


-
传感器作为智慧农业高效快速检测的核心器件,广泛应用于现代农业的众多领域。其中,基于电化学方法的传感检测技术因其高选择性、高灵敏度、快速响应且操作简单等优势备受关注[1]。该技术的核心组件是电极材料,通常用作信号识别、转换和传导的关键传感器元件。然而,传统的传感器电极材料存在价格高昂、选择性不强和响应性弱等缺陷,制约了该技术的推广应用。为了满足传感器高灵敏度和高选择性的要求,亟需开发敏感、高效且低成本的传感器电极材料。与石墨烯、碳纳米管、贵金属和导电聚合物等传统电极材料相比,生物炭材料不仅具有原材料来源广泛、成本低廉、制备工艺简单、环境友好和可持续等优势[2],还具有丰富的表面含氧官能团、良好的导电性和孔道修饰能力等特性,被广泛认为是一种极具应用潜力的传感器电极材料。
热解是最常用的生物炭制备技术,然而由生物质直接热解所得的生物炭往往存在比表面积不大和亲和性活性位点不足等缺陷,导致生物炭对重金属离子的富集能力有限,进而难以满足电化学传感器高灵敏度的实际需求[3]。为了改善这些问题,化学活化和杂原子掺杂改性被广泛应用于提高生物炭的孔隙结构和表面活性位点[4]。磷酸 (H3PO4) 、氢氧化钾 (KOH) 、氯化锌 (ZnCl2) 等作为常用的活化剂,不仅能调控孔径分布还能增大比表面积。其中,H3PO4具有丰富的活性官能团、可控性强、副反应少、对设备腐蚀性低、价格低等优势,在生物炭改性中得到广泛报道[5]。如ADHIKARI等[6]研究表明,H3PO4活化制备的纳米多孔炭比表面积和孔容分别高达1 288 m2·g−1和 1.64 cm3·g−1,并对氨表现出优异的传感检测性能。杂原子磷 (P) 的掺杂能有效丰富生物炭的孔隙结构 [7]。以H3PO4为化学活化剂调控孔结构的同时引入P基团,改善生物炭芳环结构的电子分布和表面亲和性。潘静等[8]发现,经H3PO4改性后的生物炭成功引入了含P官能团,增强了对铅离子 (Pb2+) 的吸附能力。然而,目前关于H3PO4高温炭化后生成的含P基团对Pb2+的传感检测性能影响尚不明确。另一方面,H3PO4预处理技术的选择和优化对于提高热解生物炭富集、催化以及电化学性能具有显著影响。在炭化过程中,H3PO4在生物质中的均匀分布是分级孔隙结构和高密度含P官能结构构建的关键步骤。超声空化技术是一种有效的预处理方法,通过引入气泡和涡流,能够实现高效混合、均匀浸渍,并提高反应速率。基于以上分析,可以提出如下猜想:超声空化技术辅助磷酸浸渍一步炭化技术可以实现高活性含磷官能团和多级孔隙在生物质炭中的有效生成和均匀分布,进而大幅提高生物炭对痕量重金属Pb2+的电化学传感检测性能。
为此,本研究以棉秆为原料,H3PO4为浸渍剂通过超声空化辅助H3PO4活化一步炭化制备生物质炭铅检测传感材料,探究了不同炭化温度下含P官能团对铅的检测性能和机制。通过对生物质炭传感检测材料的结构表征和传感检测性能评估,将深入探讨炭化温度对超声空化辅助H3PO4浸渍法生物炭的结构特征、比表面积、孔隙度、含P官能团结构的影响,并进一步研究其对铅传感检测性能的影响因素和机制。这将有助于深入理解超声空化辅助H3PO4浸渍一步炭化制备材料的优化策略,并为高活性铅检测传感材料的设计和应用提供数据支持。通过本研究的开展,为生物质炭的功能改性和电化学传感检测领域的研究提供新的思路和方法。
-
棉秆来源于新疆;磷酸 (H3PO4) 、氯化钾 (KCl) 均为分析纯;铁氰化钾 (K3[Fe(CN)6],≥99.5%) ;乙酸 (CH3COOH,≥99.5%) 。
主要仪器包括比表面积及孔径分析仪 (3H-2000PM2,贝士德仪器有限公司) ;电化学工作站 (CHI660E,上海辰华仪器有限公司) ;傅里叶变化红外光谱仪 (Scientific Nicolet iS5,美国赛默飞科技公司) ;X 射线光电子能谱仪 (Scientific K-Alpha,美国赛默飞科技公司) 等。
-
取适量棉秆用去离子水洗净烘干后粉碎过100目筛,称取10 g溶解于35.28 g H3PO4溶液和200 g水中,超声空化20 min,保证充分均匀混合。每2 h搅拌和超声1次,静置12 h后,在150 ℃烘箱中干燥24 h。用管式炉在100 mL·min−1 N2中以5 ℃·min−1升温至炭化温度 (700、750、800、850 ℃) 保温2 h。冷却获得材料,经去离子反复冲洗直至滤液呈中性。将所得4种棉秆炭材料分别命名为CS700、CS750、CS800和CS850。
-
预处理时,将直径3 mm的裸玻碳电极 (GCE) 依次用粒径1.0、0.3和0.05 μm的Al2O3抛光粉在麂皮上画“8”字型抛光打磨。每次抛光后洗净表面,依次用无水乙醇和去离子水分别超声清洗2~3 min。将处理后的电极在电解液为5.0 mmol·L−1 K3[Fe(CN)6]和1 mol·L−1 KCl溶液中,通过循环伏安法 (CV;扫描区间为−0.1~0.6 V;扫速为50 mV·s−1;扫描8次) 进行电化学检测,直至最后2次峰电位之差满足65~70 mV,电极合格,储存备用。
制备修饰GCE时,称取50 mg粉末过200目筛,溶入2 mL无水乙醇和 0.5 mL 0.5%的Nafion溶液中,超声空化20 min后获得均一、稳定的分散液。用移液枪取2.5 μL的分散液均匀滴涂于合格的GCE表面,自然晾干后在40 ℃干燥箱中固化15 min,得到4种修饰GCE:CS700/GCE、CS750/GCE、CS800/GCE和 CS850/GCE。
-
用比表面积分析仪测定样品在77.3 K下的N2吸附脱附量;通过BET、BJH和HK理论计算炭材料的比表面积 (SBET) 、中孔体积 (Vmes) 、微孔体积 (Vmic) 和平均孔径 (D) ;在相对压力 (p/p0) 为0.95时,计算总孔体积 (Vtot) ;用傅里叶变化红外光谱分析仪 (FTIR) 和X射线光电子能谱仪 (XPS) 分析炭材料表面官能团性质。
-
在饱和甘汞、铂丝电极和4种棉秆炭材料修饰GCE组成的三电极体系下,以5.0 mmol·L−1 K3[Fe(CN)6]和0.1 mol·L−1 KCl混合溶液作为电解液,通过CV (扫描区间为-0.1~0.6 V;扫描次数为6次;扫速为0.1 v·s−1) 和电化学阻抗谱 (EIS;初始、结束电压为1.4、0 V;电位增量为4 mV;振幅为50 mV;脉宽为0.016 7 s) 进行电化学测试。
-
选用0.2 moL·L−1的醋酸-醋酸钠 (HAC-NaAC) 作为电解液,配置不同浓度梯度的Pb2+溶液 (1~1 000 μg·L−1) ;采用阳极溶出差分脉冲伏安法 (ASDPV) 对目标分析物进行检测,根据溶出峰电流获得线性拟合曲线,计算得出传感器检测限;选用25 μg·L−1 Pb2+溶液进行ASDPV测试,验证传感器的重复性、重现性、稳定性及抗干扰性。
-
图1为4种棉秆炭材料的N2吸脱附等温曲线和DFT全孔图。由图1(a)所示,在p/p0<0.01的低压区,吸附曲线急剧上升,表明存在大量的微孔[9],其中CS800微孔含量最高。当p/p0>0.3时,吸脱附等温曲线出现了H4回滞环,说明存在中孔。因此超声空化辅助H3PO4浸渍法有助于获得微/中孔分级结构。进一步对比发现CS800具有最高的比表面积 (1 338.57 m2·g−1) 和微孔体积 (0.49 mL·g−1) ,且Vmic占比高达 (51.60%) (表1) ,这有利于电化学传感过程中电解质离子的快速扩散以及目标分析物的快速富集[10-12]。
另外,4种炭材料的平均孔径 (D) 均位于2.85~4.32 nm之间 (图1(b)) ,这与N2吸脱附分析结果相一致。其中中孔平均孔径均位于3.04~4.09 nm,而在微孔结构中还存在大量超微孔 (<0.7 nm) 和极微孔 (>0.7 nm) ,其平均孔径分别为0.44 nm和0.98 nm。另一方面,D从700 ℃到800 ℃不断下降,这可能是H3PO4在高温下形成高度聚合的聚磷酸盐,导致孔壁堵塞[13]。而当温度继续升至850 ℃时D反而增加,可能由于碳骨架的热降解和挥发性含P物质的形成,使部分中孔发展为大孔所致。
-
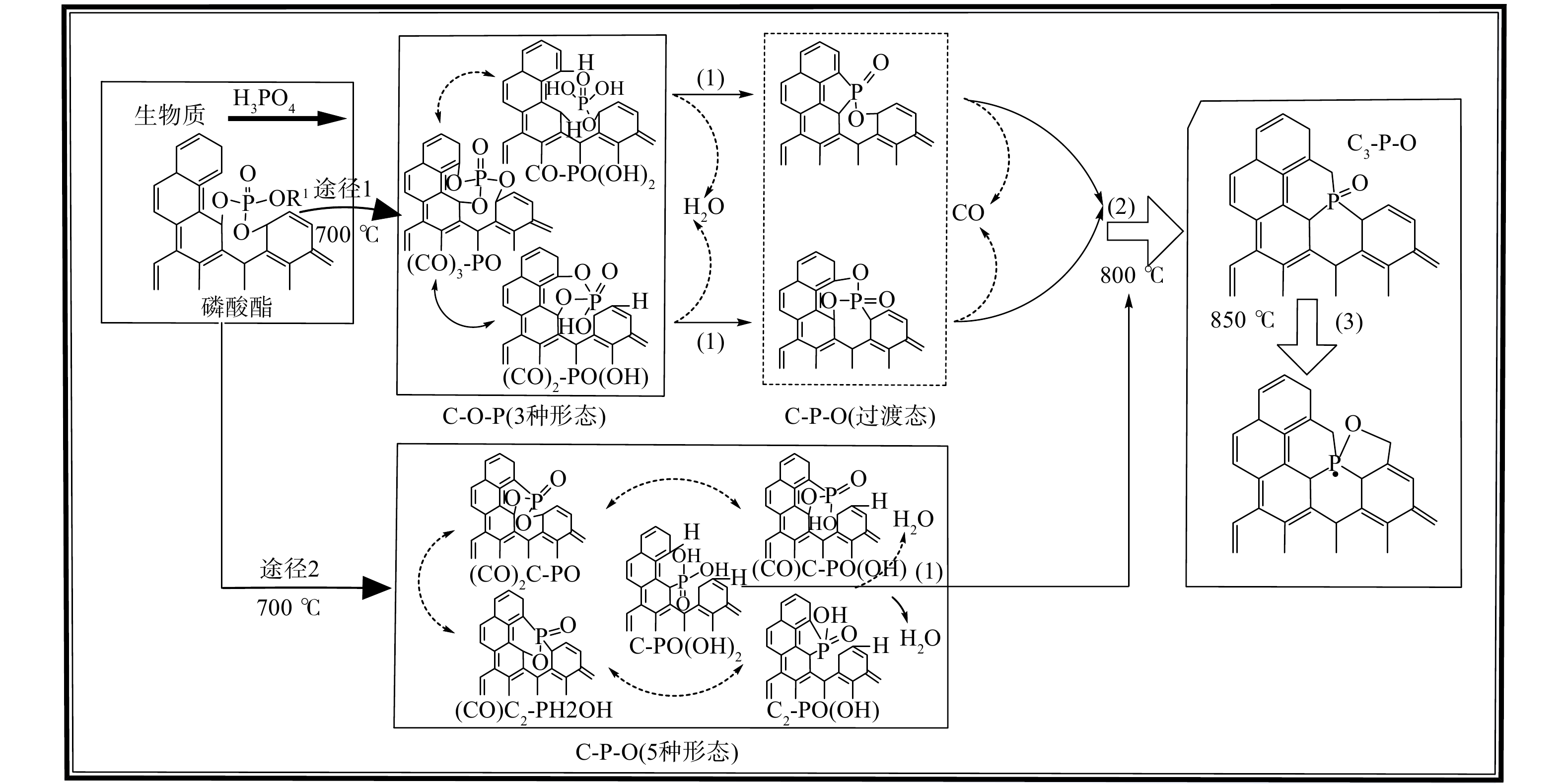
炭材料热解制备过程中,H3PO4的引入会影响生物炭表面官能团的类型。图2为4种棉秆炭材料的FTIR图谱。在峰谱带较宽的3 434 cm−1处对应O-H基团的拉伸振动,表明H3PO4活化掺杂的生物炭表面存在羟基和物理吸附水[14]。在1 634 cm−1处对应羧基C=O基团或C=C芳香环结构的拉伸振动,表明经H3PO4改性后炭材料表面含氧官能团和芳香性增加[15]。在大约1 587和1 070 cm−1处的特征峰可归因于C-O-C、C-O基团的伸缩振动和P-O基团的拉伸振动[16]。在1 171 cm−1处的吸收峰为C-O (C-O-C) 基团的伸缩振动[17]或P=O基团的拉伸振动 [18]。而在983 cm−1处吸收峰主要来源于C-O-P 基团的拉伸振动[19]。这表明P原子在生物炭中的掺杂主要是与C和O原子的结合。与其它温度相比,炭化温度为800 ℃时,在1 070、1 171 cm−1处吸收峰明显增强,表明该炭化温度下P-O和P=O基团已成功掺入碳骨架。温度上升至850 ℃时,C=O、P=O和P-O基团峰强度逐渐减弱。这些现象也表明高温炭化会促进表面官能团的分解,使光谱曲线逐渐趋于平缓。
-
利用XPS光电子能谱对生物炭表面元素组成及形态进行了定性分析。如图3(a)所示,4种炭材料均存在C1s、O1s和P2p峰,说明经H3PO4活化后,P元素成功掺杂到炭材料中。随着炭化温度升高至800 ℃时,C1s含量最高 (83.01%) (表2) ,而对应O1s含量最低。这表明高温导致C-O键的断裂,形成更多悬挂键,有助于P原子键合到碳骨架中。在P2p高分辨图谱中拟合了C-O-P ( (134.0±0.3) eV) 、C-P-O ( (133.1±0.3) eV) 和C3-P-O ( (132.2±0.3) eV) [20]3个峰 (图3(b)) 。当炭化温度在800 ℃时C3-P-O基团含量最高 (29.00%) (表2) ,表明该温度下有助于获得更多的C3-P-O结构。相反C-O-P和C-P-O基团的含量明显降低,这可能是不稳定磷化合物的蒸发和磷酸盐被碳还原[21]所致。已有文献[22]证明C-P键比C-O-P中C-O和P-O键的热稳定性高,在高温下更倾向于形成稳定的C3-P键,说明 C-O-P和C-P-O基团在800 ℃转化为C3-P-O基团。随着温度继续增至850 ℃时,C3-P-O基团含量降低,这可能是过高的炭化温度导致该基团的分解。
为了进一步了解P掺杂生物炭的形成机理,深入探讨了棉秆基炭材料含P基团的演化规律。如图4所示,H3PO4的引入会促进棉秆生物质脱水、环化、缩合等过程中化学键的断裂和交联反应[23]。具体而言,C-O-P和C-P-O基团向C3-P-O基团的转变机理可分为两个途径。途径1:随着炭化温度上升至800 ℃,C-O-P结构上的-OH基团与碳骨架上的H结合形成水分子,短暂形成过渡态C-P-O,其上 P-O键断裂并释放CO气体,最终转变为C3-P-O。途径2:C-P-O结构中的-OH基团与碳骨架上的H结合释放水分子,直接向C3-P-O基团转变。温度升高至850 ℃时,两途径中C3-P-O基团发生分解,产生部分C-O-P基团,形成了新的C3-P-O和C-O-P基团的复合结构。
-
图5 对4种棉秆炭材料进行了电化学活性表征和阻抗测试。如图5(a)所示,CS800具有最强的峰电流 (148.7 μA) ,说明CS800电化学活性最好。众所周知,峰电流与电化学活性几何面积成正相关,通过对比 4种电极的电化学活性几何面积,结果发现CS800电化学活性几何面积最大 (20.71) (图5(a)) ,进一步证实该电极材料具有优异的电催化活性。在典型的EIS图中,高频区半圆直径对应电子转移电阻 (Rct) ,而低频区直线斜率对应Warburg阻抗 (Zw) 。如图5(b)EIS所示,CS800对应的半圆直径最小 (Rct=106) ,说明电子反应界面电子传递速率较快。在低频区域,CS800对应斜率更接近于1,表明其Zw最小,电解质离子扩散速率最快,这与棉秆炭丰富的微/中孔分级结构有关。
-
4种棉秆炭材料的富集能力主要与其表面特定官能团的性质有关。引入P物种不仅可以增强炭材料表面极性,提高亲水性,同时还可以引入丰富的活性位点,增强对目标检测物的亲和力[24]。已有理论研究[25]证明丰富的活性位点能够提高对重金属离子的富集能力。H3PO4作为P掺杂源和表面功能化剂,在800 ℃下C3-P-O基团显著提高了电极表面的反应活性,为目标反应物的富集提供了更多的活性位点。图6是通过ASDPV验证了4种棉秆炭材料对Pb2+的电化学传感性能,结果发现4种棉秆炭修饰电极峰电流均有所提高。其中CS800检测峰电流最强 (38.11 μA) ,这说明丰富的C3-P-O基团提高了棉秆炭对Pb2+的富集能力,加速了Pb2+的电沉积过程。而CS850 的C3-P-O基团较少,对应的检测峰电流明显降低。显而易见,CS800/GCE具有最佳的Pb2+电化学传感性能。因此,选择CS800/GCE作为后续电化学传感平台,构建高活性的铅检测传感器。
-
以CS800/GCE为研究对象,利用ASDPV对不同浓度梯度 (1~1 000 μg·L−1) 的Pb2+进行连续测试。如图7(a),随着Pb2+浓度的递增,检测峰峰形越来越尖锐。对不同浓度Pb2+对应的峰电流值进行拟合,发现Pb2+浓度与峰电流值具有明显的线性关系。当Pb2+浓度范围为1~100 μg·L−1时,其拟合线性方程为y=0.028 31x+1.293 36 (y为峰电流μA,x为Pb2+浓度μg·L−1) ,R2为0.970 51;当Pb2+浓度范围为100~1 000 μg·L−1时,其拟合线性方程为y=0.009 95x+2.663 80 (y为峰电流μA,x为Pb2+浓度μg·L−1) ,R2为0.983 75 (图7(b)) 。说明CS800/GCE对Pb2+具有较宽的检测范围。此外,其理论检测限低至1.38 μg·L−1 (S/N=3) ,低于国家标准《农田灌溉用水水质》[26]基本控制限值0.01 mg·L−1,比世界卫生组织《饮用水水质标准》[27]甚至低约7倍。
-
为了验证CS800/GCE的电化学传感性能,对其重复性、重现性、稳定性及抗干扰性进行测试。采用同一支电极对浓度为25 μg·L−1的Pb2+溶液,进行连续6次ASDPV测试,结果发现6次检测峰电流值相对偏差 (RSD) 为3.8% (图8(a)) ,表明传感器重复性良好;类似地,同时制备6根CS800/GCE进行ASDPV测试,结果发现峰电流RSD为5.7% (图8(b)) ,表明传感器具有优异的重现性;将同一支电极置于室温环境保存14 d,发现保存前后检测峰电流RSD为7.3%,表明该传感器具有较好的稳定性。此外,继续取该浓度水样向其中加入100倍其它可能干扰离子Na+、K+、Mg2+、Cl-、NO3-,CS800/GCE检测结果显示,Pb2+峰电流并无明显变化,表明CS800/GCE具有出色的抗干扰性。上述实验结果表明由CS800/GCE构建的传感器对Pb2+的检测和分析具有较好的应用潜力。
-
1) 通过超声空化辅助磷酸浸渍一步炭化法制备了具有丰富的微/中孔分级结构、较大的比表面积和富含C3-P-O活性位的棉秆炭电极。其中,CS800比表面积高达1 338.57 m2·g−1、微孔比例接近50%、C3-P-O基团含量高达29.00%。
2) 相较CS700、CS750和CS850,CS800拥有最高的电化学活性和最低的电子转移电阻 (Rct) ,电子传递速度快,对Pb2+富集能力强,可归因于其丰富的C3-P-O基团的高活性位点和优异的分级孔隙结构。
3) 制备的CS800传感电极灵敏度高、稳定性及抗干扰性强,且对Pb2+检测线性范围宽 (1~100 μg·L−1和100~1 000 μg·L−1) ,线性相关系数R2均>0.97,检测限低至1.38 μg·L−1。
超声空化辅助磷酸浸渍一步炭化制备高活性铅检测传感器
Preparation of highly active lead detection sensor through ultrasound-assisted phosphoric acid impregnation and one-step carbonization
-
摘要: 开发高活性且低成本的生物质炭电极是构建高性能铅检测传感器的关键。以棉秆生物质为原料,通过超声空化辅助磷酸浸渍一步炭化法制备了系列不同温度 (700、750、800、850 ℃) 的磷改性棉秆炭,并基于这些炭材料构建了铅检测传感器。采用N2吸脱附、傅里叶红外光谱、X射线光电子能谱、循环伏安、电化学阻抗谱和阳极溶出差分脉冲伏安法等现代表征及测试技术,探究了不同温度棉秆炭电极孔径分布、化学结构、高活性磷官能团与电化学传感性能间的构效关系。结果表明,800 ℃制备的棉秆炭 (CS800) 具有更高的比表面积 (1 338.57 m2·g−1) 和优异的微/中孔分级结构以及更丰富的C3-P-O基团。这些独特优势的结合使CS800电极具有更高的电化学活性、最低的电子转移电阻 (Rct) 和更强的Pb2+富集能力。在1~100和100~1 000 μg·L−1 Pb2+浓度下,CS800检测峰电流与Pb2+浓度呈现良好的线性关系,线性相关系数R2均高达0.97,检测限灵敏度低至1.38 μg·L−1,并表现出优异的重复性、重现性、稳定性及抗干扰性,表明所制备的棉秆炭电极对Pb2+检测具有巨大的应用潜力。本研究结果可为该类生物质炭电极的制备及性能优化提供参考,也可为重金属监测及分析提供技术支持。Abstract: The development of biochar electrodes with high activity and low cost is essential for the construction of high-performance lead detection sensors. A series of phosphorus-modified cotton stalk carbons at different temperatures (700, 750, 800, 850 ℃) were prepared through ultrasonic cavitation-assisted phosphoric acid impregnation and one-step carbonization method using cotton stalk biomass as raw material, and a lead detection sensor was constructed based on these carbon materials. Using N2 absorption-desorption, Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy, cyclic voltammetry, electrochemical impedance spectroscopy and anode stripping differential pulse voltammetry and other modern characterization and testing techniques, the structure-activity relationship between pore size distribution, chemical structure, high active phosphorus functional groups and electrochemical sensing properties of cotton stalk carbon electrodes at different temperatures was investigated. The results showed that cotton stalk carbon (CS800) prepared at 800℃possessed higher specific surface area (1 338.57 m2·g−1), more abundant micropore/mesopore structure as well as C3-P-O groups. The combination of these unique advantages gived the CS800 electrode a higher electrochemical activity, the lowest electron transfer resistance (Rct), and a stronger Pb2+accommodation capability. At Pb2+concentrations of 1~100 μg·L−1 and 100~1 000 μg·L−1, the peak current of CS800 appeared a good linear relationship with Pb2+concentration. The linear correlation coefficients R2 were all up to 0.97, and the detection limit sensitivity was as low as 1.38 μg·L−1. Furthermore, excellent repeatability, reproducibility, consistency and anti-interference performance were shown, which indicated that the prepared cotton stalk carbon electrode had great application potential for Pb2+detection. The results of this research not only shed light on the preparation and performance optimization of biochar electrodes, but also offer technical support for the monitoring and analysis of heavy metals.
-
生态保护补偿是指采取财政转移支付或市场交易等方式,对生态保护者因履行生态保护责任所增加的支出和付出的成本,予以适当补偿的激励性制度安排。生态补偿机制最早于2005年党的十六届五中全会《中共中央关于制定国民经济和社会发展第十一个五年规划的建议》[1]提出。党的十八大以来,党中央、国务院高度重视生态补偿机制建设,党的十八大提出了建立资源有偿使用制度和生态补偿制度。中共十八届三中全会提出坚持“谁受益、谁补偿”的原则,完善重点生态功能区的生态补偿机制,推动地区间建立横向生态补偿机制。2015年4月,中共中央、国务院印发的《关于加快推进生态文明建设的意见》[2]和同年9月印发的《生态文明体制改革总体方案》[3]提出了要求探索建立多元化的生态补偿机制,加快形成受益者付费、保护者得到合理补偿的运行机制。2016年3月,中央全面深化改革领导小组第二十二次会议通过《关于健全生态保护补偿机制的意见》[4]。2017年11月,国家发改委在其出台的《关于全面深化价格机制改革的意见》[5]中,进一步要求完善生态补偿的价格机制。2021年9月,中共中央办公厅、国务院办公厅印发了《关于深化生态保护补偿制度改革的意见》[6],进一步落实生态保护权责、调动各方参与生态保护积极性、推进生态文明建设。
由于我国不同区域人口、产业结构和经济发展水平不同,危险废物的产生种类、数量以及危险废物利用处置能力存在较大差异,对于能力不足的地区,可以通过跨区域合作实现危险废物无害化利用处置[7]。危险废物跨区域转移行为发生的同时会伴随污染转移、环境风险转移、监管责任转移以及邻避效应转移等问题[8],因此,根据“谁收益,谁补偿”的原则,可以引入生态补偿机制,合理选择危险废物移出地相应生态环境利益的获得者,例如产废单位、当地生态环境受益者或能代表受益团体的地方政府等,对危险废物接受地进行补偿。
生态补偿制度在危险废物环境管理方面的应用与实践还未完善,我国已有部分省份在危险废物跨区域转移生态补偿方面进行了探索实践。如北京市财政局、北京市生态环境局于2020年3月联合印发《北京市危险废物处置生态环境补偿资金管理办法 (试行) 》,将危险废物处置设施分为医疗废物处置设施、飞灰处置设施、其它危险废物处置利用设施3类进行分类补偿,其前提条件为北京市其他工业危险废物种类较少,危险废物以医疗废物和生活垃圾焚烧飞灰为主,而对于区域危险废物种类多、产生量大的情况不适用。贵州省于2023年8月23日发布《贵州省危险废物跨省转入生态保护补偿机制试点方案》[9],规定接受省外危险废物进入贵州省利用处置的危险废物经营单位,在不违反跨省转入条件并自愿缴纳危险废物生态补偿资金后,可适当转入符合条件的危险废物进行利用或水泥窑协同处置,其主要考虑危险废物运输、贮存及利用处置过程中可能发生或次生环境污染事故时所产生的治理费用,而忽略了资源开发成本、大气治理成本及转入地保护生态环境或放弃发展机会的成本等。江苏省南通市于2019年6月印发《南通市生活垃圾及飞灰异地处置生态补偿办法》[10],规定飞灰来源地县 (市) 、区政府应当向飞灰终端处置单位所在地县 (市) 、区政府按照100 元·t-1的价格缴纳环境补偿费,用于扶持处置单位所在区域的经济发展和处置单位周边环境整治、修复、提升、补偿以及市政设施配套、周边关系协调等。北京市、贵州省、南通市进行的危险废物生态补偿实践各有其特点,且所规定的补偿主体、补偿标准、补偿范围等存在特异性。
综上,现有关于危险废物跨省转移生态补偿在区域危险废物种类多、产量大时,生态补偿如何开展尚不明晰;部分研究仅关注受偿区生态环境保护的直接成本而忽略了因邻避效应等问题造成的间接成本损失。因此,本研究通过应用收入损失模型提出危险废物跨省转移生态补偿资金计算公式,以4个临近省作为模拟对象,举例计算危险废物跨省转移生态补偿金额,制定了可能的征缴流程,以期为我国危险废物跨省转移生态补偿机制的构建提供决策参考。
1. 危险废物跨省转移生态补偿框架
本研究所假定的危险废物跨省转移生态补偿场景为,转移的危险废物在移出地处置能力充足,但在移入地处置能力基本平衡或不足,此时危险废物的移出者是最主要的受益者,而移入地政府则作为移入地收益团体的代表。围绕“谁补谁,补多少,怎么补”的思路,构建危险废物跨省转移生态补偿框架如图1所示。主要内容包括:①补偿原则的确定:本研究所构建的危险废物跨省转移生态补偿机制是依托排污权有偿使用制度探索建立的市场化交易方式。危险废物跨省转移实行排污权有偿取得,即危险废物跨省转移移出人 (以下简称“移出人”) 在缴纳危险废物跨省转移生态补偿资金后获得向危险废物跨省转移接受地 (以下简称“受偿区”) 转移危险废物的权利。②补偿主客体的识别:基于危险废物流向,移出人应当对受偿区因履行生态保护责任所增加的支出和付出的成本予以适当补偿。③补偿金额的确定:移出人应缴纳的危险废物跨省转移生态补偿资金根据危险废物跨省转移量、补偿系数和利用处置方式计算[11]。④本研究根据危险废物跨省转移工作流程初步设计生态补偿资金征缴流程与管理办法。
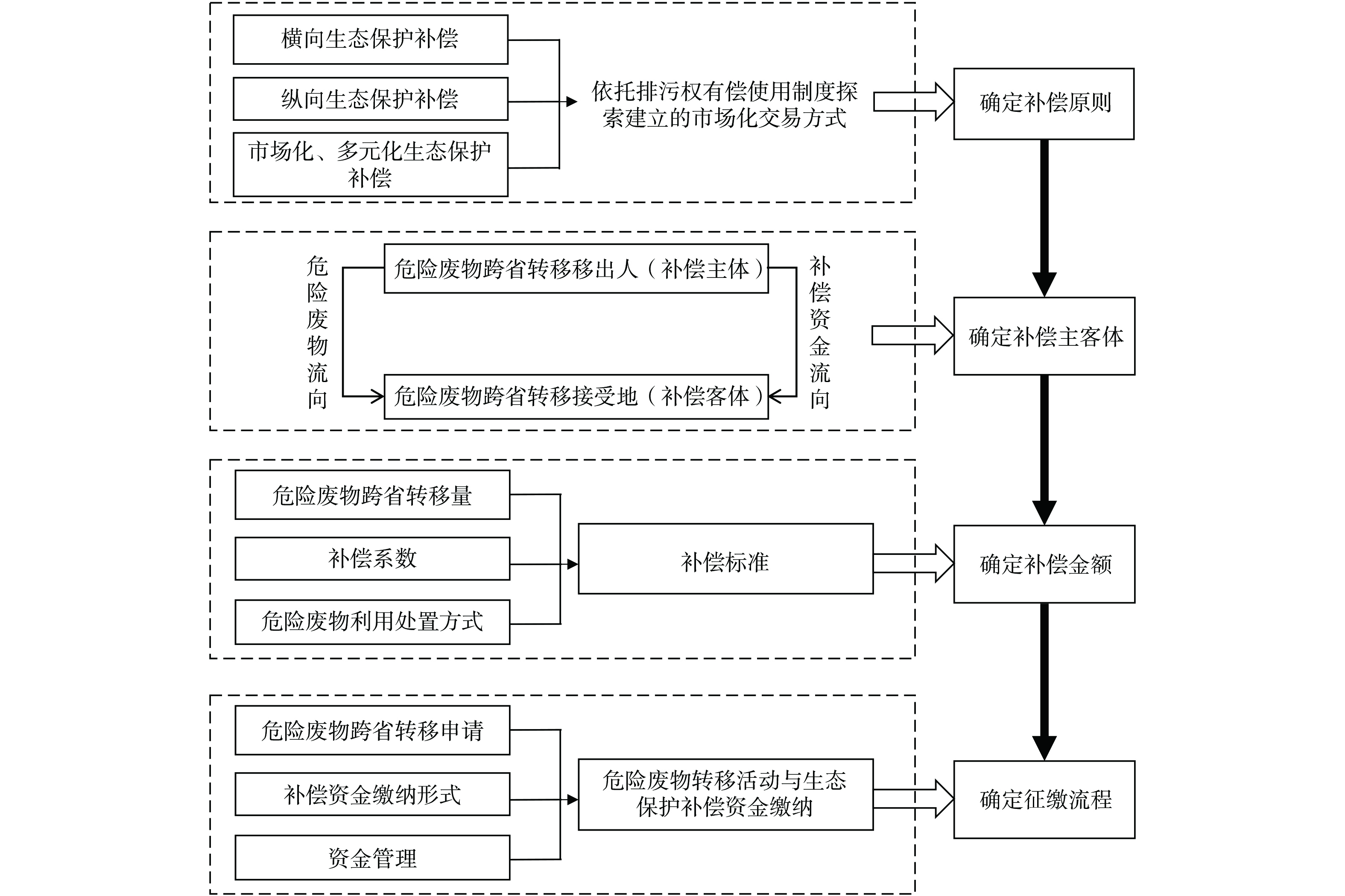 图 1 危险废物跨省转移生态补偿框架Figure 1. Ecological compensation framework for cross provincial transfer of hazardous waste
图 1 危险废物跨省转移生态补偿框架Figure 1. Ecological compensation framework for cross provincial transfer of hazardous waste2. 危险废物跨省转移生态补偿模型构建
当前国内外生态补偿标准的计算方法有支付意愿法、机会成本法、收入损失法、总成本修正模型、费用分析法等[12]。其中,支付意愿法受地区经济水平以及人们意识程度的影响较大,且往往得到补偿标准较低,如不进行细致足量的问卷调查,则可能出现重大偏差[13];机会成本法所计算出来的标准往往会高于补偿者的支付意愿,甚至超出他们的支付能力[14];费用分析法因费用的不确定性和标准的动态变化,使其在具体实施过程中存在技术难度[15]。
总成本修正模型是在收入损失法计算总成本的基础上引入修正系数,对受偿对象生态建设的各项直接成本和间接成本进行修正,使计算模型更科学、公平。如在计算新安江流域生态补偿标准时,刘玉龙等[16]将投入成本分为直接和间接投入两部分:直接投入包括林业建设与保护成本、水土流失治理投入和污染防治投入。间接投入包括发展节水投入、移民安置投入和限制产业发展的损失。水源涵养与生态保护的直接成本与间接成本共同构成了上游地区生态建设与保护的总成本,以此作为生态补偿计算的依据,在进行投入成本分担分析时,引入水量分摊系数、水质修正系数、效益修正系数,计算生态保护的综合效益。
本研究所构建的危险废物跨省转移处置生态补偿模型基于总成本修正模型,综合考虑危险废物转入地生态建设与保护的各项直接成本 (DC) 和间接成本 (IC) ,同时引入危险废物跨省转移生态补偿系数Kc、危险废物转移处置方式系数 (Km) 进行模型修正,计算危险废物跨省转移生态补偿量。
2.1 受偿区生态建设与保护的总成本分析
对于受偿区来说,用于危险废物污染防治及生态环境保护方面的直接成本 (DC) 包括:①危险废物运输、贮存及处置利用过程中因污染防治所支出的治理费用 (EGC,104元);②因危险废物填埋场建设所支出的土地资源开发补偿费用等 (LRC,104元);③危险废物焚烧所支出的年平均大气治理成本 (AGC,104元)。间接成本为因危险废物利用处置设施建设所损失的机会成本 (OC,104元)。
危险废物运输、贮存及处置利用过程中因污染防治所支出的治理费用 (EGC) 包含发生环境污染事故时所产生的治理费用和正常生产经营过程中支出的环境治理费用。其中,发生环境污染事故时所产生治理费用包括:①清除污染、修复生态环境费用;②生态环境受到损害至修复完成期间服务功能丧失导致的损失;③生态环境功能永久性损害造成的损失等合理费用[17]。具体数据可从省级生态环境损害赔偿制度改革工作领导小组办公室获取。EGC应取近3年受偿区危险废物运输、贮存及处置利用过程中因污染防治所支出的年平均治理费用进行计算。
根据土地有偿使用制度,国有土地有偿使用的法定方式包括国有土地使用权出让、国有土地租赁、国有土地使用权作价入股。危险废物填埋场建设所支出的土地资源开发补偿费用 (LRC) 可通过危险废物经营单位支付的土地使用权出让金、土地租金、土地使用费或者场地使用费等结合危险废物填埋场占地面积进行核算。同时,还应考虑危险废物经营单位所缴纳的新增建设用地有偿使用费、征地补偿安置费、耕地开垦费等土地资源开发成本。LRC应取近3年受偿区因危险废物填埋场建设所支出的年平均土地资源开发补偿费用进行计算。
危险废物焚烧过程产生大量烟气,虽经烟气净化设施处理后能够实现达标排放,但也会给受偿区大气环境带来污染,造成不必要的大气治理支出,因此还需补偿受偿区因危险废物焚烧所支出的大气治理成本 (AGC) 。
间接成本 (IC) 包括受偿区因危险废物利用处置设施建设所损失的机会成本 (OC) 。受偿区因危险废物利用处置设施的建设,可能面临产业发展受限、“邻避效应”等问题,由此造成的财政收入损失、税收损失、就业损失等经济成本也应考虑在内。
综上,受偿区因危险废物污染防治所支出的生态环境保护总成本:C=DC+IC=EGC+LRC+AGC+OC。
2.2 生态补偿系数分析
将受偿区因危险废物污染防治所支出的生态环境保护总成本C与近3年受偿区危险废物年平均利用处置量D总 (104 t) 相除,得出近3年受偿区利用处置单位质量危险废物所付出的生态环境保护成本,即危险废物跨省转移生态补偿系数Kc (元·t−1) ,见式(1)。
Kc=EGC+LRC+AGC+OCD总 (1) 移出人将危险废物跨省转移至受偿区,占据受偿区的环境容量;同时,在受偿区生态建设和保护持续投入的作用下,地区危险废物利用处置能力得以保障。基于这样的危险废物利用处置状况,确定移出人对受偿区生态建设和保护成本按转移量进行的分摊为危险废物跨省转移生态补偿系数Kc (元·t−1) 与危险废物转移量T (t) 的乘积,见式(2)。
P=T×Kc (2) 2.3 危险废物利用处置系数分析
在危险废物跨省转移利用处置过程中,不同的利用处置方式对受偿区的环境影响程度也不同,本研究引入危险废物利用处置方式系数Km对受偿区转移量分摊金额进行修正。Km的选取根据不同危险废物利用处置方式对环境的影响程度而得出。如危险废物填埋处置过程,实质上只是暂时隔离和封存,并未消除其危险特性和潜在隐患,会占用移入地的土地资源,同时存在渗滤液污染土壤和地下水的风险[18],因此危险废物若采用填埋处置,应承担较高的分摊系数。危险废物焚烧处置过程对土壤和地下水的危害风险较低,虽焚烧过程产生的残渣、飞灰以及烟气中的烟尘、重金属类、二噁英类等污染物也将给受偿区带来新的环境风险[19],但焚烧过程排气集中且较易管理,尾气达标排放后对大气的污染程度较低,因此危险废物若采用焚烧处置,承担的分摊系数应小于填埋处置。危险废物资源化利用过程能够减少危险废物排放,同时回收能再次利用的物质,降低危险废物对周边环境的危害[20],因此所承担的分摊系数最小,但值得注意的是且若资源化利用后继续产生新的危险废物则应根据新产生危险废物的处置方式重新计算分摊费用。
基于以上讨论,本研究设置转入受偿区填埋的危险废物Km取2.0,转入受偿区焚烧的危险废物Km取1.2,转入受偿区利用及以其他方式处置的危险废物Km取1.0。
综上可得,考虑危险废物转移量分摊和利用处置方式修正后移出人对受偿区的生态补偿量如式(3)所示。
P=T×Kc×Km (3) 其中,Kc的公式表示为式(4)。
Kc=EGC+LRC+AGC+OCD总 (4) 式中:P为移出人应缴纳的生态补偿资金,元;T为移出人所在地省级生态环境部门批准转移危险废物的重量,t;Kc为生态补偿系数,元·t−1;Km为危险废物利用处置方式系数;EGC为近3年受偿区危险废物运输、贮存及处置利用过程中因生态环境保护所支出的年平均治理费用,104 元;LRC为近3年受偿区因危险废物填埋场建设所支出的年平均土地资源开发补偿费用,104 元;AGC为近3年受偿区因危险废物焚烧所支出的年平均大气治理成本,104 元;OC为近3年受偿区因危险废物利用处置设施建设所损失的年平均机会成本,104 元;D总为近3年受偿区危险废物年平均利用处置量,104 t。
3. 危险废物跨省转移生态补偿结果分析
3.1 受偿区生态环境保护成本计算
生态环境保护成本测算是生态补偿机制运行的前提和保障,但由于生态补偿行为主体的多元性和过程的复杂性使得难以按照统一的标准计算生态补偿成本[21]。例如刘玉龙等[16]通过调查收集上游地区生态建设与保护的年各项直接成本构建流域生态建设与保护补偿模型,但调查收集过程具有明显主观性,调查方法的不同也会造成生态环境保护成本测算偏差。同时,从目前我国开展的横向生态补偿上看,生态补偿成本与补偿标准并没有统一的计算方法,大多由补偿双方协商制定,从出台的对口支援补偿标准上看,补偿金额也并非经过公式计算得出,而多是中央政府、补偿方和受偿方3方博弈的结果。
本研究以各省公布的近3年全省 (市) 生态环保支出及固体废物与化学品支出为依据进行受偿区因危险废物污染防治所支出的生态环境保护总成本 (即公式中EGC+LRC+AGC+OC) 初步计算。根据经验值,危险废物污染防治支出约为全省生态环保支出的1%,其中政府支出和企业支出分别约占0.5%。基于此,本研究以某4个临近省份为例进行受偿区生态环境保护成本计算。
根据4省份发布的近3年全省 (市) 节能环保支出 (表1) ,计算得出A、B、C、D省年生态环境保护成本 (即EGC+LRC+AGC+OC) 分别为19 886.7、19 716.7、20 110.0、33 080.0×104元。
表 1 近3年研究区域全省 (市) 节能环保支出Table 1. Energy conservation and environmental protection expenditure of the province (city) in the research area in the past three years108 元 年份 A省 B省 C省 D省 2022年 203.8 201.2 223.2 280.1 2021年 212.2 199.1 203.8 338.6 2020年 180.6 191.2 176.3 373.7 近3年平均值 198.9 197.2 201.1 330.8 | Show Table DownLoad:
CSV
DownLoad:
CSV
3.2 生态补偿系数计算
受偿区危险废物跨省转移生态补偿系数为近3年因危险废物污染防治所支出的年平均生态环境保护总成本 (EGC+LRC+AGC+OC) 与危险废物年平均利用处置量 (D总) 的比值。近3年该研究区域危险废物年平均利用处置量 (D总) 见表2。根据表1、表2,计算得出A、B、C、D省危险废物跨省转移生态补偿系数 (Kc) 分别为256.5、83.6、47.5、58.4元·t−1。
表 2 近3年研究区域危险废物年平均利用处置量Table 2. Annual average utilization and disposal of hazardous waste in the research area in the past three years104 t 年份 A省 B省 C省 D省 2022年 85.4 256.3 503.3 615.0 2021年 79.0 264.4 391.0 530.0 2020年 68.2 187.2 376.1 554.1 近3年平均值 77.5 236.0 423.5 566.4 | Show TableDownLoad:
CSV
3.3 危险废物跨省转移生态补偿资金计算
由2.3章节可知,危险废物跨省转移时,每吨危险废物应缴纳的生态补偿资金为Kc×Km;代入3.2章节计算结果,得出金额见表3。
表 3 每吨危险废物应缴纳的生态补偿资金Table 3. Ecological compensation funds to be paid per ton of hazardous waste利用处置方式 危险废物利用处置方式系数 受偿金/(元·t−1) A省 B省 C省 D省 填埋 2.0 513.0 167.1 95.0 116.8 焚烧 1.2 307.8 100.3 57.0 70.1 利用 1.0 256.5 83.6 47.5 58.4 其他 1.0 256.5 83.6 47.5 58.4 | Show TableDownLoad:
CSV
根据计算结果,转入A省的危险废物每吨应缴纳的生态补偿资金明显高于其余3省份,这是因为A省生态环境容量有限而造成生态环境保护总成本较高。同时A省危险废物利用处置能力存在结构上的短板[22],近3年危险废物利用处置量偏低,部分类别危险废物依赖跨省转移利用,因此计算结果明显高于其他省份。
从B、C、D 3省计算结果来看,转入受偿区填埋的危险废物每吨应缴纳95.0~167.1元的生态补偿资金,转入受偿区焚烧的危险废物每吨应缴纳57.0~100.3元的生态补偿资金,转入受偿区利用及以其他方式利用处置的危险废物每吨应缴纳47.5~83.6元生态补偿资金。根据李静等[23]的研究结果,2021年我国危险废物填埋价格为
1300 ~3150 元·t−1,焚烧价格为2 500~3 500元·t−1,协同处置价格为2 000~2170 元·t−1。基于表3,本研究所计算得出的危险废物跨省转移生态补偿资金占危废处置费用的比例为:填埋3.0%~12.9%、焚烧1.6%~2.9%、协同处置2.2%~4.2%。3.4 2022年危险废物跨省转移生态补偿资金试算
以市场化交易方式进行生态补偿时,移出人应缴纳的危险废物跨省转移生态补偿资金为每吨危险废物应缴纳的生态补偿资金与移出量的乘积。资金缴纳形式可以依托国家危险废物信息管理系统,在危险废物跨省转移申请批准后依据危险废物拟移出量、拟利用处置方式等缴纳生态补偿资金。
当前,我国部分省份危险废物利用处置设施存在“吃不饱”或“吃不完”等结构性失衡现象,省份间危险废物利用处置能力存在差距[24]。基于强化跨区域合作、开展区域联防联治等危险废物环境管理政策,各省份在转出危险废物的同时,也能成为危险废物接受地,既是危险废物生态补偿机制中的补偿主体也是补偿客体。为便于4省份间了解掌握全年危险废物生态补偿资金总额,本研究基于2022年4省份间危险废物转移总量 (图2) ,计算2022年4省需缴纳、应收取的危险废物生态补偿资金,如表4、表5所示。
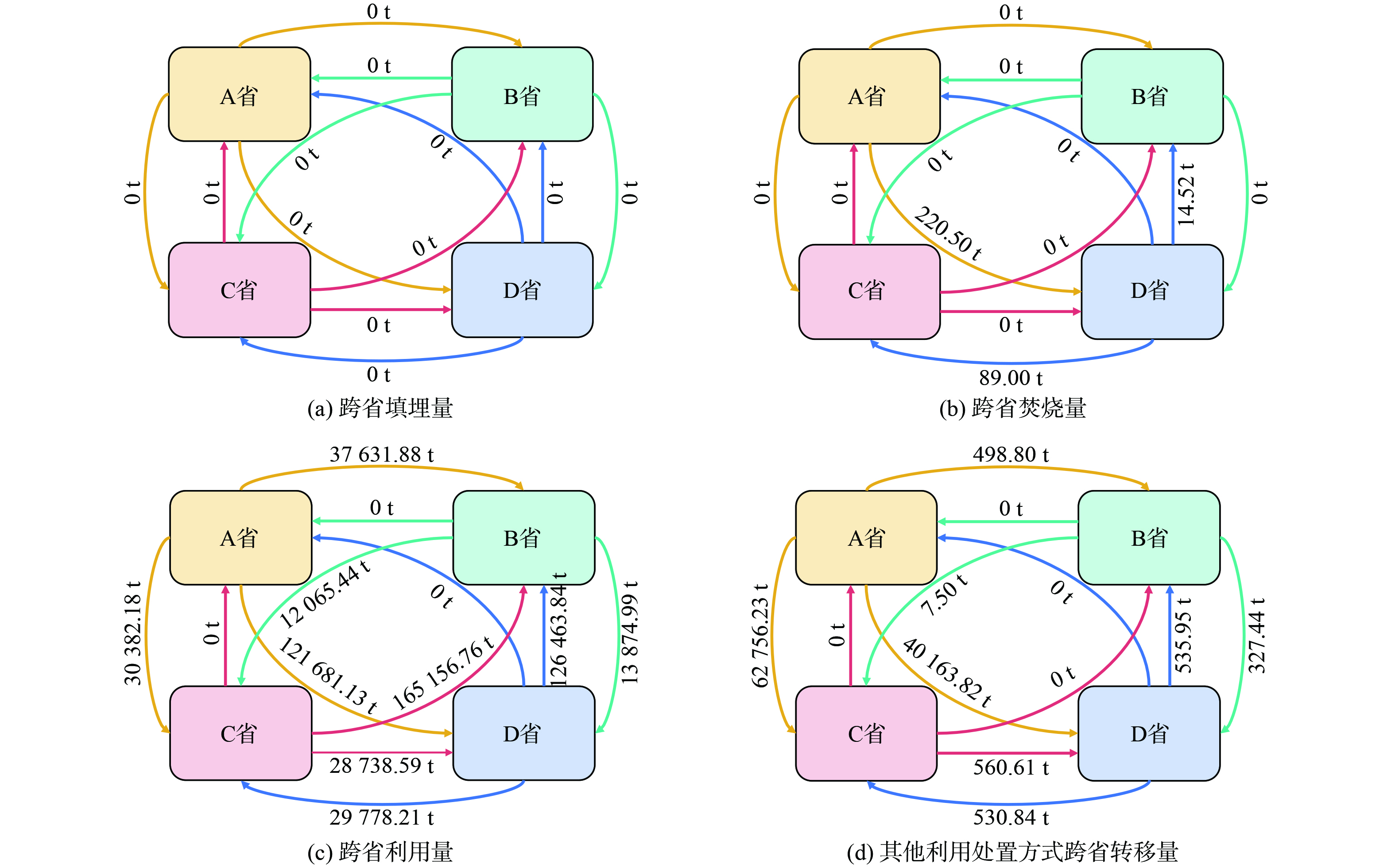 图 2 2022年4省份危险废物转移量Figure 2. Hazardous waste transfer volume (tons) in four provinces in 2022表 4 2022年应缴纳的危险废物生态补偿资金Table 4. Ecological compensation funds for hazardous waste to be paid in 2022
图 2 2022年4省份危险废物转移量Figure 2. Hazardous waste transfer volume (tons) in four provinces in 2022表 4 2022年应缴纳的危险废物生态补偿资金Table 4. Ecological compensation funds for hazardous waste to be paid in 2022元 移出地 受偿区 填埋 焚烧 利用 其他 总计 总金额 A省 B省 0 0 3 146 024.99 41 699.68 3 187 724.67 16 327 211.40 C省 0 12 568.50 5 779 853.52 1 907 781.23 7 700 203.25 D省 0 0 1 774 319.37 3 664 964.11 5 439 283.48 B省 A省 0 0 0 0 0 1 379 674.85 C省 0 0 659 062.03 15 553.40 674 615.43 D省 0 0 704 621.64 437.78 705 059.42 C省 A省 0 0.00 0 0 0 12 388 757.12 D省 0 70.10 1 739 047.46 31 001.03 1 770 118.59 B省 0 1 456.36 10 572 377.04 44 805.13 10 618 638.53 D省 A省 0 0 0 0 0 15 198 816.80 C省 0 0 1 365 083.15 26 628.79 1 391 711.94 B省 0 0 13 807 104.86 0.00 13 807 104.86 | Show TableDownLoad:
CSV
表 5 2022年应收取的危险废物生态补偿资金Table 5. Ecological compensation funds for hazardous waste to be collected in 2022元 受偿区 移出地 填埋 焚烧 利用 其他 总计 总金额 B省 A省 0 0 3 146 024.99 41 699.68 3 187 724.67 27 613 468.06 D省 0 0 13 807 104.86 0.00 13 807 104.86 C省 0 1 456.36 10 572 377.04 44 805.13 10 618 638.53 C省 A省 0 12 568.50 5 779 853.52 1 907 781.23 7 700 203.25 9 766 530.62 B省 0 0 659 062.03 15 553.40 674 615.43 D省 0 0 1 365 083.15 26 628.79 1 391 711.94 D省 A省 0 0 1 774 319.37 3 664 964.11 5 439 283.48 7 914 461.49 B省 0 0 704 621.64 437.78 705 059.42 C省 0 70.10 1 739 047.46 31 001.03 1 770 118.59 | Show TableDownLoad:
CSV
综上,经试算2022年A、B、C、D省需缴纳的危险废物生态补偿资金分别为1 632.7、138.0、1 238.9、1 519.9万元;应收取的补偿资金分别为0、2 761.3、791.4、976.7万元。
4. 生态补偿资金缴纳流程
本研究基于《危险废物转移管理办法》规定的危险废物跨省转移管理程序设计生态补偿资金缴纳流程(图3)。主要内容如下:1) 移出人应当通过国家危险废物信息管理系统 (以下简称信息系统) 填写危险废物跨省转移申请表,向危险废物移出地省级生态环境主管部门提出申请;2) 危险废物移出地省级生态环境主管部门初步审核同意移出的,通过信息系统向受偿区省级生态环境主管部门发出跨省转移商请函;3) 受偿区省级生态环境主管部门同意接受的,应通过信息系统函复移出地省级生态环境主管部门同意接受的意见。函复意见中应载明根据批准跨省转移危险废物的决定中批准的拟移出量、拟利用处置方式;4) 跨省转移危险废物的申请经批准后,移出人应当按照批准跨省转移危险废物的决定填写、运行危险废物转移联单,实施危险废物转移活动;5) 移出人缴纳危险废物跨省转移生态补偿资金的义务发生时间为发生转移危险废物活动的当日。危险废物跨省转移生态补偿资金按月计算,按季申报缴纳。移出人应当自季度终了之日起15日内,向受偿区税务部门办理申报并缴纳。
 图 3 危险废物跨省转移生态补偿资金缴纳流程和管理程序Figure 3. Payment process and management procedure for ecological compensation funds for hazardous waste transfer across provinces
图 3 危险废物跨省转移生态补偿资金缴纳流程和管理程序Figure 3. Payment process and management procedure for ecological compensation funds for hazardous waste transfer across provinces受偿区发展改革、生态环境、财政、税务等主管部门应按照各自职能负责危险废物跨省转移生态补偿征收标准的制定、征收资金核定、经费保障以及征收管理等工作,如应根据本省情况,及时核算和公布本省危险废物跨省转移生态补偿系数 (Kc) 、利用处置方式系数 (Km) ,明确外省转入危险废物生态补偿资金缴纳形式等。受偿区可根据属地实际情况,积极探索将危险废物跨省转移生态补偿资金按照政府非税收入纳入财政预算、环保基金、中华慈善基金等多种方式管理、使用和市场化运作。
5. 结论和展望
本研究基于相关法规政策,提出构建多省市间跨省转移生态补偿机制的必要性。以长江三角洲区域4个邻近省为例,通过搜集相关环保数据,应用收入损失模型,计算其危险废物跨省转移生态补偿标准,并对具体的征缴流程进行详细的方案制定,提高了实施的可能性。
开展多省市危险废物跨省转移生态补偿机制是大势所趋,需要建立和完善相关法律法规,科学界定保护者与受益者权利义务,合理制定补偿标准,加快形成受益者付费、保护者得到合理补偿的运行机制。破除跨区域合作的行政壁垒,应因地制宜建立强适应性的生态补偿体系,以政府为主导,发挥政府和市场的双重机制作用,不断培育市场主体,尽快健全市场化、多元化生态保护补偿机制,保证生态补偿机制的持续稳定运行。按照权责一致、分类分级的方式开展,做好各类型、各层级生态保护补偿政策的衔接配合,形成共同推动生态保护工作的合力。另外,还应强化信息公开,鼓励公众积极参与监督,消除公众顾虑,建立良好互信。
-
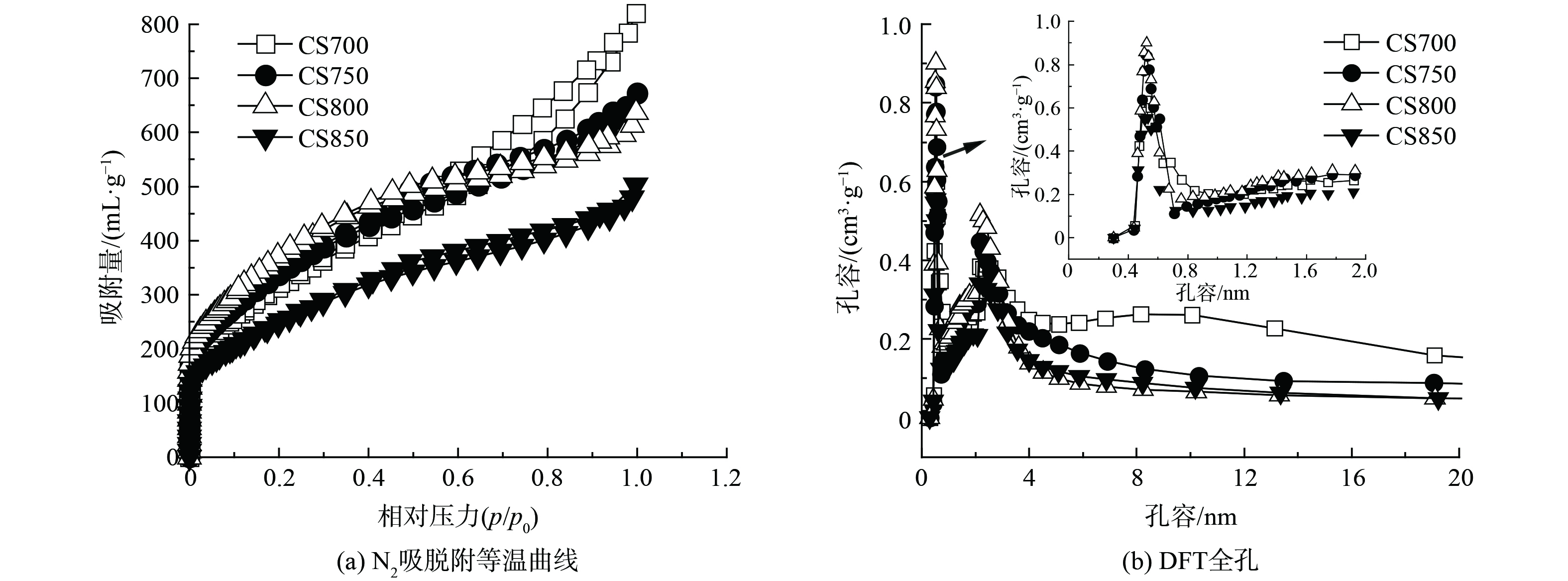
图 1 CS700、CS750、CS800和CS850的N2吸脱附等温曲线和DFT全孔分布
Figure 1. N2 adsorption-desorption isotherms and DFT total pore distribution of CS700, CS750, CS800 and CS850
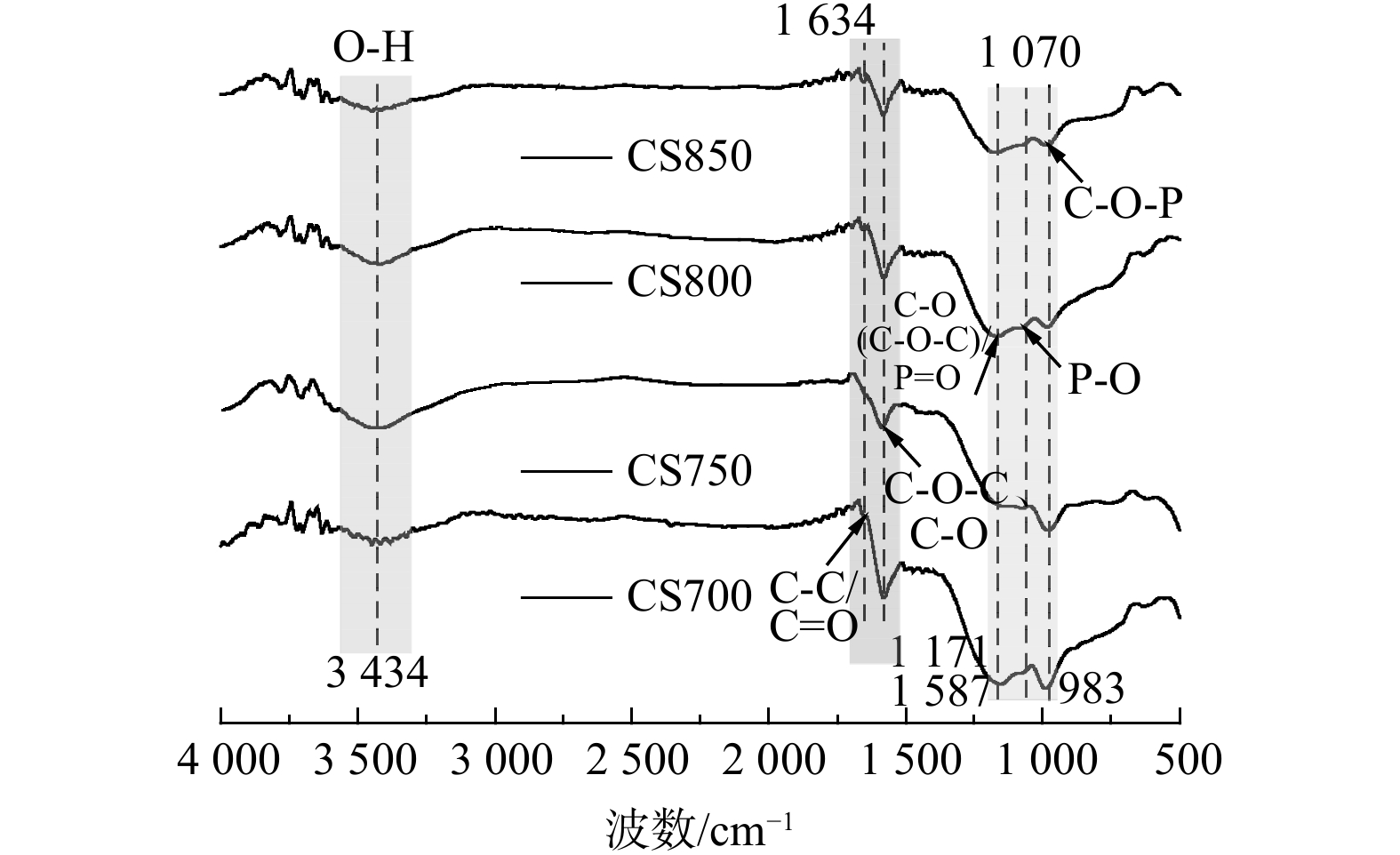
图 2 CS700、CS750、CS800和CS850的FTIR曲线图
Figure 2. FTIR curves of CS700, CS750, CS800 and CS850
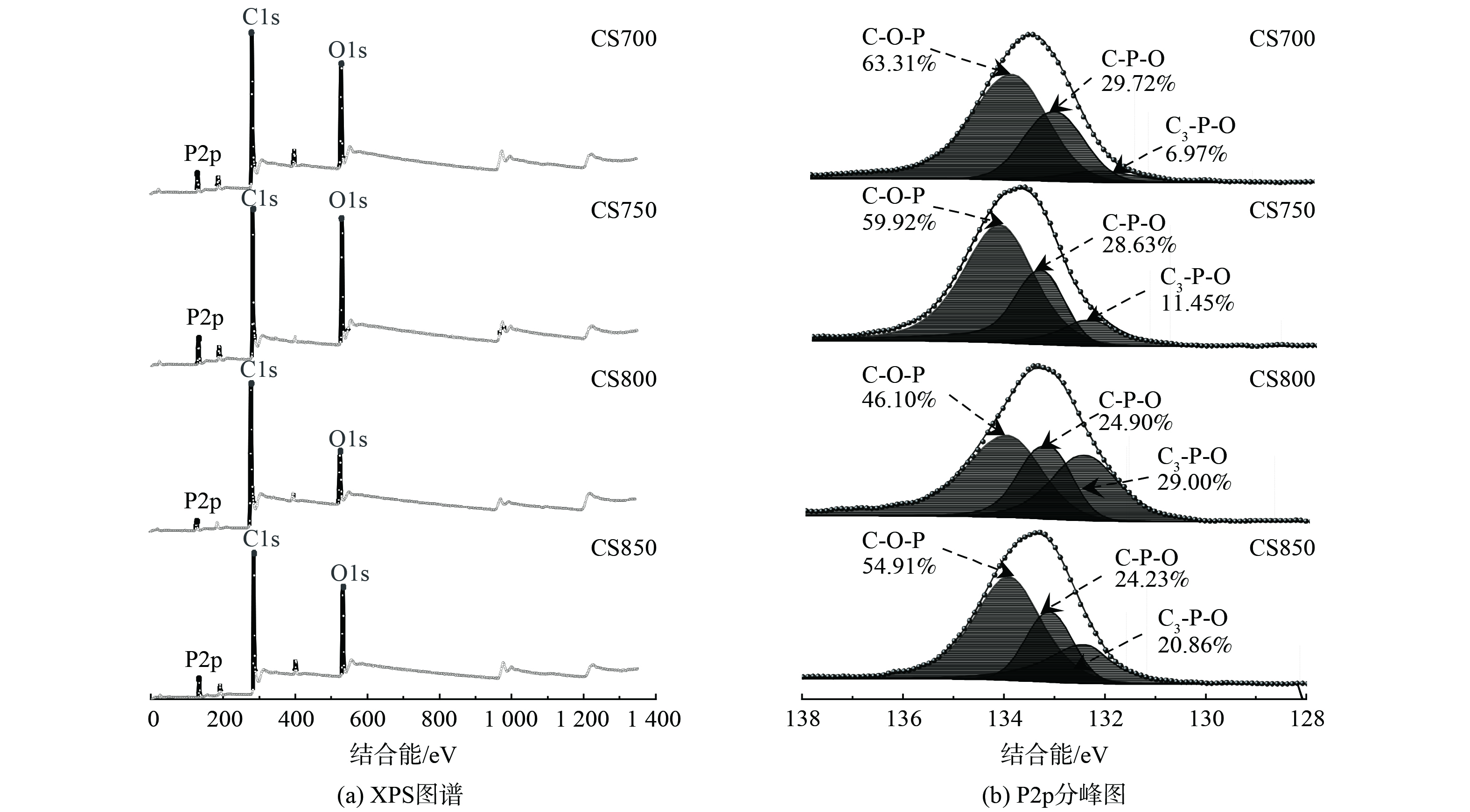
图 3 CS700、CS750、CS800和CS850的XPS图谱和P2p分峰拟合图
Figure 3. XPS spectra and P2p spectrum of CS700, CS750, CS800 and CS850
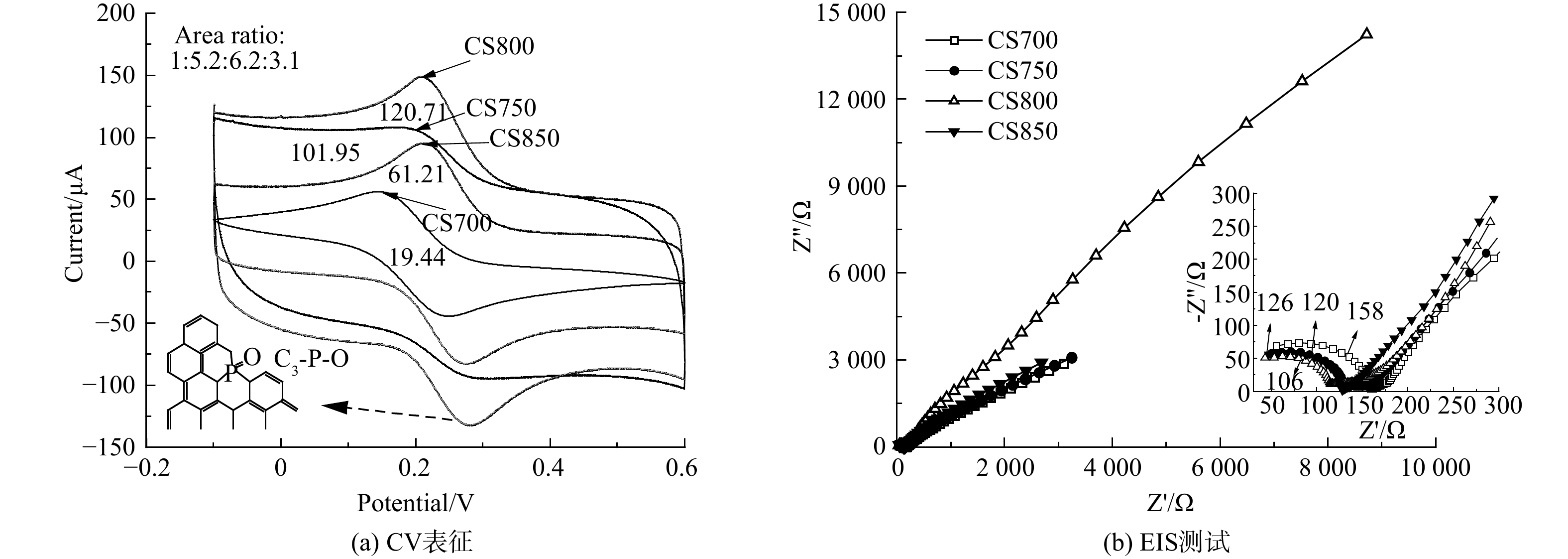
图 5 CS700、CS750、CS800和CS850 的CV曲线和EIS曲线
Figure 5. CV curves and EIS curves of CS700,CS750,CS800 and CS850
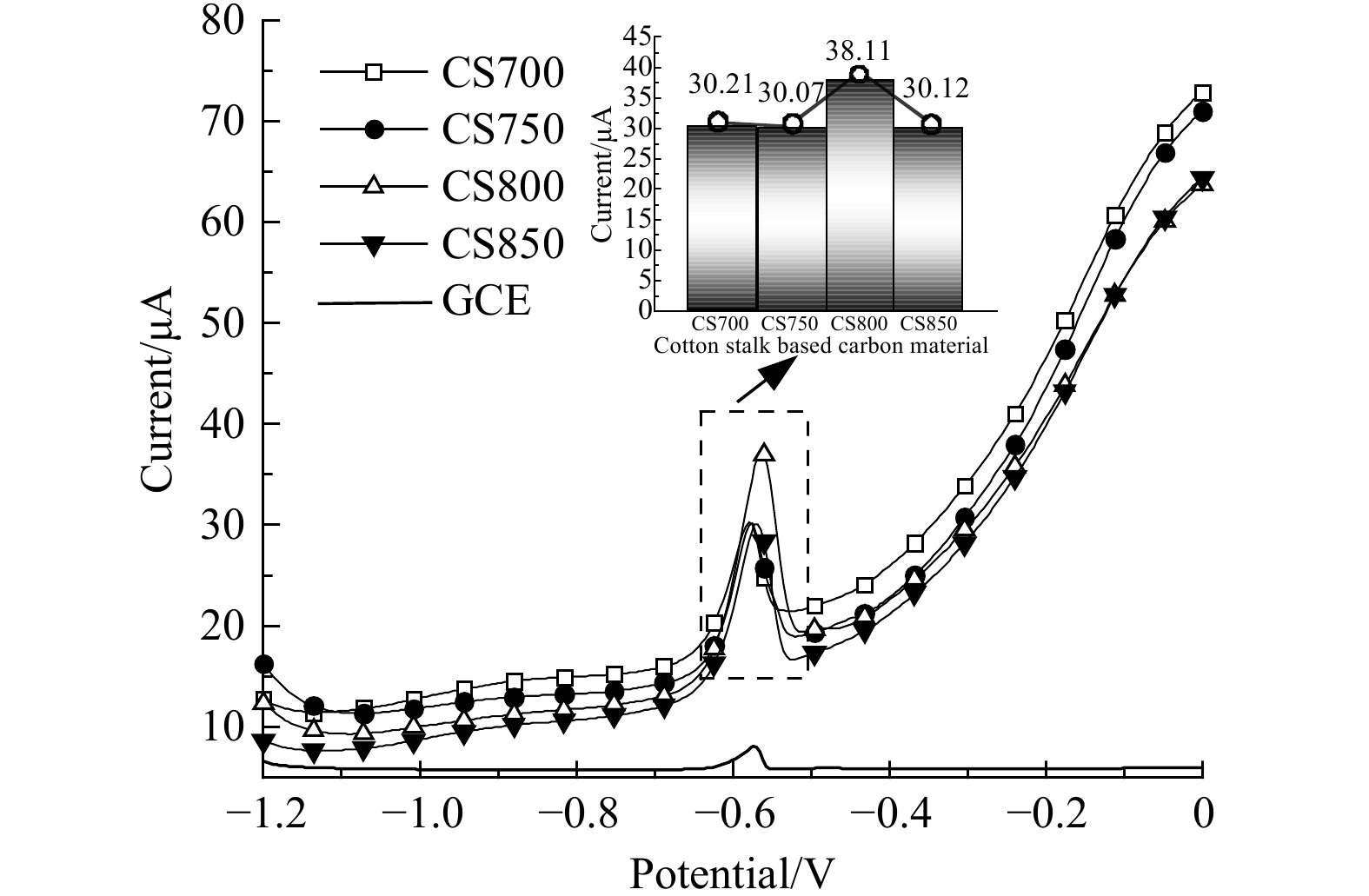
图 6 CS700、CS750、CS800、CS850和GCE在500 μg·L−1 Pb2+、 pH为3.6的HAC-NaAC缓冲溶液中的ASDPV测试曲线
Figure 6. ASDPV test curves of CS700, CS750, CS800, CS850 and GCE in 500 μg·L−1 Pb2+HAC-NaAC buffer solution with pH 3.6
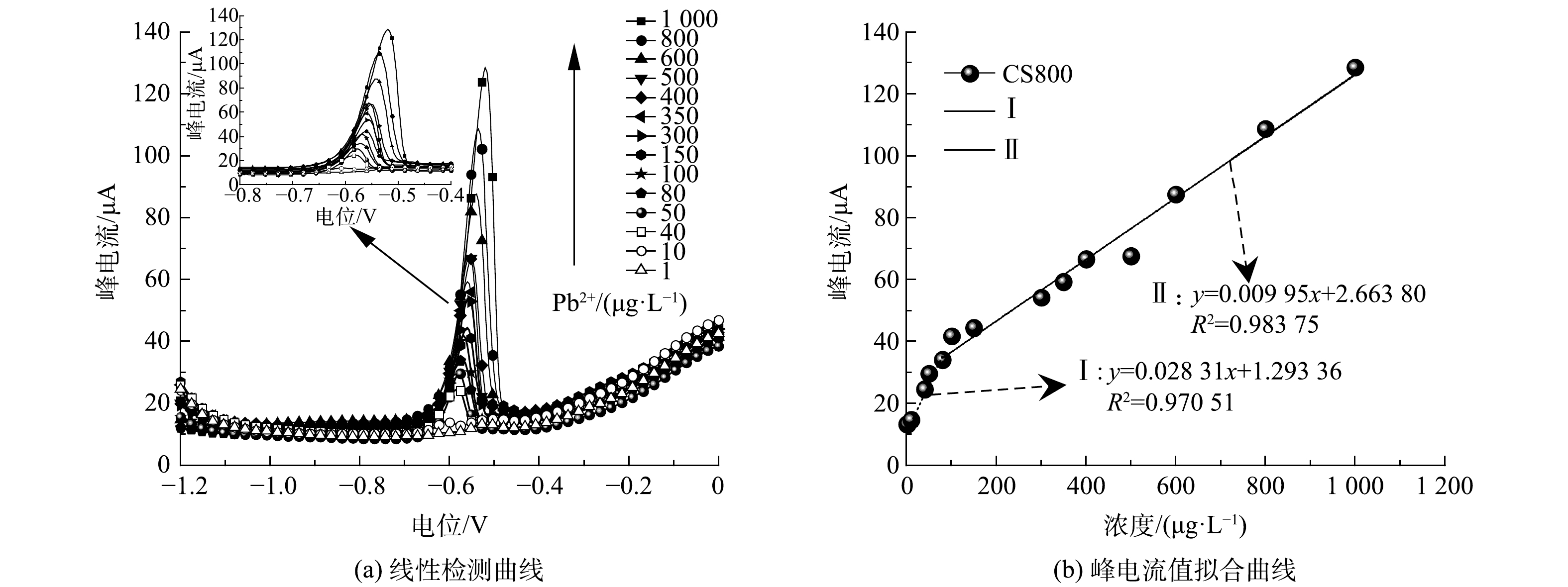
图 7 CS800/GCE对不同浓度梯度的Pb2+溶液 、pH 为4的线性检测曲线与峰电流值的拟合曲线
Figure 7. The linear detection curves of CS800/GCE with pH 4 in Pb2+solution with different concentration gradients and the fitting curves of peak current value
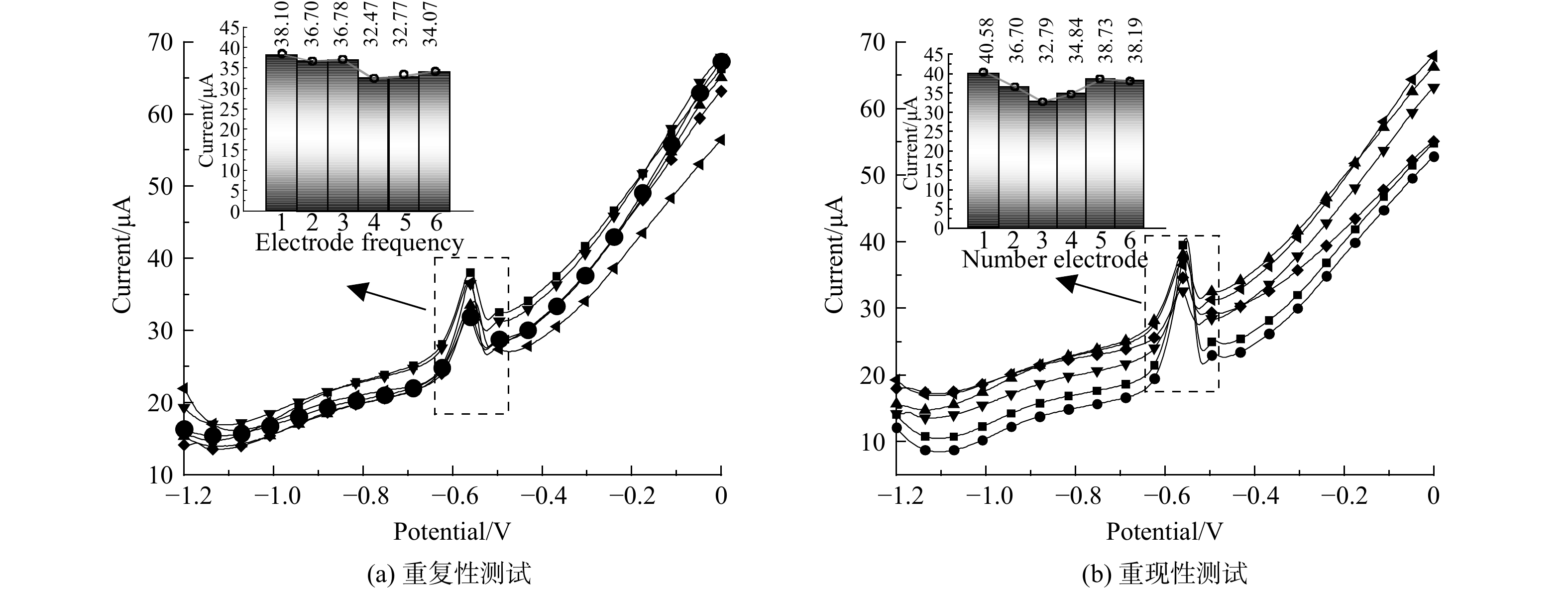
图 8 同一支CS800/GCE对浓度为25 μg·L−1的Pb2+溶液的ASDPV测试曲线与多个CS800/GCE对浓度为25 μg·L−1的Pb2+溶液的ASDPV测试曲线
Figure 8. The ASDPV test curves of the same CS800/GCE for Pb2+solution with a concentration of 25 μg·L−1 and the ASDPV test curves of multiple CS800/GCE for Pb2+solution with a concentration of 25 μg·L−1
表 1 棉秆炭材料的孔径特征
Table 1. Pore size characteristics of cotton stalk carbon materials
炭材料 比表面积/ (m2·g−1) 总孔体积/ (mL·g−1) 中孔体积/ (mL·g−1) 微孔体积/ (mL·g−1) 微孔占总孔体积/ % 平均孔径/nm CS700 1 153.74 1.25 1.22 0.42 33.60 4.32 CS750 1 238.36 1.02 0.97 0.45 44.10 3.31 CS800 1 338.57 0.95 0.88 0.49 51.60 2.85 CS850 909.09 0.75 0.71 0.33 44.00 3.30
下载: 导出CSV
表 2 CS700、CS750、CS800和CS850的表面原子比例
Table 2. Surface atomic ratio of CS700, CS750, CS800 and CS850
% 表面原子 CS700 CS750 CS800 CS850 P2p C1s 70.14 65.28 83.01 72.00 O1s 23.31 27.32 13.39 22.41 P2p 6.55 7.40 3.60 5.59 C-O-P 63.31 59.92 46.10 54.91 C-P-O 29.72 28.63 24.90 24.23 C3-P-O 6.97 11.45 29.00 20.86
下载: 导出CSV
-
[1] KANG X, WANG J, WU H, et al. A graphene-based electrochemical sensor for sensitive detection of paracetamol[J]. Talanta, 2010, 81(3): 754-759. doi: 10.1016/j.talanta.2010.01.009 [2] 鲁猷栾, 穆新伟, 黄乐舒, 等. 生物质炭材料: 构建电化学传感器的理想修饰材料[J]. 材料导报, 2022, 36(6): 5-12. [3] YANG H, CHEN P, CHEN W, et al. Insight into the formation mechanism of N, P co-doped mesoporous biochar from H3PO4 activation and NH3 modification of biomass[J]. Fuel Processing Technology, 2022, 230: 107215. doi: 10.1016/j.fuproc.2022.107215 [4] CHEN Y, ZHANG X, CHEN W, et al. The structure evolution of biochar from biomass pyrolysis and its correlation with gas pollutant adsorption performance[J]. Bioresource Technology, 2017, 246: 101-109. doi: 10.1016/j.biortech.2017.08.138 [5] ZHU G, DENG X, HOU M, et al. Comparative study on characterization and adsorption properties of activated carbons by phosphoric acid activation from corncob and its acid and alkaline hydrolysis residues[J]. Fuel Processing Technology, 2016, 144: 255-261. doi: 10.1016/j.fuproc.2016.01.007 [6] ADHIKARI M P, ADHIKARI R, SHRESTHA R G, et al. Nanoporous activated carbons derived from agro-waste corncob for enhanced electrochemical and sensing performance[J]. Bulletin of the Chemical Society of Japan, 2015, 88(8): 1108-1115. doi: 10.1246/bcsj.20150092 [7] GUO D, SHIBUYA R, AKIBA C, et al. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts[J]. Science, 2016, 351(6271): 361-365. doi: 10.1126/science.aad0832 [8] PAN J, DENG H, DU Z, et al. Design of nitrogen-phosphorus-doped biochar and its lead adsorption performance[J]. Environmental Science and Pollution Research, 2022, 29(19): 28984-28994. doi: 10.1007/s11356-021-17335-3 [9] 任洪波, 秦元成, 尚承伟, 等. 聚丙烯酰倍半硅氧烷(MPMS-SSO)有机/无机杂化气凝胶中的N2等温吸脱附特性分析[J]. 稀有金属材料与工程, 2010, 39: 475-478. [10] DAHAGHIN Z, Kilmartin P A, Mousavi H Z. Novel ion imprinted polymer electrochemical sensor for the selective detection of lead (II)[J]. Food Chemistry, 2020, 303: 125374. doi: 10.1016/j.foodchem.2019.125374 [11] SHAH A, Zahid A, Khan A, et al. Development of a highly sensitive electrochemical sensing platform for the trace level detection of lead ions[J]. Journal of the Electrochemical Society, 2019, 166(9): B3136. doi: 10.1149/2.0271909jes [12] HU J Y, LI Z, ZHAI C Y, et al. Plasmonic photo-assisted electrochemical sensor for detection of trace lead ions based on au anchored on two-dimensional gC3N4/graphene nanosheets[J]. Rare Metals, 2021, 40: 1727-1737. doi: 10.1007/s12598-020-01659-z [13] LU C, ZHANG X, GAO Y, et al. Parametric study of catalytic co-gasification of cotton stalk and aqueous phase from wheat straw using hydrothermal carbonation[J]. Energy, 2021, 216: 119266. doi: 10.1016/j.energy.2020.119266 [14] LIANG X, GUO N, ZHAO Y, et al. Rapid effectual entrapment of pesticide pollutant by phosphorus-doped biochar: Effects and response sequence of functional groups[J]. Journal of Molecular Liquids, 2022, 365: 120155. doi: 10.1016/j.molliq.2022.120155 [15] SAKA C. Phosphorus and sulphur-doped microalgae carbon as a highly active metal-free catalyst for efficient hydrogen release in NaBH4 methanolysis[J]. Fuel, 2022, 309: 122183. doi: 10.1016/j.fuel.2021.122183 [16] SHI C, HU K, NIE L, et al. Degradation of acetaminophen using persulfate activated with P-doped biochar and thiosulfate[J]. Inorganic Chemistry Communications, 2022, 146: 110160. doi: 10.1016/j.inoche.2022.110160 [17] 刘鹏, 李勇, 宁廷州, 等. 南疆棉秆能源炭制备工艺及其傅里叶红外光谱分析[J]. 江苏农业科学, 2018, 46(16): 190-193. doi: 10.15889/j.issn.1002-1302.2018.16.047 [18] OZPINAR P, DOGAN C, DEMIRAL H, et al. Activated carbons prepared from hazelnut shell waste by phosphoric acid activation for supercapacitor electrode applications and comprehensive electrochemical analysis[J]. Renewable Energy, 2022, 189: 535-548. doi: 10.1016/j.renene.2022.02.126 [19] ZHANG L, YANG Z, LI S, et al. Comparative study on the two-step pyrolysis of different lignocellulosic biomass: Effects of components[J]. Journal of Analytical and Applied Pyrolysis, 2020, 152: 104966. doi: 10.1016/j.jaap.2020.104966 [20] PUZIY A M, PODDUBNAYA O I, GAWDZIK B, et al. Phosphorus-containing carbons: Preparation, properties and utilization[J]. Carbon, 2020, 157: 796-846. doi: 10.1016/j.carbon.2019.10.018 [21] PUZIY A M, PODDUBNAYA O I, SOCHA R P, et al. XPS and NMR studies of phosphoric acid activated carbons[J]. Carbon, 2008, 46(15): 2113-2123. doi: 10.1016/j.carbon.2008.09.010 [22] IBRAHIM H, TEMERK Y. A novel electrochemical sensor based on B doped CeO2 nanocubes modified glassy carbon microspheres paste electrode for individual andsimultaneous determination of xanthine and hypoxanthine[J]. Sensors and Actuators B:Chemical, 2016, 232: 125-137. doi: 10.1016/j.snb.2016.03.133 [23] OGINNI O, SINGH K, OPORTO G, et al. Effect of one-step and two-step H3PO4 activation on activated carbon characteristics[J]. Bioresource Technology Reports, 2019, 8: 100307. doi: 10.1016/j.biteb.2019.100307 [24] LIU Z, AI J, SUN M, et al. Phosphorous-doped graphite layers with outstanding electrocatalytic activities for the oxygen and hydrogen evolution reactions in water electrolysis[J]. Advanced Functional Materials, 2020, 30(12): 1910741. doi: 10.1002/adfm.201910741 [25] SHEN Y F, XUE Y, XIA X, et al. Metallic-like boron-modified bio-carbon electrodes for simultaneous electroanalysis for Cd2+, Pb2+and Cu2+: Theoretical insight into the role of CxBOy(H)[J]. Carbon, 2023: 118350. [26] 袁鹏, 邵悦琦, 乔子茹. 长三角流域某采矿区农田土壤重金属污染研究[J]. 广东化工, 2023, 50(6): 143-146. doi: 10.3969/j.issn.1007-1865.2023.06.043 [27] FRISBIE S H, MITCHELL E J, SARKAR B. Urgent need to reevaluate the latest World Health Organization guidelines for toxic inorganic substances in drinking water[J]. Environmental Health, 2015, 14(1): 1-15. doi: 10.1186/1476-069X-14-1 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2053
- HTML全文浏览数: 2053
- PDF下载数: 67
- 施引文献: 0
