

-
随着工业的发展,石油被广泛应用于各行各业,但其带来的环境污染问题不容忽视,尤其是泄漏等生产事故诱发的土壤污染,原位化学氧化 (ISCO) 是土壤有机污染快速去除的有效方法,因具有修复效能高、成本低及操作便利等优势,常用于石油烃污染土壤修复[1-6]。近年来高锰酸钾 (KMnO4) 对石油烃的ISCO被认为是H2O2、O3/H2O2或催化H2O2氧化的良好替代品,因为其易于处理、成本低、pH范围适应性强且氧化电位 (E0=1.51 V) 相对较高[7]。然而,传统的ISCO技术在修复过程中面临着一系列挑战,特别是氧化剂的有效浓度低,难以维持长效性的问题,这使得污染物容易出现浓度反弹。
为了解决这些问题,已有报道提出了利用微囊化技术制备可控缓释材料来替代传统反应试剂,两者的主要区别在于控释材料延长了反应试剂的释放时间,降低反应物的释放速率,即控释材料具有持续释放的特点[8-10]。目前,微胶囊技术在缓释氧化材料研发领域已经取得初步的进展[11-12]。例如,有研究通过离子交联反应,将过氧化钙封装在海藻酸钙聚合物中,形成过氧化钙的释氧凝珠,为环境中的好氧微生物提供氧源用于降解1,4二恶烷,释氧凝珠可以在较长的时间内 (7 d左右) 使环境中的溶解氧保持较高的水平,并在10 d内将污染物去除近96%[13]。KAMBHU等[14]通过使用石蜡包裹KMnO4,使用模具制备了柱状KMnO4缓释材料,通过埋入表层土壤的方式对土壤多环芳烃进行降解,但是需要定期挖出去除表面石蜡,并进行再次填埋。从目前关于场地修复缓释氧化材料的研发和应用方面来看,缓释和控释技术在越来越多的研究和修复方案中得到了应用[15-16]。这些技术能够有效控制修复活性物质的释放,减少修复药剂的非选择性消耗,并提高污染物的去除效率,有望解决污染场地原位修复的长期有效性差的问题[17-19]。然而,目前所研发的控释材料缓释时长普遍处于24 h之内[19-21],其氧化活性难以维持更久,这对低渗透地层非水相污染物的原位修复功能材料提出了更大挑战。因此,需要进一步研究和开发适用于土壤修复的缓释氧化材料包覆方法,在选择包覆材料时,需要考虑其渗透性和降解性,以确保修复活性物质能够有效释放到土壤中。
本研究以硬脂酸为壳材,采用油相分离法制备了包覆型KMnO4,并通过正交实验探究最佳制备条件和实验参数。通过水相缓释实验探究缓释氧化材料的释放动力学,并根据缓释曲线拟合缓释材料的释药模型,结合表征结果探究该材料的释放机理。设计制作土柱动态淋洗实验,探究缓释氧化材料在模拟土柱中的缓释作用及其对柴油烃污染土壤的修复效果,为其在实际修复中的应用提供理论依据。
-
高锰酸钾 (KMnO4) 、硬脂酸 (C18H36O2) 、聚乙二醇4000 (PEG-4000;HO(CH2CH2O)nH) 、无水乙醇 (C2H6O) 、石油醚、正己烷 (C6H14) 均购于国药集团化学试剂有限公司;实验室用水为超纯水。
-
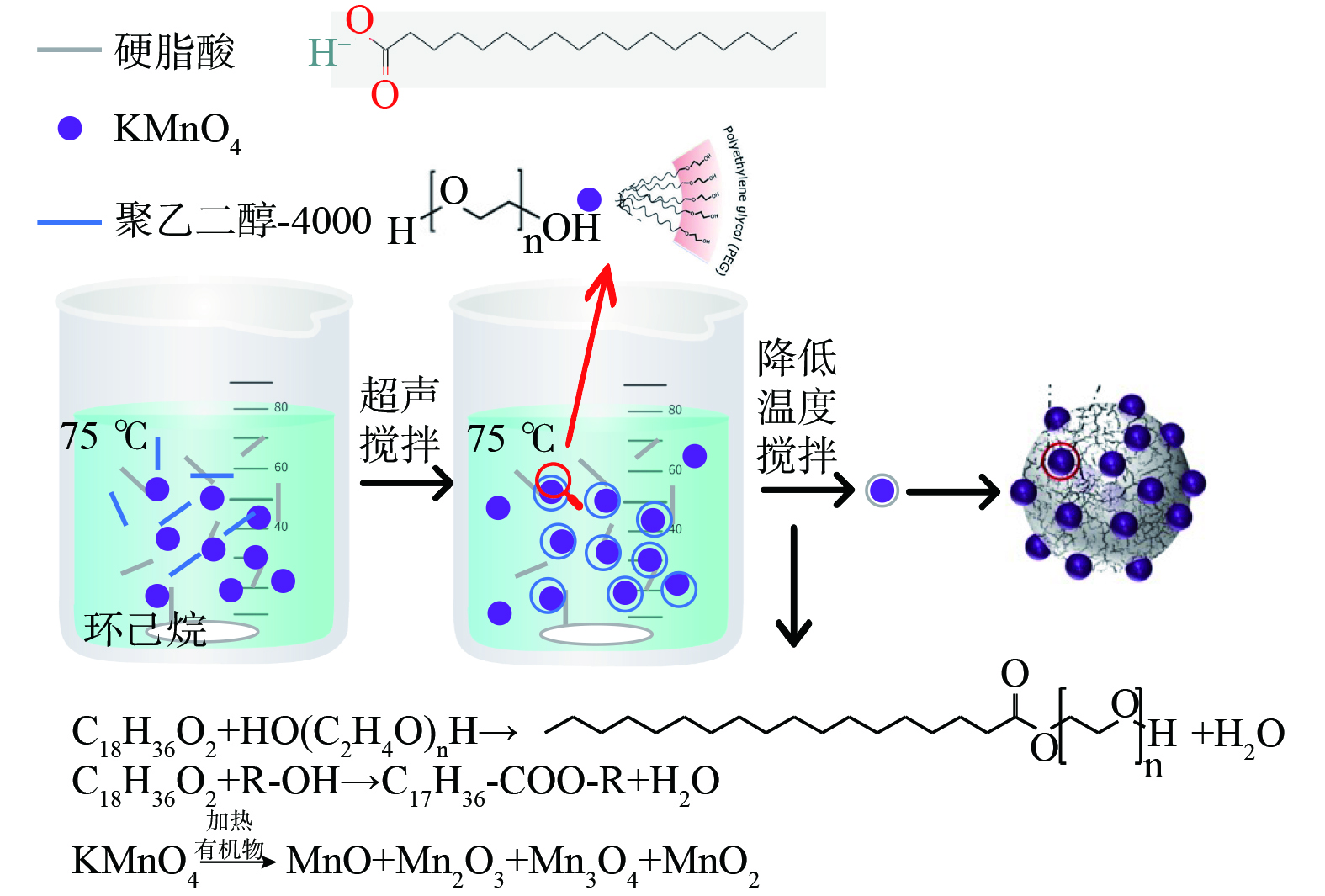
采用油相分离法制备缓释KMnO4微胶囊,取适量的壳材硬脂酸 (10、15 g) 于烧杯中,加入乙醇 (100 mL) ,水浴搅拌加热至70~75 ℃将壳材完全溶解后,按照一定的比例加入KMnO4 (5 g) ,进行超声和搅拌使KMnO4分散于壳材溶液中,然后称取适量PEG-4000 (0 、2、3 g) ,逐次少量加入体系中进行助凝以促进微胶囊化,同时降低水浴温度至25~35 ℃ (冰水浴冷却/自然冷却) ,使壳材溶解度降低,并以KMnO4为核心凝聚析出,初步形成包覆型KMnO4,然后放入40 ℃烘箱进行固化 (4 h) ,期间进行适当的搅拌和破碎防止材料凝聚结块,最后将材料进行破碎过筛,按照不同粒径保存待用,即得缓释KMnO4氧化剂 (PP@SA) 。并且通过四因素三水平L9 (34)正交表设计实验探究芯壳比、超声时间、搅拌速度和粒径对材料缓释效果的影响,芯壳比分别为1∶1、1∶2、1∶3;超声时间分别是5、10、15 min;搅拌速度分别是150、300、450 r∙min−1;粒径分别为<0.6、0.6~1.12、1.12~4 mm。
-
配置0.2000 g∙L−1的KMnO4标准溶液,分别取0.0、2.0、4.0、6.0、8.0和10.0 mL用纯水定容至10 mL,用紫外分光光度计于525 nm波长下测定吸光度并绘制浓度-吸光度标准曲线用于计算KMnO4浓度。将PP@SA材料研磨后称取0.1 g,加入适量纯水并进行超声处理使材料中的活性组分 (KMnO4) 释放并溶解在水中,然后定容至500 mL。从中取1.0 mL溶液定容至10.0 mL用紫外分光光度计测定吸光度并计算KMnO4浓度。此外,PP@SA的包封率E通过式(1)计算。其中PP@SA的实际载药量由载药量测定结果确定,理论载药量即氧化剂在壳材和芯材总含量的占比百分数。
-
按照缓释材料投加量为1 g∙L−1进行水相缓释实验,将其置于水浴磁力搅拌器中 (25 ℃,100 r∙min−1) ,取样时间设为0.5、1.0、2.0、3.0、3.5、7.5、11.5、23.5、27.5、31.5、46.5、58.5、73.5、97.5、121.5 h,取样后使其通过0.22 μm水系滤膜,取1.0 mL过滤液于10 mL比色管,并定容至10.0 mL用紫外分光光度计检测其吸光度。
采用国标HJ-1068-2019中的比重计法测定土壤的粒径和质地;采用紫外分光光度计进行柴油含量的检测;采用傅里叶红外光谱分析仪 (FTIR,美国赛默飞世尔科技公司) 测定缓释氧化材的官能团变化情况;采用X-射线衍射仪 (XRD,德国布鲁克) 对缓释前后缓释氧化材料中的物相组分进行分析;采用Gemini 300扫描电子显微镜 (SEM,德国卡尔蔡司) 和X射线能谱仪 (EDS,德国卡尔蔡司) 进行分析样品的表观形貌;采用表面张力仪 (DCAT 21,德国DataPhysics) 对其接触角信息进行测定;采用X射线光电子能谱 (XPS,赛默飞世尔ESCALAB 250xi) 对氧化剂中的特征元素的价态和成分含量进行定性和半定量分析。
-
本研究土壤样品取自上海市奉贤区某未污染场地的深层土:粉质粘土 (silty clay) 和淤泥质黏土 (chalky clay) 。粉质粘土和淤泥质黏土的pH值分别是7.95和7.8;饱和含水率分别是48%和46%;黏粒占比分别为38%和30%;粉粒占比分别为42%和33%;沙粒占比分别为20%和37%,两种土壤质地为典型的低渗透土壤。
为模拟PP@SA在实际土壤中的释放行为,利用亚克力材质柱进行柱淋洗实验,风干后的土壤使用粉碎机粉碎后过70目筛,石英砂使用前用稀盐酸清洗浸泡1 h并用去离子水冲洗至中性后待用,土柱装填采用干法装柱,自下往上依次按照以下顺序进行装填:10 cm石英砂;10 cm土壤层;1 cm石英砂;5%土壤质量的PP@SA (3.75 g) ;3 cm石英砂。使用蠕动泵进行供水,同时保持稳定的水头压力,并且从底部出水口收集渗滤液。
-
通过油相分离法制备的PP@SA在水相中具有较好的缓释效果,其释放时长达到100 h以上,图1为PP@SA材料在投加量为1 g∙L−1时高锰酸根释放曲线。结果发现添加PEG-4000的材料缓释速度较慢,硬脂酸在凝聚析出的过程中,包裹KMnO4颗粒性能较好;提高硬脂酸的用量,可以减缓初始的缓释速率和释放量;并且自然冷却方式所得材料比冰水浴冷却得到的材料释放慢,可能是由于冰水浴加快了硬脂酸的析出速度,使溶液中的硬脂酸不能均匀分散在KMnO4颗粒周围,而是原位直接析出,导致壁材的浪费。此外,当溶剂乙醇的用量为100 mL时,无论冷却方式是哪一种,都会使部分硬脂酸直接从溶液中析出。
进一步优化油相分离法的制备参数,采用环己烷作为溶剂,并且用量减少至和硬脂酸等质量的用量,使用超声和搅拌两种分散方式结合,同时考察材料粒径对缓释效果的影响。通过四因素三水平L9(34)正交表设计实验 (表1) ,探究最佳制备参数,正交试验各因素包括芯壳比、超声时间、搅拌速度和粒径。并分别以包封率和5 d的释放百分比为评价指标,进行制备参数的优化。将所得包封率和5 d释放百分比数据通过IBM SPSS Statistics 26软件进行分析 (表2) ,发现以包封率为评价指标时,影响因素对包封率的影响程度为:超声时间>芯壳比>搅拌速度>粒径。影响最大的因素是超声时间,超声过程不仅会影响KMnO4在体系中的分散,也会增加KMnO4在体系中的消耗,最佳的制备条件为:芯壳比1∶1,超声时间10 min,搅拌速度150 r∙min−1,粒径小于0.6 mm。以5 d的释放百分比为评价指标时,各因素对5 d缓释百分比的影响程度为:芯壳比>搅拌速度>粒径>超声时间。最大影响因素为芯壳比,因为KMnO4的用量越大,硬脂酸对KMnO4颗粒的包裹层越薄,从而加快释放速度;其次是搅拌速度,搅拌速度影响KMnO4在缓释颗粒中的分散;而粒径对缓释的影响主要体现在材料壳层的溶蚀速度和与水接触的表面积,粒径越小溶解速度越快。从缓释情况看,最佳制备条件为芯壳比1∶3,超声时间15 min,搅拌速度300 r∙min−1,粒径1.12~4.00 mm。
-
为探究缓释氧化材料的释放机制,将不同粒径材料的释放动力学曲线用一级、Higuchih和Ritger-Peppa模型进行拟合,一级释放模型的拟合度最高,所有样品的R2均在0.960 0以上 (图2) ,所以该模型更好地反映缓释材料中KMnO4的释放,表明氧化剂释放速率与其在环境介质中的量成正比 (随时间按恒定比例释放) 。由于硬脂酸是疏水性材料,对于水不溶性骨架结构可以通过Higuchi方程进行拟合,拟合结果如表3所示,样品1-S、2-S、3-S的拟合结果较差 (R2 < 0.900 0) ,表明粒径小于0.6 mm的材料的释放不符合骨架溶蚀机制。随材料粒径的增加,拟合结果得到改善,R2均大于0.950 0,表明随材料粒径的增大其缓释机制越来越以骨架溶蚀为主。
进一步通过Ritger-Peppas方程对缓释曲线进行了拟合,结果如表3所示,材料1-S、1-M、2-S和3-S符合Fichian定律扩散模式,即在单位时间内通过垂直于扩散方向的单位截面积的扩散物质流量 (即扩散通量Diffusion flux) 与该截面处的浓度梯度成正比。粒径小于0.6 mm的材料都符合Fickian扩散模式,可能是由于粒径较小的材料不存在骨架结构所以极易被破坏,而样品1-M的芯壳比为1∶1,硬脂酸对KMnO4的包裹效果可能相对较差,因此不存在骨架溶蚀的缓释机制。此外,其他材料的扩散指数n均在0.43~0.89之间,表明其缓释机制是溶解扩散和骨架溶蚀扩散两种机制共存的释放方式。随着粒径的增加,扩散系数n值呈现增大的趋势,表明随粒径的增加,硬脂酸对KMnO4的包封效果越好,KMnO4的释放机制逐渐由小粒径中的单一Fickian扩散模式开始转为骨架溶蚀扩散和Fickian扩散模式共存的溶解扩散方式。
-
不同芯壳比制备条件下的最小粒径材料 (即样品1-S、2-S、3-S) 的扫描电镜图如图3(a)所示。当芯壳比为1∶1时,材料表面相对比较平整,几乎没有骨架结构;芯壳比为1∶2时,材料表面变粗糙,出现较为明显的孔道结构;芯壳比为1∶3时,材料颗粒有明显的孔道结构和骨架结构。随着硬脂酸用量的增加,PP@SA的结构中骨架结构也越发达,其水相缓释机制也从单一的边界层Fickian扩散向以骨架溶蚀为主导的扩散模式转变,这与2.2部分动力学分析推测的材料结构一致。对水相缓释后的材料同样进行了SEM电镜分析 (图3(b)) ,芯壳比为1∶1的材料缓释后呈现片状结构,可能是材料结构破坏后剥落下的硬脂酸壳层;芯壳比为1∶2的材料颗粒表面出现溶蚀后的空洞;芯壳比为1∶3的颗粒整体结构比较完整,而骨架结构消失,这可能是因为硬脂酸含量增加,对KMnO4的包裹较严密,虽外层通过溶蚀造成结构破裂,但内层结构难以通过溶蚀作用破坏并释放内层的KMnO4。该结果也解释了在缓释实验中大粒径颗粒在后期仍可以保持较完整的粒状结构,并且释放浓度始终无法达到理论最大值的现象。
通过EDS表征了材料表面的元素分布和C、O、Mn、K的分布状态,该缓释材料中的C元素来源于硬脂酸和PEG-4000。不同芯壳比缓释材料的元素分布图和能量谱图如图4~6所示,芯壳比为1∶1的材料 (图4) ,C和Mn元素都均匀分布在材料表面上,表明该材料的结构不是严格的核壳结构;芯壳比为1∶2时 (图5) ,材料中出现了骨架结构,而Mn元素和C元素仍然是保持均匀分布的状态;芯壳比为1∶3时,由于硬脂酸的用量增加,在材料制备中冷凝阶段,大量的硬脂酸析出导致整个制备体系的搅拌困难,KMnO4分布不够均匀,因此出现部分区域KMnO4堆积的情况,如图6中的高亮区域。从材料的表观形貌和元素分布表明,油相分离法制备工艺能够较好地实现硬脂酸对KMnO4的包裹,但其整体的结构并不是十分规则的核壳结构,而是KMnO4在硬脂酸中形成的固体分散体[22-23]结构。
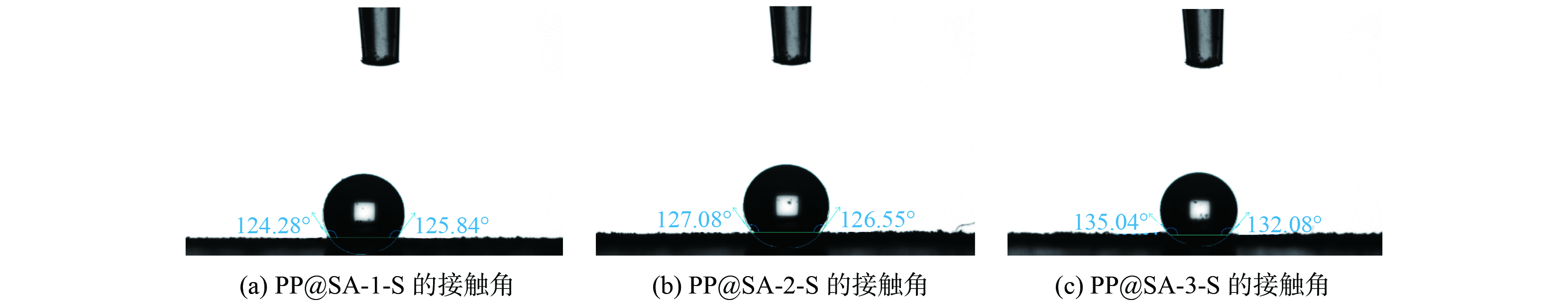
缓释材料的接触角测试结果如图7所示,结果表明该缓释材料接触角均大于120°,并且随着硬脂酸用量的增加,接触角分别达到125.84° (1-S) 、126.55° (2-S) 、132.08° (3-S) ,虽然硬脂酸结构中含有羧基基团,但是长碳链使硬脂酸表现出较强的疏水性,而材料疏水性的增强,能有效减缓材料与水的接触,降低KMnO4的释放速度,材料的亲油属性可以促进土壤环境中与污染物的接触或吸附,提高降解效率。
PP@SA材料水相缓释实验前后的红外光谱图如图8所示,通过对比硬脂酸的标准红外光谱图,2 919.35和2 853.01 cm−1为C-H键的伸缩振动吸收峰,1 701.47 cm−1为C=O键的伸缩振动吸收峰,1 457 cm−1为C-H键的变形振动吸收峰,1 164~1 362 cm−1为C-O键的伸缩振动吸收峰,915 cm−1为O-H键的面外弯曲振动峰,1 537 cm−1和1 349 cm−1处的吸收峰分别为硬脂酸中的羧基 (-COO-) 不对称振动和对称伸缩振动引起的[24],其中样品中的部分特征峰相对于硬脂酸出现了明显的减弱或消失,说明材料中游离状态的硬脂酸含量较低。同时,缓释KMnO4的样品谱图中出现了1 642和1 561 cm−1两个吸收峰,表明制备过程中产生了新的化学键,硬脂酸特征峰的减弱和新特征峰的出现表明,制备过程中硬脂酸和部分KMnO4发生反应。水相缓释后材料的红外光谱图中特征峰1 561 cm−1有较为明显的减弱,表明制备过程中产生的新的化学键在缓释过程中减少或消失,可能是由于在水中发生了溶解,或者是KMnO4溶出后与其发生了氧化还原反应。对于KMnO4的红外光谱图,可以看到在894.7 cm−1的位置有1个特征峰,而该峰在缓释实验之后消失,表明该峰应该为KMnO4中的锰氧键出峰位置。
对材料中无机组分采用XRD进一步分析,如图9所示,不同材料的成分差异主要是KMnO4和硬脂酸的比例,表面活性剂PEG-4000的用量较低 (为硬脂酸用量的5%) ,因此通过不同样品的谱图可以看到材料中有机组分和无机组分,通过物相检索主要分析了KMnO4和硬脂酸。在图9(a) 中通过谱图对比可以看到非常明显的KMnO4存在,这是因为在该样品中,KMnO4的含量相对更多,而随着硬脂酸用量的增加,在图9 (c) 中可以明显看到硬脂酸的峰强度的增加,并且粒径对于样品成分的影响较小,主要在于含量的变化,同时也说明材料的制备比较均匀。图10为材料在水相缓释实验之后测得的XRD谱图,所有样品的谱图特征类似,缓释后材料的KMnO4信号强度减弱,硬脂酸的信号更为明显,表明缓释实验之后剩余样品中主要是未溶解的硬脂酸等有机成分。
通过XPS来表征材料表面Mn元素及其化学态,缓释前后材料的XPS图谱如图11所示,Mn的3s轨道出现两个峰 (88.96 eV和83.68 eV) ,分别和KMnO4和Mn2O3、Mn3O4中的Mn的3s轨道峰位相对应,缓释反应后依然能够检测出KMnO4,且图谱峰面积有所上升。结合前文中对PP@SA形貌的分析,推测该材料在制备过程中,硬脂酸会在表面活性剂的作用下优先以KMnO4为内核进行析出,随着溶剂的挥发和反应的进行,逐渐黏结形成更大的颗粒,所以材料内部KMnO4的含量相比于外层更多。另外,Mn的2p轨道出现峰位于641.55 eV和653.78 eV的特征峰 (图12) ,与NIST谱库对比发现,它们分别对应MnO2和MnO的Mn 2p3/2和2p1/2的峰,并且在646 eV的位置可以看到有卫星峰的存在,该峰仅存在于MnO中,在Mn2O3和MnO2中则不会出现[25]。锰氧化物峰的存在说明材料制备过程中部分KMnO4与硬脂酸发生反应,缓释后MnO峰的减弱,推测可能是由于材料结构的分解。
-
从FTIR和XRD的图谱分析中可以看到乙酸钾和酯类物质,硬脂酸中具有羧基基团,而醇羟基在原料中仅存在于PEG-4000中,因此推测PEG-4000在制备中可能发生了反应,据DAVID FRIEDL等[26]的相关报道,在具备氧化条件的情况下,聚乙二醇等表面活性剂会发生自氧化反应,聚合物的氧化通常为自由基链式反应,该机理表明PEG-4000的氧化过程可能产生了乙二醇、乙酸和其他低聚物,这些产物和硬脂酸、KMnO4之间的共同作用促使硬脂酸能够包裹在KMnO4表面,并以化学键合的方式以及硬脂酸的胶结作用共同形成微胶囊结构。通过表征结果判断PP@SA中所含的物质种类,并结合缓释动力学对样品结构的分析,推测油相分离法所制备缓释KMnO4过程中的成囊机理如图13所示,该过程大致包括以下过程:水浴加热至硬脂酸溶解后,加入KMnO4并混匀,加入PEG-4000并溶解混匀,PEG-4000作为表面活性剂分散在KMnO4颗粒周围;降低温度,部分KMnO4与PEG-4000发生氧化分解反应,其分解产物与硬脂酸发生酯化等反应,使得硬脂酸围绕KMnO4颗粒形成包膜;随着温度降低、溶剂挥发,硬脂酸逐渐单独析出,并将KMnO4包裹,此时整个体系呈粘稠状,质地较软,期间不断搅拌保持KMnO4的均匀分布,直至硬脂酸自身温度降低并硬化,放入40 ℃烘箱防止硬脂酸熔融的同时挥发剩余溶剂得到最终成品。
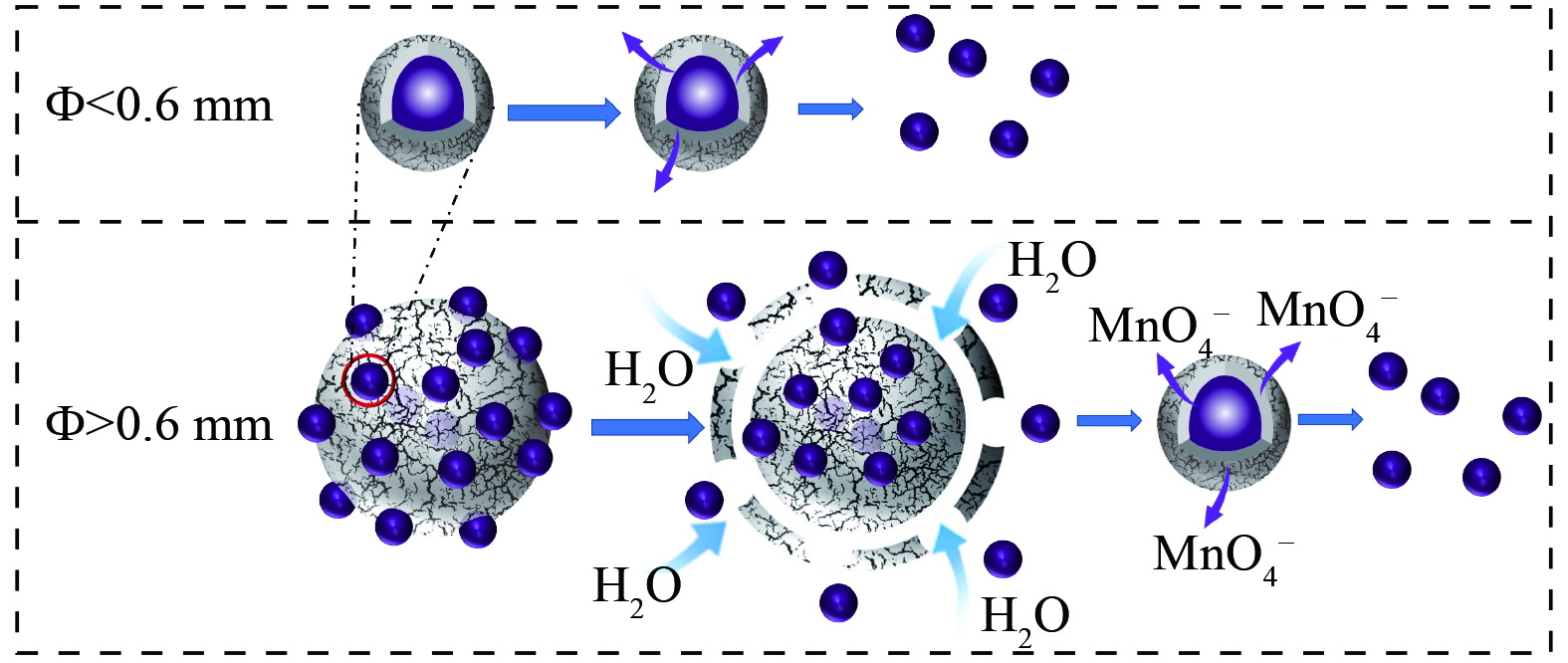
综合对不同芯壳比和不同粒径的PP@SA的缓释动力学的分析,并结合材料SEM图像推断PP@SA在的释放机理,如图14所示。粒径小于0.6 mm的颗粒直接通过硬脂酸的疏水性包膜结构溶出。而粒径大于0.6 mm的颗粒由通过硬脂酸胶结在一起的小粒径颗粒构成,含有硬脂酸骨架结构,其释放过程包括以下几个阶段:首先,颗粒最外表面的KMnO4溶解,使得硬脂酸骨架结构出现缺口,开始溶蚀并逐渐毁坏,大颗粒分解为多个小颗粒;随着小颗粒硬脂酸壳层慢慢剥落,其内部KMnO4能缓慢释放出来。因此粒径较大的缓释颗粒能够持续更长的缓释时间。当大颗粒的硬脂酸骨架结构和小颗粒的硬脂酸包膜结构完全破坏时,KMnO4能全部溶出,然而仍有小部分颗粒由于硬脂酸紧密包封而导致部分KMnO4难以在实验观测期间释放出来。
-
为了探究KMnO4在土壤环境中的传质特点,以及PP@SA在动态缓释体系中的释放行为,土柱实验设将缓释氧化材料和土壤分开放置,该过程为KMnO4先释放,再在土壤层中进行运移,最后测出水中KMnO4的浓度。由于实际土壤基质成分复杂,还原性物质和其他土壤有机质会消耗KMnO4,因此,为了更好地反映其在运移过程中的变化情况,采用石英砂 (Quartz sand) 替换土壤层作为对照组。同时为了使石英砂在渗透系数上和粘土的性质更为接近,可以更好地进行对比,根据石英砂不同粒径级别和渗透系数的对应关系和粘土渗透系数的经验值,选择120目石英砂作为土壤层的对比层。
在土柱缓释实验过程中,采用定水头供水,出水为自由出水,每次收集12 h的出水,直至出水中检测不到KMnO4,图15为3个土柱出水中KMnO4的浓度和释放百分比。初始的出水中KMnO4的浓度相对较低,并且石英砂柱在前5 d的时间内出水的KMnO4浓度始终高于土柱,在5 d之后,石英砂柱的出水浓度开始低于土柱,可能是由于石英砂颗粒较土壤颗粒大,柱体本身孔隙较为发达,水分渗透速率较快,所以在缓释氧化材料层KMnO4的释放能够始终以更大的浓度梯度进行释放,而在土柱中,缓释氧化材料层由于水分的渗透速率慢,使得KMnO4释放后向下迁移的速率较慢,使缓释层的KMnO4浓度保持在一个较高的水平,限制了KMnO4的持续释放。此外,KMnO4用于污染修复的常用浓度范围最低可以低至0.25 g∙L−1,最高可以达到饱和浓度63 g∙L−1[27-31],土柱缓释实验结果显示KMnO4在淤泥质粘土和粉质粘土中的扩散浓度保持在0.25 g∙L−1以上的天数分别达到7 d和9 d,表明该缓释氧化材料在模拟土壤中的持续性释放效果较好。通过对缓释KMnO4在不同土壤条件下的释放差异进行研究,可以更好地指导污染土壤的修复工作,选择合适的缓释氧化材料,并为相关领域的实际应用提供了理论支持。
-
通过制备包覆型缓释氧化剂PP@SA,本研究成功实现了KMnO4的缓慢释放,从而提高了原位化学氧化技术修复有机污染土壤的效率和可行性。同时揭示了PP@SA的制备条件和释放机制,为进一步优化和改进该技术提供了重要的理论依据。
1) 在最佳制备方法方面,芯壳比为1∶3,超声时间为15min,搅拌速度为300 r∙min−1,粒径范围为1.12~4.00 mm时,能够获得最佳的缓释效果。
2) PP@SA的释放机制与芯壳比和粒径有关,当芯壳比为1∶1,粒径小于0.06 mm时,缓释颗粒的释放机制符合单一的Fickian扩散模式,随着硬脂酸用量和颗粒粒径的增加,释放机制逐渐以骨架溶蚀机制为主导。
3) 与纯KMnO4相比,PP@SA在土壤中具有更好的缓释效果,这将为实际的土壤修复工作提供重要的理论依据。
包覆型缓释氧化剂的制备及其缓释性能评价
Preparation of coated slow-release oxidants and evaluation of their slow-release performance
-
摘要: 在低渗层非水相液体污染物的原位化学氧化修复过程中,土壤有机质会消耗氧化剂,导致氧化剂的有效浓度难以长期维持,从而使污染物浓度容易反弹。为了解决该问题采用硬脂酸作为壳材,通过相分离法包覆高锰酸钾制备缓释氧化材料。结果表明,硬脂酸包覆型高锰酸钾 (PP@SA) 具有良好的缓释性能,可以持续缓释120 h以上。正交实验结果显示,当芯壳比为1∶3,超声时间为15 min,搅拌速度为300 r∙min−1,粒径为1.12~4.00 mm时缓释效果最佳。并且,随着PP@SA的粒径和硬脂酸用量的增大,MnO4-的缓释机制从Fickian扩散模式向骨架溶蚀和Fickian扩散混合释放机制转变,其中骨架溶蚀释放机制逐渐起主导作用。此外,土相缓释柱实验结果表明,PP@SA在淤泥质粘土和粉质粘土两种低渗土壤中可以保持高锰酸根离子的扩散浓度在0.25 g∙L−1以上的时间分别为7和9 d,其缓释寿命远超过原高锰酸钾。Abstract: During the in-situ chemical oxidation remediation process of low-permeability non-aqueous phase liquid, the consumption of oxidants by soil organic matter is inevitable. This makes it difficult to maintain the effective concentration of oxidants in the long term, and the concentration of pollutants is prone to rebound. This study managed to prepare sustained-release oxidation materials using the phase separation method, with stearic acid as the wall material and KMnO4 as the core material. Results showed that KMnO4 coated with stearic acid (PP@SA) had good sustained-release performance, which could last for more than 120 h. The orthogonal experimental results indicated that the optimal sustained-release lifespan was achieved when the core-shell ratio was 1∶3, ultrasound time was 15 min, stirring speed was 300 r∙min−1, and the particle size ranged from 1.12 to 4.00 mm. As the particle size of PP@SA and the amount of stearic acid increased, the sustained-release mechanism shifted from the Fickian diffusion to a mixed release mechanism involving both skeleton dissolution and Fickian diffusion. Moreover, in two low permeability soils, chalky clay and silty clay, PP@SA maintained a diffusion concentration of MnO4- above 0.25 g∙ L−1 for 7 and 9 d, respectively. Its sustained release lifetime was significantly longer than that of the original potassium permanganate.
-
Key words:
- in-situ chemical oxidation /
- sustained-release /
- KMnO4 /
- low permeability soil
-
随着工业的发展,石油被广泛应用于各行各业,但其带来的环境污染问题不容忽视,尤其是泄漏等生产事故诱发的土壤污染,原位化学氧化 (ISCO) 是土壤有机污染快速去除的有效方法,因具有修复效能高、成本低及操作便利等优势,常用于石油烃污染土壤修复[1-6]。近年来高锰酸钾 (KMnO4) 对石油烃的ISCO被认为是H2O2、O3/H2O2或催化H2O2氧化的良好替代品,因为其易于处理、成本低、pH范围适应性强且氧化电位 (E0=1.51 V) 相对较高[7]。然而,传统的ISCO技术在修复过程中面临着一系列挑战,特别是氧化剂的有效浓度低,难以维持长效性的问题,这使得污染物容易出现浓度反弹。
为了解决这些问题,已有报道提出了利用微囊化技术制备可控缓释材料来替代传统反应试剂,两者的主要区别在于控释材料延长了反应试剂的释放时间,降低反应物的释放速率,即控释材料具有持续释放的特点[8-10]。目前,微胶囊技术在缓释氧化材料研发领域已经取得初步的进展[11-12]。例如,有研究通过离子交联反应,将过氧化钙封装在海藻酸钙聚合物中,形成过氧化钙的释氧凝珠,为环境中的好氧微生物提供氧源用于降解1,4二恶烷,释氧凝珠可以在较长的时间内 (7 d左右) 使环境中的溶解氧保持较高的水平,并在10 d内将污染物去除近96%[13]。KAMBHU等[14]通过使用石蜡包裹KMnO4,使用模具制备了柱状KMnO4缓释材料,通过埋入表层土壤的方式对土壤多环芳烃进行降解,但是需要定期挖出去除表面石蜡,并进行再次填埋。从目前关于场地修复缓释氧化材料的研发和应用方面来看,缓释和控释技术在越来越多的研究和修复方案中得到了应用[15-16]。这些技术能够有效控制修复活性物质的释放,减少修复药剂的非选择性消耗,并提高污染物的去除效率,有望解决污染场地原位修复的长期有效性差的问题[17-19]。然而,目前所研发的控释材料缓释时长普遍处于24 h之内[19-21],其氧化活性难以维持更久,这对低渗透地层非水相污染物的原位修复功能材料提出了更大挑战。因此,需要进一步研究和开发适用于土壤修复的缓释氧化材料包覆方法,在选择包覆材料时,需要考虑其渗透性和降解性,以确保修复活性物质能够有效释放到土壤中。
本研究以硬脂酸为壳材,采用油相分离法制备了包覆型KMnO4,并通过正交实验探究最佳制备条件和实验参数。通过水相缓释实验探究缓释氧化材料的释放动力学,并根据缓释曲线拟合缓释材料的释药模型,结合表征结果探究该材料的释放机理。设计制作土柱动态淋洗实验,探究缓释氧化材料在模拟土柱中的缓释作用及其对柴油烃污染土壤的修复效果,为其在实际修复中的应用提供理论依据。
1. 材料与方法
1.1 实验原料
高锰酸钾 (KMnO4) 、硬脂酸 (C18H36O2) 、聚乙二醇4000 (PEG-4000;HO(CH2CH2O)nH) 、无水乙醇 (C2H6O) 、石油醚、正己烷 (C6H14) 均购于国药集团化学试剂有限公司;实验室用水为超纯水。
1.2 缓释材料的制备
采用油相分离法制备缓释KMnO4微胶囊,取适量的壳材硬脂酸 (10、15 g) 于烧杯中,加入乙醇 (100 mL) ,水浴搅拌加热至70~75 ℃将壳材完全溶解后,按照一定的比例加入KMnO4 (5 g) ,进行超声和搅拌使KMnO4分散于壳材溶液中,然后称取适量PEG-4000 (0 、2、3 g) ,逐次少量加入体系中进行助凝以促进微胶囊化,同时降低水浴温度至25~35 ℃ (冰水浴冷却/自然冷却) ,使壳材溶解度降低,并以KMnO4为核心凝聚析出,初步形成包覆型KMnO4,然后放入40 ℃烘箱进行固化 (4 h) ,期间进行适当的搅拌和破碎防止材料凝聚结块,最后将材料进行破碎过筛,按照不同粒径保存待用,即得缓释KMnO4氧化剂 (PP@SA) 。并且通过四因素三水平L9 (34)正交表设计实验探究芯壳比、超声时间、搅拌速度和粒径对材料缓释效果的影响,芯壳比分别为1∶1、1∶2、1∶3;超声时间分别是5、10、15 min;搅拌速度分别是150、300、450 r∙min−1;粒径分别为<0.6、0.6~1.12、1.12~4 mm。
1.3 氧化剂浓度测定
配置0.2000 g∙L−1的KMnO4标准溶液,分别取0.0、2.0、4.0、6.0、8.0和10.0 mL用纯水定容至10 mL,用紫外分光光度计于525 nm波长下测定吸光度并绘制浓度-吸光度标准曲线用于计算KMnO4浓度。将PP@SA材料研磨后称取0.1 g,加入适量纯水并进行超声处理使材料中的活性组分 (KMnO4) 释放并溶解在水中,然后定容至500 mL。从中取1.0 mL溶液定容至10.0 mL用紫外分光光度计测定吸光度并计算KMnO4浓度。此外,PP@SA的包封率E通过式(1)计算。其中PP@SA的实际载药量由载药量测定结果确定,理论载药量即氧化剂在壳材和芯材总含量的占比百分数。
stringUtils.convertMath(!{formula.content}) (1) 1.4 缓释曲线的测定及表征方法
按照缓释材料投加量为1 g∙L−1进行水相缓释实验,将其置于水浴磁力搅拌器中 (25 ℃,100 r∙min−1) ,取样时间设为0.5、1.0、2.0、3.0、3.5、7.5、11.5、23.5、27.5、31.5、46.5、58.5、73.5、97.5、121.5 h,取样后使其通过0.22 μm水系滤膜,取1.0 mL过滤液于10 mL比色管,并定容至10.0 mL用紫外分光光度计检测其吸光度。
采用国标HJ-1068-2019中的比重计法测定土壤的粒径和质地;采用紫外分光光度计进行柴油含量的检测;采用傅里叶红外光谱分析仪 (FTIR,美国赛默飞世尔科技公司) 测定缓释氧化材的官能团变化情况;采用X-射线衍射仪 (XRD,德国布鲁克) 对缓释前后缓释氧化材料中的物相组分进行分析;采用Gemini 300扫描电子显微镜 (SEM,德国卡尔蔡司) 和X射线能谱仪 (EDS,德国卡尔蔡司) 进行分析样品的表观形貌;采用表面张力仪 (DCAT 21,德国DataPhysics) 对其接触角信息进行测定;采用X射线光电子能谱 (XPS,赛默飞世尔ESCALAB 250xi) 对氧化剂中的特征元素的价态和成分含量进行定性和半定量分析。
1.5 土样的采集与处理及模拟土柱缓释实验
本研究土壤样品取自上海市奉贤区某未污染场地的深层土:粉质粘土 (silty clay) 和淤泥质黏土 (chalky clay) 。粉质粘土和淤泥质黏土的pH值分别是7.95和7.8;饱和含水率分别是48%和46%;黏粒占比分别为38%和30%;粉粒占比分别为42%和33%;沙粒占比分别为20%和37%,两种土壤质地为典型的低渗透土壤。
为模拟PP@SA在实际土壤中的释放行为,利用亚克力材质柱进行柱淋洗实验,风干后的土壤使用粉碎机粉碎后过70目筛,石英砂使用前用稀盐酸清洗浸泡1 h并用去离子水冲洗至中性后待用,土柱装填采用干法装柱,自下往上依次按照以下顺序进行装填:10 cm石英砂;10 cm土壤层;1 cm石英砂;5%土壤质量的PP@SA (3.75 g) ;3 cm石英砂。使用蠕动泵进行供水,同时保持稳定的水头压力,并且从底部出水口收集渗滤液。
2. 结果与讨论
2.1 缓释氧化剂的制备及参数优化
通过油相分离法制备的PP@SA在水相中具有较好的缓释效果,其释放时长达到100 h以上,图1为PP@SA材料在投加量为1 g∙L−1时高锰酸根释放曲线。结果发现添加PEG-4000的材料缓释速度较慢,硬脂酸在凝聚析出的过程中,包裹KMnO4颗粒性能较好;提高硬脂酸的用量,可以减缓初始的缓释速率和释放量;并且自然冷却方式所得材料比冰水浴冷却得到的材料释放慢,可能是由于冰水浴加快了硬脂酸的析出速度,使溶液中的硬脂酸不能均匀分散在KMnO4颗粒周围,而是原位直接析出,导致壁材的浪费。此外,当溶剂乙醇的用量为100 mL时,无论冷却方式是哪一种,都会使部分硬脂酸直接从溶液中析出。
 图 1 不同条件下所制备PP@SA的MnO4-释放曲线Figure 1. Effect of different preparation conditions on the sustained release effect of PP@SA
图 1 不同条件下所制备PP@SA的MnO4-释放曲线Figure 1. Effect of different preparation conditions on the sustained release effect of PP@SA进一步优化油相分离法的制备参数,采用环己烷作为溶剂,并且用量减少至和硬脂酸等质量的用量,使用超声和搅拌两种分散方式结合,同时考察材料粒径对缓释效果的影响。通过四因素三水平L9(34)正交表设计实验 (表1) ,探究最佳制备参数,正交试验各因素包括芯壳比、超声时间、搅拌速度和粒径。并分别以包封率和5 d的释放百分比为评价指标,进行制备参数的优化。将所得包封率和5 d释放百分比数据通过IBM SPSS Statistics 26软件进行分析 (表2) ,发现以包封率为评价指标时,影响因素对包封率的影响程度为:超声时间>芯壳比>搅拌速度>粒径。影响最大的因素是超声时间,超声过程不仅会影响KMnO4在体系中的分散,也会增加KMnO4在体系中的消耗,最佳的制备条件为:芯壳比1∶1,超声时间10 min,搅拌速度150 r∙min−1,粒径小于0.6 mm。以5 d的释放百分比为评价指标时,各因素对5 d缓释百分比的影响程度为:芯壳比>搅拌速度>粒径>超声时间。最大影响因素为芯壳比,因为KMnO4的用量越大,硬脂酸对KMnO4颗粒的包裹层越薄,从而加快释放速度;其次是搅拌速度,搅拌速度影响KMnO4在缓释颗粒中的分散;而粒径对缓释的影响主要体现在材料壳层的溶蚀速度和与水接触的表面积,粒径越小溶解速度越快。从缓释情况看,最佳制备条件为芯壳比1∶3,超声时间15 min,搅拌速度300 r∙min−1,粒径1.12~4.00 mm。
表 1 正交实验方案设计 (因素) 及结果Table 1. Design and results of orthogonal test program序号 因素实验 A芯核比 B超声时间/min C搅拌速度/(r∙min−1) D粒径/mm 材料特征及缓释性能 包封率 5 d释放量 1 1-S 1∶1 5 150 <0.6 72.88%±3.2% 101.9% 2 1-M 1∶1 10 300 1.12~4 83.90%±2.00% 77.68% 3 1-L 1∶1 15 450 0.6~1.12 72.37%±8.60% 91.78% 4 2-S 1∶2 15 300 <0.6 58.91%±2.09% 74.23% 5 2-M 1∶2 10 150 0.6~1.12 73.89%±1.29% 68.84% 6 2-L 1∶2 5 450 1.12~4 45.45%±2.25% 82.70% 7 3-S 1∶3 5 300 0.6~1.12 43.60%±6.85% 52.75% 8 3-M 1∶3 10 450 <0.6 69.32%±3.25% 73.78% 9 3-L 1∶3 15 150 1.12~4 67.79%±6.33% 52.84% | Show Table DownLoad:
CSV
表 2 正交试验结果极差分析Table 2. Analysis of extreme differences of orthogonal test results
DownLoad:
CSV
表 2 正交试验结果极差分析Table 2. Analysis of extreme differences of orthogonal test resultsK 包封率指标 释放量指标 A B C D A B C D K1j 229.2 161. 3 214.6 201.1 271.3 237.2 223.5 249.8 K2j 178.3 227.1 186.4 189.9 225.8 220.3 204.7 213.4 K3j 180.7 199.1 187.1 197.1 179.4 218.9 248.3 213.2 Rj 50.9 65.2 28.6 11.3 91.9 18.4 43.6 36.6 注:表中Kij表示第j (j=A芯壳比,B搅拌时间,C搅拌速度,D粒径)列影响因素i (i=1, 2, 3)水平所对应的试验指标的总和。Rj表示第j列影响因素所对应的试验指标的极差。 | Show TableDownLoad:
CSV
2.2 缓释动力学分析
为探究缓释氧化材料的释放机制,将不同粒径材料的释放动力学曲线用一级、Higuchih和Ritger-Peppa模型进行拟合,一级释放模型的拟合度最高,所有样品的R2均在0.960 0以上 (图2) ,所以该模型更好地反映缓释材料中KMnO4的释放,表明氧化剂释放速率与其在环境介质中的量成正比 (随时间按恒定比例释放) 。由于硬脂酸是疏水性材料,对于水不溶性骨架结构可以通过Higuchi方程进行拟合,拟合结果如表3所示,样品1-S、2-S、3-S的拟合结果较差 (R2 < 0.900 0) ,表明粒径小于0.6 mm的材料的释放不符合骨架溶蚀机制。随材料粒径的增加,拟合结果得到改善,R2均大于0.950 0,表明随材料粒径的增大其缓释机制越来越以骨架溶蚀为主。
 图 2 一级释放模型对缓释实验数据拟合结果Figure 2. Fitting results of the first-order release model to the sustained release experimental data表 3 Higuchi和Ritger-Peppas方程对缓释实验数据的拟合结果Table 3. Fitting results of Higuchi and Ritger-Peppas equation for sustained release experimental data
图 2 一级释放模型对缓释实验数据拟合结果Figure 2. Fitting results of the first-order release model to the sustained release experimental data表 3 Higuchi和Ritger-Peppas方程对缓释实验数据的拟合结果Table 3. Fitting results of Higuchi and Ritger-Peppas equation for sustained release experimental data样品方程 1-S 1-M 1-L 2-S 2-M 2-L 3-S 3-M 3-L Higuchi 0.571 9 0.978 1 0.993 0 0.800 8 0.754 2 0.967 2 0.745 7 0.950 8 0.982 2 Ritger-Peppas 0.760 1 0.991 1 0.995 7 0.939 5 0.944 0 0.978 0 0.910 5 0.975 2 0.990 5 一级释放模型 0.986 6 0.986 0 0.975 5 0.989 3 0.988 2 0.997 6 0.963 6 0.996 2 0.992 5 | Show TableDownLoad:
CSV
进一步通过Ritger-Peppas方程对缓释曲线进行了拟合,结果如表3所示,材料1-S、1-M、2-S和3-S符合Fichian定律扩散模式,即在单位时间内通过垂直于扩散方向的单位截面积的扩散物质流量 (即扩散通量Diffusion flux) 与该截面处的浓度梯度成正比。粒径小于0.6 mm的材料都符合Fickian扩散模式,可能是由于粒径较小的材料不存在骨架结构所以极易被破坏,而样品1-M的芯壳比为1∶1,硬脂酸对KMnO4的包裹效果可能相对较差,因此不存在骨架溶蚀的缓释机制。此外,其他材料的扩散指数n均在0.43~0.89之间,表明其缓释机制是溶解扩散和骨架溶蚀扩散两种机制共存的释放方式。随着粒径的增加,扩散系数n值呈现增大的趋势,表明随粒径的增加,硬脂酸对KMnO4的包封效果越好,KMnO4的释放机制逐渐由小粒径中的单一Fickian扩散模式开始转为骨架溶蚀扩散和Fickian扩散模式共存的溶解扩散方式。
2.3 材料表征分析
不同芯壳比制备条件下的最小粒径材料 (即样品1-S、2-S、3-S) 的扫描电镜图如图3(a)所示。当芯壳比为1∶1时,材料表面相对比较平整,几乎没有骨架结构;芯壳比为1∶2时,材料表面变粗糙,出现较为明显的孔道结构;芯壳比为1∶3时,材料颗粒有明显的孔道结构和骨架结构。随着硬脂酸用量的增加,PP@SA的结构中骨架结构也越发达,其水相缓释机制也从单一的边界层Fickian扩散向以骨架溶蚀为主导的扩散模式转变,这与2.2部分动力学分析推测的材料结构一致。对水相缓释后的材料同样进行了SEM电镜分析 (图3(b)) ,芯壳比为1∶1的材料缓释后呈现片状结构,可能是材料结构破坏后剥落下的硬脂酸壳层;芯壳比为1∶2的材料颗粒表面出现溶蚀后的空洞;芯壳比为1∶3的颗粒整体结构比较完整,而骨架结构消失,这可能是因为硬脂酸含量增加,对KMnO4的包裹较严密,虽外层通过溶蚀造成结构破裂,但内层结构难以通过溶蚀作用破坏并释放内层的KMnO4。该结果也解释了在缓释实验中大粒径颗粒在后期仍可以保持较完整的粒状结构,并且释放浓度始终无法达到理论最大值的现象。
 图 3 样品1-S、2-S、3-S释放前后的SEM图像Figure 3. SEM images of samples 1-S, 2-S, and 3-S before and after release
图 3 样品1-S、2-S、3-S释放前后的SEM图像Figure 3. SEM images of samples 1-S, 2-S, and 3-S before and after release通过EDS表征了材料表面的元素分布和C、O、Mn、K的分布状态,该缓释材料中的C元素来源于硬脂酸和PEG-4000。不同芯壳比缓释材料的元素分布图和能量谱图如图4~6所示,芯壳比为1∶1的材料 (图4) ,C和Mn元素都均匀分布在材料表面上,表明该材料的结构不是严格的核壳结构;芯壳比为1∶2时 (图5) ,材料中出现了骨架结构,而Mn元素和C元素仍然是保持均匀分布的状态;芯壳比为1∶3时,由于硬脂酸的用量增加,在材料制备中冷凝阶段,大量的硬脂酸析出导致整个制备体系的搅拌困难,KMnO4分布不够均匀,因此出现部分区域KMnO4堆积的情况,如图6中的高亮区域。从材料的表观形貌和元素分布表明,油相分离法制备工艺能够较好地实现硬脂酸对KMnO4的包裹,但其整体的结构并不是十分规则的核壳结构,而是KMnO4在硬脂酸中形成的固体分散体[22-23]结构。
 图 4 PP@SA-1-S的元素分布图和能量谱图Figure 4. Elemental mapping distribution and EDS spectrum of PP@SA-1-S
图 4 PP@SA-1-S的元素分布图和能量谱图Figure 4. Elemental mapping distribution and EDS spectrum of PP@SA-1-S 图 6 PP@SA-3-S的元素分布图和能量谱图Figure 6. Elemental mapping distribution and EDS spectrum of PP@SA-3-S
图 6 PP@SA-3-S的元素分布图和能量谱图Figure 6. Elemental mapping distribution and EDS spectrum of PP@SA-3-S 图 5 3-20 PP@SA-2-S的元素分布图和能量谱图Figure 5. Elemental mapping distribution and EDS spectrum of PP@SA-2-S
图 5 3-20 PP@SA-2-S的元素分布图和能量谱图Figure 5. Elemental mapping distribution and EDS spectrum of PP@SA-2-S缓释材料的接触角测试结果如图7所示,结果表明该缓释材料接触角均大于120°,并且随着硬脂酸用量的增加,接触角分别达到125.84° (1-S) 、126.55° (2-S) 、132.08° (3-S) ,虽然硬脂酸结构中含有羧基基团,但是长碳链使硬脂酸表现出较强的疏水性,而材料疏水性的增强,能有效减缓材料与水的接触,降低KMnO4的释放速度,材料的亲油属性可以促进土壤环境中与污染物的接触或吸附,提高降解效率。
PP@SA材料水相缓释实验前后的红外光谱图如图8所示,通过对比硬脂酸的标准红外光谱图,2 919.35和2 853.01 cm−1为C-H键的伸缩振动吸收峰,1 701.47 cm−1为C=O键的伸缩振动吸收峰,1 457 cm−1为C-H键的变形振动吸收峰,1 164~1 362 cm−1为C-O键的伸缩振动吸收峰,915 cm−1为O-H键的面外弯曲振动峰,1 537 cm−1和1 349 cm−1处的吸收峰分别为硬脂酸中的羧基 (-COO-) 不对称振动和对称伸缩振动引起的[24],其中样品中的部分特征峰相对于硬脂酸出现了明显的减弱或消失,说明材料中游离状态的硬脂酸含量较低。同时,缓释KMnO4的样品谱图中出现了1 642和1 561 cm−1两个吸收峰,表明制备过程中产生了新的化学键,硬脂酸特征峰的减弱和新特征峰的出现表明,制备过程中硬脂酸和部分KMnO4发生反应。水相缓释后材料的红外光谱图中特征峰1 561 cm−1有较为明显的减弱,表明制备过程中产生的新的化学键在缓释过程中减少或消失,可能是由于在水中发生了溶解,或者是KMnO4溶出后与其发生了氧化还原反应。对于KMnO4的红外光谱图,可以看到在894.7 cm−1的位置有1个特征峰,而该峰在缓释实验之后消失,表明该峰应该为KMnO4中的锰氧键出峰位置。
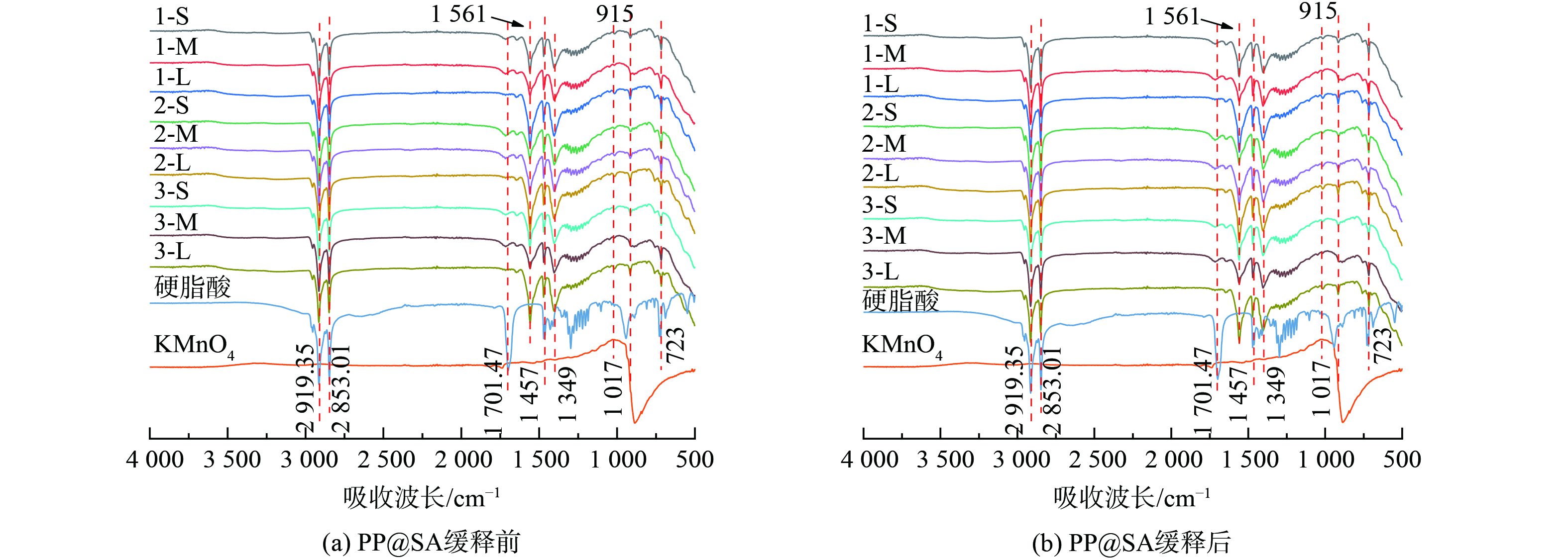 图 8 PP@SA缓释实验前后的红外光谱Figure 8. Infrared spectra of PP@SA before and after the sustained release experiment
图 8 PP@SA缓释实验前后的红外光谱Figure 8. Infrared spectra of PP@SA before and after the sustained release experiment对材料中无机组分采用XRD进一步分析,如图9所示,不同材料的成分差异主要是KMnO4和硬脂酸的比例,表面活性剂PEG-4000的用量较低 (为硬脂酸用量的5%) ,因此通过不同样品的谱图可以看到材料中有机组分和无机组分,通过物相检索主要分析了KMnO4和硬脂酸。在图9(a) 中通过谱图对比可以看到非常明显的KMnO4存在,这是因为在该样品中,KMnO4的含量相对更多,而随着硬脂酸用量的增加,在图9 (c) 中可以明显看到硬脂酸的峰强度的增加,并且粒径对于样品成分的影响较小,主要在于含量的变化,同时也说明材料的制备比较均匀。图10为材料在水相缓释实验之后测得的XRD谱图,所有样品的谱图特征类似,缓释后材料的KMnO4信号强度减弱,硬脂酸的信号更为明显,表明缓释实验之后剩余样品中主要是未溶解的硬脂酸等有机成分。
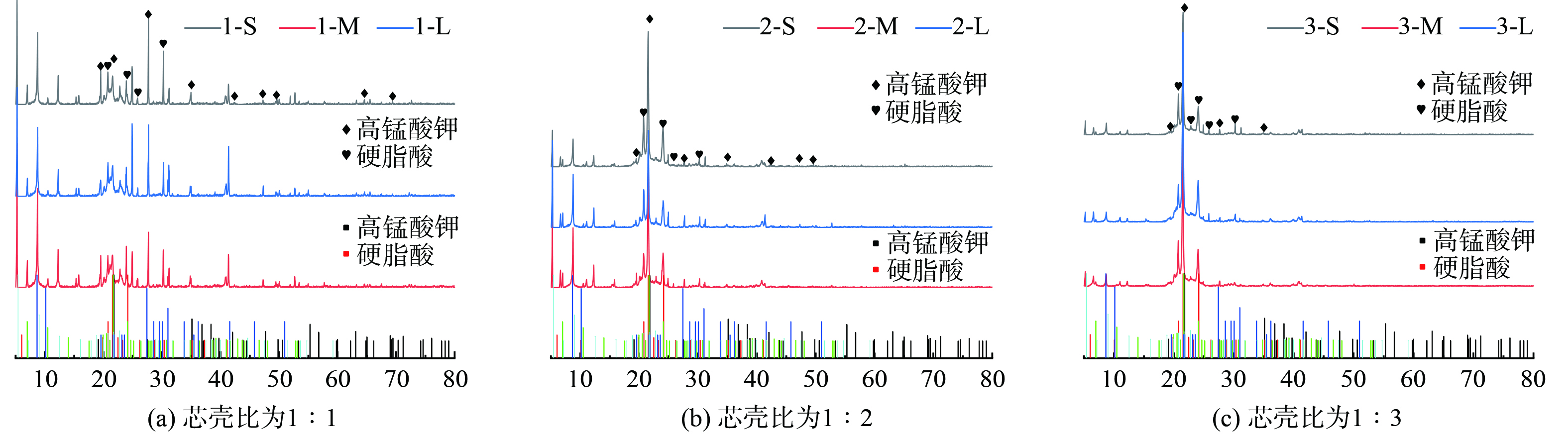 图 9 芯壳比分别为1∶1、1∶2、1∶3时所得材料的XRD图谱Figure 9. The XRD spectra of samples with core-shell ratios of 1∶1, 1∶2, and 1∶3, respectively
图 9 芯壳比分别为1∶1、1∶2、1∶3时所得材料的XRD图谱Figure 9. The XRD spectra of samples with core-shell ratios of 1∶1, 1∶2, and 1∶3, respectively 图 10 缓释实验结束后的样品XRD图谱Figure 10. XRD pattern of the sample at the end of the sustained release experiment
图 10 缓释实验结束后的样品XRD图谱Figure 10. XRD pattern of the sample at the end of the sustained release experiment通过XPS来表征材料表面Mn元素及其化学态,缓释前后材料的XPS图谱如图11所示,Mn的3s轨道出现两个峰 (88.96 eV和83.68 eV) ,分别和KMnO4和Mn2O3、Mn3O4中的Mn的3s轨道峰位相对应,缓释反应后依然能够检测出KMnO4,且图谱峰面积有所上升。结合前文中对PP@SA形貌的分析,推测该材料在制备过程中,硬脂酸会在表面活性剂的作用下优先以KMnO4为内核进行析出,随着溶剂的挥发和反应的进行,逐渐黏结形成更大的颗粒,所以材料内部KMnO4的含量相比于外层更多。另外,Mn的2p轨道出现峰位于641.55 eV和653.78 eV的特征峰 (图12) ,与NIST谱库对比发现,它们分别对应MnO2和MnO的Mn 2p3/2和2p1/2的峰,并且在646 eV的位置可以看到有卫星峰的存在,该峰仅存在于MnO中,在Mn2O3和MnO2中则不会出现[25]。锰氧化物峰的存在说明材料制备过程中部分KMnO4与硬脂酸发生反应,缓释后MnO峰的减弱,推测可能是由于材料结构的分解。
 图 11 PP@SA 样品缓释前后Mn 3s精细谱Figure 11. Fitted spectrum of Mn 3s before and after sustained release of PP@SA samples
图 11 PP@SA 样品缓释前后Mn 3s精细谱Figure 11. Fitted spectrum of Mn 3s before and after sustained release of PP@SA samples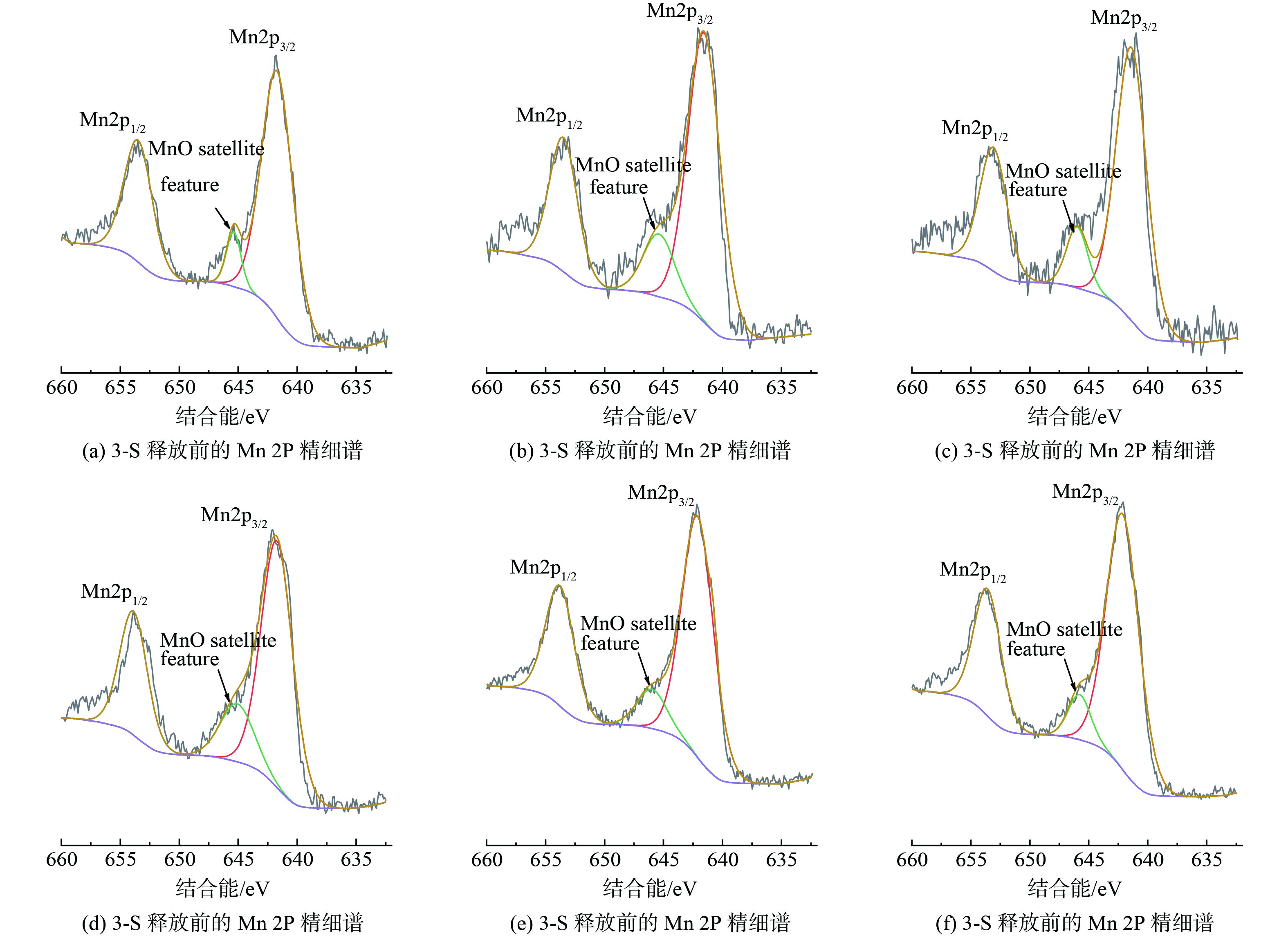 图 12 PP@SA 样品缓释前后Mn 2p精细谱Figure 12. Fitted spectrum of Mn 2p before and after sustained release of PP@SA samples
图 12 PP@SA 样品缓释前后Mn 2p精细谱Figure 12. Fitted spectrum of Mn 2p before and after sustained release of PP@SA samples2.4 材料胶囊化机制和缓释机理分析
从FTIR和XRD的图谱分析中可以看到乙酸钾和酯类物质,硬脂酸中具有羧基基团,而醇羟基在原料中仅存在于PEG-4000中,因此推测PEG-4000在制备中可能发生了反应,据DAVID FRIEDL等[26]的相关报道,在具备氧化条件的情况下,聚乙二醇等表面活性剂会发生自氧化反应,聚合物的氧化通常为自由基链式反应,该机理表明PEG-4000的氧化过程可能产生了乙二醇、乙酸和其他低聚物,这些产物和硬脂酸、KMnO4之间的共同作用促使硬脂酸能够包裹在KMnO4表面,并以化学键合的方式以及硬脂酸的胶结作用共同形成微胶囊结构。通过表征结果判断PP@SA中所含的物质种类,并结合缓释动力学对样品结构的分析,推测油相分离法所制备缓释KMnO4过程中的成囊机理如图13所示,该过程大致包括以下过程:水浴加热至硬脂酸溶解后,加入KMnO4并混匀,加入PEG-4000并溶解混匀,PEG-4000作为表面活性剂分散在KMnO4颗粒周围;降低温度,部分KMnO4与PEG-4000发生氧化分解反应,其分解产物与硬脂酸发生酯化等反应,使得硬脂酸围绕KMnO4颗粒形成包膜;随着温度降低、溶剂挥发,硬脂酸逐渐单独析出,并将KMnO4包裹,此时整个体系呈粘稠状,质地较软,期间不断搅拌保持KMnO4的均匀分布,直至硬脂酸自身温度降低并硬化,放入40 ℃烘箱防止硬脂酸熔融的同时挥发剩余溶剂得到最终成品。
综合对不同芯壳比和不同粒径的PP@SA的缓释动力学的分析,并结合材料SEM图像推断PP@SA在的释放机理,如图14所示。粒径小于0.6 mm的颗粒直接通过硬脂酸的疏水性包膜结构溶出。而粒径大于0.6 mm的颗粒由通过硬脂酸胶结在一起的小粒径颗粒构成,含有硬脂酸骨架结构,其释放过程包括以下几个阶段:首先,颗粒最外表面的KMnO4溶解,使得硬脂酸骨架结构出现缺口,开始溶蚀并逐渐毁坏,大颗粒分解为多个小颗粒;随着小颗粒硬脂酸壳层慢慢剥落,其内部KMnO4能缓慢释放出来。因此粒径较大的缓释颗粒能够持续更长的缓释时间。当大颗粒的硬脂酸骨架结构和小颗粒的硬脂酸包膜结构完全破坏时,KMnO4能全部溶出,然而仍有小部分颗粒由于硬脂酸紧密包封而导致部分KMnO4难以在实验观测期间释放出来。
2.5 缓释高锰酸钾材料在土壤修复中的应用
为了探究KMnO4在土壤环境中的传质特点,以及PP@SA在动态缓释体系中的释放行为,土柱实验设将缓释氧化材料和土壤分开放置,该过程为KMnO4先释放,再在土壤层中进行运移,最后测出水中KMnO4的浓度。由于实际土壤基质成分复杂,还原性物质和其他土壤有机质会消耗KMnO4,因此,为了更好地反映其在运移过程中的变化情况,采用石英砂 (Quartz sand) 替换土壤层作为对照组。同时为了使石英砂在渗透系数上和粘土的性质更为接近,可以更好地进行对比,根据石英砂不同粒径级别和渗透系数的对应关系和粘土渗透系数的经验值,选择120目石英砂作为土壤层的对比层。
在土柱缓释实验过程中,采用定水头供水,出水为自由出水,每次收集12 h的出水,直至出水中检测不到KMnO4,图15为3个土柱出水中KMnO4的浓度和释放百分比。初始的出水中KMnO4的浓度相对较低,并且石英砂柱在前5 d的时间内出水的KMnO4浓度始终高于土柱,在5 d之后,石英砂柱的出水浓度开始低于土柱,可能是由于石英砂颗粒较土壤颗粒大,柱体本身孔隙较为发达,水分渗透速率较快,所以在缓释氧化材料层KMnO4的释放能够始终以更大的浓度梯度进行释放,而在土柱中,缓释氧化材料层由于水分的渗透速率慢,使得KMnO4释放后向下迁移的速率较慢,使缓释层的KMnO4浓度保持在一个较高的水平,限制了KMnO4的持续释放。此外,KMnO4用于污染修复的常用浓度范围最低可以低至0.25 g∙L−1,最高可以达到饱和浓度63 g∙L−1[27-31],土柱缓释实验结果显示KMnO4在淤泥质粘土和粉质粘土中的扩散浓度保持在0.25 g∙L−1以上的天数分别达到7 d和9 d,表明该缓释氧化材料在模拟土壤中的持续性释放效果较好。通过对缓释KMnO4在不同土壤条件下的释放差异进行研究,可以更好地指导污染土壤的修复工作,选择合适的缓释氧化材料,并为相关领域的实际应用提供了理论支持。
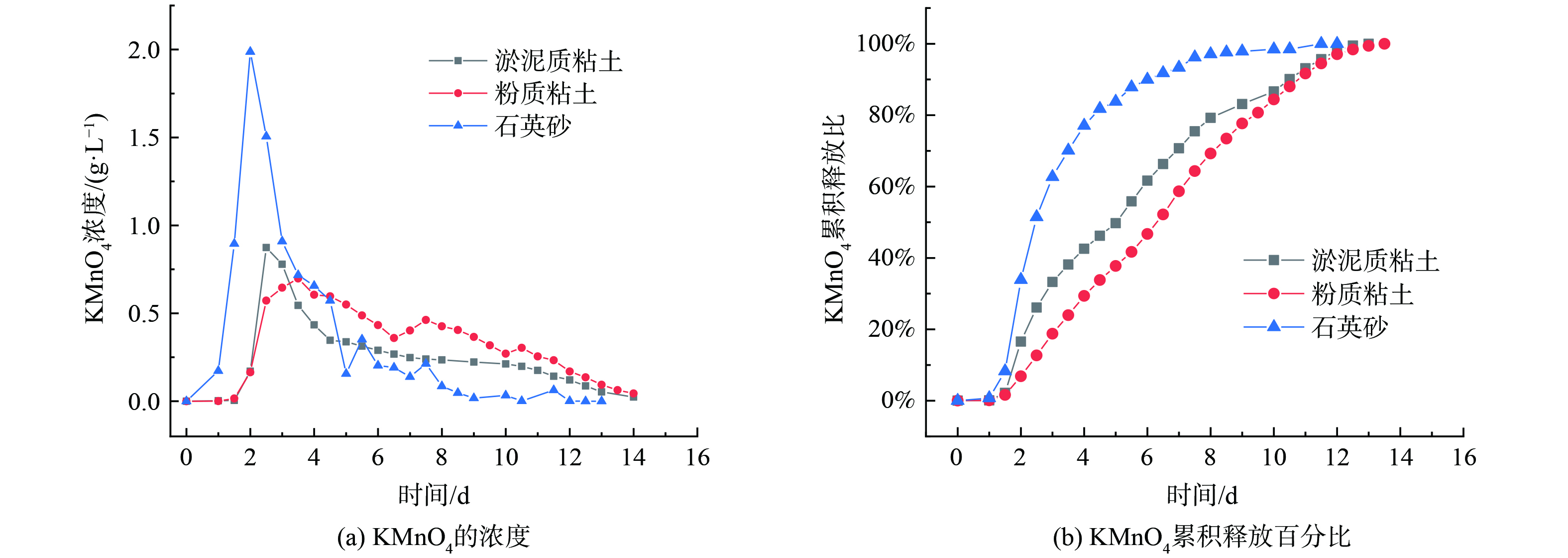 图 15 渗滤液中KMnO4的浓度和累积释放百分比Figure 15. Concentration and percentage release of potassium permanganate in the leachate
图 15 渗滤液中KMnO4的浓度和累积释放百分比Figure 15. Concentration and percentage release of potassium permanganate in the leachate3. 结论
通过制备包覆型缓释氧化剂PP@SA,本研究成功实现了KMnO4的缓慢释放,从而提高了原位化学氧化技术修复有机污染土壤的效率和可行性。同时揭示了PP@SA的制备条件和释放机制,为进一步优化和改进该技术提供了重要的理论依据。
1) 在最佳制备方法方面,芯壳比为1∶3,超声时间为15min,搅拌速度为300 r∙min−1,粒径范围为1.12~4.00 mm时,能够获得最佳的缓释效果。
2) PP@SA的释放机制与芯壳比和粒径有关,当芯壳比为1∶1,粒径小于0.06 mm时,缓释颗粒的释放机制符合单一的Fickian扩散模式,随着硬脂酸用量和颗粒粒径的增加,释放机制逐渐以骨架溶蚀机制为主导。
3) 与纯KMnO4相比,PP@SA在土壤中具有更好的缓释效果,这将为实际的土壤修复工作提供重要的理论依据。
-
图 1 不同条件下所制备PP@SA的MnO4-释放曲线
Figure 1. Effect of different preparation conditions on the sustained release effect of PP@SA
图 2 一级释放模型对缓释实验数据拟合结果
Figure 2. Fitting results of the first-order release model to the sustained release experimental data
图 3 样品1-S、2-S、3-S释放前后的SEM图像
Figure 3. SEM images of samples 1-S, 2-S, and 3-S before and after release
图 4 PP@SA-1-S的元素分布图和能量谱图
Figure 4. Elemental mapping distribution and EDS spectrum of PP@SA-1-S
图 6 PP@SA-3-S的元素分布图和能量谱图
Figure 6. Elemental mapping distribution and EDS spectrum of PP@SA-3-S
图 5 3-20 PP@SA-2-S的元素分布图和能量谱图
Figure 5. Elemental mapping distribution and EDS spectrum of PP@SA-2-S
图 8 PP@SA缓释实验前后的红外光谱
Figure 8. Infrared spectra of PP@SA before and after the sustained release experiment
图 9 芯壳比分别为1∶1、1∶2、1∶3时所得材料的XRD图谱
Figure 9. The XRD spectra of samples with core-shell ratios of 1∶1, 1∶2, and 1∶3, respectively
图 10 缓释实验结束后的样品XRD图谱
Figure 10. XRD pattern of the sample at the end of the sustained release experiment
图 11 PP@SA 样品缓释前后Mn 3s精细谱
Figure 11. Fitted spectrum of Mn 3s before and after sustained release of PP@SA samples
图 12 PP@SA 样品缓释前后Mn 2p精细谱
Figure 12. Fitted spectrum of Mn 2p before and after sustained release of PP@SA samples
图 15 渗滤液中KMnO4的浓度和累积释放百分比
Figure 15. Concentration and percentage release of potassium permanganate in the leachate
表 1 正交实验方案设计 (因素) 及结果
Table 1. Design and results of orthogonal test program
序号 因素实验 A芯核比 B超声时间/min C搅拌速度/(r∙min−1) D粒径/mm 材料特征及缓释性能 包封率 5 d释放量 1 1-S 1∶1 5 150 <0.6 72.88%±3.2% 101.9% 2 1-M 1∶1 10 300 1.12~4 83.90%±2.00% 77.68% 3 1-L 1∶1 15 450 0.6~1.12 72.37%±8.60% 91.78% 4 2-S 1∶2 15 300 <0.6 58.91%±2.09% 74.23% 5 2-M 1∶2 10 150 0.6~1.12 73.89%±1.29% 68.84% 6 2-L 1∶2 5 450 1.12~4 45.45%±2.25% 82.70% 7 3-S 1∶3 5 300 0.6~1.12 43.60%±6.85% 52.75% 8 3-M 1∶3 10 450 <0.6 69.32%±3.25% 73.78% 9 3-L 1∶3 15 150 1.12~4 67.79%±6.33% 52.84%
下载: 导出CSV
表 2 正交试验结果极差分析
Table 2. Analysis of extreme differences of orthogonal test results
K 包封率指标 释放量指标 A B C D A B C D K1j 229.2 161. 3 214.6 201.1 271.3 237.2 223.5 249.8 K2j 178.3 227.1 186.4 189.9 225.8 220.3 204.7 213.4 K3j 180.7 199.1 187.1 197.1 179.4 218.9 248.3 213.2 Rj 50.9 65.2 28.6 11.3 91.9 18.4 43.6 36.6 注:表中Kij表示第j (j=A芯壳比,B搅拌时间,C搅拌速度,D粒径)列影响因素i (i=1, 2, 3)水平所对应的试验指标的总和。Rj表示第j列影响因素所对应的试验指标的极差。
下载: 导出CSV
表 3 Higuchi和Ritger-Peppas方程对缓释实验数据的拟合结果
Table 3. Fitting results of Higuchi and Ritger-Peppas equation for sustained release experimental data
样品方程 1-S 1-M 1-L 2-S 2-M 2-L 3-S 3-M 3-L Higuchi 0.571 9 0.978 1 0.993 0 0.800 8 0.754 2 0.967 2 0.745 7 0.950 8 0.982 2 Ritger-Peppas 0.760 1 0.991 1 0.995 7 0.939 5 0.944 0 0.978 0 0.910 5 0.975 2 0.990 5 一级释放模型 0.986 6 0.986 0 0.975 5 0.989 3 0.988 2 0.997 6 0.963 6 0.996 2 0.992 5
下载: 导出CSV
-
[1] TALVENMäKI H, SAARTAMA N, HAUKKA A, et al. In situ bioremediation of Fenton’s reaction–treated oil spill site, with a soil inoculum, slow release additives, and methyl-β-cyclodextrin[J]. Environmental Science and Pollution Research, 2021, 28(16): 20273-20289. doi: 10.1007/s11356-020-11910-w [2] LOMINCHAR M A, SANTOS A, DE MIGUEL E, et al. Remediation of aged diesel contaminated soil by alkaline activated persulfate[J]. Science of the Total Environment, 2018, 622-623: 41-48. doi: 10.1016/j.scitotenv.2017.11.263 [3] MOUMED I, ARRAR J, NAMANE A, et al. Effects of surfactant and oxidant on bioremediation of contaminated soil by total petroleum hydrocarbons using indigenous bacteria[J]. International Journal of Environmental Science and Technology, 2023, 20(8): 8863-8874. doi: 10.1007/s13762-022-04600-2 [4] ZHOU Q X, SONG C L, WANG P F, et al. Generating dual-active species by triple-atom sites through peroxymonosulfate activation for treating micropollutants in complex water[J]. Proceedings of the National Academy of Sciences of the United States of America, 2023, 120(13): 2300085120. [5] LU S G, ZHANG X, XUE Y F. Application of calcium peroxide in water and soil treatment: A review[J]. Journal of Hazardous Materials, 2017, 337: 163-177. doi: 10.1016/j.jhazmat.2017.04.064 [6] XIE Y H, YANG X N, LI W W, et al. Enhanced removal of glyphosate from aqueous solution by nano-CaO2/AS composite: Oxidation and precipitation[J]. Separation and Purification Technology, 2022, 288: 120349. doi: 10.1016/j.seppur.2021.120349 [7] SUTTON N B, LANGENHOFF A A M, LASSO D H, et al. Recovery of microbial diversity and activity during bioremediation following chemical oxidation of diesel contaminated soils[J]. Applied Microbiology and Biotechnology, 2014, 98(6): 2751-2764. doi: 10.1007/s00253-013-5256-4 [8] AMERHAIDER NUAR N N, MD. JAMIL S N A, LI F, et al. Synthesis of controlled-release calcium peroxide nanoparticles coated with dextran for removal of doxycycline from aqueous system[J]. Polymers, 2022, 14(18): 3866. doi: 10.3390/polym14183866 [9] ZHANG Q F, ZUO M M, LI G H, et al. Synthesis of ammonium persulfate microcapsule with a polyaniline shell and its controlled burst release[J]. Journal of Applied Polymer Science, 2021, 138(3): 49695. doi: 10.1002/app.49695 [10] RASTINFARD A, NAZARPAK M H, MOZTARZADEH F. Controlled chemical synthesis of CaO2 particles coated with polyethylene glycol: characterization of crystallite size and oxygen release kinetics[J]. Rsc Advances, 2018, 8(1): 91-101. doi: 10.1039/C7RA08758F [11] TANG X J, YU C Y, LEI Y Y, et al. A novel chitosan-urea encapsulated material for persulfate slow-release to degrade organic pollutants[J]. Journal of Hazardous Materials, 2022, 426: 128083. doi: 10.1016/j.jhazmat.2021.128083 [12] BUI T H, LEE W, JEON S B, et al. Enhanced Gold(III) adsorption using glutaraldehyde-crosslinked chitosan beads: Effect of crosslinking degree on adsorption selectivity, capacity, and mechanism[J]. Separation and Purification Technology, 2020, 248: 116989. doi: 10.1016/j.seppur.2020.116989 [13] LEE C S, LE THANH T, KIM E J, et al. Fabrication of novel oxygen-releasing alginate beads as an efficient oxygen carrier for the enhancement of aerobic bioremediation of 1, 4-dioxane contaminated groundwater[J]. Bioresource Technology, 2014, 171: 59-65. doi: 10.1016/j.biortech.2014.08.039 [14] CHRISTENSON M, KAMBHU A, REECE J, et al. A five-year performance review of field-scale, slow-release permanganate candles with recommendations for second-generation improvements[J]. Chemosphere, 2016, 150: 239-247. doi: 10.1016/j.chemosphere.2016.01.125 [15] SONG Y, FANG G D, ZHU C Y, et al. Zero-valent iron activated persulfate remediation of polycyclic aromatic hydrocarbon-contaminated soils: An in situ pilot-scale study[J]. Chemical Engineering Journal, 2019, 355: 65-75. doi: 10.1016/j.cej.2018.08.126 [16] MEDINA R, GARA P M D, FERNáNDEZ-GONZáLEZ A J, et al. Remediation of a soil chronically contaminated with hydrocarbons through persulfate oxidation and bioremediation[J]. Science of the Total Environment, 2018, 618: 518-530. doi: 10.1016/j.scitotenv.2017.10.326 [17] FOOLADI M, MOOGOUEI R, JOZI S A, et al. Phytoremediation of BTEX from indoor air by Hyrcanian plants[J]. Environmental Health Engineering and Management Journal, 2019, 6(4): 233-240. doi: 10.15171/EHEM.2019.26 [18] SHEN H F, SHAO Z W, ZHAO Q F, et al. Facile synthesis of novel three-dimensional Bi2S3 nanocrystals capped by polyvinyl pyrrolidone to enhance photocatalytic properties under visible light[J]. Journal of Colloid and Interface Science, 2020, 573: 115-122. doi: 10.1016/j.jcis.2020.03.111 [19] CHEN J, MA H, LUO H, et al. Influencing factors and controlled release kinetics of H2O2 from PVP-coated calcium peroxide NPs for groundwater remediation[J]. Journal of Hazardous Materials, 2023, 464: 132902. [20] YUAN X H, YU S T, XUE N D, et al. Persulfate activation with sodium alginate/sulfide coated iron nanoparticles for degradation of tetrabromobisphenol a in soil[J]. Environmental Research, 2023, 221: 114820. doi: 10.1016/j.envres.2022.114820 [21] TANG X J, LI Z W, WANG Z, et al. Efficient remediation of PAHs contaminated site soil using the novel slow-release oxidant material[J]. Chemical Engineering Journal, 2023, 472: 144713. doi: 10.1016/j.cej.2023.144713 [22] KANSWAMI N, REDDY R A, LAKSHMI P K. Stable solid dispersion incorporated sustained release oral gel of 23 mg donepezil HCl for the treatment of alzheimer disease[J]. International Journal of Life Science and Pharma Research, 2020: 6: 36-42. [23] CORVIS Y, NéGRIER P, ESPEAU P. Physicochemical stability of solid dispersions of enantiomeric or racemic ibuprofen in stearic acid[J]. Journal of Pharmaceutical Sciences, 2011, 100(12): 5235-5243. doi: 10.1002/jps.22727 [24] HARIKRISHNAN S, MAGESH S, KALAISELVAM S. Preparation and thermal energy storage behaviour of stearic acid-TiO2 nanofluids as a phase change material for solar heating systems[J]. Thermochimica Acta, 2013, 565: 137-145. doi: 10.1016/j.tca.2013.05.001 [25] BIESINGER M C, PAYNE B P, GROSVENOR A P, et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni[J]. Applied Surface Science, 2011, 257(7): 2717-2730. doi: 10.1016/j.apsusc.2010.10.051 [26] DAVID FRIEDL J, WIBEL R, BURCU AKKUş-DAğDEVIREN Z, et al. Reactive oxygen species (ROS) in colloidal systems: Are “PEG-free” surfactants the answer[J]. Journal of Colloid and Interface Science, 2022, 616: 571-583. doi: 10.1016/j.jcis.2022.02.092 [27] ZHANG M Y, DONG Y, GAO S, et al. Effective stabilization and distribution of emulsified nanoscale zero-valent iron by xanthan for enhanced nitrobenzene removal[J]. Chemosphere, 2019, 223: 375-382. doi: 10.1016/j.chemosphere.2019.02.099 [28] XU Q, CHEN J J, SONG X R. Assessment of the rheological behavior of polymer-oxidant mixtures and the influence of the groundwater environment on their properties[J]. Water, 2019, 11(8): 1698. doi: 10.3390/w11081698 [29] BOULANGé M, LORGEOUX C, BIACHE C, et al. Fenton-like and potassium permanganate oxidations of PAH-contaminated soils: Impact of oxidant doses on PAH and polar PAC (polycyclic aromatic compound) behavior[J]. Chemosphere, 2019, 224: 437-444. doi: 10.1016/j.chemosphere.2019.02.108 [30] LIU D F, REN L M, WEN C Y, et al. Investigation of the compatibility of xanthan gum (XG) and calcium polysulfide and the rheological properties of XG solutions[J]. Environmental Technology, 2018, 39(5): 607-615. doi: 10.1080/09593330.2017.1309073 [31] LIU Y S, CHEN J J, WANG Q W, et al. The principle and effect of transfer agent for the removal of PCE during in situ chemical oxidation[J]. Environmental Science and Pollution Research, 2017, 24(26): 21011-21023. doi: 10.1007/s11356-017-9411-9 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1508
- HTML全文浏览数: 1508
- PDF下载数: 85
- 施引文献: 0
