,这很可能是因为脉冲灯的高光强和脉冲属性,让光在溶液中有更好的穿透性,使得溶液中更多的HOCl和OCl−接触到了光,被激发活化形成了强氧化性的氯自由基,从而促进了污染物的降解。在高流量200 mL·min−1时,3者处理下的差异并不是很大,降解效率下降到75%~80%左右,且出水状态并不稳定。实验表明,HRT在19~24 min(80~100 mL·min−1 )时达到最优的出水效果。同时,出水通过ICP-OES检测在反应过程中是否有电极的溶出,结果表明,出水中钛和钌离子浓度均在检测线以下,说明此穿透式电极反应器能够适应大流量的实际操作条件,并不会带来电极的损坏。
-
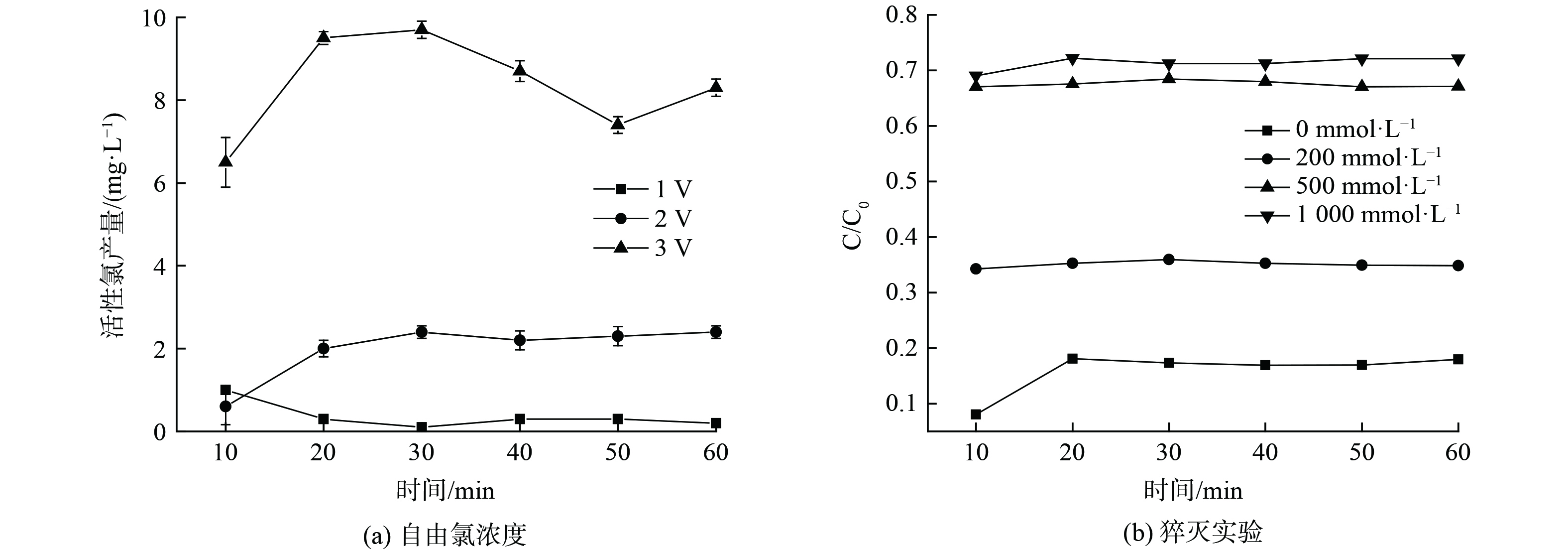
通常而言,外加电压的不同,会导致电化学过程反应速率的不同,同时直接影响了反应器的能耗。由上述实验结果也发现,本反应器体系中电催化对4-硝基酚的降解贡献明显,实验选取反应器运行性能较好的流量为100 mL·min−1时,研究0~3 V电压范围对4-硝基酚降解的影响。实验结果如图5所示,在0 V电压时,仅有光催化的作用,污染物的降解效果为5%~7%,2种光源没有表现出明显的差异,污染物降解几乎难以发生;在电压从1 V提升到3 V的3种处理中,电压和4-硝基酚降解性能呈现正相关。在施加电压下,3种电压的不同光电处理的差别较为明显,脉冲光的效果明显优于紫外。脉冲光源的光电协同处理下,2 V时,单次穿透降解污染物的效果可以达到83.1%,3 V时,降解效果可达92.1%左右。随着电压的升高,4-硝基酚被快速的降解,反应前后的溶液可以观测到明显的脱色变化。较高的电压,会促进体系中产生羟基自由基,利于间接电氧化速率,同时促进了电极表面的电子转移速率,可以有效提升污染物的直接氧化效率[16]。更为重要的是,在光电协同中,会促进HOCl的产量,而HOCl被认为是主要的去除有机污染物的活性物质[17],从而促进电化学过程的降解。然而,实验发现,在过高的电压和过长的HRT的实验中,会有少部分的钌(Ru)脱落,电压值过高会加速阳极极化并且导致大量析氧副反应的发生,降低电极效率并损坏电极,因此后续的实验中选取2 V和流量100 mL·min−1进行实验。
-
在光电协同的系统中,氯离子的含量会直接影响参与反应的活性氯物种,从而影响污染物的降解性能,实验通过改变氯化钠投加量来提升水中氯离子的浓度,并考察了溶液中氯离子浓度和pH对4-硝基酚降解性能的影响,实验结果如图6所示。通常而言,氯化钠浓度为1%以上的溶液被认为是高盐废水,基于此,实验配置了氯化钠浓度设置为25 mmol·L−1、50 mmol·L−1、171 mmol·L−1(1%)、256 mmol·L−1(1.5%)、512 mmol·L−1(3%)研究不同氯离子浓度对污染物降解的影响。如图6所示,在紫外光和脉冲光光电协同体系中,污染物降解浓度和氯离子浓度增加表现出相同的影响趋势,当氯离子浓度从25 mmol·L−1提升到50 mmol·L−1时,随着氯离子浓度的增加,污染物降解性能略有提升,出水的4-硝基酚浓度降低,降解效率分别从77.9%和79.9%提升到81.0%和83.9%;然而随着氯离子浓度的进一步增高,降解效率呈现下降的趋势。在512 mmol·L−1的处理中,降解效率分别为52.2%和59.1%。前期报道认为,当有更多的氯离子时,会大量的生产氯自由基和羟基自由基(初级自由基),同时原位电生成的Cl2可水解生成次氯酸(HOCl)和次氯酸盐离子(OCl−),增加氯离子浓度会促进一系列活性物质的产生,增加降解效率和动力学常数[18-19]。反应生成的活性氯(Cl2、HOCl和ClO−)是去除污染物的主要氧化剂,在阳极的氧化下具有较强的氧化能力,增加间接氧化效率。同时,氯离子的增加可以提高溶液中的电导率,加快电极表面的电子转移效率,提升有机物的直接氧化效果,也利于体系中产生大量羟基自由基[20]。然而,高氯化物浓度会导致大量的氯离子吸附在阳极表面,阻止了阳极上污染物的吸附和传质,从而影响了降解效率;且高浓度的氯离子可能会带来更多的副反应的发生。
溶液初始pH值会影响水中活性氯(Cl2、HOCl、ClO−)的存在形式,从而对4-硝基酚的降解有较大的影响。实验进一步研究了溶液初始pH值对污染物降解的影响,如图6所示,在pH 2~10的范围内,4-硝基酚均能够被降解去除,随着pH的增加,污染物的去除效率逐渐降低。在pH为2的极酸性条件下,污染物的降解效率最高,分别87.1%和88.8%(紫外光源和脉冲光源);在pH近似中性时,4-硝基酚的降解效率有轻微的下降,在pH为6时,降解效率分别为80.1%和83.0%;当溶液为碱性时,4-硝基酚的降解速率有大幅度的下降,分别为33.9%和36.0%。在所有的处理中,以脉冲光为光源的体系比以紫外光为光源的有略高一些的污染物降解去除率。尽管二者在污染物降解率方面差异不是很显著,但以脉冲光为光源的体系比以紫外光为光源的体系还是更具优势。因为脉冲灯光源能瞬间释放高强度光能,具有更大的穿透性(大于90%),在面向透光性较差的有色、有悬浮物的废水优势明显;其次,脉冲光是相同的能量以高峰值形式瞬间释放,同时在反应过程中,能够保持低温,两个脉冲之间也有足够的冷却时间,而紫外光主要是以热能的形式,因此光源使用寿命更长。脉冲光的间歇式发光相比于紫外光源的连续发光,如果以实际发光时间计算,对于污染物的降解和矿化性能则有显著差异。
有研究认为,不同pH下,活性中间物质活性氯的存在形式不同,从而表现出不同的降解能力,在pH>7.5时,OCl−占主导;在pH<2时,Cl2为主要的存在形式;在pH 3~7的范围内,HOCl为主要的存在形式[21]。此外,溶液pH的降解能够抑制阳极析氧副反应的产生,在酸性条件下,析氧电位较高,析氧发生更困难,从而可以减少对4-硝基酚降解的竞争作用,表现出更高的降解效率。实验表明,这种穿透式光电协同反应器适用于较广的pH范围,在酸性条件下降解效果更好。
-
由上述实验发现,脉冲光为光源的光电协同体系对污染物具有更好的降解能力,实验通过DPD法和猝灭实验测定了该体系反应过程中的氯活性物种产生,如图7所示。对于自由氯的测试发现,在低电压下,自由氯产生量较少,大约为2~3 mg·L−1,此时电直接氧化占主导,随着电压的升高,会有更多自由氯的产生,在3 V电压时,自由氯产生量大概为6~10 mg·L−1,促进污染物的降解,随着自由氯浓度的提升,DPD分光光度法测试中的显色反应也更为明显。实验通过猝灭实验研究了氯活性物种对于4-硝基酚的降解贡献,叔丁醇(TBA)被认为可以猝灭体系中的·OH、Cl·、Cl2−和ClO·[22-23]。实验发现,500 mmol L−1的猝灭剂基本可以淬灭体系中的氯活性物质,此时出水的4-硝基酚降解性能下降,约有70%的4-硝基酚未被降解。在脉冲光电协同的体系中,间接氧化反应,会产生大量的活性氯物质(·Cl、ClO−、Cl2),是去除污染物的主要氧化剂[24],进一步在脉冲光的激发下,会生成氯自由基,并通过链式反应生成很多可参与反应的氯活性物种(·ClO、·Cl2−、·ClO2),提升反应速率。通过活性氯和猝灭实验证实在光电脉冲协同体系中,氯活性物种起到了主导作用。
-
实验研究了污染物的矿化情况,如表2所示。有机物的去除率主要是指目标污染物被降解为其他的一些中间产物(包括最终产物CO2和H2O)的比例。有机物完全矿化前,会转变为一些低毒或无毒的小分子,和大量的中间产物。因此有机物的去除率高,并不完全代表目标污染物被完全转化为了CO2和H2O等无机物。矿化效率一般通过总有机碳(TOC)或化学需氧量(CODCr)衡量,本研究中以TOC为指标,衡量4-硝基酚的矿化率。如表2所示,施加不同的电压,脉冲光协同体系均表现出最好的矿化效果。在仅光照的情况下,污染物的降解性能较弱,几乎难以发生矿化,随着电压的提升,矿化效率随着提升,和污染物降解性能提升的趋势相吻合。在1 V电压时,脉冲光电协同系统矿化效率可达到45%,3 V时,矿化效率可达71%,由于脉冲光具有瞬间高强度光能释放的特点,不仅利于污染物的更快速降解,对于促进污染物深度矿化具有更明显的优势。研究认为,4-np的转化路径为4-np首先脱去有色基团亚硝基(-NO2),进而在被·Cl攻击发生亲电取代,在有羟基产生的情况下,·OH继而亲核取代或者消除·Cl,生成领苯二酚和对苯二酚,进而被氧化成苯醌。进一步在溶液中活性氯的作用下,发生开环反应,生成丁烯二酸,最后被完全矿化,生成CO2和H2O[25]。
-
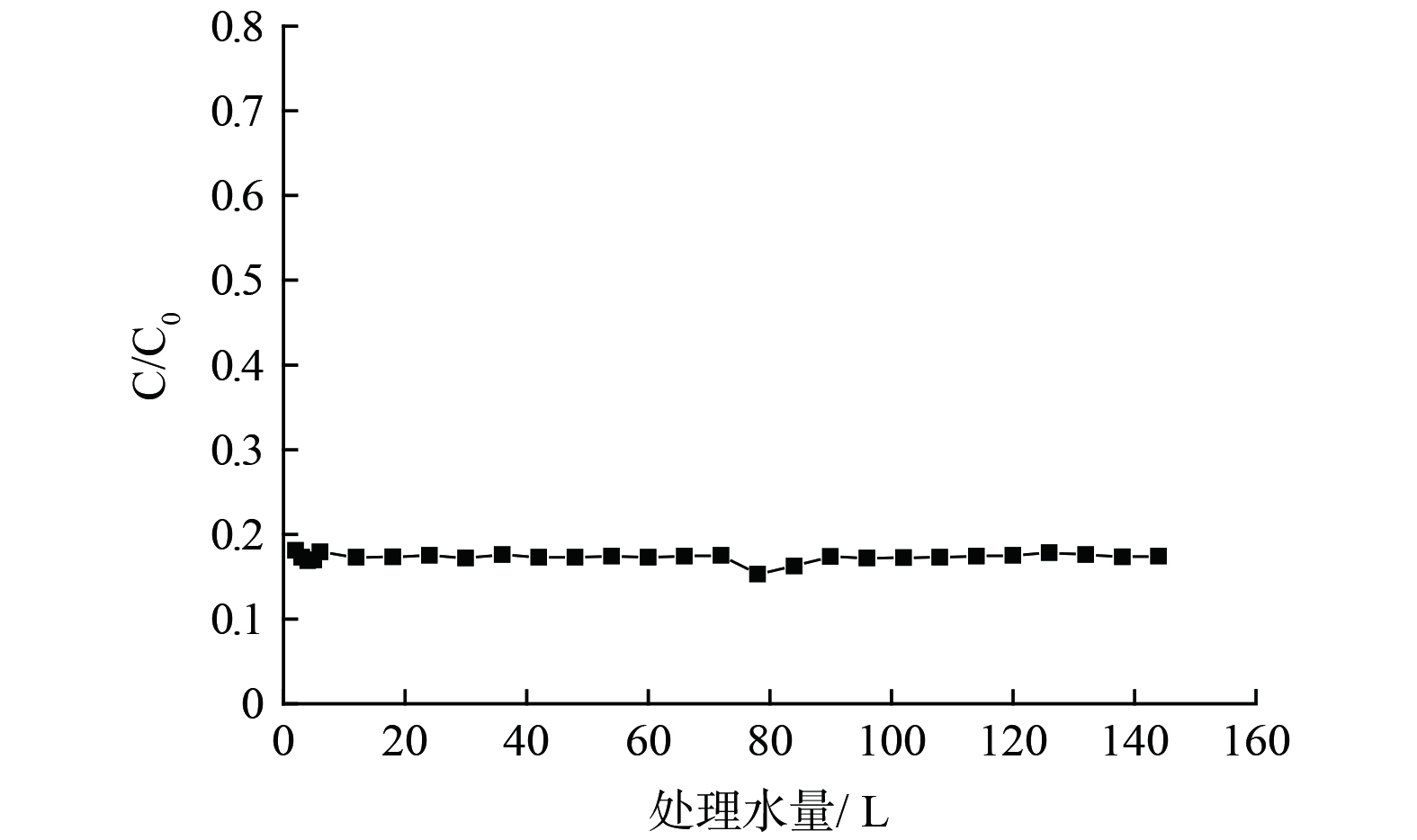
实验对反应器的长期运行稳定性和能耗进行了考察。在最优的反应条件脉冲电压作为光源, 2 V外加电压、50 mmol·L−1氯化钠浓度、19 min的停留时间下以单次穿透进行了连续运行实验。在连续运行48 h,处理水量达144 L的实验中,对污染物的去除效率有轻微波动,但仍能保持在83%以上4-硝基酚降解效率,没有明显的衰减(图8)。同时,出水中的钛和钌离子浓度均在检测线以下,没有明显的溶出,说明该反应器在较长时间的连续运行中有较好的稳定性。本研究体系中总能耗为电能耗和光能耗的总和,该反应体系的运行能耗为3.16 kwh·m−3,以平均电费0.05元·h−1计算,本体系对废水的处理成本约为0.158元·m−3,具有较好的经济性。
-
本研究构建了光电协同穿透式电极反应器,以镀钌泡沫钛作活性阳极,泡沫钛作为阴极,以4-硝基酚作为模型污染物,研究了光电协同穿透式反应器降解污染物的效果,得出的结论如下。
1)光电协同穿透式反应器可实现4-硝基酚的高效降解。在3 V的电压和19~24 min的停留时间下,目标污染物4-硝基酚在单次穿透时即可降解90%以上。
2)溶液的pH、停留时间、外加电压、氯离子浓度都会直接影响污染物的降解效率。在以强脉冲灯(IPL)为光源,2 V外加电压、50 mmol·L−1氯化钠浓度、19 min的停留时间为本研究得到的最优参数。
3)脉冲光电协同对于污染物的降解和深度矿化性能(71%)更优,在污染物降解过程中,氯活性物种占主导作用。该脉冲光电协同体系可实现长时间稳定的连续运行。
脉冲光-电协同去除含盐废水中有机污染物的研究
Removal of organic pollutants from saline wastewater by intense pulsed light (IPL)-electro reactor
-
摘要: 工业废水中本身往往含有大量的氯离子,可以在光电化学催化氧化下形成强氧化性的氯活性物种,达到净水目的。为了改进含盐废水中有机污染物在实际处理中传质效率低、光穿透性差导致的光利用效率低等问题,本研究开发了脉冲光电协同的穿透式电极反应器,并分析了其对有机污染物4-硝基酚的降解性能。研究通过脉冲光源和相同功率的常规紫外光源对比发现,强脉冲光协同电氧化对4-硝基酚的降解效率和矿化效率更高;在3 V电压协同脉冲光源,19~24 min的停留时间下,单次穿透出水可降解90%以上。反应器运行的最佳参数为脉冲光协同2 V电压,50 mmol·L−1的氯化钠浓度和19 min的水力停留时间。溶液的pH、氯离子浓度、停留时间和外加电压都会直接影响4-硝基酚的降解效能。在光电协同下,大量氯活性物质产生,对4-硝基酚降解起到了主导作用。此研究结果为含盐废水中有机污染物的去除提供了新的解决方案。Abstract: Industrial wastewater often contains a large number of chloride ions, which can form strong oxidizing reactive chlorine species (RCS) under photo-electric catalytic process for wastewater purification. In order to improve the shortcomings such as low mass transfer efficiency and poor light penetration in the practical wastewater treatment of saline organic wastewater, this study developed an intense pulsed light coupled with flow-through electrode and analyzed its degradation performance of organic pollutants 4-nitrophenol. By comparing the light source with the conventional ultraviolet light source of the same power, it was found that the degradation efficiency and mineralization efficiency of 4-nitrophenol were higher than that of intense pulse light. Under the 3 V voltage cooperative pulse light source, 19~24 min hydraulic retention time (HRT), 4-nitrophenol removal efficiency could reach up to 90% in single pass The optimum parameters for reactor operation were intense pulsed light with 2 V voltage, 50 mmol·L−1 sodium chloride concentration and HRT of 19 min. The solution pH, chloride ion concentration, HRT and applied voltage could directly affect the degradation efficiency of 4-nitrophenol. Under the photo-electric synergistic effect, reactive chlorine species were greatly produced which played a major role in the degradation of 4-nitrophenol. The results of this study provide a new solution for the removal of organic pollutants in saline wastewater.
-
减少废水中的氮化合物是改善水环境和水质的重要措施之一[1]。与传统硝化/反硝化工艺相比,短程硝化/厌氧氨氧化(partial nitrification/anaerobic ammonia oxidation, PN/A)工艺可以将脱氮需氧量降低50%,有机碳需求量降低100%,污泥产量降低90%[2-3]。因此,PN/A工艺被认为是最经济、最有前景的脱氮工艺[4]。
反应器内生物质的保留能力对厌氧氨氧化(Anammox)工艺的启动周期有着重要影响[5]。据报道,颗粒和生物膜污泥都具备良好的生物质截留能力[6-7],但已知这两种污泥形式分别单独运行时都会存在较长的启动时间[8-9]。生物膜系统形成周期短,但长期运行后载体上太厚的生物膜会导致生物质脱落并被水流冲刷[7]。Anammox颗粒污泥的形成是一个漫长的过程,但可以有效地拦截污泥流失并保持较高的生物量[10]。因此,生物膜和颗粒污泥的组合应用可能最大程度上保留反应器内生物质,从而实现PN/A的快速启动和功能菌的高效富集。然而,将好氧生物膜和厌氧颗粒相结合来启动PN/A工艺目前尚未见报道。。
不同于传统的单阶段和两阶段PN/A反应器,在多级PN/A反应器中,交替的缺氧室和好氧室不仅为 (anaerobic ammonia oxidizing bacteria(AnAOB)和ammonia oxidizing bacteria(AOB)这2种功能细菌的同时生长和富集提供了空间条件,而且在缺氧区亚硝酸盐氮(NO2−-N)和氨氮(NH4+-N)共存的环境有利于厌氧氨氧化菌的自然富集[7-8]。此外,实现PN/A工艺的关键不仅需要同时富集AOB和AnAOB,还必须尽可能抑制亚硝酸盐氧化菌(nitrite oxidizing bacteria,NOB)活性[11]。据报道,间歇曝气和pH控制等策略可以有效控制PN/A工艺中不同菌群的活性(富集AnAOB和AOB,抑制NOB)[12]。然而,具有间歇曝气、pH控制、多级反应器和生物膜/颗粒污泥系统等优点的组合PN/A反应器的运行策略仍需要研究。
本研究构建了由3个好氧反应柱和3个厌氧反应柱组成的新型多级好氧生物膜/厌氧颗粒反应器(multistage aerobic-biofilm/anaerobic-granular sludge reacto, MOBAPR),以同时促进AnAOB和AOB的富集。本研究的主要目的为:拟通过MOBAPR实现PN/A工艺的快速启动和高效运行;考察MOBAPR各反应柱的氮转化过程;探索不同MOBAPR柱中功能菌丰度的变化和微生物群落结构的差异;通过优化气液比(gas/liquid ratio, G/L),进一步提高PN/A工艺的脱氮效率(nitrogen removal efficiency, NRE)。
1. 材料与方法
1.1 进水水质与接种污泥
接种物取自中国江西省赣州市白塔生活污水处理厂的剩余污泥(普通活性污泥)。在第1天分别向每个反应柱加入100 mL接种物,其活性污泥浓度(MLSS)大约为5 100 mg·L−1。
本研究采用模拟废水(含150 mg·L−1 NH4+-N),其改编自前人研究[13]。主要成分包括0.708 g·L−1 (NH4)2SO4,1.05 g·L−1 NaHCO3, 0.02 g·L−1 KH2PO4, 0.022 g·L−1 MgSO4,0.008 g·L−1 CaCl2,1.25 mg·L−1营养液I(5 g·L−1 EDTA、0.00625 g·L−1 FeSO4)和营养液II(15 g·L−1 EDTA、0.43 g·L−1 ZnSO4·7H2O、0.25 g·L−1 CuSO4·5H2O、0.19 g·L−1 NiCl2·6H2O、0.99 g·L−1 MnCl2·4H2O、0.24 g·L−1 CoCl2·6H2O、0.22 g·L−1 NaMoO4·2H2O、0.014 g·L−1 H3BO4)。
1.2 实验装置
MOBAPR的示意图如图1所示。该反应器由6个高30 cm、直径4.5 cm的有机玻璃柱相互串联构成,总有效容积为2.5 L。从进水到出水的6个反应柱(reaction column, Rc)分别标记为 Rc1、Rc2、Rc3、Rc4、Rc5和Rc6(隔室数量可根据出水水质增减)。在好氧反应柱(Rc1、Rc3和Rc5)中添加无纺布作为填料,并采用间歇曝气。厌氧隔室(Rc2、Rc4和Rc6)采用低速搅拌装置。当PN工艺成功启动后停止搅拌。曝气量由玻璃转子流量计调节,并采用自动断电定时器电路实现间歇曝气。根据之前的报道[14],由蠕动泵(Langer,BT101L,UK)、pH 控制器(WEIPRO,pH-2010B,China)和NaOH溶液组成的pH控制系统将MOBAPR中的pH保持在8.2~8.5。在每次曝气15 min后开始检测DO(dissolved oxygen)质量浓度。
1.3 运行条件
本研究在MOBAPR中依次启动PN和PN/A工艺。第I阶段(1~7 d),为快速恢复硝化细菌(AOB和NOB)的活性,在好氧区((Rc1、Rc3和Rc5)中连续曝气,并在厌氧区(Rc2、Rc4和Rc6)连续搅拌。此外,此阶段由于污泥处于悬浮状态,会随着进水流动,因此,启动污泥回流以保证反应器内充足的生物质含量。第II阶段(8~15 d)为抑制NOB,在好氧区中使用间歇曝气。第III阶段(16~60 d)好氧区微生物已成功挂膜生长,基本没有污泥流失,停止回流。此外,为进一步抑制NOB,第29天水力停留时间(hydraulic residence time, HRT)降低至8 h(16~28 d的HRT为12 h)。第IV阶段(61~86 d)为避免搅拌影响厌氧区AnAOB富集,厌氧区停止搅拌。第V阶段(87~110 d),调整曝气量以保证反应器内充足的NO2−-N。各阶段运行参数详见表1。在第VI阶段(111~162 d),在进水NH4+-N为150 mg·L−1和好氧/厌氧时间为90 min/30 min条件下,分别调节曝气量和HRT来探讨不同G/L对NRE的影响。
表 1 实验条件及操作参数Table 1. Experimental conditions and operating parameters时期 阶段 时间/d HRT/h 曝气量/(L·min−1) 好氧(厌氧)时间/min 好氧区DO/(mg·L−1) 回流比/% 适应期 I 1~7 24 0.02 好氧 — 200 PN 启动期 II 8~15 12 0.05 80/40 0.5±0.2 100 PN运行期 III 16~60 12/8 0.05 80/40 0.5±0.2 0 PN/A 启动期 IV 61~86 8 0.08 90/30 0.0±0.2 0 PN/A运行期 V 87~110 6 0.10 90/30 0.0±0.1 0 气液调控期 VI 111~162 — — 90/30 — 0 注:表中曝气量和DO值均为好氧区(Rc1、Rc3和Rc5)的平均值;“—”表示无法检测。 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.4 分析方法
实验中反应器内NH4+-N、NO2−-N、NO3−-N的检测分析均根据《水和废水检验标准方法》中制定的方案,使用实验室规模的紫外/可见分光光度计(SQ2800,意大利UNICO)进行测定,包括纳氏试剂分光光度法(NH4+-N)(1-萘基)-乙二胺分光光度法(NO2−-N)和氨基磺酸紫外分光光度法(NO3−-N)。此外,为了更好地揭示MOBAPR中PN/A过程的氮转化机理,每天对各反应柱的氮质量浓度进行检测,并分析其亚硝酸盐积累率(nitrite accumulation rate, NAR)、氨氮去除率(ammonia nitrogen removal rate, ANR)、氮去除率(NRE)、氮负荷率(nitrogen load rate, NLR)和氮去除负荷(nitrogen removal load rate, NRR)[13-14]。
1.5 16S rRNA基因测序与微生物菌群分析
为探索MOBAPR中不同阶段微生物群落的演变,阐明连续多阶段PN/A过程中所涉及的生物学机制,分别对接种物、第56天(阶段III)和第110天(阶段V)的泥样进行微生物功能菌群分析。接种物命名为A0,第III阶段在Rc1~Rc6收集的污泥样品分别命名为A1、A2、A3、A4、A5和A6,第V阶段分别命名为B1、B2、B3、B4、B5和B6。这些样本保存在−20 ℃,直到提取DNA结束。
在成功提取样本内微生物的DNA后,使用16S rRNA基因的通用扩增引物进行PCR扩增,并且PCR产物使用 AxyPrep™ DNA凝胶提取试剂盒(Axygen Biosciences,Union City,USA)按照制造商的说明进行纯化。然后通过Illumina MiSeq测序平台(PE300)对样品高通量测序,并得到原始测序序列。为了解样本测序结果中的菌种、菌属、物种功能等信息,将在Miseq测序得到的原始序列数据利用cutadapt(version 1.18)和PRINSEQ(version 0.20.4)软件进行去除引物接头序列、拼接、识别的处理以得到各样本的有效数据。然后利用Usearch(version 11.0.667)按照97%相似性对非重复序列(不含单序列)进行OTU聚类。然后利用RDP classifier(version 2.12)等软件进行OTU物种分类,并根据得到的OTU序列进行微生物菌群分析与功能预测。
1.6 优化MOBAPR操作
由于在第V阶段DO值较低,MOBAPR的性能无法通过DO来进行控制。因此,在第VI(111~162 d),阶段,为了代替DO控制(当DO低至无法控制),本研究提出了一种新型控制参数—气液比(式(1))。分别在2、4、6和8 h的HRT条件下调控曝气量(0.05、0.1、0.15和0.2 L·min−1),从而得到G/L比为2.4、4.8、7.2、9.6、4.4、19.2、21.6、28.8和38.4。并且在每次操作条件调整后,MOBAPR连续运行3~4 d。此外,利用高斯模型预测了G/L与NRE之间的相关性(式(1))。
q=60tvV (1) 式中:q为G/L值;t为HRT,h;v为曝气速率,L·min−1;V为MOBAPR的总有效容积,L。
2. 结果与讨论
2.1 MOBAPR中PN/A工艺的脱氮性能
1)接种物驯化。阶段I(1~7 d)在进水NH4+-N为150 mg·L−1、曝气速率为0.02 L·min−1、DO为2~4 mg·L−1和HRT 24 h的条件下运行MOBAPR。如图2所示,出水NO3−-N由64.03 mg·L−1增加到122.71 mg·L−1。这说明在被重新接种后,硝化细菌(AOB和NOB)的活性在高DO水平下得到了快速恢复。此外,在阶段I中NRE大多低于零(图2(c))。这可能是一些细菌(主要是异养菌)不能适应无碳源下的MOBAPR,细菌死亡后细胞溶解释放出额外的氮源到反应器内。
2) PN工艺的启动。在阶段II(8~15 d),MOBAPR的pH为8.3,好氧区平均溶解氧为0.5 mg·L−1,间歇曝气(好氧/厌氧时间为80 min /40 min)。结果表明,NO2−-N由0 mg·L−1增加到106.89 mg·L−1(图2(b)),NO3−-N由122.71 mg·L−1减少到20.92 mg·L−1(图2(d)),这表明NOB被有效抑制的同时AOB成功富集。因此,此阶段PN工艺在MOBAPR成功启动。此外,PN工艺中的NO2−-N稳定供应是实现Anammox工艺的前提[15],其关键是高效稳定地抑制反应器中的NOB 活性[14]。有研究表明,控制pH和间歇曝气是抑制NOB活性的重要手段[13]。因此,将以上2种抑制策略的结合是实现PN过程快速启动的关键。
在阶段III(16~60 d),出水NO3−-N逐渐增加,第16~28天处于较高水平(30~46 mg·L−1)。因此,要实现PN过程的稳定运行,需要对控制条件进行调整。在第17天好氧区在已经形成稳定生物膜结构后,MOBAPR停止回流。结果NAR短暂升至81%,然后逐渐下降(图2(b))。在第20~28天,出水NO3−-N相对稳定(30~41 mg·L−1),说明综合控制策略仍能有效抑制NOB活性。第29天,HRT由12 h缩短到8 h,出水NO3−-N由29.2mg·L−1逐渐降至16.7 mg·L−1,NAR也增加到90%。因此,HRT是影响PN工艺稳定性的重要参数,HRT过长会产生额外的NO3−-N。
随着PN过程成功启动和AOB被富集积累[16-17],反应器中DO被AOB大量消耗,这导致厌氧区室中的DO质量浓度降低至0.2 mg·L−1左右,从而为厌氧菌提供了适宜的生长环境。由图2(c)可知,NRE由5.51%逐渐增加到25.52%。这表明AnAOB可能在此阶段自然富集。有研究表明,AOB是从微需氧甚至厌氧的祖先进化而来的,在亚硝酸氧化还原酶(NXR)和其他反射蛋白的形式上与AnAOB高度相似[18]。MIAO等的研究结果同样表明接种硝化污泥可以缩短Anammox的启动时间[19]。因此,基于PN工艺,AnAOB可能更容易富集。此外,高通量测序结果表明阶段III中AnAOB的增加。
3)PN/A工艺的启动与运行。有研究[20]表明,较大污泥絮凝物中的AnAOB具有更高的活性。厌氧区中污泥絮体的生长可能会受到搅拌的限制,从而抑制AnAOB的活性[20]。因此,在阶段IV(61~86 d)停止搅拌以增加AnAOB的活性。并且有研究表明,NO2−-N质量浓度越高越有利于Anammox的积累[21]。因此,延长好氧区的相对曝气时间以进一步增加反应器中NO2−-N质量浓度。在第IV阶段,厌氧区中停止搅拌,并且好氧区中好氧/厌氧时间从80 min/40 min变为90 min/30 min。在第61天后,MOBAPR的TN质量浓度逐渐下降,由126.96 mg·L−1(第61天)降低至(32.79±6.21) mg·L−1(77~86 d),NRE也从21.5%迅速增加到(78.86±4.6)%(图2(c))。这表明在本研究采用的操作策略下,61 d内成功实现PN/A工艺的快速启动。
此外,随着AnAOB成功富集,MOBAPR中脱氮速率增加,导致进水中大部分的NH4+-N在Rc1~Rc4中已经被去除,而Rc5和Rc6中功能微生物缺乏营养物质。因此,有必要适当缩短HRT以保证MOBAPR中功能微生物的进一步富集。在阶段V(87~110 d),HRT由8 h缩短至6 h。此阶段反应稳定后(102~110 d),出水NO2−-N、NO3−-N和NH4+-N质量浓度分别为(0.63±0.50)、(16.72±1.78)和(8.29±6.65) mg·L−1。其中,出水NO3−-N质量浓度(NO3−-N产生/NH4+-N去除=0.12)与Anammox的NO3−-N理论产生值(NO3−-N产生/NH4+-N去除=0.11)接近,这表明NOB被稳定抑制[21]。此外,PN/A工艺的NRE、ANR和NRR分别为(83.41±2.45)%、(97±3.61)%和(0.41±0.09) kg·(m3·d)−1(图2)。这表明该操作策略可用于MOBAPR,以实现PN/A过程的长期高效稳定运行。
有趣的是,在第IV和第V阶段(曝气量分别为0.08 L·min−1和0.10 L·min−1),所测得DO质量浓度接近0。有研究[14]表明,当AOB的耗氧速率高于曝气效率,反应器曝气后的DO质量浓度仍会保持在较低水平。因此,在MOBAPR中非曝气后,好氧区的曝气会被AOB等好氧细菌及时转化,从而维持反应器内低DO水平。此外,在MOBAPR中,AOB的富集是AnAOB快速启动的关键。AOB不仅可以为AnAOB创造环境,还提供营养物质。然而AOB主要在好氧区活性较高。因此,在整个启动期间,基本不对厌氧区进行直接调控(搅拌停止后)。
2.2 各阶段微生物群落演替分析
为了探索MOBAPR中微生物群落的变化规律,对接种物、第56天(第III阶段末期)和第110天(第V阶段)末期采集的污泥样品进行微生物群落进行分析。其中PA1为接种物A0,PA2为第III阶段各反应柱(A1、A2、A3、A4、A5和A6)内微生物丰度的平均值,PA3为第V阶段(B1、B2、B3、B4、B5和B6)微生物丰度的平均值。高通量测序得到的优质细菌序列被划分为不同的分类类别(门和属),结果如图3(a)和图3(b)所示。Proteobacteria包括具有硝化反硝化功能的细菌[16],是门水平上的主要细菌(图3(a))。Proteobacteria在接种物中的丰度为68.15%,而在阶段III和阶段V后分别下降到44.68%和40.39%(图3(a))。这表明在变形菌门中许多异养硝化或反硝化细菌由于有机物的缺乏而被淘汰。有研究表明,Planctomycete门中不仅拥有一些专性好氧菌,还包含了所有已知的AnAOB[2]。接种物(PA1)中的Planctomycete相对丰度接近第III阶段(PA2),分别为4.07%和3.71%。而到了第V阶段(PA3),Planctomycete丰度达到10.85%,这表明Planctomycete主要在第III阶段后被富集。此外,在属水平上PA1的Candidatus Kuenenia的相对丰度极低,约0.05%(图3(b))。这表明在接种物中几乎不含AnAOB。与接种物(PA1)相比,PA2和PA3中Armmonadetes和Chloroflexi的丰度显著增加(图3(a))。有研究表明,在Armarmadetes和Chloroflexi中的许多细菌含有与氮代谢相关的功能基因(Nar、NirK或Nos)[22]。因此,Armatimonadetes和Chloroflexi可能含有多种AOB和AnAOB协同细菌,为PN/A工艺的启动和运行做出了贡献。
 图 3 第I、第III和第V阶段微生物群落系统发育分析Figure 3. Phylogenetic analysis of the microbial community at stage I, stage III and stage V
图 3 第I、第III和第V阶段微生物群落系统发育分析Figure 3. Phylogenetic analysis of the microbial community at stage I, stage III and stage V图3(b)反映了PN/A工艺中所有样品在属水平上的微生物群落。在第III阶段Nitrosomonas的丰度由1.49%增加到28.20%(图3(b)),证实了该操作策略可以成功富集AOB。并且,16s结果表明Candidatus Kuenenia是MOBAPR中主要的AnAOB,其由接种物PA1(0.05%)增长到2.97%。这表明随着PN工艺的长期运行,此阶段(第56天)AnAOB开始富集。在第V阶段,PN/A工艺启动成功并长期运行后,Nitrosomonas (27.09%)和Candidatus Kuenenia(9.99%)的丰度得到了较高程度富集,这表明AOB和AnAOB在MOBAPR中可以同时富集。因此,通过第IV和第V阶段的综合运行策略,AOB和AnAOB作为优势菌被富集,并形成细菌协同关系完成脱氮。此外,NOB的主要菌属Nitrobacter、Nitrospira和Nitrospina等可能由于含量太低(<0.1),均未被检测到。
2.3 不同阶段各反应柱内的氮转化途径
1)氮转化途径分析。为了更深入地了解MOBAPR各反应柱内PN/A工艺的氮转化过程,对PN (阶段III)和PN/A 工艺(阶段V)长期运行阶段分别进行测试分析,结果如图4所示。在第III阶段,出水NO2−-N质量浓度从Rc1到Rc6逐渐增加,NH4+-N相应降低(图4(a))。这表明各反应柱内均参与到氨氧化过程中。每个反应柱内都含有NH4+-N和NO2−-N (图4(a)),这为AnAOB的富集提供了必要条件。并且由于氨氧化过程需要氧气参与,好氧区(Rc1、Rc3和Rc5)内的ANR显著高于厌氧区(Rc2、Rc4和Rc6)(图4(c))。此外,NO3−-N浓度一直处于较低水平(<10 mg·L−1)(图4(a)),这表明通过本研究采用的操作策略,NOB活性长期受到有效抑制。在这一阶段,MOBAPR的平均氮损失约为30 mg·L−1(图4(a)),证实了反硝化细菌或AnAOB的增加。特别是Rc1、Rc3和Rc5中NRE的增加也显著高于Rc2、Rc4和Rc6(图4(e)),说明好氧区内氮损失主要是反硝化细菌或AnAOB造成的。
 图 4 PN工艺(阶段III)和PN/A过程(阶段V)各反应柱内的氮转化途径Figure 4. The nitrogen conversion pathways in the reactor after the PN process (stage III) and PN/A process (stage V)
图 4 PN工艺(阶段III)和PN/A过程(阶段V)各反应柱内的氮转化途径Figure 4. The nitrogen conversion pathways in the reactor after the PN process (stage III) and PN/A process (stage V)在第V阶段,由于进水中的氨(150 mg·L−1)通过前4个反应柱被PN/A完全转化,最后2个反应柱(Rc5和Rc6)在此阶段被废弃。模拟废水在流经Rc4后被排出。如图4(b)所示,在此阶段出水NH4+-N降至较低水平。这表明经过长期运行,PN/A工艺的NRE有所提高。此外,由图4(d)可以看出,大部分氨氧化过程基本在Rc1完成,而在Rc4之后脱氮量达到最高(图4(f))。此外,在第V阶段各反应柱内NO2−-N含量低于阶段III(图4(a)),这表明由AOB产生的NO2−-N被AnAOB快速利用,即AnAOB与AOB之间形成了良好的协同脱氮效果。Rc1和Rc3中NRE和ANR均显著增加(图4(f)),因此,PN和Anammox过程主要在好氧室中进行。这是由于在好氧区内形成了内层为AnAOB和外层AOB的微生物生物膜协同脱氮系统。依赖于外部的AOB提供的NO2−-N,内部的厌氧微生物将剩余的氨转化为氮。其中,由图4(f)可知,厌氧区(Rc2和Rc4)中NRE的增加远低于好氧区(Rc1和Rc3),并且厌氧区的出水NO2−-N几乎为0(图4(b))。因此,NO2−-N的缺乏可能限制了厌氧区的NRE。此外,在MOBAPR的好氧室中添加填料形成生物膜系统,不仅有效避免了DO对AnAOB的抑制,而且还有利于AnAOB的富集。在此阶段稳定的生物膜和颗粒污泥系统分别在好氧区和厌氧区形成。一方面,在好氧区的生物膜系统中形成了分层分布的好氧外层和厌氧内层。AnAOB在厌氧环境的生物膜内层得到富集,并与外层AOB协同脱氮。另一方面,AOB产生的大部分NO2−-N被生物膜内层的AnAOB利用,剩余少量NO2−-N流出并被位于厌氧区的AnAOB颗粒污泥消耗。此阶段在好氧区的脱氮方式与单阶段PN/A工艺相似,而厌氧区脱氮方式与两阶段PN/A工艺相似。因此,生物膜和颗粒污泥结构成功将单阶段与两阶段PN/A工艺的优势结合在一起,不仅具备两阶段PN/A更快的启动速度,还具备单阶段PN/A的更高效的反应速率。
2)各反应柱中AOB和AnAOB的动态分析。为了探究在PN工艺和PN/A工艺长期运行过程中,MOBAPR中不同反应柱内微生物群落的差异,对接种污泥、阶段III和阶段V获得的污泥样品进行高通量测序。其中,接种污泥样品命名为A0,在第56天(阶段III)Rc1、Rc2、Rc3、Rc4、Rc5和Rc6采集的样品分别命名为A1、A2、A3、A4、A5和A6,在第110天(阶段V)采集的样品命名为B1、B2、B3、B4、B5和B6。高通量测序得到的相关参数如表2所示。厌氧区中的Simpson指数明显低于好氧区(表2),这说明好氧区的物种富集程度是高于厌氧区的,Shannon指数也得到了类似的结论。每个污泥样品的覆盖率超过99.70%(表2),说明高通量测序基本代表了污泥样品的实际微生物群落结构。
表 2 微生物多样性分析Table 2. Microbial diversity analysis阶段 样品 序列数 丰富度 OTU数 多样性 覆盖率/% Ace指数 Chao1指数 Simpson指数 Shannon指数 I A0 52 844 913.5 933.6 841 0.07 4.27 99.8 III A1 57 036 886 863 724 0.11 3.48 99.7 A2 64 451 1 006.4 1 024.1 877 0.05 4.13 99.7 A3 70 746 891 932.1 751 0.16 3.05 99.7 A4 54 252 949.5 953.5 801 0.08 3.78 99.7 A5 59 513 899.5 902.3 786 0.18 3.13 99.7 A6 52 884 962.5 1 003.1 842 0.07 3.99 99.7 V B1 61 553 844.7 837.2 666 0.12 3.21 99.7 B2 64 627 892.2 896.5 742 0.09 3.57 99.7 B3 75 032 845.7 863.1 692 0.11 3.2 99.8 B4 62 284 900.8 909.7 764 0.06 3.86 99.7 B5 62 838 853.3 852 703 0.21 2.84 99.7 B6 59 777 984.9 1 014.6 851 0.13 3.64 99.7 注:A1、A3、A5、B1、B3和B5是来自好氧区的污泥样品;A2、A4、A6、B2、B4和B6是来自厌氧区的污泥样品。 | Show TableDownLoad:
CSV
为深入了解好氧生物膜/厌氧颗粒污泥的微生物分布情况,在阶段III和阶段V,从MOBAPR中的6个反应柱中获得的污泥样品进行了微生物群落分析。PN工艺启动并长期运行后,A1、A3和A5中亚硝基单胞菌(Nitrosomonas)的相对丰度分别从1.49%提高到30.99%、39.77%和40.74%,A2、A4和A6中亚硝基单胞菌的相对丰度分别从1.49%提高到11.42%、22.25%和22.15%(图5(c)),这证实了AOB在每个隔间中都被富集。厌氧区中AOB的富集与好氧区出水的DO有关。因此,厌氧区的AOB丰度明显低于好氧区(图5(c))。此外,在A1、A2、A3、A4、A5和A6中,AnAOB(Candidatus Kuenenia)的丰度分别由0.05%提高到0.16%、5.25%、0.56%、5.87%、0.39%和5.06%(图5(c)),这表明AnAOB已经开始富集。AOB不仅通过消耗溶解氧为AnAOB创造厌氧环境还为AnAOB提供必需的基质(NO2−-N)。因此,此阶段Nitrosomonas富集可能为Anammox的启动奠定了基础。
 图 5 PN和PN/A(阶段III和阶段V)稳定运行后各反应柱内微生物群落结构Figure 5. The microbial community structure in each reaction column of PN and PN/A (Stage III and Stage V) under stable operation conditions
图 5 PN和PN/A(阶段III和阶段V)稳定运行后各反应柱内微生物群落结构Figure 5. The microbial community structure in each reaction column of PN and PN/A (Stage III and Stage V) under stable operation conditions在PN/A工艺成功启动和运行后(第V阶段),B1、B2、B3和B4中Candidatus Kuenenia的丰度分别增加到12.67%、19.07%、12.41%和11.43%(图5(a))。由于Rc5和Rc6中NO2−-N的缺乏,B5和B6中Candidatus Kuenenia的丰度较低(分别为0.24%和2.29%)。在各反应柱中,Nitrosomonas和Candidatus Kuenenia都得到了较高水平的积累(图5(b))。这证实了微生物之间协同脱氮系统的存在。有趣的是,Candidatus Kuenenia不仅被富集在厌氧区,而且在好氧区中也有较高的丰度,这表明好氧区已形成分层分布的生物膜系统。此外,在PN/A工艺的长期运行阶段,未分类菌数量较多(图5(c))。有研究表明,PN/A工艺微生物群落由各种功能菌和协作菌共同组成的[14]。因此,在演替过程中会出现一些未分类协作菌以促进功能菌更好的富集。
2.4 G/L对NRE的影响
在第V阶段,MOBAPR中的DO值接近于零。一方面,不能通过控制DO值来进一步优化反应器性能;另一方面,不能人为直接调控DO值,即只能通过曝气量或流量等来间接调控DO值。因此,在PN/A工艺在运行过程中,DO控制会存在一定的滞后性。此外,在此阶段发现曝气速率越高,厌氧菌活性会降低;而曝气速率越低,厌氧菌的氮转化性能同样会将低。因此,在MOBAPR中提供合适的曝气量非常重要。
第2.1节和2.2节中的结果表明可通过缩短HRT将MOBAPR的NRE进一步提高,但好氧区中AOB的需氧量随着进水氨氮浓度的增加而增加。因此,本研究通过控制G/L,一方面可以为MOBAPR中的功能微生物提供稳定适宜的NO2−-N/NH4+-N,从而促进MOBAPR的总氮去除率;另一方面,直接调控反应器参数(代替DO控制),从而简化操作。为探讨G/L对总氮去除率的影响,通过调节曝气量和HRT,在进水NH4+-N为150 mg·L−1,好氧/厌氧时间为90 min/30 min条件下,G/L参数分别设置为2.4、4.8、7.2、9.6、14.4、19.2、21.6、28.8和38.4。用高斯模型对得到的NRE和相应的G/L进行拟合(极点拟合)以得出最适G/L,拟合结果如图6所示。可以看出,PN/A工艺的NRE在G/L为0~19.2时增大,而在G/L为21.6~38.4时减小。高斯模型的相关系数(R2)为0.992 2(图6(b)),说明该模型较好地描述了NRE与G/L之间的关系。模型拟合结果表明,当G/L比值参数为20~30时,NRE可达到较高水平。
 图 6 G/L对MOBAPR氮去除和氮转化的影响Figure 6. Effect of G/L on nitrogen removal and nitrogen conversion in MOBAPR
图 6 G/L对MOBAPR氮去除和氮转化的影响Figure 6. Effect of G/L on nitrogen removal and nitrogen conversion in MOBAPR3. 结论
1)本研究构建了厌氧和好氧区共存、悬浮污泥系统与生物膜系统相结合的MOBAPR。
2)在MOBAPR中15天内成功启动PN工艺,PN/A工艺在61天内成功启动。在运行阶段,PN工艺的NAR为(87.35±2.7)%,PN/A工艺的NRE为(83.41±2.45)%。
3)高通量测序结果表明,Nitrosomonas(27.09%)和Candidatus Kuenenia(9.99%)在厌氧区和好氧区被同时富集。在长期运行阶段,PN工艺的NAR为(87.35±2.7)%,PN/A工艺的NRE为(83.41±2.45)%。
4)在DO低至无法控制时,G/L可能是一种可以代替DO控制的重要策略,并且高斯模型拟合结果表明,当G/L比值参数为20~30时,NRE可达到较高水平。
-

图 2 不同光源处理对4-硝基酚的降解性能影响
Figure 2. Degradation efficiency of 4-np under different light source

图 3 不同光电处理下反应器出水4-硝基酚的降解性能
Figure 3. Degradation efficiency of 4-np under different photo-electric condition

图 4 不同流量运行下4-硝基酚的降解效果
Figure 4. Degradation efficiency of 4-np under different flux rates
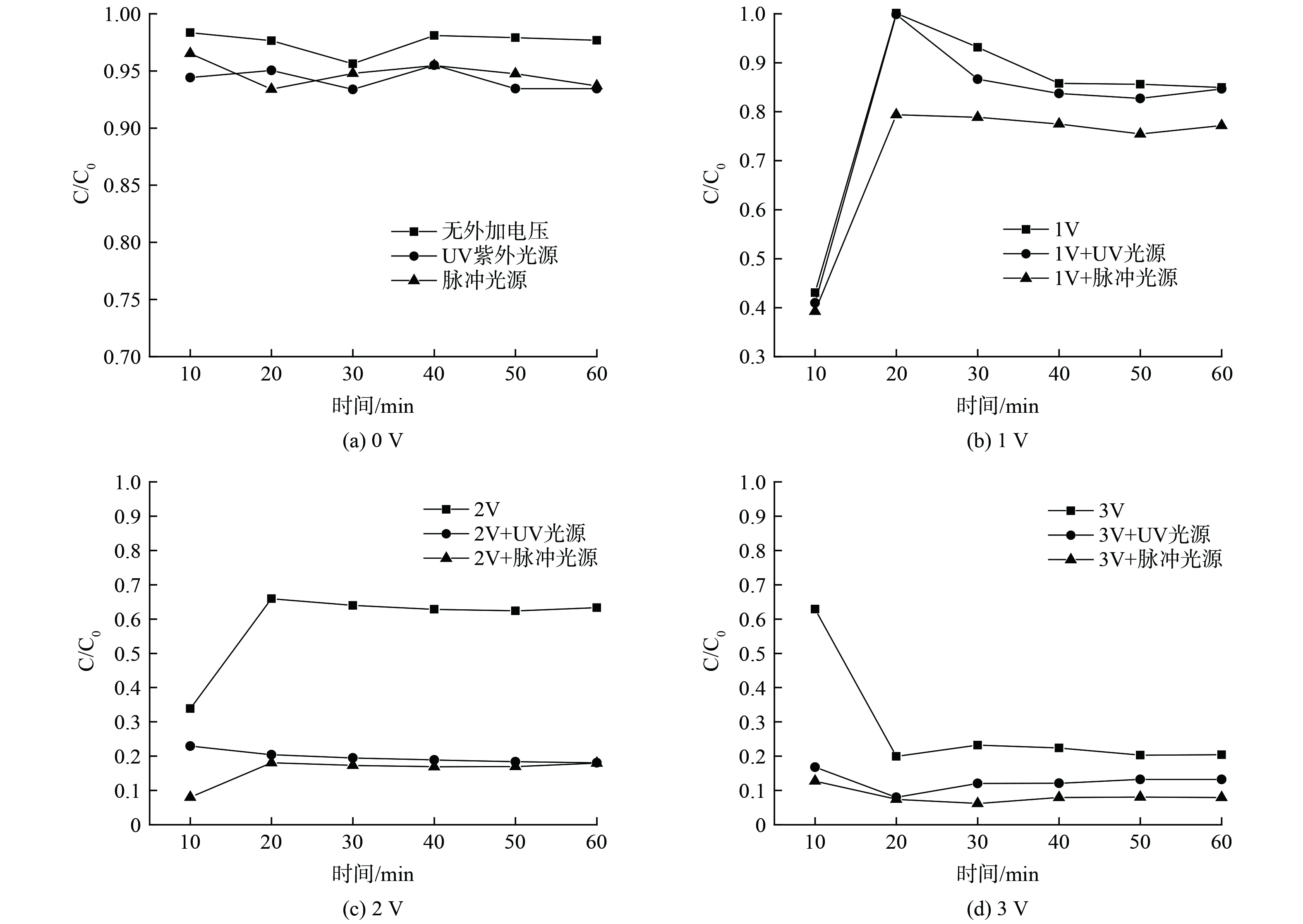
图 5 不同电压运行下4-硝基酚的降解效果
Figure 5. Degradation efficiency of 4-np under different applied voltage

图 6 氯离子浓度和pH对4-硝基酚降解影响
Figure 6. Degradation efficiency of 4-np under different Cl− concentration and pH conditions
表 1 不同流量对应的水力停留时间(HRT)
Table 1. Summary of HRT in different flow rates
编号 流量/(mL·min−1) 水力停留时间/min 1 200 9.5 2 100 19 3 80 24 4 50 38
下载: 导出CSV
表 2 不同光电处理的TOC去除率
Table 2. Summary of TOC removal efficiency under different photo-electric condition
处理/电压 0 V 1 V 2 V 3 V 仅加电 1% 9% 15% 27% 紫外光+电压 2% 34% 59% 68% 脉冲光+电压 3% 45% 65% 71%
下载: 导出CSV
-
[1] 曲久辉, 刘会娟. 水处理科学与技术: 水处理电化学原理与技术[M]. 北京: 北京科学出版社, 2007. [2] CHIANG L C, CHANG J E, WEN T C. Indirect oxidation effect in electrochemical oxidation treatment of landfill leachate[J]. Water Research, 1995, 29(2): 671-678. doi: 10.1016/0043-1354(94)00146-X [3] COMNINELLIS C, NERINI A. Anodic oxidation of phenol in the presence of NaCl for wastewater treatment[J]. Journal of Applied Electrochemistry, 1995, 25(1): 23-28. [4] RIBORDY P, PULGARIN C, KIWI J, et al. Electrochemical versus photochemical pretreatment of industrial wastewaters[J]. Water Science Techonology, 1997, 35(4): 293-302. doi: 10.2166/wst.1997.0141 [5] YUAN J, LI Y, CHEN X, et al. One electron oxidation-induced degradation of brominated flame retardants in electroactive membrane filtration system: Vital role of dichlorine radical-mediated process[J]. Journal of Hazardous Materials, 2024, 471: 134318. doi: 10.1016/j.jhazmat.2024.134318 [6] WANG Z W, ALMATRAFI E, WANG H, et al. Cobalt single atoms anchored on oxygen-doped tubular carbon nitride for efficient peroxymonosulfate activation: simultaneous coordination structure and morphology modulation[J]. Angewandte Chemie International Edition, 2022, 61(20): 202202338. [7] YANG S Q, LIU Z Q, CUI Y H, et al. Organics abatement and recovery from wastewater by a polymerization-based electrochemically assisted persulfate process: Promotion effect of chloride ion and its mechanism[J]. Journal of Hazardous Materials, 2023, 446: 130658. doi: 10.1016/j.jhazmat.2022.130658 [8] ZHOU Y J, JI Q H, LIU H J, et al. Pore structure-dependent mass transport in flow-through electrodes for water remediation[J]. Environment Science & Technology, 2018, 52(13): 7477-7485. [9] WANG S L, PEI S Z, ZHANG J N, et al. Flow-through electrochemical removal of benzotriazole by electroactive ceramic membrane[J]. Water Research, 2022, 218: 118454. doi: 10.1016/j.watres.2022.118454 [10] HAKIZIMANA I, ZHAO X, WANG C, et al. Efficient multi-stage electrochemical flow-through system for refractory organic pollutant treatment: Kinetics, mass transfer, and thermodynamic analysis[J]. Chemosphere, 2023, 344: 140405. doi: 10.1016/j.chemosphere.2023.140405 [11] WANG L L, WANG L, SHI Y W, et al. Blue TiO2 nanotube electrocatalytic membrane electrode for efficient electrochemical degradation of organic pollutants[J]. Chemosphere, 2022, 306: 135628. doi: 10.1016/j.chemosphere.2022.135628 [12] 李曈. 光化学反应中光生电子及共生自由基的调控与利用研究[D]. 北京: 中国科学院大学, 2018. [13] NATHALIE E G L, PHEBE H V L, JOHAN T P, et al. 20-fold increased limiting currents in oxygen reduction with Cu-tmpa by replacing flow-by with flow-through electrodes[J]. ACS Sustainable Chemistry & Engineering, 2024, 12: 12909-12918. [14] 刘春前. 稀土La掺杂Ti/Sb-SnO2电极电催化氧化对硝基苯酚[D]. 杭州: 浙江工业大学, 2010. [15] LIU H, VECITIS C D. Reactive transport mechanism for organic oxidation during electrochemical filtration: mass-transfer, physical adsorption, and electron-transfer[J]. The Journal of Physical Chemistry C, 2012, 116(1): 374-383. doi: 10.1021/jp209390b [16] WANG Y T, XUE Y D, ZHANG C H. Generation and application of reactive chlorine species by electrochemical process combined with UV irradiation: Synergistic mechanism for enhanced degradation performance[J]. Science of the Total Environment, 2020, 712: 136501. doi: 10.1016/j.scitotenv.2020.136501 [17] ZHANG Y, TANG W J, BAI JING, et al. Highly efficient removal of total nitrogen and dissolved organic compound in waste reverse osmosis concentrate mediated by chlorine radical on 3D Co3O4 nanowires anode[J]. Journal of Hazardous Materials, 2022, 424: 127662. doi: 10.1016/j.jhazmat.2021.127662 [18] XIANG Y Y, FANG J Y, SHANG C. Kinetics and pathways of ibuprofen degradation by the UV/chlorine advanced oxidation process[J]. Water Research, 2015, 90: 301-308. [19] YANG Y, JIEUM S, JUSTIN T J, et al. Multilayer heterojunction anodes for saline wastewater treatment: design strategies and reactive species generation mechanisms[J]. Environmental Science & Technology, 2016, 50(16): 8780-8787. [20] ZHANG J, ZHOU Y Y, YAO B, et al. Current progress in electrochemical anodic-oxidation of pharmaceuticals: Mechanisms, influencing factors, and new technique[J]. Journal of Hazardous Materials, 2021(418): 126313. [21] LI T, JIAGN Y, AN X Q, et al. Transformation of humic acid and halogenated byproduct formation in UV-chlorine processes[J]. Water Research, 2016, 102(10): 421-427. [22] WANG Z Y, LI K L, GUO J J, et al. Elimination of pesticide from high salinity wastewater by electrochlorination process: Active chlorine species and scale-up performance[J]. Separation and Purification Technology, 2023, 306: 122572. doi: 10.1016/j.seppur.2022.122572 [23] PAN Y H, CHENG S S, YANG X, et al. UV/chlorine treatment of carbamazepine: Transformation products and their formation kinetics[J]. Water Research, 2017, 116: 254-265. doi: 10.1016/j.watres.2017.03.033 [24] YANG Z C, QIAN J S, SHAN C, et al. Toward selective oxidation of contaminants in aqueous systems[J]. Environment Science & Technology, 2021(55): 14494-14514. [25] MARIE D, URS V G. Reactions of chlorine with inorganic and organic compounds during water treatment-kinetics and mechanisms: A critical review[J]. Water Research, 2008, 42: 13-51. doi: 10.1016/j.watres.2007.07.025 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 264
- HTML全文浏览数: 264
- PDF下载数: 10
- 施引文献: 0
