










 下载:
下载:










-
有机氯农药(organochlorine pesticides,OCPs)是一类人工合成的氯代芳香烃衍生物,是我国最早大规模生产使用的广谱性杀虫剂。OCPs因高毒性、生物蓄积性、半挥发性和长距离迁移性而被列为《关于持久性有机污染物的斯德哥尔摩公约》首批持久性有机污染物[1-2]。OCPs可通过食物链、呼吸道和皮肤等途径进入人体内,分布到各个器官和组织,具有慢性毒性、致癌性和遗传毒性,严重威胁人类健康。我国作为农业大国,在20世纪50—70年代时喷洒过大量的OCPs,直到1983年开始才禁止此类农药在农田中使用。虽然OCPs已禁用多年,但由于其稳定的理化性质,在我国土壤和江河湖等水体中仍大量检出,其中滴滴涕(DDT)和林丹(HCH)被广泛检出[3-4]。据估算,土壤环境中分解95%的DDT需要20 a,分解95%的HCH则需要长达30 a的时间[5],且在久受DDT污染的土壤中高频率地检出pp-DDT和4,4′-DCBP等降解产物。目前,国内外针对OCPs土壤和水体污染的修复技术主要有生物修复、物理固定和化学处理等方法。
近年来,基于
⋅SO−4 和·OH的活化过硫酸盐(PS)氧化技术是高级氧化技术中应用较为广泛的一种。与其他氧化剂相比,过硫酸盐在室温条件下性质相对比较稳定,在被污染环境修复过程中可操作性强,经活化后产生的⋅SO−4 氧化还原电位高,能够在宽pH下氧化降解难降解有机物[6],使其在有机污染物降解方面的应用开始受到关注。活化过硫酸盐的方式有加热、过渡金属离子、紫外光、碱、零价铁(ZVI)等单一或两者复合活化等方式。其中,ZVI作为活化剂具有环境友好、价格低廉和应用范围广等特点,目前已在2,4-二硝基甲苯(DNT)、双酚A、氯代苯等多种有机污染物降解方面得到应用[7-8]。ZVI粒径越小,比表面积和表面能越大,其活化效果也越好。然而,由于ZVI的还原能力,其颗粒表面总覆盖着由铁氧化物或羟基氧化物形成的钝化膜,减弱其反应活性,且纳米ZVI (nZVI)表面能过高具有磁性易发生团聚,再加上成本过高,nZVI的表面积归一化反应性并不显著大于mZVI,在实际污染环境修复中并不现实[9-10]。近年来,有学者提出对ZVI进行改性以规避上述缺陷,其中硫化改性因其独有的化学特性成为研究的热点。球磨硫化微米级零价铁(S-mZVI)是通过单质硫的硫化和球磨的机械化学混合克服mZVI制备过程中的限制因素而制备的一种硫化ZVI材料[11],目前已在三氯乙烯等氯代烃的还原上展现出比nZVI更强的催化活性[12-13]。而且,S-mZVI通过硫化形成Fe/FeS复合体取代ZVI表面的钝化膜,FeS较铁(水合)氧化物是一个更好的电子导体,可以更快地将电子从铁心传递到材料表面,即促进Fe0给出电子,更快地释放Fe2+,且制备方式简便,廉价易得。但目前,尚无关于S-mZVI活化过硫酸盐降解OCPs的报道。因此,本研究拟利用球磨S-mZVI活化PS构建S-mZVI/PS高级氧化体系,通过有机氯农药的批次降解实验,分别考察了S-mZVI的S/Fe摩尔比和pH、腐殖酸、
HCO−3 和NO−3 及溶解氧等环境因子对S-mZVI/PS体系降解有机氯农药的影响,以评估S-mZVI的实际应用性能,并优化了S-mZVI/PS体系降解有机氯农药的工艺参数,以期为S-mZVI高效活化过硫酸钠氧化降解OCPs等有机污染物的现场应用提供更为准确的操作参数。
全文HTML




-
4,4′-DCBP(99.3%)和β-HCH(99.3%),购自德国Dr. Ehrensorfer公司;微米级铁粉(98%)、硫粉(分析纯)、5,5-二甲基-1-氧化吡咯啉(DMPO;97%)和4-羟基-2,2,6,6-四甲基哌啶氧(TEMP, 99%)均购自上海麦克林公司;过硫酸钠、氢氧化钠和碳酸氢钠均为分析纯,购自上海迈瑞尔化学技术有限公司;盐酸(分析纯)购自天津科密欧公司;腐殖酸(分析纯)购自天津市精科精细化工公司;正己烷(色谱纯)购自上海安谱实验科技股份有限公司。
-
将单质硫粉和微米级铁粉按照0.05、0.10、0.125和0.25的S/Fe摩尔比进行称取并混合均匀,将其50 g混合物与200 g玛瑙球磨珠一并装入玛瑙球磨罐中,混合均匀后置于球磨机中在室温下以200 r·min−1开始球磨。球磨过程中以氮气作为保护气,球磨时间20 h。球磨结束后,在氮气氛围下分离并保存所获得的S-mZVI。为了对比硫化前后零价铁的差异,将纯微米级铁粉在相同条件下进行球磨,制得球磨mZVI。
采用热场发射扫描电子显微镜(QUANTA FEG 400,美国FEI公司)和能谱仪(GENESIS,美国伊达克斯有限公司)对mZVI和S-mZVI进行粒径尺寸、表面形貌及元素分布分析。
-
将0.224 g mZVI(S-mZVI)加入40 mL EPA瓶中,再加入40 mL 100 mmol·L−1的过硫酸钠溶液和200 μL 2 g·L−1的目标物母液,使目标物浓度达到10 mg·L−1。OCPs需利用丙酮助溶,且为避免共溶剂效应加入丙酮的量不超过0.5%。随后立即置于恒温气浴振荡箱中(25 ℃,150 r·min−1)避光反应,反应期间定时取样检测4,4′-DCBP和β-HCH的浓度;为探究硫化改性对mZVI的腐蚀速率的影响,采用邻菲啰啉法测定反应期间溶液中总铁离子和Fe2+的浓度。
-
目标物的提取:向取出的1.0 mL样品中加入1.0 mL正己烷振荡提取5 min,最后过0.45 μm有机膜置于进样小瓶中待测。4,4′-DCBP和β-HCH的提取回收率均在98%以上。
有机氯农药的检测:采用气相色谱-质谱联用仪(岛津GC-MS plus 2010,日本)。毛细色谱柱型号为HP-5MS(30 m×0.25 mm×0.25 μm)。仪器条件设置为进样口温度280 ℃,离子源温度230 ℃,进样量1 μL,EI源。柱温箱温度为35 ℃开始保持2 min,随后以15 ℃·min−1升温至150 ℃,再以3 ℃·min−1升温至290 ℃,保持2 min。溶剂延迟时间5 min。
自由基的检测方法:采用电子顺磁共振波谱仪(EPR,Bruker EMX 10/12)检测mZVI或S-mZVI活化PS反应体系中自由基。选用DMPO作为
⋅SO−4 和·OH的捕获剂,TEMP作为1O2的捕获剂。预先向40 mL EPA瓶中加入40 mL 0.1 mol·L−1的DMPO或TEMP溶液,随后加入与降解实验相同浓度的(S-)mZVI和PS开始反应。当反应进行到2 min时,采用毛细石英管迅速吸取一定量的反应溶液,并用真空硅脂封闭底端,将其装入石英测试管中并置于EPR的谐振腔内进行检测。 -
为保证数据的可靠性,本研究的数据结果采用3次重复结果的平均值±标准偏差,采用Excel 2016和Origin 9.0软件进行数据分析与绘制。采用伪一级动力学模型(式(1))对降解动力学数据进行拟合。
式中:C0为目标污染物的初始质量浓度,mg·L−1;Ct为目标污染物的瞬时质量浓度,mg·L−1;k为反应时间t的速率常数,min−1。
1.1. 实验药品
1.2. S-mZVI的制备与表征
1.3. 模拟废水降解实验
1.4. 提取与检测方法

1.5. 数据处理
-
不同S/Fe摩尔比的S-mZVI样品的SEM图谱如图1((a)~(e))所示。可以看出,球磨mZVI(图1(a))的形貌主要呈现聚合的鳞片状,而球磨S-mZVI(图1(b)~(e))的颗粒变得分散,呈不规则的球状或片状颗粒,且随S/Fe摩尔比增大,S-mZVI颗粒表面越平滑,更接近球形。这表明球磨过程中S的存在不仅可以实现对mZVI的硫化,还能使颗粒受到更大的挤压与冲蚀作用[14],从而降低mZVI颗粒的团聚。已有研究结果也证明,S-mZVI材料的比表面积会随S/Fe摩尔比的增大而增加[15],从而为后续催化反应提供大量活性位点。利用EDS测定的S-mZVI(S/Fe=0.10)中Fe和S的平均原子比为0.107(图1(g)),这接近于理论值0.10,说明S相对均匀地分散在S-mZVI颗粒表面。此外,EDS-mapping的结果(图1(h)~(i))进一步显示了Fe和S的分布的高度吻合性,再次表明经过球磨后Fe和S元素在S-mZVI颗粒表面的均匀分布。DU等[16]和RAJAJAYAVEL等[17]通过分析mZVI硫化前后的XPS图谱发现,未硫化的mZVI颗粒主要以铁氧化物和铁氢氧化物为主,而S-mZVI颗粒表面除了铁氧化物和铁氢氧化物外,还存在大量的FeS。
-
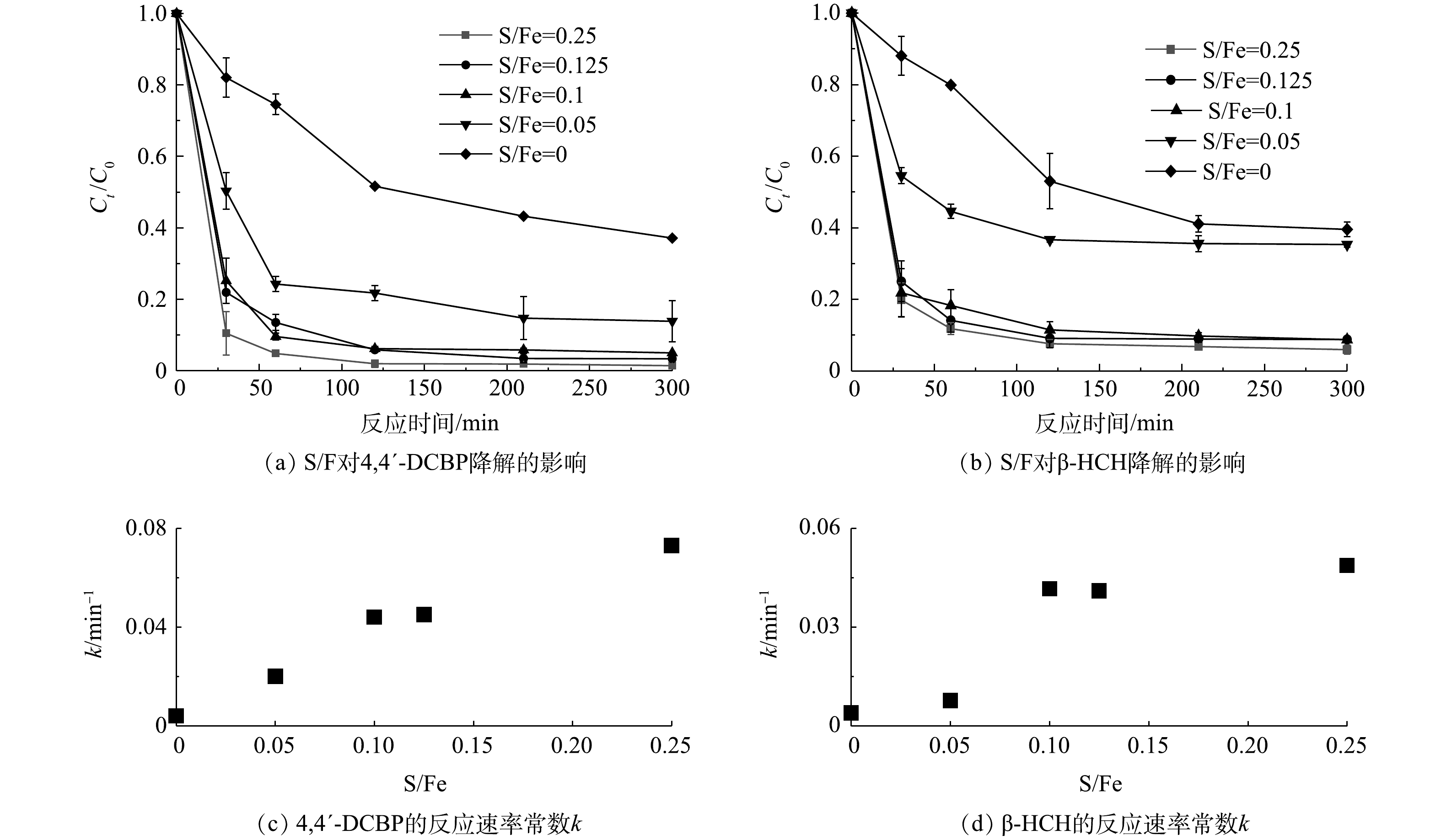
不同S/Fe摩尔比的S-mZVI活化PS氧化降解4,4′-DCBP和β-HCH的结果如图2所示。硫的加入显著促进了mZVI活化PS氧化降解4,4′-DCBP和β-HCH的效率,尤其是在反应开始后的前60 min内。其中,在5 h内,S/Fe摩尔比为0.05的S-mZVI对4,4′-DCBP和β-HCH的降解率最低,分别为86.1%和61.5%,反应速率常数k分别为0.020 min−1和0.008 min−1;当S/Fe摩尔比增加为0.10时,4,4′-DCBP和β-HCH的降解速率明显加快,反应速率常数k分别为0.044 min−1和0.042 min−1;反应2 h已趋于稳定,反应5 h后最终降解率分别为95.0%和91.3%。这主要是因为改性后mZVI表面的钝化膜(铁(氢)氧化物)被FeS取代,加快了电子从mZVI内部导出,更快地释放Fe2+[12],促进了反应的发生。而且,SEM-EDS的表征结果也表明,单质硫的加入促进了mZVI颗粒的破碎,增大其比表面积,进而提供更多的反应活性位点。此外,FeS相对于铁(氢)氧化物亲水性较低,对于疏水性污染物4,4′-DCBP和β-HCH,FeS可以有效地将电子传递到污染物而非水分子上,因而可以有效地提高材料的电子利用率[18]。但当S/Fe摩尔比继续增加到0.125和0.25时,4,4′-DCBP和β-HCH降解速率和降解率均未有显著提高。这一结果说明,在mZVI硫化改性过程中硫的含量并非越多对其活化PS越有利。KIM等[19]就曾指出,当材料中硫含量过多可能反而会阻塞零价铁表面的反应活性位点,降低其腐蚀速率。LIANG等[20]计算了S-mZVI降解TCE过程中比表面积归一化的反应速率常数(kSA),发现kSA随S/Fe摩尔比增大呈现先升高后趋于相对稳定的趋势。在本研究中,虽然S/Fe摩尔比越大,S-mZVI的比表面积越大,但S-mZVI活性位点不能被完全有效利用。此外,在同一S/Fe摩尔比的S-mZVI/PS体系中,4,4′-DCBP的反应速率常数k均高于β-HCH。这可能与2种污染物的理化性质、分子结构和降解路径等差异有关,还需要进一步研究[21-23]。因此,硫化改性虽然可以极大地提高零价铁的催化活性,但也应当探寻最佳的S/Fe摩尔比,才能将其催化活性发挥到极致。基于以上实验结果,出于对材料催化活性和经济成本等因素的考虑,后续实验均采用S/Fe摩尔比为0.10的S-mZVI进行。
-
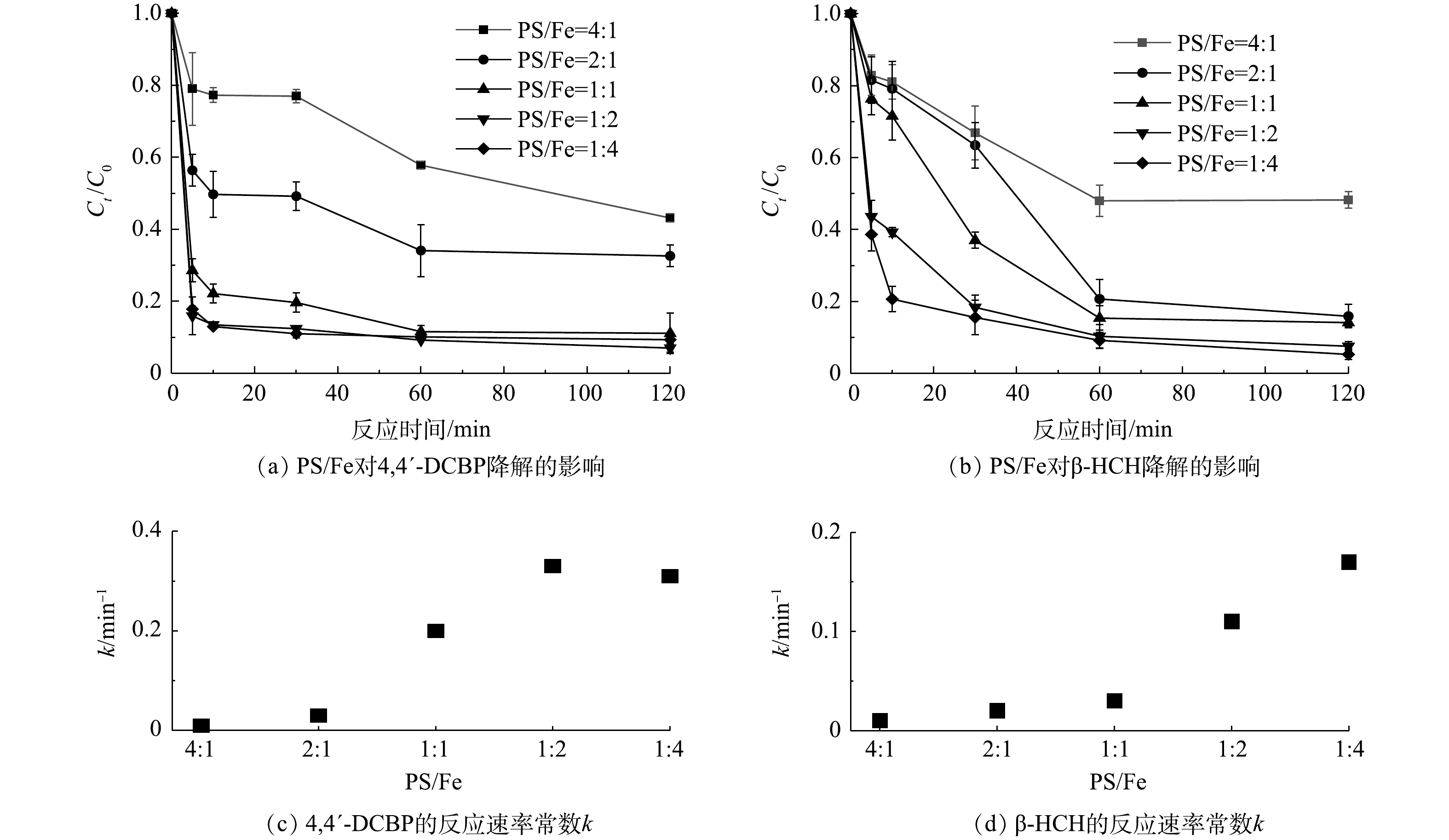
为探究S-mZVI/PS体系中PS/Fe投加量配比对目标污染物降解效果的影响,本实验在PS浓度为100 mmol·L−1的条件下,改变S-mZVI的投加量,考察PS/Fe投加量配比对S-mZVI/PS体系降解4,4′-DCBP和β-HCH的动力学影响,其结果如图3所示。在所考察的比例范围内,PS/Fe投加量配比越小,即铁源投加量越大,4,4′-DCBP和β-HCH的降解效果越好。在高PS/Fe配比(4∶1)时,S-mZVI对PS的活化能力较弱,在120 min内4,4′-DCBP和β-HCH的降解率均小于60%;当PS/Fe配比为2∶1时,4,4′-DCBP和β-HCH的降解率分别上升至67.4%和84.1%;随着S-mZVI投加量继续增加,目标污染物降解率不断提高,当PS/Fe配比为1∶2时,4,4′-DCBP在反应开始后10 min迅速达到降解平衡,降解率可达86.6%,当进行到120 min时4,4′-DCBP和β-HCH的降解率分别为93.0%和92.4%;但继续增加S-mZVI投加量时,4,4′-DCBP和β-HCH的降解率并未显著提高。与此同时,上述S-mZVI/PS体系的反应速率常数k也表现出类似的变化规律。这主要是由于随着S-mZVI投加量的增加,催化活性位点增多,可产生更多的活性自由基;但当S-mZVI增加到一定程度时,在保持PS浓度不变的情况下,多余的铁源(主要是Fe2+)没有足够的PS与其反应,故无法产生更多的自由基参与污染物的降解反应。目前已有研究[24]表明,铁活化PS体系的最佳Fe2+/PS摩尔比为2,且略小于2时Fe2+消耗量更少,污染物降解率更高。这在实际修复中具有重要意义,综合经济成本和环境友好性等因素的考量,选择一个适当的剂量显得尤为关键。因此,S-mZVI/PS体系降解4,4′-DCBP和β-HCH的最佳PS/Fe配比为1∶2,后期实验均采用PS/Fe投加量配比为1∶2进行。
-
溶液的pH是影响S-mZVI腐蚀和催化降解活性的重要环境因素。本实验考察了溶液初始pH为2.0、4.0、7.0、9.0和11.0时4,4′-DCBP和β-HCH在S-mZVI/PS体系中的降解动力学曲线。由图4可知,在初始pH为2.0的酸性条件下,4,4′-DCBP和β-HCH的降解率最高,在60 min内均达90%以上,反应速率常数k分别为0.32 min−1和0.31 min−1;初始pH为4.0时4,4′-DCBP和β-HCH的降解率也可达89.4%和88.4%。随着pH继续升高,碱性条件下的4,4′-DCBP和β-HCH降解效率逐渐降低,当初始pH为11.0时,4,4′-DCBP和β-HCH在反应时间内降解率分别降低至80.8%和83.4%,反应速率常数k降低至0.13 min−1和0.10 min−1。这是由于激活PS的关键因素Fe2+更适合在酸性环境中存在,初始pH较高时,一方面限制了零价铁的腐蚀速率,Fe2+的溶出量减少,另一方面Fe2+在pH大于5.8时开始形成沉淀,而Fe3+在pH大于4时将完全沉淀[25],从而致使零价铁表面覆盖大量的铁(氢)氧化物,阻碍高级氧化反应的反生。另外,PAN等[26]的研究也得到相似的结果,当pH为3~9时,二苯甲酮的降解率随pH的增加而降低。该文献解释为
⋅SO4− 的生成有赖于酸催化,形成较高的自由基强度,pH的降低会促进这种催化效果。此外,本研究测定了反应结束后溶液的最终pH,分别为1.78~1.80、2.13~2.15、2.38~2.40、2.75~2.77和3.04~3.06。由此可见,随着反应的进行,无论溶液初始pH为多少,S-mZVI/PS体系的pH均会随着反应的进行而降低,直至酸性。这也是溶液初始pH对4,4′-DCBP和β-HCH的降解影响偏小的主要原因。 -
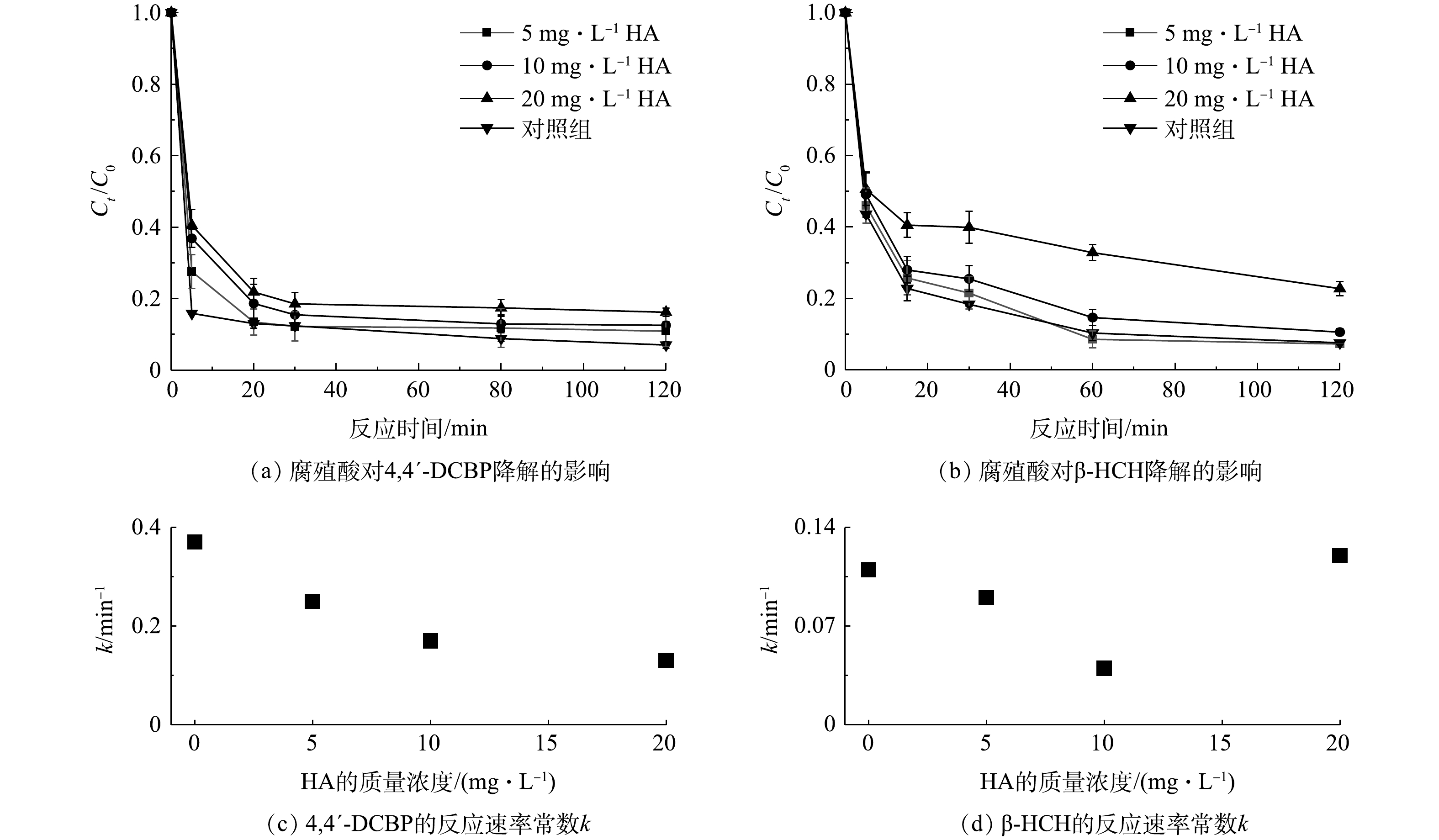
溶解性有机质(DOM)广泛存在于自然水体和土壤环境中,且以腐殖酸(HA)为主[27],本实验以HA为研究对象,考察了其对S-mZVI/PS体系氧化降解4,4′-DCBP和β-HCH的影响。如图5所示,当HA的质量浓度为5 mg·L−1时,S-mZVI/PS体系对4,4′-DCBP和β-HCH在反应60 min后的降解率分别为88.2%和89.1%,与对照组没有显著差异;而当HA的质量浓度为20 mg·L−1时,2种目标污染物在反应60 min后的降解率降低到82.6%和66.2%,显著低于对照组和HA为5 mg·L−1和10 mg·L−1的处理组。这一结果表明,HA质量浓度越高,S-mZVI/PS体系对4,4′-DCBP和β-HCH的降解率越低,且存在显著的浓度依赖性。HA对目标污染物降解的抑制作用可能存在2方面原因:一方面,HA作为有机物,可与目标污染物争夺S-mZVI/PS体系中产生的自由基;另一方面,HA具有络合作用,会与体系中的Fe2+形成稳定络合物,从而阻碍Fe2+的释放,最终阻断电子的传递和自由基的生成路径[28-29]。
-
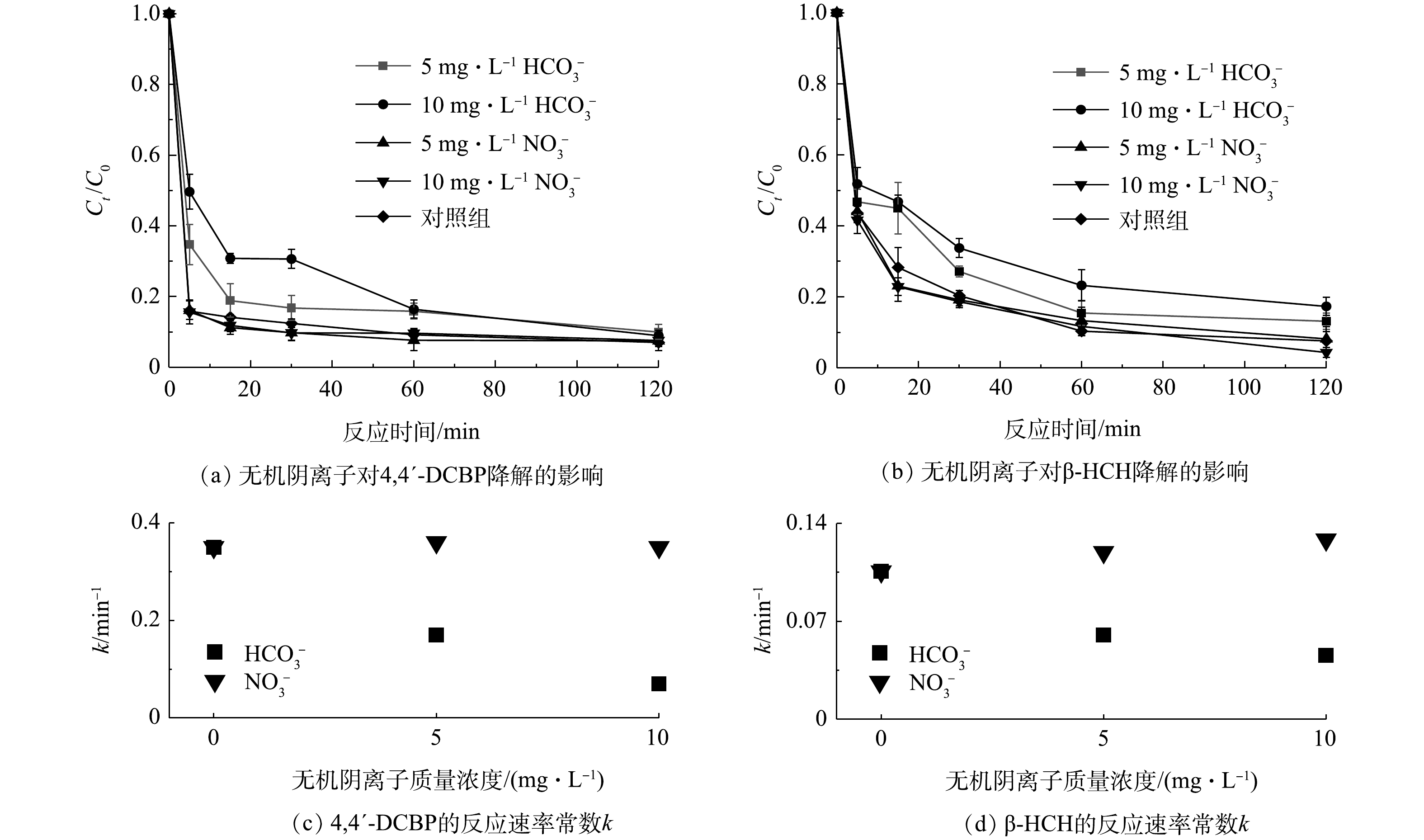
碳酸氢根(
HCO−3 )和硝酸根(NO−3 )等无机阴离子广泛存在于自然水体和土壤中,可通过自由基清除作用影响污染物的降解或与活性物种反应生成二级自由基并与污染物发生反应,从而对高级氧化反应体系产生影响[7, 30-31]。为此,本实验分别添加不同质量浓度的HCO−3 和NO−3 到S-mZVI/PS反应体系中,以探究其对4,4′-DCBP和β-HCH降解的影响。如图6所示,HCO−3 的存在对4,4′-DCBP和β-HCH的氧化降解均有明显的抑制作用,且HCO−3 浓度越高,抑制作用越明显。在反应时间60 min内,对照处理组4,4′-DCBP和β-HCH的降解率分别为90.8%和89.7%,反应速率常数k分别为0.35 min−1和0.11 min−1,且随着HCO−3 质量浓度由5 mg·L−1增加到10 mg·L−1,4,4′-DCBP的降解率由84.1%下降到75.6%,反应速率常数k由0.17 min−1下降到0.07 min−1,β-HCH的降解率由84.5%下降到76.8%,反应速率常数k由0.06 min−1下降到0.05 min−1。通常认为,HCO−3 容易吸附到零价铁颗粒表面,导致零价铁颗粒表面的活性位点减少,并且其可与⋅SO−4 和·OH发生反应,生成无机自由基⋅CO−3 (1.78 V),且⋅CO−3 的氧化还原电位低于⋅SO−4 (2.6 V)和·OH(2.7 V)[32-33]。因此,在本研究中HCO−3 的存在显著抑制了S-mZVI/PS体系对4,4′-DCBP和β-HCH的降解。然而,NO−3 也会与⋅SO−4 和·OH−等自由基反应生成·NO−3 ,且·NO−3 的氧化还原电位(2.3~2.5 V)十分接近于⋅SO−4 (2.6 V)和·OH(2.7 V)[34],能够直接氧化降解4,4′-DCBP和β-HCH。因此,在本实验体系下,NO−3 的存在并没有对目标污染物的降解表现出明显的抑制作用,其降解动力学曲线与对照处理组基本吻合。 -
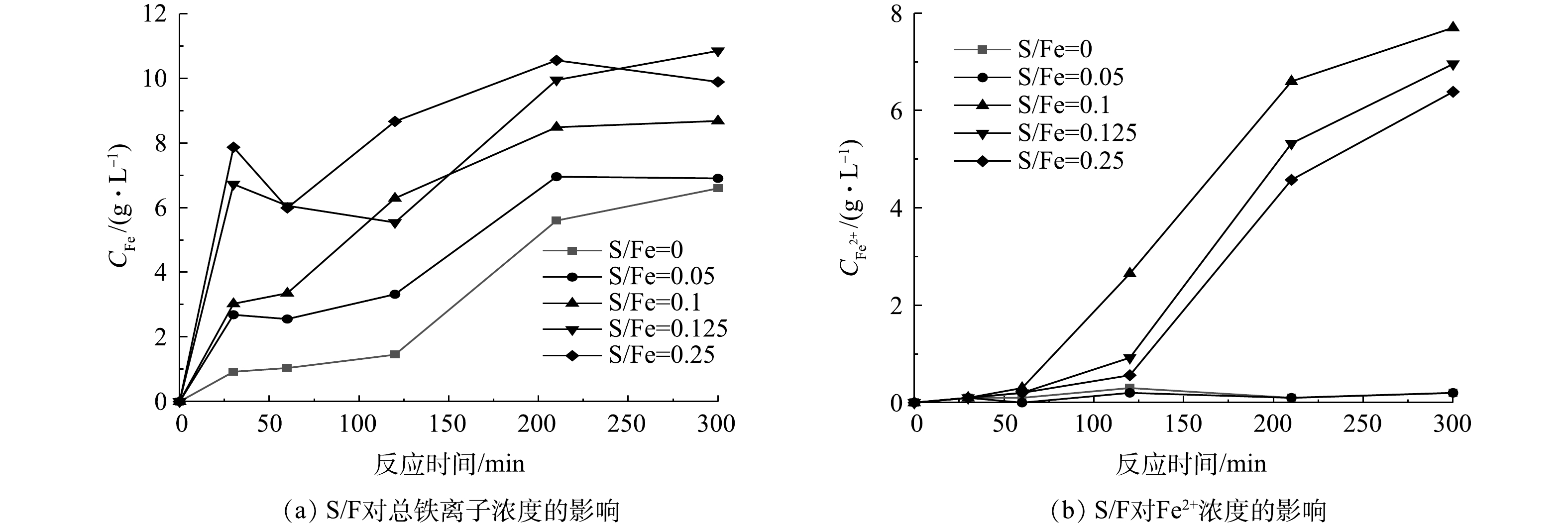
为探究不同的S/Fe摩尔比对反应体系中mZVI腐蚀的影响,本实验中检测了不同S/Fe摩尔比的S-mZVI/PS体系中总铁离子和Fe2+的浓度变化。如图7(a)所示,S/Fe摩尔比越大,S-mZVI/PS体系中的总铁离子浓度就越高。JIN等[35]的研究也得到了同样的结果,说明硫化改性对mZVI的腐蚀有着促进作用,且S/Fe摩尔比越大,促进作用越强。如图7(b)所示,反应开始的60 min内,不同S/Fe摩尔比的S-mZVI/PS体系中Fe2+浓度均较低,说明在PS浓度较高的反应前期,铁离子主要以Fe3+的形式存在。随着反应的进行,在S/Fe摩尔比为0和0.05的体系中铁离子仍主要以Fe3+形式存在,而在S/Fe摩尔比为0.1、0.125和0.25的体系中Fe2+浓度远远高于S/Fe摩尔比为0和0.05的体系,且在300 min时其Fe2+浓度分别占铁离子总浓度的64.1%、64.1%和64.6%。这可能是因为,硫化改性提高了mZVI表面的FeS含量,FeS可以取代mZVI表面的铁氧化物钝化膜,而且FeS较铁氧化物是一种更好的电子导体,可以加速电子从mZVI内部传递到表面,即促进Fe0给出电子,更快地释放Fe2+[12]。因此,硫化改性可通过促进铁离子溶出来提高mZVI的催化活性,且当S/Fe摩尔比为0.10时其Fe2+溶出浓度最高。
-
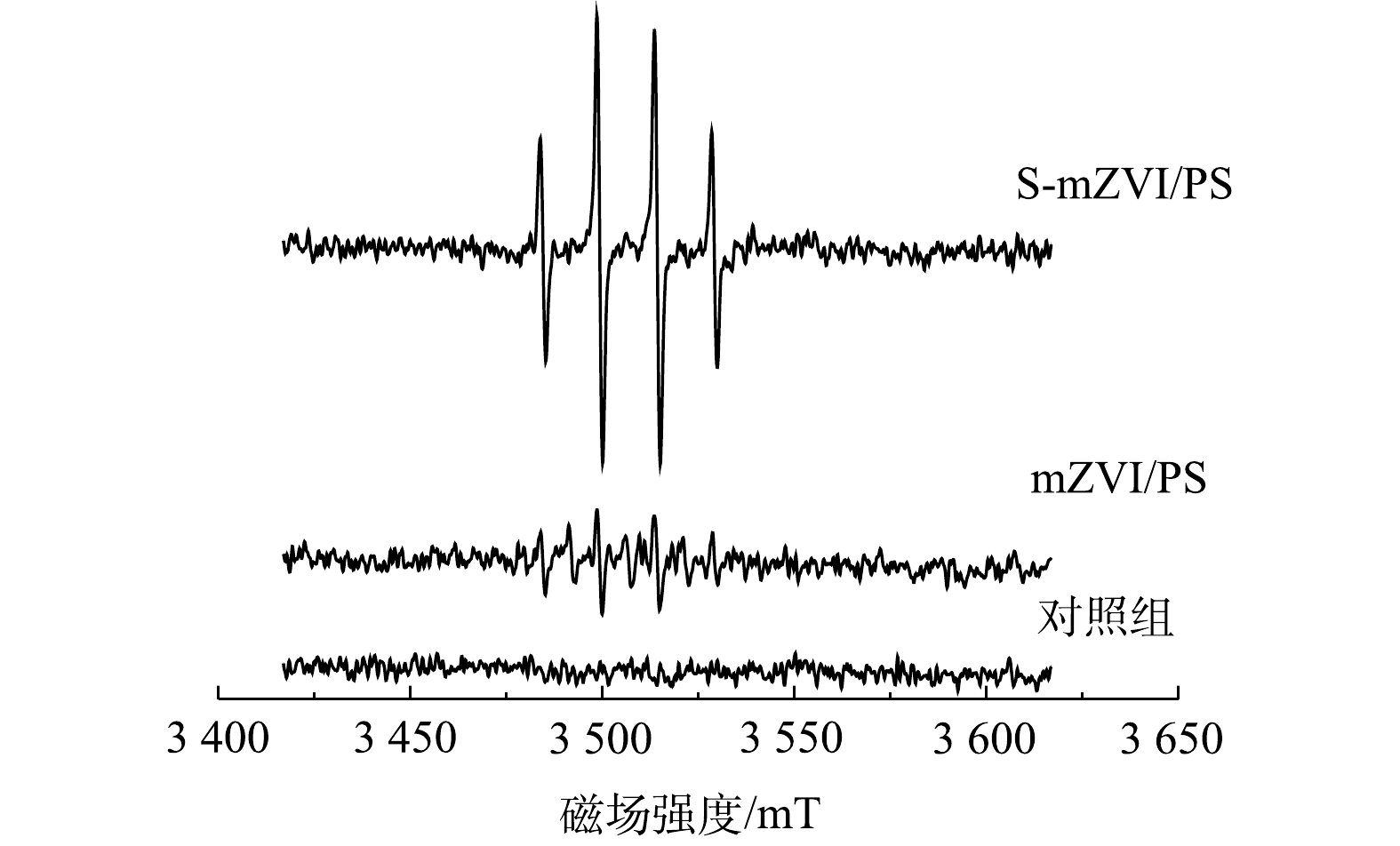
为了鉴定S-mZVI/PS体系中自由基的种类,通过添加DMPO和TEMP对体系中的自由基进行捕获。如图8所示,S-mZVI/PS和mZVI/PS体系中均能检测到
⋅SO−4 和·OH,但在S-mZVI/PS体系中⋅SO−4 和·OH的信号更强,加入TEMP后反应体系中均未捕获到TEMP-1O2信号。已有研究表明,ZVI作为Fe2+的缓慢释放源,并将生成的Fe3+还原成Fe2+,即Fe0既可作为过硫酸盐的活化剂,又可作为Fe3+的还原剂,其反应机理如式(2)~式(6)所示[36-38]。硫化改性显著提高mZVI释放Fe2+的速率,加快式(3)、式(4)和式(6)等反应的进行,从而加速了⋅SO−4 和·OH的产生和对污染物的降解。同时,反应产生的H+提高了反应体系的pH。
2.1. 扫描电子显微镜-能谱(SEM-EDS)分析结果
2.2. S/Fe摩尔比对4,4′-DCBP和β-HCH降解的影响
2.3. PS/Fe投加量配比对4,4′-DCBP和β-HCH降解的影响
2.4. 溶液初始pH对4,4′-DCBP和β-HCH降解的影响

2.5. 腐殖酸对4,4′-DCBP和β-HCH降解的影响
2.6. 无机阴离子对4,4′-DCBP和β-HCH降解的影响



















2.7. S/Fe摩尔比对反应体系中mZVI腐蚀的影响
2.8. 催化机制



-
1)球磨过程中硫的存在不仅可以实现mZVI的硫化,还能提高mZVI的球磨效率。与团聚状球磨mZVI相比,球磨S-mZVI的颗粒变得分散,呈不规则球状,且S/Fe摩尔比越大,颗粒表面越平滑,更接近球形。
2)硫的加入使S-mZVI/PS体系较mZVI/PS体系表现出对有机氯农药更高的催化降解效率,且S/Fe摩尔比越高,mZVI的腐蚀速率越高,活化效果越好,产生的
⋅SO−4 和·OH越多。当S/Fe=0.10、PS/Fe投加量配比为1/2时,4,4′-DCBP和β-HCH的降解效果最好,适合用作有机氯农药污染水体的修复。3)溶液初始pH为2.0~11.0时,随着pH的升高,4,4′-DCBP和β-HCH降解速率逐渐降低,但降解效果之间的差异较小。腐殖酸和
HCO−3 的存在会降低4,4′-DCBP和β-HCH的降解速率,且随着浓度的增加,对其降解效果的抑制作用增强;而NO−3 的存在及浓度变化对S-mZVI/PS体系中4,4′-DCBP和β-HCH的降解影响较小。



