



-
硝酸盐(NO3−)是一种在水体中广泛存在的无机污染物。近几十年来,由于人类的工业和农业活动,水体中的NO3−浓度不断升高,已逐渐成为世界各国密切关注的问题[1-2]。我国30个省71条主要河流样品的硝酸盐数据表明,这些样品中有约7.83%的硝酸盐含量超过45 mg·L−1[1]。一般而言,污水厂二沉池出水中的硝酸盐浓度在10 mg·L−1,约占我国污水厂一级A标准中规定出水总氮浓度要求(15 mg·L−1)的67%左右,而一些水环境敏感地区(如滇池、太湖等)的污水厂出水总氮浓度的内控指标低至5 mg·L−1(以氮计),硝态氮去除对污水厂总氮控制造成极大压力。污水厂出水中硝酸盐氮浓度过高导致的总氮超标是城镇污水处理厂中最常见的问题之一[3]。目前,污水中的NO3−主要通过生物反硝化去除,需要外加碳源、延长反硝化停留时间或增大回流比强化脱氮,能耗和药耗高。化学还原法具有还原效率高、操作简单、成本低等优点,如果能利用化学还原方法强化NO3−脱除,将有助于降低厌氧生物脱氮负荷,进一步提高污水中的总氮去除效率。
近年来,基于二氧化碳阴离子自由基 (CO2·−)的高级还原技术逐步引起人们关注。CO2·−分子结构中存在一个未成对的电子,具有很强的还原特性,还原电位E0(CO2/CO2·−)为−1.9 V[4]。CO2·−作为电子供体,通过电子传递、亲核攻击等途径还原降解污染物[5-6]。研究表明,CO2·−能有效去除水中卤代有机物、全氟化合物、溴酸盐和六价铬等污染物[7-10]。近期研究发现,基于CO2·−的高级还原技术能将NO3−定向还原为气态氮,并且水中共存的溶解性有机物对CO2·−驱动的NO3−还原去除影响很小,仅在有机物浓度30 mg·L−1时的NO3−还原去除略有下降[11-12]。但是,目前基于污水体系的CO2·−的高级还原研究较少,其实际应用仍存在诸多挑战[11]。
紫外消毒已在污水处理中广泛应用,截至2016年我国已建成使用的5 000多座城镇污水厂中超过50%采用了中压紫外消毒。硝酸盐在波长250~400 nm具有吸光性,特别是在302 nm处具有特征吸收峰,摩尔吸光系数为7.2 L·(mol·cm)−1。同时,甲酸盐(HCOO−)可作为一种新型绿色碳源被投入到二级生物反应池中促进生物脱氮过程[13-14],未被生物利用的甲酸盐则进入紫外消毒阶段。污水中NO3−在紫外辐照下光解产生含氮活性物种(RNS)(式(1))和羟基自由基(HO·)(式(2))[15],HO·与甲酸盐或者甲酸反应生成CO2·−(式(3)~(4))。CO2·−的还原性可以促进进一步硝酸盐还原(式(5))。
基于此,本研究将围绕污水处理的紫外消毒环节构建中压紫外/甲酸盐反应体系,解析此体系中的NO3−还原效果和关键影响因素;围绕硝酸盐光解以及CO2·−与硝酸盐的反应活性揭示硝酸盐降解原理,并进一步探索硝酸盐强化脱除方法。研究结果将深化CO2·−高级还原体系的基础数据和反应机制,为污水的深度脱氮提供新思路。
-
硝酸钠、甲酸钠、甲酸均为分析纯,采购于上海安谱璀世有限公司;过氧化氢为纯度30%,购于广州牌药剂公司。95%~98%浓硫酸(H2SO4)为分析纯,购于福晨(天津)化学试剂有限公司;氢氧化钠(NaOH)为分析纯,购于国药集团化学试剂有限公司。所有试剂在使用前均未经过纯化或其他处理步骤,实验用水均来自Merck-Millipore系统产生的超纯水。
-
硝酸盐的光还原降解实验在平行光紫外辐照装置中进行,由中压紫外光源(500 W,PLS-LAM500,Beijing PerfectLight)、反应石英皿和磁力搅拌装置组成。中压汞灯在225~425 nm的发射光谱,主波长在320 nm和360 nm,与污水处理厂的中压汞灯消毒的特征波长一致,所有实验未采用滤光手段控制光源辐照的波长。由于中压汞灯的特征波长的复杂性,本研究以254 nm处的紫外光强作为指示光强,其值为0.5 mW·cm−2。在研究紫外光强影响时,通过改变中压紫外灯与水样的间距,将光强划分为低、中、高3个水平,其在254 nm处的光强值分别为0.04、0.5和1.3 mW·cm−2。
实验水样中初始NO3−浓度为0.1 mmol·L−1,甲酸或者甲酸盐为0~10 mmol·L−1。此实验中初始硝酸盐浓度低于实际污水中的硝酸盐存在水平,其原因是为了与实验中较低光强的紫外光源相匹配。除研究pH影响的实验外,水样初始pH为7.0±0.2,不另加pH缓冲溶液。在研究pH影响时,水样中加入10 mmol·L−1磷酸盐缓冲溶液,采用硫酸或氢氧化钠调节初始pH。石英皿置于紫外装置下,以加入甲酸/甲酸盐(0~10 mmol·L−1)为反应起始点,反应过程中采用磁力搅拌器搅拌,确保溶液和试剂混合均匀。在一定时间间隔下取样测定硝酸盐及产物浓度。
-
采用激光闪光光解技术测定CO2·−与硝酸盐的二级反应速率常数。激光闪光光解系统(LKS80,Applied Photophysics Ltd., United Kingdom)的激发波长为266 nm,激光束截面为0.5 cm2,脉冲激光能量为(25±5) mJ,脉冲持续时间为4~6 ns;使用150 W氙灯作为检测光源。实验水样中含H2O2(50 mmol·L−1),HCOO−(500 mmol·L−1),竞争参考物甲基紫精(MV2+)浓度为100 μmol·L−1,向溶液中投加不同浓度的硝酸盐(0~100 mmol·L−1)。激光实验前,水样由氩气吹脱30 min (溶解氧<0.2 mg·L−1)并调节pH为7.0。测定MV2+反应产物MV·+在600 nm特征吸收峰信号。
-
硝酸盐、亚硝酸盐、甲酸盐浓度利用离子色谱仪(ThermoFisher, ICS-600)进行测定。离子色谱仪的主要组成部分为:RFC-30淋洗液自动发生器,ChromeLeon色谱工作站,Dionex IonPacTM AG-19保护柱(4 mm×50 mm),Dionex IonPacTM AS-19分析柱(4 mm×250 mm)。KOH淋洗液由RFC-30淋洗液自动发生器在线自动产生,流速为1.0 mL·min−1。样品检测前需经过0.22 μm滤膜过滤。采用纳氏试剂分光光度法测定氨氮的浓度。pH采用pH计(S400,Mettler Toledo)测定。反应溶液所受的中压汞灯辐照光强通过UV254光强测定仪UVC-254A(Lutron electronic enterprise Co; LTD)初步量化,以确定辐照光强的相对大小。
-
1)甲酸盐浓度对硝酸盐光还原降解的影响。图1所示为不同HCOO−投加量(0~10 mmol·L−1)条件下硝酸盐的光还原降解速率。当体系中不存在甲酸盐时,紫外辐照120分钟后硝酸盐去除率仅为13.7%,NO3−拟一级降解速率kobs为1.0×10−3 min−1。HCOO−投加量为0.1、0.5、1、5和10 mmol·L−1时,反应120min的硝酸盐去除率分别为20.6%、39.4%、44.1%、45.9%和43.9%。由此可见,甲酸盐在0.1~1 mmol·L−1条件下,硝酸盐降解速率kobs由2.41×10−3 min−1逐步增大到5.98×10−3 min−1。随着甲酸浓度继续增大(1~10 mmol·L−1),NO3−的一级降解速率趋于稳定。可见,硝酸盐紫外光解产生的HO·可以促进甲酸盐转化为CO2·−,进而强化硝酸盐的还原降解(式(1)~式(4))。然而,受到硝酸盐光解的HO·生成量限制,高浓度的甲酸盐(大于1 mmol·L−1)不能形成更多的CO2·−,使得硝酸盐不能进一步降解。因此,硝酸盐和甲酸盐的摩尔浓度比在1:5~1:10的促进效果较好。
2)紫外光强对硝酸盐光还原降解的影响。图2为紫外光强对MPUV/HCOO−体系降解硝酸盐的影响结果。将甲酸盐投加浓度设为2 mmol·L−1,以防止高光强导致的甲酸盐快速消耗而影响实验结果。通过调节紫外灯与水样间距选定3种光强,其在254 nm处指示的低、中、高光强分别为0.04、0.5和1.3 mW·cm−2。单独紫外光解(无甲酸盐)时,硝酸盐降解速率随着紫外光强增大而缓慢增大,在低、中、高光强条件下,kobs分别为1.8×10−4、1.0×10−3 和1.2×10−3 min−1,表明紫外光强增大能促进硝酸盐光解。当2 mmol·L−1甲酸盐存在时,MPUV/HCOO−体系中硝酸盐降解速率随紫外光强的增加而显著增加,kobs分别为9.9×10−4、3.6×10−3和6.9×10−3 min−1。MPUV/HCOO−体系对紫外强化的依赖性更高,说明增加紫外光强不仅促进了硝酸盐分解,也增大了硝酸盐光解介导的CO2·−生成,从2个途径加速了硝酸盐的光还原降解。因此,紫外光强是重要的硝酸盐光还原影响因素。
3)pH对硝酸盐光还原降解的影响。图3反映了pH对MPUV/HCOO−体系降解硝酸盐的影响。甲酸盐投加量设为0.5 mmol·L−1,以规避硝酸盐去除能力上限造成的干扰。溶液pH的增大可促进硝酸盐光降解。在pH为2.5、5.0和8.0条件下,MPUV/HCOO−体系中硝酸盐降解的kobs分别1.5×10−3、2.6×10−3和3.6×10−3 min−1。可见,碱性条件更利于MPUV/HCOO−体系的硝酸盐降解。溶液的pH对硝酸盐和甲酸盐的存在形态以及还原性自由基与硝酸盐的反应速率均存在影响。对于甲酸的存在形态,HCOOH/HCOO−的pKa为3.8,pH=2.5时的kobs最低,说明HCOO−形式更助于硝酸盐的光还原降解。对于硝态氮,硝酸盐光敏化生成一种吸光度极高的中间体过氧亚硝酸盐(ONOO−),ONOOH/ONOO−的pKa为6.6,pH=8.0时的kobs高于pH=5.0,意味着以ONOO−形式存在更助于硝酸盐的光解,并促进自由基还原降解途径。以上多重因素叠加下,MPUV/HCOO−体系以去质子化形式存在的CO2·−、HCOO−和ONOO−相比于其质子化形式的HCO2·、HCOOH和HOONO,更有助于硝酸盐的降解,碱性条件下MPUV/HCOO−体系降解硝酸盐效果更佳。
-
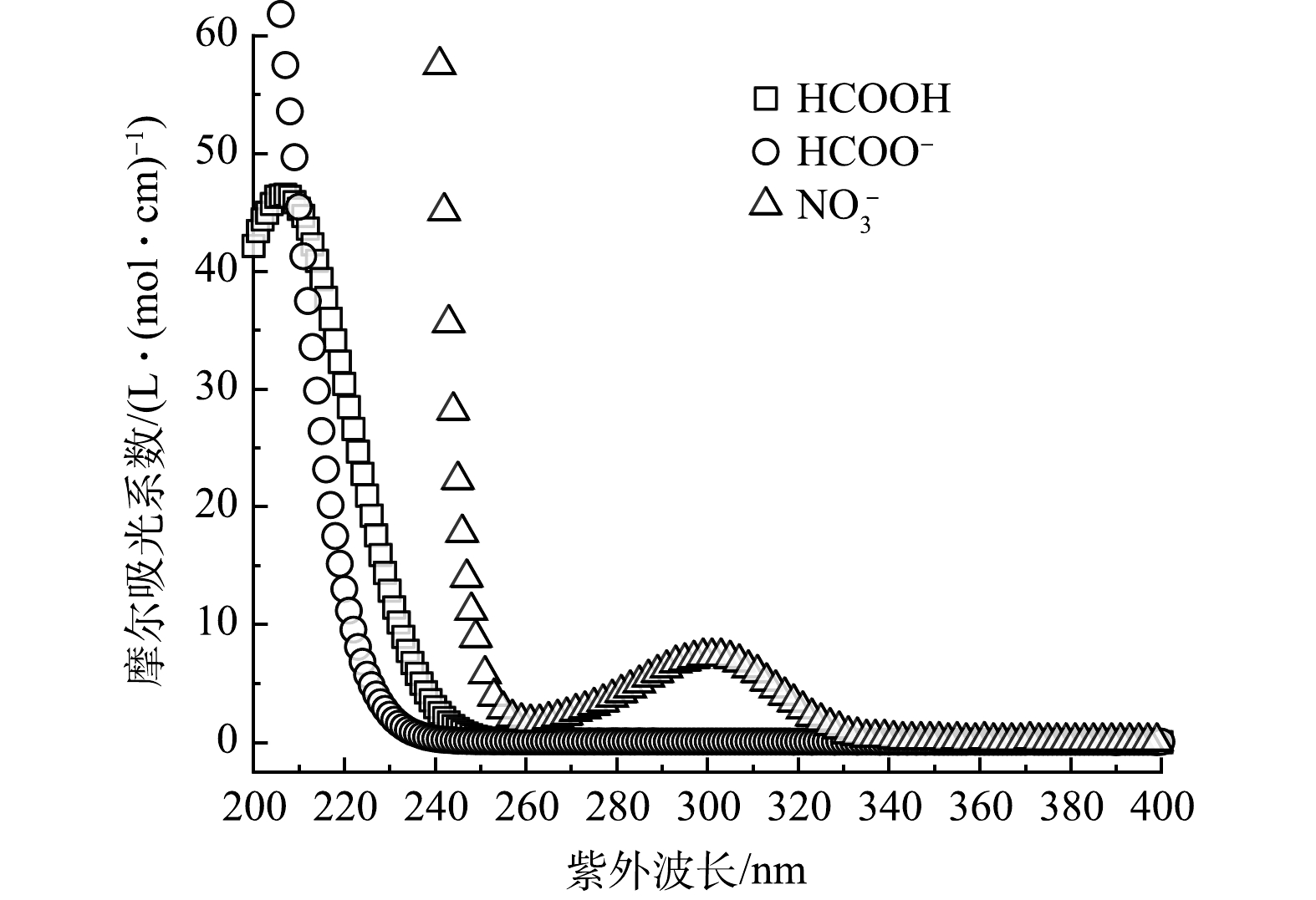
1)硝酸盐的紫外光解。图4为硝酸盐、甲酸和甲酸盐的紫外吸收光谱。可见硝酸盐在300 nm处存在特征吸收峰,而甲酸和甲酸盐不能直接光解。辐照波长(λ)>280 nm条件下,硝酸盐光解主要产生自由基(·NO2和HO·)(式(1)和式(2))。在波长<280 nm的区间内,NO3−异构化形成过氧亚硝酸盐(ONOO−)。并且,·NO2和HO·也可以重组形成过氧亚硝酸(HOONO)通过去质子化生成ONOO− [16]。过氧亚硝酸(盐)被认为是硝酸盐光还原降解过程中的重要活性中间体[17],ONOO−在300 nm处存在强吸收峰,摩尔吸光系数约为1 600 L·(mol·cm)−1。但ONOO−的反应活性极高,在pH<6.6时迅速异构化为硝酸盐(ONOOH→NO3−+H+, k=0.9 s−1),而在pH>6.6时分解为一氧化氮和超氧阴离子(ONOO−→·NO+O2·−, k=0.023 s−1)[18],导致ONOO−仅为一种瞬态反应活性中间体,可能参与了硝酸盐降解,但反应过程难以捕捉。
2)CO2·−与硝酸盐的二级反应动力学常数。基于激光闪光光解技术,建立了以MV2+为竞争参考物的CO2·−与硝酸盐二级反应速率常数(
k″CO⋅−2−NO−3 )的测定方法。266 nm激光辐射实验水样产生的CO2·−在340 nm处呈现特征吸收峰。但由于硝酸盐的单电子还原产物(NO32−)的特征吸收峰与CO2·−的吸收信号存在重叠,不能采用CO2·−衰减动力学方法直接测定。鉴于甲基紫精(MV2+)与CO2·−具有较高的反应活性(式(6))[19],以MV2+(100 μmol·L−1)为竞争参考物,采用竞争动力学法间接测定k″CO⋅−2−NO−3 。具体而言,实验溶液中被激光激发产生的CO2·−主要被硝酸盐、MV2+和H2O2反应消耗(式(6)~(8))[20]。固定实验溶液中MV2+浓度为100 μmol·L−1,改变硝酸盐浓度(0~100 mmol·L−1),测定MV·+在600 nm特征吸收峰的信号的变化,带入式(9),即可计算得到k″CO⋅−2−NO−3 。式中
:A0 和A分别表示无硝酸盐和硝酸盐浓度为10~100 mmol·L−1时MV·+在600 nm处的吸光度值;[MV2+]和[H2O2]分别表示溶液中MV2+和H2O2的浓度,mol·L−1;kS1 和kS2 分别表示CO2·−与MV2+和H2O2的二级反应速率常数,L·(mol·s)−1。图5(a)反映了在不同NO3−浓度(0~100 mmol·L−1)下,0~2 500 ns内竞争物与CO2·−反应生成的中间体自由基MV·+在600 nm的吸光信号值。可见,随着硝酸盐浓度的逐渐升高,MV·+的信号逐渐减弱,证明了一部分CO2·−被硝酸盐反应消耗,使MV·+生成速率降低。图5(b)为基于式(9)的线性拟合方程,通过拟合曲线得到斜率值
k''CO·−2−NO−3 为1.57×106 L·(mol·s)−1。另外,采用自由基衰减动力学方法测定了CO2·−与亚硝酸盐的二级反应速率常数为9.12×107 L·(mol·s)−1,表明亚硝酸盐相较于硝酸盐更容易被CO2·−还原。同时也有文献报道了CO2·−与一些活性氮中间体的化学反应及其二级反应速率常数(式(10)~(12))[21]。基于以上分析,硝酸盐向亚硝酸盐转化的过程是CO2·−诱导的硝酸盐还原的限速步骤,CO2·−的主要贡献为还原硝酸盐紫外光解过程中产生的活性氮中间体。3)还原产物及反应机理分析。进一步分析了MPUV/HCOO−体系降解硝酸盐的主要产物。由图6可见,在初始HCOO−浓度0.5 mmol·L−1时,在pH为2.5、5.0、8.0的条件下,硝酸盐减少量与亚硝酸盐增加量的比值接近1:1,并且未检测到氨氮的生成,说明MPUV/HCOO−体系主要将硝酸盐转化为亚硝酸盐。由二级反应速率常数可知,如果体系中CO2·−充足,可以实现亚硝酸盐的进一步还原。图7为MPUV/HCOO−体系促进硝酸盐的降解过程:步骤1,紫外光解硝酸盐生成含氮活性物种和羟基自由基;步骤2,羟基自由基与甲酸盐反应生成CO2·−;步骤3,CO2·−促进了活性氮中间体还原(主要过程)且其可与硝酸盐直接发生还原反应(次要过程)。因此,硝酸盐的紫外光解及CO2·−主导的高级还原过程共同促进了硝酸盐还原降解。
-
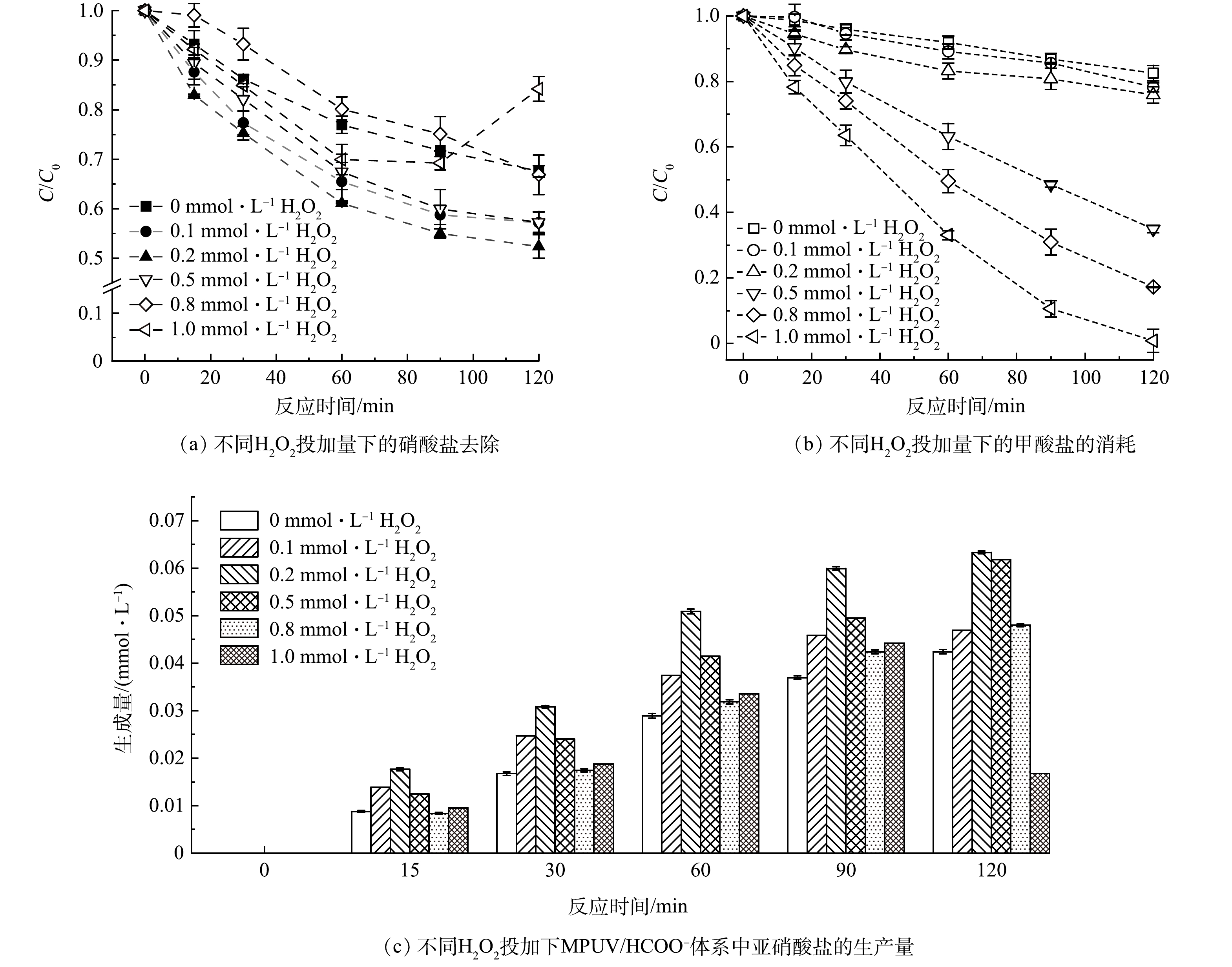
由于硝酸盐光解的HO·生成量较低,限制了CO2·−的转化生成和硝酸盐的CO2·−还原路径。因此,向MPUV/HCOO−体系中投加过氧化氢(H2O2)提升HO·生成量,从而进一步强化生成CO2·−以促进硝酸盐的脱除。如图8(a)所示,在HCOO−浓度为2 mmol·L−1、光照反应120 min后,H2O2投量为0、0.1、0.2、0.5、0.8和1 mmol·L−1时的硝酸盐去除率分别为32.5%、42.8%、47.7%、42.8%、33.2%和15.8%,在H2O2投量为0.2 mmol·L−1时硝酸盐去除率最高,而H2O2投量增大到1 mmol·L−1反而抑制了硝酸盐降解。相比不加H2O2的MPUV/HCOO−体系降解硝酸盐(k=3.6×10−3 min−1),投加0.2 mmol·L−1的H2O2使硝酸盐降解速率(k=6.2×10−3 min−1)提高了1.7倍。在H2O2投加量为0.2 mmol·L−1时,通过紫外光解H2O2促进HO·产生,进而提升了体系中CO2·−浓度水平并强化了硝酸盐还原。由图8(b)可见,甲酸盐消耗量随着H2O2投量的增大而增大,证明了在中压汞灯/甲酸盐体系中补充H2O2会促进甲酸盐向CO2·−的转化。并且,当体系中存在剩余甲酸盐时可以通过适量投加H2O2淬灭,如1 mmol·L−1 H2O2反应120 min后的甲酸盐基本全部去除。图8(c)显示了H2O2投加量为0.2 mmol·L−1时反应120 min后的硝酸盐向亚硝酸盐的转化率为63.3%且无氨氮被检出,说明36.7%的硝态氮转化为气态氮(如氧化亚氮、氮气等)排出,与CHEN等[21]的研究结果一致。而紫外光解过量的H2O2 (1.0 mmol·L−1)形成了以HO·主导的氧化体系,导致硝酸盐光解产生的活性氮中间体被重新氧化为硝态氮,使硝酸盐浓度在90~120 min反而增加(图8(a))。在本研究紫外条件下,甲酸盐和H2O2的摩尔比为10:1时,硝酸盐还原降解效果最佳。
-
1)相较于单独中压紫外(MPUV)光解,MPUV/HCOO−体系促进了硝酸盐的还原降解,CO2·−对促进硝酸盐降解具有重要的贡献。
2)在MPUV/HCOO−体系中,甲酸盐与硝酸盐摩尔浓度比在5:1~10:1条件下的硝酸盐去除效果最好。增大紫外光强和弱碱性(pH=8.0)环境能可有效促进硝酸盐的还原降解。
3)利用激光闪光光解技术量化了CO2·−与NO3−和NO2−反应的二级反应速率常数,分别为1.57×106 L·(mol·s)−1和9.12×107 L·(mol·s)−1。虽然CO2·−与硝酸盐反应活性较低,但可以促进硝酸盐光解产生的活性氮中间体的还原,中压紫外光解和CO2·−的介导还原共同促进了硝酸盐还原效能。
4)在MPUV/HCOO−体系中投加H2O2能进一步促进硝酸盐降解,投加0.2 mmol·L−1的H2O2使硝酸盐降解速率提高了1.7倍,并将36.7%的硝态氮转化为气态氮排出。
中压紫外/甲酸盐还原体系对硝酸盐的去除效能及机制
Removal efficiency and mechanism of nitrate in the medium pressure ultraviolet/formate system
-
摘要: 研究了中压紫外/甲酸盐(MPUV/ HCOO−)还原体系对硝酸盐(NO3−)的去除效能和机制。结果表明,MPUV/HCOO−体系的NO3−去除效果显著高于单独MPUV光解。当NO3−初始浓度为0.1 mmol·L−1,HCOO−投加量为1 mmol·L−1时,MPUV/HCOO−体系中NO3−的伪一级动力学降解速率(5.7×10−3 min−1)是单独MPUV(1.0×10−3 min−1)光解的5.7倍,且HCOO−的投加使得120 min后NO3−还原率由13.7%提升至44.1%。增大紫外光强和弱碱性pH环境均有利于NO3−降解。该研究认为,中压紫外直接光解NO3−以及由HCOO−转化生成的二氧化碳阴离子自由基(CO2·−)介导的还原过程被认为共同促进了NO3−的还原效率。利用激光闪光光解技术,首次定量了CO2·−与NO3−和NO2−的二级反应速率常数分别为1.57×106 L·(mol·s)−1和9.12×107 L·(mol·s)−1。MPUV/HCOO−体系将硝酸盐转化为亚硝酸盐,补充投加过氧化氢促进CO2·−的生成,CO2·−与亚硝酸盐反应进一步生成气态氮(氮气、氧化亚氮等)从水中脱除。在污水处理中,借助中压紫外消毒和生物处理系统中剩余碳源甲酸盐构建中压紫外/甲酸盐体系,有望强化污水深度脱氮,具有良好的应用前景。
-
关键词:
- 硝酸盐 /
- 深度脱氮 /
- 中压紫外 /
- 甲酸盐 /
- 二氧化碳阴离子自由基(CO2·)
Abstract: In this study, the nitrate removal efficiency and mechanism in the combination of medium pressure ultraviolet and formate (MPUV/HCOO−) system was investigated. The results showed that the nitrate removal efficiency in the MPUV/HCOO− system was significantly higher than that of MPUV photolysis alone. When the initial NO3− concentration was 0.1 mmol·L−1 and the HCOO− dosage was 1 mmol·L−1, the pseudo-first-order degradation rate of NO3− in the MPUV/HCOO− system (5.7×10−3 min−1) was 5.7 times higher than that of MPUV photolysis alone (1.0×10−3 min−1). The addition of HCOO− increased the NO3− reduction efficiency from 13.7% to 44.1% after 120 minutes. Increasing MPUV intensity and weakly alkaline pH condition were conducive to nitrate degradation. This study indicated that the reduction efficiency of NO3− was collectively enhanced by the direct photolysis of NO3− by MPUV and the reduction process mediated by carbon dioxide radical anion (CO2·−) generated from HCOO−. Using the laser flash photolysis technology, the second order rate constants of the reaction between CO2·− and nitrate or CO2·− and nitrite were first quantified as 1.57×106 L·(mol·s)−1 or 9.12×107 L·(mol·s)−1, respectively. The MPUV/HCOO− system could lead to the transformation from NO3− to NO2−, supplemental hydrogen peroxide promoted the formation of CO2·−, which could react with NO2− and produce gaseous nitrogen (such as nitrogen gas and nitrous oxide) discharged from water. In wastewater treatment, the MPUV/HCOO− system could be constructed with the medium pressure ultraviolet disinfection and the residual formate as the carbon source of biological treatment, which is expected to strengthen the deep denitrification of sewage and has a good application prospect. -
由于人类活动的影响,大量氮磷等营养元素进入水体,导致水体富营养化[1]。水体富营养化严重破坏水生生态系统,导致蓝藻爆发,鱼类大量死亡,生物多样性丧失[2]。根据2020年《中国生态环境状况公报》,我国主要江河监测的1 614个水质断面中,Ⅴ类水占1.5%,劣Ⅴ类水占0.2%。尽管我国流域水质状况总体变好,但仍需要改善。
目前,富营养水体的治理技术主要分为物理技术、化学技术和生物技术[3-4]。物理技术包括人工曝气、截污、调水冲污和底泥疏浚等措施,而这些措施存在一些缺陷,如底泥疏浚只是将污染物转移,并没有从根本上解决问题。化学技术主要是添加化学药剂和吸附剂改变水体中氧化还原电位、pH、吸附沉淀水体中悬浮物质和有机质,但成本昂贵,不具有可持续性,还可能会造成二次污染。生物技术主要是利用水生生物(植物、动物和微生物)的代谢活动去除富营养水体中氮、磷等营养元素,有利于恢复水生生态系统的健康与稳定,其具有较好的发展前景,但仍需要研究最佳环境因子[5-6]。
水体富营养化的治理是一个系统工程,单一的治理技术效果并不显著,探究综合治理水体富营养化的技术模式十分必要。邓泓等[7]利用截污清淤、调水冲污、异位生物处理、种植水生植物和放养螺、蚌和鱼类等技术对丽娃河进行了综合整治,NH3-N、TN和TP从治理前的劣Ⅴ类达到地表水Ⅴ类标准。闫飞[8]通过引清入河、水下种植沉水植物、混养鲢鳙等水生动物、安装以生物膜为基础的原位修复装置形成综合生态修复系统,使水体水质除TN外,其他指标均达到地表水Ⅲ类标准。四川省广元市王家沟的黑臭水体采用SCEU强化环保生态方法,在不清淤疏浚的条件下,水体黑臭消除,水质由劣Ⅴ类水变为地表水Ⅳ类水[9]。但在实际应用中,根据水体污染情况、水文状况和当地环境条件,选择恰当的技术进行综合匹配,才能快速恢复生态系统、提升水质,同时降低治理成本,从而实现可持续发展。
在我国,因考虑河道的防洪、排涝等问题,目前主要采用的河岸类型是硬质河岸[10]。硬质河岸阻断了河岸土壤与水体之间的物质交换和能量流动,破坏了其原有的生态群落结构,使河岸对水体的净化能力完全消失,降低了河流的水环境容量[11-13]。城市现存硬质驳岸生态化改造的方法主要分为3种:一是利用种植箱,种植攀援植物覆盖原有的硬质护岸;二是采用石笼护岸并覆盖土质护坡,再铺设草皮;三是利用保水剂、粘合剂、抗蒸腾剂、团粒剂、植物纤维、泥炭土、腐殖土、缓释复合肥等材料制成的客土覆盖到很缓的护岸上,并种植水生和湿生植物[14]。例如,英格兰威尔特郡马登河在所有垂直河边挡土墙的地基内,运用石灰岩石板以一致的角度铺设,创造出墙体建在天然岩石上的外观,种植边缘水生植物,并且在铺路砖之间播撒草种,打造整体景观环境[15]。威海某河道原设计采用浆砌石挡墙,对河岸进行生态化改造时,拆除浆砌石挡墙,改为自然原型驳岸,斜坡入水,丛状点缀不同的水生植物,并铺设天然圆石或鹅卵石[16]。目前的硬质驳岸生态改造技术存在成本较高,景观效果较为单一,对水质净化效果有限等缺点。考虑原有河道的排洪能力和护岸的不同倾斜程度,在不破坏原有硬质护岸的基础上,将生态护岸与水体直接连接起来,同时配备适宜的植被,不仅能提升河道景观效果,而且有利于提高水体自净能力。
柴桑河位于四川省眉山市仁寿县,流经天府新区眉山片区后,进入天府新区成都片区并汇入锦江。天府新区成都片区和天府新区眉山片区共同组成天府新区,是四川省的国家级新区,是成渝经济建设的重要部分[17]。天府新区的经济发展需要大量的水资源供给,但天府新区的河流总体水质较差,加之水生态流量不足,水环境容量没有扩容空间,这成为天府新区面临的环境挑战之一[18]。柴桑河的治理对天府新区的水环境状况有着重要意义。由于柴桑河两岸工业的开发以及人口的增加,流域水质逐渐变差,河流的污染问题引发了社会的高度关注。2018年,眉山天府新区投入28.9亿元,对柴桑河进行治理,打造城市公园,构建生态湿地系统,大大提升了当地的环境状况。但是,柴桑河水体水质仍然存在不达标的情况,水体浑浊且泥沙较多,水面有大量油膜覆盖,河道两岸均为浆砌石硬质河岸,无植被生长,河流生态系统破坏严重。因此,眉山天府新区管委会要求对位于天府新区眉山片区中心城区的柴桑河河段进行水质提升和环境改善。
本研究以位于四川省天府新区眉山片区中心城区的柴桑河河段为例,在不破坏原有河岸的基础上,对硬质河岸进行生态重建,改造周围景观环境,实现低成本处理;同时设计了使水质长期稳定在地表水Ⅳ类标准的综合治理技术。以恢复水生生态系统的自净能力为基础,通过各项综合措施,匹配生态系统构成所需的要素,建立强大的生态系统,实现水体长效改善。
1. 研究区域
1.1 治理河段介绍
柴桑河治理河段位于四川省天府新区眉山片区,紧邻眉山天府新区管委会和天府大道,是天府新区眉山片区的重要水环境之一(图1)。柴桑河上游流经清水镇后,流入柴桑河治理河道,然后经过天府新区眉山片区,进入视高镇区域。柴桑河治理区域介于2个拦水坝之间,河道长度约为1.5 km,水面宽为80~100 m,平均水深约为1.85 m。柴桑河两岸为浆砌石护岸,无植被生长,水体浑浊,水面漂浮有大量垃圾且有大量油膜,部分排污点发现黑灰色水体并伴有恶臭气味。
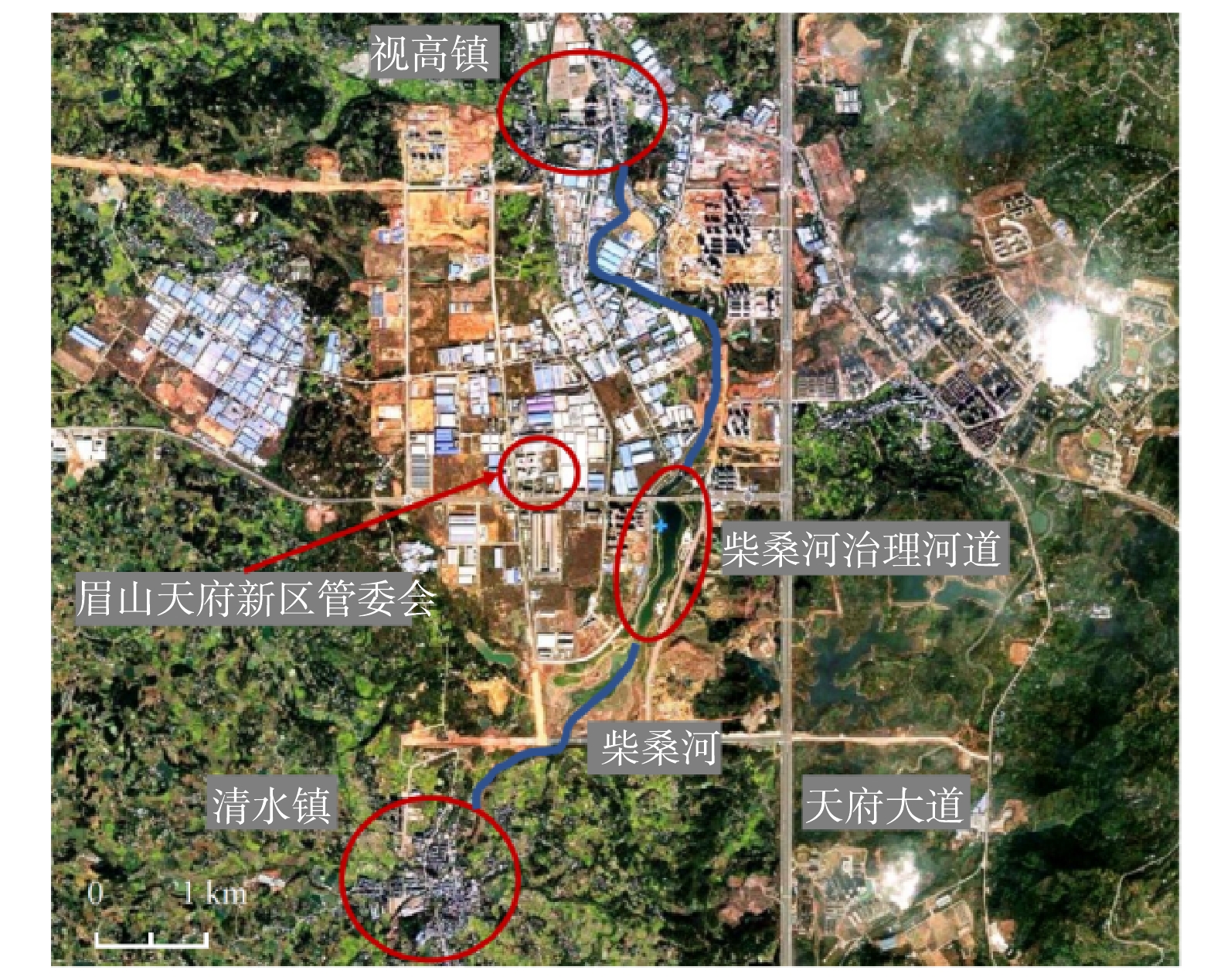 图 1 柴桑河治理河道地理位置图Figure 1. The geographical location of the treated section of the Chaisang River
图 1 柴桑河治理河道地理位置图Figure 1. The geographical location of the treated section of the Chaisang River1.2 水体污染成因分析
1)上游来水水质较差。前些年柴桑河上游清水镇有近5 万人的居民生活污水和城镇工业生产污水直接排入,致使柴桑河污染严重。
2)治理河段未有效拦截污染物。调查发现,柴桑河治理河段存在排污口,周围建设用水及工人生活用水可能直接排入河中。
3)河流两岸均为浆砌石硬质河岸,阻断了土壤与水体之间的物质交换,水体自净能力差。河段上下游均设置拦水坝,水体流速缓慢,水体长期滞留,污染物无法及时被稀释扩散。
1.3 水体检测及评价
根据当地政府提供的2020年1—4月,清水镇出境和视高镇入境的监测断面水质监测数据(表1),同时参照GB 3838-2002《地表水环境质量标准》进行评价,柴桑河水质为劣Ⅴ类水,水体为重度富营养化状态。
表 1 柴桑河水质状况(2020年1—4月)Table 1. The water quality of Chaisang River (January to April 2020)水质及标准 TP/(mg·L−1) TN/(mg·L−1) NH3-N/(mg·L−1) CODMn/(mg·L−1) GB 3838-2002 V类水标准 ≤0.4 — ≤2.0 ≤15 柴桑河水质 0.16~9.35 10.9~27.6 2.02~25.8 7.3~36 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.4 “一河一策”的治理方案
柴桑河治理河段的主要问题是浆砌石硬质河岸降低了水体自净能力,水体氮磷污染超标,两岸有排污口排入污水,两端有大坝拦截造成水流速度缓慢,不利于污染物的稀释扩散(全年平均风速为1.3 m·s−1)。浆砌石硬质河岸破坏了水体和两岸的物质交换和能量流动,因此,成本低、简单易操作且不破坏原有硬质河岸行洪能力的生态重建方式对水体恢复是至关重要的。本研究选择直接铺设护岸专用基料种植适宜植物,配备滴灌系统为植物提供水分,不仅能够在不破坏原有浆砌石硬质河岸的基础上对河岸进行生态修复,恢复水体自净能力,而且能够改善河岸景观,缓冲岸上氮磷来源对水体的影响。
治理河段在水质方面的主要问题是水体中氮磷等营养物质的浓度超标,因此,去除水体中氮磷的含量是恢复河流生态系统的关键。因除磷剂质地轻,无需外加设备,能够长期悬浮在水体中,故通过施用除磷剂,可以快速高效地去除水体中磷的含量;微生物菌剂是目前水体去除氮磷的常用方法,施用土著微生物菌剂,可以充分利用微生物的生长繁殖消耗水体中的氮磷。另外,治理河段周围为公园环境,水体治理也应考虑与周围景观相契合。设置生态浮床,利用植物生长消耗水体中氮磷,为微生物提供附着的场所,同时还可以改善水面景观环境,与当地公园景观相匹配。
针对治理河段两端设置拦水坝,当地风速小,水流速度缓慢从而导致水体污染物稀释扩散难的问题,本研究在河道中放置水车式增氧机,一方面为水体增氧,使污染物更易稀释扩散,另一方面能够扰动水体表面,使水面油膜等漂浮物沉降下来。同时,为了给滴灌系统和水车式增氧机供能,考虑采用光伏太阳能发电,使系统自运行,降低运行成本,方便后期的运营管理。
2. 材料和方法
2.1 硬质河岸生态重建
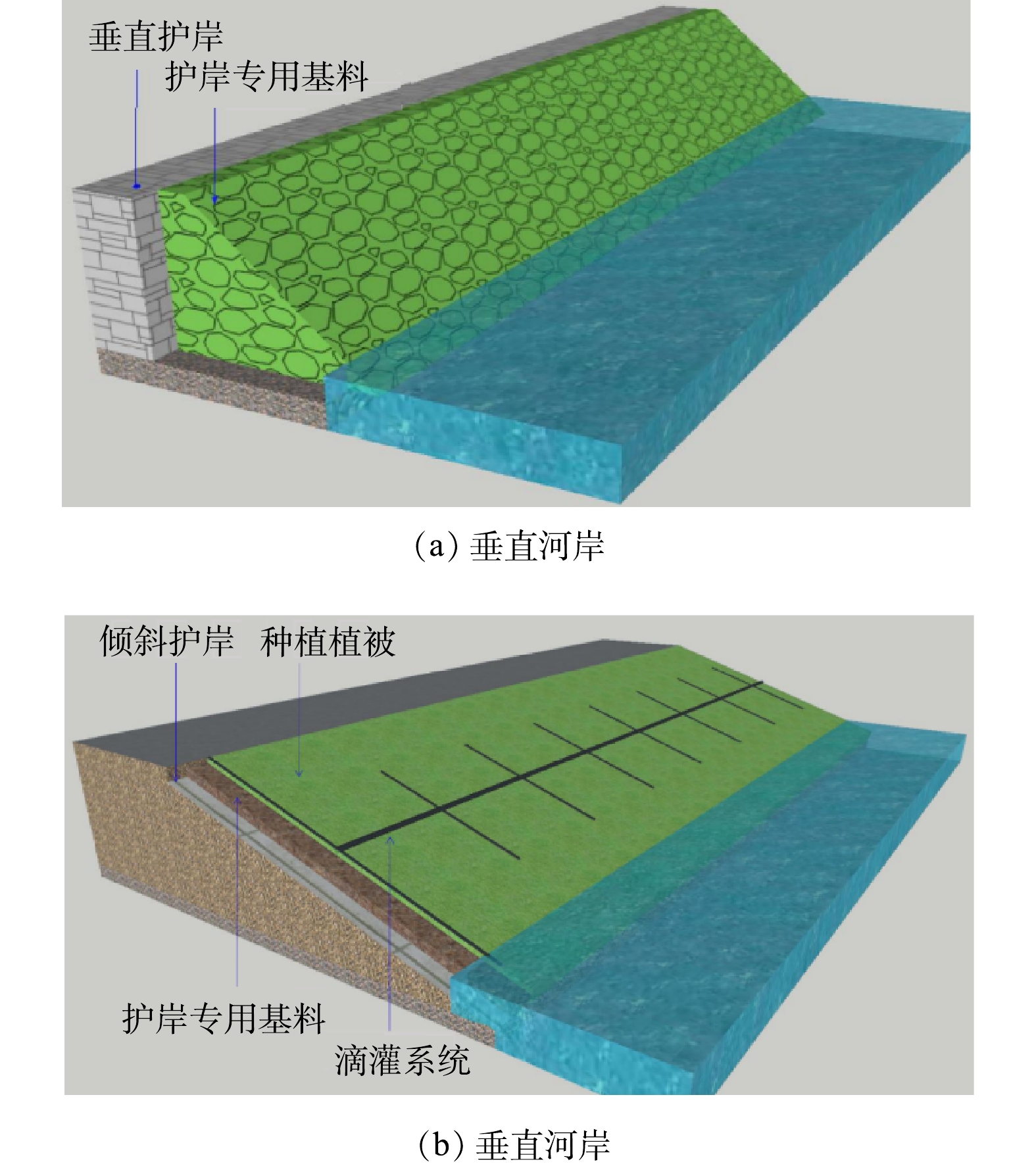
选用质量分数为30%的膨润土和70%的腐熟污泥组成的护岸专用基料进行硬质河岸的生态重建[19]。护岸专用基料养分含量丰富,阳离子交换量高,有利于生物生长,同时能有效拦截入河污染物。河道上游段河岸倾角约90°,在水上和水下部分使用装有护岸专用基料并混有植物种子的生态袋堆砌铺设为一个梯形(图2(a));下游段河岸倾角约为25°~30°,在水上和水下部分设置挡板,直接将护岸专用基料铺设在河岸表面,铺设厚度为15~30 cm,同时在铺设好的护岸基料表面播撒狗牙根、三叶草、波斯菊和硫化菊4种植物(图2(b))。为了满足河岸植被生长的水分要求,安装滴灌系统,利用河水对河岸植被进行灌溉。滴灌系统设计总长度750 m,沿河道两岸布置。水泵参数:Q为5 m3·h−1,H为30 m,配合自控系统实现自动运行,同时可根据干旱程度调节滴灌量。
2.2 富营养水体治理
1)除磷剂。除磷剂是根据《一种高效去除富营养水体中磷的方法》专利技术制备而成,具体是利用80%普通硅酸盐水泥与20%膨润土混合均匀[20]。除磷剂为粉末状,难溶于水,可直接加入水体中,加入后可均匀分散于水中并与水反应,无需任何外加设施设备的辅助。根据柴桑河治理河段的总蓄水量,按照5 g·m−3的剂量投加除磷剂,总投入1 250 kg除磷剂。
2)微生物菌剂。本项目使用的微生物菌剂是由水体中能够高效去除氮磷污染物,以硝化菌和反硝化菌为主的土著微生物分离制备而成。土著微生物能够更好地适应当地的水生生态环境,有效地发挥降解污染物的作用。按20 g·m−3的剂量计算微生物菌剂的投加量,柴桑河治理河段一次性投入5 000 kg微生物菌剂。将难溶于水、比表面积大且微生物易附着的特殊材料固定投放在水体中,形成云膜系统,为微生物提供栖息之地,增加微生物的生物量,让微生物菌剂能够长期发挥作用。根据水域面积,每1 000 m2配置1套云膜系统,另外在两岸每个排污口同时放置1套云膜系统,总共145 套。
3)生态浮床。本项目以环保PE材料为浮床框架,同时种植目前在生态浮床方面常用的紫色鸢尾、美人蕉和狐尾藻。在污染严重的入河排口处设置小型生态浮床,在中游和下游河道中间分别设置生态浮床,柴桑河河段共设置生态浮床800 m2(图3(a))。
 图 3 生态浮床、水车式增氧机和太阳能光伏系统Figure 3. Ecological floating bed, paddlewheel aerator and solar photovoltaic system
图 3 生态浮床、水车式增氧机和太阳能光伏系统Figure 3. Ecological floating bed, paddlewheel aerator and solar photovoltaic system4)水车式增氧机。柴桑河治理河段选用12个水车式增氧机(图3(a)),利用太阳能供电对水体进行曝气,每小时工作3~5 min。
2.3 太阳能光伏系统
柴桑河治理河段共安装2 套功率为12 kW的太阳能光伏发电系统,配合PLC自动控制程序,为整个柴桑河综合治理系统供电(图3(b))。太阳能光伏系统能够直接利用太阳能为水体中水车式增氧机和滴灌系统提供能源,以降低系统的运行成本。
2.4 水质监测
为了验证富营养水体的综合治理效果,对柴桑河治理河段的治理过程进行了水质监测,频度为每月1次,共进行了为期6个月的跟踪监测。分别在柴桑河治理河段上游、中游和下游设置监测点位,同时在河段中间的明显的排污口处设置1个监测点位,共4个监测点位。2020年12—2021年1月,项目施工阶段,各项综合治理措施已基本设置完毕。2021年2月受新冠疫情影响,无法监测水质状况。在2021年3—6月,连续每月对河段设置的监测点位进行水质监测。
总磷(TP)的测定参照GB 11893-1989《水质 总磷的测定 钼酸铵分光光度法》,总氮(TN)的测定参照HJ 636-2012《水质 总氮的测定 碱性过硫酸钾消解紫外分光光度法》,氨氮(NH3-N)的测定参考HJ 538-2009《水质 氨氮的测定 纳氏试剂分光光度法》,高锰酸盐指数(CODMn)的测定参照GB 11892-1989《水质 高锰酸盐指数的测定》,pH的测定参照GB 6920-1986《水质 pH 值的测定 玻璃电极法》,溶解氧(DO)的测定参考GB 11913-1989《水质溶解氧的测定 电化学探头法》。使用ZEISS Axioskop40立式显微镜对水体中的微生物进行观察。
3. 结果与讨论
3.1 水质提升效果
根据前期当地政府提供的监测数据,治理前柴桑河属于劣Ⅴ类水,治理后TP、TN、NH3-N和CODMn指标下降明显(图4)。2020年12月—2021年1月,河道中水体受项目施工影响,水质状况不稳定。2021年3—6月,水体TP和NH3-N符合地表水Ⅲ类水标准,CODMn符合地表水Ⅳ水标准并接近地表水Ⅲ类水标准,TN符合地表水湖泊、水库Ⅴ类水标。根据GB 3838-2002《地表水环境质量标准》,河流水体不考核TN,因此,项目施工完成后,柴桑河治理河段水体稳定达到地表水Ⅳ水标准。
在2020年12月—2021年6月的连续监测中,排污口间歇性的排入高氮、磷的污水,排入的污水TP最高达1.493 mg·L−1,TN最高达21.952 mg·L−1,NH3-N最高达18.651 mg·L−1(表2)。本研究使用的综合治理技术不仅处理了水体中原有的污染物,同时容纳了一定程度的污水排入,增加了河道水体的环境容量,使河道水体保持稳定。
表 2 排污口处连续监测数据Table 2. Continuous monitoring data at the sewage outlet采样日期 pH DO/(mg·L−1) CODMn/(mg·L−1) TP/(mg·L−1) TN/(mg·L−1) 2020-12 7.93 8.8 8.30 0.057 2.472 2021-01 7.90 6.8 11.80 1.493 21.952 2021-03 8.29 11.3 3.49 0.141 1.712 2021-04 8.05 8.8 6.99 1.033 9.324 2021-05 7.73 6.1 6.00 0.213 2.518 2021-06 7.83 6.1 6.05 0.236 1.860 | Show TableDownLoad:
CSV
3.2 生态景观效益
柴桑河治理前水体浑浊,表面有大量油膜覆盖,河岸两侧均为浆砌石硬质河岸,无植被覆盖,整体感官效果较差。治理后水体为浅绿色,河岸两侧全部被植被覆盖,与周围公园景观融为一体。在前期施工过程中,河岸两侧仅播种三叶草、波斯菊和硫化菊4种植物的种子。通过为期6个月的观察发现,河岸通过自然演替生长出大量的空心莲子草、鬼针草、稗草和狗尾草等其他植物,植物种类有数十种。河流经过治理后,观察发现,有大量鸟类觅食,水体中鱼类的数量大大增加,同时发现大量水草。通过光学显微镜观察发现,水体中藻类生物多样性和生物活跃性增加,发现了大量的绿藻、裸藻、甲藻和硅藻。浮游藻类与水生生态环境息息相关,能够从种类和数量上直接反应水质的变化[21-23]。柴桑河河段的藻类生物多样性和活跃性的增加一定程度上也说明了水生生态系统恢复健康和活力。
3.3 经济社会效益
底泥疏浚作为目前富营养水体治理过程中的常用方法,投入产出比及其可能对生态治理的负面影响都是值得重视的问题[24]。疏浚项目通常涉及的投资费用较大,且在消减沉积物中污染物含量的同时可能会导致污染物向水体中释放,对底栖生物造成损害,降低生物多样性,降低生态系统对外界物理干扰的抵抗能力[25-26]。生态环保且成本更低的新型技术是目前社会发展所需要的富营养水体治理技术。
柴桑河治理河段全长为1.5 km,水面宽为80~100 m,平均水深约为1.85 m,利用综合治理技术对其进行水质提升和环境改善,项目总投资约为320 万元。采用硬质河岸生态重建、微生物菌剂、除磷剂、生态浮床、水车式增氧机和太阳能光伏系统等技术综合治理柴桑河河段,匹配生态系统构成所需的要素,可在提升水质的同时打造优美的景观环境,为周围居民提供一个良好的休闲环境。与目前常用的治理技术相比,该项综合治理技术成本更低,无需外加能源,无需大量机械且能够长期维持水质等优点,符合社会发展的需求。
3.4 运营管理建议
河道治理后的运营维护对水体水质长效保持十分重要。后期运营维护可分为以下4点:第一,因柴桑河有较多排口,所以需特别注意突发的大量污水排入破坏整个河流生态系统平衡的情况,应根据情况及时补充微生物菌剂或者除磷剂;第二,雇佣专人巡视,以确保各项综合措施如水车式增氧机和滴灌系统等正常运行;第三,定期监测水质和底泥状况,同时对水体中微生物进行显微镜观察,便于了解水体和水生生物的实际状况,根据监测结果制定相应的治理控制措施;第四,若冬季水温低,应格外注意水体水质状况,根据情况补充适量除磷剂。治理后6个月内的监测频率可为1月1次,待系统稳定后可适当延长监测周期。
4. 结论
1)在浆砌石硬质河岸上铺设护岸专用基料后,播撒植物种子,配合滴灌系统为植物提供水分,对硬质河岸进行生态重建,不仅可改善1河流两岸的景观环境,而且将水体与河流两岸连接起来,增加河流生态系统的生物多样性,增强河道的自净能力。
2)施用除磷剂和微生物菌剂,安装生态浮床、水车式增氧机和太阳能光伏系统对柴桑河水体进行综合治理,能够有效地降低TP、TN、NH3-N和CODMn等指标含量,水体水质由前期劣Ⅴ类水,TP和NH3-N提升至地表水Ⅲ类水标准,CODMn提升至地表水Ⅳ水标准,TN提升至地表水Ⅴ类水标准。
3)本研究采用的综合治理技术不清淤不疏浚,成本低且操作简单,可为我国富营养水体的治理提供参考。
致 谢 感谢项目合作方四川省环境保护治理工程有限公司的大力支持。感谢成都纺织专科高等技术学校税永红教授团队水质监测的技术支持。感谢中国科学院成都山地灾害与环境研究所邱敦莲研究员对文章撰写和修改提出建议。
-
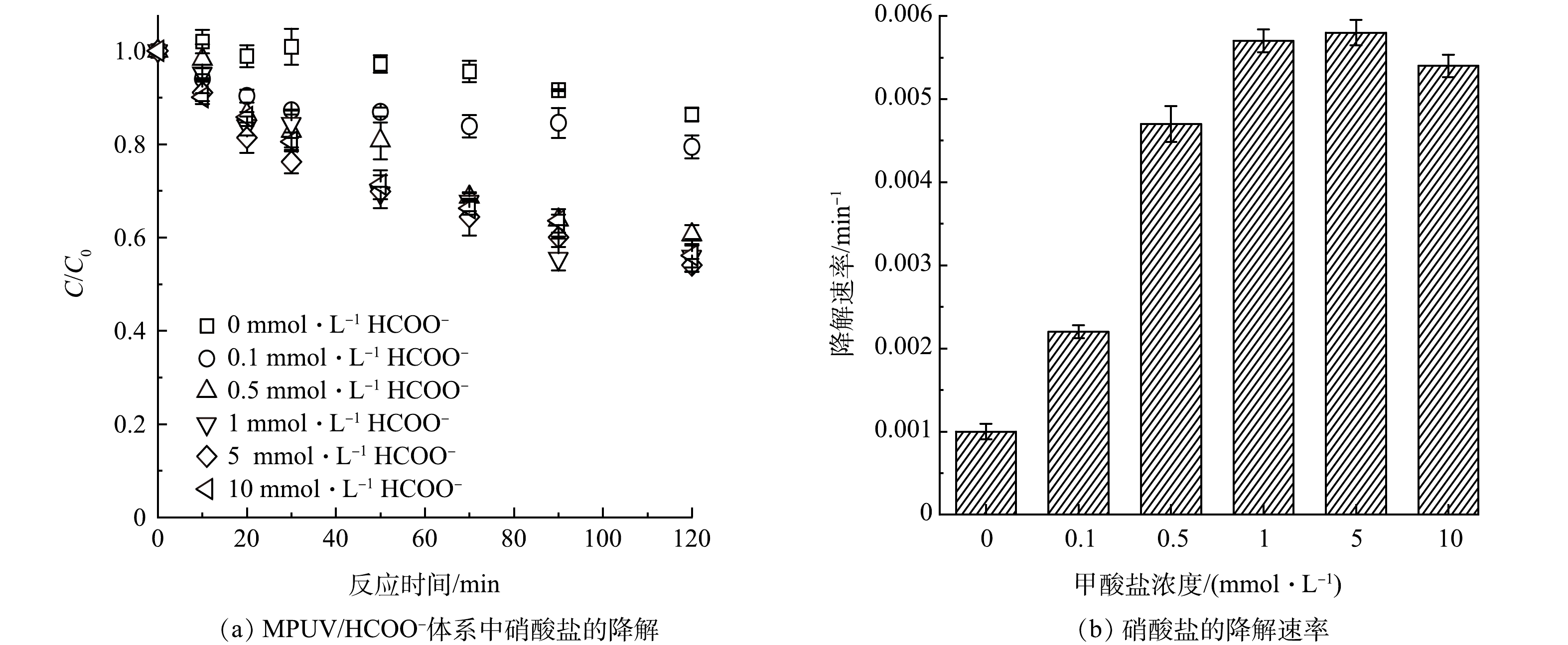
图 1 甲酸盐浓度对MPUV/HCOO−体系降解硝酸盐的影响
Figure 1. Effects of formate concentrations on nitrate degradation in MPUV/HCOO− system
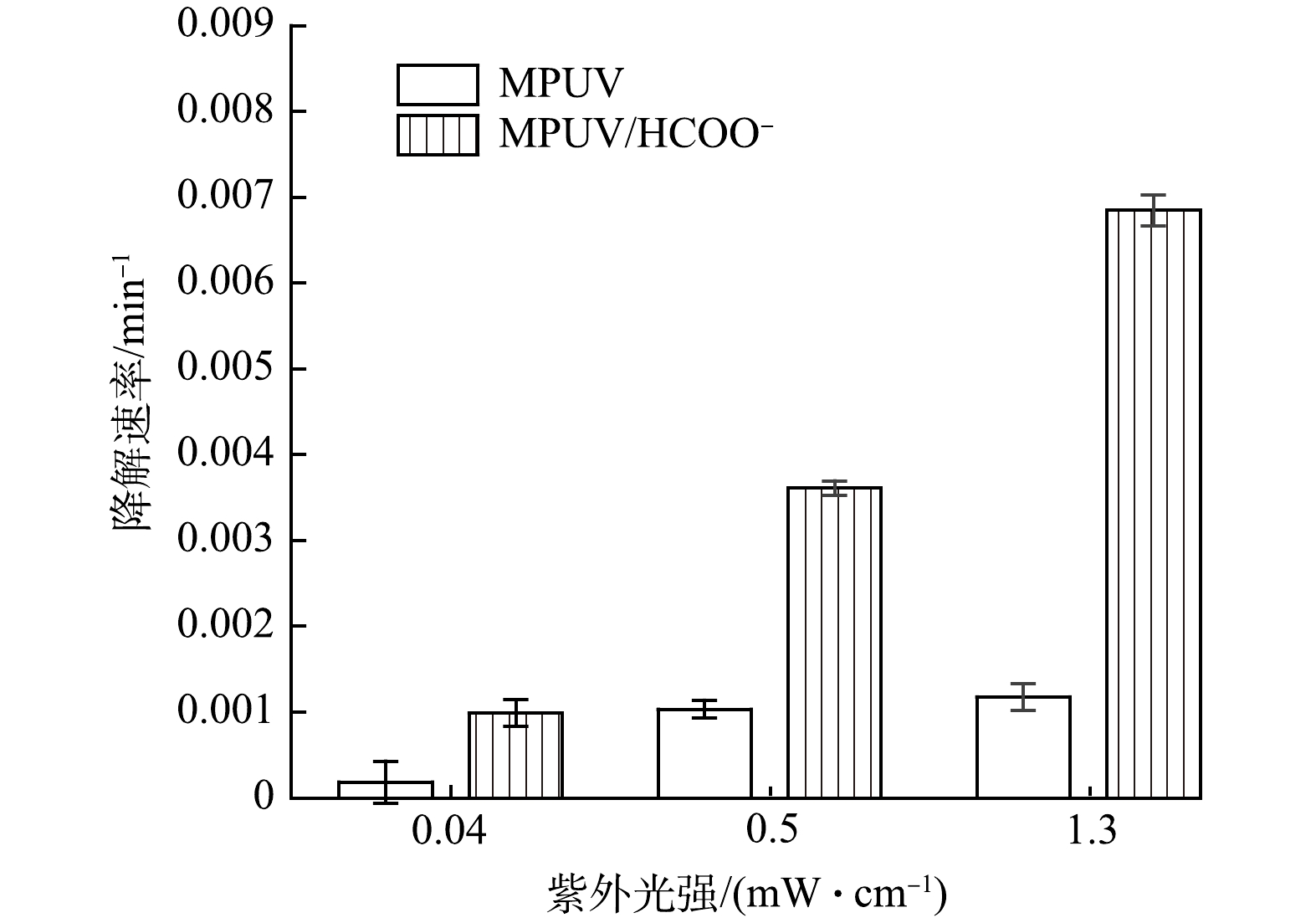
图 2 紫外光强对MPUV/HCOO−体系降解硝酸盐的影响
Figure 2. Effects of MPUV intensities on nitrate degradation in MPUV/HCOO− system

图 3 pH条件对MPUV/HCOO−体系降解硝酸盐的影响
Figure 3. Effects of pH conditions on nitrate degradation in MPUV/HCOO− system

图 5 MV2+竞争动力学法测定CO2·−与NO3−的二级反应速率常数
Figure 5. Determination of second-order reaction rate constant of CO2·− with NO3− using MV2+ competition kinetics method
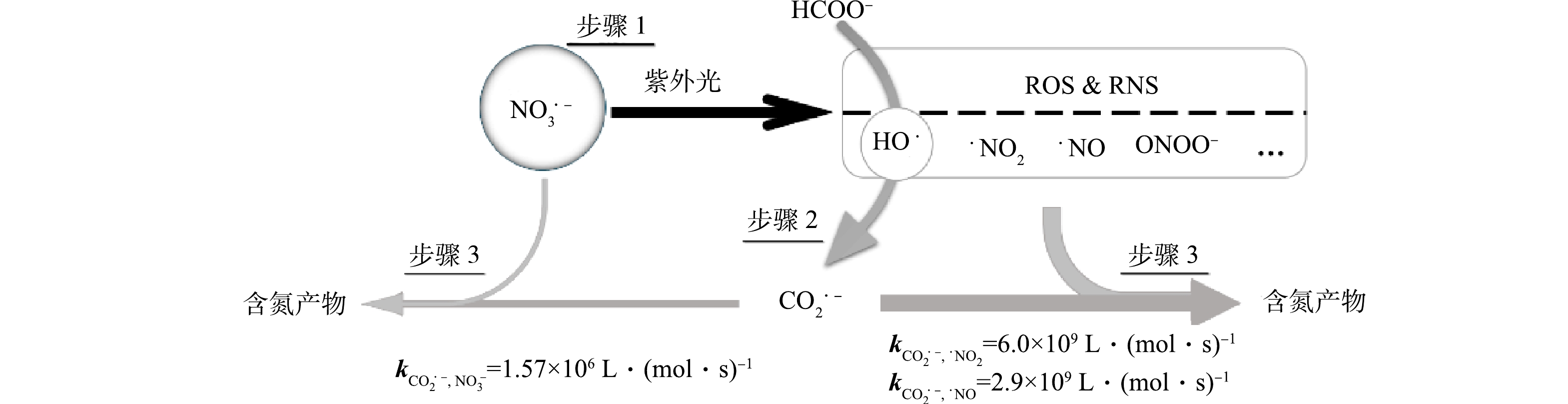
图 7 MPUV/HCOO−体系降解硝酸盐的反应机制
Figure 7. Mechanisms of nitrate degradation in the MPUV/HCOO− system
-
[1] ZHANG X, DAVIDSON E A, MAUZERALL D L, et al. Managing nitrogen for sustainable development[J]. Nature, 2015, 528(7580): 51-59. doi: 10.1038/nature15743 [2] PENNINO M J, COMPTON J E, LEIBOWITZ S G. Trends in drinking water nitrate violations across the United States[J]. Environmental Science & Technology, 2017, 51(22): 13450-13460. [3] 石岩, 郑凯凯, 邹吕熙, 等. 城镇污水处理厂总氮超标逻辑分析方法及应用[J]. 环境工程学报, 2020, 14(5): 1412-1420. doi: 10.12030/j.cjee.201811049 [4] SCHWARZ H, DODSON R. Reduction potentials of CO2 and the alcohol radicals[J]. The Journal of Physical Chemistry, 1989, 93: 409-414. doi: 10.1021/j100338a079 [5] ALSALKA Y, AL-MADANAT O, CURTI M, et al. Photocatalytic H2 evolution from oxalic acid: Effect of cocatalysts and carbon dioxide radical anion on the surface charge transfer mechanisms[J]. ACS Applied Energy Materials, 2020, 3(7): 6678-6691. doi: 10.1021/acsaem.0c00826 [6] TANG P, JIANG W, LYU S, et al. Application of glutamate to enhance carbon tetrachloride (CT) degradation by Fe(II) activated calcium peroxide in the presence of methanol: CT removal performance and mechanism[J]. Separation and Purification Technology, 2020, 236: 116259. doi: 10.1016/j.seppur.2019.116259 [7] LIU X, ZHONG J, FANG L, et al. Trichloroacetic acid reduction by an advanced reduction process based on carboxyl anion radical[J]. Chemical Engineering Journal, 2016, 303: 56-63. doi: 10.1016/j.cej.2016.05.130 [8] GU X, LU S, FU X, et al. Carbon dioxide radical anion-based UV/S2O82−/HCOOH reductive process for carbon tetrachloride degradation in aqueous solution[J]. Separation and Purification Technology, 2017, 172: 211-216. doi: 10.1016/j.seppur.2016.08.019 [9] 李炳智. 二氧化碳阴离子自由基还原降解水溶液中全氟辛烷磺酸盐研究[J]. 上海环境科学, 2019, 38(2): 47-52. [10] 秦宝雨, 唐海, 严律, 等. 紫外活化过硫酸盐/甲酸体系还原水中Cr(Ⅵ)机理及影响因素[J]. 环境工程学报, 2019, 13(9): 105-113. doi: 10.12030/j.cjee.201812139 [11] TUGAOEN H O, GARCIASEGURA S, HRISTOVSKI K, et al. Challenges in photocatalytic reduction of nitrate as a water treatment technology[J]. Science of the Total Environment, 2017, 599: 1524-1551. [12] CHEN G, HANUKOVICH S, CHEBEIR M, et al. Nitrate removal via a formate radical-induced photochemical process[J]. Environmental Science & Technology, 2019, 53(1): 316-324. [13] DONG N, ZENG Z, RUSSENBERGER M, et al. Investigating cake layer development and functional genes in formate- and acetate-driven heterotrophic denitrifying AnMBRs[J]. Chemical Engineering Journal, 2024, 485: 149623. doi: 10.1016/j.cej.2024.149623 [14] YISHAI O, LINDNER S N, GONZALEZ DE LA CRUZ J, et al. The formate bio-economy[J]. Current Opinion in Chemical Biology, 2016, 35: 1-9. doi: 10.1016/j.cbpa.2016.07.005 [15] MACK J, BOLTON J R. Photochemistry of nitrite and nitrate in aqueous solution: A review[J]. Journal of Photochemistry and Photobiology A, 1999, 128(1): 1-13. [16] MARK G, KORTH H-G, SCHUCHMANN H-P, et al. The photochemistry of aqueous nitrate ion revisited[J]. Journal of Photochemistry and Photobiology A: Chemistry, 1996, 101(2): 89-103. [17] GOLDSTEIN S, RABANI J. Mechanism of nitrite formation by nitrate photolysis in aqueous solutions: The role of peroxynitrite, nitrogen dioxide, and hydroxyl radical[J]. Journal of the American Chemical Society, 2007, 129(34): 10597-10601. doi: 10.1021/ja073609+ [18] BENEDICT K B, MCFALL A S, ANASTASIO C. Quantum yield of nitrite from the photolysis of aqueous nitrate above 300 nm[J]. Environmental Science & Technology, 2017, 51(8): 4387-4395. [19] NESHVAD G, HOFFMAN M Z. Reductive quenching of the luminescent excited state of tris (2, 2'-bipyrazine) ruthenium (2+) ion in aqueous solution[J]. The Journal of Physical Chemistry, 1989, 93(6): 2445-2452. doi: 10.1021/j100343a044 [20] BUXTON G V, GREENSTOCK C L, HELMAN W P, et al. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O− in aqueous solution)[J]. Journal of Physical and Chemical Reference Data, 1988, 17(2): 513-886. doi: 10.1063/1.555805 [21] CHEN J, LIU J, ZHOU J, et al. Reductive removal of nitrate by carbon dioxide radical with high product selectivity to form N2 in a UV/H2O2/HCOOH system[J]. Journal of Water Process Engineering, 2020, 33: 101097. doi: 10.1016/j.jwpe.2019.101097 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1637
- HTML全文浏览数: 1637
- PDF下载数: 63
- 施引文献: 0







