

 下载:
下载:

-
“十三五”规划把重金属污染防治列为环境治理工作的重心之一。然而,微/痕量重金属污染物具有污染面广、种类多、持久性及毒性大的特点,逐步引起人们的重视[1-2]。微量污染物,或称痕量污染物,包括传统型微污染物和新型微污染物,如微量重金属、类激素、多抗生素、氯联苯(PCBs)、有机氯类杀虫剂(DDT)、多环芳烃(PAHs)等[3]。由于微污染物浓度很低,只有ng·L−1~μg·L−1水平,且重金属与有机污染物不同,难以通过生物、化学降解转化途径去除。目前,重金属微/痕量污染物主要通过化学沉淀法、氧化还原法和电化学处理等方法去除,如臭氧氧化和活性炭吸附,但是臭氧氧化成本高、活性炭吸附选择性差的缺点阻碍了其工程推广应用[4-5]。
电镀行业、含镉矿山开采/冶炼以及镍镉电池生产过程中会产生一定量的含镉废水,镉污染物进入水体和土壤后容易富集且不被微生物降解,进入食物链后易对人体造成伤害,如痛痛病。目前,镉污染治理技术包括化学沉淀、氧化还原、离子交换、电化学、膜分离和吸附法等。其中常用的去除方法是化学沉淀、吸附以及膜分离法。然而,化学沉淀法存在污泥量大、难以沉降、易形成二次污染等缺点,膜分离法存在运行成本高、膜通量小、膜易污染等问题。目前,吸附剂主要为活性炭,活性炭存在pH适用范围小、分离困难等缺点。《生活饮用水卫生标准》(GB 5749-2006)对镉的限制量为0.005 mg·L−1。传统的重金属处理方法存在着难以克服的弊端,很难达到污水排放标准。同时重金属在水体中具有富集效应,ng·L−1或μg·L−1级的重金属经富集放大后,对人体的生理系统仍会造成极大伤害。
碳纳米材料具有比表面积大、机械强度高、结构可控、耐高温、表面基团丰富等特征,具有独特的物理或化学优势,是目前研究的热点和难点之一[6-9]。通过软/硬模板法、选择性蚀刻法、自组装等方法可合成独特结构的碳纳米材料[10-13],碳纳米材料表面含有丰富的羟基和羧基[14-15],可与重金属形成共价键或络合结构;独特的纳米孔道结构,甚至具有选择性吸附重金属离子的潜力[16-17],在环境领域具有潜在的应用价值[18]。
针对难以去除的微量/痕量镉重金属微污染物,强化混凝/吸附具有特有的优势。吸附法具有处理效率高、运行费用低、可操作性强、原材料来源丰富、价格便宜、易解吸等优点,是含镉废水处理工程实际应用首选方法之一。同时,高分子微球具有合成简单、价格便宜、比表面积大的特点,其表面含有大量的羟基,通过组装成空心高分子微球,可有效提高高分子微球的选择吸附性能,在微量/痕量重金属吸附领域具有自身独特的应用前景和研究价值[19-21]。本研究针对重金属微量/痕量污染物难于达标去除的难题,采用高分子微球为吸附剂,考察其对微/痕量重金属污染物的吸附性能,为微量/痕量污染物的去除提供新的技术选择和参考。
-
实验主要试剂:葡萄糖(A.R.)、盐酸(A.R.)购于天津市科密欧化学试剂有限公司,硝酸镉(A.R.)购于阿拉丁学试剂有限公司,硝酸铅(A.R.)、氟化钠(工业纯)购于国药集团化学试剂有限公司,氢氧化钠(A.R.)购于天津市风船化学试剂科技有限公司。
实验仪器:电子天平(AL104,Mettler-Toledo公司)、数显强力电动搅拌机(JB90-S,上海标本模型厂)、台式pH计(FE20,Mettler-Toledo公司)、电热恒温鼓风干燥器(DGG-9053A,上海森信实验仪器有限公司)、紫外可见分光光度计(UV-2450,日本Shimadz公司)、X射线衍射分析仪(XTRA,瑞士ARL公司)、数显恒温水浴锅(HH-2,国华电器有限公司)、镉离子电极(DX312,立德科技有限公司)。
-
空心高分子微球的合成。采用改进的高分子微球合成方法[22]:使用水热方法,以葡萄糖为碳源,氨水(28%)为软模板,每10 mL水加1 g葡萄糖、0.1 mL氨水,水热温度分别为170、180、190、200、210 ℃,水热时间分别为2、3、4、5、6 h。
镉微污染物吸附。CdNO3浓度20 mg·L−1(40 mL),不同温度、不同时间的高分子微球0.05 g,磁力搅拌20 min,静止30 min或离心,测上清液Cd浓度。
起始pH对镉微污染物吸附的影响。CdNO3浓度20 mg·L−1(40 mL),高分子微球投加量为0.05 g,pH为2~11;磁力搅拌20 min,静止30 min或离心,测上清液Cd浓度。
投加量对镉微污染物吸附的影响。取镉微污染吸附实验中的最佳温度和时间的高分子微球,CdNO3浓度20 mg·L−1(40 mL),高分子微球投加量为0.01、0.02、0.05、0.1和0.2 g;磁力搅拌20 min,静止30 min或离心,测上清液Cd浓度。
温度对镉微污染物吸附性能的影响。CdNO3浓度20 mg·L−1 (40 mL),反应温度为10、15、20和25 ℃,高分子微球投加量为0.05 g,pH为4,磁力搅拌20 min,静止30 min或离心,测上清液Cd离子浓度。
干扰离子对镉微污染物竞争吸附的影响。采用酸洗对高分子空心微球(180 ℃、4 h镉微污染物吸附后的微球)进行再生,盐酸浓度为0.1~2 mg·L−1,酸洗时间为10~60 min,酸洗后样品在60 ℃烘箱烘干,采用最佳参数重新吸附镉微污染物,测上清液镉离子浓度。
-
镉离子浓度测量采用镉离子选择电极法。
-
不同温度条件下合成的高分子微球XRD、SEM结果如图1所示。XRD结果表明,不同温度条件下合成的高分子微球在2θ=23°有一驼峰,没有发现石墨化的衍射峰,表明高分子以无定型碳形式存在。SEM结果表明,合成的高分子微球粒径为150~600 nm。在水热温度为180 ℃时,高分子微球的粒径均一性较210 ℃的微球稍差,但210 ℃条件下合成的高分子微球出现粘连现象,不利于高分子微球的分散。
以氨水为起泡剂,葡萄糖为碳源,合成的高分子微球为空心微球,根据已报道的采用类似方法合成的高分子微球及研究结果[23-24],可以证明合成的高分子微球为空心微球。
不同温度条件下合成的高分子微球镉微污染物吸附性能如图2所示。结果表明,水热反应时间和水热温度对镉微污染物吸附有相似的影响规律。随着水热反应时间的延长,Cd2+去除率先升高后降低,当水热反应时间为5 h (170 ℃)时,Cd2+去除率达到最大值,约60%;当升高水热温度时,Cd2+去除率先升后降,当水热温度为180 ℃(4 h)时,Cd2+去除率达到最高值,为67%。
SEM结果表明,水热温度的升高会造成高分子微球的粘连,但水热温度的升高同时会提高高分子微球的均一性。因此,合适的水热温度和水热时间有助于合成性能一致且分散性良好的高分子微球。从节约能源角度及高分子微球粘连与温度呈正比例关系角度考虑,本实验取最佳水热温度为180 ℃,水热时间为4 h。
-
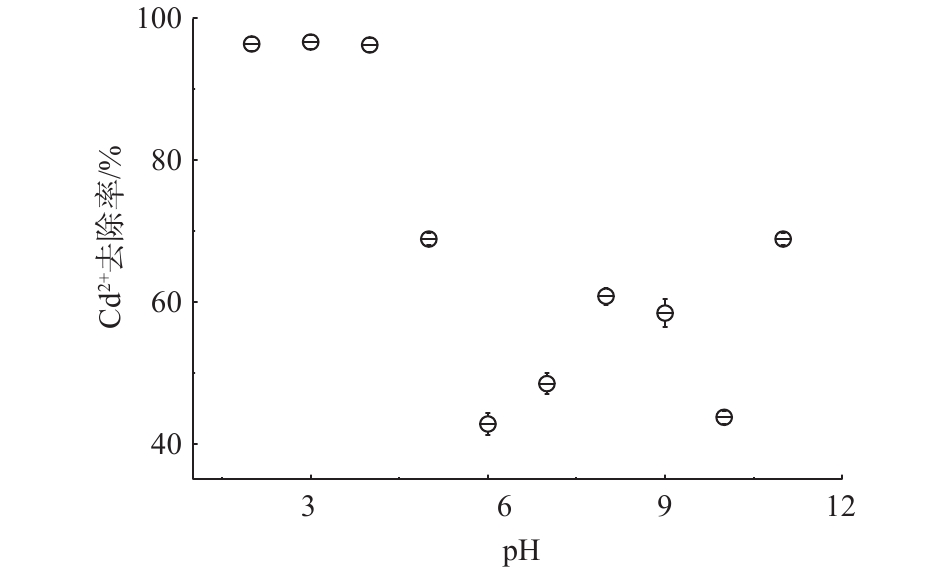
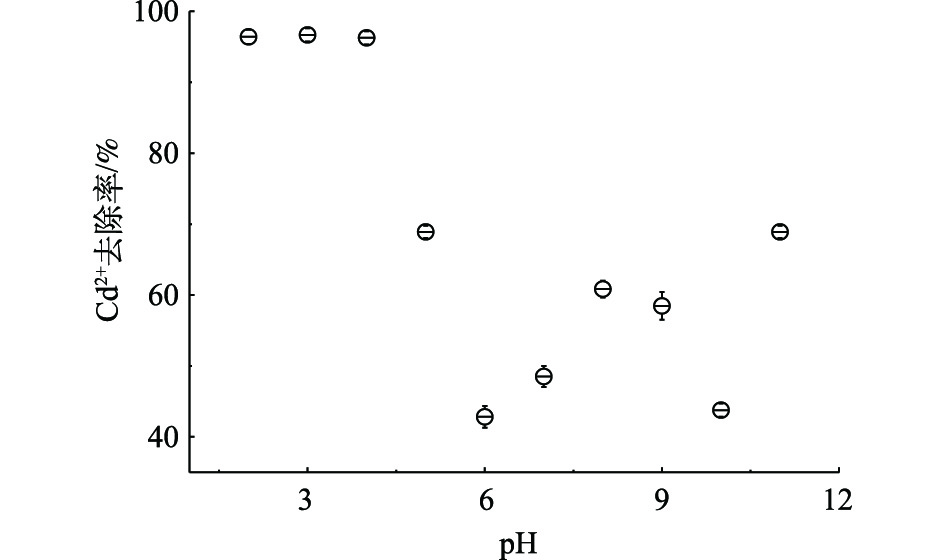
pH是金属微污染物吸附法关键的影响因素之一。pH会影响金属离子在水体中的存在形态,在酸性条件下,部分金属离子与水分子形成水合离子团体;而在碱性条件下,大部分金属离子会形成沉淀或聚合物。图3表明,高分子空心碳微球在酸性条件下镉微污染物的吸附效率高于中碱性条件,在pH<4时,镉微污染物的去除率超过95%,随着pH的升高,镉微污染物去除率先急速下降,然后缓慢微升;pH为6时,去除率接近40%;而当pH>7时,镉微污染物去除率约为60%。研究结果与碳材料吸附结果[25]不同。
在酸性条件下,镉离子会与水分子形成较大的表面带正电荷的水合离子团体,理论上会降低镉离子的吸附效率及速率。而当pH升高时,由于羟基浓度上升,水合镉离子团体表面张力下降,镉离子被释放出来,与羟基结合,形成带负电荷的中间体。而高分子微球表面含有大量的—OH、—COOH、—CO基团,这些基团中的氢离子会被较大的阳离子交换,阳离子质量越大,直径越大,越容易交换氢离子。因此,在酸性条件下,水合镉离子团体更易交换高分子微球表面的氢离子。而当pH升高后,镉聚合物表面的电性由正极转为负极,与高分子表面基团的电荷相同,降低了镉微污染物的吸附效率;当pH继续升高,在碱性条件下,镉离子会形成氢氧化镉沉淀,提高了镉离子的去除率。
-
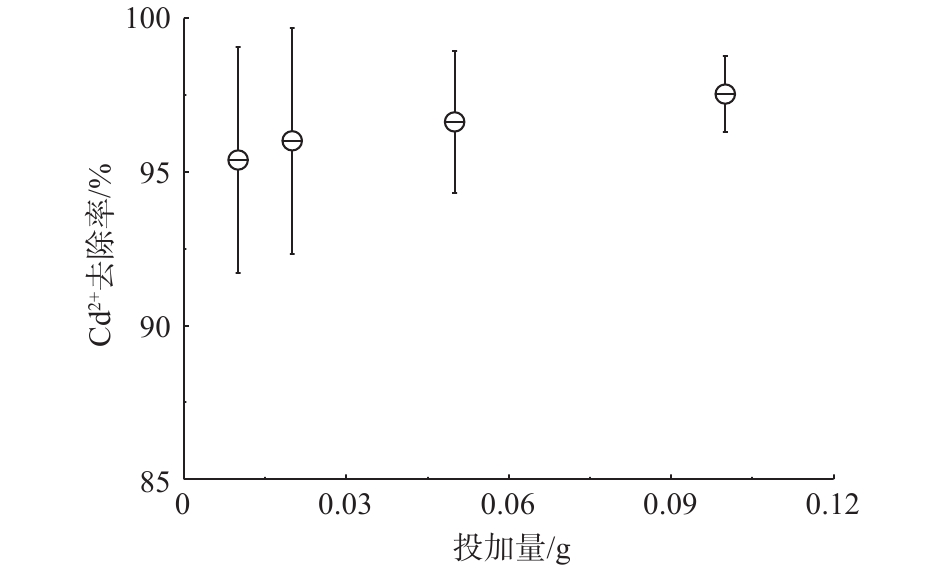
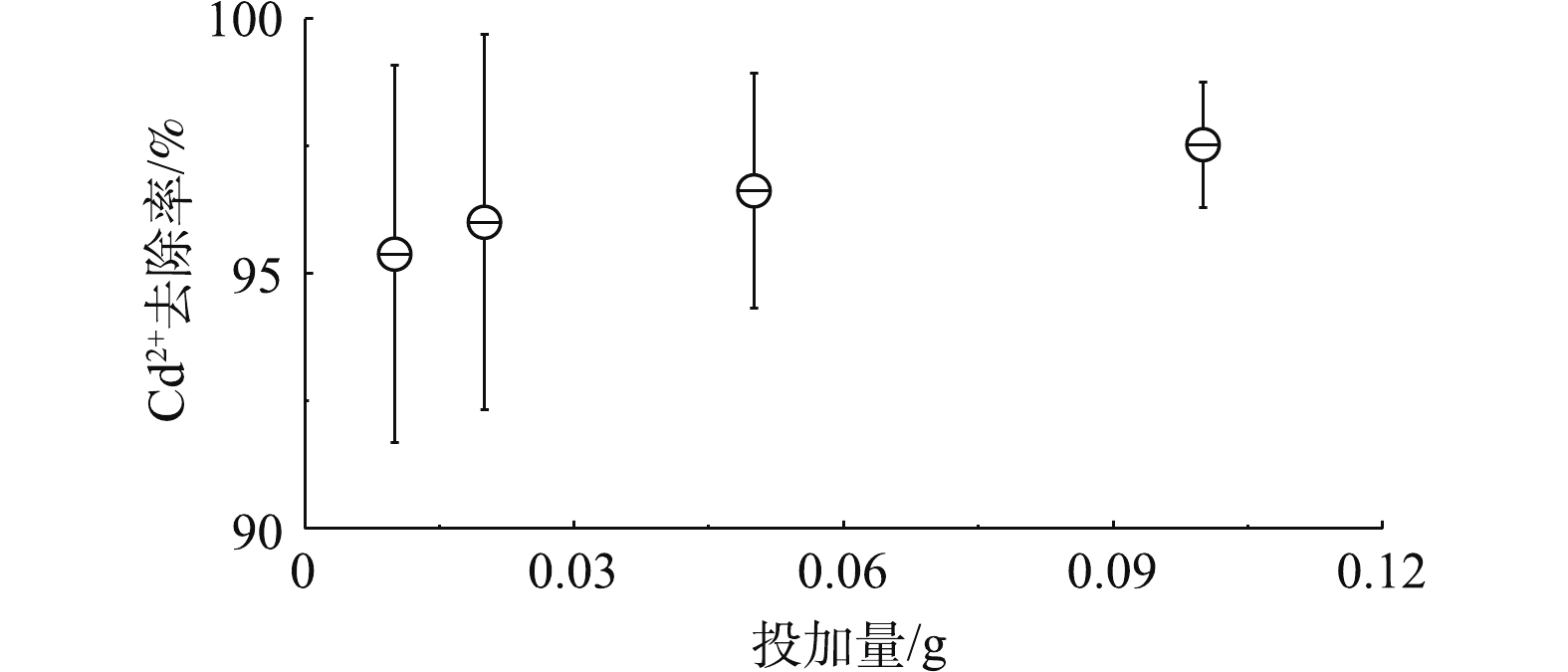
吸附剂投加量决定废水的起始浓度,过高或过低都会影响镉微污染物的去除效果。实验中镉离子的浓度为20 mg·L−1,取40 mL水,也就是说样品中镉离子的质量为0.8 mg。图4表明,当高分子空心微球的投加量为0.01 g时,镉离子去除率达到95%以上,继续提高高分子空心微球的投加量,镉离子去除率略有上升,但上升幅度不大。高分子空心微球的投加量为0.01 g时,镉离子大部分被吸附,此时镉离子的吸附量大于76 mg·g−1,高分子空心微球对镉离子的最大吸附量大于76 mg·g−1。
-
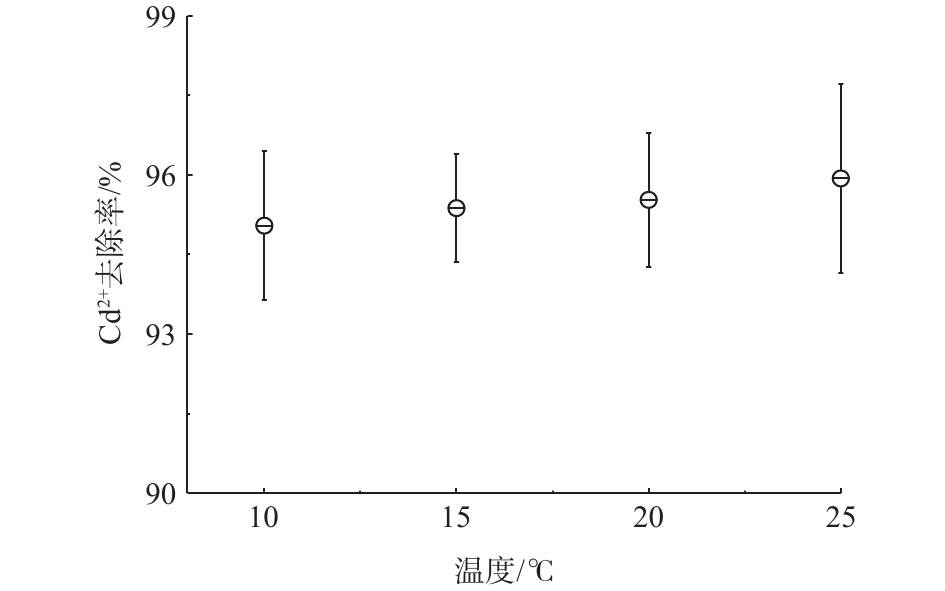
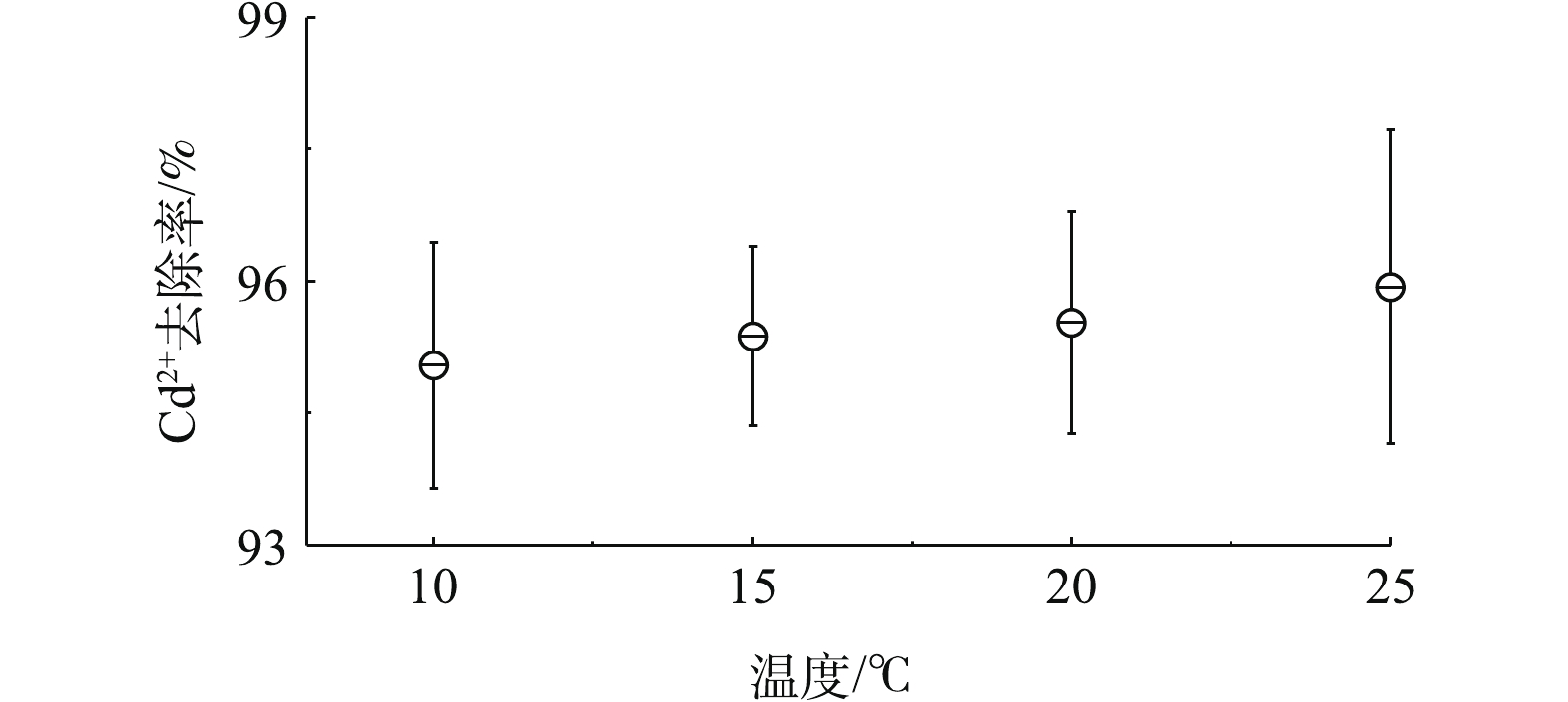
温度会改变吸附的热力学和动力学,也会改变镉离子在水体中的存在状态;同时可以根据温度的变化考察镉离子的吸附热力学过程。图5表明,随着温度的升高,镉离子的去除率呈上升趋势,从10 ℃时的最低约95%升到25 ℃时的约96%。为了准确测量温度对高分子空心微球吸附镉离子的影响,重复做3次实验,其变化趋势基本一致,呈缓慢上升过程。由此推测镉离子吸附过程不是明显的放热反应。同时,在吸附过程中,静电吸附和化学吸附同时存在。随着静电吸附温度的降低,吸附效率不断提高。而实验结果与此相反,表明除了静电吸附外,还存在着化学吸附,也就是阳离子交换反应。
-
吸附剂的循环利用可有效降低工程应用成本,高分子空心微球的再生吸附结果如图6所示。随着盐酸浓度的升高,镉微污染物吸附效率呈下降趋势;而随着再生时间的延长,在低盐酸浓度再生条件下,镉离子吸附效率先升后降,由此推测,在酸性条件下,有可能会改变高分子微球表面的官能团性质或电荷性质,当酸洗时间过长时,微球表面官能团性质出现改变,从而影响镉离子再生效率。而在高盐酸浓度再生条件下,镉离子吸附效率呈下降趋势。经酸洗再生后,盐酸浓度为0.1 mol·L−1,再生时间为30 min时,高分子空心微球吸附镉离子的效率最高达到96%以上,其吸附率没有下降。
在高分子空心微球再生过程中,其结构有可能被破坏。当酸浓度较低时,水体中pH降低,氢离子浓度上升,氢离子会反向和镉离子交换,重新置换镉离子,达到脱附的目的。但当酸浓度过高时,高分子空心微球表面基团有可能被破坏,或者高分子空心微球的结构被破坏,从而造成再生吸附时率下降。
-
1)高分子空心微球的合成温度和保温时间会影响其表面的基团及微球的粘连,当水热温度为180 ℃、水热时间为4 h时,镉微污染物去除率最佳。
2)与碳材料吸附镉离子不同,高分子空心微球在酸性条件下更易吸附镉离子,当pH<4时,镉离子去除率达到95%以上。
3)高分子空心微球吸附镉离子不存在明显放热过程,最大吸附量超过75 mg·g−1。
4)用盐酸再生时,盐酸浓度为0.1 mol·L−1,再生时间为30 min时,镉离子的吸附率达到96%以上。
高分子空心微球的合成及其对废水镉微污染物的吸附性能
Formation of polymer with hollow microspheres and its adsorption efficiency of cadmium micropollutants
-
摘要: 为考察高分子纳米微球对微污染物镉的吸附性能,以高分子空心微球为吸附剂,以废水镉微污染物为吸附对象,探讨高分子空心微球的重金属吸附性能,重点考察水热时间和水热温度对高分子微球表面基团的影响。结果表明:水热温度升高,高分子微球出现粘连;当水热温度为180 ℃,水热时间为4 h时,镉微污染物去除率最佳。强酸性条件有利于高分子微球吸附镉微污染物,当pH<4时,镉微污染物去除率超过95%;而中碱性条件的去除率不超过70%。高分子空心微球镉离子吸附过程不属于放热过程,最大吸附量超过75 mg·g−1。经盐酸再生利用时,高分子空心微球的镉吸附率没有出现下降,超过95%以上。高分子微球对微污染物镉具有良好的吸附性能。Abstract: The adsorption efficiency of Cd2+ by polymer were investigated. Compared to traditional carbon nanospheres, polymer with hollow microspheres were used as adsorbent to adsorb cadmium micropollutants in this study, the adsorption efficiency of cadmium micropollutants by the hollow microspheres were investigated. The effect of hydrothermal temperature and holding time on surface groups of the polymer were studied. The results showed that the groups on the surface of the polymer were affected by hydrothermal temperature and holding time, the particles would be stick together with higher hydrothermal temperature. The removal rate of cadmium micropollutants was at the best at 180 ℃ for 4 h. When pH was below 4, the removal rate of cadmium micropollutants was above 95%, it showed that it benefit to adsorption cadmium micropollutants at acid condition. When pH was above 7, the removal rate was below 70%. The maximal adsorption capacity of the hollow microspheres polymer was above 75 mg·g−1, and it was not exothermic process during adsorption. The recycling efficiency of the polymer was above 95% by the recovery technique of hydrochloric acid. The Cd2+ ions can be well adsorbed by the polymer.
-
Key words:
- polymer with hollow microspheres /
- micropollutants /
- adsorption
-
“十三五”规划把重金属污染防治列为环境治理工作的重心之一。然而,微/痕量重金属污染物具有污染面广、种类多、持久性及毒性大的特点,逐步引起人们的重视[1-2]。微量污染物,或称痕量污染物,包括传统型微污染物和新型微污染物,如微量重金属、类激素、多抗生素、氯联苯(PCBs)、有机氯类杀虫剂(DDT)、多环芳烃(PAHs)等[3]。由于微污染物浓度很低,只有ng·L−1~μg·L−1水平,且重金属与有机污染物不同,难以通过生物、化学降解转化途径去除。目前,重金属微/痕量污染物主要通过化学沉淀法、氧化还原法和电化学处理等方法去除,如臭氧氧化和活性炭吸附,但是臭氧氧化成本高、活性炭吸附选择性差的缺点阻碍了其工程推广应用[4-5]。
电镀行业、含镉矿山开采/冶炼以及镍镉电池生产过程中会产生一定量的含镉废水,镉污染物进入水体和土壤后容易富集且不被微生物降解,进入食物链后易对人体造成伤害,如痛痛病。目前,镉污染治理技术包括化学沉淀、氧化还原、离子交换、电化学、膜分离和吸附法等。其中常用的去除方法是化学沉淀、吸附以及膜分离法。然而,化学沉淀法存在污泥量大、难以沉降、易形成二次污染等缺点,膜分离法存在运行成本高、膜通量小、膜易污染等问题。目前,吸附剂主要为活性炭,活性炭存在pH适用范围小、分离困难等缺点。《生活饮用水卫生标准》(GB 5749-2006)对镉的限制量为0.005 mg·L−1。传统的重金属处理方法存在着难以克服的弊端,很难达到污水排放标准。同时重金属在水体中具有富集效应,ng·L−1或μg·L−1级的重金属经富集放大后,对人体的生理系统仍会造成极大伤害。
碳纳米材料具有比表面积大、机械强度高、结构可控、耐高温、表面基团丰富等特征,具有独特的物理或化学优势,是目前研究的热点和难点之一[6-9]。通过软/硬模板法、选择性蚀刻法、自组装等方法可合成独特结构的碳纳米材料[10-13],碳纳米材料表面含有丰富的羟基和羧基[14-15],可与重金属形成共价键或络合结构;独特的纳米孔道结构,甚至具有选择性吸附重金属离子的潜力[16-17],在环境领域具有潜在的应用价值[18]。
针对难以去除的微量/痕量镉重金属微污染物,强化混凝/吸附具有特有的优势。吸附法具有处理效率高、运行费用低、可操作性强、原材料来源丰富、价格便宜、易解吸等优点,是含镉废水处理工程实际应用首选方法之一。同时,高分子微球具有合成简单、价格便宜、比表面积大的特点,其表面含有大量的羟基,通过组装成空心高分子微球,可有效提高高分子微球的选择吸附性能,在微量/痕量重金属吸附领域具有自身独特的应用前景和研究价值[19-21]。本研究针对重金属微量/痕量污染物难于达标去除的难题,采用高分子微球为吸附剂,考察其对微/痕量重金属污染物的吸附性能,为微量/痕量污染物的去除提供新的技术选择和参考。
1. 材料与方法
1.1 实验原料
实验主要试剂:葡萄糖(A.R.)、盐酸(A.R.)购于天津市科密欧化学试剂有限公司,硝酸镉(A.R.)购于阿拉丁学试剂有限公司,硝酸铅(A.R.)、氟化钠(工业纯)购于国药集团化学试剂有限公司,氢氧化钠(A.R.)购于天津市风船化学试剂科技有限公司。
实验仪器:电子天平(AL104,Mettler-Toledo公司)、数显强力电动搅拌机(JB90-S,上海标本模型厂)、台式pH计(FE20,Mettler-Toledo公司)、电热恒温鼓风干燥器(DGG-9053A,上海森信实验仪器有限公司)、紫外可见分光光度计(UV-2450,日本Shimadz公司)、X射线衍射分析仪(XTRA,瑞士ARL公司)、数显恒温水浴锅(HH-2,国华电器有限公司)、镉离子电极(DX312,立德科技有限公司)。
1.2 实验方法
空心高分子微球的合成。采用改进的高分子微球合成方法[22]:使用水热方法,以葡萄糖为碳源,氨水(28%)为软模板,每10 mL水加1 g葡萄糖、0.1 mL氨水,水热温度分别为170、180、190、200、210 ℃,水热时间分别为2、3、4、5、6 h。
镉微污染物吸附。CdNO3浓度20 mg·L−1(40 mL),不同温度、不同时间的高分子微球0.05 g,磁力搅拌20 min,静止30 min或离心,测上清液Cd浓度。
起始pH对镉微污染物吸附的影响。CdNO3浓度20 mg·L−1(40 mL),高分子微球投加量为0.05 g,pH为2~11;磁力搅拌20 min,静止30 min或离心,测上清液Cd浓度。
投加量对镉微污染物吸附的影响。取镉微污染吸附实验中的最佳温度和时间的高分子微球,CdNO3浓度20 mg·L−1(40 mL),高分子微球投加量为0.01、0.02、0.05、0.1和0.2 g;磁力搅拌20 min,静止30 min或离心,测上清液Cd浓度。
温度对镉微污染物吸附性能的影响。CdNO3浓度20 mg·L−1 (40 mL),反应温度为10、15、20和25 ℃,高分子微球投加量为0.05 g,pH为4,磁力搅拌20 min,静止30 min或离心,测上清液Cd离子浓度。
干扰离子对镉微污染物竞争吸附的影响。采用酸洗对高分子空心微球(180 ℃、4 h镉微污染物吸附后的微球)进行再生,盐酸浓度为0.1~2 mg·L−1,酸洗时间为10~60 min,酸洗后样品在60 ℃烘箱烘干,采用最佳参数重新吸附镉微污染物,测上清液镉离子浓度。
1.3 分析方法
镉离子浓度测量采用镉离子选择电极法。
2. 结果与讨论
2.1 镉微污染物吸附
不同温度条件下合成的高分子微球XRD、SEM结果如图1所示。XRD结果表明,不同温度条件下合成的高分子微球在2θ=23°有一驼峰,没有发现石墨化的衍射峰,表明高分子以无定型碳形式存在。SEM结果表明,合成的高分子微球粒径为150~600 nm。在水热温度为180 ℃时,高分子微球的粒径均一性较210 ℃的微球稍差,但210 ℃条件下合成的高分子微球出现粘连现象,不利于高分子微球的分散。
 图 1 不同合成条件下高分子微球的XRD和SEMFigure 1. XRD and SEM of polymer nano-spheres at different condition
图 1 不同合成条件下高分子微球的XRD和SEMFigure 1. XRD and SEM of polymer nano-spheres at different condition以氨水为起泡剂,葡萄糖为碳源,合成的高分子微球为空心微球,根据已报道的采用类似方法合成的高分子微球及研究结果[23-24],可以证明合成的高分子微球为空心微球。
不同温度条件下合成的高分子微球镉微污染物吸附性能如图2所示。结果表明,水热反应时间和水热温度对镉微污染物吸附有相似的影响规律。随着水热反应时间的延长,Cd2+去除率先升高后降低,当水热反应时间为5 h (170 ℃)时,Cd2+去除率达到最大值,约60%;当升高水热温度时,Cd2+去除率先升后降,当水热温度为180 ℃(4 h)时,Cd2+去除率达到最高值,为67%。
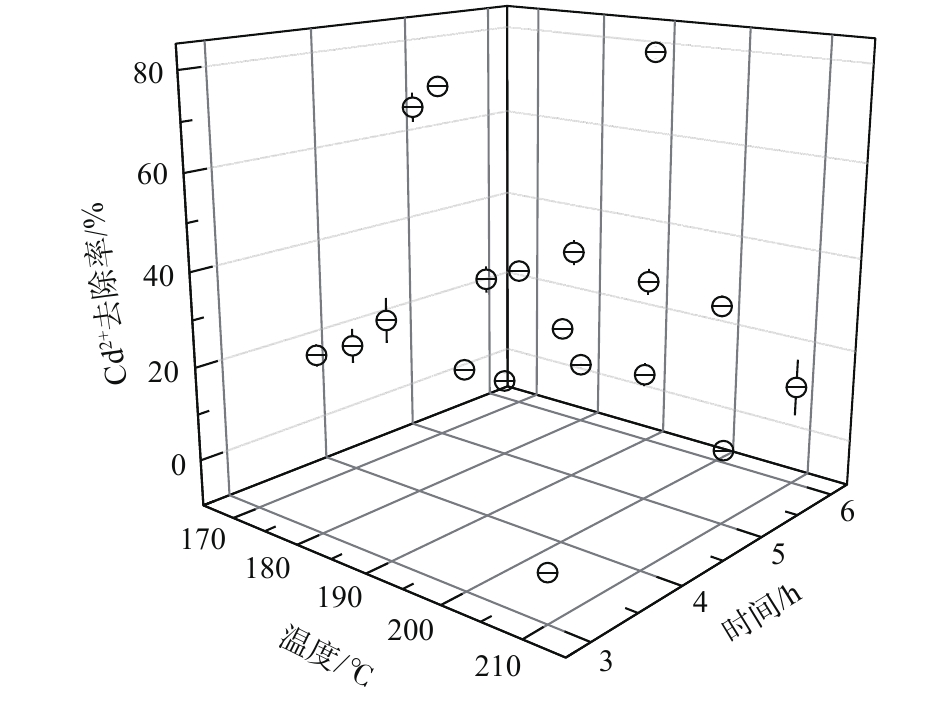 图 2 不同合成条件下高分子微球镉离子吸附性能Figure 2. Cd2+ removal rate by polymer nano-spheres atdifferent synthesis condition
图 2 不同合成条件下高分子微球镉离子吸附性能Figure 2. Cd2+ removal rate by polymer nano-spheres atdifferent synthesis conditionSEM结果表明,水热温度的升高会造成高分子微球的粘连,但水热温度的升高同时会提高高分子微球的均一性。因此,合适的水热温度和水热时间有助于合成性能一致且分散性良好的高分子微球。从节约能源角度及高分子微球粘连与温度呈正比例关系角度考虑,本实验取最佳水热温度为180 ℃,水热时间为4 h。
2.2 起始pH对镉微污染物吸附的影响
pH是金属微污染物吸附法关键的影响因素之一。pH会影响金属离子在水体中的存在形态,在酸性条件下,部分金属离子与水分子形成水合离子团体;而在碱性条件下,大部分金属离子会形成沉淀或聚合物。图3表明,高分子空心碳微球在酸性条件下镉微污染物的吸附效率高于中碱性条件,在pH<4时,镉微污染物的去除率超过95%,随着pH的升高,镉微污染物去除率先急速下降,然后缓慢微升;pH为6时,去除率接近40%;而当pH>7时,镉微污染物去除率约为60%。研究结果与碳材料吸附结果[25]不同。
在酸性条件下,镉离子会与水分子形成较大的表面带正电荷的水合离子团体,理论上会降低镉离子的吸附效率及速率。而当pH升高时,由于羟基浓度上升,水合镉离子团体表面张力下降,镉离子被释放出来,与羟基结合,形成带负电荷的中间体。而高分子微球表面含有大量的—OH、—COOH、—CO基团,这些基团中的氢离子会被较大的阳离子交换,阳离子质量越大,直径越大,越容易交换氢离子。因此,在酸性条件下,水合镉离子团体更易交换高分子微球表面的氢离子。而当pH升高后,镉聚合物表面的电性由正极转为负极,与高分子表面基团的电荷相同,降低了镉微污染物的吸附效率;当pH继续升高,在碱性条件下,镉离子会形成氢氧化镉沉淀,提高了镉离子的去除率。
2.3 投加量对镉微污染物吸附的影响
吸附剂投加量决定废水的起始浓度,过高或过低都会影响镉微污染物的去除效果。实验中镉离子的浓度为20 mg·L−1,取40 mL水,也就是说样品中镉离子的质量为0.8 mg。图4表明,当高分子空心微球的投加量为0.01 g时,镉离子去除率达到95%以上,继续提高高分子空心微球的投加量,镉离子去除率略有上升,但上升幅度不大。高分子空心微球的投加量为0.01 g时,镉离子大部分被吸附,此时镉离子的吸附量大于76 mg·g−1,高分子空心微球对镉离子的最大吸附量大于76 mg·g−1。
2.4 不同温度条件下镉微污染物吸附性能
温度会改变吸附的热力学和动力学,也会改变镉离子在水体中的存在状态;同时可以根据温度的变化考察镉离子的吸附热力学过程。图5表明,随着温度的升高,镉离子的去除率呈上升趋势,从10 ℃时的最低约95%升到25 ℃时的约96%。为了准确测量温度对高分子空心微球吸附镉离子的影响,重复做3次实验,其变化趋势基本一致,呈缓慢上升过程。由此推测镉离子吸附过程不是明显的放热反应。同时,在吸附过程中,静电吸附和化学吸附同时存在。随着静电吸附温度的降低,吸附效率不断提高。而实验结果与此相反,表明除了静电吸附外,还存在着化学吸附,也就是阳离子交换反应。
2.5 镉微污染物再生循环吸附
吸附剂的循环利用可有效降低工程应用成本,高分子空心微球的再生吸附结果如图6所示。随着盐酸浓度的升高,镉微污染物吸附效率呈下降趋势;而随着再生时间的延长,在低盐酸浓度再生条件下,镉离子吸附效率先升后降,由此推测,在酸性条件下,有可能会改变高分子微球表面的官能团性质或电荷性质,当酸洗时间过长时,微球表面官能团性质出现改变,从而影响镉离子再生效率。而在高盐酸浓度再生条件下,镉离子吸附效率呈下降趋势。经酸洗再生后,盐酸浓度为0.1 mol·L−1,再生时间为30 min时,高分子空心微球吸附镉离子的效率最高达到96%以上,其吸附率没有下降。
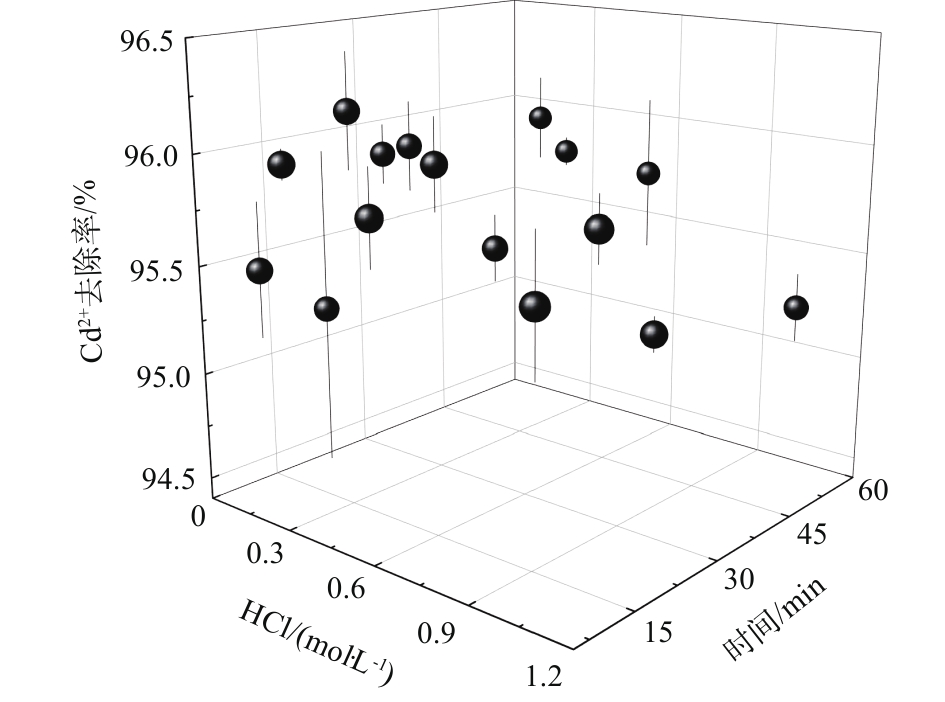 图 6 不同HCl浓度和浸泡时间对Cd2+离子去除率的影响Figure 6. Effect of Cd2+ removal rate at different HCl concentraten and soak time
图 6 不同HCl浓度和浸泡时间对Cd2+离子去除率的影响Figure 6. Effect of Cd2+ removal rate at different HCl concentraten and soak time在高分子空心微球再生过程中,其结构有可能被破坏。当酸浓度较低时,水体中pH降低,氢离子浓度上升,氢离子会反向和镉离子交换,重新置换镉离子,达到脱附的目的。但当酸浓度过高时,高分子空心微球表面基团有可能被破坏,或者高分子空心微球的结构被破坏,从而造成再生吸附时率下降。
3. 结论
1)高分子空心微球的合成温度和保温时间会影响其表面的基团及微球的粘连,当水热温度为180 ℃、水热时间为4 h时,镉微污染物去除率最佳。
2)与碳材料吸附镉离子不同,高分子空心微球在酸性条件下更易吸附镉离子,当pH<4时,镉离子去除率达到95%以上。
3)高分子空心微球吸附镉离子不存在明显放热过程,最大吸附量超过75 mg·g−1。
4)用盐酸再生时,盐酸浓度为0.1 mol·L−1,再生时间为30 min时,镉离子的吸附率达到96%以上。
-

图 1 不同合成条件下高分子微球的XRD和SEM
Figure 1. XRD and SEM of polymer nano-spheres at different condition
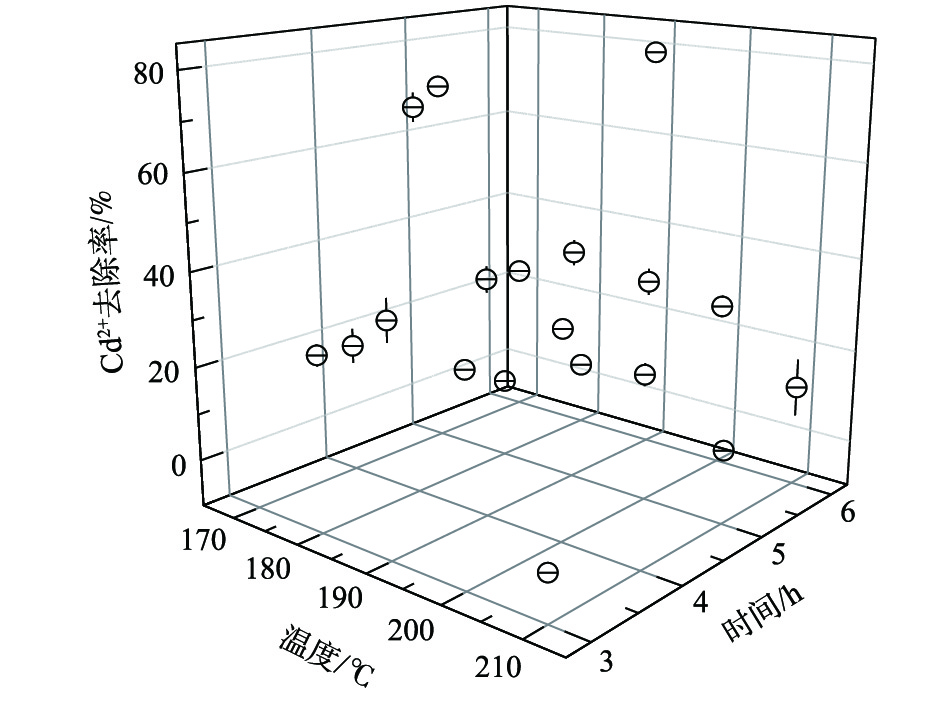
图 2 不同合成条件下高分子微球镉离子吸附性能
Figure 2. Cd2+ removal rate by polymer nano-spheres atdifferent synthesis condition
-
[1] ZHOU J L, HONG H, ZHANG Z, et al. Multi-phase distribution of organic micropollutants in Xiamen Harbour, China[J]. Water Research, 2000, 34(7): 2132-2150. doi: 10.1016/S0043-1354(99)00360-7 [2] ADEWATE G, ABDALLAH D, JOANNA K. Membrane bioreactors and electrochemical processes for treatment of wastewaters containing heavy metal ions, organics, micropollutants and dyes: Recent developments[J]. Journal of Hazardous Materials, 2019, 370(15): 172-195. [3] 许国栋, 张婧怡, 陈珺, 等. 城市污水处理微污染物的挑战与对策[J]. 给水排水, 2016, 52(9): 40-44. doi: 10.3969/j.issn.1002-8471.2016.09.009 [4] JONAS M, CORNELIA K, ANOŸS M, et al. Treatment of micropollutants in municipal wastewater: Ozone or powdered activated polymer[J]. Science of the Total Environment, 2013, 461-462(1): 480-498. [5] WALASZEK M, BOIS P, LAURNT J, et al. Micropollutants removal and storage efficiencies in urban stormwater constructed wetland[J]. Science of the Total Environment, 2018, 645(15): 854-864. [6] TANG J, YAMAUCHI Y. Polymer materials: MOF morphologies in control[J]. Nature Chemistry, 2016, 8: 638-639. doi: 10.1038/nchem.2548 [7] XU F, TANG Z W, HUANG S Q, et al. Facile synthesis of ultrahigh-surface-area hollow polymer nanospheres for enhanced adsorption and energy storage[J]. Nature Communication, 2015, 6: 7221. doi: 10.1038/ncomms8221 [8] PEI F, AN T H, ZANG J, et al. From hollow polymer spheres to N-doped hollow porous polymer bowls: Rational design of hollow polymer host for Li-S batteries[J]. Advance Energy Materials, 2016, 6(8): 1502539. doi: 10.1002/aenm.201502539 [9] ZHENG G Y, LEE S W, LIANG Z, et al. Interconnected hollow polymer nanospheres for stable lithium metal anodes[J]. Nature Nanotechnology, 2014, 9: 618-623. doi: 10.1038/nnano.2014.152 [10] LIU J, WICKRAMARATNE N P, QIAO S Z, et al. Molecular-based design and emerging applications of nanoporous polymer spheres[J]. Nature Materials, 2015, 14: 763-774. doi: 10.1038/nmat4317 [11] ZHOU L, ZHUANG Z C, ZHAO H H, et al. Intricate hollow structures: controlled synthesis and applications in energy storage and conversion[J]. Advance Materials, 2017, 29(20): 1602914. doi: 10.1002/adma.v29.20 [12] GRZELZAK M, VERMANT J, FURST E M, et al. Directed self-assembly of nanoparticles[J]. ACS Nanotechnology, 2010, 4(7): 3591-3605. [13] PARK H, AFZALI A, HANS J, et al. High-density integrationof carbonnanotubes via chemical self-assembly[J]. Nature Nanotechnology, 2012, 7: 787-791. doi: 10.1038/nnano.2012.189 [14] ZHANG P, QIAO Z A, DAI S. Recent advances in carbonnanospheres: Synthetic routes and applications[J]. Chemistry Communication, 2015, 51: 9246-9256. doi: 10.1039/C5CC01759A [15] SANG Y, HUANG Y, LI W, et al. Bioinspired design of Fe3+ doped mesoporous polymer nanospheres for enhanced nanozyme activity[J]. Chemistry: A European Journal, 2018, 24(28): 7259-7263. doi: 10.1002/chem.v24.28 [16] ZHAO G, LI J, REN X, et al. Few-layered graphene oxide nanosheets as superior sorbents for heavy metal ion pollution management[J]. Environment Science & Technology, 2011, 45(24): 10454-10462. [17] YU S, WANG X, TAN X, et al. Sorption of radionuclides from aqueous systems onto grapheme oxide-based materials: A review[J]. Inorganic Chemistry Frontiers, 2015, 2: 593-612. doi: 10.1039/C4QI00221K [18] SHALINI T, AHIN R, SAJITHA N, et al. Removal of U(VI) from aqueous solution by adsorption onto synthesized silica and zinc silicate nanotubes: Equilibrium and kinetic aspects with application to real samples[J]. Environmental Nanotechnology, Monitoring & Management, 2018, 10: 127-139. [19] ZHANG X, WU M, DONG H, et al. Simultaneous oxidation and sequestraten of As(III) from water by using redox polymer-based Fe(III) oxide nanocomposite[J]. Environmental Science & Technology, 2017, 51(11): 6326-6334. [20] QIN Q, WANG Q, FU D, et al. An efficient approach for Pb(II) and Cd(II) removal using manganese dioxide formed in situ[J]. Chemical Engineering Journal, 2011, 172(1): 68-74. doi: 10.1016/j.cej.2011.05.066 [21] LIU Y, WANG L, WANG X, et al. Highly efficient removal of trace thallium from contaminated source waters with ferrate: Role of in situ formed ferric nanoparticle[J]. Water Research, 2017, 124(1): 149-157. [22] 叶靓雯. 空心高分子微球的制备及其氧还原电催化性能研究[D]. 北京: 北京化工大学, 2018. [23] 余荣台, 刘健聪, 马湘, 等. 基于磁流体组装的空心磁性碳微球的合成及其重金属污染去除性能[J]. 陶瓷学报, 2018, 39(6): 753-757. [24] 冯珊珊. 基于磁流体组装的空心磁性碳微球及其功能性复合体[D]. 哈尔滨: 黑龙江大学, 2011. [25] AZOUAOU N, SADAOUI Z, DJAAFRI L, et al. Adsorption of cadmium from aqueous solution onto untreated coffee grounds: Equilibrium, kinetics and thermodynamics[J]. Journal of Hazardous Materials, 2010, 184(1/2/3): 126-134. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3377
- HTML全文浏览数: 3377
- PDF下载数: 110
- 施引文献: 0
