


















































-
甲苯是重要的化工原料,也是一种常见的VOCs污染物,广泛来源于石油化工、涂装印染等行业,对呼吸道、神经、心脑血管具有较强的毒害作用,亟需得到治理[1-3]。在众多VOCs去除技术中,催化氧化法以净化效率高、无二次污染等优点受到广泛关注[4],该技术核心为催化剂的开发。常见VOCs催化剂分为贵金属催化剂和过渡金属氧化物催化剂[5-6]。常见贵金属催化剂有Pd[7]、Pt[8]、Ru[9]等,但贵金属催化剂往往价格较高,同时在高温时易烧结,极大影响了贵金属催化剂应用效果。因此,价格低廉的过渡金属氧化物催化剂受到更多关注。彭新宇等[10]以ZSM-5为载体采用浸渍法制备了一系列Cu、Mn、Fe、Ti等过渡金属氧化物催化剂,发现负载量为5% (质量分数) 的Cu/ZSM-5表现出优异的甲苯催化活性,在空速24 000 h−1下T90为250 ℃。YUE等[11]以高锰酸钾、乙酸锰、硝酸锰、氯化锰和硫酸锰为前驱体制备了MnO2催化剂,发现以高锰酸钾制备的MnO2催化剂具有最高的活性,300 ℃即可将甲苯完全转化,这与高比表面积、高氧空位浓度和吸附氧含量有关。ZHANG等[12]通过酸处理对MnOx催化剂进行改性,结果表明低浓度酸处理可提高Mn4+浓度,通过酸浸使得Mn3+发生歧化形成空位缺陷,提高催化剂表面活性氧迁移率,进而提高催化剂活性。但过渡金属氧化物催化剂完全催化氧化VOCs温度较高,低温活性较贵金属催化剂还有较大距离,故可通过添加助剂以提高过渡金属氧化物催化剂低温活性。稀土元素Ce因其独特的4f层电子结构被越来越多用作催化剂助剂。LI等[13]通过氧化还原沉淀法制备了Ce-Mn固溶体催化剂,相较于MnO2,Ce1Mn3催化剂在空速60 000 mL·(h·g)−1,180 ℃时即可将12 321 mL·(h·g)−1的甲苯完全氧化,这主要得益于Ce1Mn3催化剂具有大量的Ce3+、Mn3+和表面吸附氧。杨玉玲等[14]采用草酸沉淀法同样合成了Ce1Mn3固溶体催化剂,结果表明Ce的加入不但增加了CeMn氧化物表面氧空位浓度,而且提高了活性氧物种在催化剂表面的流动性,Ce-Mn间的协同作用提高了催化剂的低温活性。LUO等[15]将MnO2纳米颗粒包裹在Ce-Mn固溶体球中制备Ce1Mn2催化剂,发现封装结构有利于提高催化剂比表面积及活性氧迁移率,将MnO2和CeO2间的协同作用最大化。GENG等[16]采用沉淀法制备Mn9Ce1Ox催化剂,在70 ℃时对苯酚具有88.1%的TOC转化率,这表明高Mn4+浓度有利于苯酚的吸附与氧化,Mn3+浓度高则有利于氧的活化,而Ce的引入优化了Mn4+/Mn3+的比例,从而提高了催化剂活性。因此,过渡金属与稀土元素Ce的比例是影响催化剂活性的重要因素。
本研究通过浸渍法合成了Mn-Ce/γ-Al2O3催化剂,考察不同负载量及Mn/Ce摩尔比对催化剂催化氧化甲苯活性的影响,通过XRD、BET、TEM、H2-TPR、O2-TPD和XPS表征手段考察CeO2的引入对MnO2催化剂形貌特征及催化活性的影响,以期阐明Ce对MnOx体系催化剂的作用机制。
-
按摩尔比将一定体积的0.1 mol·L−1 Ce(NO3)3溶液和Mn(NO3)2溶液溶于去离子水中;超声分散1 h,然后加入γ-Al2O3粉末;在60 °C水浴中继续超声搅拌3 h至粘稠状;之后在110 °C下干燥4 h;于550°C下焙烧3 h后,研磨至20~40目备用。
-
采用德国Bruker D8 Advance 型X射线衍射仪对催化剂样品进行结构分析。采用Cu Ka辐射源 (35 kV, 35 mA) ,扫描速度为5°·min−1, 扫描步长为0.02°,扫描范围为5°~80°。采用日本 JEM-2100 型高分辨透射电镜 (HRTEM) 在200 kV加速电压下测量催化剂样品的微观形貌特征和金属氧化物晶面分布。
采用美国Micromeritics ASAP 2050型物理吸附仪对催化剂孔道结构分析。测试前,样品先在90 ℃下预处理1 h, 再以10 ℃·min−1升温速率加热至240 ℃,并保持2 h,自然冷却至室温后进行测试。以高纯氮气为动力源,采用低温氮气物理吸附法进行测试,分别以Brunauer-Emmet-Teller(BET)法和Barrett-Joyner-Halenda(BJH)法计算催化剂样品的比表面积和孔容孔径。
采用美国美国Micromeritics Auto Chem Ⅱ 2920型化学吸附仪对催化剂进行程序升温还原 (H2-TPR) 测试。100 mg样品在空气气氛下以10 ℃·min−1升温速率加热至300 ℃,保持1后,用氦气吹扫冷却至室温,在通入10%H2/He进行催化剂还原测试,并以10 ℃·min−1升温速率由室温升温至850 ℃,TCD信号检测。氧气程序升温脱附 (O2-TPD) 通过使用相同的装置进行。样品 (0.1g) 在300 ℃下用氦气处理1 h,冷却至室温后,将20%O2/He气体通入1 h。待O2吸附饱和后,用He吹扫1 h,去除物理吸附的O2。然后将样品在He气氛下,从50 ℃加热至900 ℃,加热速率10 ℃·min−1。
采用美国Thermo Scientific Esca Lab 250XI 型X射线光电子能谱仪 (XPS) 对催化剂样品金属元素及表面氧物种价态进行分析,以Al Ka为离子源,C 1s=284.8 eV结合能进行校正,测定了催化剂样品中Mn 2p、Ce 3d、Al 2p、O1s和C1s的XPS光谱。
-
采用自制常压固定床反应器对催化剂甲苯催化活性进行测评,并假设反应器的空气泄漏率为零。将4mL催化剂置于反应器的恒温区,不进行任何预处理,热电偶置于催化剂床的顶部,反应器温度由温度控制器控制,加热速率为5 °C·min−1。在标准大气压下,使用清洁空气作为载气 (即O2体积分数为21%,N2体积分数为79%) ,通过吹扫甲苯发生器瓶产生甲苯饱和蒸汽,获得质量浓度为3 000 mg·m−3的甲苯废气,总流量为2 L min−1,空速为30 000 h−1。为消除甲苯吸附对催化剂催化效果的影响,将催化剂的床温升至120 ℃并保持1 h,以确保反应器入口和出口的甲苯质量浓度相同,然后测试活性。通过Agilent GC7890A气相色谱法测定反应器出口和入口的甲苯质量浓度。T10、T50和T90用于表示催化剂的催化效率分别为10%、50%和90%的温度。催化剂的催化效率如式 (1) 计算。
式中:η表示催化剂催化效率,%;
Cin 表示反应器入口气体甲苯质量浓度,mg·m−3;Cout 表示反应器出口气体甲苯质量浓度,mg·m−3。 -
图1为不同Mn/Ce (摩尔比) 对催化剂催化氧化甲苯活性的影响及局部放大图。图1 (a) 表明,当活性组分含量固定时,催化剂对甲苯的催化活性随Ce浓度的升高呈现先增大后降低的趋势, 当Mn/Ce为3∶2时, 即Mn0.6Ce0.4/γ-Al2O3催化剂具有最高的甲苯催化活性, 其T10、T50和T90分别为162 ℃、180 ℃和220 ℃, 较MnO2/γ-Al2O3分别降低20 ℃、30 ℃和40 ℃。图1 (b) 表明,添加Ce可显著提高MnO2/γ-Al2O3催化剂的低温活性, 在180 ℃时其活性大小为Mn0.4Ce0.6(38%) > Mn 0.6Ce0.4(35%) > Mn 0.8Ce0.2(15%) > Mn 0.2Ce0.8(13%) > MnO 2(0.9%), Mn0.4Ce0.6与Mn0.6Ce0.4的低温活性相差不大, 但在200 ℃后, Mn0.6Ce0.4的催化活性最高, 在240 ℃即可将甲苯完全催化氧化。这表明Ce的添加可显著提高MnO2对甲苯的催化活性。本研究所制备的Mn0.6Ce0.4/γ-Al2O3优催化剂与其他Mn-Ce催化剂性能对比如表1所示。
-
图2为Mn0.6Ce0.4/γ-Al2O3催化剂不同活性组分含量对催化剂催化氧化甲苯活性的影响。催化活性随着活性组分浓度升高呈现先增大后降低的趋势, 20%为最优负载含量。由于过渡金属氧化物催化活性较贵金属较差, 故活性组分含量较少时, 催化剂表面的活性位点不足以将污染物分子快速反应, 而活性组分含量过高时, 则可能造成催化剂表面活性位点覆盖, 并造成催化剂孔道堵塞, 影响污染物分子在催化剂表面的吸附, 进而影响了催化活性[17-18]。
-
为探究稀土元素Ce的引入与Mn基化剂催化氧化甲苯性能提高的原因, 对MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3催化剂进行了详细表征, 包括晶相结构 (XRD) 、孔结构 (BET) 、颗粒形貌 (TEM) 、还原性能 (H2-TPR) 及表面元素组成、化学态及表面氧物种 (XPS、O2-TPD) 的分析。
-
MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3催化剂的XRD图谱如图3所示。2个样品在20.4°, 32.2°, 37.1°, 39.8°, 45.7°和66.9°观察到的衍射峰分别对应于具有立方尖晶石结构的γ-Al2O3 (JCPDS PDF#10-0425) 。在32.9°, 38.2°, 55.1°和65.7°观察到Mn2O3的衍射峰 (PDF#71-0635) , 在28.7°, 56.6°和72.2°观察到MnO2的衍射峰 (PDF#82-2169) ,表明催化剂同时存在Mn3+和Mn4+, 有利于化学吸附氧的形成,促进甲苯催化[19-20]。对于Mn0.6Ce0.4/γ-Al2O3催化剂, 在27.3°, 34.4°, 51.6°, 57.3°和59.1°处观察到衍射峰归属于具有立方萤石结构的CeO2 (JCPDS PDF#44-1001) ,CeO2具有较强的储放氧能力,与Mn3+共同作用可促进甲苯氧化还原反应进程[21]。
图4 (a) 表明, Mn-Ce催化剂均表现出IV型吸脱附曲线和H3型滞后环, 并且在相对压力为0.6~0.7的 (P/P0) 处闭合, 为介孔材料[22-23], 其比表面积、孔容和孔径分别在140~161 m2·g−1、0.49~0.61 cm3·g−1和10.5~10.9 nm (表2) 。图4 (b) 表明, Mn-Ce催化剂的孔径主要分布在50 nm以下, 处于介孔范围, 以6~10nm的孔为主, 这表明Ce的添加会在一定程度上降低催化剂的比表面积和, 但孔径变化较小[24-25]。
图5为MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3催化剂的TEM图。添加Ce后, MnOx颗粒粒径有所降低, 在Mn0.6Ce0.4/γ-Al2O3催化剂的HRTEM图 (图5 (f) ) 上出MnO2 (110) 晶面外, 还观察到Mn2O3的 (222) 晶面与CeO2的 (004) 晶面, 其晶格间距分别为0.40 nm、0.27 nm和0.26 nm, 与XRD测试结果相符。这表明Mn和Ce元素成功负载到γ-Al2O3载体上, 且负载Ce后, MnOx暴露出更多的Mn3+。 Mn3+的存在促进化学吸附氧的形成,进而促进Mn3+向Mn4+的转化,提高催化剂对甲苯的催化氧化性能[26-27]。
-
图6为MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3催化剂的H2-TPR曲线图。图6 (a) 表明出, MnO2/γ-Al2O3催化剂在340 ℃, 403 ℃和450 ℃处出现3个还原峰, 分别对应Mn4+向Mn3+[28]、Mn3+向Mn2+[6]和Mn2+向Mn0[29]的还原及表面化学吸附氧的移除。Mn0.6Ce0.4/γ-Al2O3催化剂在296 ℃和365 ℃处出现一个强且宽的峰, 还原峰向低温区偏移近50 ℃。这表明Ce与Mn之间可能存在较强的金属间相互作用[30-31],得益于CeO2的存在为催化剂表面引入了更多的氧空位,有利于化学吸附氧的形成,并增强了催化剂表面氧的析出,进而促进Mn3+向Mn4+的转化, 加强了催化剂催化氧化甲苯性能[32-33]。为了更好地比较样品的低温还原性,计算了初始H2消耗速率[34-35], 结果如图6 (b) 所示。与不含CeO2的催化剂相比, Mn-Ce催化剂具有更强的低温还原性。将具有萤石结构的CeO2负载到MnO2/γ-Al2O3催化剂体系中, 不仅由于Ce3+的存在而产生了许多表面氧空位,而且还诱导了MnO2和CeO2之间的相互作用[36], 使得与MnO2相邻的Ce-O键更容易断裂[37],激活了CeO2的表面氧和晶格氧,促进了氧空位的形成,并增强了表面氧的吸附[38]。此外,催化剂的表面反应速率和催化活性也得到了提高。
采用O2-TPD技术对Mn-Ce催化剂进行表面氧物种分析, 结果如图7(a)所示。2种催化剂在100 ℃左右出现均出现1个强峰, 这主要是物理吸附氧的脱附造成的[39]。100 ℃后, MnO2的脱附峰强度缓慢下降, 至302 ℃和377 ℃时出现2个较弱氧脱附峰;而Mn0.6Ce0.4的脱附氧在200 ℃后开始增高, 并在279 ℃出现氧的脱附峰。这表明Ce的引入提高了催化剂表面化学吸附氧的含量[40-41], 且Mn0.6Ce0.4的氧脱附量 (35.62 mmol·g−1) 大于MnO2的氧脱附量 (27.93 mmol·g−1) , 如表3所示。这表明Ce的引入提高了催化剂表面化学吸附氧含量,促进了氧的吸附与析出。由于VOCs催化反应多发生于500 ℃之前, 因此对O2-TPD曲线进行峰峰拟合分析, 如图7 (b) 所示。在200~300 ℃内Mn0.6Ce0.4有3个拟合峰, 而MnO2只有1个相对较弱的拟合峰, 再一次证明了Mn0.6Ce0.4催化剂由于表面具有更多的化学吸附氧, 且活性氧在较低温度下即可与析出参与催化反应,甲苯催化活性远高于MnO2催化剂,这与实验结果相吻合。
-
图8为MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3两种催化剂的Mn 2p、O 1s和Ce 3d的XPS光谱。图8 (a) 为Mn 2p的 XPS测试谱图, 结合能位于644.9 eV和655.4 ev归属于Mn4+物种, 结合能位于642.1 eV和653.5 eV归属于Mn3+物种。这表明催化剂表面存在多种氧化态的MnOx物种, 在添加Ce元素后, Mn 2p的结合能发生明显的正向偏移。这是由于CeO2具有较强的储氧能力,因此会吸引与之相邻的Mn2O3中的晶格氧,造成Mn3+失去带负电的氧原子造成Mn 2p结合能正向偏移。这表明催化剂更容易释放电子, 不但有利于表面氧物种的吸附与释放,更能促进甲苯分子氧化[17, 42]。如表2所示, MnO2/γ-Al2O3和Mn0.6Ce0.4Ce/γ-Al2O3催化剂表面Mn4+/Mn3+摩尔比分别为0.49和0.39。这表明催化剂表面形成更多Mn3+物种, 有利于催化剂表面氧空穴的形成[43-44], 这与其他表征结果相吻合。如图8 (b) 所示, 在530.6 eV处的特征峰属于表面晶格氧 (Olatt) , 另一个在531.4 eV处的特征峰则可归因于表面吸附氧种类 (Oads) 的贡献[12,45-46]。MnO2/γ-Al2O3和Mn0.6Ce0.4Ce/γ-Al2O3催化剂表面的Oads/Olatt (摩尔比) 分别为0.55和1.74 (如表2所示) 。这表明负载Ce后催化剂表面产生更多的吸附氧, 有利于活性氧物种 (O2−, O22−或O−等) 从CeO2附近向MnOx活性位点附近转移, 提高催化剂活性[27, 47],这从H2-TPR结果已得到印证。催化剂Mn0.6Ce0.4/γ-Al2O3的Ce 3d轨道 XPS测试光谱如图8 (c) 所示。前4个分别位于882.5 eV (U1) , 885.9 eV (U2) , 889.1 eV (U3) 和898.4 eV (U4) 的峰可归属于Ce 3d5/2轨道特征峰, 而后4个位于901.1 eV (V1) , 903.7 eV (V2) , 907.2 eV (V3) 和916.9 eV (V4) 的峰则可归属于Ce 3d3/2轨道特征峰[48-50]。此外, Ce 3d光谱中885.9 eV (U2) 和901.1 eV (V1) 处特征峰归因于Ce3+物种的贡献, 其他特征峰则归因于Ce4+物种的贡献[8, 51],其表面Ce4+/Ce3+ (摩尔比) 为2.32。正是由于Ce3+和Ce4+同时存在,提高了催化剂表面吸附氧及氧空位含量,强化了催化剂催化活性,从而提高了甲苯转化率。
-
1) 通过浸渍在MnO2/γ-Al2O3催化剂上负载CeO2可有效地提高催化剂对甲苯的催化活性。特别是在低温区域,与单一金属氧化物催化剂MnO2/γ-Al2O3相比,双金属氧化物催化剂Mn0.6Ce0.4/γ-Al2O3的T10和T90分别降低20 ℃和40 ℃。2) CeO2添加到MnO2/γ-Al2O3后,催化剂孔道结构未发生显著变化,保持介孔结构不变,而催化剂表面存在的Ce3+,增加了催化剂表面活性氧物种和氧空位数量,在Ce3+向Ce4+的转化过程中,为O2和MnO2提供了电子。这不仅加速了活性氧物种的形成,而且加速了Mn4+向Mn3+的转化,促进了甲苯分子的吸附与活化反应,最终提高了甲苯催化氧化反应活性。
Mn-Ce复合金属氧化物催化氧化甲苯性能
Catalytic oxidation of toluene over Mn-Ce composite metal oxide catalyst
-
摘要: 通过等体积浸渍法制备了一系列Mn-Ce/γ-Al2O3催化剂, 并考察不同CeO2负载量对MnO2/γ-Al2O3催化剂催化氧化甲苯性能的影响。利用XRD、N2吸脱附曲线、TEM、H2-TPR、XPS和O2-TPD等方法表征催化剂比表面积、表面形貌及氧化还原性能。结果表明,CeO2的负载一定程度上降低了MnO2/γ-Al2O3催化剂的比表面积, 且催化剂仍保持介孔结构。CeO2的存在增加了催化剂表面的化学吸附氧含量,其良好的储放氧能力促进了Mn3+向Mn4+的转化;Mn和Ce之间存在较强的协同作用, 与MnOx相邻的CeO2更容易打开Ce—O键释放活性氧, 加速氧化还原进程,Mn0.6Ce0.4/γ-Al2O3催化剂T10和T90与MnO2/γ-Al2O3催化剂相比分别降低20和40 ℃。本研究可为VOCs催化氧化技术中低成本金属催化剂的开发提供参考。Abstract: A series of Mn-Ce/γ-Al2O3 catalysts were prepared by the method of equal volume impregnation, and the effect of different CeO2 loading on the catalytic oxidation performance of MnO2/γ-Al2O3 catalysts was investigated. The specific surface area, surface morphology and oxidation-reduction performance of the catalysts were characterized by XRD, N2 adsorption-desorption curve, TEM, H2-TPR, XPS and O2-TPD. The results showed that CeO2 loading reduced the specific surface area of MnO2/γ-Al2O3 catalysts to some extent, and the catalysts still maintained mesoporous structure. The presence of CeO2 increased the chemical adsorption oxygen content on the surface of the catalyst, and its good oxygen storage capacity promoted the transformation of Mn3+ to Mn4+. There was a strong synergistic effect between Mn and Ce, and the CeO2 adjacent to MnOx was more likely to break the Ce-O bond to release active oxygen and accelerate the oxidation-reduction process. Compared with MnO2/γ-Al2O3 catalyst, T10 and T90 of Mn0.6Ce0.4/γ-Al2O3 catalyst reduced 20 and 40 ℃, respectively.
-
Key words:
- catalytic oxidation /
- MnO2 /
- CeO2 /
- toluene /
- synergistic effect
-
酸雨是全球重要环境问题之一,对生态环境和经济发展造成很大不利影响[1]。研究指出,除无机酸外,有机酸对雨水酸化的贡献不可忽视[2-3]。我国东南部地区降水研究发现,有机酸对降水酸度贡献为5%—66.3%[4-5];在非工业区、远离人类活动区,降水有机酸对降水酸度的贡献甚至高达80%—90%[6]。降水中有机酸多为低分子有机酸,主要包括甲酸、乙酸、丙酸、草酸等[7-8],其中甲、乙酸是最重要的两种有机酸,约占有机酸总量的75%;草酸是重要的二元有机酸[9],含量不高,但对降水酸化具有重要影响[10-11]。
近年来,研究者们围绕区域降水有机酸的分布与来源开展相关研究并取得一定成果。徐刚等[12]研究贵阳地区降水有机酸的季节变化为:春>冬>秋>夏;章炎麟等[13]研究指出,安顺市降水有机酸浓度的季节变化特征为:冬>春>夏>秋。降水有机酸被认为直接来源于植被释放和人类活动(生物质燃烧、机动车尾气和烹饪油烟等),或间接来源于气态前体物的光化学反应过程[7]。目前对有机酸的来源分析主要采用比值法或统计分析法[14]。龙晓娟等[15]应用降水中甲酸/乙酸(F/A)比值分析鼎湖山降水有机酸主要来自植物排放,以自然源为主,而秋冬表现出一定人为污染的影响。李一兰等[16]采用F/A比值法分析得到天津市降水有机酸主要来源于植物、蚂蚁的直接源排放和不饱和烃、醛类的间接转化。Sun 等[10]采用F/A比值法得到庐山降水中有机酸以直接来源为主。何晓欢[17]采用因子分析法解析淮安和郴州降水来源,发现区域降水中甲酸来源与酸性气溶胶有关。龙晓娟[15]采用因子分析法发现鼎湖山降水中甲、乙酸以植物排放源为主。
当前,降水有机酸的相关研究多是针对整场降雨中有机酸的分布研究,而鲜有对同场降水事件的不同段降水样品中有机酸的分布研究;其次,关于降水样品中有机酸的来源解析,现有研究尚未定量分析云下冲刷过程和云水对降水样品中有机酸的贡献;此外,在不同的区域,降水有机酸的分布受局地或区域源的影响,因此对不同区域降水有机酸的分布与来源进行解析追踪十分有必要。
南昌是长江中下游中心城市之一,属亚热带湿润季风气候区,气候湿润温和,降水量大,太阳辐射强烈,大气光化学反应活跃,酸雨频率高。本研究针对南昌前湖区域的降水,对同一降水事件不同时段的降水样品分段采集,研究分段降水样品中低分子有机酸的浓度分布;基于分段降水样品中有机酸的分布,计算云下冲刷大气污染物和云水对降水样品中有机酸的贡献度;利用有机酸与其他水溶性离子的相关性分析,结合不同有机酸根的比值分析,解析降水有机酸的来源。本研究有助于丰富降水有机酸的分布与来源的研究方法和成果,对于区域大气污染成因分析和污染控制具有重要意义。
1. 实验部分(Experimental section)
1.1 采样点
采样点位于南昌大学前湖校区环境楼的楼顶平台(离地面高约25 m,E115°47′33″、N28°39′47″),附近无典型工业污染源,500 m范围内无高于采样点的自然或人工物体,是一个混合受体点,受道路交通源、生活排放源、建筑扬尘和城市其他源的混合影响。
1.2 降水样品的采集
2020年5—9月,利用APS-3A降水采样器(长沙湘兰科学仪器有限公司)采集了20个降水事件的88个分段降水样品。图1是APS-3A降水采集器示意图。同一降水事件的分段降水样品根据预设条件(每4 mm降水量为一段)通过仪器自动采集。同一降水事件中,分段降水样品的最大数量为8,降水量0—4 mm的降水收集为第1段降水样品,降水量4—8 mm的降水为第2段样品,依次类推第3—7段降水样品。若整场降水的降水量大于28 mm,则所有大于28 mm以上的降水为第8段降水样品。仪器内配套有8个1000 mL的聚乙烯瓶为降水样品收集瓶。聚乙烯瓶放置于仪器自带的冰箱内。每场降水事件后,从仪器内置的冰箱中取出装有降水样品的聚乙烯瓶,带回实验室,将每段降水样品从聚乙烯瓶中倒出,分别转置到实验室干净的储存瓶,冰箱保存至待测。腾空的聚乙烯瓶用去离子水洗净后放回降水采样器,复位等待下一个降水事件。
1.3 降水中有机酸等组分的测定
1.3.1 测定方法
降水中3种低分子有机酸(甲酸HCOO−、乙酸CH3COO−、草酸
C2O2−4 NO−2 NO−3 SO2−4 表 1 淋洗液梯度淋洗程序Table 1. Gradient eluting procedure of eluent时间/min Time 流速/(mL·min−1) Velocity of flow 淋洗液NaOH浓度/(mmol·L−1) Eluent concentration of NaOH 0 1 2 0—1 1 2 1—35 1 13 35—45 1 35 45—45.1 1 12 45.1—48 1 2 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.3.2 质量控制
样品测定前,对5个浓度梯度的有机酸标准溶液进行测定,做出标准工作曲线;每次更换淋洗液后,重新测标样,做标准曲线。各组分标准曲线的线性相关系数(r)大于0.999,再进行样品的测定。样品检测前进行灌注、走基线、测量纯水、检查仪器压力等操作,确保仪器运行正常。已知浓度样品的回标检测,误差小于5%。
2. 结果与讨论(Results and discussion)
2.1 降水中有机酸与无机阴离子浓度的对比
本研究20个降水事件的88个分段降水样品中有机酸和无机酸根组分的测定结果见表2。本课题组前期研究[18]发现南昌前湖区域降水的pH值小于6,为酸性降雨;龚娴等[19]对南昌地区降水pH值的研究也表明区域降水pH值小于6。由表2,采样期间南昌前湖区域降水中有机酸和无机酸的浓度差别较大,南昌各月份有机酸浓度的差异大,可能与各月的降雨量、降雨强度和气温的差异有关[20-21]。3种有机酸的总平均浓度为3.12 μmol·L−1,占所测有机酸、无机阴离子总量的16%,其中,甲酸、乙酸、草酸的平均浓度分别为3.81、4.56、1.13 μmol·L−1;4种无机阴离子总平均浓度为10.84 μmol·L−1,占所测阴离子含量的84%,其中,Cl−、
NO−2 NO−3 SO2−4 表 2 本研究降水中有机酸和无机阴离子的月均浓度水平(μmol·L−1)Table 2. Monthly average concentrations of organic acids and inorganic anions in precipitation in this study(μmol·L−1)项目Project HCOO− CH3COO− C2O2−4 Cl- NO2- NO−3 SO2−4 5月 2.85 2.02 1.17 8.32 2.39 30.68 33.59 6月 2.13 6.16 0.88 6.07 2.45 9.48 17.41 7月 3.56 6.07 1.51 5.34 3.18 24.53 1.42 8月 3.26 6.94 1.09 6.53 3.04 42.26 1.02 9月 7.27 1.62 1.01 2.62 1.87 13.57 0.95 平均 3.81 4.56 1.13 5.78 2.58 24.10 10.88 | Show TableDownLoad:
CSV
图2为本研究降水中3种有机酸和4种无机酸含量的月变化情况。根据图2,5—9月间,降水中有机酸含量与无机酸含量的月变化有相反的趋势。5—6月降水有机酸浓度先上升,到7—8月维持较高浓度,9月浓度下降,分析原因:南昌地区在5月开始增温,极高温度一般出现在每年的 7、8 月,7—8月气温高、辐射强,大气光化学反应有助于二次有机物的生成,间接导致7—8月降水中有机酸浓度偏高;另外,植物在6—8月份处于生长旺盛期,直接释放有机酸较多,经雨水冲刷进入降水样品中[24],使降水有机酸在温度较高的月份出现浓度的高值,与深圳[4]等、鼎湖山[15]等地的研究结论相似。5—7月降水无机酸浓度先下降,8月上升,9月下降,分析原因:7月气温高,大气颗粒物中硝酸盐不稳定易分解,间接导致降水中硝酸盐浓度低。
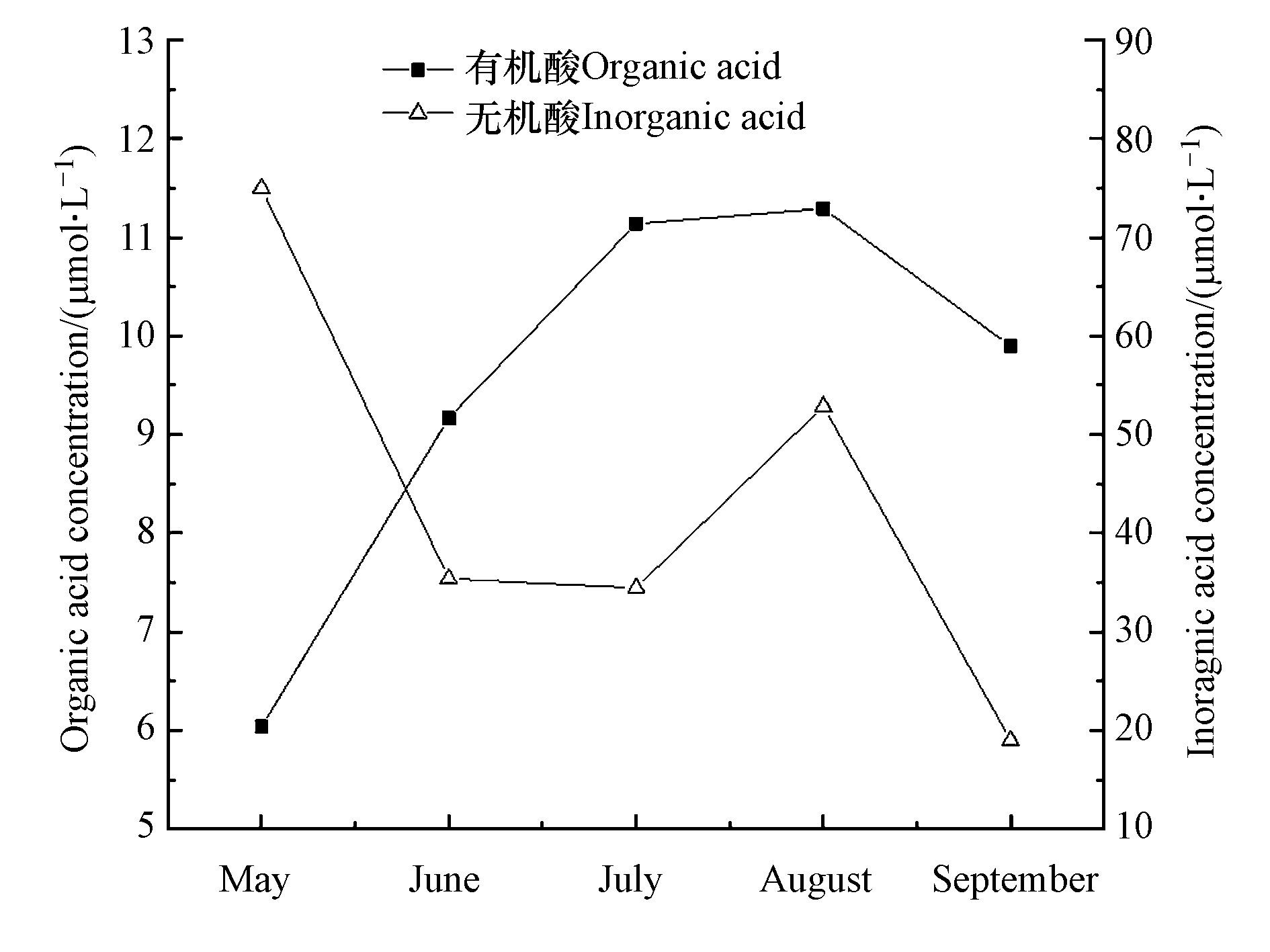 图 2 采样期间降水有机酸、无机酸含量的月变化Figure 2. Monthly variation of organic and inorganic acid concentrations in precipitation during sampling period
图 2 采样期间降水有机酸、无机酸含量的月变化Figure 2. Monthly variation of organic and inorganic acid concentrations in precipitation during sampling period2.2 分段降水中有机酸在降水进程中的分布特征
从20个降水事件中选择降水量大于24 mm(分段降水样品超过6段)的长降水事件,对长降水事件的分段降水样品中3种低分子有机酸随降水进程的变化进行分析。图3为长降水事件的分段降水有机酸随降水进程的浓度分布图。
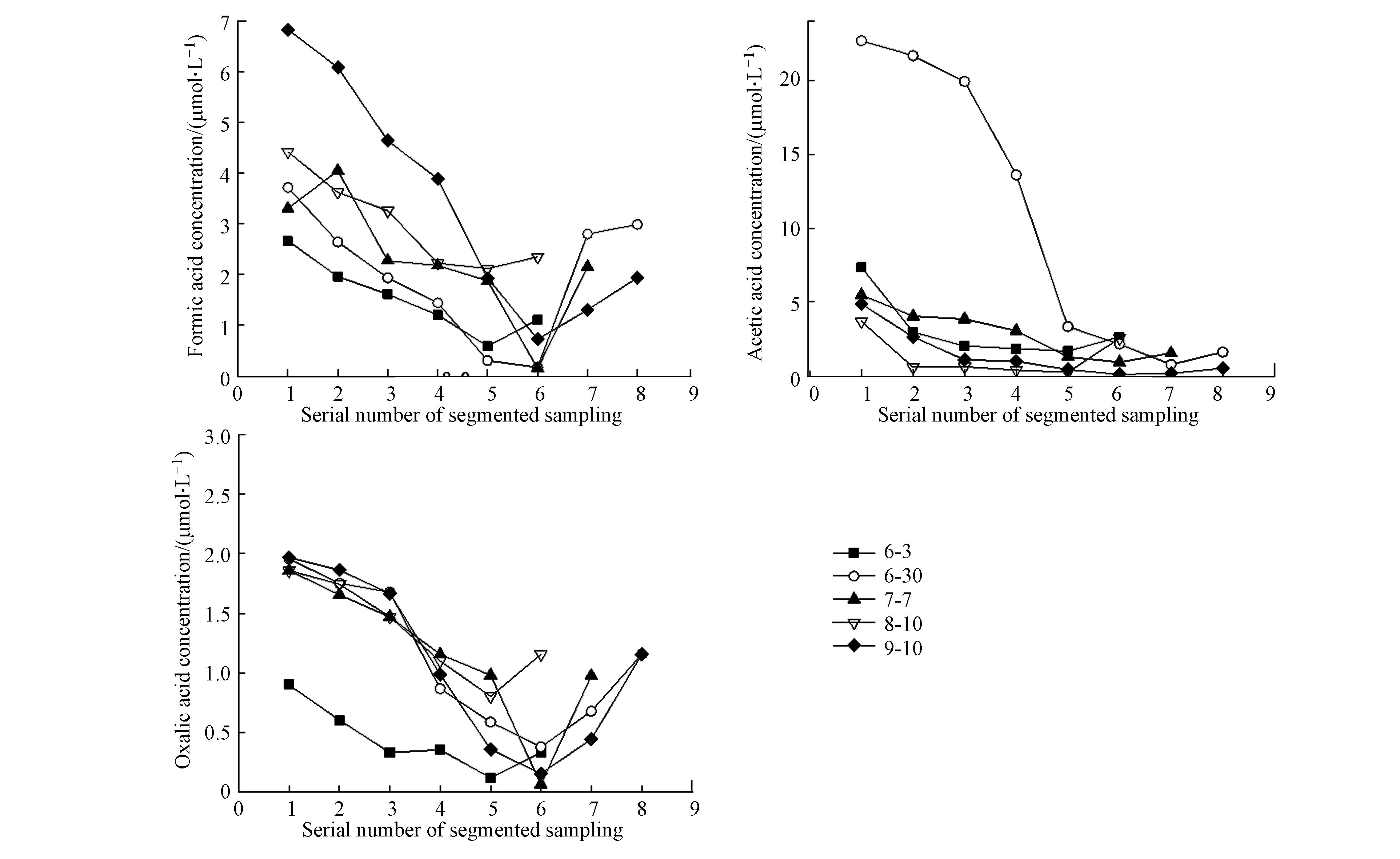 图 3 长降水事件的分段降水有机酸随降水进程的浓度分布图Figure 3. Concentration distribution of organic acids with precipitation process in segmented precipitation of long precipitation events
图 3 长降水事件的分段降水有机酸随降水进程的浓度分布图Figure 3. Concentration distribution of organic acids with precipitation process in segmented precipitation of long precipitation events分段降水样品中甲酸、乙酸、草酸的浓度范围分别为0.15—6.83、0.12—22.67、0.07—1.97 μmol·L−1,同一降水事件的分段降水样品中组分浓度具有较大的范围跨度,分段收集降水样品比整场降水单一样品的研究方法更能科学准确地反映降水有机酸的分布特征。
由图3,长降水事件的分段降水样品中甲酸、乙酸、草酸在降水进程中的变化特征相似:先逐渐降低,到降水进程末期(6—8段)趋于平稳或稍稍反升。降水初期,大气环境中污染物(颗粒相和气相)浓度较高,雨滴在下落过程中对大气污染物的冲刷较多,含有机酸组分的气溶胶颗粒和气相有机酸被雨滴冲刷而进入降水样品中,因此初期降水样品中有机酸浓度最高。随着降水进程的持续,大气污染物逐渐被冲刷到降水样品中,在大气环境中的量逐渐减少,因此降水进程的中后期,降水对大气污染物的冲刷作用有限,中后期降水样品中有机酸浓度逐渐降低并趋于平稳。但有时,降水进程的末期,长降水事件中倒数1—3段的分段降水样品中有机酸浓度会出现稍稍反升的情况。研究表明,粒径较大且吸湿性强的颗粒比粒径较小吸湿性弱的颗粒具有更低的临界过饱和度,饱和率和颗粒尺寸的关系可由传统的Köhler理论解释,即临界饱和度大则临界半径大,水溶性也越强[25]。当降水即将结束时,大气相对湿度比降水前、降水初期(100%)稍低,约为90%,此时,大气中残余的积聚模态(0.1—1 μm)颗粒可凝结成粗颗粒,这些粗颗粒在降水进程末期被冲刷到降水样品中,导致降水末期分段降水样品中有机酸浓度的稍稍反升。
图3中长降水事件进程中3种有机酸的变化趋势表现为:不同的降水事件中,分段降水样品中同种有机酸浓度的分布趋势高度相似。稍有特殊的是,6月30日的降水事件中,降水前中期样品中乙酸浓度异常高,间接反映6月30日降水事件之前的大气环境中颗粒相和气相污染物中乙酸浓度高。不同降水事件中分段降水样品中甲酸和草酸的分布趋势相似,大气环境中乙酸组分的分布变化大,而甲酸和草酸组分的分布相对稳定。
2.3 与其它地区降水中有机酸浓度的对比
据文献调研,尚未见其它地区分段降水样品中有机酸分布的报道,因此取本研究2.1节降水有机酸平均浓度与其它地区降水有机酸浓度进行对比分析(见表3)
表 3 本研究与其它地区降水中有机酸浓度的对比(μmol·L−1)Table 3. Comparison of organic acid concentration in precipitation between this study and other areas(μmol·L−1)地区/区域Region 对象Object 甲酸Formic acid 乙酸Acetic acid 草酸Oxalic acid 3种有机酸总含量Total concentration 参考文献Reference 南昌 雨水 3.81 4.56 1.13 9.48 本研究 临安 雨水 9.58 3.89 — 13.47 Niu [11] 安顺 雨水 8.77 6.90 2.84 18.51 章炎麟[13] 鼎湖山 雨水 4.12 3.39 2.51 10.02 龙晓娟[15] 天津 雨水 12.53 3.45 0.64 16.62 李一兰[16] 庐山 云水 10.83 9.29 9.90 30.02 Sun [10] 雨水 10.21 11.20 5.07 26.48 厦门 雨水 0.15 0.18 0.14 0.47 Du [8] 上海城区 雨水 0.81 1.34 0.86 3.01 马琳[26] 崇明东滩 雨水 0.87 2.48 0.29 3.64 郑州 雪水 42.82 8.13 1.67 52.62 殷美雪[27] 杭州 23.91 — 2.00 25.91 武汉 10.43 12.78 6.67 29.88 石家庄 15.87 0.33 2.56 18.76 南京 0.11 0.12 0.33 0.56 成都龙泉驿区 雨水 0.7826 2.123 0.3 3.2056 蒋贤栋[28] 济南 雨水 5.32 3.80 1.19 10.31 王秀秀[29] 北京 雨水 4.62 4.60 1.17 10.39 Xu [30] 泰山 雨水 4.98 3.85 1.65 10.48 Wang [31] 贵阳 雨水 14.24 9.35 5.58 29.17 Xu [32] 衡山 雨水 14.30 16.46 3.31 34.07 Wang [33] 圣保罗 雨水 17.00 8.90 — 25.90 Fornaro [34] 巴西郊区 雨水 10.20 29.87 — 40.07 洛杉矶 雨水 6.50 5.60 7.80 19.90 Kawamura [35] 西班牙 雨水 0.5474 0.8816 0.5016 1.9306 PEñA [36] | Show TableDownLoad:
CSV
根据表3,南昌前湖区域降水有机酸浓度低于临安、安顺、鼎湖山、天津、庐山、郑州、杭州、武汉、石家庄、济南、北京、泰山、贵阳、衡山等地区的报道结果,但高于厦门、上海、成都等地区的报道结果,说明南昌地区大气中有机酸排放源有限,间接反映南昌地区的大气环境质量中等偏优,但仍有优化的潜力。南昌地区降水中不同有机酸浓度的高低顺序(乙酸>甲酸>草酸)与国内大部分其它城市区域降水的基本一致。
与国外圣保罗、巴西、洛杉矶以及西班牙等城市区域的降水相比,南昌前湖区域降水中总有机酸浓度更低,说明南昌地区大气中有机酸的排放源有限,对降水有机酸贡献较小。圣保罗、巴西、洛杉矶以及西班牙等城市区域降水中有机酸与当地的植物生长释放和汽车尾气排放密切相关。国外城市降水有机酸浓度的高低顺序与南昌地区相比也略有不同,如洛杉矶降水有机酸浓度排序为草酸>乙酸>甲酸。草酸是一种二元羧酸,反映该区域大气中二次有机物的贡献明显。不同地区降水有机酸浓度的差异反映有机酸与各地的排放源有着密切的联系。
2.4 云下冲刷和云水对降水有机酸的贡献
雨滴离开云层落到地面的过程中,会因多种机制与大气颗粒物和气体分子发生碰撞,被碰撞的颗粒和气体分子大部分会被雨滴捕集,降水样品组分不仅包括云水本身组分,还包括被冲刷的颗粒组分和气态污染物[37]。
一般认为前期降水组分受云下冲刷和云水的共同影响,同场降水事件的分段降水样品组分浓度最小值表示云水对降水组分贡献,前期降水样品中组分浓度与云水组分浓度的差值表示云下冲刷对降水组分的贡献。本研究云下冲刷对降水组分贡献的计算采用式1,云水对降水组分的贡献等于1减云下冲刷贡献的差值[38]。
Fi=(Ci−rw+ro−Ci−min)/Ci−rw+ro×100% 式中,Fi表示云下冲刷对降水组分i的贡献,%;Ci-rw+ro是前期降水中组分i的浓度,表示云下冲刷、云水加和贡献给降水组分i的浓度;Ci-min是组分i在同场降水的分段降水样品中的最低浓度,表示云水中组分i的浓度。
选择分段降水样品超过5段的降水事件,基于分段降水样品中有机酸的浓度分布采用式1计算得到云下冲刷和云水对降水中甲酸、乙酸、草酸的贡献率,见图4。
 图 4 云下冲刷、云水对分段降水样品中有机酸的贡献率Figure 4. Contribution of below-cloud scavenging and cloud water to organic acids in segmented precipitation samples
图 4 云下冲刷、云水对分段降水样品中有机酸的贡献率Figure 4. Contribution of below-cloud scavenging and cloud water to organic acids in segmented precipitation samples云下冲刷对第1—5段分段降水样品中甲酸的平均贡献率分别为73.5%、68.1%、46.0%、38.8%、22.8%,对分段降水中乙酸的贡献率分别为78.5%、61.3%、48.3%、33.6%、32.4%,对分段降水中草酸的贡献率分别为72.5%、59.8%、50.0%、24.1%、17.7%。云水对第1—5段分段降水样品中甲酸的平均贡献率分别为 26.5%、31.9%、54.0%,61.2 %、77.2%,对分段降水中乙酸的贡献率分别为21.5%、38.7%、51.7%、66.4%、67.6%,对分段降水中草酸的贡献率分别为27.6%、40.2%、50.0%、75.9%、82.3%。随着降水进程的持续,云下冲刷对降水中3种有机酸(甲酸、乙酸、草酸)的贡献率逐渐减小,而云水对降水有机酸的贡献率逐渐增大,降雨前期云下冲刷为降水中有机酸根的主要来源,随着降雨的持续进行,大气中的污染物逐渐被清洁,降雨后期,降水中有机酸以云水来源为主。
2.5 降水中有机酸的来源分析
大气中有机酸的来源较为广泛,主要包括:植物直接释放、机动车尾气排放、生物质燃烧和大气中醛类和烯烃等不饱和有机物的大气氧化等。
2.5.1 降水中有机酸来源初步分析
大气中甲、乙酸的比值(F/A)g常用于判断大气中有机酸的来源[34],结合气液平衡亨利定律可采用液相(F/A)aq来判断降水有机酸的来源。一般来说,低F/A比值反映一次排放源,而高F/A比值反映光化学反应二次源[39];(F/A)aq<1用于判断降水有机酸主要来源于人类活动直接排放或生物质燃烧,(F/A)aq>1用于判断降水有机酸主要来源于不饱和碳氢化合物的光化学氧化[40],文献报道不同源的F/A比值见表4。表5为本研究分段降水样品中F/A比值的月均值。
表 4 不同源的F/A比值Table 4. F/A ratio of different sources来源种类Sources 甲酸/乙酸比Formic acid / Acetic acid 源文献Literature 一次源 <1 生物质燃烧 0.1—0.5 Talbot [41] 0.2—0.4 Hartmann [42] 机动车排放 0.4—0.6 Talbot [41] 0.3—0.5 Grosjean [43] 植被(热带雨林) 0.6 Talbot [44] 一般植被排放 0.4 Servant [45] 二次源 >1 Talbot [41] | Show TableDownLoad:
CSV
表 5 本研究分段降水样品中F/A比值的月均值Table 5. The monthly mean value of F/A ratio in segmented precipitation samples in this study月份 Month F/A 5月 1.0817 6月 0.2652 7月 0.4498 8月 0.3606 9月 3.4454 平均 1.1205 | Show TableDownLoad:
CSV
根据表5,本研究采样期间降水样品中F/A比值平均1.1205,大于1,这与济南[29]、北京[30]、泰山[31]等北方观测点和南方观测站点贵阳[32]的研究结果相似,但与南方清洁站点衡山[33]的研究结果(0.8688)不同。南昌前湖区域6月的F/A比值在0.2—0.4的范围内,说明6月降水中的有机酸主要来源于生物质燃烧,7月的F/A比值为0.4498,说明7月降水中的有机酸主要来源于生物质燃烧、机动车排放和植被排放,8月的F/A比值为0.3606,说明8月降水中的有机酸也主要来源于生物质燃烧和机动车排放。5、9月降水中F/A比值大于1,而6、7、8月的F/A比值小于1,说明5、9月降水有机酸受不饱和碳氢化合物的氧化的影响较大,5月(毕业季)和9月(开学季)校园及周边的人流和车流量较大,人为活动和机动车尾气排放增多,经过光化学反应后产生碳氢化合物,进而导致二次有机物的生成;6、7、8月植物生长旺盛,直接排放有机酸对降水组分带来影响。
为了能进一步说明鼎湖山降水中有机酸与无机离子成分的相互关系及其差异,本研究降水中有机酸与无机阴离子的相关性见表6、表7和表8。
表 6 前两段分段降水样品中有机酸与无机阴离子的相关系数Table 6. Correlation coefficients of organic acids and inorganic anions in the first two segmented precipitation samples组分 Components HCOO− CH3COO− C2O2−4 Cl− NO2− NO−3 SO2−4 HCOO− 1.000 CH3COO− −0.015 1.000 C2O2−4 −0.032 −0.101 1.000 Cl− 0.156 0.128 −0.024 1.000 NO−2 0.129 0.088 0.093 −0.035 1.000 NO−3 0.080 0.035 0.042 0.628** −0.029 1.000 SO2−4 0.229 0.221 0.145 0.418* −0.009 0.515** 1.000 注:**,在0.01级别(双尾),相关性显著;*,在0.05级别(双尾),相关性显著. | Show TableDownLoad:
CSV
表 7 末两段分段降水样品中有机酸与无机阴离子的相关系数Table 7. Correlation coefficients of organic acids and inorganic anions in the last two segmented precipitation samples组分 Components HCOO− CH3COO− C2O2−4 Cl− NO2− NO−3 SO2−4 HCOO− 1.000 CH3COO− 0.562** 1.000 C2O2−4 0.033 0.123 1.000 Cl− 0.220 0.666** 0.361 1.000 NO−2 0.524** 0.420* 0.351 0.308 1.000 NO−3 0.570** 0.524** 0.046 0.434* 0.257 1.000 SO2−4 0.571** 0.513** 0.202 581** 0.475* 0.691** 1.000 注:**,在0.01级别(双尾),相关性显著;*,在0.05级别(双尾),相关性显著. | Show TableDownLoad:
CSV
表 8 所有分段降水样品中有机酸与无机阴离子的相关系数Table 8. Correlation coefficients of organic acids and inorganic anions in all segmented precipitation samples组分 Components HCOO− CH3COO− C2O2−4 Cl− NO2− NO−3 SO2−4 HCOO− 1.000 CH3COO− 0.266* 1.000 C2O2−4 0.046 0.021 1.000 Cl− 0.157 0.321** 0.213* 1.000 NO−2 0.118 0.080 0.156 0.095 1.000 NO−3 0.163 0.206 0.171 0.541** 0.129 1.000 SO2−4 0.234* 0.458** 0.300** 0.526** 0.151 0.577** 1.000 注:**,在0.01级别(双尾),相关性显著;*,在0.05级别(双尾),相关性显著. | Show TableDownLoad:
CSV
根据表6和表7,前期降水样品中有机酸与无机离子的相关性弱,后期降水样品中,甲酸与
NO−2 NO−3 SO2−4 NO−3 SO2−4 根据表8,所有分段降水样品中甲酸与乙酸的相关性强于甲酸与草酸的相关性和乙酸与草酸的相关性,表明南昌前湖区域降水中的甲酸和乙酸更具有相似源,草酸的来源较甲、乙酸更为复杂,草酸主要来源于大气光化学氧化过程和汽车尾气。降水中草酸与
SO2−4 SO2−4 SO2−4 2.5.2 降水有机酸来源的因子分析
因子分析是将多个实测变量转换为少数几个不相关的综合变量的一种降维多元统计分析方法[46-47],表9是南昌前湖区域降水中有机酸和无机阴离子组分的因子分析载荷矩阵。
表 9 南昌前湖区域降水组分因子分析载荷矩阵Table 9. The factor loading matrix of precipitation components in Qianhu area of Nanchang变量 Variables 因子1 Factor 1 因子2 Factor 2 因子3 Factor 3 CH3COO− 0.921 0.100 0.028 HCOO− 0.051 0.027 0.911 Cl− −0.313 0.702 0.085 NO−2 −0.378 −0.839 0.046 NO−3 −0.001 0.226 0.143 SO2−4 0.273 0.025 0.008 C2O2−4 0.290 0.114 −0.427 累计贡献率/% 59.396 70.330 80.260 | Show TableDownLoad:
CSV
根据因子分析,筛选出3个特征因子,第1个因子中CH3COO−载荷(0.921)高,反映降水对酸性气溶胶颗粒的冲刷;第2个因子中Cl−载荷(0.702)高,反映燃烧源的影响;第3个因子中HCOO−载荷(0.911)高,反映植被排放源的影响。因此,因子分析表明区域降水中有机酸等组分的主要来源是酸性颗粒的冲刷、燃烧源和植被排放源。
2.5.3 降水中有机酸来源的PMF分析
基于 PMF 模型对南昌前湖区域降水样品进行源解析,图5为降水中有机酸和无机阴离子的PMF分析结果。
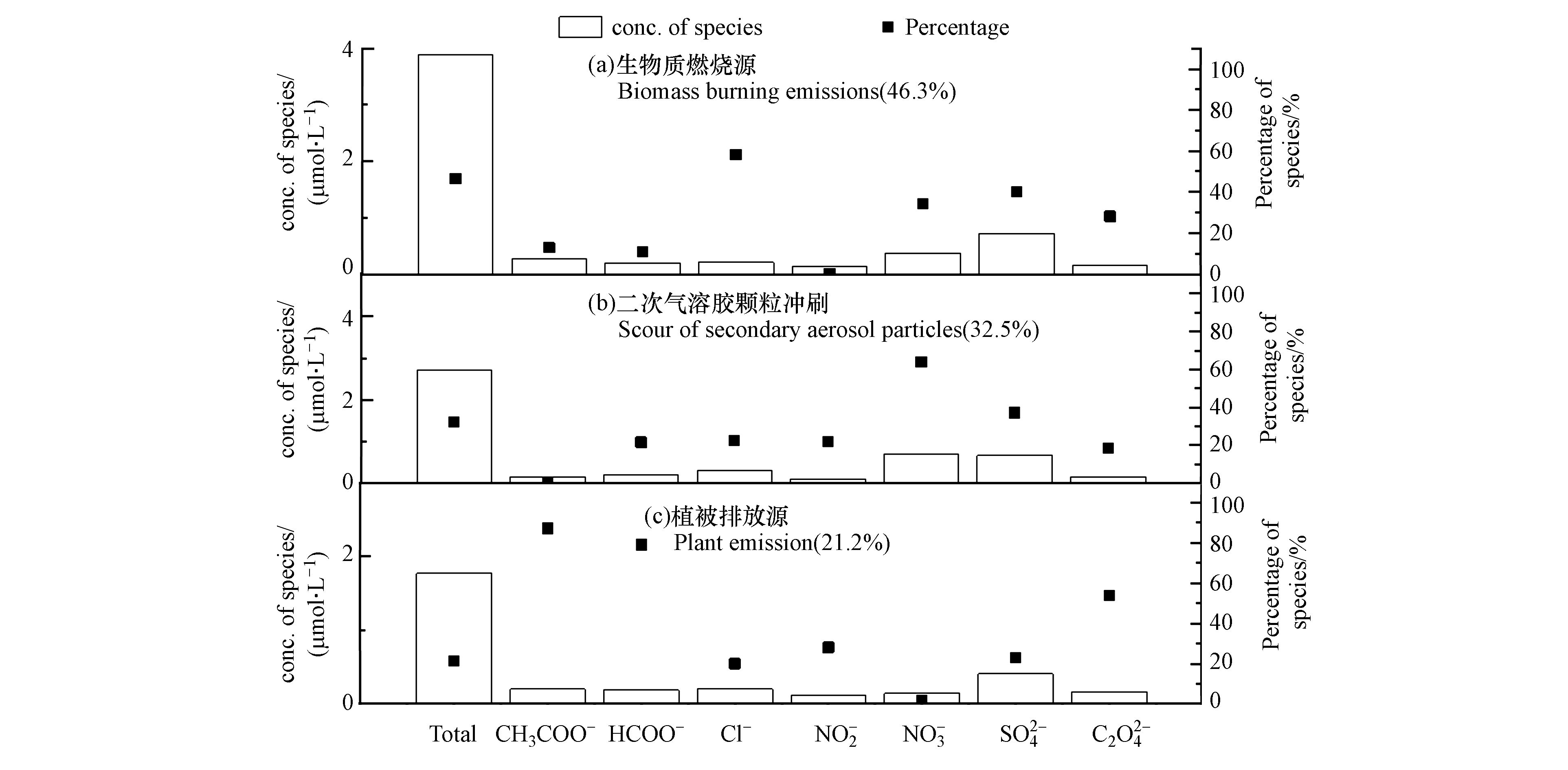 图 5 南昌前湖区域降水中有机酸、无机阴离子的PMF分析结果Figure 5. PMF analysis results of organic acids and inorganic anions in precipitation of Qianhu area of Nanchang
图 5 南昌前湖区域降水中有机酸、无机阴离子的PMF分析结果Figure 5. PMF analysis results of organic acids and inorganic anions in precipitation of Qianhu area of Nanchang根据图5,PMF分析得到南昌前湖区域降水中有机酸、无机阴离子组分的3个贡献因子,因子1(贡献率46.3%)中Cl−载荷值高(图5a),反映生物质燃烧源[48]的贡献;因子2(贡献率32.5%)中
NO−3 SO2−4 3. 结论(Conclusion)
(1)采样期间,南昌前湖区域降水中甲酸、乙酸和草酸的平均浓度分别3.81 μmol·L−1、4.56 μmol·L−1、1.13 μmol·L−1,占所测有机酸、无机阴离子总量的16 %;降水有机酸浓度月变化特征为:5—6月上升,到7—8月维持较高浓度,9月下降。
(2)长降水事件中,降水有机酸浓度在降水进程中呈现先逐渐降低,到降水进程末期趋于平稳或稍稍反升的变化特征,表明随着降雨进程的持续进行,降水对大气污染物的冲刷作用逐渐减弱。
(3)国内外不同地区降水有机酸浓度呈现差异,南昌地区降水有机酸浓度反映区域大气环境质量中等偏优。
(4)随着降水进程的持续,云下冲刷对降水中甲酸、乙酸、草酸的贡献率逐渐减小,云水贡献逐渐增大,降雨前期云下冲刷为降水中有机酸根的主要来源,降雨后期,降水中有机酸以云水来源为主。
(5)前期降水中3种有机酸两两之间的相关性比末期降水中的弱,降水中3种有机酸两两之间的相关性随降雨进程增强,草酸与
SO2−4 SO2−4 -
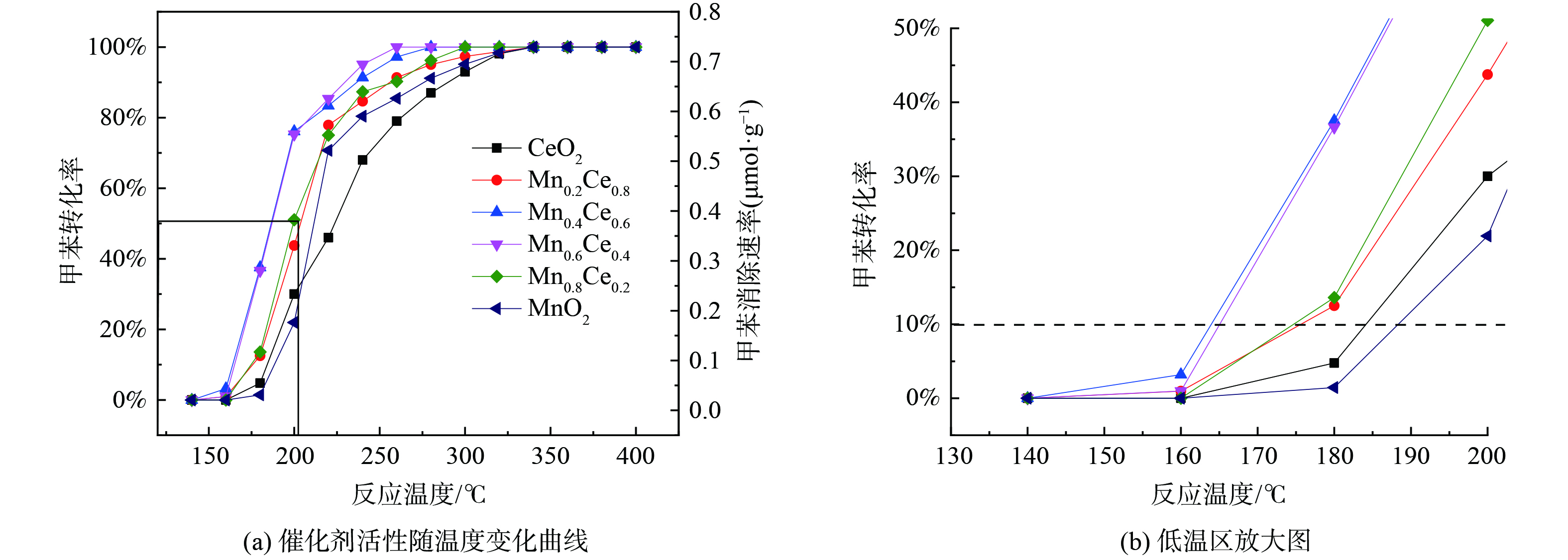
图 1 Mn/Ce摩尔比对催化剂催化氧化甲苯性能影响
Figure 1. Effect of Mn/Ce molar ratios on the catalytic performance of toluene oxidation
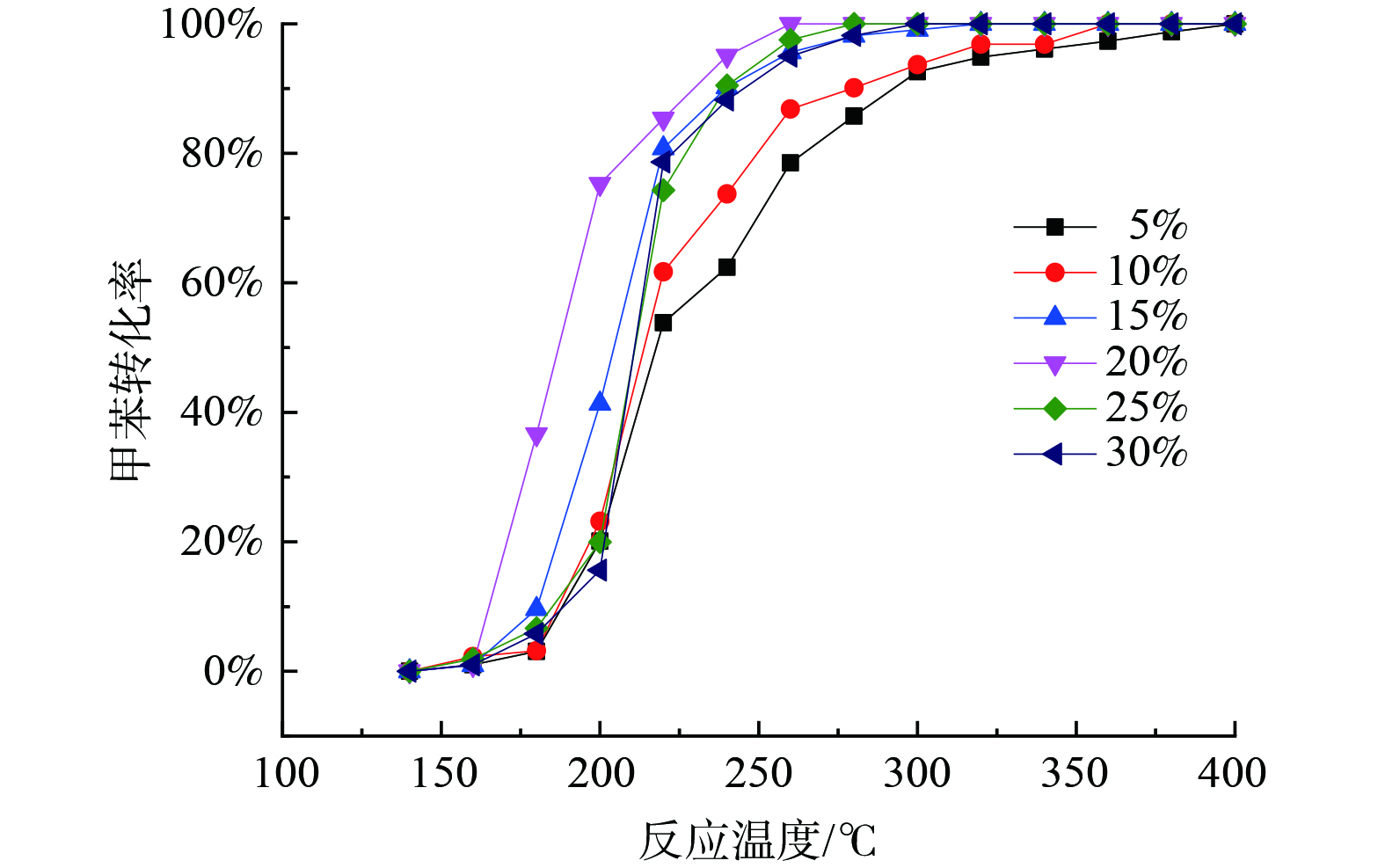
图 2 活性组分含量对催化剂催化氧化甲苯活性影响
Figure 2. Effect of active components content on the catalytic performance of toluene oxidation
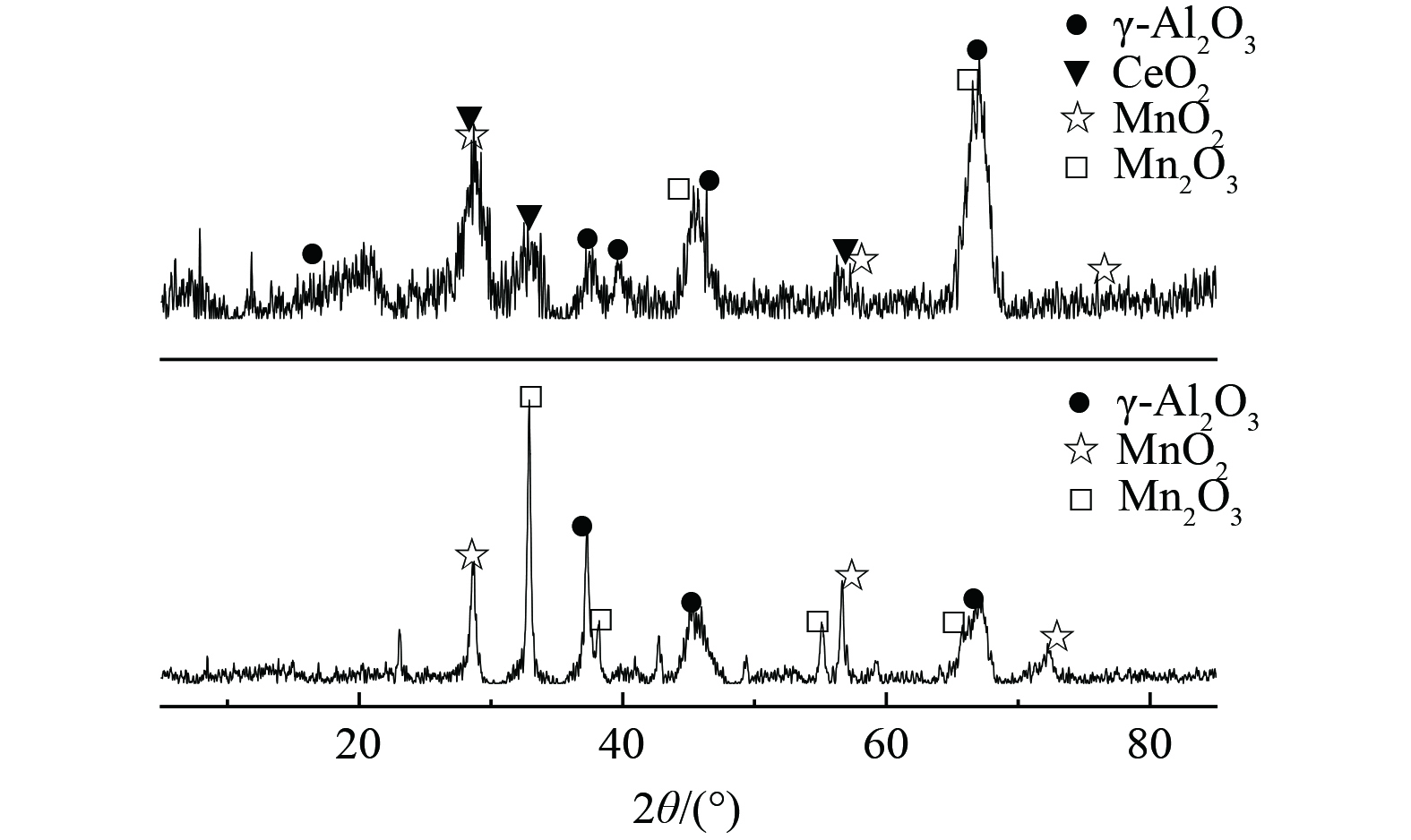
图 3 MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3 XRD曲线
Figure 3. XRD curves of MnO2/γ-Al2O3 and Mn0.6Ce0.4/γ-Al2O3
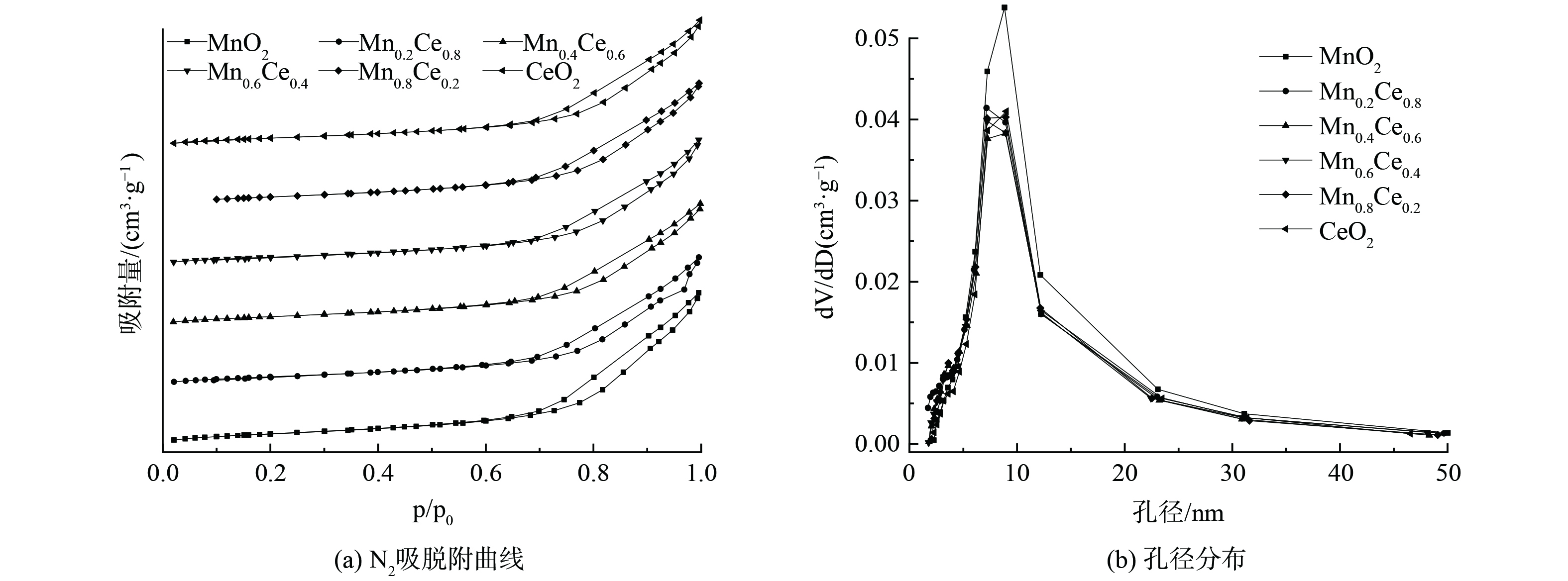
图 4 MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3的BET测试图
Figure 4. BET test diagram of MnO2/γ-Al2O3 and Mn0.6Ce0.4/γ-Al2O3

图 5 MnO2/γ-Al2O3 催化剂和Mn0.6Ce0.4/γ-Al2O3催化剂的TEM图
Figure 5. TEM images of MnO2/γ-Al2O3and Mn0.6Ce0.4/γ-Al2O3
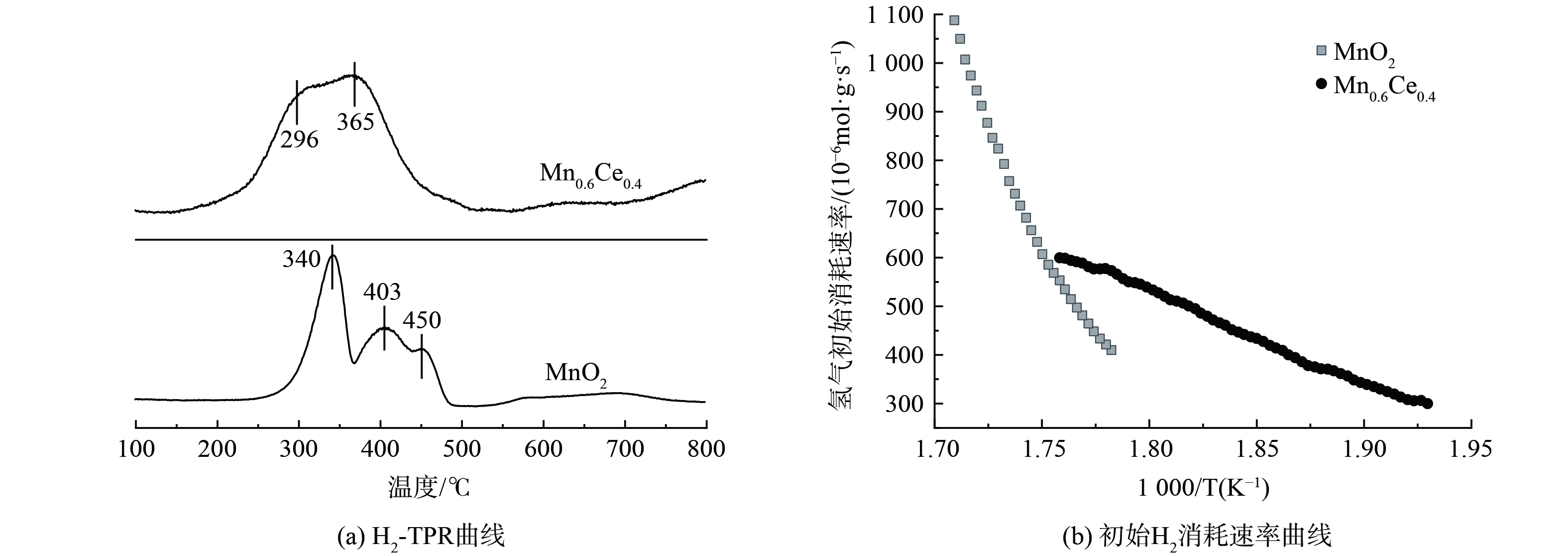
图 6 MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3催化剂的H2-TPR测试
Figure 6. H2-TPR test of MnO2/γ-Al2O3 and Mn0.6Ce0.4/γ-Al2O3
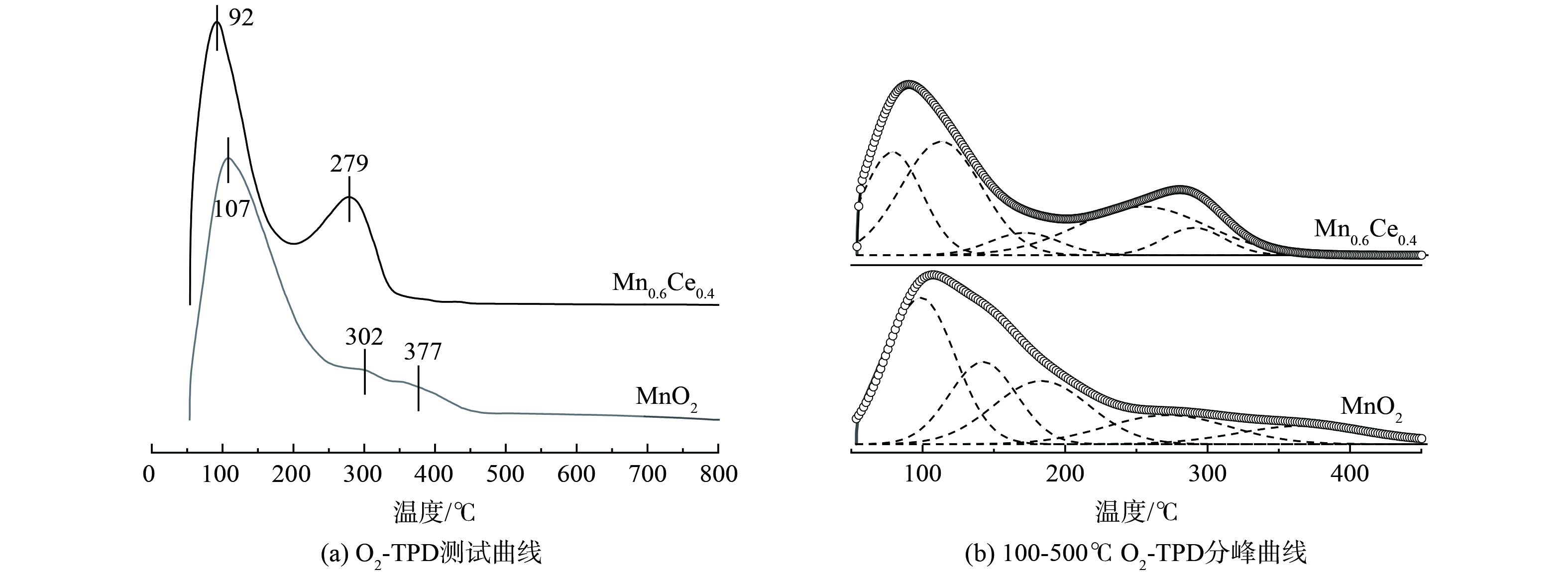
图 7 MnO2/γ-Al2O3和Mn0.6Ce0.4/γ-Al2O3的O2-TPD测试结果
Figure 7. O2-TPD profiles for MnO2/γ-Al2O3 and Mn0.6Ce0.4/γ-Al2O3

图 8 MnO2/γ-Al2O3 and Mn0.6Ce0.4/γ-Al2O3的XPS谱图
Figure 8. XPS spectra of MnO2/γ-Al2O3 and Mn0.6Ce0.4/γ-Al2O3.
表 2 催化剂织构性质和元素组成
Table 2. Textural and structural properties and chemical composition of catalyst
催化剂 孔道结构参数 表面元素摩尔比 比表面积/(m2·g−1) 孔容/ (cm3·g−1) 孔径/nm Mn4+/Mn3+ Ce4+/Ce3+ Oads/Olatt MnO2 151.43 0.61 10.9 0.49 − 0.55 Mn0.6Ce0.4 146.68 0.49 10.5 0.39 2.32 1.74
下载: 导出CSV
表 3 催化剂活性、H2消耗量和O2脱附量
Table 3. Reaction activity, H2 consumption and O2 desorption
催化剂 反应温度 /( ℃) 氢气消耗量/(mmol·g−1) 氧气脱附量/(mmol·g−1) T10 T50 T90 峰1+峰2 峰3+峰4 总量 MnO2 186 210 260 0.32 0.19 0.52 27.93 Mn0.6Ce0.4 162 180 220 1.12 1.61 2.73 35.62
下载: 导出CSV
-
[1] GAO G Q, LIAO Y, LI W W, et al. Active surface RuO species originating from size-driving self-dispersion process for toluene catalytic combustion[J]. Chemical Engineering Journal, 2022, 441: 136127. doi: 10.1016/j.cej.2022.136127 [2] IAMAIL A, LI M Y, ZAHID M, et al. Effect of strong interaction between Co and Ce oxides in CoxCe1-xO2-δ oxides on its catalytic oxidation of toluene[J]. Molecular Catalysis, 2021, 502: 111356. doi: 10.1016/j.mcat.2020.111356 [3] WANG J, WANG P, YOSHIDA A, et al. Mn-Co oxide decorated on Cu nanowires as efficient catalysts for catalytic oxidation of toluene[J]. Carbon Resources Conversion, 2020, 3: 36-45. doi: 10.1016/j.crcon.2020.02.001 [4] LIN Y, SUN J, LI S J, et al. An Efficient Pt/CeyCoOx Composite Metal Oxide for Catalytic Oxidation of Toluene[J]. Catalysis Letters, 2020, 150(11): 3206-3213. doi: 10.1007/s10562-020-03217-9 [5] ZHANG X J, ZHAO J G, SONG Z X, et al. Cooperative Effect of the Ce-Co-Ox for the Catalytic Oxidation of Toluene[J]. Chemistry Select, 2019, 4: 8902-8909. [6] LI Y F, XIAO L J, LIU F Y, et al. Core-shell structure Ag@Pd nanoparticles supported on layered MnO2 substrate as toluene oxidation catalyst[J]. Journal of Nanoparticle Research, 2019, 21: 28. doi: 10.1007/s11051-019-4467-8 [7] REN S D, LIANG W J, LI Q L, et al. Effect of Pd/Ce loading on the performance of Pd-Ce/γ-Al2O3 catalysts for toluene abatement[J]. Chemosphere, 2020, 251: 126382. doi: 10.1016/j.chemosphere.2020.126382 [8] HOU Z Y, ZHOU X Y, LIN T, et al. The promotion effect of tungsten on monolith Pt/Ce0.65Zr0.35O2 catalysts for the catalytic oxidation of toluene[J]. New Journal of Chemistry, 2019, 43: 5719-5726. doi: 10.1039/C8NJ06245E [9] 梁文俊, 朱玉雪, 石秀娟, 等. Ce掺杂对Ru/TiO2催化氯苯性能的影响[J]. 化工学报, 2020, 71(8): 3585-3593. [10] 彭新宇, 刘丽君, 沈伯雄, 等. M-ZSM-5(M=Cu、Mn、Fe、Ce、Ti)催化氧化甲苯性能研究[J]. 燃料化学学报, 2023, 51(6): 1-11. [11] LYU Y, LI C, DU X Y, et al. Catalytic oxidation of toluene over MnO2 catalysts with different Mn(II) precursors and the study of reaction pathway[J]. Fuel, 2020, 262: 116610. doi: 10.1016/j.fuel.2019.116610 [12] YANG X Q, YU X L, LIN M Y, et al. Enhancement effect of acid treatment on Mn2O3 catalyst for toluene oxidation[J]. Catalysis Today, 2019, 327: 254-261. doi: 10.1016/j.cattod.2018.04.041 [13] LI X L, NIU Y F, ZHANG C W, et al. Catalytic combustion of toluene over broccoli-shaped Ce1Mn3Ox solid solution[J]. ChemCatChem, 2021, 13(19): 4223-4236. doi: 10.1002/cctc.202100974 [14] 杨玉玲, 周家斌, 张天磊, 等. CeMn氧化物催化剂的制备及其对甲苯的催化降解性能[J]. 化工环保, 2021, 41(2): 223-228. doi: 10.3969/j.issn.1006-1878.2021.02.016 [15] LUO Y J, LIN D F, ZHENG Y B, et al. MnO2 nanoparticles encapsuled in spheres of Ce-Mn solid solution: Efficient catalyst and good water tolerance for low-temperature toluene oxidation[J]. Applied Surface Science, 2020, 504: 144481. doi: 10.1016/j.apsusc.2019.144481 [16] GENG L L, CHEN B B, YANG J H, et al. Synergistic effect between Mn and Ce for active and stable catalytic wet air oxidation of phenol over MnCeOx[J]. Applied Catalysis A:General, 2020, 604: 117774. doi: 10.1016/j.apcata.2020.117774 [17] SHU Y, XU Y, HUANG H, et al. Catalytic oxidation of VOCs over Mn/TiO2/activated carbon under 185nm VUV irradiation[J]. Chemosphere, 2018, 208: 550-558. doi: 10.1016/j.chemosphere.2018.06.011 [18] LI N, CHENG J, XING X, et al. Hydrotalcite-derived Pd/Co3MnxAl1-xO mixed oxides as efficient catalysts for complete oxidation of toluene[J]. Catalysis Today, 2019, 327: 382-388. doi: 10.1016/j.cattod.2018.03.009 [19] YAOSI M, YANG C H, LI X, et al. Clean synthesis of RGO/Mn3O4 nanocomposite with well-dispersed Pd nanoparticles as a high-performance catalyst for hydroquinone oxidation[J]. Journal of Colloid Interface Science, 2019, 552: 72-83. doi: 10.1016/j.jcis.2019.05.009 [20] ZHENG Y, ZHOU J, ZENG X, et al. Template and interfacial reaction engaged synthesis of CeMnOx hollow nanospheres and their performance for toluene oxidation[J]. RSC Advances, 2022, 12(40): 25898-25905. doi: 10.1039/D2RA04678D [21] HUANG Z Z, ZHAO J, SONG Z, et al. Controllable construction of Ce-Mn-Ox with tunable oxygen vacancies and active species for toluene catalytic combustion[J]. Applied Organometallic Chemistry, 2020, e5958. [22] PEI J, PENG B, LIN H, et al. Single-Atom Ru on Al2O3 for highly active and selective 1, 2-Dichloroethane catalytic degradation[J]. ACS Applied Materials & Interfaces, 2021, 13(45): 53683-53690. [23] WANG T, YANG S, SUN K, et al. Preparation of Pt/beta zeolite-Al2O3/cordierite monolith for automobile exhaust purification[J]. Ceramics International, 2011, 37: 621-626. doi: 10.1016/j.ceramint.2010.09.035 [24] ZHANG C, CHU W, CHEN F, et al. Effects of cerium precursors on surface properties of mesoporous CeMnO catalysts for toluene combustion[J]. Journal of Rare Earths, 2020, 38(1): 6. [25] XIAO M, YANG X, PENG Y, et al. Confining shell-sandwiched Ag clusters in MnO2-CeO2 hollow spheres to boost activity and stability of toluene combustion[J]. Nano Research, 2022, 15(8): 7042-7051. doi: 10.1007/s12274-022-4360-0 [26] TIAN M, JIANG Z, CHEN C, et al. Engineering Ru/MnCo3Ox for 1, 2-Dichloroethane benign destruction by strengthening C-Cl cleavage and chlorine desorption: Decisive role of H2O and reaction mechanism[J]. ACS Catalysis, 2022, 12(15): 8776-8792. doi: 10.1021/acscatal.2c02304 [27] LIU H, LI X, DAI Q, et al. Catalytic oxidation of chlorinated volatile organic compounds over Mn-Ti composite oxides catalysts: Elucidating the influence of surface acidity[J]. Applied Catalysis B:Environmental, 2021, 282: 119577. doi: 10.1016/j.apcatb.2020.119577 [28] ZHANG C, HUANG H, LI G, et al. Zeolitic acidity as a promoter for the catalytic oxidation of toluene over MnO/HZSM-5 catalysts[J]. Catalysis Today, 2019, 327: 374-381. doi: 10.1016/j.cattod.2018.03.019 [29] XIAO J, WANG M, WANG Y, et al. Rational design of Bimetal Mn-Ce nanosheets anchored on porous nano-sized ZSM-5 zeolite for adsorption-enhanced catalytic oxidation of toluene[J]. Industrial & Engineering Chemistry Research, 2022, 61(50): 18382-18389. [30] CHEN J, CHEN X, CHEN X, et al. Homogeneous introduction of CeOy into MnOx-based catalyst for oxidation of aromatic VOCs[J]. Applied Catalysis B-Environmental, 2018, 224: 825-835. doi: 10.1016/j.apcatb.2017.11.036 [31] WAN J, TAO F, SHI Y, et al. Designed preparation of nano rod shaped CeO2-MnO catalysts with different Ce/Mn ratios and its highly efficient catalytic performance for chlorobenzene complete oxidation: New insights into structure–activity correlations[J]. Chemical Engineering Journal, 2022, 433: 133788. doi: 10.1016/j.cej.2021.133788 [32] ZHANG X, WU D. Ceramic monolith supported Mn-Ce-M ternary mixed-oxide (M=Cu, Ni or Co) catalyst for VOCs catalytic oxidation[J]. Ceramics International, 2016, 42(15): 16563-16570. doi: 10.1016/j.ceramint.2016.07.076 [33] CAO L, HUANG X, FENG Y. Preparation of CuMnOx Composite Oxide Catalyst Doped with CeO2 and Its Catalytic Performance for Toluene[J]. Journal of Xi’an University of Architecture & Technology. (Natural Science Edition), 2010, 42(5): 729-733. [34] DAI H, BELL A T, IGLESIA E. Effects of molybdena on the catalytic properties of vanadia domains supported on alumina for oxidative dehydrogenation of propane[J]. Journal of Catalysis, 2004, 221: 491-499. doi: 10.1016/j.jcat.2003.09.020 [35] XUE L, ZHANG C, HE H, et al. Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst[J]. Applied Catalysis B:Environmental, 2007, 75: 167-174. doi: 10.1016/j.apcatb.2007.04.013 [36] 李安明, 卫广程, 郝乔慧, 等. Mn含量对CeO2-ZrO2-MnOx化剂甲苯氧化净化性能的影响[J]. 燃料化学学报, 2020, 48(2): 231-239. [37] CAO Y, ZHANG C, XU D, et al. Low-temperature oxidation of toluene over MnOx-CeO2 nanorod composites with high sinter resistance: Dual effect of synergistic interaction on hydrocarbon adsorption and oxygen activation[J]. Inorgnic Chemistry, 2022, 61(38): 15273-15286. doi: 10.1021/acs.inorgchem.2c02738 [38] VAYSSILOV G N, LYKHACH Y, MIGANI A, et al. Support nanostructure boosts oxygen transfer to catalytically active platinum nanoparticles[J]. Nature Materials, 2011, 10: 310-315. doi: 10.1038/nmat2976 [39] AZALIM S, FRANCO M, BRAHMI R, et al. Removal of oxygenated volatile organic compounds by catalytic oxidation over Zr-Ce-Mn catalysts[J]. Journal of Hazardous Materials, 2011, 188(1/2/3): 422-427. [40] TANG W X, WU X F, LIU G, et al. Preparation of hierarchical layer-stacking Mn-Ce composite oxide for catalytic total oxidation of VOCs[J]. Journal of Rare Earths, 2015, 33(1): 62-69. doi: 10.1016/S1002-0721(14)60384-7 [41] ZHANG X J, ZHAO J G, SONG Z X, et al. The catalytic oxidation performance of toluene over the Ce-Mn-Ox catalysts: Effect of synthetic routes[J]. Journal of Colloid and Interface Science, 2020, 562: 170-181. doi: 10.1016/j.jcis.2019.12.029 [42] ZHOU C X, ZHANG H L, ZHANG Z, et al. Improved reactivity for toluene oxidation on MnOx/CeO2-ZrO2 catalyst by the synthesis of cubic-tetragonal interfaces[J]. Applied Surface Science, 2021, 539: 148188. doi: 10.1016/j.apsusc.2020.148188 [43] YE Z P, GIRAUDON J M, DE GEYTER N, et al. The design of MnOx based catalyst in post-plasma catalysis configuration for toluene abatement[J]. Catalysts, 2018, 8(2): 91. doi: 10.3390/catal8020091 [44] LI L, ZHANG C Y, YAN J L, et al. Distinctive Bimetallic Oxides for Enhanced Catalytic Toluene Combustion: Insights into the Tunable Fabrication of Mn-Ce Hollow Structure[J]. ChemCatChem, 2020, 12(10): 2872-2879. doi: 10.1002/cctc.202000038 [45] PENG Y X, ZHANG L, JIANG Y W, et al. Fe-ZSM-5 supported palladium nanoparticles as an efficient catalyst for toluene abatement[J]. Catalysis Today, 2019, 332: 195-200. doi: 10.1016/j.cattod.2018.05.032 [46] WU M D, CHEN S Y, SOOMRO A, et al. Investigation of synergistic effects and high performance of La-Co composite oxides for toluene catalytic oxidation at low temperature[J]. Environmental Science and Pollution Research, 2019, 26: 12123-12135. doi: 10.1007/s11356-019-04672-7 [47] YANG M, SHEN G L, WANG Q, et al. Roles of oxygen vacancies of CeO2 and Mn-Doped CeO2 with the same morphology in benzene catalytic oxidation[J]. Molecules, 2021, 26(21): 15. [48] FENG Z T, ZHANG M Y, REN Q M, et al. Design of 3-dimensionally self-assembled CeO2 hierarchical nanosphere as high efficiency catalysts for toluene oxidation[J]. Chemical Engineering Journal, 2019, 369: 18-25. doi: 10.1016/j.cej.2019.03.051 [49] PENG R S, LI S J, SUN X B, et al. Size effect of Pt nanoparticles on the catalytic oxidation of toluene over Pt/CeO2 catalysts[J]. Applied Catalysis B:Environmental, 2018, 220: 462-470. doi: 10.1016/j.apcatb.2017.07.048 [50] SHE W, QI T Q, CUI M X, et al. High Catalytic Performance of a CeO2-Supported Ni Catalyst for Hydrogenation of Nitroarenes, Fabricated via Coordination-Assisted Strategy[J]. ACS Applied Materials & Interfaces, 2018, 10(17): 14698-14707. [51] LIU W, WANG S N, CUI R Y, et al. Enhancement of catalytic combustion of toluene over CuMnOx hollow spheres prepared by oxidation method[J]. Microporous and Mesoporous Materials, 2021, 326: 111370. doi: 10.1016/j.micromeso.2021.111370 期刊类型引用(1)
1. 张敬东,杨娇羽,揭翠,李鸿鹄,彭喜燕,安淼. Mn-Fe改性碳基磁性吸附剂去除Hg~0性能及机制. 中国环境科学. 2024(12): 6628-6640 .  百度学术
百度学术
其他类型引用(1)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2903
- HTML全文浏览数: 2903
- PDF下载数: 86
- 施引文献: 2


