




























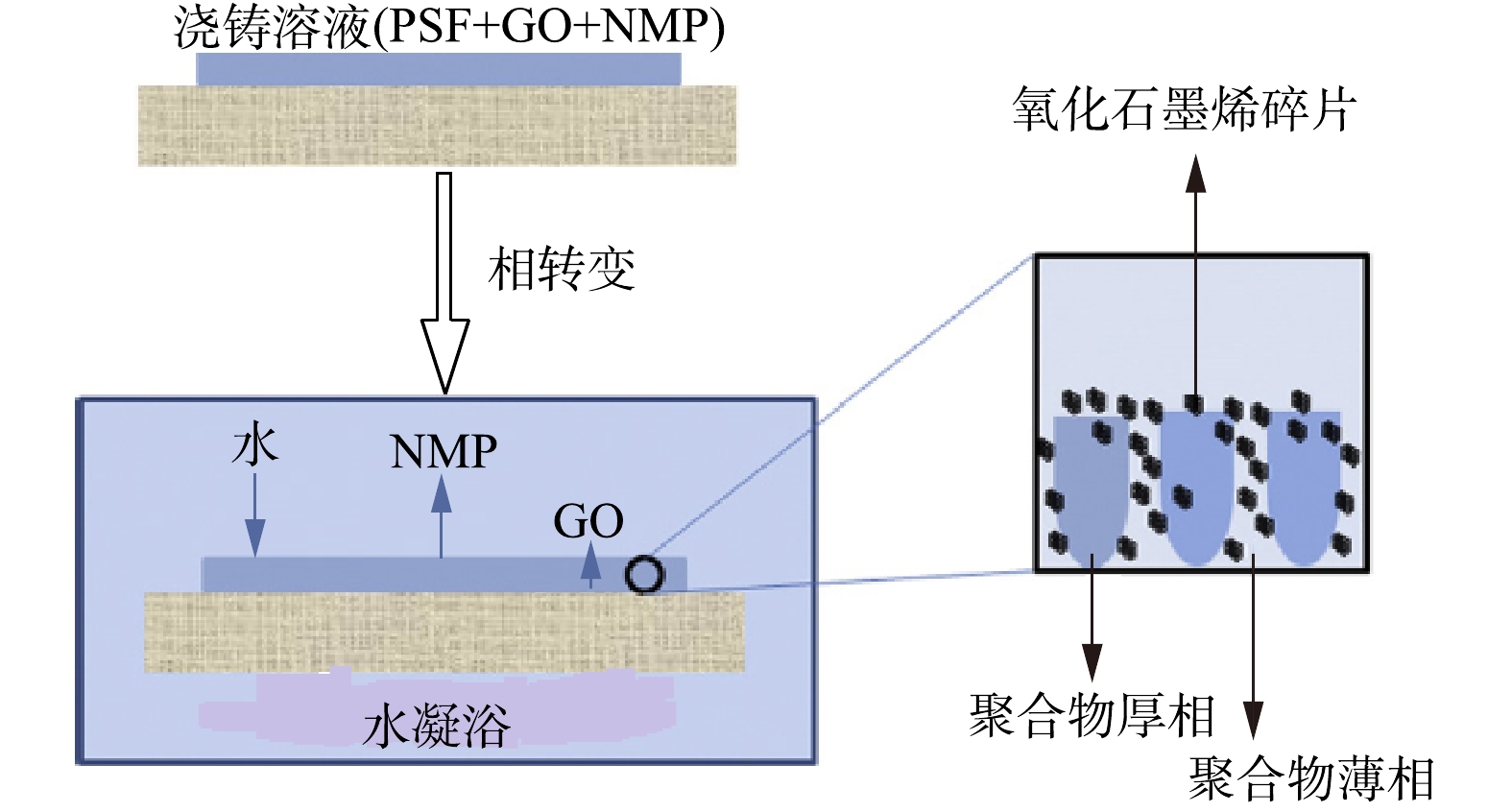
 下载:
下载:
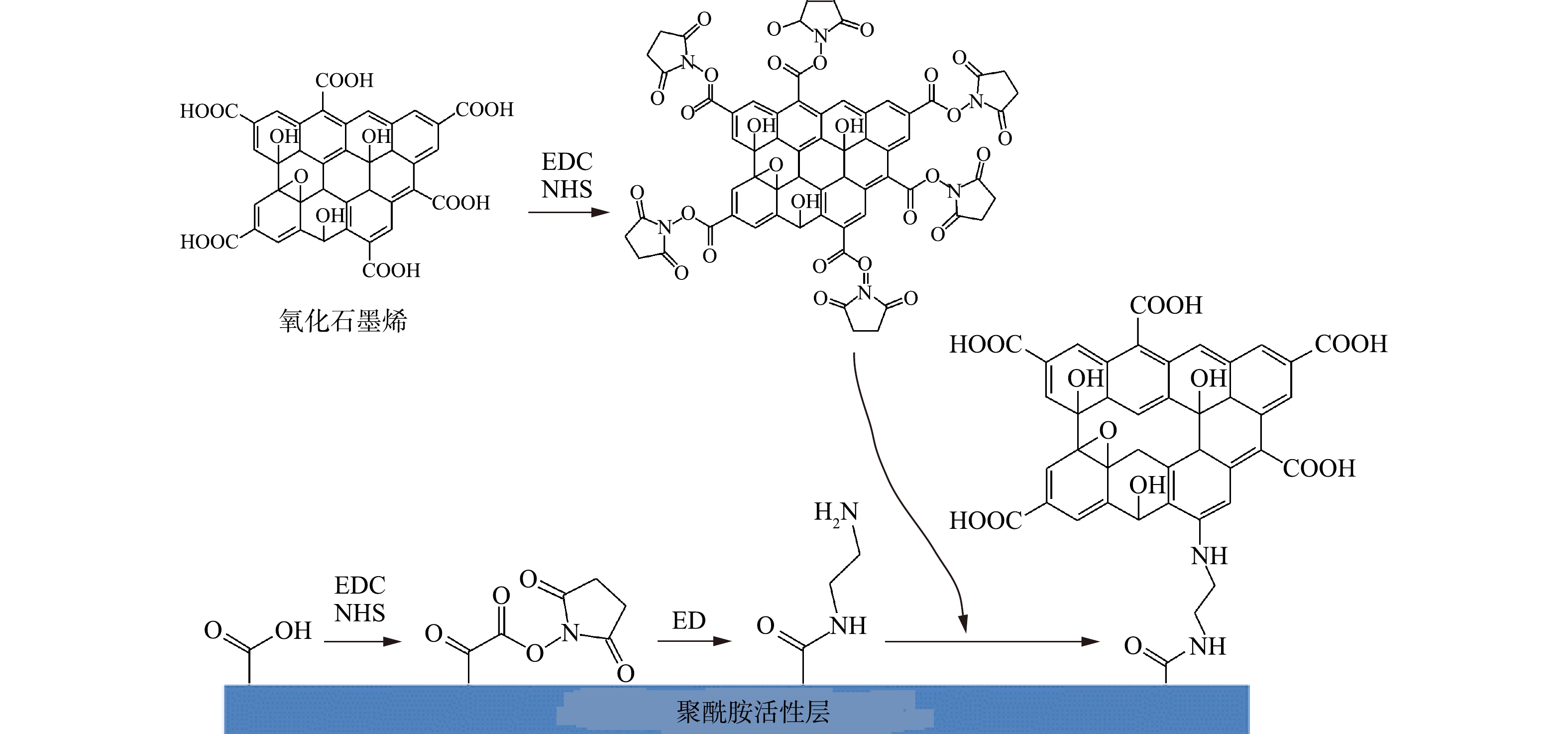
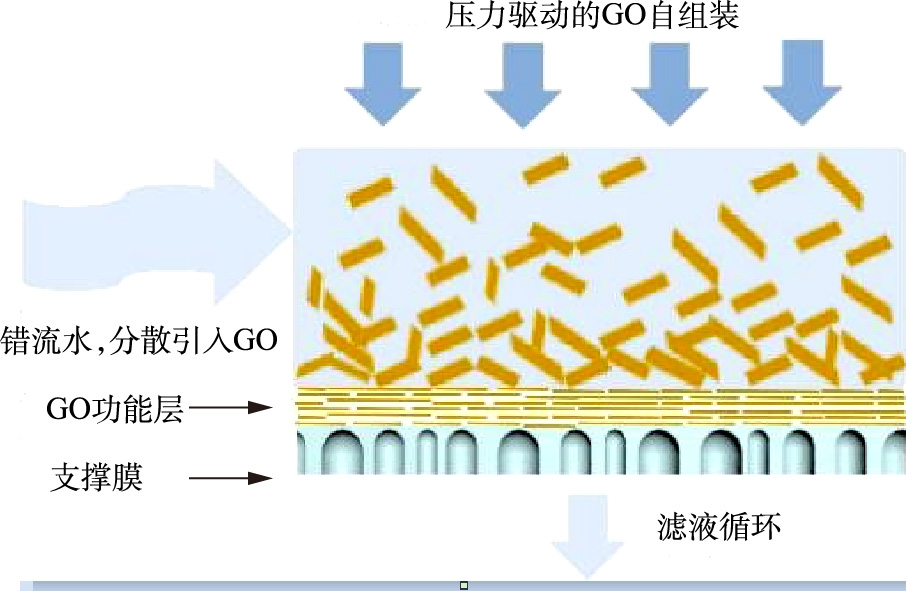
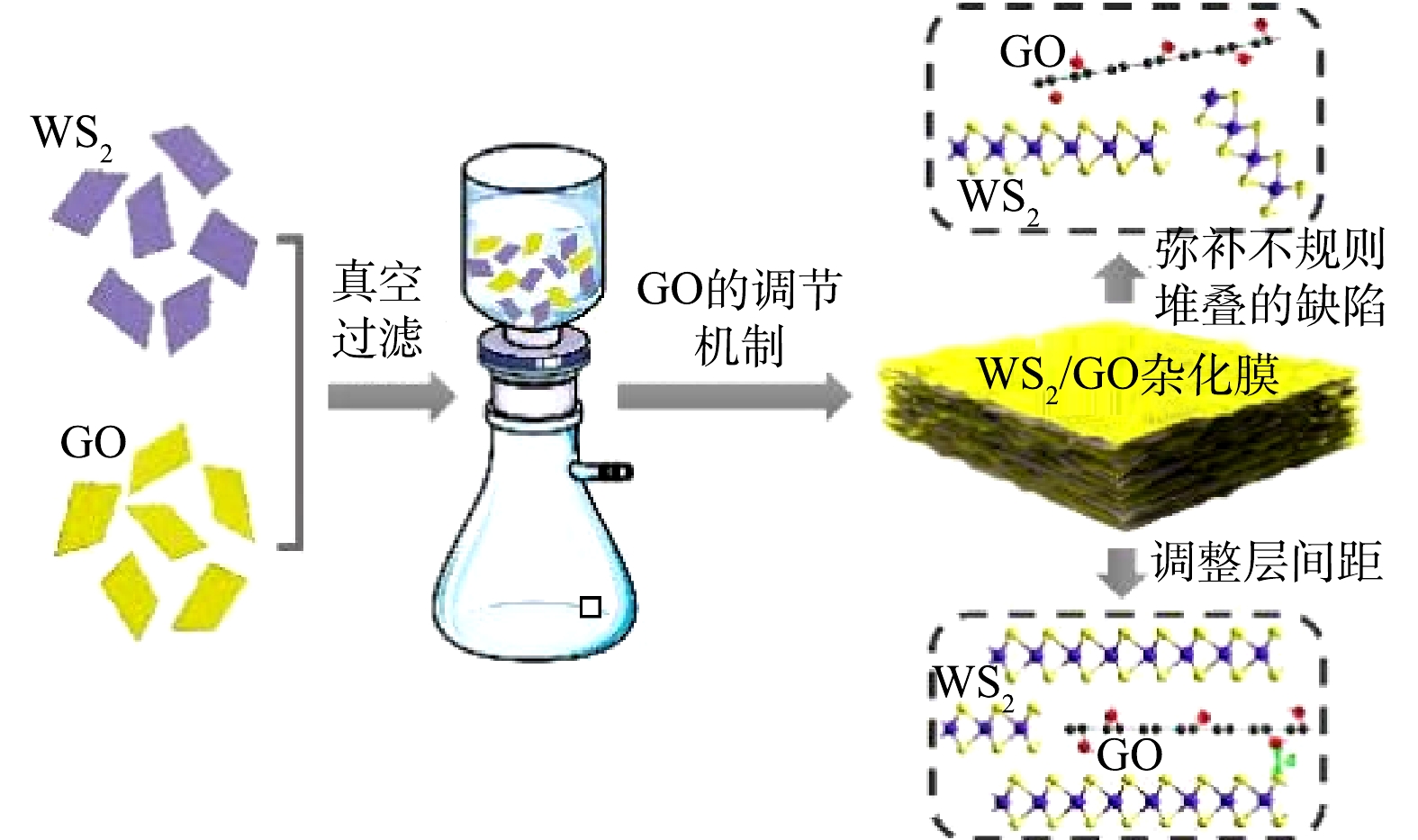

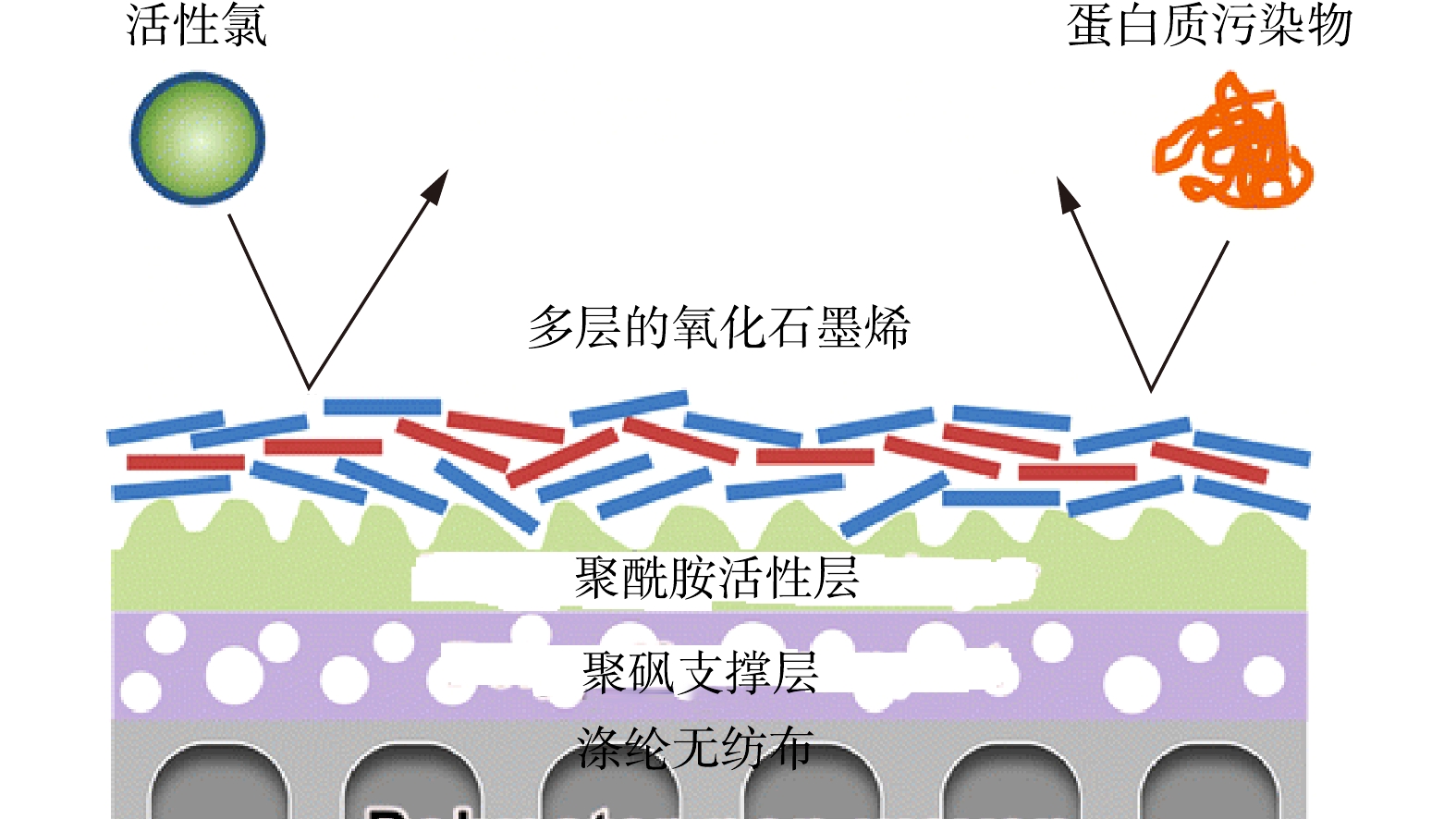
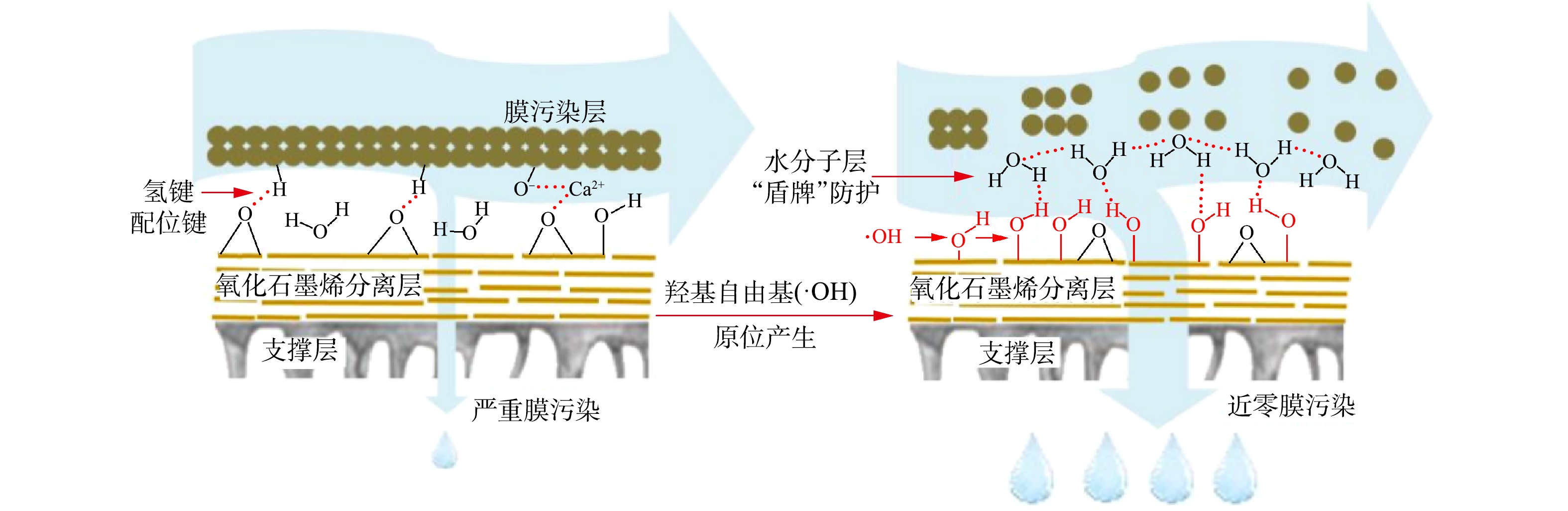
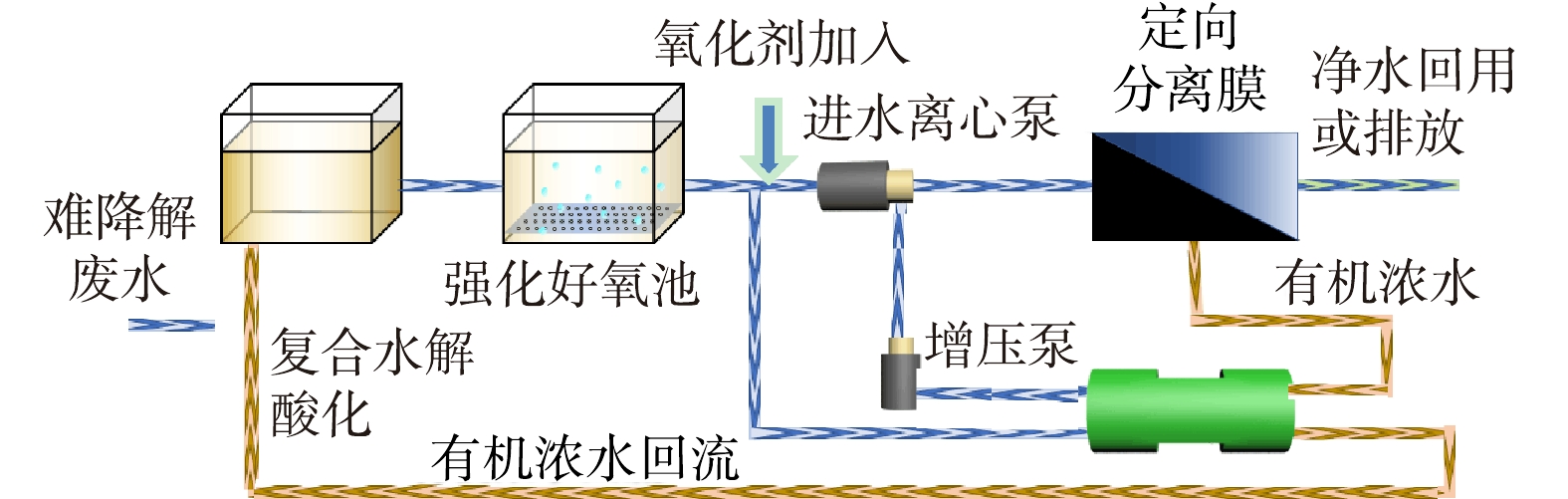

































-
工业废水中的氮、磷如果处理不当或直接排放,会导致水体富营养化,威胁生态系统的安全[1-3]。工业废水中的氨氮(
NH+4 -N)转换成硝态氮(NO−3 -N)或亚硝态氮(NO−2 -N)后,毒性会更大,对水生生物的生存造成威胁,并可能危害人类健康[4]。磷广泛应用于工农业,但在自然界中,单向循环导致磷的不可再生性,未来将面临着严重的磷短缺[5]。因此,废水中氮的无害化去除,同时实现磷资源的回收是当前工业废水处理中的热点。工业废水领域常用的对氮、磷的处理方法有结晶法[6-7]、吸附法[8]、生物法[9-10]以及化学沉淀法[11]等。依靠单一的沉淀法或者氧化法,很难实现废水同时脱氮除磷。近年来,以过一硫酸盐(peroxymonosulfate,PMS)为基础的高级氧化工艺(advanced oxidation process,AOPs)处理废水中难降解污染物的研究受到关注[12]。利用超声波、紫外线辐射、加热、金属离子或金属氧化剂和非金属催化剂等方法,可有效活化PMS,并产生多种活性自由基[13-15]。亚铁离子(Fe2+)可以有效活化PMS产生活性物种,实现罗丹明B的高效降解[16]。另外,研究者还发现活化PMS产生的
SO⋅−4 可以进一步将水中Cl−氧化成为氯自由基(Cl·)[17]。而污水中的NH+4 在Cl·作用下可转化成氮气(N2),实现无害化去除[18]。本研究以
NH+4 、PO3−4 为目标污染物,通过向溶液中投加FeCl2和PMS,将废水中的NH+4 选择性氧化成N2,以实现NH+4 的无害化去除,同时生成的Fe3+在一定条件下与PO3−4 可生成磷酸铁(FePO4)沉淀实现磷的回收;为详细探讨NH+4 、PO3−4 同步去除及磷回收的过程机制,进一步考察PMS初始浓度、Fe2+/Cl−比、pH、共存CO2−3 和HA等多种因素对反应效果的影响,以期为工业废水中氮、磷的处理与资源化利用提供参考。
全文HTML
-
硫酸铵((NH4)2SO4)、磷酸二氢钾(KH2PO4)、氯化亚铁(FeCl2)、氯化钠(NaCl)、甲醇(CH4O)、叔丁醇(C4H10O)、硝基苯(C6H5NO2)、硫酸(H2SO4)、氢氧化钠(NaOH)、碳酸钠(Na2CO3)和腐殖酸(HA)购自国药集团化学有限公司。过一硫酸盐(2KHSO5·KHSO4·K2SO4,PMS)购自Sigma-Aldrich西格玛奥德里奥奇(上海)贸易有限公司。5,5-二甲基-1-氧化吡咯啉(DMPO)购自东仁化学科技(上海)有限公司。药品均为分析纯,实验用水为超纯水。
-
反应体积250 mL。溶液中
NH+4 (以N计)和PO3−4 (以P计)初始浓度为10 mg·L−1。反应开始前,加入定量的PMS和NaCl,溶液pH用H2SO4或NaOH调节并保持恒定;在磁力搅拌器上持续搅拌(300 r·min−1),加入FeCl2启动反应;分别在0、5、10、15和30 min进行取样;样品经过0.22 μm的滤膜后,采用紫外可见分光光度计进行分析NH+4 和PO3−4 的残余浓度。实验过程中,pH用稀H2SO4或稀NaOH保持恒定。反应时需用水浴锅保持溶液恒温,除考察温度影响外,其他体系温度均在室温(25±3) ℃下进行。在以上反应体系中,分别考察了PMS浓度(0、5、10、15、20 mmol·L−1)、Fe2+/Cl−(0、1/82、1/42、1/22、1/15、1/12)、pH(2、4、6、8、10、12)、温度(20、30、40、50、60 ℃)、Na2CO3浓度(0、1、3、5 mmol·L−1)和HA浓度(0、5、10、20 mg·L−1)对
NH+4 和PO3−4 去除的影响。在进行自由基淬灭时,分别加入0.1或4 mmol·L−1甲醇作为
SO⋅−4 和·OH淬灭剂;0.1或4 mmol·L−1叔丁醇作为·OH和Cl·淬灭剂;0.1或4 mmol·L−1硝基苯作为·OH淬灭剂。在相同的实验条件下,测定NH+4 浓度的变化来确定其去除情况。 -
溶液中剩余
NH+4 浓度采用国标纳氏试剂分光光度法测定;剩余PO3−4 浓度采用国标钼酸铵分光光度法测定。溶液的pH用pH计(PHS-3C,上海雷磁仪器厂)进行检测。自由基的测定采用电子自旋共振仪(ESR,Bruker A300-10/12,德国)完成。沉淀物形貌采用场发射扫描电子显微镜(SEM,SU-8020,日本日立有限公司)进行表征。晶体结构通过X射线衍射(XRD,X/Pert Pro MPD,帕纳科分析仪器有限公司,荷兰)进行表征。傅里叶变换红外光谱(FT-IR)采用Nicolet 5700型光谱仪进行测试。
1.1. 实验试剂
1.2. 实验步骤
1.3. 分析测定方法
-
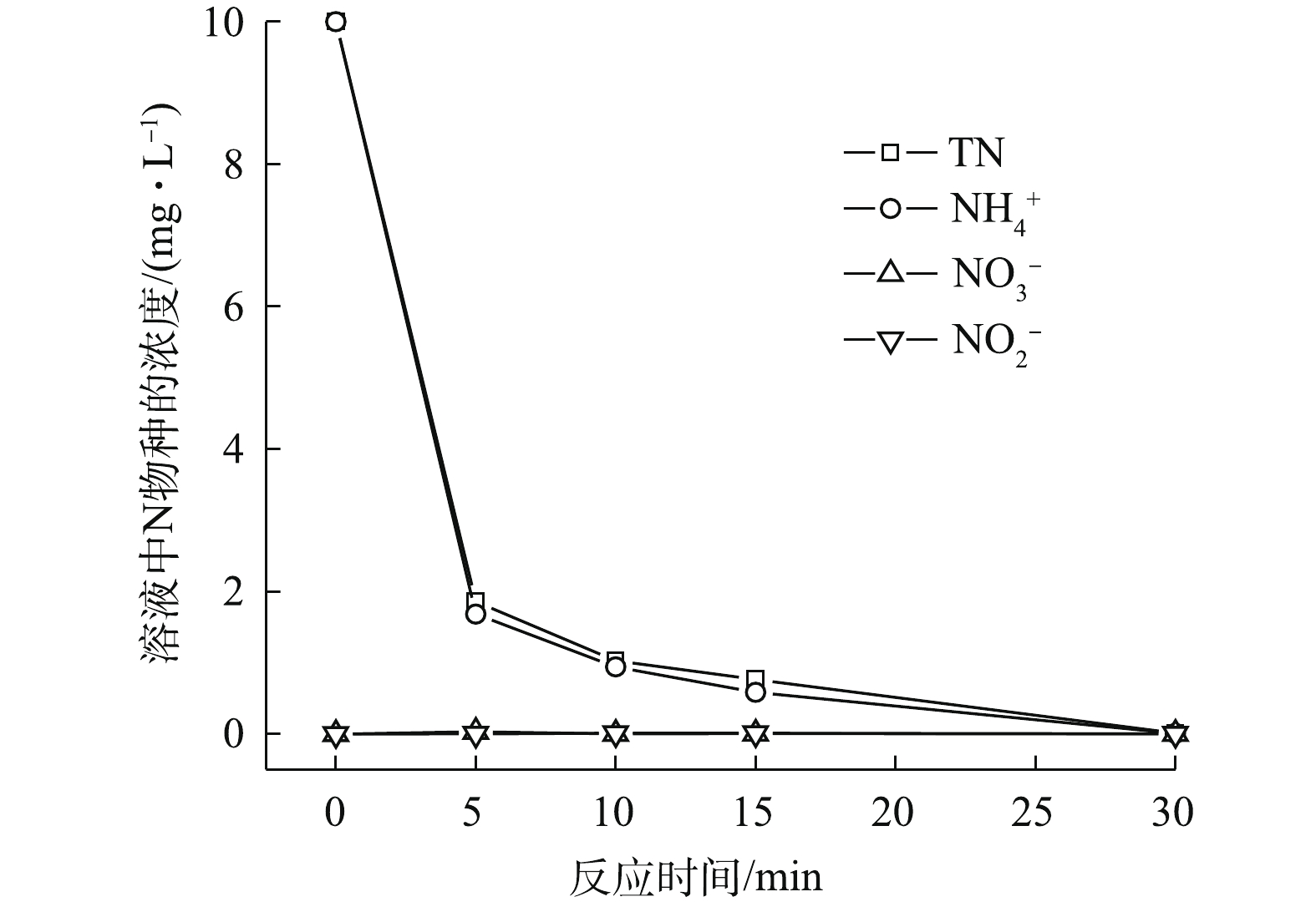
图1显示了不同反应体系中
NH+4 、PO3−4 的去除效果。反应30 min时,单独投加PMS和单独投加H2O2,NH+4 的去除率约为7.5%,PO3−4 没有得到去除。单独投加FeCl2可将PO3−4 全部去除,但NH+4 基本没有去除;在FeCl2/H2O2体系和FeCl2/PMS体系中,PO3−4 的去除率均可达到100%,但NH+4 的去除率仅有19.9%和23%。为保证体系中Cl‒量充足,在FeCl2/PMS体系中额外投加NaCl,形成Fe2+/PMS/Cl−体系后,NH+4 、PO3−4 的去除率可达到100%。另外,在PO3−4 去除效果明显的体系中,如单独投加FeCl2、FeCl2/H2O2体系、FeCl2/PMS体系以及Fe2+/PMS/Cl−体系。溶液中均产生了沉淀物。分析原因,可能是由于这些体系中存在的二价铁或三价铁能与PO3−4 发生反应,生成沉淀。为研究Fe2+/PMS/Cl−体系中
NH+4 的去除情况,对反应过程中的氮平衡进行了分析(图2)。随着反应的进行,溶液中的NO−2 和NO−3 均未检测到。而其中的NH+4 和总氮(TN)均随时间不断减少,且二者量基本保持一致。已有文献报道,Fe2+活化PMS产生SO⋅−4 [16],SO⋅−4 可进一步将Cl−氧化成为Cl·[17];而在酸性条件下,NH+4 受Cl·作用可转化成N2,实现无害化去除[18-19]。溶液pH是影响三价铁和正磷生成不同类型沉淀的主要影响因素[20]。由环境水化学平衡软件Visual MINTEQ模拟分析(图3)可知,当溶液pH不同时,三价铁和正磷在溶液中的存在形态也不同。当溶液pH为2~7时,正磷在溶液中主要以
H2PO−4 存在;当溶液pH为2.4~4.3时,三价铁在溶液中主要以Fe(OH)2+存在。当H2PO−4 与Fe(OH)2+发生沉淀反应,结合形成(Fe(OH)2+)(H2PO−4 ),该物质脱水后可生成FePO4沉淀物。而当溶液pH大于4.3时,溶液中的三价铁形成Fe(OH)3进而析出,影响FePO4沉淀物的形成。为进一步探究Fe2+/PMS/Cl−体系中沉淀产物的组成,采用XRD、SEM以及FT-IR等手段对沉淀产物进行了表征(溶液pH为4、反应30 min后生成的沉淀物),结果如图4所示。XRD结果表明,测试结果中的衍射峰符合FePO4的结构,符合JCPDS(No. 30-0659)标准卡片[21];同时,SEM显示出沉淀物高度团聚,呈近似椭圆形或球形,与FePO4的特征形貌一致[22]。FTIR结果显示,1 000 cm−1为
PO3−4 基团不对称的弯曲振动峰[23],而在1 642 cm−1和3 371 cm−1处的峰分别对应水分子中H—O—H和O—H基团的弯曲振动[24]。这说明在Fe2+/PMS/Cl−体系中,当溶液pH为4时,反应30 min后生成的沉淀物为FePO4,故PO3−4 最终以FePO4形式被回收。以上结果表明,在Fe2+/PMS/Cl−体系中,当溶液pH为4,反应30 min后,溶液中的
NH+4 几乎全部转化成N2,实现无害化去除。溶液中的PO3−4 则最终可以生成FePO4沉淀被全部回收。 -
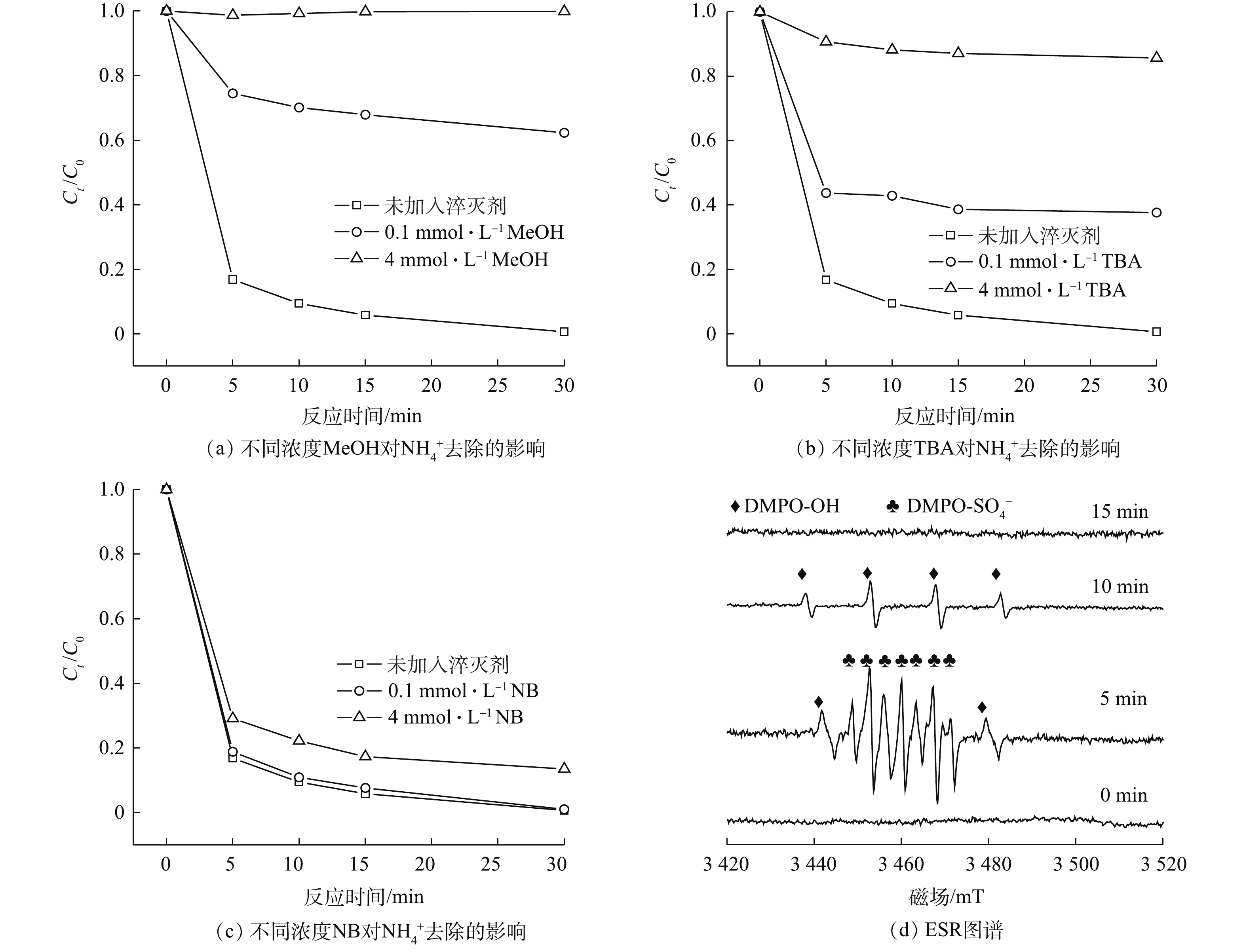
为进一步确定该体系的反应机理,向溶液中分别加入甲醇(MeOH)、叔丁醇(TBA)和硝基苯(NB) 3种自由基淬灭剂,通过观察对
NH+4 去除效果的影响来判定体系中主要存在的有效自由基。MeOH可淬灭SO⋅−4 (kSO⋅−4 =1.6×109 mol·(L·s)‒1)和·OH(k·OH=1.9×109 mol·(L·s)‒1);TBA可淬灭·OH(k ·OH=6×108 mol·(L·s)‒1)和Cl·(k Cl·=3×108 mol·(L·s)‒1);NB可淬灭·OH(k·OH=3.9×109 mol·(L·s)‒1)[25]。如图5(a)~图5(c)所示,当在体系中加入4 mmol·L−1 MeOH时,溶液中的NH+4 基本没有被去除;加入4 mmol·L−1 TBA时,NH+4 的去除率下降到14.3%;而加入4 mmol·L−1NB时,NH+4 的去除受到较小的抑制作用。这是由于当体系中SO4·−被淬灭后,Cl−无法被氧化为Cl·,NH+4 也并不能得到有效去除;当体系中的Cl·被淬灭后,NH+4 的去除受到影响;而·OH被淬灭时,NH+4 的去除影响较小。同时,在反应过程中生成的Fe3+与PO3−4 发生反应,生成FePO4沉淀实现磷回收。由此可知,SO⋅−4 和Cl·在Fe2+/PMS/Cl−体系中实现NH+4 、PO3−4 的同步无害化去除及磷回收中起到重要作用。反应机理[18, 26-28]如式(1)~式(12)。采用ESR进一步测定体系中的自由基,实验过程中以DMPO为捕获剂,对体系中产生的自由基进行捕获。由图5(d)可知,体系中出现了DMPO-
SO⋅−4 和DMPO-·OH加合物的典型特征峰[29]。该结果证实,在Fe2+/PMS/Cl−体系中产生了SO⋅−4 和·OH。 -
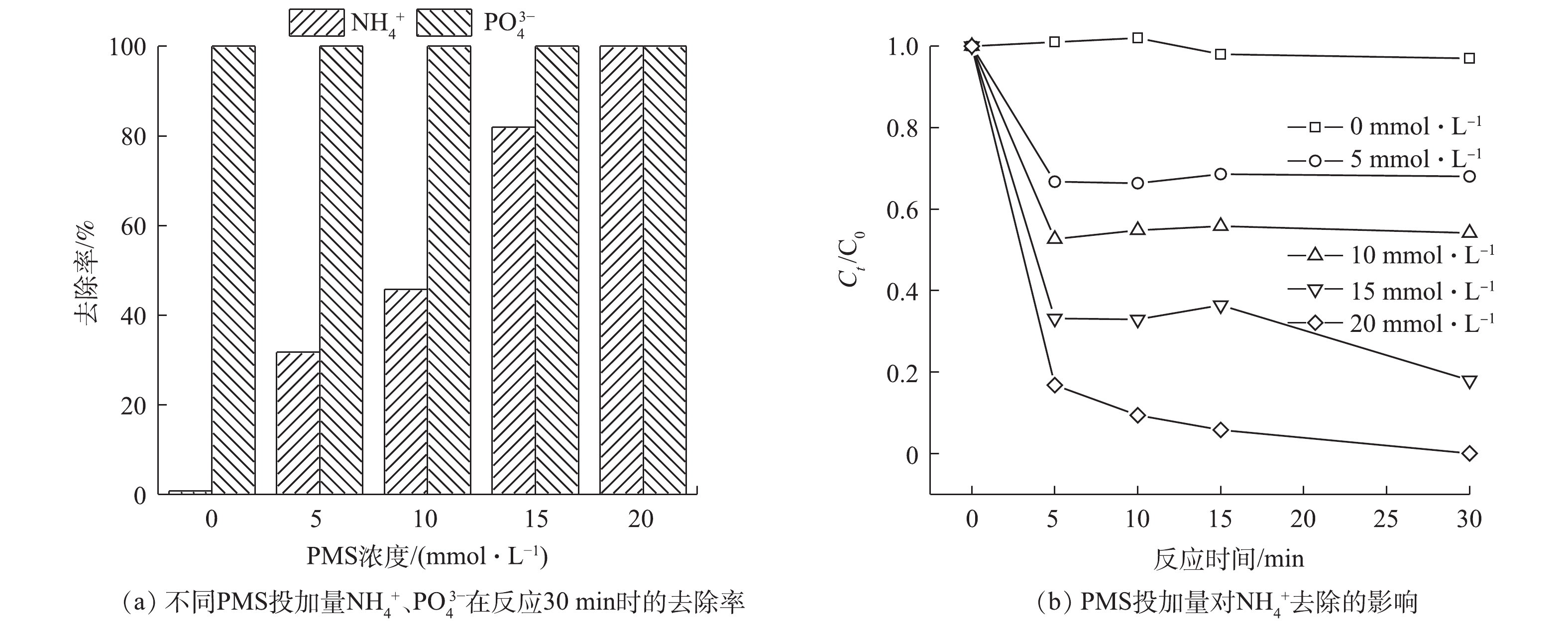
体系中的PMS浓度会影响SO4·−的产生量,进而影响
NH+4 的去除速率和PO3−4 的回收率。图6显示了不同PMS投加量对NH+4 去除以及PO3−4 回收情况的影响。当PMS浓度为5 mmol·L−1时,反应30 min时即可将PO3−4 全部去除,但此时NH+4 的去除率仅有31.7%。分析其原因,是因为此时体系中产生的SO⋅−4 较少,导致SO⋅−4 氧化Cl−产生的Cl·量较少,无法充分氧化NH+4 。随着PMS浓度的增加,SO⋅−4 的产量逐渐增大,NH+4 的去除率逐渐提高。当PMS的浓度为20 mmol·L−1时,可实现溶液中NH+4 、PO3−4 的全部同步无害化去除及磷的回收。 -
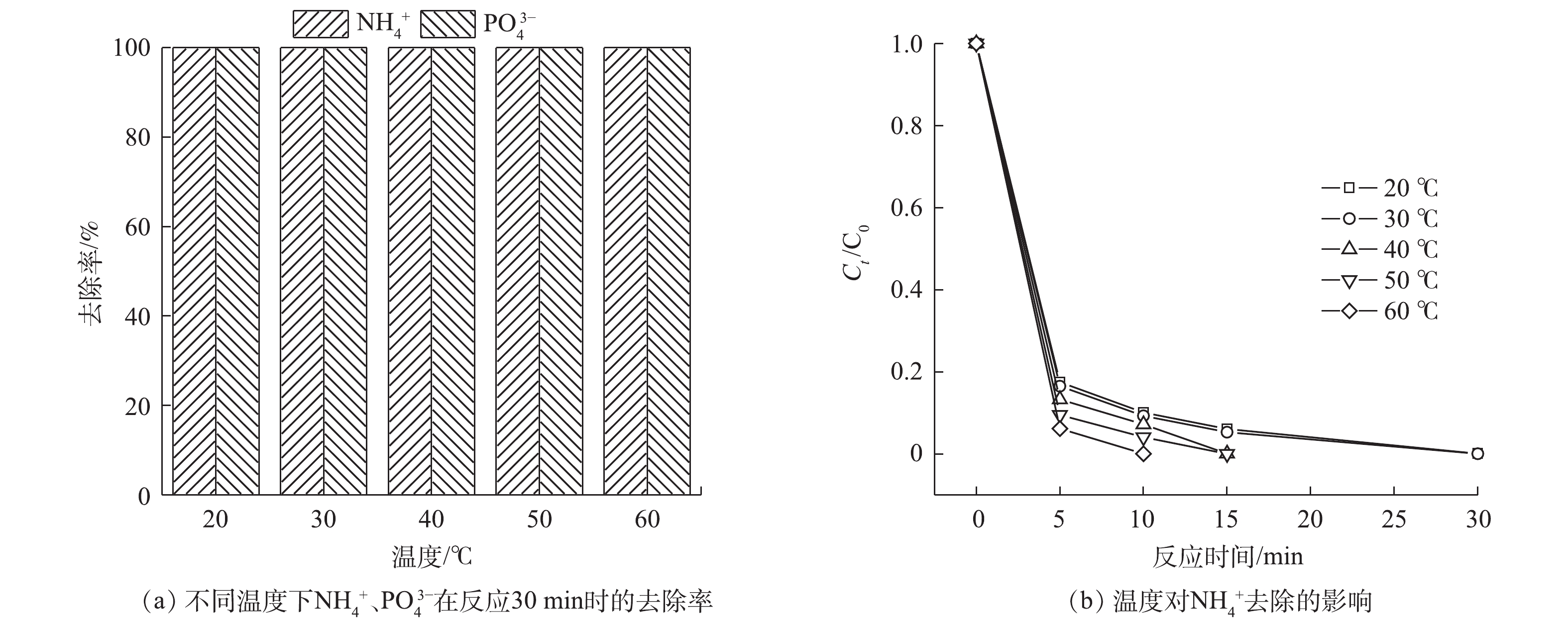
考察了温度为20~60 ℃,
NH+4 、PO3−4 的去除情况,结果如图7所示。随着温度的升高,去除显著加快。当温度升至60 ℃时,NH+4 可在10 min内去除。分析原因可能有:一方面,温度的升高增加了PMS的活化效率,增加了体系中活性自由基的产生量[26];另一方面,反应速率也随着温度的升高而变大[30]。 -
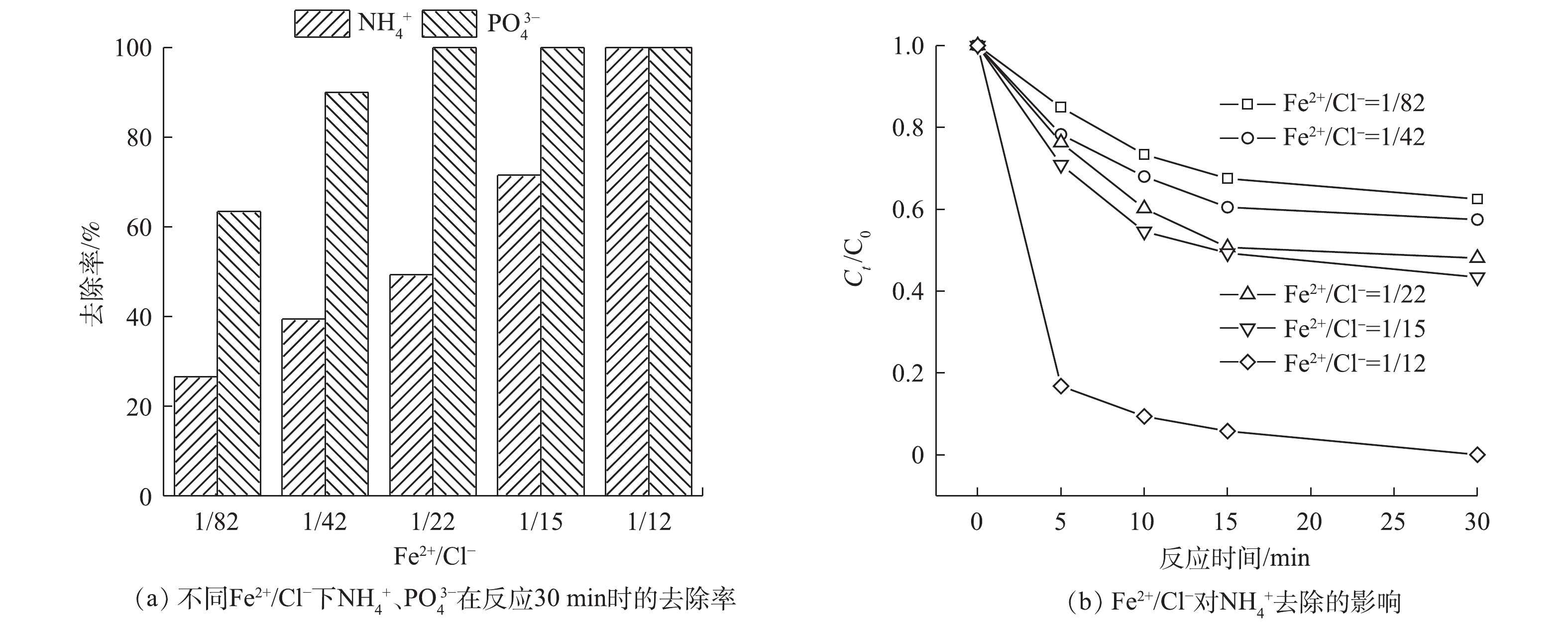
不同Fe2+/Cl−对SO4·−的产生起着非常重要的作用。为研究不同Fe2+/Cl−对反应过程的影响,选取Fe2+/Cl−分别为0、1/82、1/42、1/22、1/15、1/12,结果如图8所示。随着Fe2+/Cl−的增大,
NH+4 的去除率和PO3−4 的回收率逐渐提高。当Fe2+/Cl−为1/22,PO3−4 基本完全回收,但此时NH+4 的去除率只有47.8%。这主要是由于此时溶液中Fe2+浓度较低,产生的自由基较少[31],不足以将NH+4 氧化。当Fe2+/Cl−增加到1/12时,NH+4 的去除率可达到为100%。这可能是由于,当增加体系中的FeCl2时,体系中SO⋅−4 和Cl·的产量也随之增加,促使NH+4 的去除率逐渐提高。 -
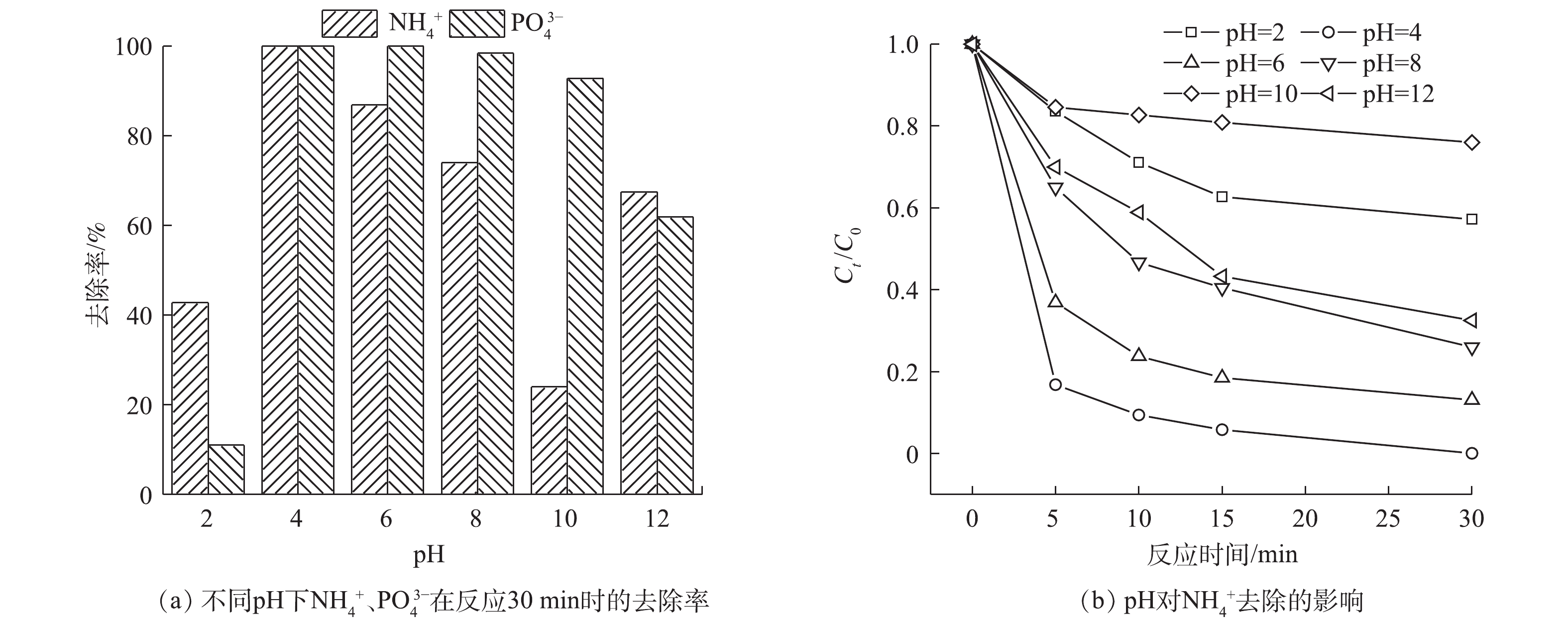
不同pH对
NH+4 、PO3−4 去除效果的影响如图9所示。当pH为4时,溶液中的NH+4 和PO3−4 基本上可以完全去除和回收。当pH过高或过低时,NH+4 的去除和PO3−4 的回收都会受到影响。这是由于溶液中PMS在强酸条件下主要以H2SO5的形式存在,SO4·−不能有效生成[32]。在pH为8和10的偏碱性环境时,NH+4 的去除率降低。这可能是由于Fe2+转化成Fe3+,生成沉淀导致催化剂减少[33];HSO−5 发生了非自由基途径的自分解[33]。而当pH升高到12时,NH+4 的去除率反而较pH为8和10的情况有所提高。这是由于在此时的强碱环境下,NH+4 的主要存在形式为氨气;氨气从溶液中溢出,导致NH+4 的去除率有所提高。当pH为2时,PO3−4 的回收率较低的原因可能有:1)生成的磷酸铁沉淀有一部分溶解到溶液中,使其不能生成沉淀去除;2)溶液中的H+可以与SO4·−和·OH反应,消耗体系中的自由基,反应[34]见式(13)~式(14);3)可能生成了(Fe(Ⅱ)(H2O))2+,导致游离的Fe2+减少[35]。当pH过高时,由于水中存在大量的OH−,而且氢氧化铁的溶度积(4×10−36)远小于磷酸铁(9.91×10−16)的溶度积,所以会优先生成氢氧化铁沉淀,导致PO3−4 仍存在于溶液中。 -
由于水体中存在着各种无机阴离子,而本研究的反应过程中存在
PO3−4 与Cl−,所以考察了水中共存的CO2−3 对反应的影响,即Fe2+/PMS/Cl−体系在水中含CO2−3 的情况下,以及CO2−3 浓度发生变化时对NH+4 、PO3−4 去除的影响。由图10可知,CO2−3 的加入及浓度变化对PO3−4 的去除基本没有影响,而对NH+4 的去除有明显影响;当溶液中不存在CO2−3 时,溶液中NH+4 可完全去除;当CO2−3 的投加量为1、3、5 mmol·L−1时,NH+4 的去除率分别为76.8%、73.6%、63.9%。分析原因可能有:1)CO2−3 在溶液中可部分水解成HCO−3 ,这2种离子会与SO⋅−4 发生副反应,如式(15)~式(17)所示,减少了SO⋅−4 的有效浓度[36];2)CO2−3 可以与·OH发生反应[25](式(18)),进而影响了NH+4 的去除。 -
腐殖酸(humic acid,HA)是一种广泛存在于地表水和土壤中的天然有机质(natural organic matter,NOM),对水处理过程影响较大[37]。因此,考察了初始HA浓度为0、5、10、20 mg·L−1,对体系的
NH+4 、PO3−4 处理效果影响,结果如图11所示。随着HA浓度的增加,NH+4 的去除受到一定影响,PO3−4 的去除并没有受到影响。这可能是由于,HA作为一种大分子有机物质,可以和NH+4 竞争溶液中的自由基[38-39],导致体系中NH+4 的去除受到抑制,且这种影响随HA含量的增加而增加。
2.1.
不同体系中NH+4![]()
![]()
、PO3−4![]()
![]()
的去除效果
2.2. 机理分析
2.3. PMS浓度对去除效果的影响
2.4. 温度对去除效果的影响
2.5. Fe2+/Cl−对去除效果的影响
2.6. pH对去除效果的影响
2.7. 阴离子对去除效果的影响
2.8. 腐殖酸(HA)对去除效果的影响
-
1)采用Fe2+/PMS/Cl−体系实现了
NH+4 、PO3−4 的同步无害化去除并回收磷,结果证明NH+4 以N2的形式无害化去除,PO3−4 以FePO4沉淀的形式得到资源化回收。自由基淬灭实验和ESR证明,该体系中产生了·OH、SO⋅−4 和Cl·,而SO⋅−4 和Cl·在反应中起主要作用。2)
NH+4 的去除率随PMS浓度、Fe2+/Cl−、温度的提高而得到增加,PO3−4 的回收几乎不受影响。溶液pH对实验的影响比较显著,强酸强碱不利于NH+4 的去除和PO3−4 的回收。3)水中共存
CO2−3 和HA对NH+4 的去除有不利影响,而对PO3−4 回收无明显影响。
