

 下载:
下载:
-
化学镀铜工艺是指在无外加电流的条件下,利用合适的还原剂(常用次磷酸盐),使溶液中的铜离子在具有催化活性的基体表面还原沉积出金属铜,形成铜镀层的一种工艺[1-3]。近年来,化学镀铜工艺在表面处理行业中所占的地位在不断上升,在机械工业、航空航天、电子工业等各行各业都有着越来越广泛的应用[4]。在化学镀铜工艺中,会产生大量的化学镀铜废水,将这些废水进行处理和回收,对保护生态环境,变废为宝,提高经济效益,尤为重要[5]。
化学镀铜废水主要来源于清洗零部件时所产生的清洗废水,因此,也可以称为化学镀铜清洗废水,其中主要含有铜离子和次磷酸盐等污染物[6]。过量的铜会刺激人类的消化系统,引起腹痛、呕吐等,严重时可造成中毒。而含铜废水进入水体后,成为持久性污染物,危机植物生长,影响水产养殖。当进入土壤时,会在土壤和作物中富集,经过一系列的环境迁移转化最终进入食物链,对人类健康产生威胁[7]。与正磷酸盐比,次磷酸盐由于其溶解度大且难与沉淀剂反应形成沉淀,导致水体富营养化严重的同时亦造成磷资源的流失[8]。故次磷的去除通常需氧化成正磷,再加入沉淀剂将正磷彻底去除或回收[9]。因此,对化学镀铜清洗废水的处理并回收磷和铜成为当前研究热点之一。
目前,含铜废水处理方法有很多,例如物化沉淀法、膜分离法、吸附法、混凝法和电解法等[10-11]。其中电解法可以使铜离子以金属铜的形式沉积在阴极上,实现了金属铜的回收[12]。除电解法外,其他方法只改变了铜离子存在形态,使铜离子发生迁移,但污染并没有彻底消除。而采用电解法时,当溶液为偏碱性条件下,铜离子很容易水解生成铜的氧化物,累积在阳极或生成沉淀物,使其难以在阴极进行电化学沉积回收[13]。因此,在电解法沉积铜离子实现阴极回收金属铜时,控制溶液的pH较为重要。
光电催化法是一种将光化学和电化学法相结合的方法,通过对半导体光催化剂施加外加偏压作用实现光生电子和光生空穴的有效分离,有效促进自由基的生成,提升污染物的降解效果[14]。光电催化作为高级氧化技术研究热点之一,是一种不仅能产生强氧化性活性物种实现污染物氧化降解,同时也能利用光生电子的还原能力实现阴极还原回收重金属的有效方法[15]。具有运行成本较低、温度和压力适应范围广、可实现有机物矿化且无二次污染等优点,在环境保护水处理领域越来受到关注[16]。二氧化钛(TiO2)纳米管光电极具有高度有序、比表面积大、电池容量高及量子化学效应强等优点,被广泛应用于纳米微电子、光伏器件、水分解产氢、环境污染物降解等领域[17-19]。有研究[20]表明,利用TiO2纳米管电极作为光阳极可有效实现铜氰络合物的氧化破络合同时电还原回收金属铜。
本研究采用电化学阳极氧化法制得的TiO2纳米管电极为光阳极和钛片(Ti)为阴极,在模拟太阳光(AM 1.5)照射进行光电催化处理次磷酸根离子(H2PO2−)和重金属铜离子(Cu2+)同时回收金属铜(Cu)。对TiO2纳米管电极进行了表征分析;对比分析了光电催化(PEC)、电催化(EC)和光催化(PC)体系对次磷氧化和Cu回收效果;考察了电压、初始PH、电解质种类对PEC体系下次磷氧化和Cu回收效率的影响,并进一步探讨该体系的反应机理。本研究结果可为含次磷和重金属铜的工业废水资源化处理提供参考。
-
电极材料钛片购自北京恒力钛工贸公司。实验用次磷酸钠(NaH2PO2·H2O)、亚磷酸钠(Na2HPO3·5H2O)、硫酸铵((NH4)2SO4)、氟化铵(NH4F)、丙三醇(C3H8O3)、硫酸铜(CuSO4)、硝酸(HNO3)、氢氟酸 (HF)、氢氧化钠(NaOH)、硫酸(H2SO4)、过硫酸钾(K2S2O8)、抗坏血酸(C6H8O6)、钼酸铵((NH4)6Mo7O24·4H2O)、酒石酸锑钾(KSbC4H4O7·1/2H2O)、硫酸钠(Na2SO4)、高氯酸钠(NaClO4)、氯化钠(NaCl)、叔丁醇(C4H10O)等均购自国药集团化学试剂公司,均为分析纯。
-
光电催化氧化装置如图1所示,其中包括石英反应器(长5.0 cm,宽5.0 cm,高6.0 cm),150 W的氙灯(Zolix instruments Co,China),直流电源(DH1718E-4,北京大华仪器公司,中国),磁力搅拌器(MS-H380-Pro,北京大龙兴创实验仪器有限公司,中国)。在氙灯光源处安装了一个AM 1.5滤光片,使其照射到反应器内阳极的光为模拟的太阳光。阳极为TiO2纳米管电极,阴极为钛片(长5.0 cm,宽3.0 cm,厚0.2 mm)。
-
1) TiO2纳米管电极的制备。采用阳极氧化法制备电极,制备方法参考文献[20]。钛片预处理:将钛片分别在无水乙醇和丙酮中超声清洗,后用不同目数金相砂纸(200、400、600、1000 目)依次打磨,去离子水清洗,将清洗后的钛片置于HF/HNO3/H2O体积比为1∶4∶5 的混合溶液中浸泡1 min,使钛片化学抛光。电解质制备方法:配制100 g质量比为0.5% NH4F + 1% (NH4)2SO4+ 90% C3H8O3的混合水溶液,即为所需电解质电解质溶液。TiO2纳米管电极制备方法:阳极为预处理钛片,阴极为铂丝,两级间距为20 mm,垂直插入电解质中,电压为20 V,室温下阳极氧化10 h,将氧化后的电极放入马弗炉中450 ºC热处理2 h,升温程序为5 ºC·min−1。
2)降解实验。含次磷酸根离子和重金属铜离子的化学镀铜模拟废水制备方法如下:配制1.0 mmol·L−1的NaH2PO2溶液;将CuSO4溶于其中使Cu2+浓度为0.5 mmol·L−1,即为所需化学镀铜模拟废水。取上述溶液120 mL置于反应器中,开启直流电源在两极间施以一定的电压,同时开启氙灯,反应时间为180 min,取样时间为0、30、60、90、120、150、180 min。反应液以10 mmol·L−1的Na2SO4为电解质。光催化反应时只开启氙灯,两极之间不施加电压。电化学反应时只开启直流电源。
3)表征及分析方法。电极表面形态通过场发射扫描电镜(SEM,SU-8010,日本日立公司)进行观察;晶体结构通过X射线衍射(XRD,XPert Pro MPD,荷兰帕纳科分析仪器有限公司)进行表征,所用的仪器是配有石墨晶体单色器的Rigaku D/max-B衍射仪,2θ扫描范围为10°~90°,扫描速率为0.5°·min−1,加速电压和工作电流分别为30 kV和30 mA;阴极回收Cu价态通过X射线光电子能谱仪(XPS,PHI Quantera SXM,日本ULVAC-PHI 公司)进行测定;电子自旋共振波谱仪(ESR,A300−10/12,德国布鲁克有限公司)用来检测自由基的生成。总磷的测定方法为采用国标过硫酸钾氧化-钼酸铵分光光度法;正磷的测定方法为采用国标钼酸铵分光光度法;次磷和亚磷采用离子色谱(IC,ICS-1500,美国戴安公司)测定,所用色谱柱为AS23分析柱和AG23保护住,淋洗液为4.5 mmol·L−1的Na2CO3和0.8 mmol·L−1的NaHCO3溶液,流速为1.0 mL·min−1;重金属Cu含量采用电感耦合等离子体发射光谱仪(ICP-OES,P700,美国安捷伦科技公司)来测定。
-
图2(a)和图2(b)分别是Ti基底和TiO2纳米管电极的SEM正面图像。可见,Ti基底表面平整,经阳极氧化法制备的TiO2纳米管电极上,纳米管阵列高度有序、管径均匀、排列整齐的在Ti基底上呈现。图2(c)是TiO2纳米管电极的SEM截面图像,可以发现电极截面呈现明显的管状结构。由图2(d)可见,TiO2纳米管电极在2θ在25.3°和48°处出现明显的衍射峰。这表明TiO2呈现锐钛矿结构。
-
图3(a)分别对比了光电催化(PEC)、电催化(EC)和光催化(PC)体系对次磷氧化和Cu回收效果的影响。结果表明:PEC体系效果最好,当电压为2.0 V,反应时间180 min,PEC、EC、PC 3个体系对次磷的氧化率分别为100%、11%和0,Cu的回收率分别是97%、7%和0。图3(b)反映了反应180 min时Cu在溶液、阳极、阴极的分布情况。结果证明,回收的Cu均沉积在阴极上。通过对不同体系进行比较,单独EC或者单独PC均不能实现高效率的次磷氧化和Cu回收;当电化学作用和光催化作用联合即PEC体系时,可以产生很好的协同作用。这是因为通过光激发TiO2半导体产生光生空穴与电子,外加偏压促进了空穴和电子的高效分离,大大提高了反应的氧化还原作用。
图4反映了TiO2纳米管电极作为光阳极在2.0 V下PEC体系次磷氧化过程中间产物的生成和P元素的平衡过程。可以看出,随着反应的进行,总磷的浓度基本保持不变,而亚磷酸盐的浓度随反应时间延长先升高后降低,正磷酸盐的浓度则一直呈现升高的趋势。由此可见,在次磷氧化过程中,次磷(P为+1价)先被氧化成为亚磷(P为+3价),进而最终被氧化成为正磷(P为+5价),且随着反应的进行,总磷浓度基本不变。
-
不同电压条件下TiO2纳米管电极作为光阳极的PEC体系对次磷氧化和Cu回收效率的影响如图5所示。随着电压的增大及反应时间的延长,次磷氧化和Cu回收的效率逐渐升高。当电压为2.0 V、反应时间为180 min时,1 mmol·L−1的次磷全部被氧化,其中,84%以正磷形式存在,剩下的16%以亚磷形式存在并且呈现继续下降趋势,同时0.5 mmol·L−1的Cu 全部以金属形式在阴极沉积回收。而当电压增加至2.5 V时,效果反而变差。其原因可能是,随着电压的增加,阳极析氧和阴极析氢等副反应越来越剧烈,从而抑制了污染物在电极表面的迁移,导致电极表面电流效率的降低以及能量的大量损耗[21]。
-
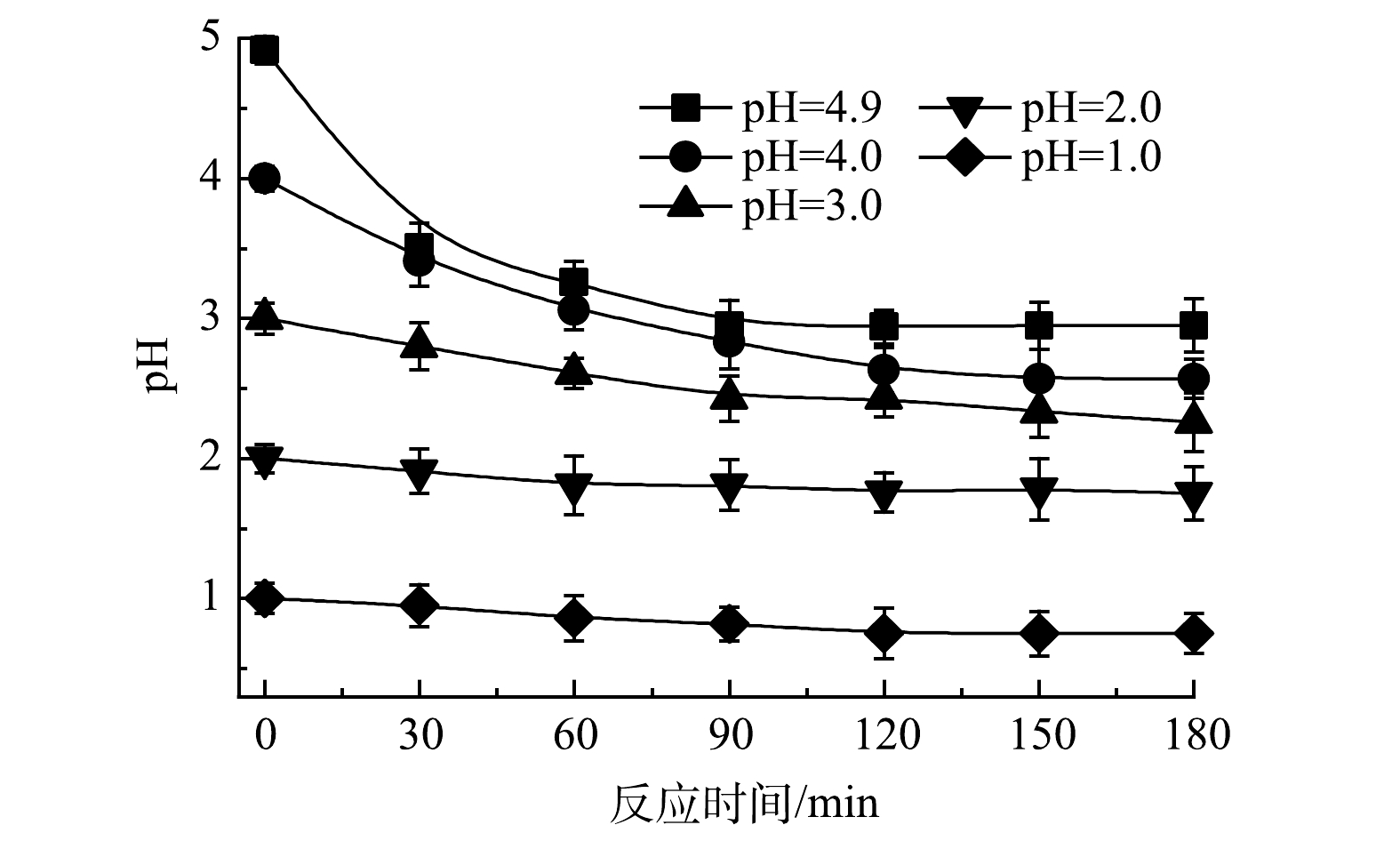
反应溶液的初始pH为4.9,由于Cu2+存在,Cu(OH)2的溶度积为2.2×10−20,由此计算得出,在本研究中pH大于5.8后会产生Cu(OH)2沉淀。因此,本研究利用H2SO4的稀释溶液调节溶液的初始pH为4.0、3.0、2.0、1.0进行对比实验。如图6所示,当溶液初始pH为4.9,即不用H2SO4调节时,对次磷氧化和Cu回收效果最好;当溶液初始pH降低后,次磷氧化和Cu回收效果均受到明显的抑制,同时,次磷氧化过程中生成的亚磷以及正磷也受到了抑制。由图7可以看出,随着反应的进行,pH均会下降。这说明反应过程中有大量氢离子释放,推测是因为在次磷最终氧化成为正磷的过程中,均会有氢离子释放[8]。pH能影响水中溶解氧(DO)含量[22],随着pH降低,DO含量减少,从而影响了·OH的生成,且在酸性条件下,·OH更易反应生成活性较弱的·OOH[23],因此,会影响体系次磷氧化的效率。此外,在单独电沉积Cu2+时由于阴极析发生氢反应造成溶液偏碱性,Cu2+水解生成铜的氧化物在阳极沉积生成沉淀物[13],而次磷氧化的同时在溶液中释放氢离子可以降低溶液pH,克服了Cu2+难以在阴极进行电化学沉积回收金属Cu的问题,使Cu2+有效沉积在阴极回收为金属Cu。
-
如图8所示,溶液采用的电解质不同时对次磷氧化和Cu回收效果也有影响。当电解质为Na2SO4和NaClO4时,反应效率基本无明显差异,但当电解质为NaCl时,反应效率明显提高,1 mmol·L−1次磷全部被氧化为正磷。进一步详细探讨了NaCl电解质在反应中的作用。如图9所示,在不同浓度NaCl对次磷氧化和Cu回收效果影响实验中,NaCl浓度越高,效果越高。其原因为,在PEC体系中,反应中的氯离子可以通过一系列反应生成活性氯(式(1)~式(3))[24]。在阳极表面生成的活性氯以氯气(Cl2)、次氯酸(HClO)和次氯酸根(ClO−)等形式在溶液中存在。图10是在不同NaCl浓度反应体系中,活性氯浓度的测定结果,NaCl浓度越高时,活性氯生成量越多。此外,有文献报道,在紫外光照射下,活性氯有利于进一步产生羟基自由基(·OH)和氯自由基(Cl·)(式(4))[25-26]。因此,当采用NaCl作为电解质时,氯离子的加入最终会促进以上自由基的产生,强化了反应效率。
-
为了探究反应过程中PEC体系下存在的主要活性物种,通过加入不同浓度的·OH自由基淬灭剂叔丁醇(TBA)来探究·OH对次磷氧化的作用。如图11(a)所示,考察了TBA对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效率的影响。结果表明:TBA对PEC体系中次磷氧化有明显的抑制作用,当TBA浓度为10 mmol·L−1时,次磷的去除率降低到60%,而TBA的加入对Cu回收无明显影响。这一结果表明,·OH 自由基对次磷氧化起重要作用。ESR检测结果也进一步验证了这一结果。如图11(b)所示,使用DMPO为捕获剂,在反应过程中观测到了特征的 DMPO-·OH络合物的信号,并随反应时间的延长而显著增强。以上结果表明,·OH自由基是次磷氧化的主要活性物种。
图12是反应过程中阴极钛片上Cu沉积物的XPS谱图。当反应时间分别为1、2、3 h时,Cu2p3/2的峰值在932.68 eV处出现,主要对应金属Cu的特征峰,且随反应的进行峰值强度有所增强,且并未检测到其他价态的Cu。由此证明在本研究中阴极钛片上回收的是金属Cu。
-
1)通过PEC、EC、PC 3个体系对次磷氧化和Cu回收效率比较,发现单独EC或者PC均不能实现高效率的次磷氧化和Cu回收,当电化学和光催化联合(PEC体系)时,可以产生很好的协同效果。
2)对于PEC体系中次磷氧化和Cu的回收效果,在电压为2.0 V、反应时间为180 min时1 mmol·L−1的次磷全部被氧化且84%以正磷形式存在,另外16%以亚磷形式存在并且呈现继续下降趋势,同时0.5 mmol·L−1的Cu 100%以金属形式在阴极沉积回收。当溶液初始pH为4.9时,采用NaCl作为电解质,可促进·OH的产生,1 mmol·L−1次磷全部被氧化为正磷,提高了反应效率。
3)在单独电沉积Cu2+时,由于阴极发生析氢反应造成溶液偏碱性,Cu2+水解生成铜的氧化物在阳极沉积生成沉淀物,而次磷氧化的同时在溶液中释放氢离子可以降低溶液pH,克服了Cu2+难以在阴极进行电化学沉积回收金属Cu的问题,使Cu2+有效沉积在阴极回收为金属Cu。
4) TBA对PEC体系中次磷的氧化有明显的抑制作用,且对Cu回收无明显影响,表明·OH为实现次磷氧化的主要活性物种。
5)采用光电催化技术处理含次磷和重金属铜废水,废水中的Cu2+在阴极电沉积生成金属Cu回收,而废水中次磷氧化后形成正磷,然后加入钙盐或铁盐等与正磷反应生成沉淀将磷在废水中去除同时回收磷。
TiO2纳米管阳极光电催化氧化次磷酸盐同时阴极回收金属铜
Photoelectrocatalytic oxidation of hypophosphite with TiO2 nanotube arrays anode and simultaneous recovery of copper on the cathode
-
摘要: 采用电化学阳极氧化法制备了高度有序阵列的二氧化钛(TiO2)纳米管电极,采用此电极作为光阳极在模拟太阳光(AM 1.5)照射下进行了光电催化氧化次磷酸盐且同时回收金属铜(Cu)的研究,比较了光电催化、电催化和光催化体系对次磷氧化和Cu回收效率的差异,详细探究了电压、初始pH、电解质种类对光电催化体系下次磷氧化和Cu回收效率的影响。结果表明,当电压为2.0 V,初始pH为4.9,电解质为10 mmol·L−1 NaCl,在180 min内,1 mmol·L−1次磷全部被氧化为正磷,同时0.5 mmol·L−1的Cu 100%以金属形式在阴极沉积回收。在该光电催化体系中,在一定范围内,外加偏压的增加、溶液初始pH的升高和NaCl浓度的提高有利于次磷氧化和Cu回收。自由基淬灭实验和电子自旋共振实验结果证明,该体系中羟基自由基为氧化次磷的主要活性物种。以上研究结果可为含次磷和重金属铜废水的资源化处理提供参考。Abstract: Highly ordered TiO2 nanotube arrays electrode fabricated using Ti plate by electrochemical anodization method was used as a photoanode under simulated sunlight (AM 1.5G, 100 mW cm−2) irradiation for hypophosphite (H2PO2−) oxidation and Cu recovery. The performance for H2PO2− oxidation and Cu recovery in the photoelectrocatalytic (PEC) process was evaluated compared with the individual electrocatalytic (EC) or photocatalytic (PC) process. The effects of applied bias, initial solution pH and electrolyte type were investigated in detail. The results show that for 1 mmol·L−1 H2PO2− and 0.5 mmol·L−1 Cu2+, 100 % H2PO2− was oxidized to PO43−, 100 % Cu2+ was recovered as metallic Cu within 180 min at an applied bias of 2.0 V, initial solution pH of 4.9 and electrolyte of 10 mmol·L−1 NaCl. In this constructed PEC system, the increase of applied bias, initial solution pH or NaCl concentration within a certain range was proved to be favorable in the efficiency of H2PO2− oxidation and Cu recovery. The radical quenching experiments and electron spin resonance results indicated that hydroxyl (·OH) radicals were the major active species for H2PO− oxidation. This study can provide a reference for the resource treatment of wastewater containing H2PO2− and heavy metal Cu2+ ions.
-
化学镀铜工艺是指在无外加电流的条件下,利用合适的还原剂(常用次磷酸盐),使溶液中的铜离子在具有催化活性的基体表面还原沉积出金属铜,形成铜镀层的一种工艺[1-3]。近年来,化学镀铜工艺在表面处理行业中所占的地位在不断上升,在机械工业、航空航天、电子工业等各行各业都有着越来越广泛的应用[4]。在化学镀铜工艺中,会产生大量的化学镀铜废水,将这些废水进行处理和回收,对保护生态环境,变废为宝,提高经济效益,尤为重要[5]。
化学镀铜废水主要来源于清洗零部件时所产生的清洗废水,因此,也可以称为化学镀铜清洗废水,其中主要含有铜离子和次磷酸盐等污染物[6]。过量的铜会刺激人类的消化系统,引起腹痛、呕吐等,严重时可造成中毒。而含铜废水进入水体后,成为持久性污染物,危机植物生长,影响水产养殖。当进入土壤时,会在土壤和作物中富集,经过一系列的环境迁移转化最终进入食物链,对人类健康产生威胁[7]。与正磷酸盐比,次磷酸盐由于其溶解度大且难与沉淀剂反应形成沉淀,导致水体富营养化严重的同时亦造成磷资源的流失[8]。故次磷的去除通常需氧化成正磷,再加入沉淀剂将正磷彻底去除或回收[9]。因此,对化学镀铜清洗废水的处理并回收磷和铜成为当前研究热点之一。
目前,含铜废水处理方法有很多,例如物化沉淀法、膜分离法、吸附法、混凝法和电解法等[10-11]。其中电解法可以使铜离子以金属铜的形式沉积在阴极上,实现了金属铜的回收[12]。除电解法外,其他方法只改变了铜离子存在形态,使铜离子发生迁移,但污染并没有彻底消除。而采用电解法时,当溶液为偏碱性条件下,铜离子很容易水解生成铜的氧化物,累积在阳极或生成沉淀物,使其难以在阴极进行电化学沉积回收[13]。因此,在电解法沉积铜离子实现阴极回收金属铜时,控制溶液的pH较为重要。
光电催化法是一种将光化学和电化学法相结合的方法,通过对半导体光催化剂施加外加偏压作用实现光生电子和光生空穴的有效分离,有效促进自由基的生成,提升污染物的降解效果[14]。光电催化作为高级氧化技术研究热点之一,是一种不仅能产生强氧化性活性物种实现污染物氧化降解,同时也能利用光生电子的还原能力实现阴极还原回收重金属的有效方法[15]。具有运行成本较低、温度和压力适应范围广、可实现有机物矿化且无二次污染等优点,在环境保护水处理领域越来受到关注[16]。二氧化钛(TiO2)纳米管光电极具有高度有序、比表面积大、电池容量高及量子化学效应强等优点,被广泛应用于纳米微电子、光伏器件、水分解产氢、环境污染物降解等领域[17-19]。有研究[20]表明,利用TiO2纳米管电极作为光阳极可有效实现铜氰络合物的氧化破络合同时电还原回收金属铜。
本研究采用电化学阳极氧化法制得的TiO2纳米管电极为光阳极和钛片(Ti)为阴极,在模拟太阳光(AM 1.5)照射进行光电催化处理次磷酸根离子(H2PO2−)和重金属铜离子(Cu2+)同时回收金属铜(Cu)。对TiO2纳米管电极进行了表征分析;对比分析了光电催化(PEC)、电催化(EC)和光催化(PC)体系对次磷氧化和Cu回收效果;考察了电压、初始PH、电解质种类对PEC体系下次磷氧化和Cu回收效率的影响,并进一步探讨该体系的反应机理。本研究结果可为含次磷和重金属铜的工业废水资源化处理提供参考。
1. 实验部分
1.1 实验材料及主要试剂
电极材料钛片购自北京恒力钛工贸公司。实验用次磷酸钠(NaH2PO2·H2O)、亚磷酸钠(Na2HPO3·5H2O)、硫酸铵((NH4)2SO4)、氟化铵(NH4F)、丙三醇(C3H8O3)、硫酸铜(CuSO4)、硝酸(HNO3)、氢氟酸 (HF)、氢氧化钠(NaOH)、硫酸(H2SO4)、过硫酸钾(K2S2O8)、抗坏血酸(C6H8O6)、钼酸铵((NH4)6Mo7O24·4H2O)、酒石酸锑钾(KSbC4H4O7·1/2H2O)、硫酸钠(Na2SO4)、高氯酸钠(NaClO4)、氯化钠(NaCl)、叔丁醇(C4H10O)等均购自国药集团化学试剂公司,均为分析纯。
1.2 实验装置及主要仪器
光电催化氧化装置如图1所示,其中包括石英反应器(长5.0 cm,宽5.0 cm,高6.0 cm),150 W的氙灯(Zolix instruments Co,China),直流电源(DH1718E-4,北京大华仪器公司,中国),磁力搅拌器(MS-H380-Pro,北京大龙兴创实验仪器有限公司,中国)。在氙灯光源处安装了一个AM 1.5滤光片,使其照射到反应器内阳极的光为模拟的太阳光。阳极为TiO2纳米管电极,阴极为钛片(长5.0 cm,宽3.0 cm,厚0.2 mm)。
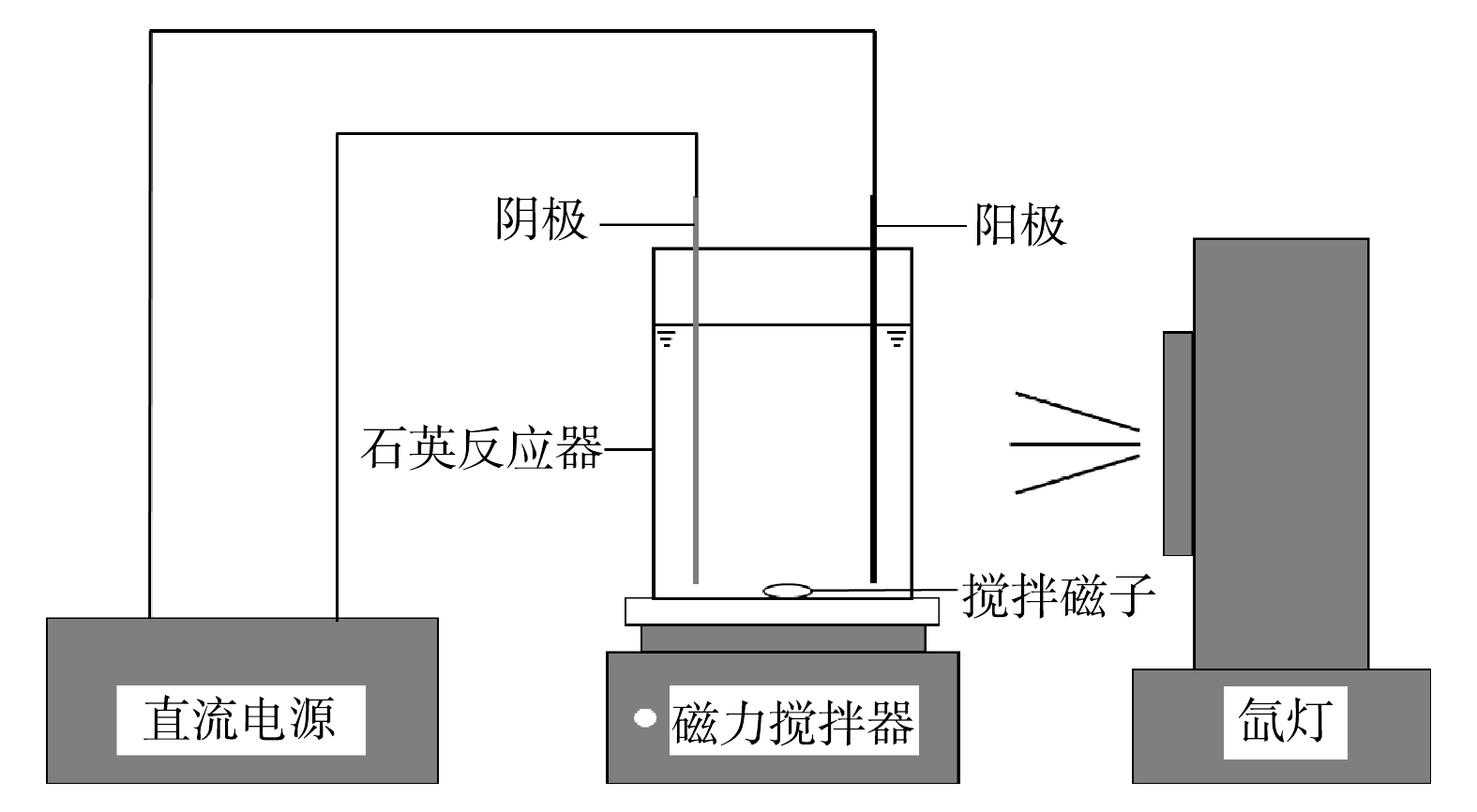 图 1 光电催化氧化实验装置示意图Figure 1. Schematic diagram of the photoelectrocatalytic oxidation system
图 1 光电催化氧化实验装置示意图Figure 1. Schematic diagram of the photoelectrocatalytic oxidation system1.3 实验方法
1) TiO2纳米管电极的制备。采用阳极氧化法制备电极,制备方法参考文献[20]。钛片预处理:将钛片分别在无水乙醇和丙酮中超声清洗,后用不同目数金相砂纸(200、400、600、1000 目)依次打磨,去离子水清洗,将清洗后的钛片置于HF/HNO3/H2O体积比为1∶4∶5 的混合溶液中浸泡1 min,使钛片化学抛光。电解质制备方法:配制100 g质量比为0.5% NH4F + 1% (NH4)2SO4+ 90% C3H8O3的混合水溶液,即为所需电解质电解质溶液。TiO2纳米管电极制备方法:阳极为预处理钛片,阴极为铂丝,两级间距为20 mm,垂直插入电解质中,电压为20 V,室温下阳极氧化10 h,将氧化后的电极放入马弗炉中450 ºC热处理2 h,升温程序为5 ºC·min−1。
2)降解实验。含次磷酸根离子和重金属铜离子的化学镀铜模拟废水制备方法如下:配制1.0 mmol·L−1的NaH2PO2溶液;将CuSO4溶于其中使Cu2+浓度为0.5 mmol·L−1,即为所需化学镀铜模拟废水。取上述溶液120 mL置于反应器中,开启直流电源在两极间施以一定的电压,同时开启氙灯,反应时间为180 min,取样时间为0、30、60、90、120、150、180 min。反应液以10 mmol·L−1的Na2SO4为电解质。光催化反应时只开启氙灯,两极之间不施加电压。电化学反应时只开启直流电源。
3)表征及分析方法。电极表面形态通过场发射扫描电镜(SEM,SU-8010,日本日立公司)进行观察;晶体结构通过X射线衍射(XRD,XPert Pro MPD,荷兰帕纳科分析仪器有限公司)进行表征,所用的仪器是配有石墨晶体单色器的Rigaku D/max-B衍射仪,2θ扫描范围为10°~90°,扫描速率为0.5°·min−1,加速电压和工作电流分别为30 kV和30 mA;阴极回收Cu价态通过X射线光电子能谱仪(XPS,PHI Quantera SXM,日本ULVAC-PHI 公司)进行测定;电子自旋共振波谱仪(ESR,A300−10/12,德国布鲁克有限公司)用来检测自由基的生成。总磷的测定方法为采用国标过硫酸钾氧化-钼酸铵分光光度法;正磷的测定方法为采用国标钼酸铵分光光度法;次磷和亚磷采用离子色谱(IC,ICS-1500,美国戴安公司)测定,所用色谱柱为AS23分析柱和AG23保护住,淋洗液为4.5 mmol·L−1的Na2CO3和0.8 mmol·L−1的NaHCO3溶液,流速为1.0 mL·min−1;重金属Cu含量采用电感耦合等离子体发射光谱仪(ICP-OES,P700,美国安捷伦科技公司)来测定。
2. 结果与讨论
2.1 TiO2纳米管电极表征分析
图2(a)和图2(b)分别是Ti基底和TiO2纳米管电极的SEM正面图像。可见,Ti基底表面平整,经阳极氧化法制备的TiO2纳米管电极上,纳米管阵列高度有序、管径均匀、排列整齐的在Ti基底上呈现。图2(c)是TiO2纳米管电极的SEM截面图像,可以发现电极截面呈现明显的管状结构。由图2(d)可见,TiO2纳米管电极在2θ在25.3°和48°处出现明显的衍射峰。这表明TiO2呈现锐钛矿结构。
 图 2 Ti基底和TiO2纳米管电极的SEM图像和XRD谱图Figure 2. SEM images and XRD patterns of Ti substrate and TiO2 nanotube arrays electrode
图 2 Ti基底和TiO2纳米管电极的SEM图像和XRD谱图Figure 2. SEM images and XRD patterns of Ti substrate and TiO2 nanotube arrays electrode2.2 光电催化(PEC)、电催化(EC)和光催化(PC)体系对次磷氧化和Cu回收效果对比
图3(a)分别对比了光电催化(PEC)、电催化(EC)和光催化(PC)体系对次磷氧化和Cu回收效果的影响。结果表明:PEC体系效果最好,当电压为2.0 V,反应时间180 min,PEC、EC、PC 3个体系对次磷的氧化率分别为100%、11%和0,Cu的回收率分别是97%、7%和0。图3(b)反映了反应180 min时Cu在溶液、阳极、阴极的分布情况。结果证明,回收的Cu均沉积在阴极上。通过对不同体系进行比较,单独EC或者单独PC均不能实现高效率的次磷氧化和Cu回收;当电化学作用和光催化作用联合即PEC体系时,可以产生很好的协同作用。这是因为通过光激发TiO2半导体产生光生空穴与电子,外加偏压促进了空穴和电子的高效分离,大大提高了反应的氧化还原作用。
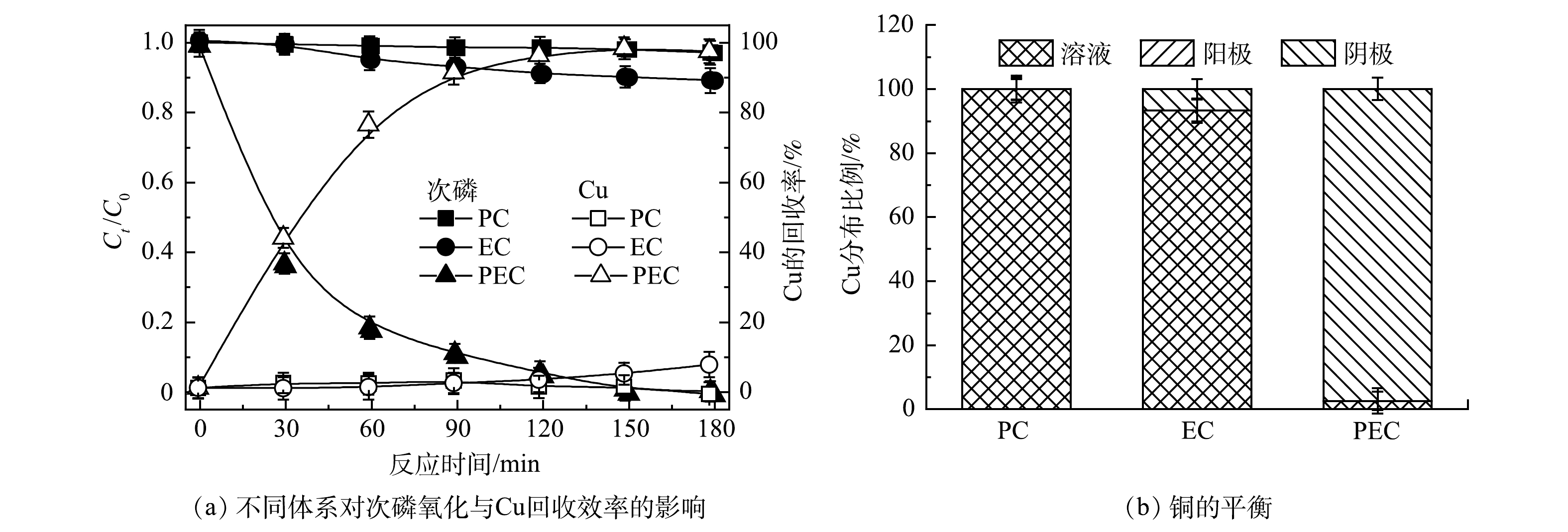 图 3 TiO2纳米管光阳极在光电催化(PEC)、电催化(EC)和光催化(PC)下对次磷氧化与Cu回收效果对比Figure 3. Hypophosphite oxidation and Cu2+ ions recovery via photoelectrocatalytic (PEC), electrocatalytic (EC), and photocatalytic (PC) processes using TiO2 nanotube arrays photoanode
图 3 TiO2纳米管光阳极在光电催化(PEC)、电催化(EC)和光催化(PC)下对次磷氧化与Cu回收效果对比Figure 3. Hypophosphite oxidation and Cu2+ ions recovery via photoelectrocatalytic (PEC), electrocatalytic (EC), and photocatalytic (PC) processes using TiO2 nanotube arrays photoanode图4反映了TiO2纳米管电极作为光阳极在2.0 V下PEC体系次磷氧化过程中间产物的生成和P元素的平衡过程。可以看出,随着反应的进行,总磷的浓度基本保持不变,而亚磷酸盐的浓度随反应时间延长先升高后降低,正磷酸盐的浓度则一直呈现升高的趋势。由此可见,在次磷氧化过程中,次磷(P为+1价)先被氧化成为亚磷(P为+3价),进而最终被氧化成为正磷(P为+5价),且随着反应的进行,总磷浓度基本不变。
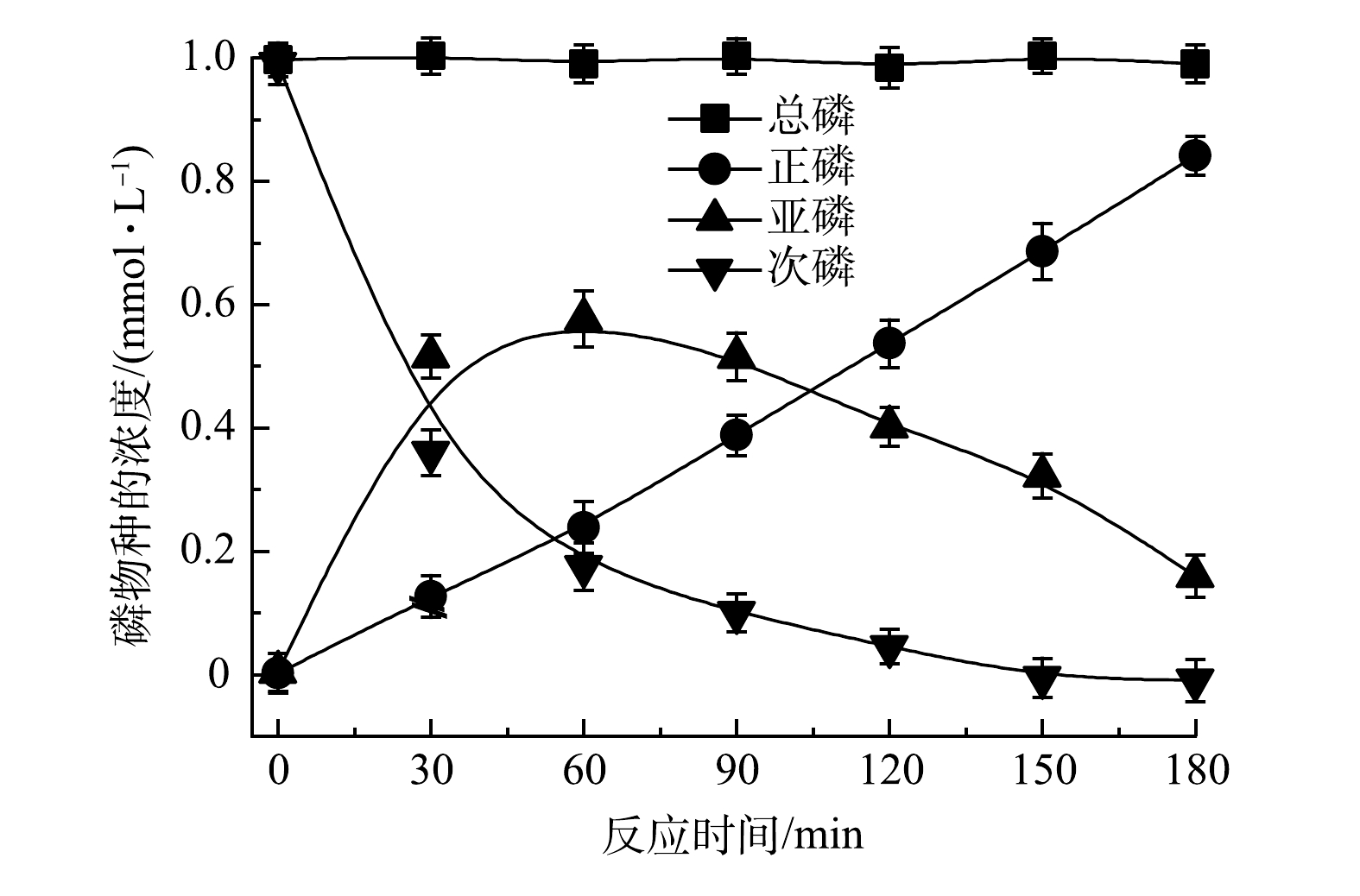 图 4 在电压为2.0 V条件下PEC体系次磷氧化过程中间产物的生成和P元素平衡Figure 4. Generated intermediates and P mass balance in the PEC process for hypophosphite oxidation at 2.0 V bias potential
图 4 在电压为2.0 V条件下PEC体系次磷氧化过程中间产物的生成和P元素平衡Figure 4. Generated intermediates and P mass balance in the PEC process for hypophosphite oxidation at 2.0 V bias potential2.3 电压对次磷氧化和Cu回收效果的影响
不同电压条件下TiO2纳米管电极作为光阳极的PEC体系对次磷氧化和Cu回收效率的影响如图5所示。随着电压的增大及反应时间的延长,次磷氧化和Cu回收的效率逐渐升高。当电压为2.0 V、反应时间为180 min时,1 mmol·L−1的次磷全部被氧化,其中,84%以正磷形式存在,剩下的16%以亚磷形式存在并且呈现继续下降趋势,同时0.5 mmol·L−1的Cu 全部以金属形式在阴极沉积回收。而当电压增加至2.5 V时,效果反而变差。其原因可能是,随着电压的增加,阳极析氧和阴极析氢等副反应越来越剧烈,从而抑制了污染物在电极表面的迁移,导致电极表面电流效率的降低以及能量的大量损耗[21]。
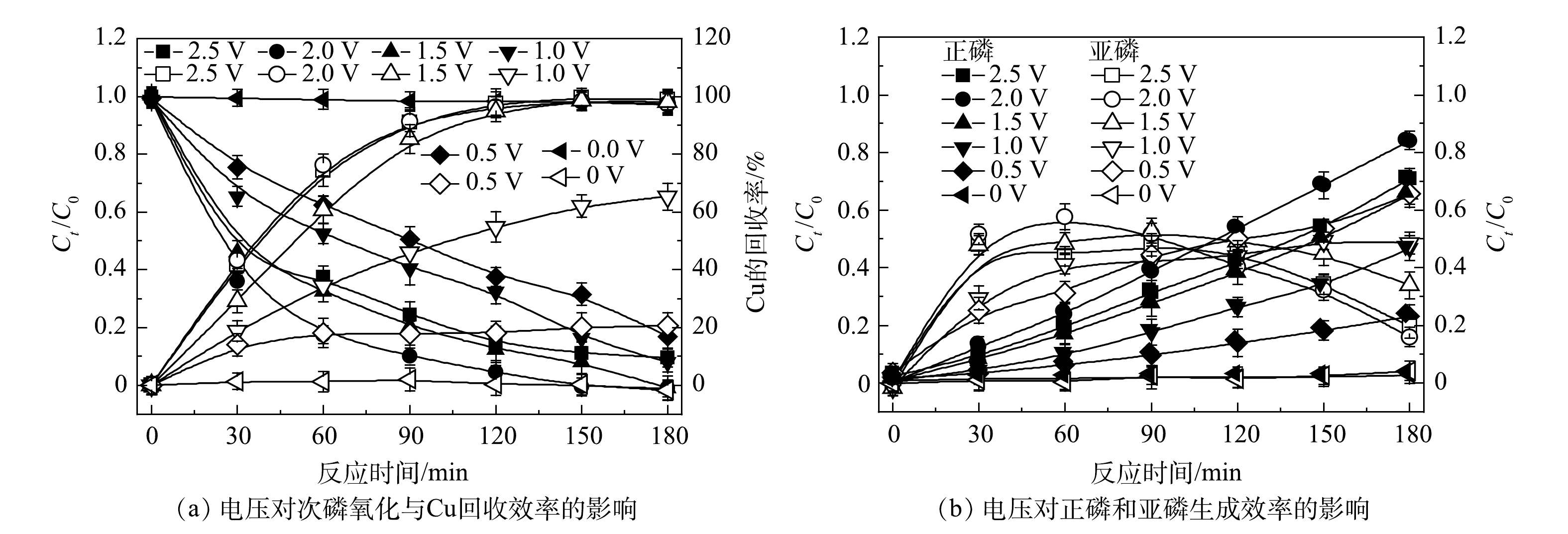 图 5 外加偏压对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响Figure 5. Effect of applied voltages on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode
图 5 外加偏压对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响Figure 5. Effect of applied voltages on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode2.4 初始pH对次磷氧化和Cu回收效果的影响
反应溶液的初始pH为4.9,由于Cu2+存在,Cu(OH)2的溶度积为2.2×10−20,由此计算得出,在本研究中pH大于5.8后会产生Cu(OH)2沉淀。因此,本研究利用H2SO4的稀释溶液调节溶液的初始pH为4.0、3.0、2.0、1.0进行对比实验。如图6所示,当溶液初始pH为4.9,即不用H2SO4调节时,对次磷氧化和Cu回收效果最好;当溶液初始pH降低后,次磷氧化和Cu回收效果均受到明显的抑制,同时,次磷氧化过程中生成的亚磷以及正磷也受到了抑制。由图7可以看出,随着反应的进行,pH均会下降。这说明反应过程中有大量氢离子释放,推测是因为在次磷最终氧化成为正磷的过程中,均会有氢离子释放[8]。pH能影响水中溶解氧(DO)含量[22],随着pH降低,DO含量减少,从而影响了·OH的生成,且在酸性条件下,·OH更易反应生成活性较弱的·OOH[23],因此,会影响体系次磷氧化的效率。此外,在单独电沉积Cu2+时由于阴极析发生氢反应造成溶液偏碱性,Cu2+水解生成铜的氧化物在阳极沉积生成沉淀物[13],而次磷氧化的同时在溶液中释放氢离子可以降低溶液pH,克服了Cu2+难以在阴极进行电化学沉积回收金属Cu的问题,使Cu2+有效沉积在阴极回收为金属Cu。
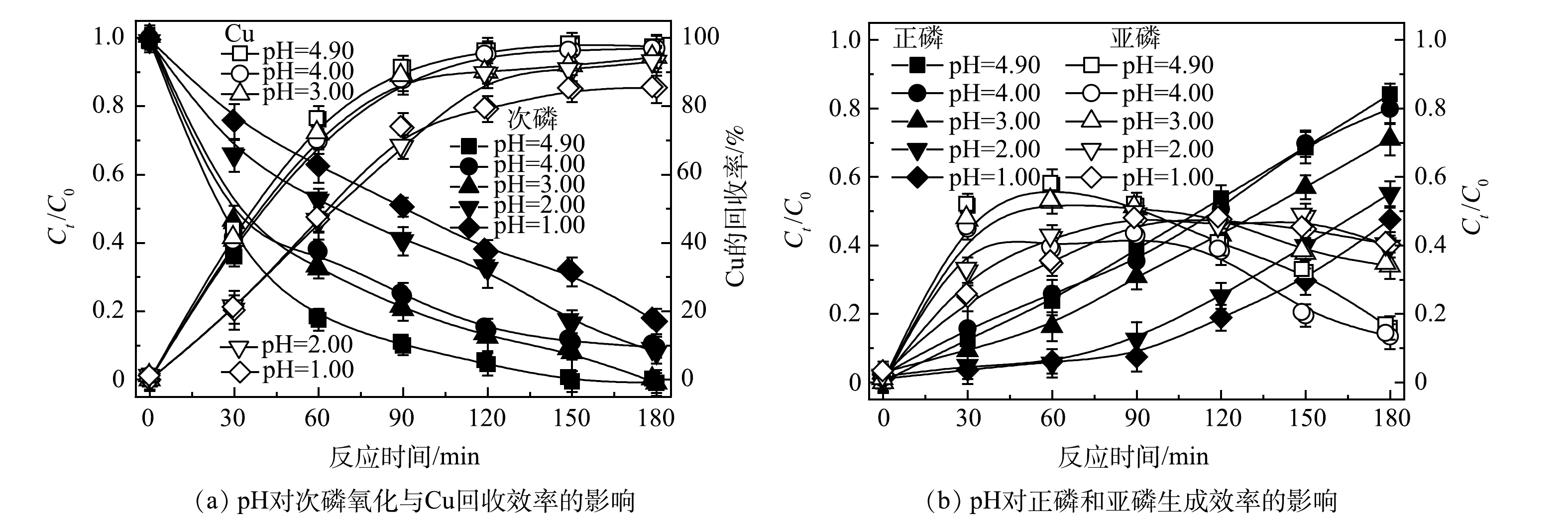 图 6 溶液初始pH对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响Figure 6. Effect of initial pH on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode
图 6 溶液初始pH对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响Figure 6. Effect of initial pH on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode2.5 电解质种类对次磷氧化和Cu回收效果的影响
如图8所示,溶液采用的电解质不同时对次磷氧化和Cu回收效果也有影响。当电解质为Na2SO4和NaClO4时,反应效率基本无明显差异,但当电解质为NaCl时,反应效率明显提高,1 mmol·L−1次磷全部被氧化为正磷。进一步详细探讨了NaCl电解质在反应中的作用。如图9所示,在不同浓度NaCl对次磷氧化和Cu回收效果影响实验中,NaCl浓度越高,效果越高。其原因为,在PEC体系中,反应中的氯离子可以通过一系列反应生成活性氯(式(1)~式(3))[24]。在阳极表面生成的活性氯以氯气(Cl2)、次氯酸(HClO)和次氯酸根(ClO−)等形式在溶液中存在。图10是在不同NaCl浓度反应体系中,活性氯浓度的测定结果,NaCl浓度越高时,活性氯生成量越多。此外,有文献报道,在紫外光照射下,活性氯有利于进一步产生羟基自由基(·OH)和氯自由基(Cl·)(式(4))[25-26]。因此,当采用NaCl作为电解质时,氯离子的加入最终会促进以上自由基的产生,强化了反应效率。
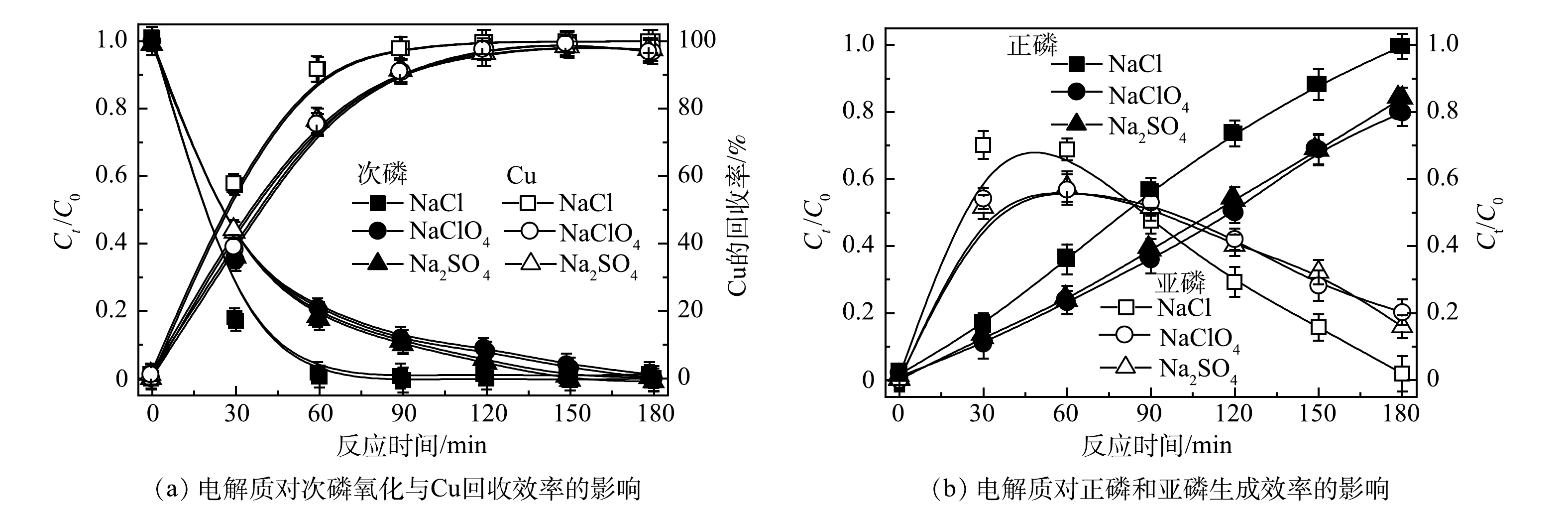 图 8 电解质种类对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响Figure 8. Effect of electrolyte type on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode
图 8 电解质种类对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响Figure 8. Effect of electrolyte type on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode 图 9 不同浓度NaCl对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响Figure 9. Effect of different NaCl concentrations on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode
图 9 不同浓度NaCl对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响Figure 9. Effect of different NaCl concentrations on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode 图 10 不同浓度NaCl在反应过程中活性氯的产量Figure 10. The amount of reactive chlorine produced in the PEC process with different concentrations of NaCl
图 10 不同浓度NaCl在反应过程中活性氯的产量Figure 10. The amount of reactive chlorine produced in the PEC process with different concentrations of NaCl2Cl−→Cl2+2e− (1) Cl2+H2O→HClO+H++Cl− (2) HClO→H++ClO− (3) HClO+hν→⋅OH+Cl⋅ (4) 2.6 反应机理及阴极回收铜的效果
为了探究反应过程中PEC体系下存在的主要活性物种,通过加入不同浓度的·OH自由基淬灭剂叔丁醇(TBA)来探究·OH对次磷氧化的作用。如图11(a)所示,考察了TBA对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效率的影响。结果表明:TBA对PEC体系中次磷氧化有明显的抑制作用,当TBA浓度为10 mmol·L−1时,次磷的去除率降低到60%,而TBA的加入对Cu回收无明显影响。这一结果表明,·OH 自由基对次磷氧化起重要作用。ESR检测结果也进一步验证了这一结果。如图11(b)所示,使用DMPO为捕获剂,在反应过程中观测到了特征的 DMPO-·OH络合物的信号,并随反应时间的延长而显著增强。以上结果表明,·OH自由基是次磷氧化的主要活性物种。
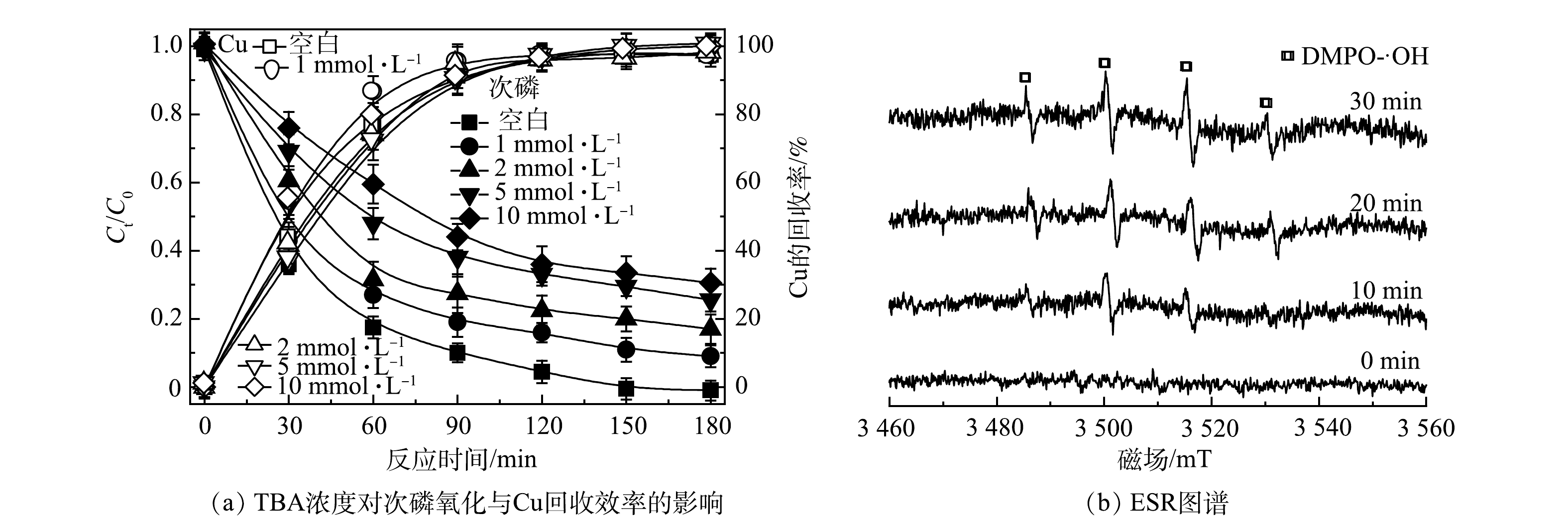 图 11 (a) 不同浓度TBA对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响以及反应过程中的DMPO−•OH的ESR图谱Figure 11. Effect of different TBA concentrations on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode, and ESR signals of DMPO−•OH
图 11 (a) 不同浓度TBA对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响以及反应过程中的DMPO−•OH的ESR图谱Figure 11. Effect of different TBA concentrations on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode, and ESR signals of DMPO−•OH图12是反应过程中阴极钛片上Cu沉积物的XPS谱图。当反应时间分别为1、2、3 h时,Cu2p3/2的峰值在932.68 eV处出现,主要对应金属Cu的特征峰,且随反应的进行峰值强度有所增强,且并未检测到其他价态的Cu。由此证明在本研究中阴极钛片上回收的是金属Cu。
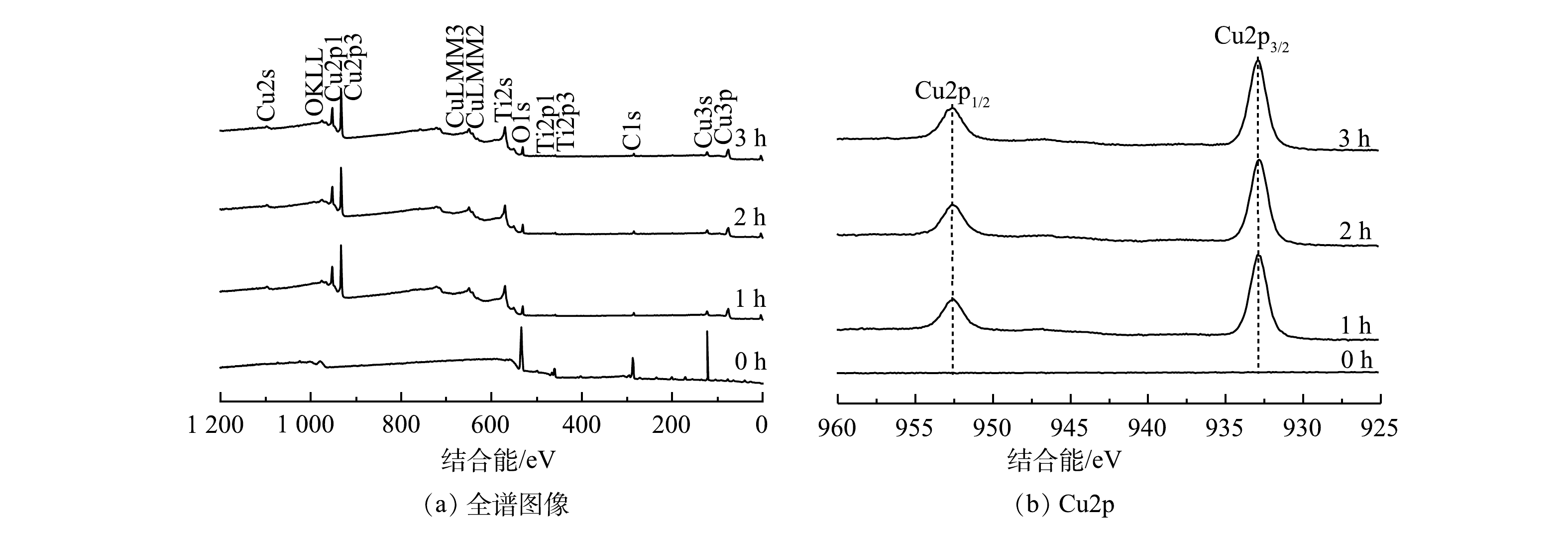 图 12 不同反应时间阴极沉积物的XPS图谱Figure 12. XPS spectra of deposits on cathode films at different reaction time
图 12 不同反应时间阴极沉积物的XPS图谱Figure 12. XPS spectra of deposits on cathode films at different reaction time3. 结论
1)通过PEC、EC、PC 3个体系对次磷氧化和Cu回收效率比较,发现单独EC或者PC均不能实现高效率的次磷氧化和Cu回收,当电化学和光催化联合(PEC体系)时,可以产生很好的协同效果。
2)对于PEC体系中次磷氧化和Cu的回收效果,在电压为2.0 V、反应时间为180 min时1 mmol·L−1的次磷全部被氧化且84%以正磷形式存在,另外16%以亚磷形式存在并且呈现继续下降趋势,同时0.5 mmol·L−1的Cu 100%以金属形式在阴极沉积回收。当溶液初始pH为4.9时,采用NaCl作为电解质,可促进·OH的产生,1 mmol·L−1次磷全部被氧化为正磷,提高了反应效率。
3)在单独电沉积Cu2+时,由于阴极发生析氢反应造成溶液偏碱性,Cu2+水解生成铜的氧化物在阳极沉积生成沉淀物,而次磷氧化的同时在溶液中释放氢离子可以降低溶液pH,克服了Cu2+难以在阴极进行电化学沉积回收金属Cu的问题,使Cu2+有效沉积在阴极回收为金属Cu。
4) TBA对PEC体系中次磷的氧化有明显的抑制作用,且对Cu回收无明显影响,表明·OH为实现次磷氧化的主要活性物种。
5)采用光电催化技术处理含次磷和重金属铜废水,废水中的Cu2+在阴极电沉积生成金属Cu回收,而废水中次磷氧化后形成正磷,然后加入钙盐或铁盐等与正磷反应生成沉淀将磷在废水中去除同时回收磷。
-
图 1 光电催化氧化实验装置示意图
Figure 1. Schematic diagram of the photoelectrocatalytic oxidation system
图 2 Ti基底和TiO2纳米管电极的SEM图像和XRD谱图
Figure 2. SEM images and XRD patterns of Ti substrate and TiO2 nanotube arrays electrode
图 3 TiO2纳米管光阳极在光电催化(PEC)、电催化(EC)和光催化(PC)下对次磷氧化与Cu回收效果对比
Figure 3. Hypophosphite oxidation and Cu2+ ions recovery via photoelectrocatalytic (PEC), electrocatalytic (EC), and photocatalytic (PC) processes using TiO2 nanotube arrays photoanode
图 4 在电压为2.0 V条件下PEC体系次磷氧化过程中间产物的生成和P元素平衡
Figure 4. Generated intermediates and P mass balance in the PEC process for hypophosphite oxidation at 2.0 V bias potential
图 5 外加偏压对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响
Figure 5. Effect of applied voltages on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode
图 6 溶液初始pH对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响
Figure 6. Effect of initial pH on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode
图 8 电解质种类对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响
Figure 8. Effect of electrolyte type on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode
图 9 不同浓度NaCl对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响
Figure 9. Effect of different NaCl concentrations on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode
图 10 不同浓度NaCl在反应过程中活性氯的产量
Figure 10. The amount of reactive chlorine produced in the PEC process with different concentrations of NaCl
图 11 (a) 不同浓度TBA对TiO2纳米管电极作为光阳极下PEC对次磷氧化与Cu回收效果的影响以及反应过程中的DMPO−•OH的ESR图谱
Figure 11. Effect of different TBA concentrations on hypophosphite oxidation and Cu2+ ions recovery via PEC process using TiO2 nanotube arrays photoanode, and ESR signals of DMPO−•OH
-
[1] 赵大伟, 张鹏. 化学镀铜的应用与发展[J]. 黑龙江冶金, 2010, 31(1): 38-39. [2] 田庆华, 闫剑锋, 郭雪益. 化学镀铜的应用与发展概况[J]. 电镀与涂饰, 2007, 26(4): 38-41. doi: 10.3969/j.issn.1004-227X.2007.04.012 [3] LIN Y M, YEN S C. Effects of additives and chelating agents on electroless copper plating[J]. Applied Surface Science, 2001, 178(1-4): 116-126. doi: 10.1016/S0169-4332(01)00306-3 [4] YUAN X L, GAO J, YANG Z F, et al. New electroless copper plating bath using sodium hypophosphite as reductant[J]. Surface Engineering, 2012, 28(5): 377-381. doi: 10.1179/1743294411Y.0000000049 [5] 黄万抚, 胡昌顺, 曹明帅, 等. 难处理含铜废水处理技术研究[J]. 应用化工, 2018, 47(10): 2248-2253. doi: 10.3969/j.issn.1671-3206.2018.10.047 [6] 李立清, 冯罗, 吴盼旺, 等. 新型次磷酸钠体系化学镀铜添加剂及其对镀液和镀层性能的影响[J]. 表面技术, 2020, 49(7): 329-337. [7] CHANG Y, DENG L, MENG X Y, et al. Closed-loop electrochemical recycling of spent copper(II) from etchant wastewater using a carbon nanotube modified graphite felt anode[J]. Environmental Science & Technology, 2018, 52: 5940-5948. [8] LIU P, LI C L, LIANG X G, et al. Advanced oxidation of hypophosphite and phosphite using a UV/H2O2 process[J]. Environmental Technology, 2013, 34(15): 2231-2239. doi: 10.1080/09593330.2013.765917 [9] LIU P, Li C, LIANG X, et al. Recovery of high purity ferric phosphate from a spent electroless nickel plating bath[J]. Green Chemistry, 2014, 16(3): 1217-1224. doi: 10.1039/C3GC41779D [10] 郭燕妮, 方增坤, 胡杰华, 等. 化学沉淀法处理含重金属废水的研究进展[J]. 工业水处理, 2011, 31(12): 9-12. doi: 10.11894/1005-829x.2011.31(12).9 [11] 王蓬勃, 李金花, 周保学, 等. 电镀含铜废水的资源化回收利用[J]. 环境科学与技术, 2020, 43(S2): 184-187. [12] 张惠灵, 杨瑾, 吴健, 等. 电沉积法回收离子交换再生液中Cu2+的研究[J]. 电镀与精饰, 2014, 36(3): 43-46. doi: 10.3969/j.issn.1001-3849.2014.03.011 [13] ZHAO X, ZHANG J J, QIAO M, et al. Enhanced photoelectrocatalytic decomposition of copper cyanide complexes and simultaneous recovery of copper with a Bi2MoO6 electrode under visible light by EDTA/K4P2O7[J]. Environmental Science & Technology, 2015, 49(7): 4567-4574. [14] 陈佩仪, 李彦旭, 孙楹煌, 等. 光电催化水处理技术研究进展[J]. 工业水处理, 2005, 25(12): 13-17. doi: 10.3969/j.issn.1005-829X.2005.12.004 [15] JEON T H, KOO M S, KIM H, et al. Dual-functional photocatalytic and photoelectrocatalytic systems for energy- and resource-recovering water treatment[J]. ACS Catalysis, 2018, 8(12): 11542-11563. doi: 10.1021/acscatal.8b03521 [16] PARK H, PARK Y, KIM W, et al. Surface modification of TiO2 photocatalyst for environmental applications[J]. Journal of Photochemistry and Photobiology C-Photochemistry Reviews, 2013, 15: 1-20. doi: 10.1016/j.jphotochemrev.2012.10.001 [17] MACAK J M, TSUCHIYA H, TAVEIRA L, et al. Smooth anodic TiO2 nanotube[J]. Angewandte Chemie International Edition, 2005, 44: 7463-7465. doi: 10.1002/anie.200502781 [18] MOR G K, SHANKAR K, PAULOSE M, et al. Use of highly-ordered TiO2 nanotube arrays in dye-sensitized solar cells[J]. Nano Letters, 2006, 6: 215-218. doi: 10.1021/nl052099j [19] WANG J, LIN ZQ. Anodic Formation of Ordered TiO2 Nanotube Arrays: Effects of electrolyte temperature and anodization potential[J]. Journal of Physical Chemistry C, 2009, 113: 4026-4030. doi: 10.1021/jp811201x [20] ZHAO X, ZHANG J J, QU J H. Photoelectrocatalytic oxidation of Cu-cyanides and Cu-EDTA at TiO2 nanotube electrode[J]. Electrochimica Acta, 2015, 180: 129-137. doi: 10.1016/j.electacta.2015.08.103 [21] ÖĞÜTVEREN Ü B, TÖRÜ E, KOPARAL S. Removal of cyanide by anodic oxidation for wastewater treatment[J]. Water Research, 1999,33(8): 1851-1856. [22] CHEN W S, HUANHG C P. Mineralization of aniline in aqueous solution by electrochemical activation of per-sulfate[J]. Chemosphere, 2015, 125: 175-181. doi: 10.1016/j.chemosphere.2014.12.053 [23] MILLER J S, OLEJNIK D. Photolysis of polycyclic aromatic hydrocarbons in water[J]. Water Research, 2001, 35(1): 233-243. doi: 10.1016/S0043-1354(00)00230-X [24] HERNLEM B J, Electrolytic destruction of urea in dilute chloride solution using DSA electrodes in a recycled batch cell[J]. Water Research, 2005, 39(11): 2245-2252. [25] NAM S W, YOON Y, CHOI D J, et al. Degradation characteristics of metoprolol during UV/chlorination reaction and a factorial design optimization[J]. Journal of Hazardous Materials, 2015, 285: 453-463. doi: 10.1016/j.jhazmat.2014.11.052 [26] XIAO S, QU J, ZHAO X, LIU H, et al. Electrochemical process combined with UV light irradiation for synergistic degradation of ammonia in chloride-containing solutions[J]. Water Research, 2009, 43(5): 1432-1440. doi: 10.1016/j.watres.2008.12.023 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4777
- HTML全文浏览数: 4777
- PDF下载数: 78
- 施引文献: 0
