

 下载:
下载:
-
砷是一种剧毒的类金属,与镉、铬、铅、汞并称为土壤重金属污染的“五毒”,具有生物积累效应和“三致” (致癌、致畸、致突变) 作用,长期的砷暴露或通过饮用水、食物链过量摄入砷会严重损害人体健康[1-2]。砷在土壤中主要来源于含砷矿物风化、大气沉降、微生物分解以及工农业污染,其中采矿、冶炼和工农业生产排放是砷污染最主要的来源[3]。据2014年《全国土壤污染状况调查公报》显示,我国土壤砷污染的点位超标率高达2.7%,已对生态环境和人类健康构成严重威胁[4]。
砷在土壤中通常以亚砷酸盐 (三价) 和砷酸盐 (五价) 形式存在[5]。与五价砷相比,三价砷在土壤中不易被吸附或生成沉淀,因此迁移性更强,且其毒性约为五价砷的25~60倍[6]。铁盐作为经济实用的稳定化药剂,常被用于砷污染土稳定化修复中,铁盐或溶解后生成的铁氧化物会与砷发生吸附-沉淀反应,具有较高的砷结合能力[7-8],因为其对三价和五价砷都有较好的稳定化效果,所以目前用铁盐处理砷污染土时一般不区分砷的原始价态。BAGHERIFAM等[9]研究了添加量为5%的铁氧化物对砷的质量分数为50.0 mg·kg−1污染土的稳定化效果,发现铁氧化物可将生物有效性砷的质量分数从40.8 mg·L−1降至12.6 mg·L−1,并降低植物对砷的吸收;ZHAO等[10]向2 117 mg·kg−1的砷污染土中加入3倍铁砷比的FeCl3进行稳定化修复,使其浸出浓度降至17.9 mg·L−1;但发现过量的铁盐 (水解呈酸性) 引起土壤酸化。修复不彻底或修复后土壤酸化都不利于土地资源化利用,因此科学的修复方法应当区分砷的价态,将三价砷氧化为易被稳定化的五价砷进而提高修复效率,且在一定程度上减少铁盐用量。
常见的氧化剂有臭氧、高锰酸钾、过硫酸钠、过氧化钙和芬顿试剂等[11],其中芬顿试剂能够生成强氧化性的羟基自由基 (·OH) ,·OH拥有很高的氧化还原电位以及电负性,此外还有环境友好、反应速率快、可操作性强和可控等优点,目前已被广泛用于有机污染和砷污染废水治理[12]。KATSOYIANNIS等[13]研究发现,芬顿试剂可快速将废水中的三价砷氧化为五价砷,且处理后环境友好;JAAFARZADEH等[14]通过使用芬顿试剂预处理含三价砷的废水,发现芬顿试剂预处理能有效提高火山灰对砷的吸附;QIN等[15]通过研究天然雨水中微量的过氧化氢及其产生的芬顿效应对无机砷和有机砷的修复效果,发现芬顿效应一定程度上减少了水稻中的砷累积和毒害效应。类似于污水处理,在砷污染土的修复中,采用芬顿试剂将三价砷氧化为五价,将显著提高后续铁盐对于砷的固定效果[16],然而目前在砷污染土修复中却未见有使用芬顿试剂进行氧化的研究。
基于不同价态砷的迁移性和毒性差异,本研究提出先使用芬顿试剂将土壤中的三价砷氧化为毒性低、弱迁移性的五价砷,再施加氯化铁稳定化五价砷的协同修复方法。对总砷的质量分数为1 500 mg·kg−1 (其中三价砷为1 330 mg·kg−1) 的污染土进行芬顿-氯化铁协同修复试验,通过合成沉降淋滤试验 (SPLP) 、生物可利用性验证修复效果;同时,通过pH测试、连续萃取试验、光谱分析研究修复前后土壤的矿物成分、砷的赋存形态和价态,以期揭示其协同修复机理。
-
为了保证平行试验中各试样砷的质量分数相同,通过向未污染的土壤中添加亚砷酸钠 (NaAsO2) 溶液的方法人工配制砷污染土。试验原土取自广东汕头某工地,土壤风干后过2 mm筛,原土的pH为7.45,为砂质土,其中细砂粒组含量占55.5%,中砂占25.5%,强酸消解后用原子吸收法 (ICP-OES) 测得土中金属的质量分数分别为:铝 67 700 mg·kg−1、铁 13 100 mg·kg−1、钙 9 650 mg·kg−1、镁 2 470 mg·kg−1、锰 679 mg·kg−1、砷 8.90 mg·kg−1。
向试验原土中加入定容后浓度为0.132 mol·L−1的NaAsO2溶液,制备砷的质量分数为1 500 mg·kg−1的砷污染土,充分搅拌后密封焖土 (老化) 120 d[17]。老化后测得污染土pH为7.76,三价砷的质量分数为1 330±20 mg·kg−1,说明制样过程中小部分三价砷被 (土壤中铁锰氧化物或氧气) 氧化成五价[18]。本试验所用溶液均用去离子水配制,所用试剂均为分析纯。
-
针对前述污染土壤,本研究单独使用氯化铁进行稳定化修复与芬顿-氯化铁协同修复进行了对比,另外单独使用芬顿进行了土壤氧化处理,以便揭示相关的效果和机理。
所用芬顿试剂按照H2O2∶Fe2+ (摩尔比) 10∶1配制,同时加入稀硫酸调节pH为5,以促进更多·OH产生从而提高氧化能力[19]。单独使用芬顿试剂氧化处理污染土时,芬顿试剂添加量 (按未处理污染土的干重计算) 为1%~5%,加入去离子水使固液比为1∶1,充分混合后静置24 h,使氧化完成 (后续分析表明24 h已足够)。
单独使用氯化铁稳定化修复时,向污染土中加入不同添加量 (铁砷摩尔比1~6) 的氯化铁,然后加入离子水,控制固液比为1∶1并混合均匀,静置3 d。
芬顿-氯化铁协同修复时,芬顿试剂添加量为1%,按上述氧化处理步骤操作后,再加入铁砷比1~6的氯化铁,按上述稳定化步骤操作。每组试验重复3次,结果取平均值。
-
本研究采用SPLP评价污染土修复后的浸出毒性,试验参照《固体废物 浸出毒性浸出方法 硫酸硝酸法》 (HJ/T299-2007) ,采用硫酸与硝酸的混合溶液 (质量比6∶4;pH=3.20±0.05) 作为浸提液,用于模拟酸雨淋滤条件下砷在土壤中的浸出[20]。操作步骤为:修复后的污染土破碎后过9.5 mm筛,取50 g置于锥形瓶中,加入500 mL浸提液充分混合后,于翻转振荡器 (FZ-12,常州市亿能实验仪器厂) 中以 (30±2) r·min−1振荡18 h,静置30 min后过0.45 um微孔滤膜,使用ICP-OES测定滤液中砷浓度。
为了评价修复后砷污染土对环境中动植物的毒性,进行了生物有效性试验,提取剂为0.05 mol·L−1的碳酸氢钠 (NaHCO3) 溶液[21]。具体操作为:取1 g修复后的污染土置于50 mL离心管中,按固液比1∶10与提取剂混合后,于水浴恒温振荡器 (HSY-B,常州金坛精达仪器制造有限公司) 中以200 r·min−1振荡2 h,然后于台式离心机 (TDL-5,常州中捷实验制造有限公司) 中以4 000 r·min−1离心10 min,过0.45 um滤膜后测定上清液中砷的浓度。
土壤pH测试操作步骤为:取10 g过2 mm筛的土壤置于锥形瓶中,加入去离子水使固液比为1∶5,在180 r·min−1下水平振荡3 min,静置30 min后用pH计 (FE28-Standard,瑞士METTLER TOLEDO公司) 测定上清液pH。
为了揭示修复前后砷在土壤中的形态稳定性,进行分步萃取试验分析砷的赋存形态。参照Wenzel的连续萃取法[22],按每步采用的萃取剂,将提取的重金属形态分为5种,分别是非专性吸附态 (F1) 、专性吸附态 (F2) 、无定型铁铝氧化物结合 (F3) 、晶质铁铝氧化物结合态 (F4) 和残渣态 (F5) ,从F1到F5生物可利用性逐渐降低,即稳定性逐渐升高。
采用X射线衍射 (XRD,DX-2700,丹东浩元仪器有限公司) 对土壤中矿物进行晶型测试,使用Cu Kɑ射线以2θ=10°·min−1的速度从2θ=10°扫描至2θ=80°,通过High Score Plus v3.0软件分析矿物晶体。采用X射线光电子能谱仪 (XPS,Axis-Ultra DLD,英国Kratos公司) 对砷在土壤中的价态进行分析,以Al Kɑ射线作为激发源,测试电压为12 kV,测试电流为6 mA,以C 1s=284.8 eV作为标准结合能进行校对,通过XPSPEAK41软件进行曲线拟合后,根据结合能峰值确定成分。
-
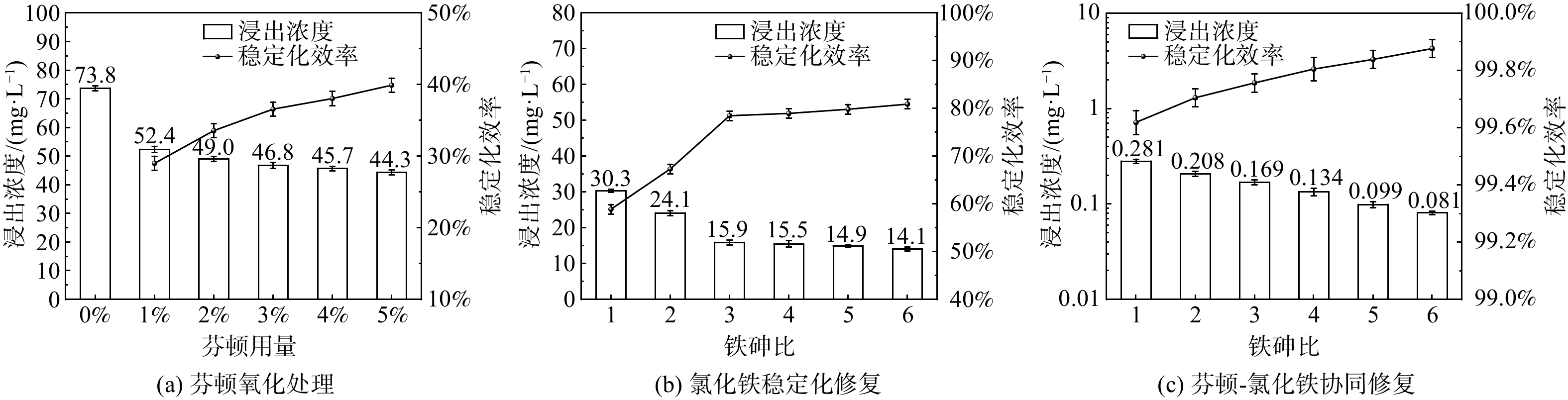
各修复方法对应的SPLP浸出浓度如图1所示。图1(a)中未经处理的污染土 (CK) 砷浸出浓度为73.8 mg·L−1。使用1%芬顿氧化处理后浸出浓度降至52.4 mg·L−1,说明氧化处理有一定效果但效果有限;随芬顿用量由1%增至5%,浸出浓度略有降低。因此,芬顿氧化处理主要改变了砷的价态,降低浸出浓度的效果非常有限。
使用氯化铁进行稳定化处理后 (图1(b)) ,浸出浓度随铁盐用量增加而降低,但铁砷比超过3以后降低不明显,且铁砷比为5时浸出浓度仍有14.1 mg·L−1,稳定化效率仅为79.8%,说明仅增加氯化铁用量不能达到预期修复效果。
使用1%芬顿氧化后,再用氯化铁进行协同修复,浸出浓度显著降低 (图1(c)) ,铁砷比为1时浸出浓度降至0.281 mg·L−1,已满足低于《污水排入城镇下水道水质标准》CJ343-2010)中规定的0.3 mg·L−1[23],稳定化效率高达99.6%。在此情况下,继续增加铁盐用量虽然会减少浸出浓度,但已超过实际需要。综上可知,芬顿氧化大幅提高了后续稳定化处理效果,也可在达到某浸出浓度标准的前提下一定程度上减少铁盐用量。
-
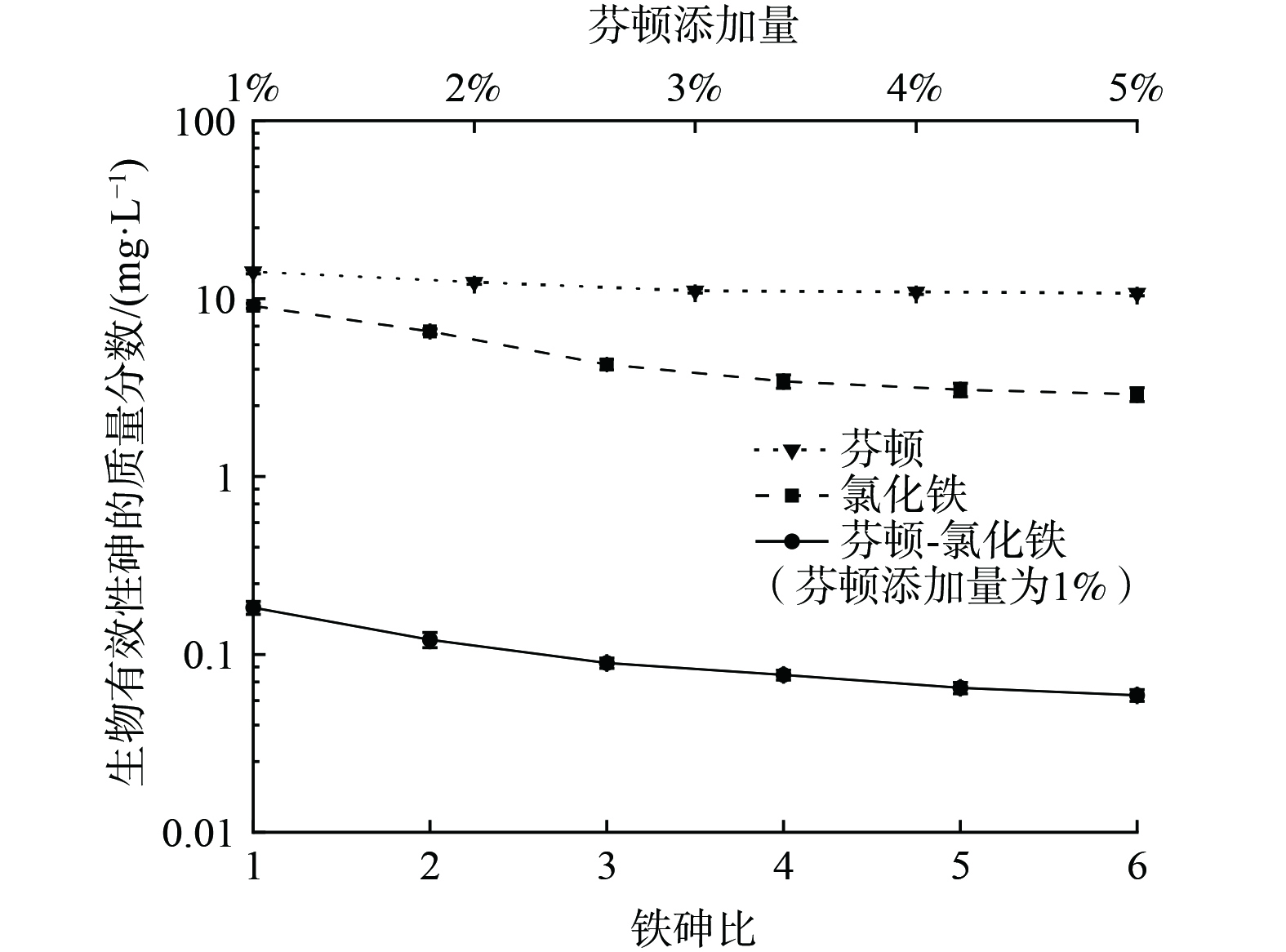
生物有效性是衡土壤中污染物的迁移性以及对生态环境影响的重要指标,是评估污染土修复后是否适合再利用的重要依据[24]。未处理污染土中生物可利用性砷的质量分数为30.8 mg·L−1,单独用芬顿氧化时,随芬顿添加量由1%增至5%,生物可利用性砷的质量分数仅由14.2 mg·L−1降至10.7 mg·L−1,说明芬顿氧化不能显著降低砷的生物有效性;使用氯化铁进行稳定化修复时,土壤中生物有效性砷的质量分数降低 (图2) ,铁砷比为6时降至2.89 mg·L−1;芬顿-氯化铁协同修复后,生物有效性呈数量级减小,如铁砷比为1时生物可利用性砷含量即降至为0.183 mg·L−1,协同修复后的潜在生态环境风险极大降低。
-
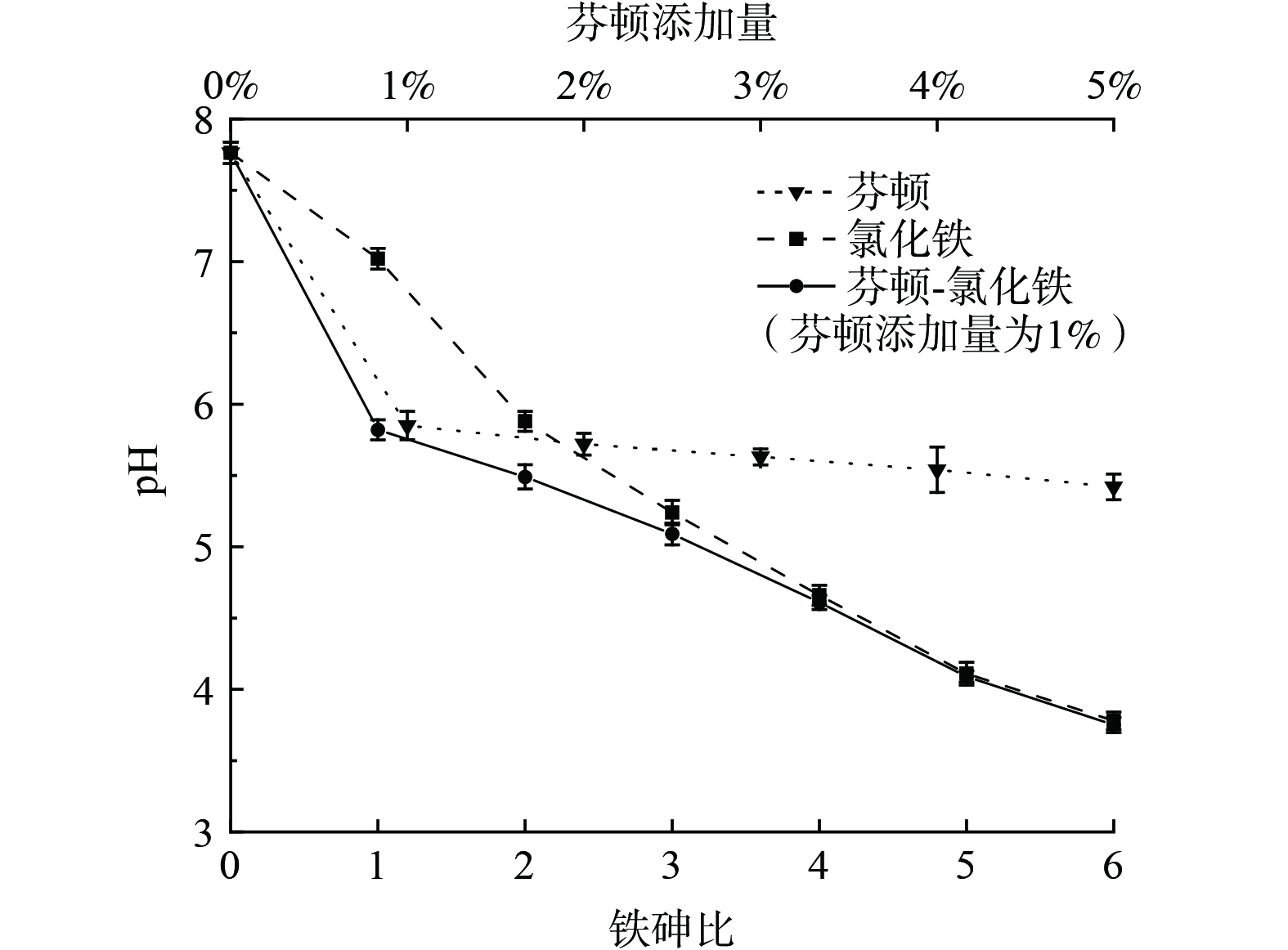
未经处理的污染土pH为7.76,由于芬顿试剂呈酸性 (初始pH=5) ,添加1%芬顿后pH为5.85,随添加量增至5%,pH降低至5.22;氯化铁修复后土壤pH随氯化铁掺量增加而不断降低 (图3) ,当添加量为6倍铁砷比时,土壤pH降低至3.78,这是因为铁盐水解过程中产生了大量H+;芬顿-氯化铁协同修复时,pH也随铁砷比增大而减小,但在1倍铁砷比 (已具有较好修复效果) 下土壤pH为5.82,明显高于氯化铁单独修复时6倍铁砷比下的pH (3.78) ,说明芬顿-氯化铁协同修复在满足一定浸出浓度标准的同时,可避免修复后土壤的过度酸化。
-
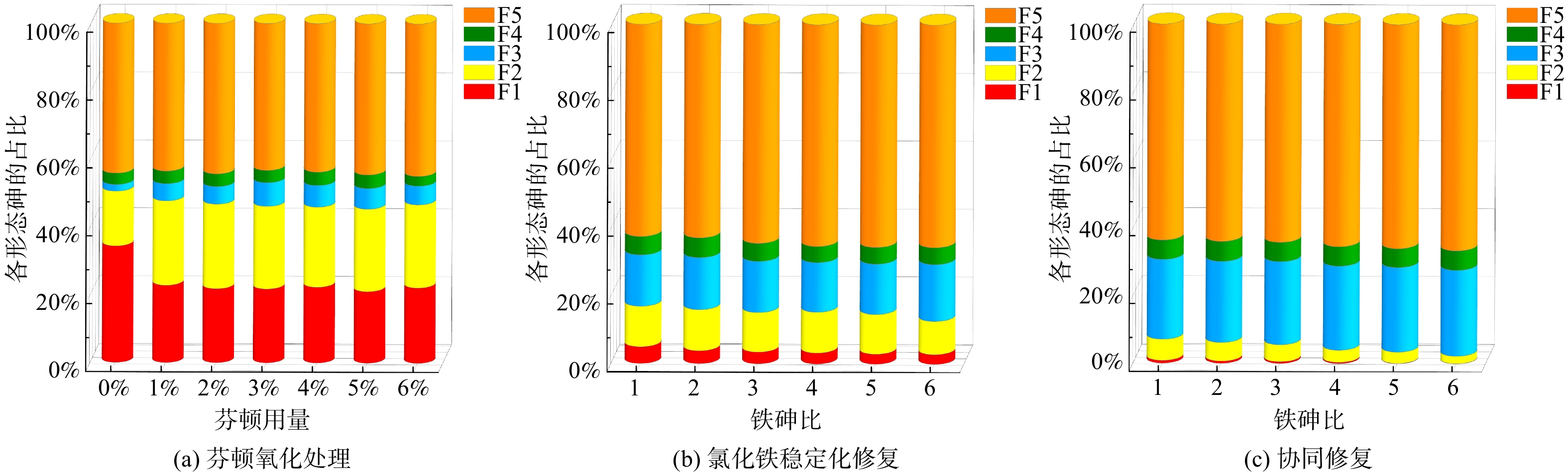
按Wenzel连续萃取法得到修复后砷的形态如图4所示,图中每种形态的砷表示为每步提取的砷占总提取量的百分比。F1、F2都是不稳定形态,未处理污染土的F1、F2占比分别为34.4%和16.2%,具有较高的潜在环境风险[25]。1%芬顿氧化处理后,F1的占比下降到22.8%,而F2的占比却增加至24.9%,说明氧化处理并不能有效处理专性吸附态砷。
使用1倍铁砷比的氯化铁稳定化修复后,F1明显下降到4.95%,同时F3、F5的占比分别增加到15.1%、62.5%,这是因为铁盐水解后与砷发生吸附反应并将砷包裹[26],这说明氯化铁修复可以在一定程度上提高砷的稳定性,但随氯化铁用量增加,各形态占比无明显的降低,这说明仅用氯化铁并不能有效地降低砷的潜在环境风险。
使用1%芬顿和1倍铁砷比的氯化铁协同修复后,F1、F2的占比分别减少至0.813%和6.23%,同时F3和F5的占比增加,表明土壤中的大部分砷被转化为稳定的形态,砷的潜在环境风险明显降低。随着铁砷比增加,F5的占比增加不明显,说明1倍铁砷比的氯化铁水化产生的铁氧化物/氢氧化物足以将土壤中的大部分砷转化为残渣态砷,这与前述SPLP浸出浓度的结果一致。
-
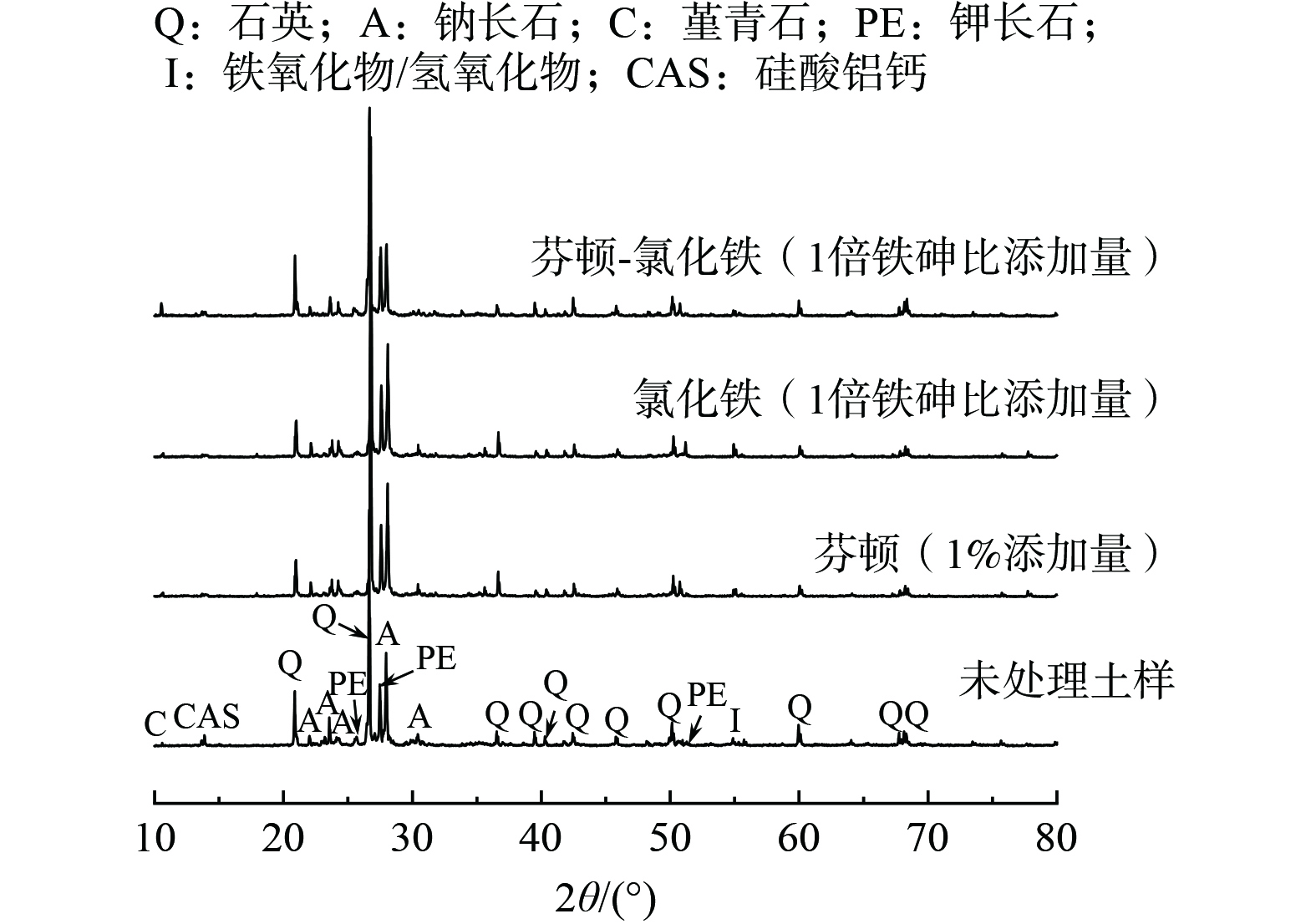
图5为不同方法处理前后土壤的XRD图谱。未经处理的污染土中含有少量铁锰氧化物/氢氧化物,能对砷起到一定程度的稳定化作用[17]。当添加了1%芬顿试剂氧化处理后,2θ为50°~51°处钾长石的特征峰发生了微弱的偏移,略微往低角度移动,这可能是由于芬顿的强氧化性和酸性导致钾长石内部晶体发生缺陷,使得砷被包裹在钾长石内部,从而形成残渣态砷。此外并没有观察到其他新的特征峰出现,这可能表明仅通过氧化处理后并不会生成新的矿物晶体或化合物。使用氯化铁对污染土进行稳定化处理时,观察到铁氧化物/氢氧化物的特征峰强度略有升高,但变化并不明显,这表明铁盐在水解过程中生成了水合铁氧化物/氢氧化物,但其结晶度较低,这与连续萃取试验中的分析结果一致。研究表明,三价铁可以与砷形成砷酸铁从而降低砷的迁移性[27-28],然而在XRD图谱中并未发现相关物质的特征峰,说明这部分铁砷物相的结晶度较差,这与前述连续萃取试验发现的修复后F3 (无定型铁铝氧化物结合态) 明显增加的结果一致。
-
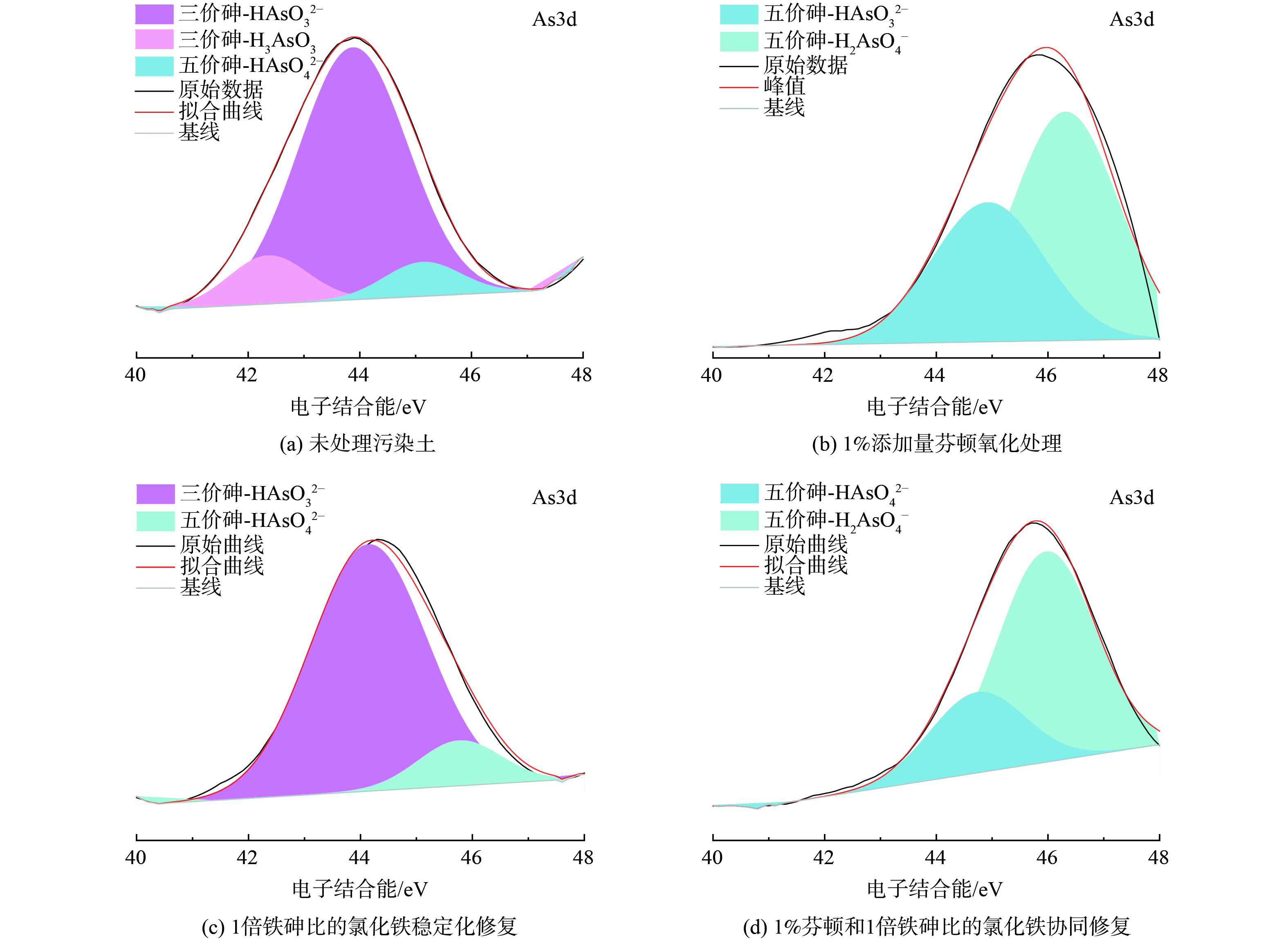
图6为污染土经过不同修复后的XPS图谱。未处理污染土中三价砷 (电子结合能为42.2和44.1 eV) 占91.3%,五价砷 (45.2 eV) 的占比为9.70%。1%芬顿氧化处理后 (图6(b)) ,土壤中砷形态仅有五价 (电子结合能为44.9和46.1 eV) ,这说明1%芬顿能将污染土中三价砷全部氧化成五价。研究表明芬顿试剂产生的·OH使三价砷氧化成五价砷的过程中,会先经历产生四价砷的短暂过程,而四价砷更易被氧化物质 (如:·OH、O2等) 氧化为五价砷[29-30]。芬顿氧化处理后的浸出浓度随添加量增加而降低可归因于芬顿中含有的少量铁盐 (氯化铁) 。1倍铁砷比的氯化铁修复后 (图6(c)) ,三价砷的占比仍高达89.1%,说明氯化铁不能改变土壤中砷的主要价态,但五价砷的电子结合能往更高方向移动,说明铁盐水解与砷结合形成了更稳定的形态[25],因此氯化铁具有一定稳定化作用。芬顿-氯化铁协同修复后土壤中只有五价砷,同时含砷物质的电子结合能往更高方向移动,说明形成了更加稳定的物质从而降低了毒性和迁移性。因此芬顿-氯化铁协同修复后的浸出浓度更低,稳定化效率得到提升。综上所述,协同修复通过先将迁移性强的三价砷氧化为迁移性低的五价砷,氯化铁水解产物再对五价砷通过共沉淀和内外球络合作用进行固定[31],从而有效降低了砷的潜在环境风险。
-
1) 芬顿-氯化铁协同修复可有效降低砷的浸出浓度和生物有效性。1%用量的芬顿和1倍铁砷比的氯化铁协同修复下可使浸出浓度降至0.281 mg·L−1,稳定化效率高达99.6%,已满足《污水排入城镇下水道水质标准》CJ343-2010)要求;生物可利用性砷的质量分数由30.8 mg·L−1降至0.183 mg·L−1,降低了99.4%,潜在环境风险显著降低。
2) 在满足浸出标准条件下,芬顿-氯化铁协同修复可显著减少铁盐的用量,从而一定程度上避免了土壤酸化。
3) 通过协同修复,芬顿将土壤中的三价砷完全氧化为五价,随后被氯化铁的水解产物通过吸附、沉淀反应进行固定,从而有效将砷从不稳定态转化为稳定形态。
芬顿和氯化铁协同修复三价砷污染土的稳定化效果
Stabilizing effects of Fenton and FeCl3 in remediation of arsenite contaminated soil
-
摘要: 由于铁盐稳定化修复砷污染土通常不考虑砷价态对毒性和迁移性的影响,导致了修复效果欠佳。针对三价砷污染土提出了预氧化-稳定化协同修复方法,先使用芬顿将三价砷氧化为低迁移性的五价砷,再利用氯化铁将其稳定化。通过合成沉降淋滤试验、生物可利用性、pH测试、连续萃取试验和光谱分析研究了协同修复的稳定化效果和机理。结果表明,与未经处理的污染土相比,1%用量的芬顿和1倍铁砷比的氯化铁进行协同修复的稳定化处理可使浸出浓度降低99.6%,生物可利用性砷的质量分数降低99.4%,修复效率得到提高;芬顿氧化和氯化铁稳定化都使土壤pH降低,但1倍铁砷比下协同修复后的pH为5.82,高于6倍铁砷比下只用氯化铁修复的pH (3.78) ,由此可一定程度上避免过量使用铁盐造成的土壤酸化;连续萃取试验表明协同修复后更多不稳定态的砷转变为稳定形态;光谱分析发现协同修复可以将土壤中三价砷全部氧化为五价,并通过铁氧化物/氢氧化物吸附和砷酸铁沉淀的形式固定砷。本研究结果可为三价砷污染土修复提供理论和技术支持。Abstract: The effect of different arsenic valences on toxicity and mobility was usually not considered in stabilization using ferric salt for remediation of arsenic contaminated soil, resulting in an inefficient treatment. A combined treatment of oxidization-stabilization was proposed aiming at arsenite contaminated soil, which included oxidizing arsenite to low mobile arsenate by Fenton, and subsequently stabilizing arsenate by FeCl3. Synthetic precipitation leaching procedure (SPLP), bioavailability, pH measurement, sequential extraction procedure, and spectral analysis were carried out to investigate the effect and mechanism of the proposed treatment method. The results showed that compared with the untreated contaminated soil, the leaching concentration was reduced by 99.6% with 1% dosage of Fenton and FeCl3 at an iron/arsenic ratio of 1, and the bioavailability As mass fraction was decreased by 99.4%. Soil pH was reduced by both Fenton oxidation and FeCl3 stabilization. However, the pH (5.82) after 1% dosage of Fenton and 1-molar-ratio of FeCl3 was obviously higher than that treated by 6-molar-ratio of FeCl3 (3.78), indicating that soil acidification caused by the excessive use of FeCl3 could be alleviated by the proposed treatment. The sequential extraction procedure results showed that a lot of unstable-state arsenic was converted to stable states by the combined treatment. The spectral analysis revealed that arsenite was converted to arsenate after the combined treatment and was subsequently adsorbed by iron oxide/hydroxide or precipitates as ferric arsenate. This study provides theory and technical support for effective remediation of arsenite contaminated soil.
-
砷是一种剧毒的类金属,与镉、铬、铅、汞并称为土壤重金属污染的“五毒”,具有生物积累效应和“三致” (致癌、致畸、致突变) 作用,长期的砷暴露或通过饮用水、食物链过量摄入砷会严重损害人体健康[1-2]。砷在土壤中主要来源于含砷矿物风化、大气沉降、微生物分解以及工农业污染,其中采矿、冶炼和工农业生产排放是砷污染最主要的来源[3]。据2014年《全国土壤污染状况调查公报》显示,我国土壤砷污染的点位超标率高达2.7%,已对生态环境和人类健康构成严重威胁[4]。
砷在土壤中通常以亚砷酸盐 (三价) 和砷酸盐 (五价) 形式存在[5]。与五价砷相比,三价砷在土壤中不易被吸附或生成沉淀,因此迁移性更强,且其毒性约为五价砷的25~60倍[6]。铁盐作为经济实用的稳定化药剂,常被用于砷污染土稳定化修复中,铁盐或溶解后生成的铁氧化物会与砷发生吸附-沉淀反应,具有较高的砷结合能力[7-8],因为其对三价和五价砷都有较好的稳定化效果,所以目前用铁盐处理砷污染土时一般不区分砷的原始价态。BAGHERIFAM等[9]研究了添加量为5%的铁氧化物对砷的质量分数为50.0 mg·kg−1污染土的稳定化效果,发现铁氧化物可将生物有效性砷的质量分数从40.8 mg·L−1降至12.6 mg·L−1,并降低植物对砷的吸收;ZHAO等[10]向2 117 mg·kg−1的砷污染土中加入3倍铁砷比的FeCl3进行稳定化修复,使其浸出浓度降至17.9 mg·L−1;但发现过量的铁盐 (水解呈酸性) 引起土壤酸化。修复不彻底或修复后土壤酸化都不利于土地资源化利用,因此科学的修复方法应当区分砷的价态,将三价砷氧化为易被稳定化的五价砷进而提高修复效率,且在一定程度上减少铁盐用量。
常见的氧化剂有臭氧、高锰酸钾、过硫酸钠、过氧化钙和芬顿试剂等[11],其中芬顿试剂能够生成强氧化性的羟基自由基 (·OH) ,·OH拥有很高的氧化还原电位以及电负性,此外还有环境友好、反应速率快、可操作性强和可控等优点,目前已被广泛用于有机污染和砷污染废水治理[12]。KATSOYIANNIS等[13]研究发现,芬顿试剂可快速将废水中的三价砷氧化为五价砷,且处理后环境友好;JAAFARZADEH等[14]通过使用芬顿试剂预处理含三价砷的废水,发现芬顿试剂预处理能有效提高火山灰对砷的吸附;QIN等[15]通过研究天然雨水中微量的过氧化氢及其产生的芬顿效应对无机砷和有机砷的修复效果,发现芬顿效应一定程度上减少了水稻中的砷累积和毒害效应。类似于污水处理,在砷污染土的修复中,采用芬顿试剂将三价砷氧化为五价,将显著提高后续铁盐对于砷的固定效果[16],然而目前在砷污染土修复中却未见有使用芬顿试剂进行氧化的研究。
基于不同价态砷的迁移性和毒性差异,本研究提出先使用芬顿试剂将土壤中的三价砷氧化为毒性低、弱迁移性的五价砷,再施加氯化铁稳定化五价砷的协同修复方法。对总砷的质量分数为1 500 mg·kg−1 (其中三价砷为1 330 mg·kg−1) 的污染土进行芬顿-氯化铁协同修复试验,通过合成沉降淋滤试验 (SPLP) 、生物可利用性验证修复效果;同时,通过pH测试、连续萃取试验、光谱分析研究修复前后土壤的矿物成分、砷的赋存形态和价态,以期揭示其协同修复机理。
1. 材料与方法
1.1 污染土制备
为了保证平行试验中各试样砷的质量分数相同,通过向未污染的土壤中添加亚砷酸钠 (NaAsO2) 溶液的方法人工配制砷污染土。试验原土取自广东汕头某工地,土壤风干后过2 mm筛,原土的pH为7.45,为砂质土,其中细砂粒组含量占55.5%,中砂占25.5%,强酸消解后用原子吸收法 (ICP-OES) 测得土中金属的质量分数分别为:铝 67 700 mg·kg−1、铁 13 100 mg·kg−1、钙 9 650 mg·kg−1、镁 2 470 mg·kg−1、锰 679 mg·kg−1、砷 8.90 mg·kg−1。
向试验原土中加入定容后浓度为0.132 mol·L−1的NaAsO2溶液,制备砷的质量分数为1 500 mg·kg−1的砷污染土,充分搅拌后密封焖土 (老化) 120 d[17]。老化后测得污染土pH为7.76,三价砷的质量分数为1 330±20 mg·kg−1,说明制样过程中小部分三价砷被 (土壤中铁锰氧化物或氧气) 氧化成五价[18]。本试验所用溶液均用去离子水配制,所用试剂均为分析纯。
1.2 修复方法对比
针对前述污染土壤,本研究单独使用氯化铁进行稳定化修复与芬顿-氯化铁协同修复进行了对比,另外单独使用芬顿进行了土壤氧化处理,以便揭示相关的效果和机理。
所用芬顿试剂按照H2O2∶Fe2+ (摩尔比) 10∶1配制,同时加入稀硫酸调节pH为5,以促进更多·OH产生从而提高氧化能力[19]。单独使用芬顿试剂氧化处理污染土时,芬顿试剂添加量 (按未处理污染土的干重计算) 为1%~5%,加入去离子水使固液比为1∶1,充分混合后静置24 h,使氧化完成 (后续分析表明24 h已足够)。
单独使用氯化铁稳定化修复时,向污染土中加入不同添加量 (铁砷摩尔比1~6) 的氯化铁,然后加入离子水,控制固液比为1∶1并混合均匀,静置3 d。
芬顿-氯化铁协同修复时,芬顿试剂添加量为1%,按上述氧化处理步骤操作后,再加入铁砷比1~6的氯化铁,按上述稳定化步骤操作。每组试验重复3次,结果取平均值。
1.3 效果评价和机理分析的试验方法
本研究采用SPLP评价污染土修复后的浸出毒性,试验参照《固体废物 浸出毒性浸出方法 硫酸硝酸法》 (HJ/T299-2007) ,采用硫酸与硝酸的混合溶液 (质量比6∶4;pH=3.20±0.05) 作为浸提液,用于模拟酸雨淋滤条件下砷在土壤中的浸出[20]。操作步骤为:修复后的污染土破碎后过9.5 mm筛,取50 g置于锥形瓶中,加入500 mL浸提液充分混合后,于翻转振荡器 (FZ-12,常州市亿能实验仪器厂) 中以 (30±2) r·min−1振荡18 h,静置30 min后过0.45 um微孔滤膜,使用ICP-OES测定滤液中砷浓度。
为了评价修复后砷污染土对环境中动植物的毒性,进行了生物有效性试验,提取剂为0.05 mol·L−1的碳酸氢钠 (NaHCO3) 溶液[21]。具体操作为:取1 g修复后的污染土置于50 mL离心管中,按固液比1∶10与提取剂混合后,于水浴恒温振荡器 (HSY-B,常州金坛精达仪器制造有限公司) 中以200 r·min−1振荡2 h,然后于台式离心机 (TDL-5,常州中捷实验制造有限公司) 中以4 000 r·min−1离心10 min,过0.45 um滤膜后测定上清液中砷的浓度。
土壤pH测试操作步骤为:取10 g过2 mm筛的土壤置于锥形瓶中,加入去离子水使固液比为1∶5,在180 r·min−1下水平振荡3 min,静置30 min后用pH计 (FE28-Standard,瑞士METTLER TOLEDO公司) 测定上清液pH。
为了揭示修复前后砷在土壤中的形态稳定性,进行分步萃取试验分析砷的赋存形态。参照Wenzel的连续萃取法[22],按每步采用的萃取剂,将提取的重金属形态分为5种,分别是非专性吸附态 (F1) 、专性吸附态 (F2) 、无定型铁铝氧化物结合 (F3) 、晶质铁铝氧化物结合态 (F4) 和残渣态 (F5) ,从F1到F5生物可利用性逐渐降低,即稳定性逐渐升高。
采用X射线衍射 (XRD,DX-2700,丹东浩元仪器有限公司) 对土壤中矿物进行晶型测试,使用Cu Kɑ射线以2θ=10°·min−1的速度从2θ=10°扫描至2θ=80°,通过High Score Plus v3.0软件分析矿物晶体。采用X射线光电子能谱仪 (XPS,Axis-Ultra DLD,英国Kratos公司) 对砷在土壤中的价态进行分析,以Al Kɑ射线作为激发源,测试电压为12 kV,测试电流为6 mA,以C 1s=284.8 eV作为标准结合能进行校对,通过XPSPEAK41软件进行曲线拟合后,根据结合能峰值确定成分。
2. 结果与讨论
2.1 浸出浓度
各修复方法对应的SPLP浸出浓度如图1所示。图1(a)中未经处理的污染土 (CK) 砷浸出浓度为73.8 mg·L−1。使用1%芬顿氧化处理后浸出浓度降至52.4 mg·L−1,说明氧化处理有一定效果但效果有限;随芬顿用量由1%增至5%,浸出浓度略有降低。因此,芬顿氧化处理主要改变了砷的价态,降低浸出浓度的效果非常有限。
使用氯化铁进行稳定化处理后 (图1(b)) ,浸出浓度随铁盐用量增加而降低,但铁砷比超过3以后降低不明显,且铁砷比为5时浸出浓度仍有14.1 mg·L−1,稳定化效率仅为79.8%,说明仅增加氯化铁用量不能达到预期修复效果。
使用1%芬顿氧化后,再用氯化铁进行协同修复,浸出浓度显著降低 (图1(c)) ,铁砷比为1时浸出浓度降至0.281 mg·L−1,已满足低于《污水排入城镇下水道水质标准》CJ343-2010)中规定的0.3 mg·L−1[23],稳定化效率高达99.6%。在此情况下,继续增加铁盐用量虽然会减少浸出浓度,但已超过实际需要。综上可知,芬顿氧化大幅提高了后续稳定化处理效果,也可在达到某浸出浓度标准的前提下一定程度上减少铁盐用量。
2.2 生物有效性
生物有效性是衡土壤中污染物的迁移性以及对生态环境影响的重要指标,是评估污染土修复后是否适合再利用的重要依据[24]。未处理污染土中生物可利用性砷的质量分数为30.8 mg·L−1,单独用芬顿氧化时,随芬顿添加量由1%增至5%,生物可利用性砷的质量分数仅由14.2 mg·L−1降至10.7 mg·L−1,说明芬顿氧化不能显著降低砷的生物有效性;使用氯化铁进行稳定化修复时,土壤中生物有效性砷的质量分数降低 (图2) ,铁砷比为6时降至2.89 mg·L−1;芬顿-氯化铁协同修复后,生物有效性呈数量级减小,如铁砷比为1时生物可利用性砷含量即降至为0.183 mg·L−1,协同修复后的潜在生态环境风险极大降低。
2.3 修复后土壤的pH
未经处理的污染土pH为7.76,由于芬顿试剂呈酸性 (初始pH=5) ,添加1%芬顿后pH为5.85,随添加量增至5%,pH降低至5.22;氯化铁修复后土壤pH随氯化铁掺量增加而不断降低 (图3) ,当添加量为6倍铁砷比时,土壤pH降低至3.78,这是因为铁盐水解过程中产生了大量H+;芬顿-氯化铁协同修复时,pH也随铁砷比增大而减小,但在1倍铁砷比 (已具有较好修复效果) 下土壤pH为5.82,明显高于氯化铁单独修复时6倍铁砷比下的pH (3.78) ,说明芬顿-氯化铁协同修复在满足一定浸出浓度标准的同时,可避免修复后土壤的过度酸化。
2.4 修复后砷的形态分布
按Wenzel连续萃取法得到修复后砷的形态如图4所示,图中每种形态的砷表示为每步提取的砷占总提取量的百分比。F1、F2都是不稳定形态,未处理污染土的F1、F2占比分别为34.4%和16.2%,具有较高的潜在环境风险[25]。1%芬顿氧化处理后,F1的占比下降到22.8%,而F2的占比却增加至24.9%,说明氧化处理并不能有效处理专性吸附态砷。
使用1倍铁砷比的氯化铁稳定化修复后,F1明显下降到4.95%,同时F3、F5的占比分别增加到15.1%、62.5%,这是因为铁盐水解后与砷发生吸附反应并将砷包裹[26],这说明氯化铁修复可以在一定程度上提高砷的稳定性,但随氯化铁用量增加,各形态占比无明显的降低,这说明仅用氯化铁并不能有效地降低砷的潜在环境风险。
使用1%芬顿和1倍铁砷比的氯化铁协同修复后,F1、F2的占比分别减少至0.813%和6.23%,同时F3和F5的占比增加,表明土壤中的大部分砷被转化为稳定的形态,砷的潜在环境风险明显降低。随着铁砷比增加,F5的占比增加不明显,说明1倍铁砷比的氯化铁水化产生的铁氧化物/氢氧化物足以将土壤中的大部分砷转化为残渣态砷,这与前述SPLP浸出浓度的结果一致。
2.5 土壤矿物成分分析
图5为不同方法处理前后土壤的XRD图谱。未经处理的污染土中含有少量铁锰氧化物/氢氧化物,能对砷起到一定程度的稳定化作用[17]。当添加了1%芬顿试剂氧化处理后,2θ为50°~51°处钾长石的特征峰发生了微弱的偏移,略微往低角度移动,这可能是由于芬顿的强氧化性和酸性导致钾长石内部晶体发生缺陷,使得砷被包裹在钾长石内部,从而形成残渣态砷。此外并没有观察到其他新的特征峰出现,这可能表明仅通过氧化处理后并不会生成新的矿物晶体或化合物。使用氯化铁对污染土进行稳定化处理时,观察到铁氧化物/氢氧化物的特征峰强度略有升高,但变化并不明显,这表明铁盐在水解过程中生成了水合铁氧化物/氢氧化物,但其结晶度较低,这与连续萃取试验中的分析结果一致。研究表明,三价铁可以与砷形成砷酸铁从而降低砷的迁移性[27-28],然而在XRD图谱中并未发现相关物质的特征峰,说明这部分铁砷物相的结晶度较差,这与前述连续萃取试验发现的修复后F3 (无定型铁铝氧化物结合态) 明显增加的结果一致。
2.6 砷的价态分析
图6为污染土经过不同修复后的XPS图谱。未处理污染土中三价砷 (电子结合能为42.2和44.1 eV) 占91.3%,五价砷 (45.2 eV) 的占比为9.70%。1%芬顿氧化处理后 (图6(b)) ,土壤中砷形态仅有五价 (电子结合能为44.9和46.1 eV) ,这说明1%芬顿能将污染土中三价砷全部氧化成五价。研究表明芬顿试剂产生的·OH使三价砷氧化成五价砷的过程中,会先经历产生四价砷的短暂过程,而四价砷更易被氧化物质 (如:·OH、O2等) 氧化为五价砷[29-30]。芬顿氧化处理后的浸出浓度随添加量增加而降低可归因于芬顿中含有的少量铁盐 (氯化铁) 。1倍铁砷比的氯化铁修复后 (图6(c)) ,三价砷的占比仍高达89.1%,说明氯化铁不能改变土壤中砷的主要价态,但五价砷的电子结合能往更高方向移动,说明铁盐水解与砷结合形成了更稳定的形态[25],因此氯化铁具有一定稳定化作用。芬顿-氯化铁协同修复后土壤中只有五价砷,同时含砷物质的电子结合能往更高方向移动,说明形成了更加稳定的物质从而降低了毒性和迁移性。因此芬顿-氯化铁协同修复后的浸出浓度更低,稳定化效率得到提升。综上所述,协同修复通过先将迁移性强的三价砷氧化为迁移性低的五价砷,氯化铁水解产物再对五价砷通过共沉淀和内外球络合作用进行固定[31],从而有效降低了砷的潜在环境风险。
3. 结论
1) 芬顿-氯化铁协同修复可有效降低砷的浸出浓度和生物有效性。1%用量的芬顿和1倍铁砷比的氯化铁协同修复下可使浸出浓度降至0.281 mg·L−1,稳定化效率高达99.6%,已满足《污水排入城镇下水道水质标准》CJ343-2010)要求;生物可利用性砷的质量分数由30.8 mg·L−1降至0.183 mg·L−1,降低了99.4%,潜在环境风险显著降低。
2) 在满足浸出标准条件下,芬顿-氯化铁协同修复可显著减少铁盐的用量,从而一定程度上避免了土壤酸化。
3) 通过协同修复,芬顿将土壤中的三价砷完全氧化为五价,随后被氯化铁的水解产物通过吸附、沉淀反应进行固定,从而有效将砷从不稳定态转化为稳定形态。
-
[1] LI J S, WANG L, TSANG D C W, et al. Dynamic leaching behavior of geogenic As in soils after cement-based stabilization/solidification[J]. Environmental Science and Pollution Research, 2017, 24(36): 27822-27832. doi: 10.1007/s11356-017-0266-x [2] PHUNG D, CONNELL D, RUTHERFORD S, et al. Cardiovascular risk from water arsenic exposure in Vietnam: Application of systematic review and meta-regression analysis in chemical health risk assessment[J]. Chemosphere, 2017, 177: 167-175. doi: 10.1016/j.chemosphere.2017.03.012 [3] BEIYUAN J Z, LI J S, TSANG D C W, et al. Fate of arsenic before and after chemical-enhanced washing of an arsenic-containing soil in Hong Kong[J]. Science of the Total Environment, 2017, 599: 679-688. [4] 环境保护部, 国土资源部. 全国土壤污染状况调查公报[J]. 中国环保产业, 2014, 36(5): 10-11. [5] BALOCH M Y J, TALPUR S A, TALPUR H A, et al. Effects of arsenic toxicity on the environment and its remediation techniques: a review[J]. Journal of Water and Environment Technology, 2020, 18(5): 275-289. doi: 10.2965/jwet.19-130 [6] OSUNA-MARTÍNEZ C C, ARMIENTA M A, BERGÉS-TIZNADO M E, et al. Arsenic in waters, soils, sediments, and biota from Mexico: An environmental review[J]. Science of the Total Environment, 2021, 752: 142062. doi: 10.1016/j.scitotenv.2020.142062 [7] 赵述华, 张太平, 陈志良, 等. 金矿区高浓度砷污染土壤的稳定化处理[J]. 环境工程学报, 2016, 10(10): 5987-5994. doi: 10.12030/j.cjee.201505193 [8] 余志元, 李二平, 杨厅, 等. 基于单纯形重心设计法的含砷废渣稳定剂设计与开发[J]. 环境工程, 2023, 41(5): 84-91. doi: 10.13205/j.hjgc.202305012 [9] BAGHERIFAM S, LAKZIAN A, FOTOVAT A, et al. In situ stabilization of As and Sb with naturally occurring Mn, Al and Fe oxides in a calcareous soil: Bioaccessibility, bioavailability and speciation studies[J]. Journal of Hazardous Materials, 2014, 273: 247-252. doi: 10.1016/j.jhazmat.2014.03.054 [10] ZHAO H M, ZHANG H Z, TANG M, et al. In situ chemical stabilization of arsenic-contaminated soils using ferrous sulfate[C]//2010 4th International Conference on Bioinformatics and Biomedical Engineering. IEEE, 2010: 1-5. [11] 张易旻, 陈铮铮, 陈昆柏, 等. 氯代有机物污染土壤高级化学氧化修复技术研究进展[J]. 环境化学, 2019, 38(3): 480-493. doi: 10.7524/j.issn.0254-6108.2018050203 [12] CHEN M, CHEN Z, WU P, et al. Simultaneous oxidation and removal of arsenite by Fe (III) /CaO2 Fenton-like technology[J]. Water Research, 2021, 201: 117312. doi: 10.1016/j.watres.2021.117312 [13] KATSOYIANNIS I A, RUETTIMANN T, HUG S J. pH dependence of Fenton reagent generation and As (III) oxidation and removal by corrosion of zero valent iron in aerated water[J]. Environmental Science & Technology, 2008, 42(19): 7424-7430. [14] JAAFARZADEH N, AMIRI H, AHMADI M. Factorial experimental design application in modification of volcanic ash as a natural adsorbent with Fenton process for arsenic removal[J]. Environmental Technology, 2012, 33(2): 159-165. doi: 10.1080/09593330.2011.554887 [15] QIN J H, LI Y, FENG M, et al. Fenton reagent reduces the level of arsenic in paddy rice grain[J]. Geoderma, 2017, 307: 73-80. doi: 10.1016/j.geoderma.2017.07.039 [16] WANG L, LUO Y, PANG J, et al. Fe-biochar for simultaneous stabilization of chromium and arsenic in soil: Rational design and long-term performance[J]. Science of the Total Environment, 2023, 862: 160843. doi: 10.1016/j.scitotenv.2022.160843 [17] HAN Y, PARK J, AHN J. Aging effects on fractionation and speciation of redox-sensitive metals in artificially contaminated soil[J]. Chemosphere, 2021, 263: 127931. doi: 10.1016/j.chemosphere.2020.127931 [18] 张钦喜, 张雪冬, 张志红. SP-MOC固化砷污染土的浸出试验研究[J]. 岩土工程技术, 2020, 34: 238-242. [19] BURBANO A A, DIONYSIOU D D, SUIDAN M T, et al. Oxidation kinetics and effect of pH on the degradation of MTBE with Fenton reagent[J]. Water Research. 2005, 39 (1) : 107-118. [20] TOWNSEND T, DUBEY B, TOLAYMAT T. Interpretation of synthetic precipitation leaching procedure (SPLP) results for assessing risk to groundwater from land-applied granular waste[J]. Environmental engineering science 2006, 23 (1) : 239-251. [21] YAN X L, ZHANG M, LIAO X Y, et al. Influence of amendments on soil arsenic fractionation and phytoavailability by Pteris vittata L[J]. Chemosphere, 2012, 88(2): 240-244. doi: 10.1016/j.chemosphere.2012.03.015 [22] WENZEL W W, KIRCHBAUMER N, PROHASKA T, et al. Arsenic fractionation in soils using an improved sequential extraction procedure[J]. Analytica Chimica Acta, 2001, 436(2): 309-323. doi: 10.1016/S0003-2670(01)00924-2 [23] YU B W, DU Y J, LIU R, et al. Reuse of stabilized contaminated soils with heavy metals as greening soils: leaching, physicochemical, and phytotoxicity characterization[M]//Geo-Chicago 2016. 2016: 557-571. [24] MIRANDA L S, AYOKO G A, EGODAWATTA P, et al. Physico-chemical properties of sediments governing the bioavailability of heavy metals in urban waterways[J]. Science of the Total Environment, 2021, 763: 142984. doi: 10.1016/j.scitotenv.2020.142984 [25] 林龙勇, 阎秀兰, 杨硕. 铁铈氧化物对土壤As (Ⅴ) 和P的稳定化效果[J]. 环境科学, 2019, 40(8): 3785-3791. [26] YANG K, KIM B C, NAM K, et al. The effect of arsenic chemical form and mixing regime on arsenic mass transfer from soil to magnetite[J]. Environmental Science and Pollution Research, 2017, 24(9): 8479-8488. doi: 10.1007/s11356-017-8510-y [27] LIN Y, WU B, NING P, et al. Stabilization of arsenic in waste slag using FeCl2 or FeCl3 stabilizer[J]. RSC Advances, 2017, 7(87): 54956-54963. doi: 10.1039/C7RA10169D [28] LI Y, WANG J, PENG X, et al. Evaluation of arsenic immobilization in red mud by CO2 or waste acid acidification combined ferrous (Fe2+) treatment[J]. Journal of Hazardous Materials, 2012, 199: 43-50. [29] CHEN Z H, JIN J Y, SONG X J, et al. Redox conversion of arsenite and nitrate in the UV/quinone systems[J]. Environmental Science & Technology, 2018, 52(17): 10011-10018. [30] KOPPENOL W H, STANBURY D M, BOUNDS P L. Electrode potentials of partially reduced oxygen species, from dioxygen to water[J]. Free Radical Biology and Medicine, 2010, 49(3): 317-322. doi: 10.1016/j.freeradbiomed.2010.04.011 [31] WANG L, CHEN L, GUO B, et al. Red mud-enhanced magnesium phosphate cement for remediation of Pb and As contaminated soil[J]. Journal of Hazardous Materials 2020, 400: 123317. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2222
- HTML全文浏览数: 2222
- PDF下载数: 60
- 施引文献: 0
