




 下载:
下载:










-
近年来,由于高级氧化技术可以产生大量活性氧组分(ROS,如·OH、
SO⋅−4 、1O2等),从而有效促进痕量微污染物的降解,故而该技术成为降解高风险微量有机物的重要手段,而且逐渐成为研究热点[1-2]。虽然单一的臭氧氧化法能有效去除不饱和芳香族和脂肪族化合物,但其对饱和有机化合物的去除率很低[3]。为了高效去除难降解的饱和的持久性有机污染物,本文通过选用一种高性能的催化剂催化臭氧产生更多的活性物质[4-6],以达到彻底去除污染物的目的。锰酸铜(CuMn2O4)尖晶石是一种密度较大的空心六面体,其晶体结构主要是由Mn4+与Cu2+搭建的,还有较少的Mn3+与Cu+增加了结构的缺陷程度。CuMn2O4可以催化臭氧产生具有氧化能力强、无选择性的·OH,从而有效提高臭氧对水体中难降解污染物的去除效果。有研究[7]指出,臭氧与CuMn2O4的结合对二苯甲酮-3的降解有明显的协同作用。然而,在实际使用过程中,CuMn2O4的密度大、容易团聚、不易分散的特性使其利用率很低。为了提高其利用率,需要选用另外一种催化剂进行耦合,弥补其在使用过程中的缺陷。研究表明,二维层状碳材料在催化臭氧氧化领域中有很好的效果[8]。因为二维层状碳材料不仅在平面内的热运输和电荷运输过程中具有突出的物理化学特性,而且与其他材料复合后可以产生良好的耦合效应[9-10]。石墨烯/还原氧化石墨烯(rGO)是其中一种极具吸引力的二维材料,具有卓越的化学稳定性、导电性和表面体积比[11-12]。此外,石墨相氮化碳(g-C3N4)是一种具有2.7 eV带隙的二维非金属聚合物半导体[13],在化学、热和光照射过程中具有较好的稳定性。同时,g-C3N4还是一种有效的催化剂载体[14-15],在其中掺杂选定的杂原子,通过电荷转移可以形成络合的复合材料[16-17]。因此,可以考虑将rGO和g-C3N4与CuMn2O4进行复合,应用于催化臭氧氧化过程中。与大多数亲脂性的有机防晒剂不同的是,二苯甲酮-4(BP-4)是一种亲水性的紫外线吸收剂,因其质地更轻、油性更少,被广泛应用于洗发水、剃须凝胶、止汗剂、化妆品和牙膏等日用品中[18]。但是,由于BP-4化学稳定性好、不易降解,因而被认为是一种伪持久性有机污染物,越来越受到人们的关注[19]。在目前的废水处理领域中常见的水处理方法并不能将其完全去除[20]。此外,由于rGO和g-C3N4在催化臭氧氧化过程中对臭氧氧化副产物溴酸盐有很好的抑制效果[8],因此,本研究将rGO和g-C3N4与CuMn2O4复合,来探究他们在催化臭氧氧化过程中对BP-4的降解效果以及溴酸盐生成的影响。
全文HTML
-
一水合硫酸锰(MnSO4·H2O,≥99.0%)、三水合硝酸铜(Cu(NO3)2·3H2O,≥99.0%)、碳酸钠(Na2CO3,≥99.8%)、尿素(CO(NH2)2,99%)、硝酸钠(NaNO3,≥99.0%)、高锰酸钾(KMnO4,≥99.0%)、二苯甲酮-4(C14H12O6S,98%)、硫酸(H2SO4,≥70%)、石墨粉、过氧化氢(H2O2,≥27.5%)和盐酸(HCl,38%)。
X射线衍射仪(XRD-7000,日本岛津公司);比表面积测试仪(SSA-7000,北京博德电子科技公司);X射线光电子能谱仪(AXIS Ultra,日本岛津公司);高效液相色谱仪(Waters 2695,沃特世科技(上海)有限公司);智能箱式高温炉(DC-B06/02,北京独创科技有限公司);恒温水浴振荡器(SHA-B,常州市金坛友联仪器研究所);鼓风干燥箱(DGA-9073 B-2,上海福玛实验设备有限公司);电子天平(AL 104,梅特勒-托利多仪器有限公司);pH计(S210 Seven Compact,梅特勒-托利多仪器有限公司);离子色谱仪(ICS-3000,上海普迪生物技术有限公司);电化学工作站(CHI660E,上海辰华仪器有限公司);磁力搅拌器(RH Digita,德国艾卡IKA公司);电子扫描显微镜(S4800,日本Hitachi公司)。
-
采用共沉淀法制备CuMn2O4。具体操作步骤如下:将3.561 7 g Cu(NO3)2·3H2O溶于120 mL去离子水中,得到溶液1;将4.969 2 g MnSO4·H2O溶于120 mL去离子水中,得到溶液2;将6.359 4 g Na2CO3溶于60 mL去离子水中,得到1 mol·L−1的Na2CO3溶液。磁力搅拌溶液2,使用塑料滴管将溶液1逐滴加入溶液2中(用时20 min)。溶液1加完后,使用pH计测定混合溶液的初始pH,此时的溶液显酸性。将溶液2使用水浴锅加热至80 ℃,磁力搅拌。将Na2CO3溶液逐滴加入溶液2中,每滴加一滴,停止滴加,搅拌均匀后,继续滴加,使用pH计测定溶液的pH,在pH=10时停止滴加(用时20 min),在水浴条件下继续搅拌20 min后停止搅拌,停止水浴加热,静置1 h。使用砂芯漏斗对沉淀进行真空抽滤,并使用去离子水洗涤,取滤液测定其pH,直到pH基本不变,再用无水乙醇洗涤5次。将沉淀转移到表面皿,将有残留样品的滤纸和表面皿在烘箱干燥,温度为120 ℃,时间为15 h。将干燥后的样品收集,置于马弗炉中900 ℃煅烧,升温速率为1 ℃·min−1,保温时间为6 h。
热缩聚的方法制备g-C3N4。称量10 g尿素,研磨成粉末,置于马弗炉中350 ℃煅烧,升温速率为5 ℃·min−1,保温时间为1 h。使用Hummers法[21]制备氧化石墨烯(GO)。
两步煅烧法制备CuMn2O4/rGO和CuMn2O4/g-C3N4。将共沉淀法制备的CuMn2O4和GO按照质量比例1∶1进行混合,研磨均匀,将粉末转移到坩埚中,置于马弗炉中煅烧,温度为350 ℃,升温速率为5 ℃·min−1,保温时间为1 h。在反应结束后,冷却至室温,得到复合的CuMn2O4/rGO粉末。将10 g尿素与0.2 g的CuMn2O4研磨均匀后,每次取定量研磨好的粉末置于马弗炉中,加热到350 ℃,加热时间为120 min,加热速率为10 ℃·min−1。在反应结束后,冷却至室温,得到复合的CuMn2O4/g-C3N4粉末。
-
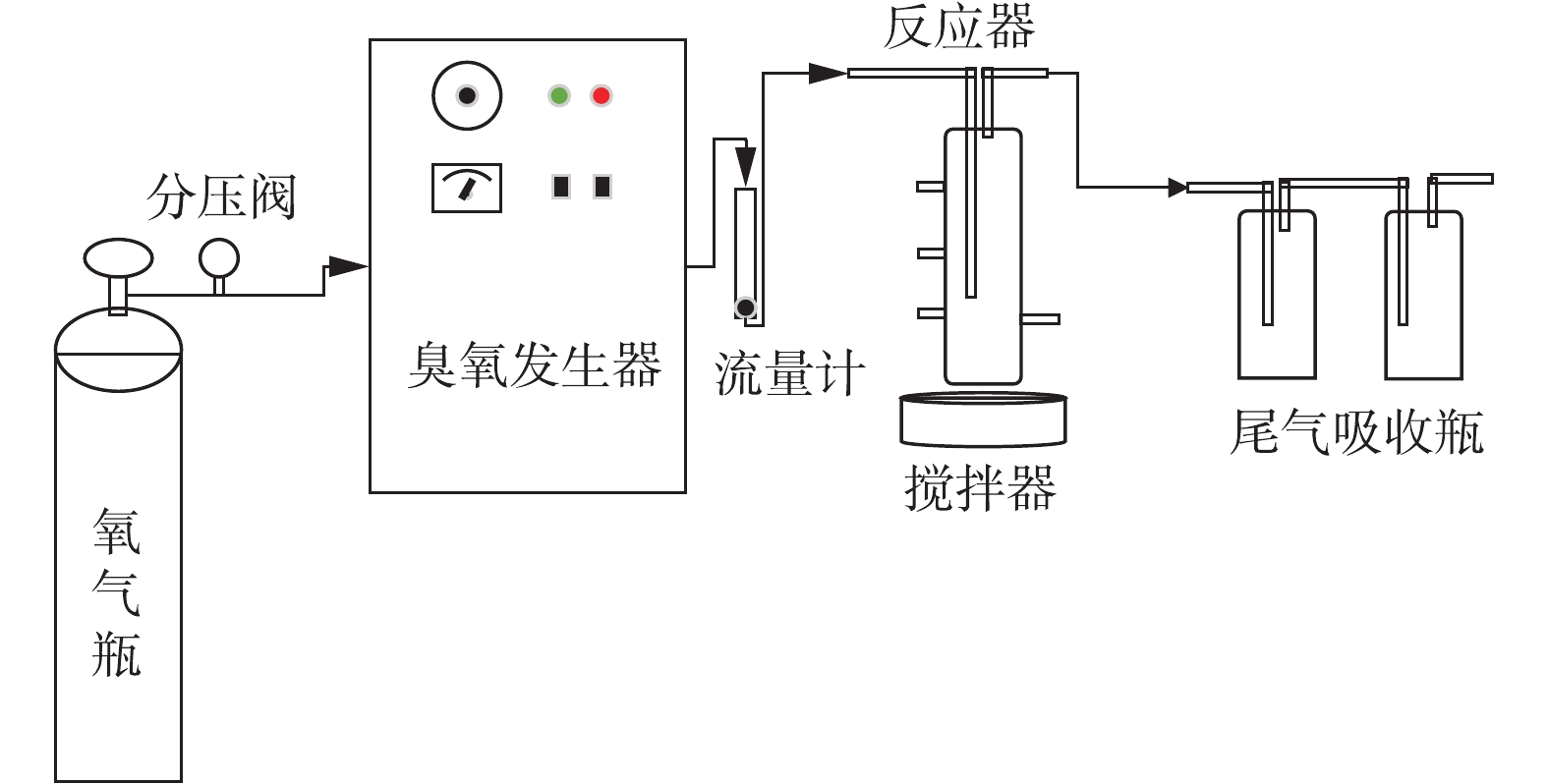
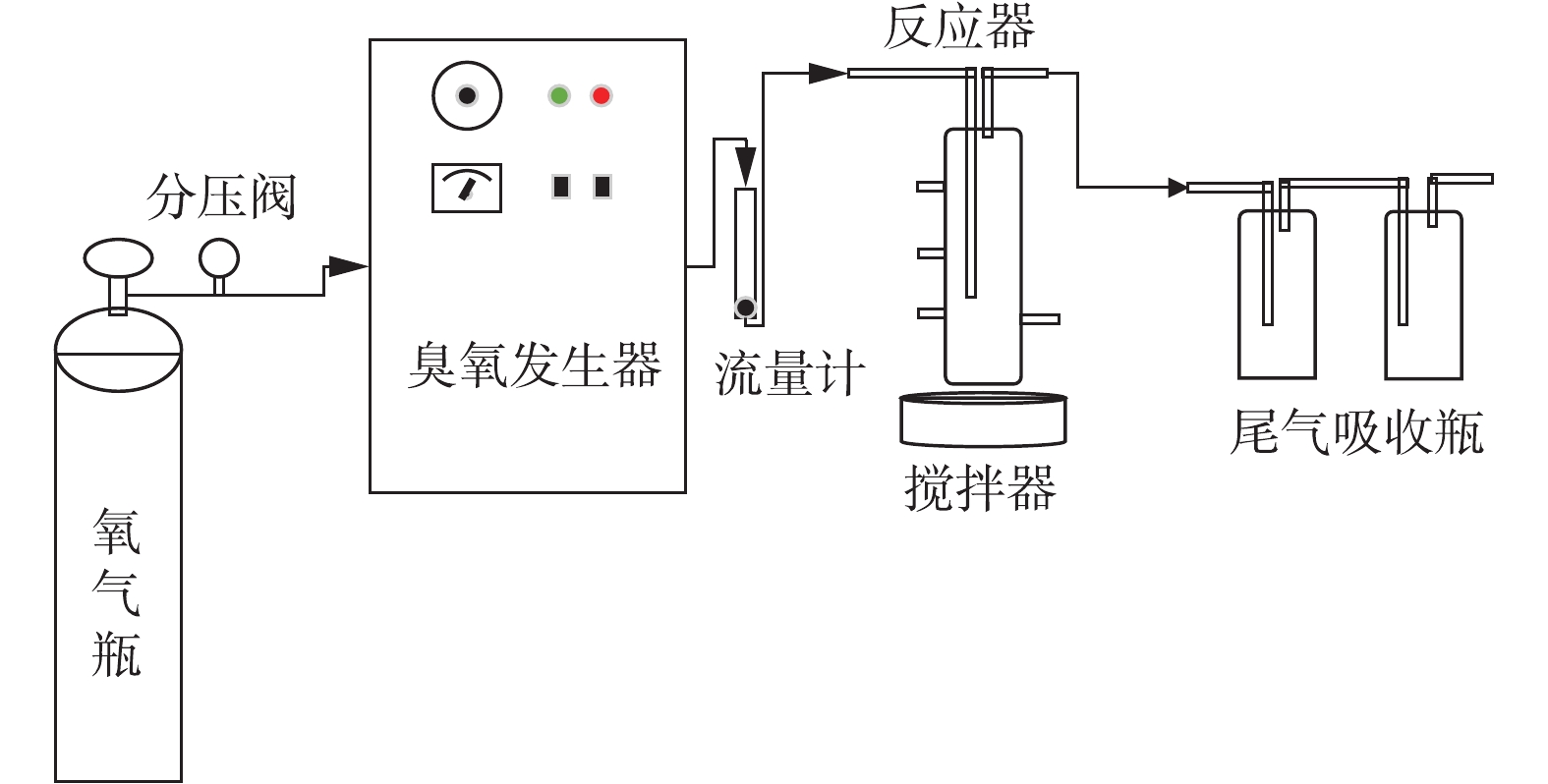
催化臭氧氧化BP-4的效能实验采用间歇反应模式进行。反应器为圆柱形的玻璃容器,直径为6.2 cm,高为26.5 cm,有效容积为300 mL。实验使用北京同林高科技有限责任公司生产的3S-A5型臭氧发生器(臭氧产量为0~1 g·h−1),以高纯氧气为气源,本实验所用的实验装置如图1所示。实验中每次处理的水样为300 mL,所有的溶液均用去离子水配制。臭氧进气浓度通过臭氧发生器的放电功率来调节。在每次实验开始之前,用纯氧进行吹扫,并预臭氧化处理。打开臭氧发生器,调节气流量为400 mL·min−1,臭氧发生器电流为0.025 A,预热时间为60 min,预臭氧时间为30 min。
-
在反应器中加入290 mL超纯水,O3曝气30 min,搅拌器的转速为800 r·min−1。加入10 mL BP-4母液(反应器中的浓度是0.084 mmol·L−1(25.91 mg·L−1));100 μL Br−母液(母液浓度为300 mg·L−1,反应器中的浓度是100 μg·L−1),开始反应并计时。在反应时间为0、1、2、5、7、10、15、30 min时分别取样,并使用浓度为10 mmol·L−1的亚硫酸钠溶液还原残留臭氧;使用0.22 μm的水系滤膜过滤粉体催化剂后待分析。
-
使用X-射线衍射仪(XRD)对制得的粉体催化剂的矿物组成与结晶结构进行分析;使用比表面积分析仪对制得的粉末催化剂的比表面积及表面孔结构进行表征;使用X射线光电子能谱仪(XPS)表征粉末状催化剂中各元素的价态;使用电化学工作站分析粉末催化剂的阻抗。使用扫描电镜(SEM)对制得的粉末催化剂进行表观形貌的分析。
BP-4的浓度由高效液相色谱仪Waters 2695测定。使用的色谱柱为Symmetry C18,柱温为30 ℃。流动相为0.3%甲酸缓冲液和甲醇的混合溶液,其中0.3%甲酸缓冲液的体积分数为35%,流速为1 mL·min−1,检测波长为286 nm,进样量为10 μL,保留时间为8.5 min。
Br−和
BrO−3 的浓度由离子色谱仪ICS-3000测定。样品先经过AG19保护柱,然后进入分析柱AS19进行分析。取样量为500 μL,流速为1 mL·min−1,柱温为35 ℃。淋洗条件为梯度淋洗(0~18.0 min:10.0 mmol·L−1 NaOH;18.1~26.0 min:35.0 mmol·L−1 NaOH;26.1~40.0 min:10.0 mmol·L−1 NaOH)。催化臭氧氧化过程中溴酸盐的生成速率可根据式(1)进行计算。式中:v为生成速率,min−1;(
CBrO−3 )max为整个催化臭氧氧化过程中的BrO−3 的最大浓度,mol·L−1;(CBr− )0为溴离子的初始浓度,mol·L−1,本研究中该值为1.25 μmol·L−1;t为BrO−3 达到最大浓度时的反应时间,min。
1.1. 实验原料及仪器
1.2. 催化剂的制备
1.3. 实验装置
1.4. 实验方法
1.5. 分析方法
-
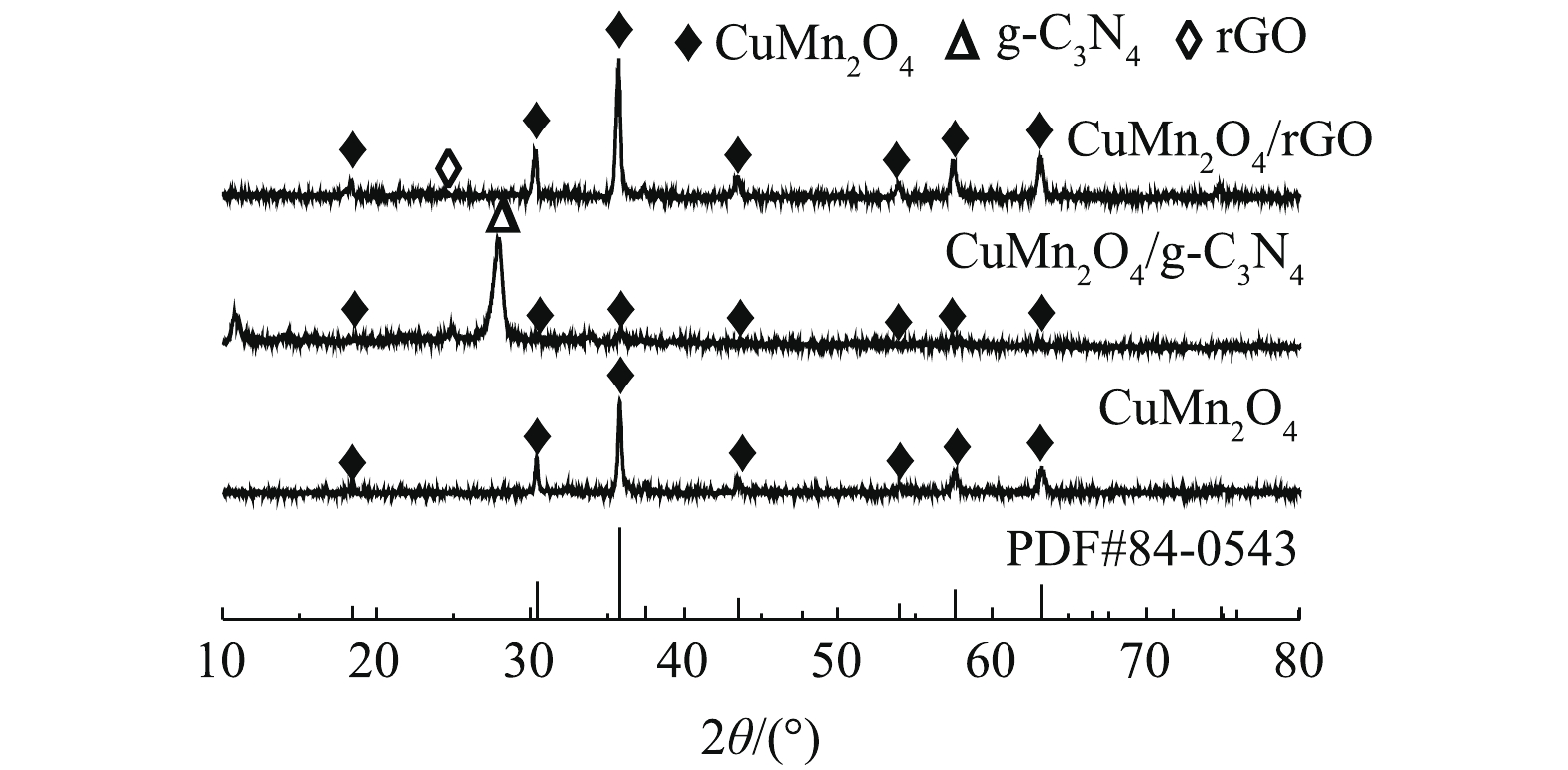
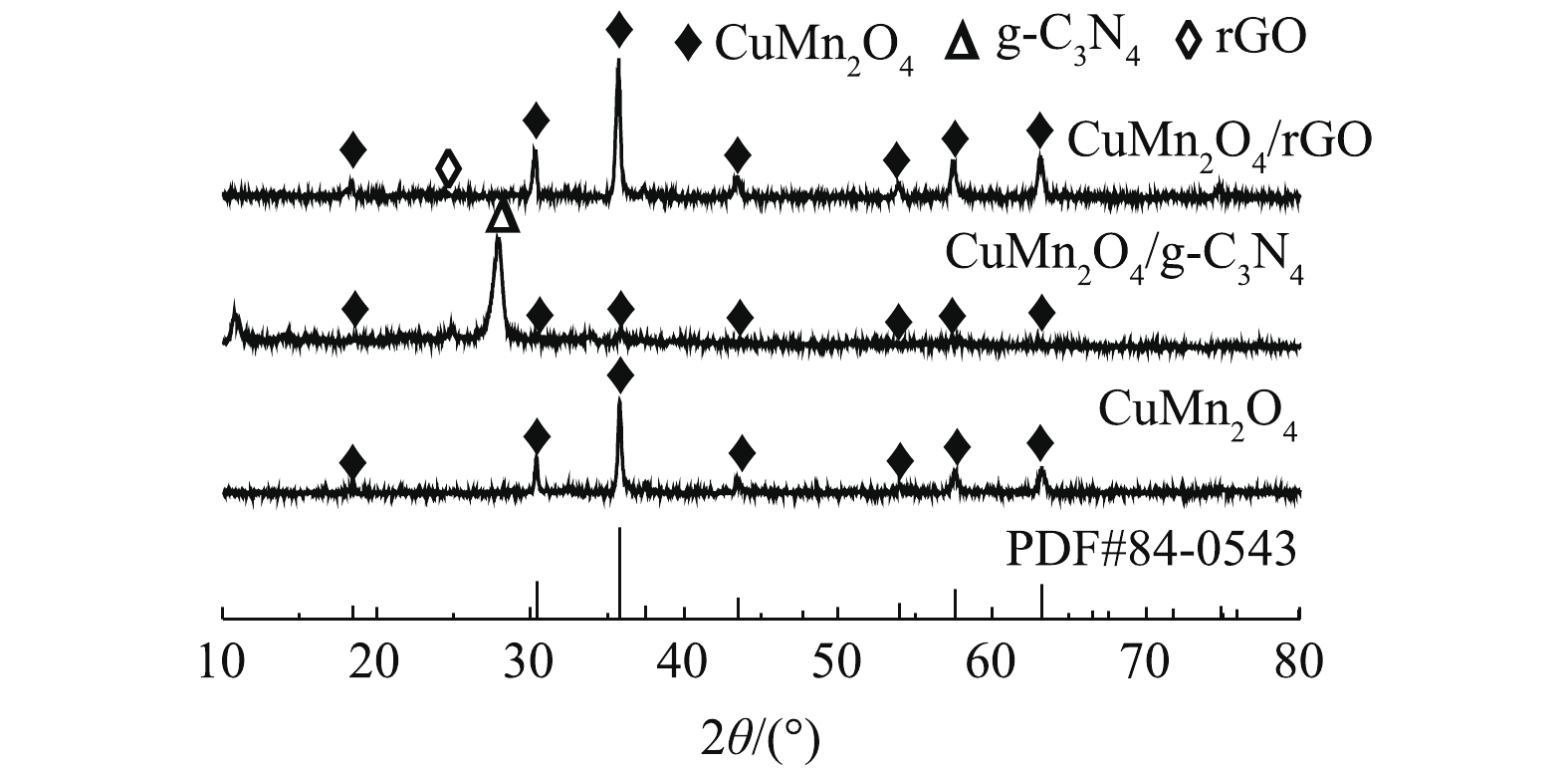
1)矿物组成与晶体结构。如图2所示,CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4具有相似的XRD谱图。分别在2θ=18.4°、30.2°、35.6°、43.5°、54.1°、57.5°、63.3°和77.5°处出现的衍射峰与CuMn2O4的标准卡片(JCPDS#84-0543)中特征衍射峰的位置是一致的。这些衍射峰分别对应着CuMn2O4的(111)、(220)、(311)、(222)、(400)、(422)、(511)、(440)和(533)晶格面。此外,CuMn2O4/rGO的谱图中在2θ=25.4°处对应的是rGO的特征衍射峰;CuMn2O4/g-C3N4的谱图中在2θ=27.3°处对应的是g-C3N4的特征衍射峰。可见,通过两步煅烧的方法已经将CuMn2O4与g-C3N4和rGO成功复合在一起。值得注意的是,与CuMn2O4/rGO相比,CuMn2O4/g-C3N4中CuMn2O4的衍射峰强变弱。ZHU等[22]将NiFe2O4与g-C3N4复合后,通过XRD分析,同样发现NiFe2O4的峰强减弱。可见,rGO的掺入不会影响CuMn2O4的物相结构和结晶度;g-C3N4的掺入虽然不会影响CuMn2O4的物相结构,但会降低CuMn2O4的结晶度。
2)催化剂形貌分析。通过SEM对CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4的形貌进行分析,结果如图3所示。如图3(a)所示,CuMn2O4是表面光滑的不规则立方体,平均粒径为2~3 μm。将其与rGO复合后,SEM表征结果如图3(b)所示,与CuMn2O4相比,催化剂颗粒的体积变小,这可能是因为rGO的存在阻碍了CuMn2O4的团聚。还可以发现,层状rGO基本全部包覆在CuMn2O4的外表面。将CuMn2O4与g-C3N4复合后,形貌如图3(c)所示。与CuMn2O4相比,催化剂颗粒的体积减小,这可能也是因为g-C3N4阻碍了CuMn2O4的团聚。此外,还可以发现,与CuMn2O4/rGO不同的是,在CuMn2O4/g-C3N4样品中,碎片状的g-C3N4一部分覆盖在CuMn2O4的外表面,另一部分独立分散在CuMn2O4颗粒之间。据此可以初步推测,与g-C3N4相比,rGO可能更容易与CuMn2O4结合,CuMn2O4/rGO的结构更稳定。
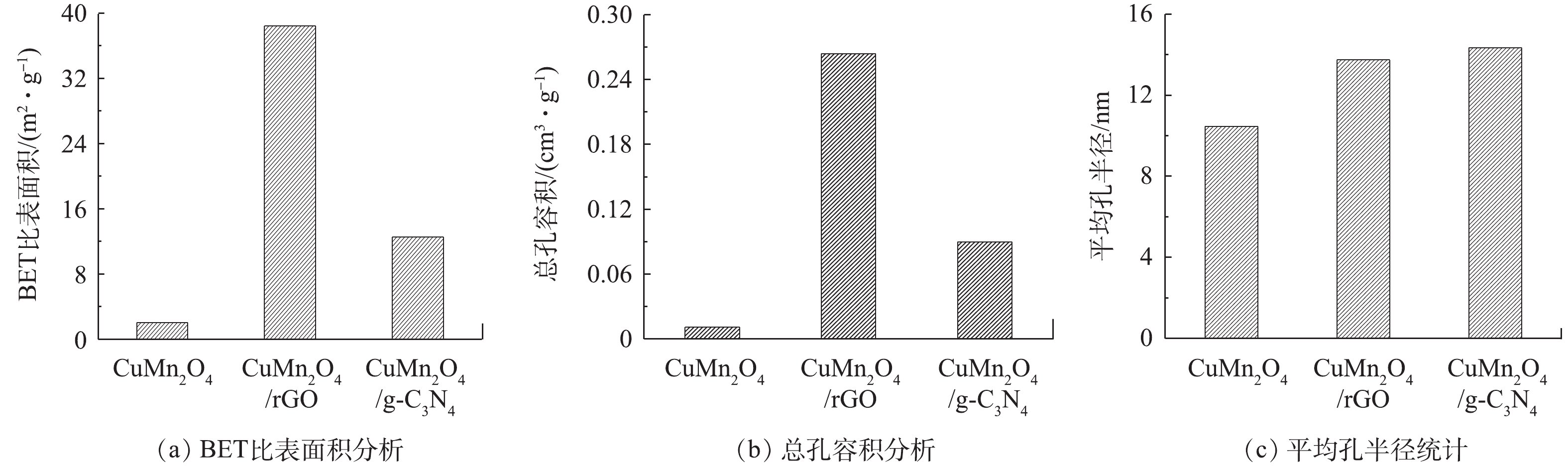
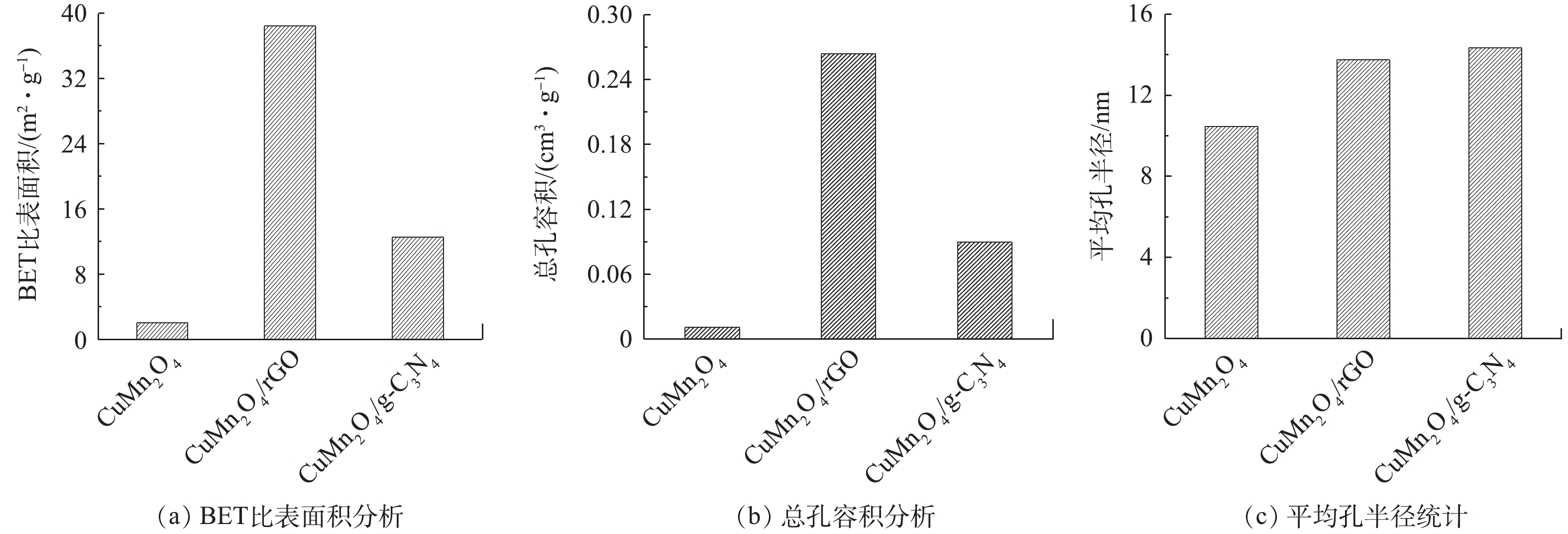
3)比表面积分析。使用比表面积分析仪分析催化剂CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4的孔隙结构特征,结果如图4所示。与CuMn2O4相比,CuMn2O4/rGO和CuMn2O4/g-C3N4的比表面积、总孔容积和平均孔半径均有所增加。与rGO和g-C3N4分别复合后,CuMn2O4的比表面积从2.058 m2·g−1分别增加到38.438 m2·g−1和12.553 m2·g−1;总孔容积从0.011 cm3·g−1分别提升到0.264 cm3·g−1和0.09 cm3·g−1;平均孔半径从10.47 nm分别增至13.74 nm和14.35 nm。DEVI等[23]将rGO与CoFe2O4复合到一起后,也得到了相似的结果:在掺入rGO后,催化剂的比表面积从6.202 m2·g−1增加到了25.18 m2·g−1,总孔容积从0.003 1 cm3·g−1增至0.022 cm3·g−1。他们认为,比表面积和总孔容积的增加是由于CoFe2O4在rGO片层中随意堆积导致的,这也极有可能是CuMn2O4/rGO和CuMn2O4/g-C3N4的比表面积和总孔容积的增加的原因。此外,CuMn2O4/rGO的比表面积是CuMn2O4/g-C3N4的3.06倍,总孔容积为2.93倍,平均孔径为0.98倍。由此可见,rGO的掺入对增大催化剂比表面积和总孔容积的效果更好。
4)表面元素价态分析。为了进一步确定CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4中Cu、Mn与O元素的价态,对3种催化剂分别进行XPS表征,并对测试结果进行解卷积分析,结果如表1所示。Cu、Mn这2种元素在复合前后的价态分布有明显的变化。CuMn2O4中Cu2+的含量为12.89%,当其与层状碳材料复合后,Cu2+的含量分别增加到了70.92%(g-C3N4)和53.96%(rGO);CuMn2O4中Mn4+的含量为28.41%,当其与层状碳材料复合后,Mn4+的含量分别增至34.30%(rGO)和34.30%(g-C3N4)。这说明层状碳材料rGO和g-C3N4都可以氧化CuMn2O4中的Cu+与Mn3+,使他们失去电子导致其化合价升高,失去的电子通过层状碳材料表面的官能团(如羟基)进行转移。与rGO相比,当g-C3N4掺入CuMn2O4后催化剂中Cu2+的含量更高,可见g-C3N4对CuMn2O4中的Cu+有更强的氧化能力,这也意味着CuMn2O4/g-C3N4的表面会有更多的转移电子。
此外,在3种催化剂中均可以观察到晶格氧(Olatt)、表面羟基氧(OOH)和表面氧空位(OV)[24]。粉末催化剂中的氧空位是催化臭氧氧化重要的潜在催化位点[25],CuMn2O4中OV的含量为13.6%,CuMn2O4/rGO与CuMn2O4/g-C3N4中OV的含量分别增加至50.8%和47.7%。这一结果归因于层状碳材料rGO和g-C3N4与CuMn2O4晶格结合后,导致其晶格膨胀或畸变,产生了更多的缺陷位,从而形成更多氧空位[25]。与CuMn2O4/rGO相比,CuMn2O4/g-C3N4中OV的含量更多。
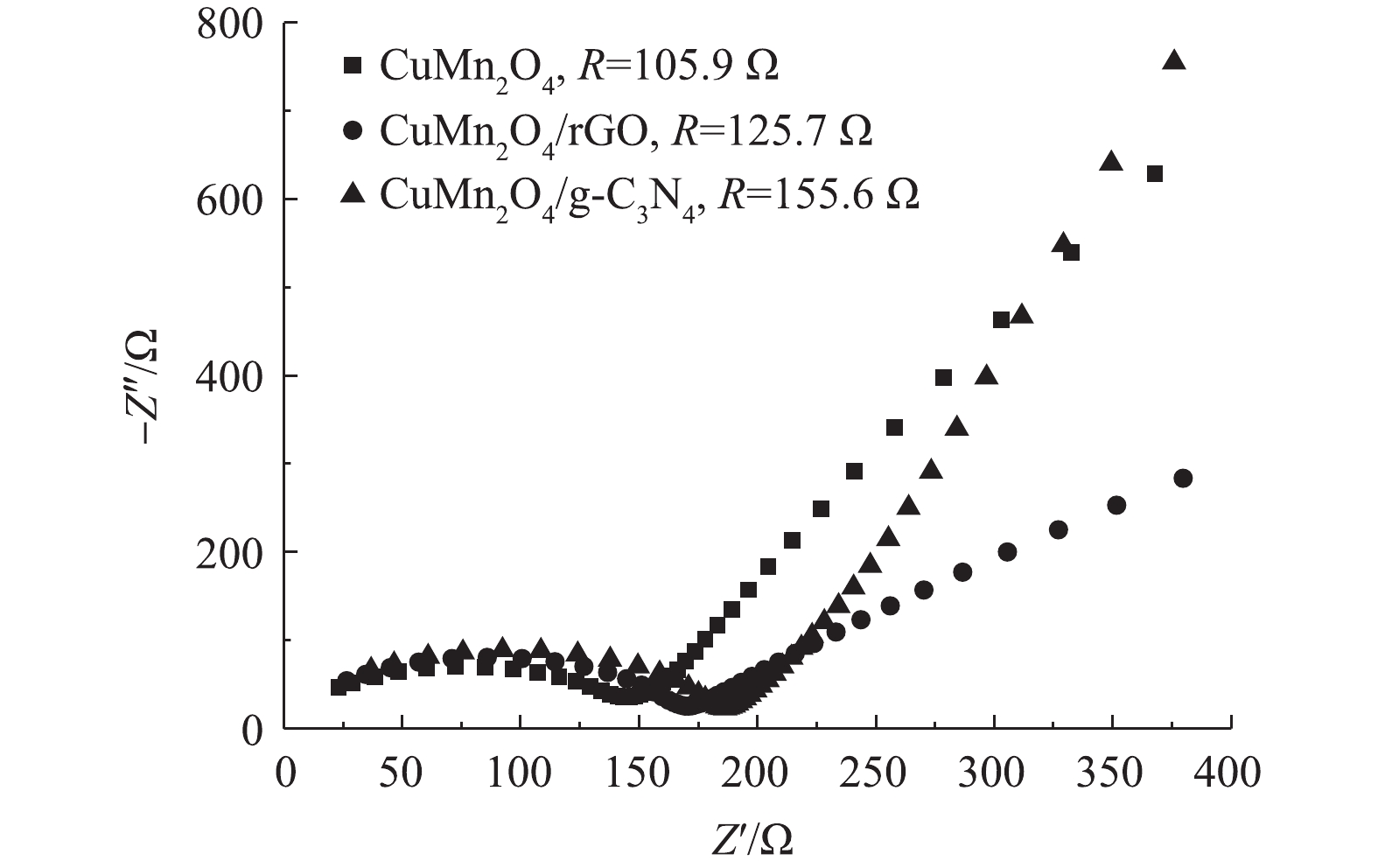
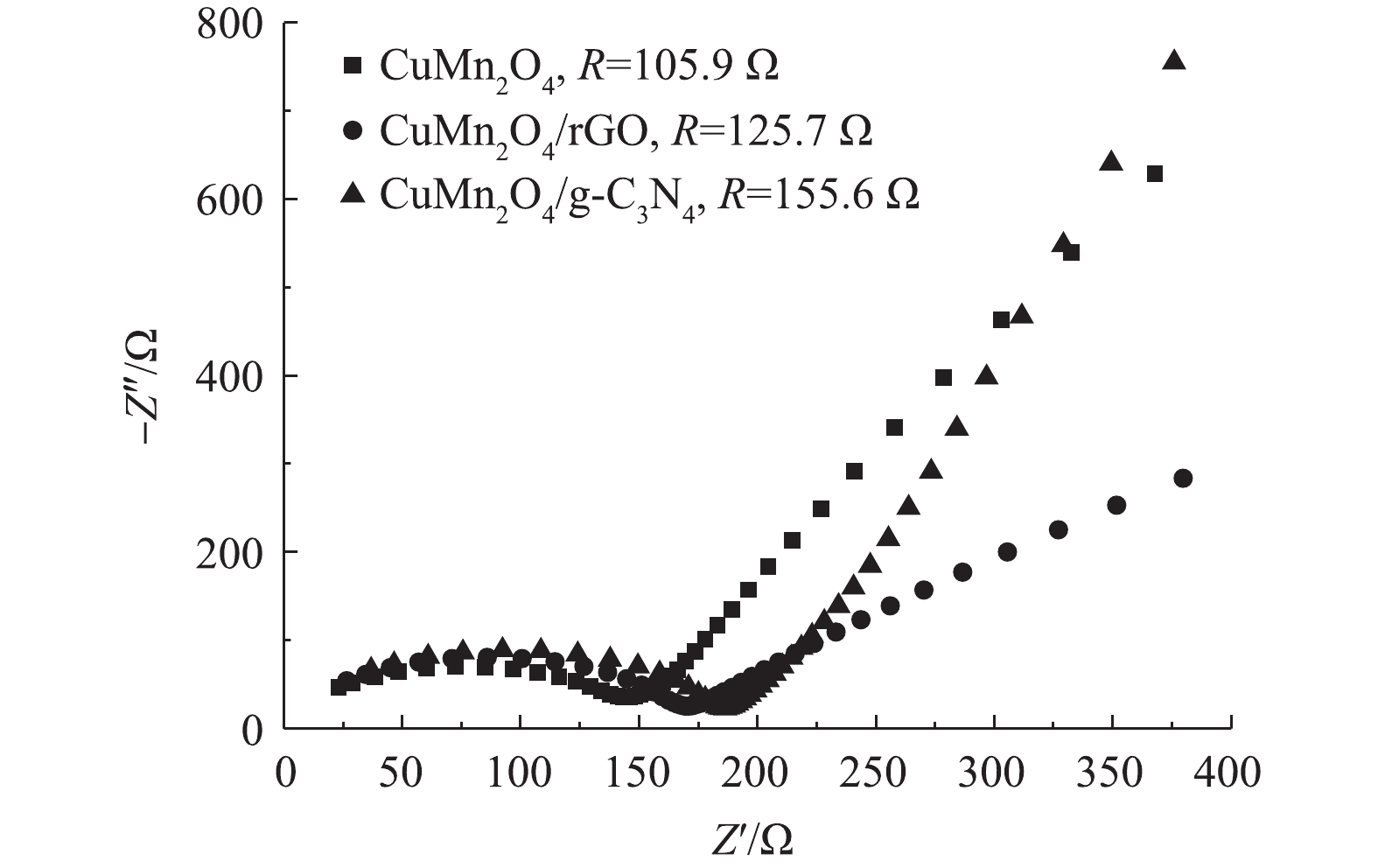
5)阻抗分析。CuMn2O4,CuMn2O4/rGO和CuMn2O4/g-C3N4的Nyquist曲线如图5所示。与CuMn2O4相比,CuMn2O4/rGO和CuMn2O4/g-C3N4的半圆弧形半径增大。这说明,复合催化剂CuMn2O4/rGO和CuMn2O4/g-C3N4的电荷转移电阻变大。通过进一步使用模拟电路进行拟合计算,CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4的阻抗值分别为105.9、125.7和155.6 Ω。这与LI等[26]的实验结果不一致,他们将石墨烯纳米片与CuMn2O4复合后,CuMn2O4/rGO的阻抗小于CuMn2O4。CHEN等[27]将LiMn2O4与rGO通过水热法和直接将2种物质混合的方法分别进行复合后,也得到了相似的结果:2种方法得到的LiMn2O4@rGO的阻抗分别为56.79 Ω和178.10 Ω,而复合前LiMn2O4的阻抗为287.0 Ω。CHEN等[27]认为,与LiMn2O4相比,复合催化剂阻抗的减小主要归因于rGO自身优越的导电性;与直接将2种材料混合的方法相比,水热法制备的复合催化剂阻抗更小则是因为借由水热反应,rGO与LiMn2O4之间的结合方式更有利于电荷转移。据此可以推测,本研究中复合材料CuMn2O4/rGO和CuMn2O4/g-C3N4的阻抗比CuMn2O4大的原因极有可能是,因为通过两步煅烧的方法制备的复合催化剂中CuMn2O4与rGO和g-C3N4之间的结合方式不利于电荷转移。此外,与CuMn2O4/rGO相比,CuMn2O4/g-C3N4的阻抗更大。可见,rGO掺入CuMn2O4后催化剂的导电能力增强。
-
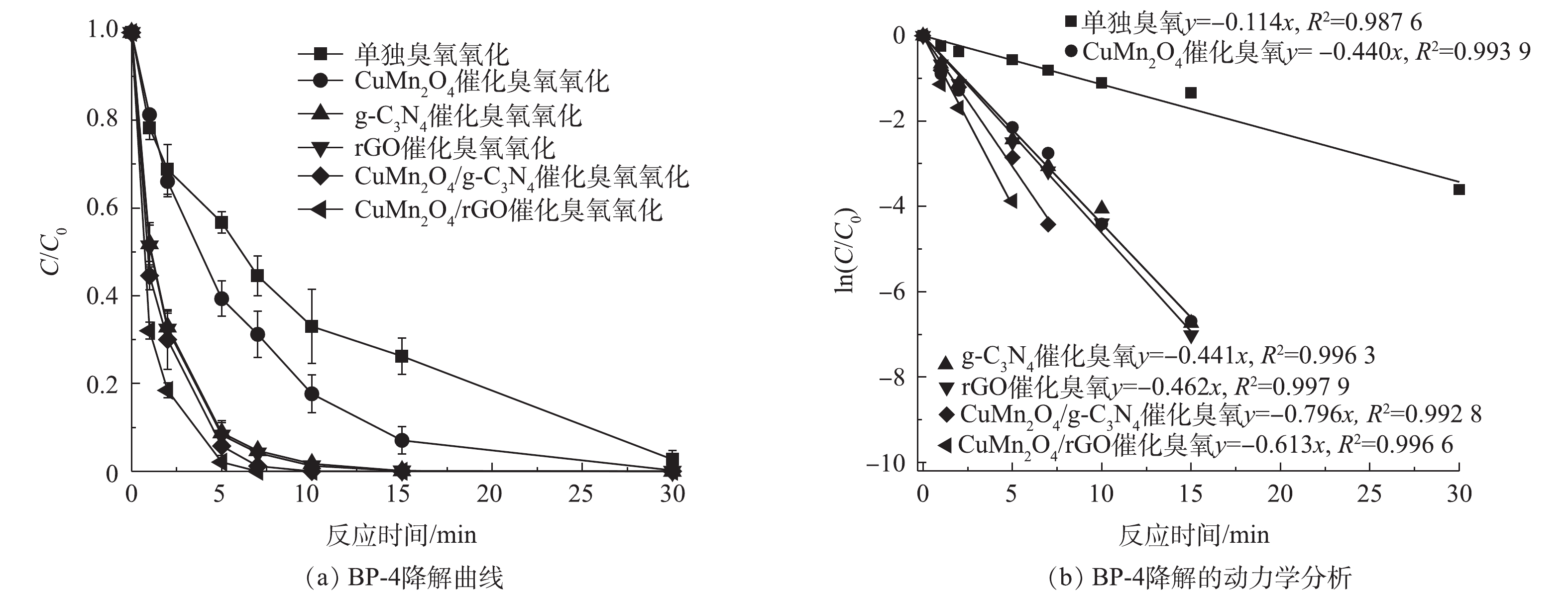
1) rGO 和g-C3N4改性的CuMn2O4催化臭氧降解BP-4的速率对比。单独臭氧氧化、CuMn2O4、rGO、g-C3N4、CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧氧化对BP-4的降解效果如图6所示。如图6(a)所示,单独臭氧氧化和CuMn2O4催化臭氧氧化降解BP-4时,反应30 min后BP-4才能被完全降解;g-C3N4、rGO、CuMn2O4/g-C3N4和CuMn2O4/rGO催化臭氧氧化则可以在10 min后将BP-4完全降解。对BP-4的降解进行拟一级动力学分析,结果如图6(b)所示,单独臭氧氧化、CuMn2O4、g-C3N4、rGO、CuMn2O4/g-C3N4和CuMn2O4/rGO催化臭氧氧化降解BP-4的动力学常数k分别为0.114、0.440、0.441、0.462、0.613和0.796 min−1,可以发现,与g-C3N4和rGO相比,CuMn2O4/g-C3N4和CuMn2O4/rGO催化臭氧氧化降解BP-4的速率得到进一步提升。AKHUNDI等[28]将g-C3N4纳米片(g-C3N4-NS)与CuCr2O4复合后,将其应用于光催化降解RhB、MB染料和苯酚,也得到了相似的结果,他们发现,复合催化剂g-C3N4-NS/CuCr2O4降解RhB的速率分别是块状g-C3N4和g-C3N4-NS单独光催化的11.8倍和4.8倍。此外,进一步对比可以发现,与CuMn2O4/g-C3N4相比,CuMn2O4/rGO催化臭氧氧化降解BP-4的速率更快。结合之前对于2种复合材料结构的分析与比较,可以推测导致以上结果的原因归为以下3点:其一,尽管CuMn2O4/g-C3N4表面有较多的转移电子和氧空位,但是CuMn2O4/rGO的结晶度更高、更稳定;其二,CuMn2O4/rGO的比表面积和总孔容积更大,与O3分子的接触面积更大;其三,CuMn2O4/rGO的导电性更好,更有利于反应过程中的电荷转移,故而催化臭氧氧化降解BP-4的速度更快。
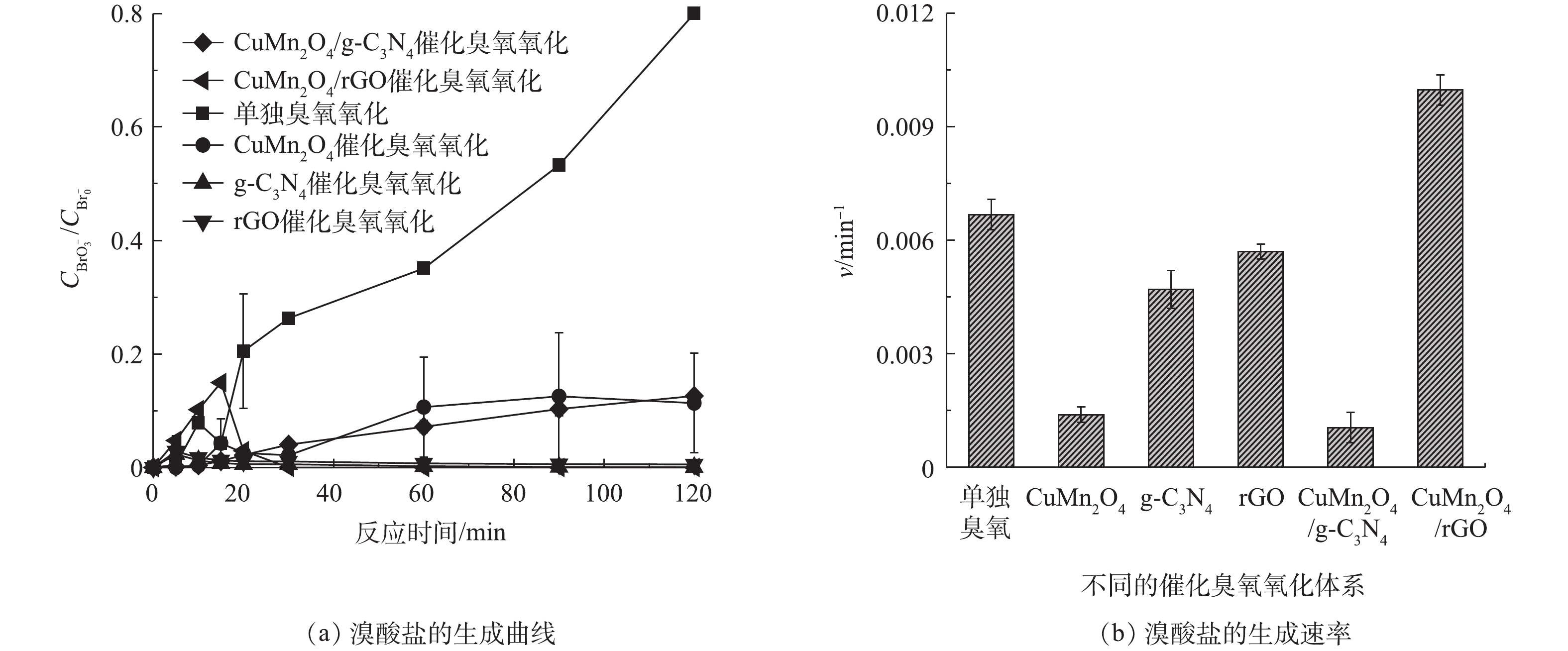
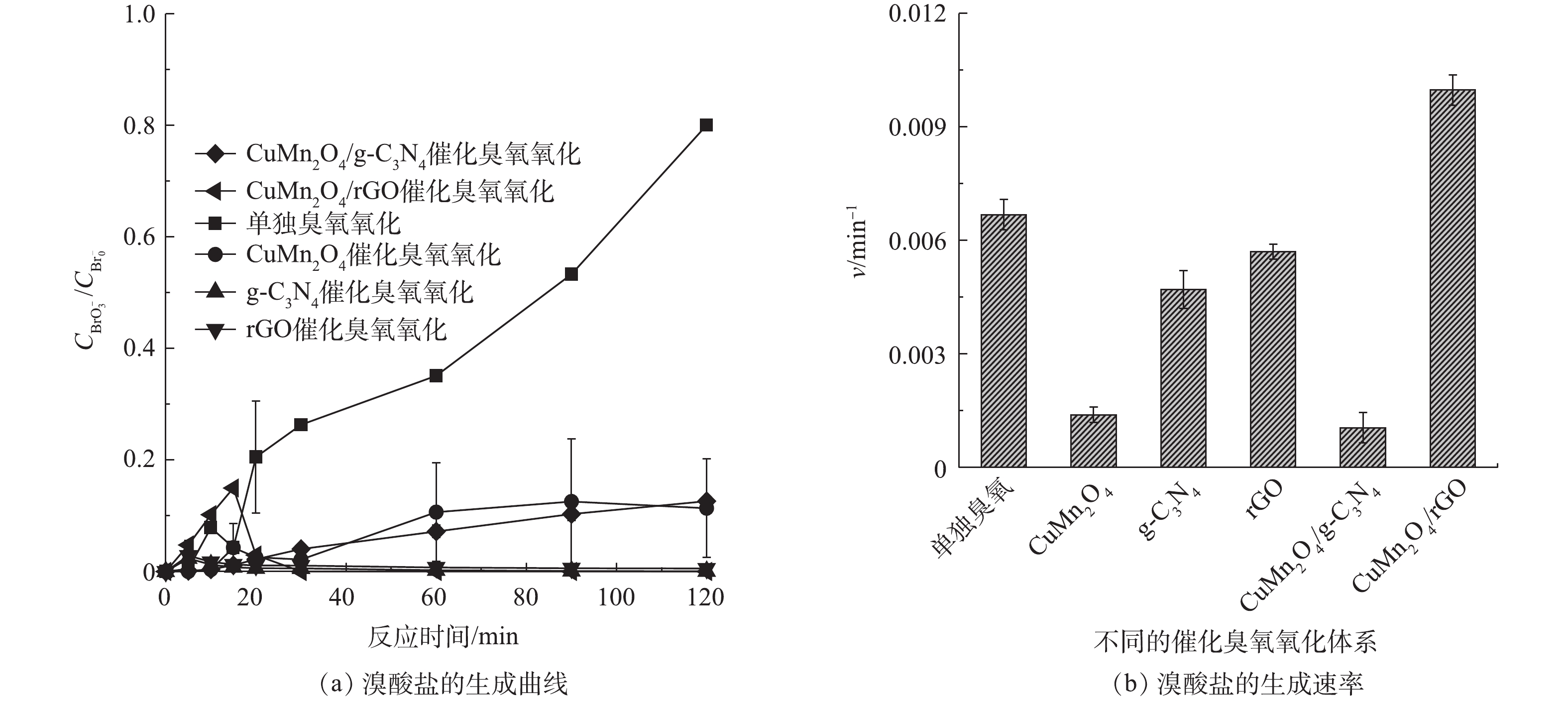
2) rGO 和g-C3N4改性的CuMn2O4催化臭氧对溴酸盐的抑制效果的比较。单独臭氧氧化、CuMn2O4、rGO、g-C3N4、CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧氧化过程中溴酸盐的生成情况如图7所示。如图7(a)所示,在单独臭氧氧化过程中,溴酸盐的含量随着反应时间延长显著增多;而在催化臭氧氧化过程中,溴酸盐的生成量得到有效控制。值得注意的是,不同催化臭氧氧化体系中溴酸盐含量的变化趋势存在差异:在CuMn2O4和CuMn2O4/g-C3N4这2种催化臭氧氧化体系中,溴酸盐的生成量随着反应的进行缓慢增加,与单独臭氧氧化相比,溴酸盐的生成量分别减少了86.30%和84.23%;而在g-C3N4、rGO和CuMn2O4/rGO这3种催化臭氧氧化体系中,溴酸盐的生成量迅速增加后又逐渐减少,具体变化过程如下:在反应前20 min溴酸盐的生成量迅速增加,在20 min时溴酸盐的含量最高;反应20 min后,溴酸盐的含量开始减少,反应120 min后溴酸盐的含量接近于0,与单独臭氧氧化相比,溴酸盐的生成量减少了100%。溴酸盐含量的第一种变化过程与之前大部分[29-31]的研究结果是一致的,在他们的实验过程中,溴酸盐的含量也是在不断增加并最终保持不变。这些研究中对于溴酸盐的消除机理有2种不同的解释,其中一种观点认为溴酸盐的减少得益于β-FeOOH/Al2O3和Fe-Al-LDH/Al2O3[29-30]表面上的Fe2+对溴酸盐的还原,另一种观点则认为催化剂加速了O3的分解,生成更多的·OH,减少了可用于氧化Br−的O3[31]。溴酸盐生成量的第2种变化过程与ZHANG等[32-33]先后使用LaFeO3和LaCoO3与LaCoO3/g-C3N4催化臭氧氧化降解BZA过程中溴酸盐的变化趋势是一致的,溴酸盐的含量均是先增加后减少,他们认为溴酸盐的消除主要归因于H2O2对溴酸盐的还原。此前,SONG等[8]在rGO和g-C3N4催化臭氧氧化降解BZA及去除溴酸盐的研究中指出,rGO表面的含氧官能团和碳层结构中的π电子可以促进臭氧分解形成H2O2;g-C3N4表面的含氧官能团(如—C=O)也会促进H2O2的产生。结合本研究中的实验结果可以推测,在CuMn2O4和CuMn2O4/g-C3N4这2种催化臭氧氧化体系中,溴酸盐的消除有2个方面的原因:其一,催化剂加速了O3的分解,生成更多的·OH,减少了可用于氧化Br−的O3;其二,催化剂可以催化臭氧产生H2O2来还原溴酸盐。在g-C3N4、rGO和CuMn2O4/rGO这3种催化臭氧氧化体系中,溴酸盐的减少极有可能是因为生成的H2O2促进了溴酸盐的还原。对实验结果进一步分析可以发现,与CuMn2O4/g-C3N4相比,CuMn2O4/rGO催化臭氧氧化过程对于溴酸盐的控制效果更好。
为了进一步分析反应过程中溴酸盐的生成规律,通过式(1)计算了不同反应体系中溴酸盐的生成速率v,结果如图7(b)所示,在不同催化臭氧氧化体系中,溴酸盐的生成速率依次是CuMn2O4/rGO>rGO>g-C3N4>CuMn2O4>CuMn2O4/g-C3N4,可见CuMn2O4/rGO催化臭氧体系中溴酸盐的生成速率要明显快于CuMn2O4/g-C3N4,这与二者对BP-4的降解速率的快慢是一致的。据此可以推测,CuMn2O4/rGO催化臭氧生成溴酸盐的速率更快的原因主要是因为CuMn2O4/rGO的结晶度更高、更稳定、比表面积和总孔容积更大、导电性更好。
-
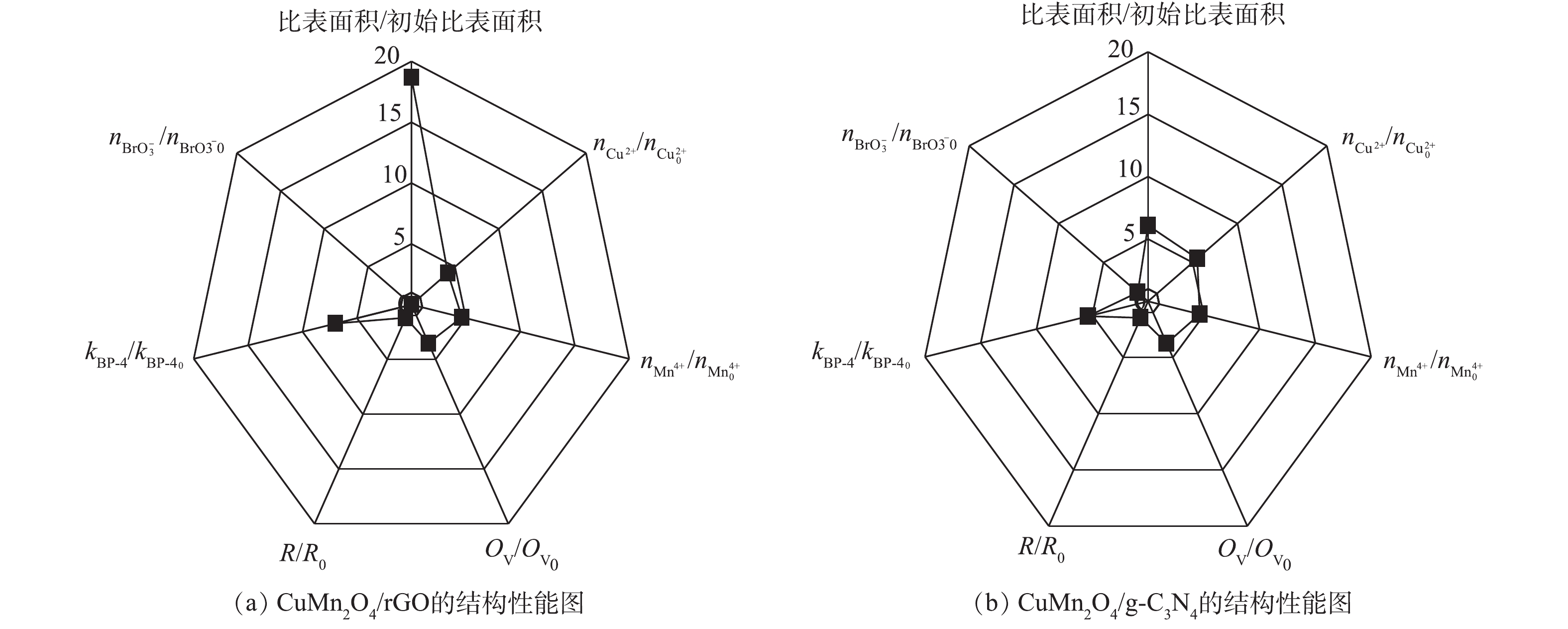
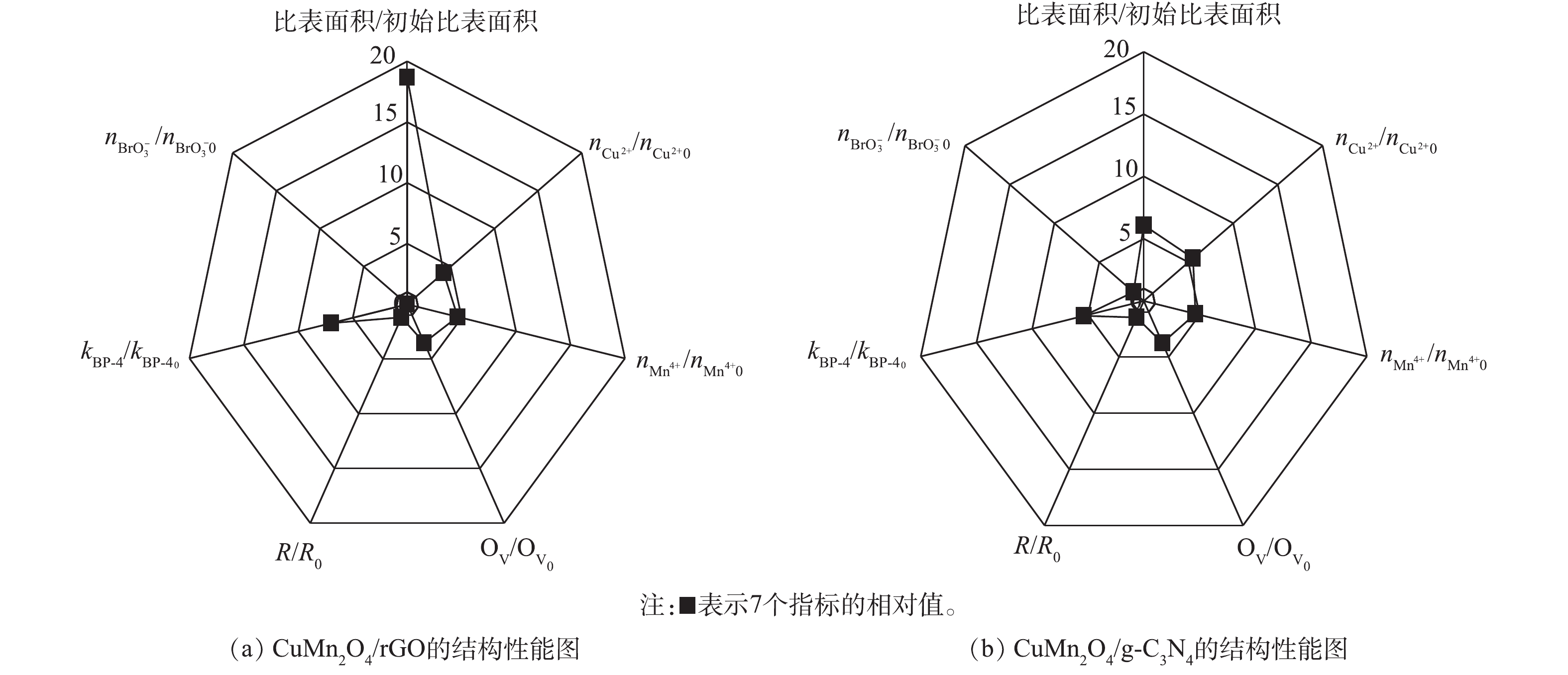
为了进一步确定CuMn2O4/rGO和CuMn2O4/g-C3N4的结构与催化性能之间的关系,使用雷达图对2种催化剂的结构和性能进行了比较,结果如图8所示。图8中7个指标的初始值均为CuMn2O4的相关参数,结果表示的分别是CuMn2O4/rGO和CuMn2O4/g-C3N4中相应指标的相对值。从结构上看,rGO和g-C3N4的掺入对CuMn2O4的比表面的影响程度最大。与CuMn2O4相比,CuMn2O4/rGO的比表面积增大了17.68倍,CuMn2O4/g-C3N4的比表面积增大了5.09倍。rGO和g-C3N4的掺入对Cu2+、Mn4+和OV的相对含量的影响次之,CuMn2O4/rGO中Cu2+和Mn4+的相对含量分别增加了3.17倍和3.64倍,CuMn2O4/g-C3N4中Cu2+和Mn4+的相对含量分别增加了4.50倍和3.64倍;CuMn2O4/rGO中OV的相对含量增加了2.51倍,CuMn2O4/g-C3N4中OV的相对含量增加了2.74倍。rGO和g-C3N4的掺入对阻抗R的影响最小,CuMn2O4/rGO的阻抗增大了18.70%,CuMn2O4/g-C3N4的阻抗增大了46.93%。rGO和g-C3N4掺入CuMn2O4后带来的结构上的显著变化可能是因为CuMn2O4中的金属离子化学掺杂到rGO和g-C3N4的骨架中形成复合物,其作为催化中心改变了rGO和g-C3N4的表面性质和电子分布。
从降解效能上看,rGO和g-C3N4的掺入均能显著加快BP-4的降解,与CuMn2O4相比,CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧降解BP-4的速率分别提高了5.98倍和5.37倍。但是,rGO和g-C3N4的掺入对溴酸盐生成量的抑制效果有显著差异。与CuMn2O4相比,CuMn2O4/rGO催化臭氧使
BrO−3 的含量降低了100%,而CuMn2O4/g-C3N4催化臭氧生成BrO−3 的含量没有进一步减少。结合rGO和g-C3N4掺入CuMn2O4后结构上发生的变化,可以推测,反应速率的增强主要得益于3个方面:其一,具有平面片状结构的rGO和g-C3N4阻碍了CuMn2O4的团聚,同时可以作为一个比表面积大、导电性高的框架来维持CuMn2O4纳米颗粒在催化臭氧氧化过程中电子的转移,产生更多的活性物种;其二,rGO和g-C3N4的掺入,可以促进金属离子的电荷转移,使催化剂表面有更多的转移电子,加速反应的进行;其三,rGO和g-C3N4掺入CuMn2O4后,催化剂中含有更多的氧空位,这为催化臭氧氧化提供了更多潜在的反应位点。综合比较CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧氧化降解BP-4的效果和对溴酸盐的控制效果,CuMn2O4/rGO更适合应用于催化臭氧氧化降解微污染物及控制溴酸盐生成的反应过程中。
2.1. CuMn2O4、CuMn2O4/rGO与CuMn2O4/g-C3N4的结构与表面特性分析
2.2. rGO和g-C3N4改性的CuMn2O4催化臭氧氧化性能研究
2.3. 结构与性能之间关系分析
-
1)使用两步煅烧法成功制备出了CuMn2O4/rGO与CuMn2O4/g-C3N4。通过XRD表征、BET比表面积分析、XPS分析以及电化学交流阻抗测试分析发现,尽管CuMn2O4/g-C3N4比CuMn2O4/rGO电子转移速率更快、氧空位更多,但是CuMn2O4/rGO比CuMn2O4/g-C3N4的结晶度更高、比表面积更大、导电性更好。
2)通过催化臭氧氧化降解BP-4的实验结果表明,rGO和g-C3N4的掺入均能有效提升CuMn2O4催化臭氧氧化降解BP-4的速率。但是,二者的掺入对于溴酸盐生成的控制效果有显著差异。在掺入rGO后,溴酸盐的生成量能进一步减少;而g-C3N4的掺入对溴酸盐生成的控制效果没有提升。
3)进一步比较CuMn2O4/rGO与CuMn2O4/g-C3N4的结构和性能发现,rGO和g-C3N4掺入CuMn2O4后可以阻碍了CuMn2O4的团聚的同时,还可以作为一个高导电性的框架,促进CuMn2O4在催化臭氧氧化过程中电子的转移,此外,由于其具有高导电性和大表面积而提高了催化效率。综合考虑2种复合催化剂对BP-4的降解效果与对溴酸盐的控制效果,CuMn2O4/rGO更适用于催化臭氧氧化。
