



 下载:
下载:



-
土壤和地下水中铬污染是我国当前亟待解决的环境问题。铬污染主要来源于铬盐生产、电镀、制革等工业废水及固体废弃物的排放[1]。历史上,我国曾有大量露天堆积的铬渣,虽然经整治与处理后大部分铬渣已得到安全处置[2],但铬渣堆场附近仍存在大面积受Cr(Ⅵ)污染土壤[3]。这些受Cr(Ⅵ)污染的土壤,由于淋溶作用,极易溶出Cr(Ⅵ),导致场地周围地下水在相当长的时间内受到Cr(Ⅵ)潜在污染的威胁[4-6]。因此,开展土壤-地下水Cr(Ⅵ)污染修复具有重要的实际意义。
自然条件下,铬在土壤-地下水中主要以Cr(Ⅲ)和Cr(Ⅵ) 2种氧化状态存在[7]。其中,Cr(Ⅲ)在pH=6~12时,由于能够形成稳定的氢氧化物沉淀析出[8],导致其迁移性大大降低;同时Cr(Ⅲ)毒性较低,故仅在很高浓度条件下才表现出生物毒性[9]。与此相反,Cr(Ⅵ)具有致癌性,因易溶于水,故在地下水中具有很强的迁移性[10-11]。因此,对受Cr(Ⅵ)污染的地下水常采用原位还原固定化技术,通过向地下含水层中注入还原剂将Cr(Ⅵ)还原固定为Cr(Ⅲ)降低其迁移性和毒性,从而实现修复的目的。
常用的Cr(Ⅵ)原位修复技术主要包括可渗透反应墙(PRB)、生物修复、电动修复和原位氧化还原控制墙技术(in situ redox manipulation, ISRM)等。传统的PRB技术通过在污染源下游地下含水层中开挖沟槽、回填活性反应材料以实现修复目的。但实际应用中该技术一般用于受污染的浅层地下水,不适于深部地下水的修复。ISRM技术是近年来提出的一类新型的原位还原技术。该技术通过注入井向地下污染区域注入还原剂以建立还原性环境,从而实现污染物的还原修复[12-15]。与PRB技术相比,ISRM对较深含水层的污染修复具有明显的优势。
地下水修复中常用的还原剂包括硫类化合物、铁基及有机类还原剂[16]。其中硫基还原剂因具有优异的反应活性,在地下水Cr(Ⅵ)还原固定化修复中受到广泛的关注。采用ISRM技术修复受Cr(Ⅵ)污染地下水中研究最多的2种硫化物为连二亚硫酸钠和多硫化物。虽然连二亚硫酸钠具有很强的还原能力,但由于其在中性水溶液中不稳定,在实际修复中需要外加碱性物质(如碳酸钾/碳酸氢钾缓冲)[14],因此,在实际应用中受到一定限制。有研究表明,多硫化物(Sx2-)性能稳定、还原能力强,可以在多孔介质中形成相对持久的还原性区域,是EPA推荐的修复试剂,近年来,在地下水修复中引起了广泛的关注[12,17]。连二亚硫酸钠与多硫化物的修复机理类似。以ISRM中采用多硫化物还原固定化Cr(Ⅵ)为例,通过注入井向含水层注入的多硫化物首先还原孔隙水中的Cr(Ⅵ)使其以三价铬的形式固定下来;而剩余的多硫化物能进一步与含水层中含铁矿物中的Fe(Ⅲ)反应生成Fe(Ⅱ)。SHI等[18]的研究表明,多硫化物可以与纤铁矿反应生成含二价铁的次生还原性产物;所产生的Fe(Ⅱ)以固相和强吸附态形态为主,而溶解态的相对较少。这也解释了多硫化物具有在受污染含水层中建立持久还原性区域能力的原因,使其在地下水还原修复中得到广泛研究和应用[13,19]。
现场实验结果也表明,多硫化物对受铬污染土壤地下水的修复具有优异的效果[1,20-23]。一项有关采用多硫化钙对英国格拉斯哥某受铬渣污染地下水进行的中试研究结果表明:在所选用的3种修复方法中,以直接向含水层注入多硫化钙的方案效果最佳,可以将Cr(Ⅵ)浓度从800 μg·L−1降至30 μg·L−1。MESSER等[17]对美国亚利桑那某电镀污染现场的中试结果表明,经多硫化物处理,可使包气带土壤和孔隙水中Cr(Ⅵ)分别从处理前的2 190 mg·kg−1和3 600 mg·L−1降至10%以下,而含水层中的Cr(Ⅵ)迅速由240 mg·L−1降至1 mg·L−1。进一步的检测表明,多硫化物在含水层中所形成的还原性区域能维持数月,对地下水中Cr(Ⅵ)污染具有长期修复的潜力。
由于褐土和红壤是我国2种重要的土壤类型,其分别在河南省和云南省有广泛的分布,而且在两省均曾有铬渣的堆放,因此,本研究选择褐土和红壤为研究对象,在室内通过土壤柱实验模拟研究了采用ISRM技术还原固定化修复受Cr(Ⅵ)污染的土壤-地下水的过程,考察了多硫化物对土壤样品的硫化处理及硫化后对Cr(Ⅵ)的还原能力,以及还原Cr(Ⅵ)柱实验过程中多硫化物的电子利用效率等。在柱实验过程中,同时对出水pH、氧化还原电位进行实时监测,以考察柱体内是否有效地建立了还原区域。研究结果可对以多硫化物为还原剂采用ISRM技术现场修复Cr(Ⅵ)污染的土壤-地下水具有一定的参考价值。
全文HTML
-
将0.1 mol·L−1硫化钠溶液与等摩尔数的单质硫加入500 mL血清瓶中,转移至恒温振荡箱中,在65 ℃和100 r·min−1条件下振荡16 h[24-25]。多硫化物和硫化钠浓度采用碘量法[26]测定。
-
实验所用的红壤和褐土样品分别采自云南和河南。样品采集使用棋盘法,首先在采样现场选取7~10个采样点,在地表下10~30 cm的土层,每个采样点取2~3 kg样品。采集的土壤样品在实验室通风阴凉处风干并剔除土壤中杂质,用木锤敲打风干的大块样品,压碎并混匀后研磨过2 mm尼龙筛,筛下物样品用于土柱实验。
-
图1(a)为所搭建的实验室土壤柱实验装置。柱实验过程中采用自下而上的入水方式。实验中所有溶液均采用经氮气吹脱的Milli-Q超纯水(18.2 MΩ)配制,以保证整个实验过程在厌氧条件下进行。
土柱装填采用干堆法[27-28]。在装填过程中,每次加入等量的样品,用橡胶锤轻轻击打柱体并均匀压实,以免颗粒间产生明显缝隙,从而使实验中所使用柱子性质基本保持一致。在实验运行过程中,若土壤的黏性较大可能会导致柱内土壤无法均匀润湿、柱压过高,从而造成土柱中的优先流或柱子堵塞等现象。此时可在土壤中掺杂质量比1∶1的石英砂(0.1~0.4 mm)[29],以避免上述现象的出现。图1(b)为按上述方法所装填好的土柱,土柱内径和装填高度分别为2.5 cm和20 cm。装填好的红壤柱和褐土柱的孔隙率分别为0.50和0.49,所装填土壤质量分别为130.3 g和136.3 g。
土壤柱实验包括土柱预处理、土柱硫化以及硫化土柱对Cr(Ⅵ) 还原固定化3个过程。在土柱预处理中,向柱体通入模拟地下水以充分润湿土壤并使孔隙饱和。实验中所用模拟地下水组成[30]为10 mmol·L−1 Na+、0.8 mmol·L−1 Ca2+、6.6 mmol·L−1 Cl−、3 mmol·L−1
HCO−3 、1 mmol·L−1SO2−4 。在土壤硫化过程中,向上述经水饱和的土柱以1.15 mL·min−1流速通入初始浓度为50 mmol·L−1多硫化物,并在设定时间内对出水口处溶液的pH、氧化还原电位和多硫化物浓度进行测定。当多硫化物浓度接近50 mmol·L−1时,停止通入含多硫化物的地下水。在接下来的实验中,向硫化后土柱以同样流速通入含有10 mg·L−1 Cr(Ⅵ)的模拟地下水,以观测硫化土柱对Cr(Ⅵ)的持续还原能力。在实验中,在既定时间取出水口处样品记录溶液的pH和氧化还原电位,同时分析多硫化物和Cr(Ⅵ)的浓度。基于我国地下水分类标准(GB/T 14848-2017),针对Cr(Ⅵ)这一指标,Ⅳ类和Ⅴ类的界限为0.1 mg·L−1,本实验将出水中Cr(Ⅵ)的浓度达0.1 mg·L−1时定义为穿透点,达到穿透点后,继续通入Cr(Ⅵ) 模拟地下水,当出水浓度接近10 mg·L−1时,停止柱实验。
土壤柱实验过程中多硫化物浓度的测定采用亚甲基蓝分光光度法[31],Cr(Ⅵ)浓度的测定采用《水质 六价铬的测定 二苯碳酰二肼分光光度法》(GB/T 7467-1987),pH的测定使用pH电极(InLab Expert Go-ISM, METTLER TOLEDO),氧化还原电位的测定使用氧化还原电极(ORION 9678BNWP, Thermo)。
1.1. 多硫化物的制备
1.2. 土壤样品的采集和预处理
1.3. 室内土壤柱实验模拟研究
-
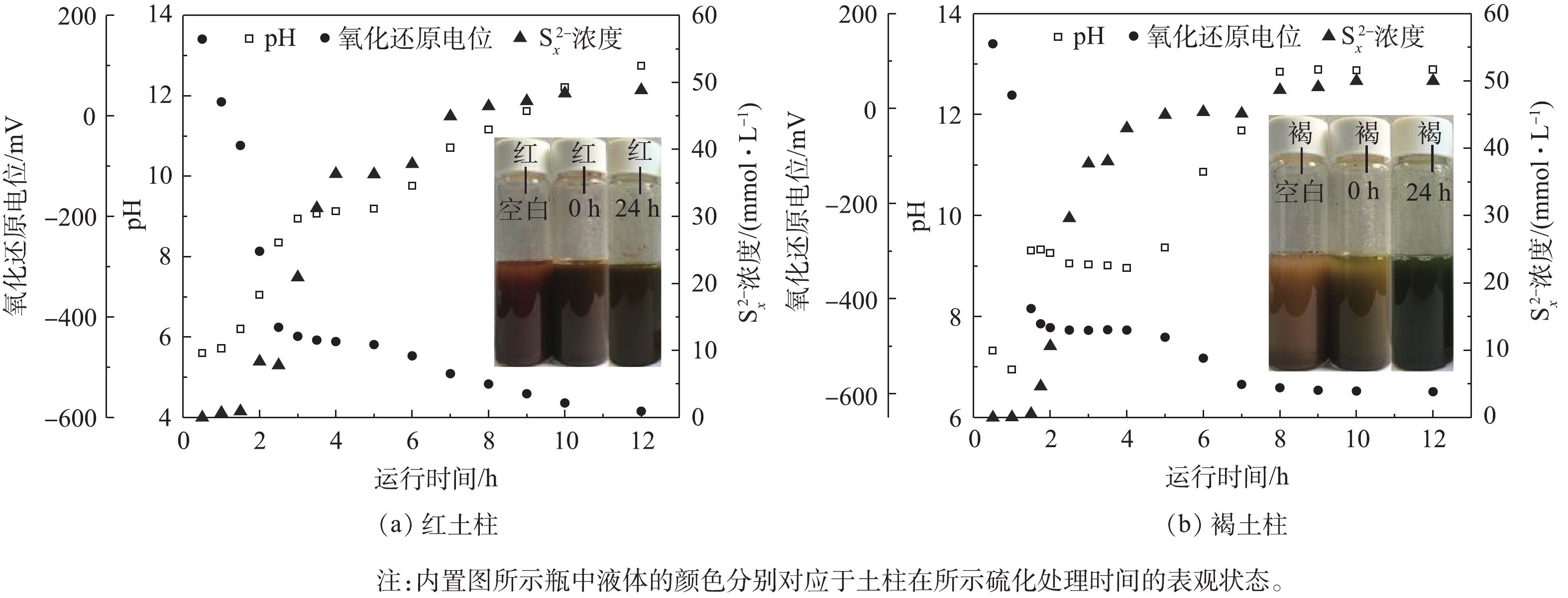
图2(a)和图2(b)分别为红壤和褐土柱子在多硫化物硫化处理过程中,出水溶液的pH、氧化还原电位以及多硫化物浓度随时间的变化规律。实验中所观察到的红壤和褐土柱硫化现象类似。
在2种土柱硫化过程中均观察到出水和柱体颜色随时间发生了变化。对于红壤实验,当向红壤柱中通入约1~2个PV多硫化物后,出水溶液开始呈现乳白色,这可能是由于单质硫的析出所造成的。之后出水颜色逐渐加深,并最终呈深墨绿色,而柱内的土壤由初始的红色自下而上渐渐呈现出深墨绿色。在褐土柱硫化过程中,颜色随时间的变化与红壤柱变化规律类似,出水溶液的颜色逐渐加深并最终变为草绿色;土柱内土壤从初始的棕褐色由下至上最终全部变为绿色。2种土柱及其出水颜色随时间发生变化可能是由于土壤中含铁物质与多硫化物反应,生成了亚铁以及硫铁化合物所造成[12,18,32-33]。
在红壤柱和褐土柱的硫化过程中也观察到了出水多硫化物浓度随时间的变化(图2(a)和图2(b))。在红壤柱硫化过程中,在最初2 h内没有检出多硫化物,随后多硫化物浓度逐渐提高,在12 h时基本达到初始多硫化物入水浓度(50 mmol·L−1)。在褐土柱硫化过程中,多硫化物浓度随时间的变化与红壤柱类似,经过1.5 h后,开始检出多硫化物,并在12 h时达到平衡。
上述2个土柱的硫化过程中也观察到出水溶液的pH、氧化还原电位随时间的变化。对红壤柱,出水溶液pH变化从初始的5.6逐渐上升,12 h后达到12.7;出水溶液的氧化还原电位从初始的152.7 mV降至−582.5 mV。褐土柱出水溶液初始pH为7.3,经12 h硫化后,逐步升高至12.9;溶液氧化还原电位则从初始的136.4 mV逐渐下降至−595.8 mV。2种土壤硫化过程中pH和氧化还原电位的变化是由多硫化物所造成的。多硫化物是强碱性的物质,可导致出水溶液pH升高;同时由于其具有很强的还原特性,从而使得出水溶液氧化还原电位大大降低,并最终在柱体内建立有效的还原性区域。
-
图3(a)和图3(b) 为红壤和褐土经硫化后还原固定化Cr(Ⅵ)的性能随时间变化的柱实验结果。由图3可知,在还原固定化过程中,柱体颜色、出水溶液的颜色、pH、氧化还原电位以及出水溶液中Cr(Ⅵ)和多硫化物的浓度均随时间发生变化。2种硫化土柱具有相似的变化趋势,但也存在一定的差异。
当红壤柱和褐土柱还原固定化Cr(Ⅵ)时,柱体与出水液体颜色均随时间发生了变化。在柱实验过程中,随着对Cr(Ⅵ)的还原固定化,硫化红壤柱的颜色自下而上由深墨绿色逐渐变成红色,而其出水溶液颜色则由深墨绿色逐渐变淡,直至柱体还原能力消失,并最终变为淡黄色。硫化褐土柱在与Cr(Ⅵ)反应过程中,反应初始阶段柱体的颜色为绿色,随着反应时间的增加,其颜色自下而上逐渐变为黄褐色,而反应后出水颜色由初始的草绿色逐渐变为柱实验接近结束时的淡黄色。在2个硫化柱实验中,柱体中的绿色可能缘于硫化过程中所产生的亚铁与硫铁化合物。随着还原性物质的反应其颜色逐渐消失。淡黄色的出水接近入水Cr(Ⅵ)的本色。这主要是因为硫化过程中柱体内所产生的还原性物质随着与Cr(Ⅵ)反应,浓度逐渐降低;当还原性物质被彻底消耗后,入水和出水中Cr(Ⅵ)浓度接近,从而使得出水颜色呈淡黄色。
在硫化红壤柱和褐土柱还原固定化Cr(Ⅵ)的过程中,也观察到了其出水溶液pH和氧化还原电位类似的变化情况(图3(a)和图3(b))。对硫化红壤柱,出水溶液初始pH为12.6,之后随反应的进行,pH逐步下降,最终达到8.9;出水溶液氧化还原电位从−574.9 mV逐渐上升至117 mV。硫化褐土柱的出水溶液pH从12.9下降至9.4,氧化还原电位则从−571.9 mV逐渐上升至38.5 mV。柱实验中pH和氧化还原电位的变化是由于多硫化物和Cr(Ⅵ)造成的。多硫化物是强碱性的物质,随着与Cr(Ⅵ)的反应,其浓度逐渐降低,造成了pH和氧化还原电位的上述变化。
实验中观察到Cr(Ⅵ)浓度随时间发生变化。向硫化红壤柱持续通入含10 mg·L−1Cr(Ⅵ)地下水27 h(约38PV)后,土柱达到穿透点(0.1 mg·L−1)。由于已穿透的土柱仍然具有一定的还原能力,因此,可以进一步还原固定化Cr(Ⅵ),但其还原固定化能力的降低,导致出水中Cr(Ⅵ)的浓度逐渐升高,并最终在155 h (约218PV)时达到平衡。通过积分可得,硫化红壤柱在整个实验过程中共去除1.21 mmol Cr(Ⅵ),由于Cr(Ⅵ)被还原固定化为Cr(Ⅲ),即反应过程中需净利用3.63 mmol电子。硫化褐土柱也经历了类似的过程,在通入10 mg·L−1 Cr(Ⅵ) 15 h (约22PV)后,土柱达到穿透点。之后该土柱仍能继续还原Cr(Ⅵ),并在反应96 h (约138PV)时丧失还原能力。同样由积分可知,硫化褐土柱在整个反应过程中共去除0.77 mmol Cr(Ⅵ),即2.31 mmol净利用电子量。柱实验中,土柱累积去除Cr(Ⅵ)的量可根据式(1)计算。
式中:Ci为取样时间ti时出水中Cr(Ⅵ)浓度,mg·L−1;C0为入水中Cr(Ⅵ)浓度,mg·L−1;q为入水流速,取值1.15 mL·min−1;n0为取样数据点数。
在2个硫化土柱还原Cr(Ⅵ)过程中,反应初期在出水中还检测到多硫化物存在。向硫化红壤通入Cr(Ⅵ)溶液,出水中多硫化物的浓度在2 h内由初始的50 mmol·L−1迅速降低到约10 mmol·L−1,并最终在10 h后接近0。对褐土柱来说,出水中多硫化物浓度变化的趋势与上述红壤中观察到的变化趋势类似:多硫化物浓度在经历了2 h快速下降后,并在反应7.5 h后,浓度逐渐接近0。经过硫化的土柱孔隙中仍存在未反应的多硫化物,这些多硫化物在后续通入Cr(Ⅵ)溶液时被取替而流出,因此造成了多硫化物仍出现在出水中。考虑到扣除流出的这部分多硫化物后,是实际消耗的多硫化物,即为硫化土柱所持有的净还原能力,因此,硫化红壤柱所持有的净还原能力相当于7.89 mmol多硫化物。由于
S2−x 在氧化过程中的产物以S0为主,而1 molS2−x 在氧化还原反应中能提供2 mol电子,因此,硫化红壤所持有的净还原能力相当于15.78 mmol电子。对褐土柱来讲,柱体所持有的净还原能力相当于4.65 mmol多硫化物或9.3 mmol电子。综合以上实验结果,可以看出,由多硫化物在柱体内所形成的理论还原能力与还原Cr(Ⅵ)中实际消耗的还原能力存在偏差。如果定义柱实验过程中的电子利用效率为Cr(Ⅵ)还原固定化所需理论电子数与柱体所持有总还原能力对应的电子数之比,基于柱实验还原固定化Cr(Ⅵ)的结果可知,硫化后的红壤和褐土的电子利用效率分别为23.0%和24.8%,相当于去除等当量的Cr(Ⅵ)所需理论还原能力的4.35倍和4.03倍。柱实验与理论值的偏差可能是由于土壤复杂性所造成的。因此,以多硫化物为还原剂,采用ISRM修复技术还原固定化Cr(Ⅵ)时,需要加入过量还原试剂。基于对红壤柱和褐土柱实验结果,可以看出,柱实验中所用的多硫化物是所需理论值的4~5倍。这一结果对受Cr(Ⅵ)污染的土壤-地下水修复具有一定的实际意义。
2.1. 土柱的硫化处理
2.2. 硫化土柱对Cr(Ⅵ)的还原固定化
-
1)经硫化处理的2种土壤均具有还原固定化Cr(Ⅵ)的能力,但其还原容量不同。在实验条件下,向硫化土柱中通入10 mg·L−1的Cr(Ⅵ)溶液,红壤柱和褐土柱分别在38PV和22PV穿透(基于GB/T 14848-2017,穿透浓度设为0.1 mg·L−1)。2种土柱在穿透后虽然其还原能力降低但仍能持续的还原固定化Cr(Ⅵ),并最终在218PV和138PV左右丧失对Cr(Ⅵ)的还原能力。
2)多硫化物对红壤和褐土的硫化会使孔隙水的pH和氧化还原电位发生变化。在经硫化处理后,红壤柱和褐土柱的出水pH分别升高至12.7和12.9,而对应的氧化还原电位则逐渐降低至−582.5 mV和−595.8 mV,这表明多硫化物在土壤中已建立有效的还原区域。向2种硫化土柱中通入Cr(Ⅵ)溶液会降低出水的pH,同时伴随氧化还原电位的升高:反应结束时,红壤和褐土系统的pH分别为8.9和9.4,对应的氧化还原电位分别为117 mV和38.5 mV。
3)经多硫化物硫化后的红壤和褐土在还原Cr(Ⅵ)过程中实际电子利用效率分别为23.0%和24.8%,即实际所需多硫化物的量是理论值的4~5倍。考虑到污染现场地下介质的非均质性和复杂性,在实际Cr(Ⅵ)修复中多硫化物的用量可能会更高。
