









-
挥发性有机化合物(volatile organic compounds,VOC)是室内最常见的环境污染物之一,而甲醛(HCHO)是其中一种典型污染物[1]。当人体处在甲醛质量浓度大于0.1 mg·m-3环境中时,即可能引发呼吸系统受损,而长期暴露会对人体其他器官造成损害[2]。甲醛已被列为第一类致癌物[3-4]。目前,防治室内甲醛污染的方法有物理法、生物法和化学法,其中物理法包括自然通风和吸附法等,生物法包括植物生态净化和微生物降解等,化学法包括等离子体法、催化氧化技术等[5-7]。物理法成本低、效率高,但不能彻底解决甲醛污染问题,亦可能造成二次污染[8];生物法绿色环保,处理成本低,无二次污染,但存在受环境温/湿度、菌种选择等条件限制,效率低等问题[9];等离子体技术虽受环境影响小,但需施加外部条件,耗能高,安全性差[10-11];催化氧化技术已被证明是一种行之有效的甲醛去除方法,其具体包括热催化、光催化、常温催化、臭氧净化等[12-14]。其中,常温催化氧化技术具有操作简单、耗能低、无二次污染、净化效率高等优点而备受广泛关注,其在实现去除室内低浓度气态甲醛方面有着重要应用价值。
常温催化氧化催化剂可分为两大类:一是贵金属催化剂;二是过渡金属氧化物催化剂。贵金属如Pt、Pd、Au和Ag等,通常以TiO2、Al2O3、SiO2、Co3O4、CeO2等高比表面积为载体制备而成,其活性高、降解性能优越[15-17],但是贵金属资源稀缺、价格昂贵,限制了其广泛范围。过渡金属氧化物因资源丰富、氧化活性高、稳定性良好等优势而具备替代贵金属的潜力,且过渡金属锰氧化物(MnOx)在降解甲醛性能方面表现优异,氧化能力强、CO2选择性高,且晶型、价态、形貌可调,应用前景广泛[18-23]。LU等[24]通过水热法制备了石墨烯-MnO2复合纳米材料,相较于单金属催化剂MnO2,复合催化剂完全催化甲醛所需温度降低了75 ℃,催化性能提高,并且石墨烯增强了Mn3+和Mn4+间的电荷转移,其催化剂表面产生了丰富的氢氧自由基,提升了甲醛催化氧化中间产物-甲酸盐的降解活性。ZHU等[25]通过氧化还原法及铈改性制备了掺杂型MnO2,研究发现随着Ce掺杂量的增加,MnO2结晶度降低,催化剂比表面积增大。当Ce-MnO2中铈锰摩尔比达1∶10时,催化剂常温催化性能稳定,且在100 ℃下可将低浓度甲醛完全氧化为CO2和H2O,这是由于Ce掺杂的MnO2具有更多的表面吸附氧,减小了CeO2纳米团簇与MnO2间的晶界间距,提升了表面氧和晶格氧的氧化还原能力和迁移速率。LI等[26]利用Pechini法制备了MnxCe1-xO2 (x=0.3~0.9) 催化剂,其中Mn0.5Ce0.5O2催化剂活性最佳,且随着质量分数为5% CuOx的掺入,催化剂可进一步降低甲醛100%转化时所需的反应温度。氧化铜的引入增强了氧化物间的协同作用,提高了材料分散性和氧化能力,促进催化材料中氧的释放,进而优化了催化氧化甲醛性能。
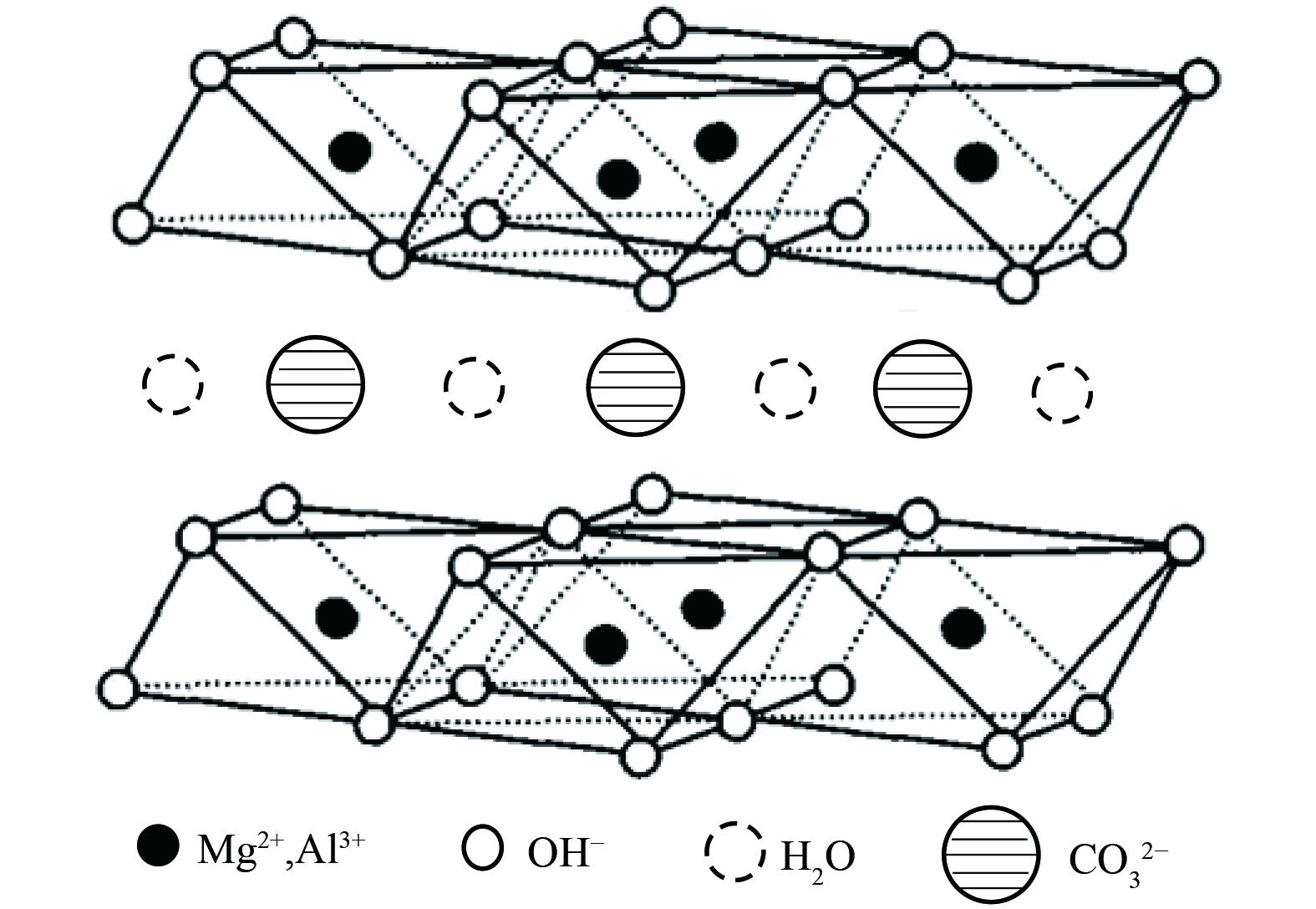
如今催化氧化技术已取得长足进展,但探求高效稳定、抗水蒸气能力强、低成本、常温条件去除低浓度甲醛催化剂仍面临挑战。类水滑石化合物(LDHs)是近年来发展迅速的阴离子型粘土材料[27],其通式表示为[M2+1-xM3+x(OH)2]x+[An-x/n]·mH2O,结构如图1所示。其中,M2+是二价金属阳离子,M3+是三价金属阳离子,An-是层间阴离子,通常为无机阴离子Cl-、CO32-等。因其具有耐酸碱性、热稳定性、吸附性、层间阴离子可交换性及高比表面积等特点,在吸附、催化、阻燃、医药等领域已被广泛应用[28-29]。为此本研究提出采用共沉淀法制备了MnOx/类水滑石催化剂,以类水滑石为载体、片状氧化锰为活性组分,探究载体各组分、煅烧温度、氧化锰负载量等对催化剂常温催化氧化性能的影响,运用XRD、SEM、BET、EPR、XPS等技术表征分析探究催化剂微观结构,考察并分析其常温催化氧化甲醛活性和稳定性,以克服锰基氧化物常温条件下因大量吸附水蒸气而稳定性降低的缺点,研制一种高效稳定的过渡金属氧化物催化剂,为常温催化氧化催化剂的研制提供参考。
-
1) 类水滑石载体的制备。准确称取一定量六水氯化镁和九水合硝酸铝,并将其溶解于100 mL去离子水中,充分搅拌得溶液A;并按照一定比例配制碳酸钠和氢氧化钠的混合溶液B;再将溶液B缓慢滴加到溶液A中,继续搅拌直至pH达10为止,继续搅拌3 h;再将上述溶液离心分离,并用去离子水和无水乙醇各清洗3次,将所得水凝胶在烘箱80 ℃干燥过夜,再置于马弗炉中100 ℃煅烧4 h,根据n(Mg)/n(Al)命名为Mg1Al1、Mg4Al1等类水滑石载体。
2) MnOx/类水滑石负载催化剂的制备。采用共沉淀法制备MnOx/类水滑石催化剂,具体方法如下:准确称量4.0 g Mg1Al1类水滑石载体,将其浸泡于50 mL去离子水中,充分搅拌,并向其中加入一定量硝酸锰溶液,获得悬浊液;再配制0.2 mol·L−1氢氧化钠溶液,并将其缓慢滴加至上述悬浊液中直至pH=10,继续搅拌3 h。再将上述溶液离心分离,并用去离子水和无水乙醇各清洗3次,将得沉淀物在烘箱80 ℃干燥过夜和马弗炉100 ℃、4 h煅烧,即制得负载量为10%的MnOx/Mg4Al1催化剂,其他不同n(Mg)/n(Al)、煅烧温度、MnOx负载量以及类水滑石组分催化剂均采用上述方法制得。
-
实验在50 cm×50 cm×50 cm的密闭玻璃反应器中室温条件下进行。首先在该反应器内采用甲醛溶液挥发方式配制初始低浓度气态甲醛(1.0~1.1 mg·m−3),并将1.0 g的催化剂粉末均匀铺设于一培养皿中(Φ=6 cm),并将其置于反应器底部,并迅速密封该反应器,其反应器内甲醛质量浓度采用甲醛分析仪(PPM-400ST)测定,每12 h连续测定3次,取平均值,连续观察其48 h甲醛质量浓度变化情况,其甲醛去除率按下式计算。
式中:Ct表示每12 h后反应器内甲醛质量浓度,mg·m−3;C0表示反应器内初始甲醛质量浓度,mg·m−3。
催化剂稳定性测试在上述氧化活性测试基础上进行。催化剂经48 h测试后,直接采用注入甲醛溶液方式再次使其在玻璃反应器内挥发至设定的甲醛质量浓度(1.0~1.1 mg·m-3),然后迅速取出甲醛溶液并密封该反应器,并测定其浓度作为第二次活性测试的起始浓度,过程中催化剂保持固定不变,反复操作进行即可。
-
为探究催化剂微观结构,采用X射线衍射仪(XRD, D/teX Ultra 250 detector, Shimadzu XRD-6100, 日本)对催化剂进行XRD分析,检测过程使用Cu-K α辐射(λ=0.154 06 nm),并在40 kV和40 mA下进行;表面形貌采用德国蔡司公司生产的Gemini SEM扫描电镜检测分析,工作电压5.0 kV;比表面积、孔径大小及分布分别基于Brunauer-Emmett-Teller (BET)和Barrett-Joyner-Halenda (BJH)方法(Micromeritics ASAP 2460)在77 K下对催化剂进行N2吸附脱附条件下进行检测;室温下采集了电子顺磁共振图谱(EPR),其X波段微波频率为9.43 GHz,功率为1.5 mW。运用X射线光电子能谱(Thermo ESCALAB 250XI) 分析材料表面的元素组成、化学状态及元素种类, 放射源为Al-Kα (hv=1486.8 eV) ,所得能谱采用C 1s (284.6 eV) 的结合能校准,使用15 kV和300 W的Al-Kα X射线源分析元素化学结合能。
-
1) n(Mg)/n(Al)对催化剂氧化性能的影响。为对比分析,研究考察了空白反应器以及等量活性炭的吸附性能 (图2) 。空白反应器内甲醛48 h略有下降,下降率仅为16.9%,其可能为吸附及分解等所致。这表明该反应器较为稳定,不存在大量吸附及漏气等情况。等量活性炭因具有较高比表面积而展示出较优异的吸附性能,48 h甲醛吸附率为55.0%。但其氧化活性远低于锰氧化物,这是由于锰氧化物具有更优异的吸附+常温催化性能。载体可对催化剂氧化性能产生显著深刻影响。为此,考察了n(Mg)/n(Al)对MnOx/类水滑石常温催化氧化甲醛性能的影响。在同等测试条件下,纯水滑石Mg4Al1-300性能最差,其48 h甲醛去除率仅为52.3%,仅显示出一定吸附性能。这表明类水滑石Mg4Al1主要作为载体而存在,而纯氧化锰粉末展示出良好的吸附+常温催化氧化性能,其甲醛去除率明显高于比表面积更高的Mg4Al1水滑石(58.2 m2·g−1),表明其可能发生了催化氧化反应。还将MnOx负载至水滑石载体上,其催化剂氧化活性较MnOx显著提升。当n(Mg)∶n(Al)=4∶1、MnOx负载量为40%和煅烧温度为300 ℃时,MnOx/Mg4Al1(比表面积:81.4 m2·g−1)催化剂氧化性能最优,48 h内可将甲醛质量浓度由1.049 mg·m−3降至0.028 mg·m−3,去除率高达97.3%,其前24 h甲醛去除率高达92.5%。而随着时间推移其甲醛去除率逐渐降低,反应受反应物扩散速率控制。相比其他MnOx/MgAl,MnOx/Mg1Al1催化剂性能最差,其比表面积(79.0 m2·g−1)、孔容(0.187 cm3·g−1)和孔径(6.5 nm)亦最低。虽MnOx/Mg1Al4比表面积最高,达156.7 m2·g−1,但其孔径明显低于MnOx/Mg4Al1,其一定程度上限制了甲醛的吸附和反应产物脱附,致使其氧化活性与MnOx/Mg4Al1相当。3种催化剂的氧化活性均高于MnOx(22.6 m2·g−1),这表明其材料的比表面积和孔容孔径是影响催化剂氧化活性的因素之一。
2) 煅烧温度对催化剂氧化性能的影响。图3为不同煅烧温度条件对MnOx/Mg4Al1催化剂氧化性能的影响。随着煅烧温度的提升,其甲醛去除率先升高后持续降低。当煅烧温度为200 ℃时,催化剂表现出最佳的氧化活性,甲醛去除率高达98.5%,其质量浓度由1.000 mg·m−3降至0.015 mg·m−3,明显优于其他同类催化剂。其中,经500 ℃高温煅烧所制催化剂性能最差,48 h甲醛去除率仅为73.7%。这表明其氧化锰晶体及催化剂表面结构可能发生了显著变化,这一结果与XRD和SEM相一致。
3) MnOx负载量对催化剂氧化性能的影响。为进一步提升催化剂氧化性能、降低成本,考察了MnOx负载量对催化剂常温催化氧化甲醛的性能影响 (图4) 。随着MnOx负载量由40%降至10%,其氧化性能持续降低。当MnOx负载量为10%时,48 h内甲醛去除率为79.5%。而随着MnOx负载量提升至50%时,其氧化性能亦未得到显著提升,反而有所降低,其可能与催化剂比面积下降有关。这表明其MnOx负载量并非越多越佳,存在最佳比例。上述结果表明,MnOx负载量在20%~40%均可接受,其催化剂氧化性能良好,为此选择MnOx负载量为20%进行后续研究。
4) 类水滑石组分对催化剂氧化性能的影响。考虑到载体组成成分亦可能会对催化剂降解甲醛能力产生差异,考察了5种组分的类水滑石对催化剂氧化性能的影响 (图5) 。不同组分对催化剂催化效果影响差异较小,48 h内MnOx/Mg4Al1、MnOx/Mg4Fe1、MnOx/Mg4Cr1、MnOx/Co4Al1和MnOx/Ni4Al1催化剂的甲醛转化率依次为98.5%、97.5%、93.7%、97.0%和96.9%。相比之下,MnOx/Mg4Cr1催化剂活性较低,而MnOx/Mg4Al1催化活性最佳。结果表明,不同组分的类水滑石对催化剂的氧化性能影响较小,且对负载有MnOx的MnOx/类水滑石催化剂氧化性能均有显著提升。
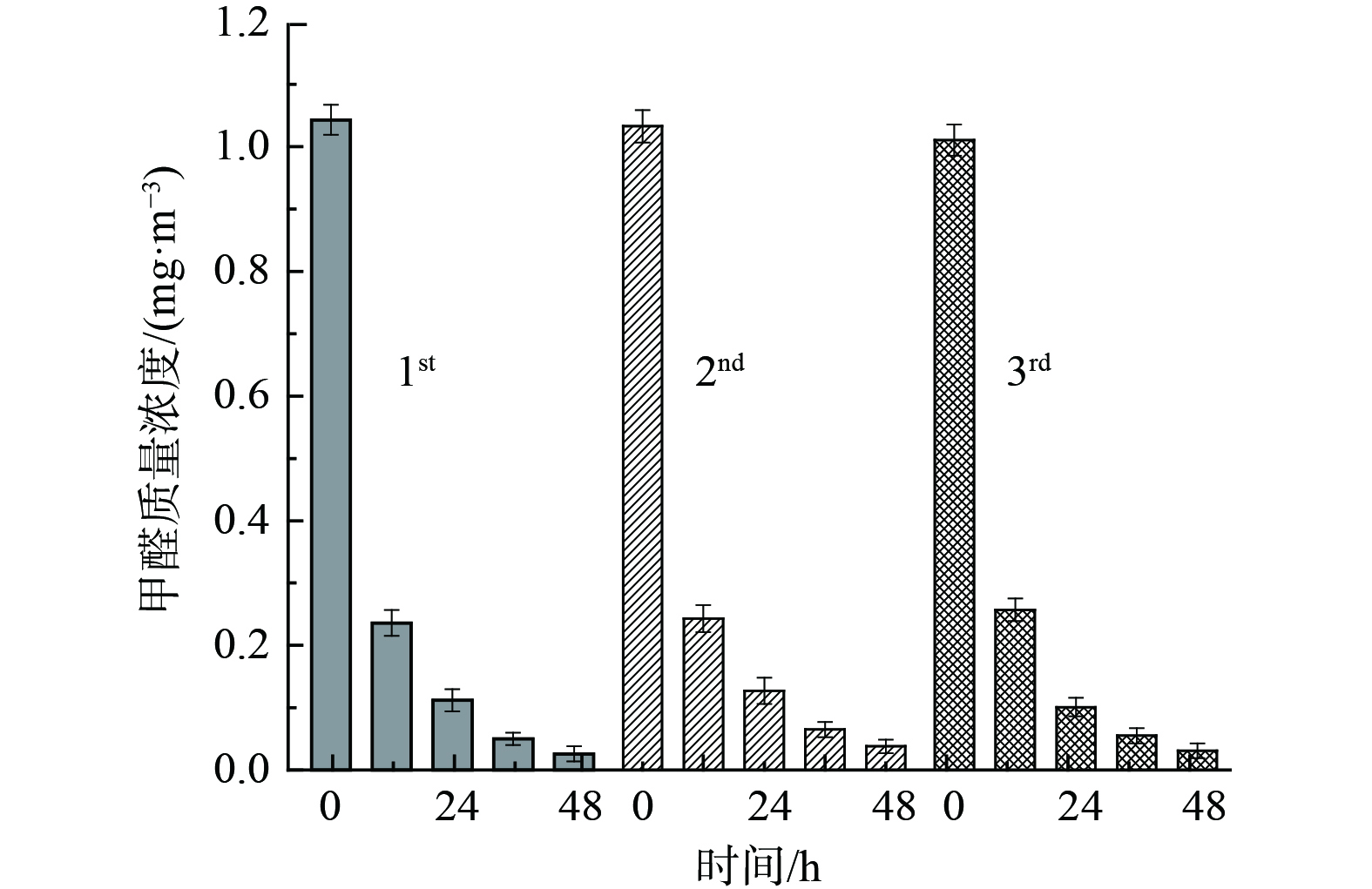
5) 催化剂稳定性研究。图6为氧化性能最佳的20% MnOx/Mg4Al1-200催化剂常温催化氧化甲醛的稳定性研究结果。在相同条件下对该催化剂进行了连续3次活性测试,其48 h甲醛去除率依次为97.5%、96.3%和96.9%,催化剂稳定性良好,未见明显的因吸附大量水蒸气而导致的吸附和氧化活性下降的趋势。这表明,催化剂抗水蒸气性能良好,可反复使用,保持着较高的甲醛去除效率,对实际应用有参考价值。
-
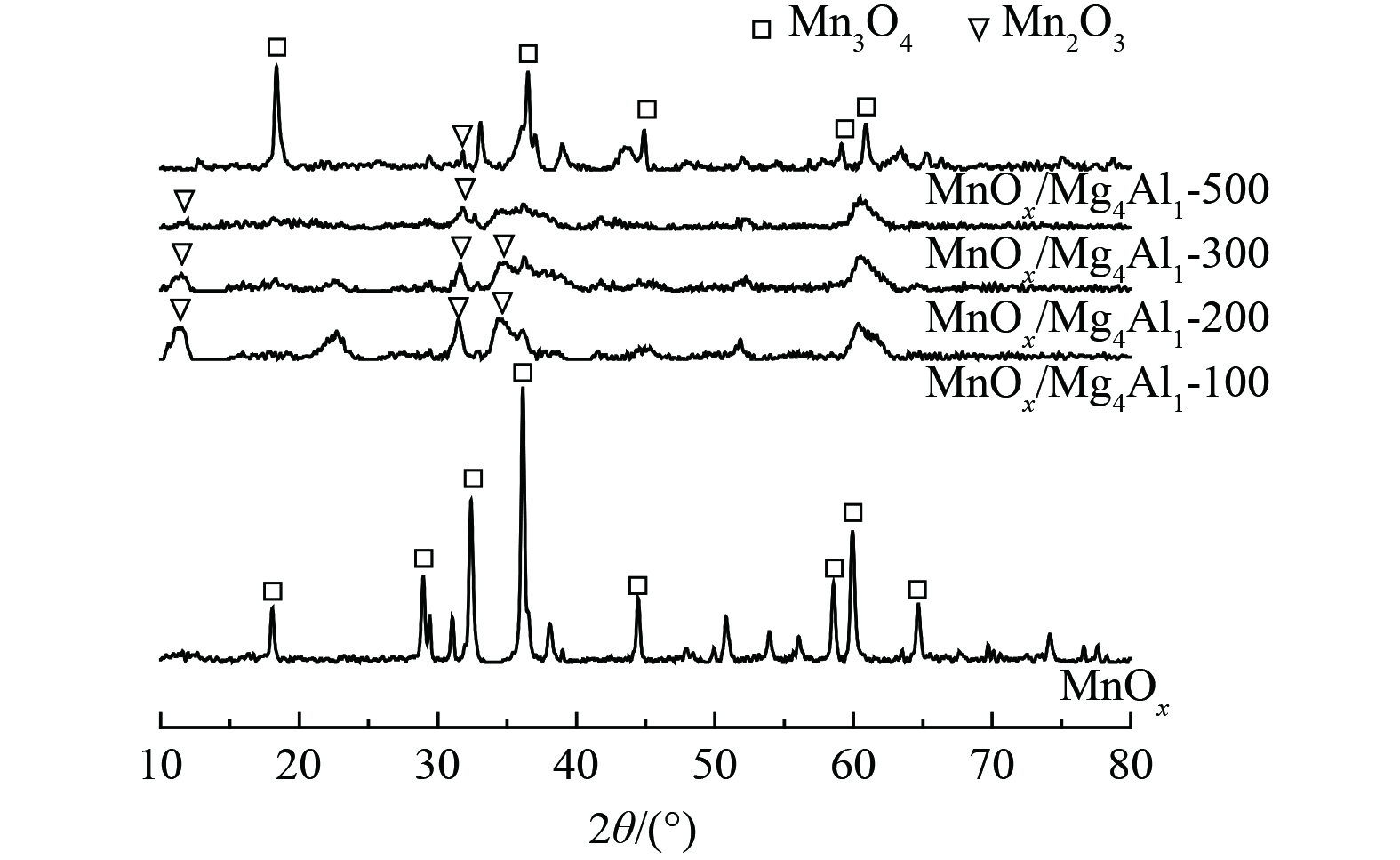
1) 结构形态分析。为探究催化剂结构对催化剂性能的影响,对经过不同温度煅烧后的MnOx/Mg4Al1催化剂及MnOx进行了XRD分析,如图7所示。对于MnOx,所显现的特征峰均为Mn3O4的特征衍射峰(PDF#80-0382),而未发现有其他氧化锰的特征衍射峰。对于MnOx/Mg4Al1催化剂,当煅烧温度为100、200和300 ℃时,MnOx/Mg4Al1催化剂中观察到少量Mn2O3 (PDF#73-1826)和Mn3O4相关晶体衍射峰,衍射峰较宽,晶体粒径较小。这说明晶体高度分散于类水滑石载体上,且衍射峰强度较小,亦表明Mn2O3和Mn3O4部分可能仍以无定形晶体形式存在。在2θ=11.2°、22.4°、34.5°和60.5°处的特征峰与镁铝类水滑石的(003)、(006)、(012)和(110)晶面相匹配[30]。这表明形成了与类水滑石结构相似的片层结构,随着温度升高,催化剂中类水滑石结构的特征峰逐渐消失,可能是由于温度的升高导致层间水损失。而典型的片层结构的峰消失,则说明类水滑石结构在煅烧过程中坍塌。当煅烧温度达到500 ℃时,Mn3O4和Mn2O3衍射峰明显变为尖峰,晶体粒径增大,其不利于催化剂氧化性能的发挥,这一结果与氧化活性结果相一致。
为探究煅烧温度对催化剂的表面结构形态的影响,利用SEM对MnOx/Mg4Al1催化剂进行了表观分析 (图8) 。图8 (a)为经100 ℃煅烧的MnOx/Mg4Al1催化剂SEM图,该催化剂显示为颗粒物块状结构,凹凸不平,这可能是由于催化剂煅烧不充分未能较好的形成片状Mg4Al1所致;当煅烧温度升高至200 ℃和300 ℃时(图8 (b, c)),催化剂表面呈现明显层状结构,即Mg4Al1类水滑石,且表面含有棒状结构物质。随着煅烧温度的提升,其棒状结构物质分布和含量降低。当煅烧温度达到500 ℃时(图8 (d)),催化剂表面结构松散,即Mg4Al1类水滑石片状结构遭到破坏,亦发现大量小片状或棒状结构物质,孔径、孔体积增大,比表面积降低致使催化剂氧化活性降低。这与BET和活性测试结果相一致。
2) 比表面积分析。为探究煅烧温度对MnOx/MgAl催化剂的比表面积和及孔容孔径的影响,对经200 ℃煅烧的MnOx、Mg4Al1及分别经100、200、300和500 ℃煅烧的MnOx/Mg4Al1催化剂进行了BET分析,如表1所示。相较于MnOx,Mg4Al1载体显现出较MnOx更高的比表面积和更低的孔容孔径,其吸附及降解性能应优于MnOx。但结果与事实相反,MnOx展示出更佳的吸附及降解性能。这表明甲醛在MnOx上发生催化降解作用,而Mg4Al1载体仅显现出吸附甲醛性能,无降解作用。研究亦发现,MnOx/类水滑石催化剂比表面积和孔容孔径较MnOx和Mg4Al1显著增大,且比表面积呈现出先增加后降低趋势,其与催化剂氧化活性次序相一致。相较于氧化性能最佳的MnOx/Mg4Al1-200催化剂(78.8 m2·g−1),MnOx/Mg1Al4-300催化剂比表面积更高,达156.7 m2·g−1,但其氧化活性未见显著提升。这表明比表面积是影响催化剂氧化性能的因素之一,但不是唯一决定性因素,其组成亦深刻影响催化剂性能。
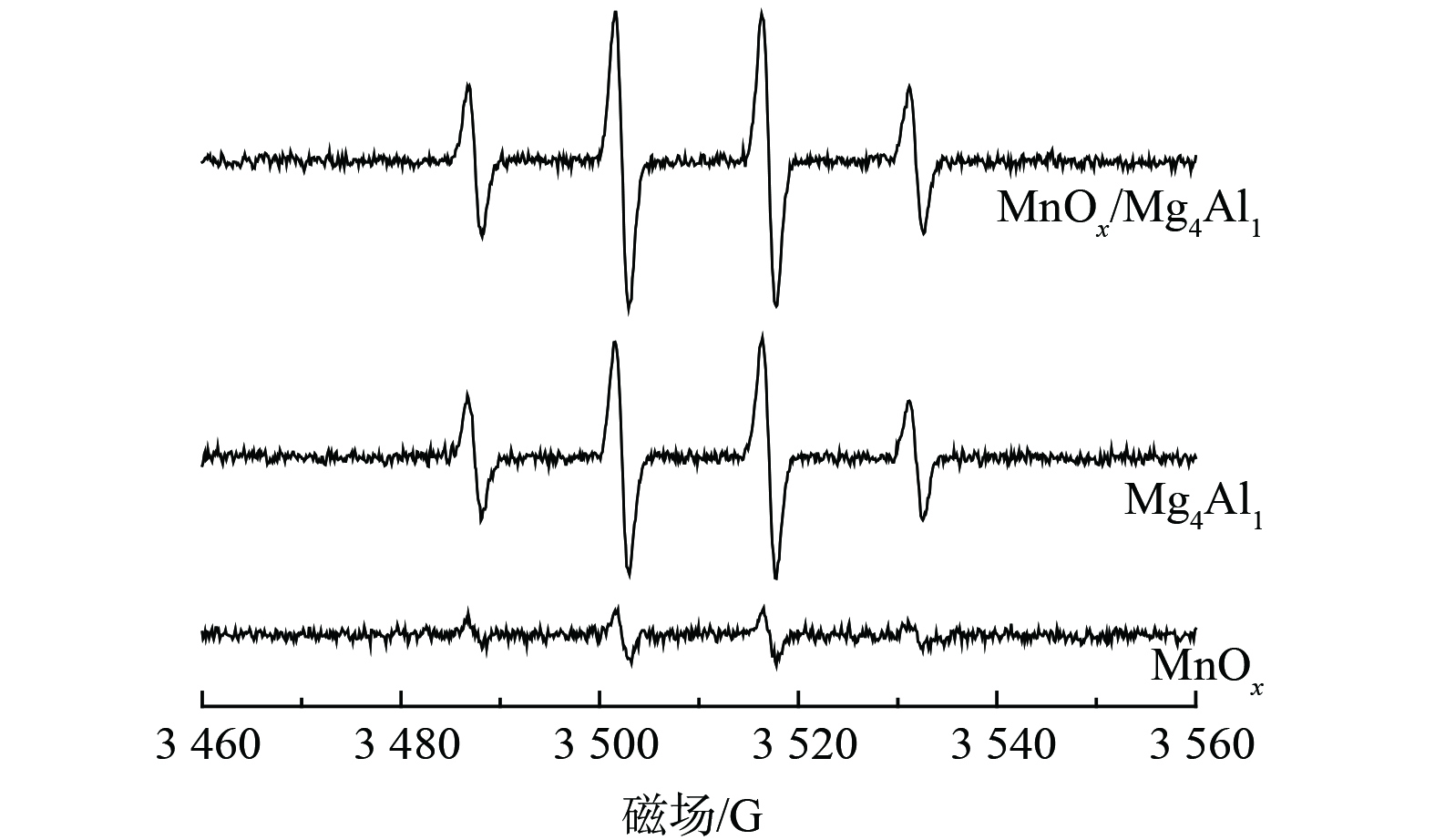
3) EPR分析。为探究催化反应后催化剂表面反应成分组成情况,对MnOx、Mg4Al1、MnOx/Mg4Al1进行了EPR分析 (图9) 。图中显示出4个明显的特征峰,从左到右,其特征峰的比例为1∶2∶2∶1,对应羟基自由基(·OH)的EPR谱图。这表明MnOx/Mg4Al1催化剂中的大量羟基来源于类水滑石载体,而非活性组分MnOx。这些存在于催化剂表面的羟基可有效吸附气态甲醛及中间产物[31],促进催化剂的氧化活性,进而提升催化剂对甲醛的催化降解速率。
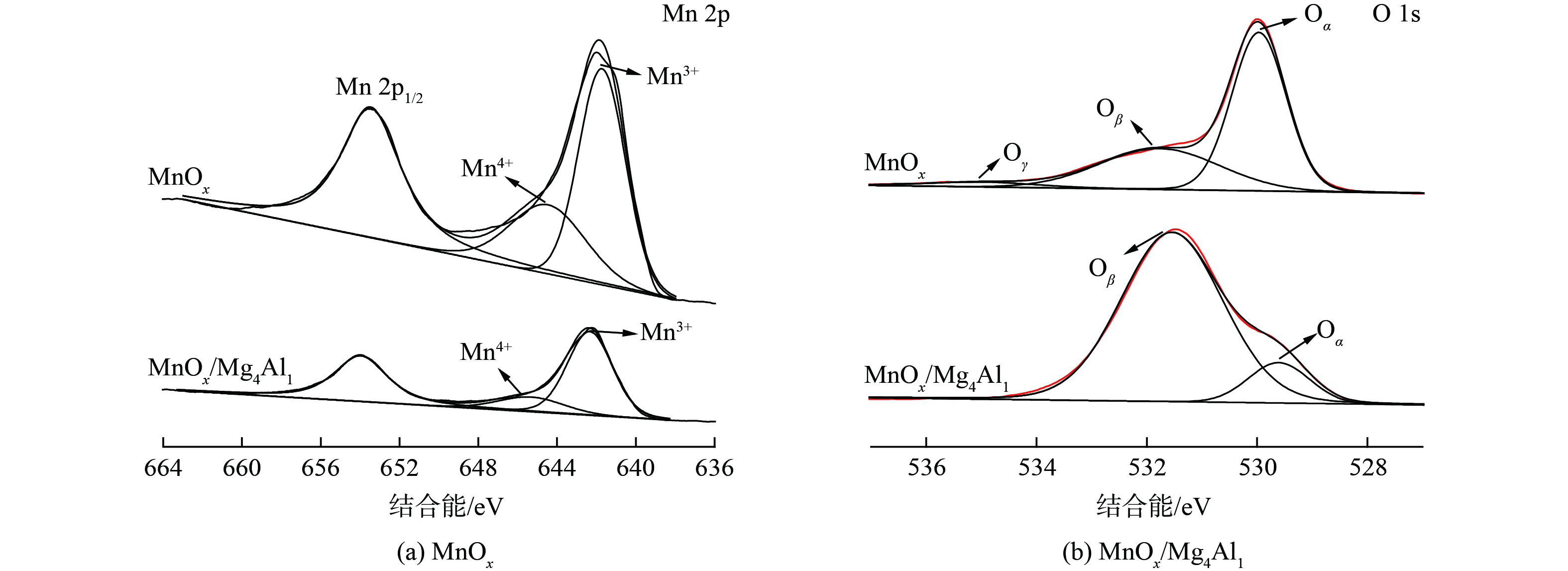
4) XPS分析。为确定催化剂中Mn和O元素的组成及化学结合状态,对MnOx和MnOx/Mg4Al1催化剂进行了XPS分析 (图10) 。图10 (a) Mn 2p 光谱表明,结合能值在653.5 eV和641.9 eV处的特征峰归属为MnO2的Mn 2p1/2和Mn 2p3/2 自旋轨道峰,且Mn 2p3/2反褶积为2个特征峰[32],结合能值为644.5 eV和642 eV处分别归属于Mn4+和Mn3+,且MnOx和MnOx/Mg4Al1催化剂中Mn3+/(Mn3++Mn4+)相对含量比例分别为63.9%和79.8%,可见在MnOx/Mg4Al1催化剂中,Mn3+占比更大。这表明MnOx/Mg4Al1催化剂具有更多氧空位,有利于催化氧化反应的进行。图10 (b) O 1s 光谱表明,结合能值在535.3、531.8和529.6 eV处分别归属于羟基或吸附水(Oγ),表面氧(Oβ)和晶格氧(Oα),而在MnOx/Mg4Al1催化剂中,活性氧的数量和种类发生了显著变化,结合能值在531.5 eV和529.6 eV处分别归属为表面氧(Oβ)和晶格氧(Oα),不存在羟基或吸附水(Oγ)的特征峰[33]。2种催化剂相比,MnOx/Mg4Al1催化剂中的表面氧(Oβ)比例明显上升,其Oβ/(Oα+Oβ)相对含量比例分别为35.7%和88%。由于表面氧(Oβ)在常温催化氧化反应中起着重要作用,且在MnOx/Mg4Al1催化剂中占比更大。这表明MnOx/Mg4Al1催化剂具有较MnOx更高的氧化性能可能是与表面氧(Oβ)的含量高有关。
-
1) 当n(Mg)∶n(Al)为4∶1、煅烧温度200 ℃、MnOx负载量为40%时,催化剂常温催化氧化活性最佳,48 h可将质量浓度为1.000 mg·m−3甲醛降、至0.015 mg·m−3,去除率达98.5%,且展示出良好的催化稳定性。2) 负载型MnOx/类水滑石催化剂催化降解甲醛性能优异,其主要是由于类水滑石载体表面存有大量羟基基团,有利于吸附低浓度甲醛及中间产物,且无明显的与水蒸气竞争吸附而导致的活性下降趋势,同时丰富的表面氧(Oβ)和氧空位、较大的比表面积和较小的颗粒物粒径均利于催化剂氧化性能提升,具有重要的实际应用潜力。
高表面氧MnOx/类水滑石催化剂常温催化低浓度甲醛的性能
Performance of high surface oxygen MnOx/ hydrotalcite-like catalyst for the catalysis of low concentration formaldehyde at ambient temperature
-
摘要: 常温催化氧化技术是去除室内低浓度甲醛(HCHO)的一种有效方法,其中过渡金属氧化物锰基催化剂在催化降解方面性能优异,相较于贵金属催化剂,具有成本低、活性高、稳定性良好、应用前景广等优点。采用共沉淀法制备了类水滑石载体和锰基/类水滑石催化剂,将类水滑石与锰基氧化物相结合用于常温催化氧化,克服了锰基氧化物常温条件下因大量吸附水蒸气而导致稳定性降低的缺点,同时研究考察了载体组成、煅烧温度、氧化锰负载量等对催化剂常温催化氧化性能的影响,并通过XRD、SEM、BET、EPR、XPS等技术对催化剂进行了微观表征。结果表明,当类水滑石中n(Mg)∶n(Al)=4∶1,氧化锰负载量 (质量分数) 为40%,煅烧温度为200 ℃时,所述MnOx/Mg4Al1催化剂性能最佳,其48 h甲醛去除率高达98.5%,甲醛质量浓度由1.000 mg·m-3降至0.015 mg·m−3。类水滑石载体表面存有大量羟基基团,有利于吸附低浓度甲醛及中间产物进而利于催化剂催化氧化性能提升。通过XPS分析可知,相较于MnOx催化剂,MnOx/Mg4Al1催化剂中Oβ/(Oα+Oβ)相对含量比例由35.7%提升至88.0%,表面氧含量(Oβ)大幅提升,其在甲醛常温催化氧化过程中发挥着极其重要作用。本研究结果可为常温催化氧化甲醛提供参考。Abstract: Catalytic oxidation at ambient temperature is an effective method for the removal of low-concentrated formaldehyde (HCHO). The transition metal oxide manganese based catalyst has excellent catalytic degradation performance. Compared with the precious metal catalyst, it has the advantages of low cost, high activity, good stability and wide application prospect. In this study, the hydrotalcite-like and manganese oxides/ hydrotalcite-like catalysts were synthesized by the co-precipitation method. The combination of hydrotalcite-like and manganese oxides for catalytic oxidation of HCHO at ambient temperature overcomed the disadvantage of stability reduction caused by large amount of water vapor adsorption of mangan-based oxides at room temperature. Meanwhile, the effects of carrier composition, calcination temperature and manganese oxides supporting, etc. on the catalytic oxidation at ambient temperature were investigated and characterized by XRD, SEM, BET, EPR and XPS. The results demonstrated that the highest performance of the catalyst was MnOx/Mg4Al1 with n(Mg)∶n(Al)=4∶1, 40 % manganese oxides supporting and calcining at 200 °C. The removal rate of HCHO was up to 98.5% within 48 h, and the concentration was decreased from 1.000 mg·m-3 to 0.015 mg·m-3. The presence of a large number of hydroxyl groups on the surface of the hydrotalcite-like was beneficial to the adsorption of low concentration of HCHO and intermediate by-products, which also improved the oxidation activity of the catalysts. According to XPS analysis, compared to the MnOx catalyst, the Oβ/(Oα+Oβ) ratio of the MnOx/Mg4Al1 was increased from 35.7% to 88.0% and the surface oxygen content (Oβ) also enhanced significantly. The results suggested that the high surface oxygen content played an important role in the catalytic oxidation of HCHO at ambient temperature. This study can provide a reference for the reduction for the catalytic oxidation of HCHO at ambient temperature.
-
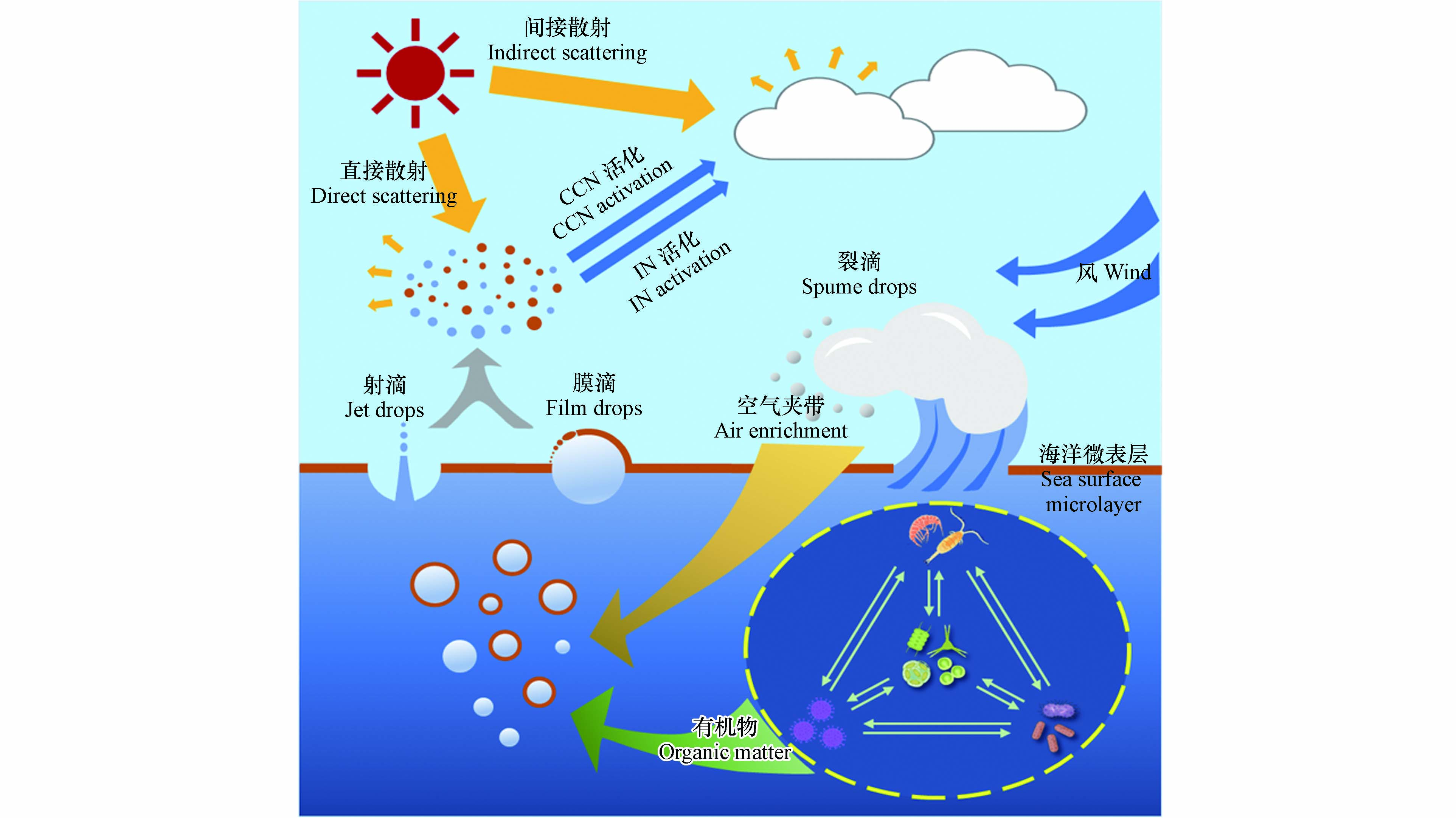
海洋飞沫气溶胶(SSA)是指直接从海洋表面向大气喷射的悬浮颗粒[1-2]. 当其进入海洋边界层(marine boundary layer,MBL)后不仅可以通过吸收和散射太阳辐射而直接影响全球辐射平衡,也可以作为云凝结核(cloud condensation nuclei, CCN)和冰核(ice nuclei, IN)间接影响全球气候[3-6]. 据统计,海洋每年向大气输送的SSA可达100—300亿t[7],且远高于沙尘气溶胶[8]. 因此SSA被普遍认为是影响全球气候系统的重要调节因子. 研究发现SSA排放量的增加有助于全球气候的冷却,进而能缓解或抵消由温室气体所引发的全球变暖[9-10]. 此外,SSA在海-气界面水分、盐分、有机物和微生物的传输过程中也发挥着至关重要的作用[11]. 随着全球经济迅速发展,人为活动引起的海洋环境问题日益突出,海洋中的持久性有机污染物[12]、微塑料[13]、过渡金属[14]、藻毒素[15]、细菌和病毒[16]等均可在SSA上富集并远距离传输. 这不仅会影响SSA的化学组成,还会对大气环境和人类健康产生重大影响. 如何准确评估SSA在全球环境和气候变化中的作用成为了当前大气-海洋交叉学科亟需解决的关键科学问题.
自1953年Woodcock等将风速确定为SSA产生的主要驱动力之后[17],一系列将SSA产生与风速相联系的研究接踵而至[18-19]. 虽然风速是目前已知控制SSA产生的关键,但SSA的产生也会受到其它环境因素(海表温度、海水盐度、表面活性剂和浮游微生物活动等)的影响[20]. 由于对这些环境因素的潜在影响机制认识尚不完全,围绕SSA产生过程和相关影响因素的不确定性使其成为一个持续活跃的研究领域. 外场观测虽然可以对SSA进行原位测量,但是存在一定的时空局限性,且难以明确地分离和量化单个环境因素的影响. 针对上述的研究“短板”,近年来科研工作者在实验室中开展了大量模拟工作,并在SSA产生技术方面取得了重大进展. 同时,实验室模拟研究可实现在受控条件下深入探究不同环境因素对SSA产生过程的影响及作用机制,这对SSA的产生机制研究和全球气候效应预测都至关重要. 因此,本文系统总结了近十年来该领域模拟研究的相关进展,重点介绍SSA的产生、粒径分布、化学组成、物质富集以及影响SSA产生的关键环境因素.
1. SSA产生(SSA Formation)
1.1 SSA自然产生
风驱动产生的SSA粒径大小可跨越几个数量级,一般按其产生机制分为三类:膜滴、射滴和裂滴(图1).
当风速大于5 m·s−1时,波浪破碎使上表层海水夹带大量气泡. 随后气泡上浮至海洋表面,其露出水面的气泡膜不断变薄直至破裂. 气泡破裂过程中气泡膜通过液条断裂[21-23]或快速抖动[24]而产生数百个膜滴. 而后,周围海水将回填气泡破裂后留下的空腔而激起若干垂直喷射的射滴[25]. 当风速大于10 m·s−1时,风可以直接吹裂波峰而产生几十微米至几毫米的裂滴[26],其大气停留时间从几秒到几分钟不等. 由于粒径较大的裂滴大气停留时间短,因此当前研究一般忽略其在大气化学和气候效应中的贡献[2]. 一般而言,气泡破裂产生的SSA粒径范围在0.01—25 μm之间[1],其中膜滴粒径较小,主要集中在亚微米(粒径<1 μm)范围内[24];而射滴粒径较大,贡献了大部分的超微米(粒径>1 μm)颗粒[20]. 膜滴和射滴的产生数量与气泡尺寸大小密切相关. 研究发现气泡尺寸越大产生的膜滴越多. 例如,气泡直径从2 mm增加到6 mm,膜滴数量可从100个增加到1000个,增加一个数量级[27]. 相反,射滴数量随着气泡尺寸的增加而减少. 例如,直径在100—300 μm内的气泡可以产生5—6个射滴,而直径为3 mm的气泡只能产生1个射滴[28]. 此外,膜滴粒径受气泡膜厚度影响. 研究表明气泡膜厚度会随着气泡直径增加而增加,进而容易产生更大的膜滴[23, 29];而射滴粒径约为其破裂气泡直径的1/40[20]. 总之,气泡的破裂过程及尺寸大小共同控制着膜滴与射滴的产生,最终将影响SSA的粒径分布和化学组成[1, 30-32].
1.2 SSA模拟产生
迄今为止,有关SSA的研究已经做了大量的外场观测工作,但仍然存在两个重要问题亟需解决:(1)原位SSA的产生容易受到多种环境因素耦合的影响,且无法识别各因素在SSA产生过程中的具体作用机制;(2)海洋气溶胶来源复杂,如何区分SSA对海洋气溶胶的贡献依然是个难题. 在这些问题的驱使下,一系列模拟SSA产生的装置应运而生,很好地弥补了外场观测的研究短板. 目前常用的模拟装置有加压雾化器、鼓泡式发生器、跌落式发生器、“Sea Sweep”发生器和波浪破碎式发生器等(图2),具体介绍如下.
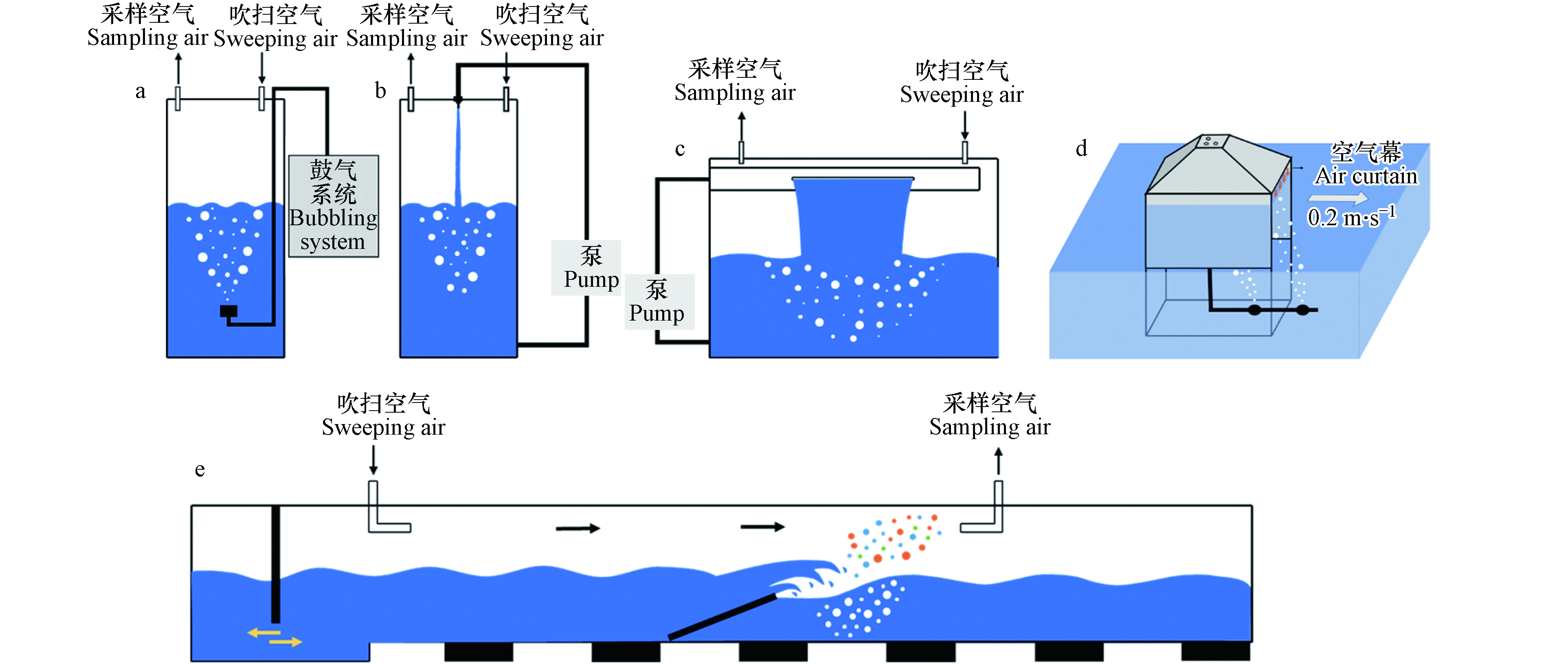 图 2 典型的SSA模拟发生装置Figure 2. Typical SSA simulation generators(a.鼓泡式;b.射流式;c.飞瀑式;d. “Sea Sweep”;e. 波浪破碎式)(a. Bubbling;b. Plunging jet;c. Plunging waterfull;d. “Sea Sweep”;e. Breaking waves)
图 2 典型的SSA模拟发生装置Figure 2. Typical SSA simulation generators(a.鼓泡式;b.射流式;c.飞瀑式;d. “Sea Sweep”;e. 波浪破碎式)(a. Bubbling;b. Plunging jet;c. Plunging waterfull;d. “Sea Sweep”;e. Breaking waves)(1)加压雾化器:利用伯努利原理将压缩空气直接作用于封闭容器内的海水,高压海水经喷出雾化后形成SSA. 雾化器凭其结构简单、生产效率高等优点在很多陆源气溶胶的实验室模拟中得到广泛应用. 再次强调SSA的产生与气泡的产生与破裂密切相关,但在一些吸湿性增长[33]、N2O5吸收[34]和CCN活性[35]的研究中仍延用雾化发生器来产生SSA. 实验研究表明,该装置产生的SSA粒径分布只有两个模态(约35 nm和约100 nm),与其它模拟方法和外场观测结果不一致[36]. 因此加压雾化器不再适合于SSA的研究.
(2)鼓泡式发生器:将洁净空气通入浸没在水下的烧结玻璃过滤器来产生气泡羽流以模拟膜滴和射滴的形成. 产生的SSA在吹扫空气作用下被载入分析检测设备. 当前设计的鼓泡式发生器形式多样[37-50],有小至容积为500 mL的五颈圆底烧瓶[41],也有大至高为2.1 m的空心圆柱体[37]. 鼓泡式发生器主要通过鼓气流量[51]、多孔过滤器孔径[52]及其水下深度[46]来调节SSA的生成. 例如,研究发现细孔径过滤器产生的SSA质量浓度和数量浓度分别是粗孔径过滤器的57倍和194倍[40]. 虽然鼓泡法一直被用于模拟SSA的产生,但是近年来有越来越多的学者发现它并不是一种理想的模拟产生方法. 这主要是因为其产生的气泡大小更多地取决于所使用过滤器的孔径大小,并不会重现波浪破碎时的气泡尺寸分布,无法实现SSA的仿真模拟.
(3)跌落式发生器:利用水流撞击水面,在水下一定深度内产生气泡羽流,气泡上浮至水面破裂后产生SSA. 按照水流入水的方式可为射流式[36, 43, 53-56]和飞瀑式[31, 57]. 射流式发生器利用喷嘴产生的线状入射水柱撞击水面来产生气泡羽流. 其射流速度和射流高度会改变水下气泡羽流的状态,进而影响SSA的数浓度和中心粒径[53-54]. 飞瀑式发生器以Stokes等[31]在2013年设计的MART(marine aerosol reference tank, MART)为代表. 与射流式发生器的区别在于喷嘴被狭缝替代而形成瀑布状的水帘. 跌落式发生器中的气泡羽流是由水柱/水帘夹带的空气所产生,其空气夹带速率受到冲击速度、流体特性、喷嘴/狭缝设计和下跌高度等因素的影响[54, 58]. 总之,跌落式发生器可以很好地模拟波浪破碎后的气泡尺寸分布[31, 58],而且该装置结构简单、操作便捷,因此在很多模拟研究中都获得了广泛应用.
(4)“Sea Sweep”发生器:由一个0.91 m×0.61 m×0.91 m(长×宽×高)的不锈钢框架构成,如图2(d)所示. 顶部高出不锈钢框架0.3 m,4个锥面由不锈钢板封闭. 该装置由左右两侧的浮筒来维持箱体漂浮状态(图中未展示). 前后两侧保持开放均与海面保持1 cm的距离,并利用1 m·s−1垂直向下的空气“幕”来阻止外界颗粒物对模拟体系的干扰. 在海面以下0.75 m处放置有两个多孔过滤器以产生气泡羽流,随后在锥顶采样口处收集新生的SSA. 同时,“Sea Sweep”发生器随船以0.2 m·s−1的速度前进,以确保海水的实时更新[59]. “Sea Sweep”发生器是外场观测与实验室模拟生成有机结合的典型代表,能在原位产生SSA的同时最大限度的减少外界因素的干扰.
(5)波浪破碎式发生器:加州大学斯克里普斯海洋研究所开发的波浪通道是一个33 m×0.5 m×0.8 m(长×宽×高)的狭长水道,其内部可容纳超104 L的海水. 位于通道中部有一个与底面呈16°的斜坡. 通道一侧在桨叶推动下产生波浪,波浪途经斜坡而发生破碎[60],由此产生的气泡羽流可达液面下0.15 m[57]. 相比前几种发生装置,波浪通道所产生的波浪、气泡尺寸和SSA粒径分布可以更大程度的贴近真实环境[21, 30]. 该通道已被用于研究海洋-大气复杂系统的围隔实验,包括了生物活动对新生SSA颗粒的影响、挥发性有机物(VOCs)的产生以及光化学老化对SSA的影响等研究[60]. 虽然波浪通道是研究SSA的理想装置,但是其巨大的体积容量和高昂的运行成本限制了其广泛应用.
总之,由于生成方法和设计思路的不同,SSA发生器在结构上存在着较大的差异. 但因目前缺少更多的公认参数,只有加压雾化器能够从原理上被排除于SSA的实验室生成,而其余方法均有应用. 不同方法产生的SSA可能在理化性质上存在较大差异,导致一些不同甚至是矛盾的实验结果出现. 因此,基于海浪产生原理开发一种可操作性强、仿真度高的标准化SSA生成方法是当下相关领域仍需解决的技术难题之一.
2. SSA的粒径分布与化学组成(Particle size distribution and chemical composition of SSA)
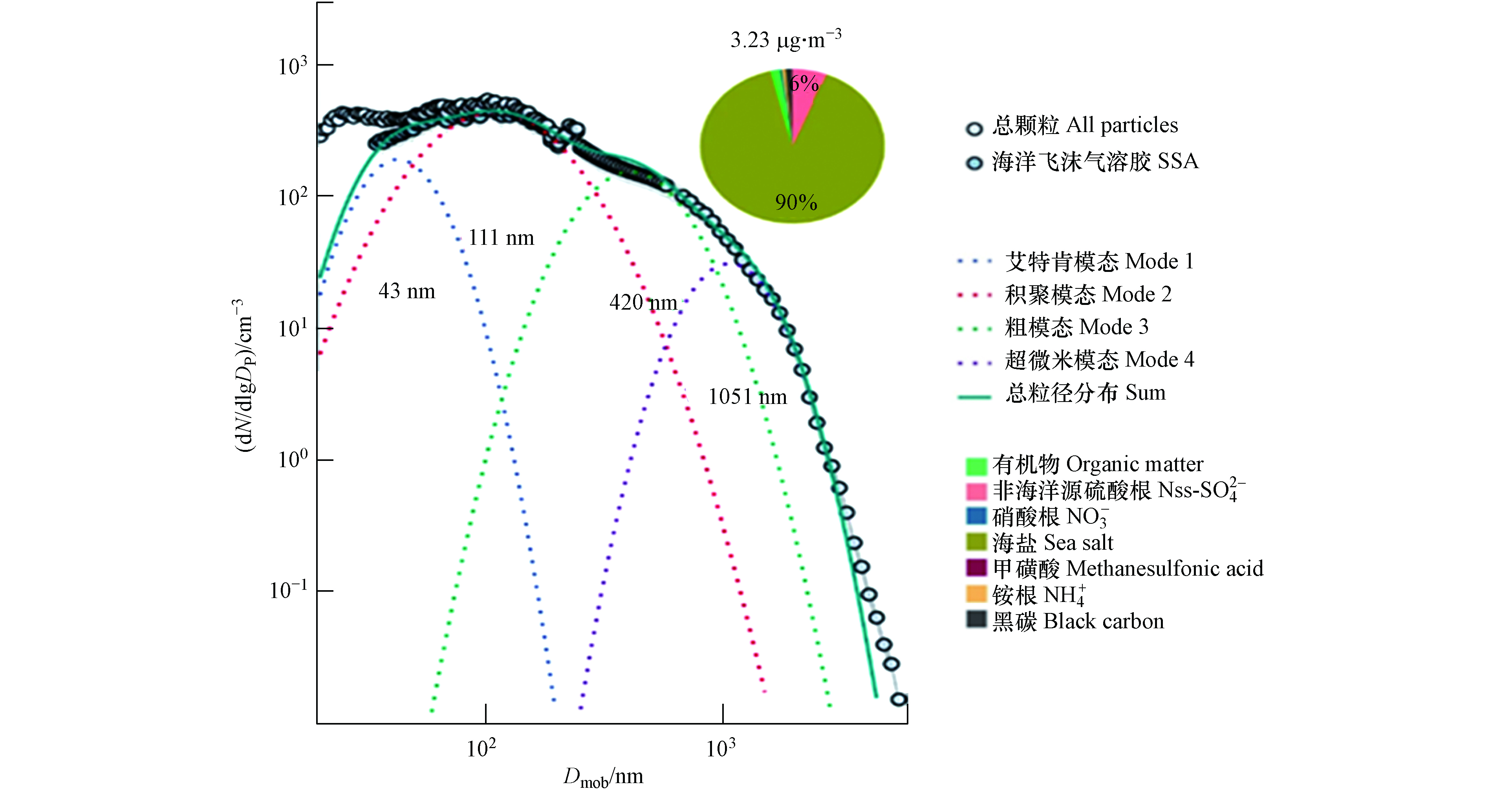
SSA在MBL中常以液滴的形式存在,其直径取决于自身含水量和周围环境相对湿度. 在MBL中环境的相对湿度一般被认为是80%. 从海面刚产生的SSA进入MBL后,会快速与相对湿度为80%的环境到达平衡,此时SSA直径会缩小为初始直径的二分之一;而当相对湿度低于50%后,水分不断蒸干而引发SSA从液滴向固体颗粒的相态转变,其直径将缩小为初始直径的四分之一[1]. 外场观测和实验室研究均表明,对于粒径小于10 μm的颗粒,所得到的SSA粒径分布呈现为多峰分布(图3):(1)在亚微米级分为以25—85 nm为中心的艾特肯模态(Mode 1)、以100—250 nm为中心的积聚模态(Mode 2)和大于250 nm的粗模态(Mode 3);(2)在超微米区域以1—3 μm为中心的超微米模态(Mode 4)[7, 36, 43, 59, 61]. SSA的这种多模态分布表明其产生过程将涉及多种机制,而传统的膜滴和射滴产生机制似乎仍不足以解释[5]. 在不同粒径尺寸的SSA中,亚微米颗粒对SSA数浓度的贡献占绝对优势[7],是大洋上空CCN的主要来源[59]. 与超微米颗粒相比,亚微米颗粒具有更大的比表面积和更复杂的化学组成,所以亚微米颗粒对大气非均相氧化反应和成核过程的影响尤为重要[62]. 随着海洋气溶胶在全球气候效应中的作用日益凸显,亚微米颗粒的相关研究已成为当前该领域的关注热点.
SSA的化学组分极为复杂,与海洋表面的化学组成密切相关. 海洋含有多种多样的无机物和有机物,在上浮气泡的顶托作用下易向海洋表面富集,进而发生从液相向颗粒相的转移. 由此产生的SSA将是无机物与有机物的混合物[63]. 其中无机组分源于海盐,包括Na+、K+、Mg2+、Ca2+和Cl-等,但又与海水中的比例不完全相同. 在一般情况下,从海洋转移到SSA的过程中仅有Na+浓度保持恒定,其它的无机离子则存在一定的富集和损耗[37, 64-66]. 在SSA中有机组分种类繁多,包括脂肪族、糖类、蛋白质、氨基酸和生物组分等[67-71],其对SSA的质量贡献从4%到80%不等[42, 59, 72-73]. 外部混合和内部混合从两个角度描述了SSA的混合状态. 外部混合是指SSA由多种不同化学性质的气溶胶颗粒混合而成. Prather等[30]根据物理性状和化学特性定义了四种主要的颗粒类型,包括海盐(sea salt,SS)、含有机碳海盐(sea salt with organic carbon,SS-OC)、有机物(organic carbon,OC)和生物(biological,Bio). 内部混合是指SSA的单颗粒内部是由不同化学成分组成的混合物. 例如,在SS颗粒中Na+和Cl-的贡献十分显著,而海水中的其他无机离子贡献较小;在SS-OC颗粒中虽仍以NaCl为主,但有机物的比例明显提高;OC颗粒内部则由有机碳主导;在Bio颗粒上则富集了尺寸足够小的细菌或病毒 [14, 57, 74]. 不同类型颗粒的相对丰度与粒径之间存在明显的依赖性[25, 75]. SS颗粒和Bio颗粒主要集中在超微米范围内;而SS-OC和OC颗粒则主要集中在亚微米区域,且随着颗粒物粒径减小,其有机占比不断提高[25, 30, 37]. 扫描电子显微镜结果显示(图4),SS颗粒呈现出明亮且规则的晶体状、OC颗粒一般为深色圆形颗粒、SS-OC颗粒则呈现有机组分包裹无机内核的核壳结构[76]. 有机壳层不仅会改变SSA界面的物质传输,也会改变SSA颗粒的理化性质(如吸湿性[77]、表面张力[78-79]、光散射能力[80]和CCN活化能力[77-78, 81]),进而影响海洋环境乃至全球气候.

3. SSA中的物质转移(Substance transfer in SSA)
研究SSA的化学组成对于理解其对大气化学和气候变化的影响至关重要,而不同物质从海水转移到SSA时常表现出富集或损耗,导致无法仅依靠海水的化学组成来预测SSA的化学组成. 通常用富集因子EFc(i)来量化某种物质在SSA上的富集程度[53, 64-65, 79],其被定义为:
stringUtils.convertMath(!{formula.content}) (1) 其中,
c(i)SSA i c(Na+)SSA c(i)sea i c(Na+)SSA i 3.1 无机离子
近年来,有越来越多的研究报道了无机离子在SSA颗粒中的富集. Kenee等在北大西洋海水的26个单独鼓泡实验中发现,有24个结果显示了Ca2+的EF中位数为1.2[37]. Cochran等在人工海水中加入羧酸盐后观察到Ca2+的EF可高达5,且EF随着颗粒物粒径的减小而增加[82]. Ca2+作为最容易在SSA上富集的无机离子[65, 83-84],因其易与有机物络合密切相关. 若SSA颗粒中的Ca2+不是以有机络合物的形式存在,那么它可能与碳酸盐或碳酸氢盐结合,这将对SSA的pH及其与pH相关的化学过程产生影响[37, 65]. 另一些研究发现,Mg2+与阴离子表面活性剂的优先结合也会促进Mg2+在SSA上的富集[85-86]. 此外,还有研究发现极地海冰形成过程中会发生Na2SO4·10H2O等盐类沉淀,抑制Na+从海水向SSA的转移,因而促进SSA中Mg2+/Na+和K+/Na+比值增加[87]. 这种无机离子在SSA上的富集随海冰季节性变化,将有助于加强对极地冰芯形成的理解. 虽然已有研究证实无机组分会影响SSA颗粒的吸湿性[65, 88-89],但对无机组分的转移机制以及其对SSA理化特性的影响仍需要更深入的研究.
3.2 有机物
有机物从海水转移到SSA的过程具有选择性. 首先,这种选择性会直观地体现在SSA的粒径上. 在糖类富集实验中,观察到细SSA颗粒(<2.5 μm)上的葡萄糖富集程度是粗SSA颗粒(2.5—10 μm)的3倍[64],亚微米SSA中的糖类富集程度比超微米SSA高出1个数量级[67]. 在氨基酸富集实验中,观察到氨基酸在亚微米颗粒上的富集能力比超微米SSA高10—20倍[68]. 这种粒径上的选择性可能是由SSA的产生机制引起的. 气泡在海洋微表层上破裂,由此产生的粒径较小的膜滴将富集大量海洋微表层中的疏水性有机物;而粒径较大的射滴主要来自于气泡的内部气-液界面,主要富含海水中的无机盐和少量的溶解有机碳与颗粒有机碳[47]. 其次,这种选择性体现在有机物的结构性质上. 具有极性酸侧链的游离氨基酸[68]、短链脂肪酸[90-91]和具有表面活性的糖类[92]会优先富集到SSA上. 此外,二价阳离子也会对有机物的选择性富集产生重要影响. 例如,Ca2+可以桥接并稳定海藻酸盐的聚合物链,进而增加海藻酸盐在SSA上的富集[92]. 阴离子可溶性糖可通过二价阳离子的桥接作用共吸附到不溶性表面活性剂棕榈酸微层上,从而增强可溶性糖类在SSA颗粒中的富集[93-94].
3.3 微生物
除无机离子和有机物外,尺寸足够小的细菌和病毒也可以在SSA中富集. 当它们进入海洋大气后,不仅可以在保持传染性前提下进行长距离输送[15, 95],还可以充当CCN和诱导冰成核来影响气候[32, 96-97]. 细菌和病毒在海-气交换过程中发生的相对富集可以通过计算每个基因组的气溶胶化因子(aerosolization factor, AF)来确定,A、B和S分别指在气溶胶、海水和海洋微表层(SSML)中某一基因组在总基因组中的占比[16],公式如下:
stringUtils.convertMath(!{formula.content}) (2) Jennifer等[16]的研究表明,大多数同纲或同目的细菌和病毒在SSA上的富集存在分类群相似性,而部分具有选择性基因组的细菌和病毒可能表现出分类群的特异性,例如,疏水性外膜有助于增强细菌和病毒的富集能力. 相比于病毒,细菌更容易富集到SSA上且不易受环境条件的影响.
4. 影响SSA生成的重要因素(Important factors affecting SSA formation)
4.1 海面风速
风对海洋表面施压会导致波浪的形成与破裂. 随着风速的增加,SSA产量将非线性增加. Staniec等的研究表明当风速从15 m·s−1增加到32 m·s−1时,海洋飞沫液滴的体积通量从58 mm3· s−1·m−2 增加到3300 mm3· s−1·m-2[98],增幅可达两个数量级. Andreas等的函数模型结果显示,风速增加1倍会引起SSA产量至少增加50倍[99]. 截至现在,源函数是描述SSA产量的最常见方法. 它被定义为在单位时间和面积内,某一粒径范围的SSA通过参考平面(通常为高于海洋表面10 m处的平面)的数量、体积或质量[100]. 目前大多源函数的一般表达式为[101]:
stringUtils.convertMath(!{formula.content}) (3) 式中,W(U10)是白浪(因夹带大量气泡而具有高反照率的波浪)覆盖率[102],它通常被认为仅依赖于10 m处的风速(U10). FN作为一个形状函数,给出了某粒径(Dp)增量下对应的SSA数量、体积或质量. 比较不同的源函数计算结果发现,SSA年产量存在1—2个数量级的差异[2]. 造成这种差异的原因至少有两个:其一,风驱动SSA形成的动力学机制复杂(如:大气湍流运动、波浪破碎动力学、空气夹带动力学和气泡破裂动力学等[103-104]);其二,仅考虑风速而忽略其它因素的扰动. 虽然从大尺度范围来看风速是决定SSA产生的关键因子,但是在局部过程中其它环境因素对SSA产生的影响也是不容忽视的[103].
4.2 海表温度
温度是影响SSA生成的重要因素之一. 温度通过改变海水的理化性质,进而影响气泡羽流、破裂和SSA的产生[36, 50, 105-106]. 整理汇总相关研究进展发现SSA的产生对温度的依赖性结论存在较大争议. Hultin等[107]、Salter等[58]、Zábori等[49-50]和Christiansen等[54]利用射流式发生装置所获得的研究结果表明SSA的数浓度与水温存在一种非线性关系. 当温度从0 ℃增加到约10 ℃时,SSA数浓度显著减少;而温度继续增加到约25 ℃时,SSA数浓度略有增加或几乎不变. 相反,利用MART/miniMART发生装置的模拟结果显示SSA数浓度随着温度的升高而线性增加[108]. 此外,一些鼓泡式发生器的实验结果显示SSA的中心粒径随着温度升高而减小,小粒径分布模态(<300 nm)的SSA数浓度随着温度的升高而降低,而大粒径分布模态(>350 nm)则表现出相反的变化趋势[43, 54, 109]. 目前对这一争议结论的普遍解释是由不同SSA发生装置引发的不同空气夹带率(单位时间内被夹带到海水中的空气体积)所造成的. 鼓泡式发生器的空气夹带率由额定的鼓气流量控制,不受温度影响;射流式发生器的空气夹带率则与水温呈负线性相关[58];而MART的空气夹带率随着温度升高而升高[110]. 此外,一些研究还发现温度会对气泡的羽流特征和尺寸分布等产生影响. 例如,Thorpe等指出水温升高会降低气体溶解度和水体粘度,导致气泡数量和上升速率增加,但这种增加在一定程度上被分子扩散速率的增加而缓解[111]. Nielsere等认为水温变化会显著影响表面气泡的寿命,从而影响SSA的产生[41]. Grythe等[101]统筹分析大量的全球观测数据发现海表温度的升高会引起SSA数浓度增加,这与Liu等[112]在太平洋与大西洋上空的飞机航测结果一致. 但是外场观测无法排除海温与风速之间的强相关性. 综合来看,目前海表温度对SSA生成的影响机制的解释还未达成统一共识.
4.3 海水盐度
海水盐度作为海洋重要的环境参数之一会对SSA的产生带来一定影响. 纵观全球海洋,大部分海域表层海水盐度在33‰—37‰的范围内变化[51],但在少数海域中盐度可能高于(红海盐度约40 ‰)或低于(波罗的海盐度约8 ‰)该范围. Tyree等[51]和Lv等[40]分别对不同盐度的人工海水进行了鼓泡实验,均得出了SSA数浓度随着盐度增加而增加的结论. Park等采集河流入海区域不同盐度梯度下射流产生的SSA并对其颗粒进行了表征,结果发现随着盐度从0 ‰增加到34 ‰,SSA数浓度增加8—17倍,且中心粒径也随之变大[113]. 研究认为高盐水体容易产生更多更小的气泡,从而产生更多数量的SSA[107, 114]. 但从全球范围来讲,除个别海域外,海洋微小的盐度变化对SSA全球排放通量的影响有限.
4.4 表面活性物质
已有研究发现海洋表层含有各种各样的表面活性物质,包括生物产生的脂肪酸类[69]和微囊藻毒素[115]等,人为排放的全氟烷基酸类[116]和二棕榈酰磷脂酰胆碱(医用气溶胶疗法中的赋形剂)[117]等. 这类物质特有的亲水和疏水基团会使其在海洋表面和海水气泡表面定向排列而显著降低海水的表面张力. 研究发现表面活性物质的种类、浓度和聚合程度等均会对SSA的生成产生不同程度的影响. 例如,丙二酸、果糖和丙二酸钠在一定浓度范围内都会促进SSA的生成,但它们对SSA粒径分布、混合状态和形貌特征的影响不尽相同[40]. 当油酸浓度从0.1 mg·L−1增加至1 mg·L−1时,SSA数浓度明显增加,而继续添加至10 mg·L−1时,数浓度几乎不再发生变化[51]. 乙二醇和不同分子量的聚乙二醇会不同程度地抑制SSA的生成,但会增加颗粒的几何直径. 值得注意的是,随着它们浓度的增加会削弱这种抑制作用,高浓度的聚乙二醇甚至会促进SSA的生成[53]. 此外,表面活性物质还会影响水面气泡的寿命和气泡膜的脆性. Sellegri等的研究发现加入过量的十二烷基硫酸钠会导致水面出现一层泡沫层延长气泡的破裂时间,极大限度抑制了SSA的产生[43]. 虽然表面张力在一定程度上可以解释表面活性物质对SSA生成的影响,但究竟是促进还是抑制SSA的产生在很大程度上是未知的.
4.5 浮游生物
浮游生物作为海洋有机物的主要生产者,在SSA的形成过程中起到关键作用. 高生产力或发生藻华的水体常伴有高的浮游生物量. 叶绿素-a是衡量生物量大小的常用指标. 模拟研究常采用叶绿素-a浓度来预测SSA上有机物的富集[118-120],但二者相关性的结果存在差异. 一些研究认为SSA中有机物占比与叶绿素-a浓度相关[121-123],而另一些研究认为两者之间没有相关性[79, 124-125]. 浮游植物作为初级生产者能够产生大量有机物,包括溶解有机碳和颗粒有机碳. 不同有机物在SSA上表现出的选择性富集,进而导致并不是所有海水中的有机物均能及时体现在SSA上. 在MART的模拟水华实验中,Santander等推断水华期间海洋有机物向SSA中转移的过程可能是由胞外聚合物主导[126]. 浮游植物的生长导致海水中的溶解有机碳浓度升高,促进SSA颗粒上有机物的富集[127]. Wang等的水华实验表明亚微米和超微米SSA中有机物的富集种类不同,且富集程度取决于有机物源(生物产生)-汇(细菌消耗)过程的动态平衡[32]. 此外,研究发现SSA中腐殖质(HULIS)荧光信号相比于海水而言发生了20 nm的红移,这说明浮游植物虽然不是HULIS从海洋转移到大气的直接驱动因素,但会间接诱导HULIS化学成分的变化[126]. 有趣的是最新的航测结果表明海洋上空SSA数浓度存在昼夜循环规律,这与浮游植物的昼夜活动规律一致,进一步佐证了浮游生物对SSA的产生过程具有重要的调节作用[52, 128-129]. 尽管浮游植物的大量繁殖会导致海洋生物群落结构和生物量的显著变化,但在水华期间SSA的化学组成与浮游生物活动并未建立起明确的内在联系[130].
5. 结论与展望(Conclusion and prospective)
SSA的相关研究是当前全球气候变化研究的重要前沿领域,已成为国际上层海洋-低层大气研究计划(SOLAS)的核心内容之一. 海洋具有自身特有的生物地球化学循环系统,并与陆地-大气多圈层相互作用,进而导致SSA的形成机制、传输条件和理化特性与陆源气溶胶相比具有显著性差异. 尽管近些年已在该领域开展了大量的研究,尤其是在SSA的实验室模拟方面,但仍有诸多具有挑战性的科学问题有待回答与解决:
(1)研究方法:SSA的产生受到气泡尺寸、羽流特征和气泡破裂过程等参数的共同控制,能否准确还原这些参数是模拟真实SSA产生的关键. 由于当前不同SSA发生器的设计思路和装置结构存在显著差异,导致目前无法对SSA的理化性质得到更多的共识. 因此,研发并普及一种标准化的SSA发生器仍是当前和未来实验室模拟SSA研究亟需解决的难题. 突破这一挑战才能实现对参数的精细调控,从而对SSA的粒径分布、化学组成、表面形貌等理化性质进行表征和定量,最终揭示多模态分布下SSA的具体产生机制.
(2)环境行为:SSA的化学组成与形貌特征决定其大气化学命运. 因此亟需开展以SSA为媒介研究物质在海-气界面迁移与转化的全链条综合研究. 聚焦海洋有机物在SSA的产生、迁移与转化行为对多种环境因素耦合影响的响应机制,识别影响该过程的关键调控因子. 另外,如何结合对现有数据的分析,同时开展多海域的大规模调查和模拟,完善SSA的排放源函数关系是今后在SSA环境行为研究方面需要着重思考的问题. 未来还需要着重考虑海洋浮游生物对SSA产生与转化的影响,科学评估浮游生物在SSA形成中所扮演的“角色”.
(3)气候效应:SSA作为CCN引起云辐射强迫的变化是气候评估中最大的不确定因素. SSA的迁移与转移以及对其“汇”过程的定量研究仍然不足,导致当前SSA的研究无法准确实现其环境效应评估. 解决这一研究“瓶颈”的关键在于要量化环境因素变化对海盐气溶胶理化性质的影响,同时精确定量不同环境体系下SSA对CCN贡献的绝对值. 这将有助于全面且系统地认识SSA的形成与转化及其气候效应,并大大提升其对全球气候模型预测的准确性.
-
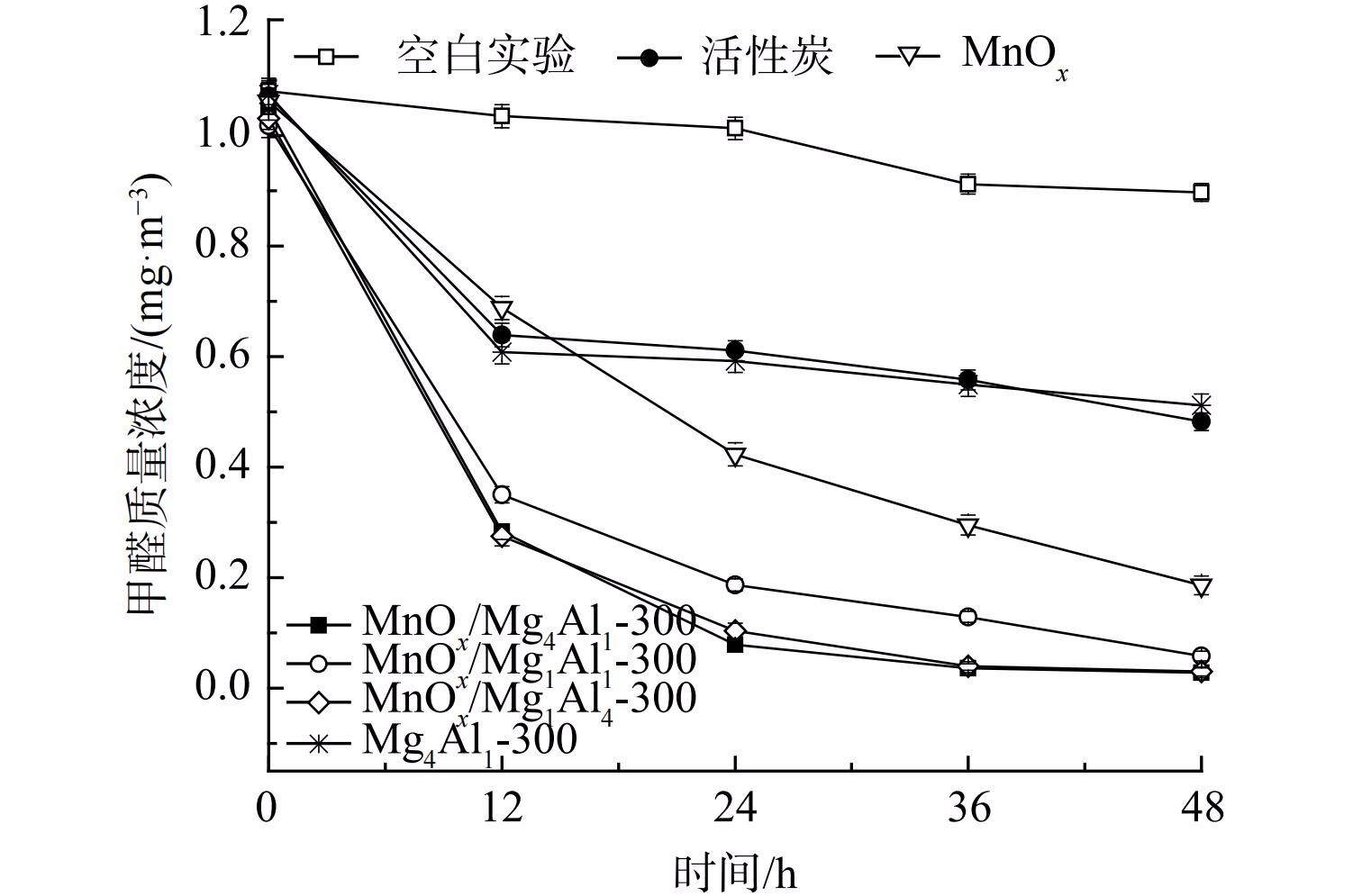
图 2 n(Mg)∶n(Al)对催化剂氧化性能的影响
Figure 2. The effect of n(Mg)∶n(Al) molar ratio on the oxidation performance of catalysts
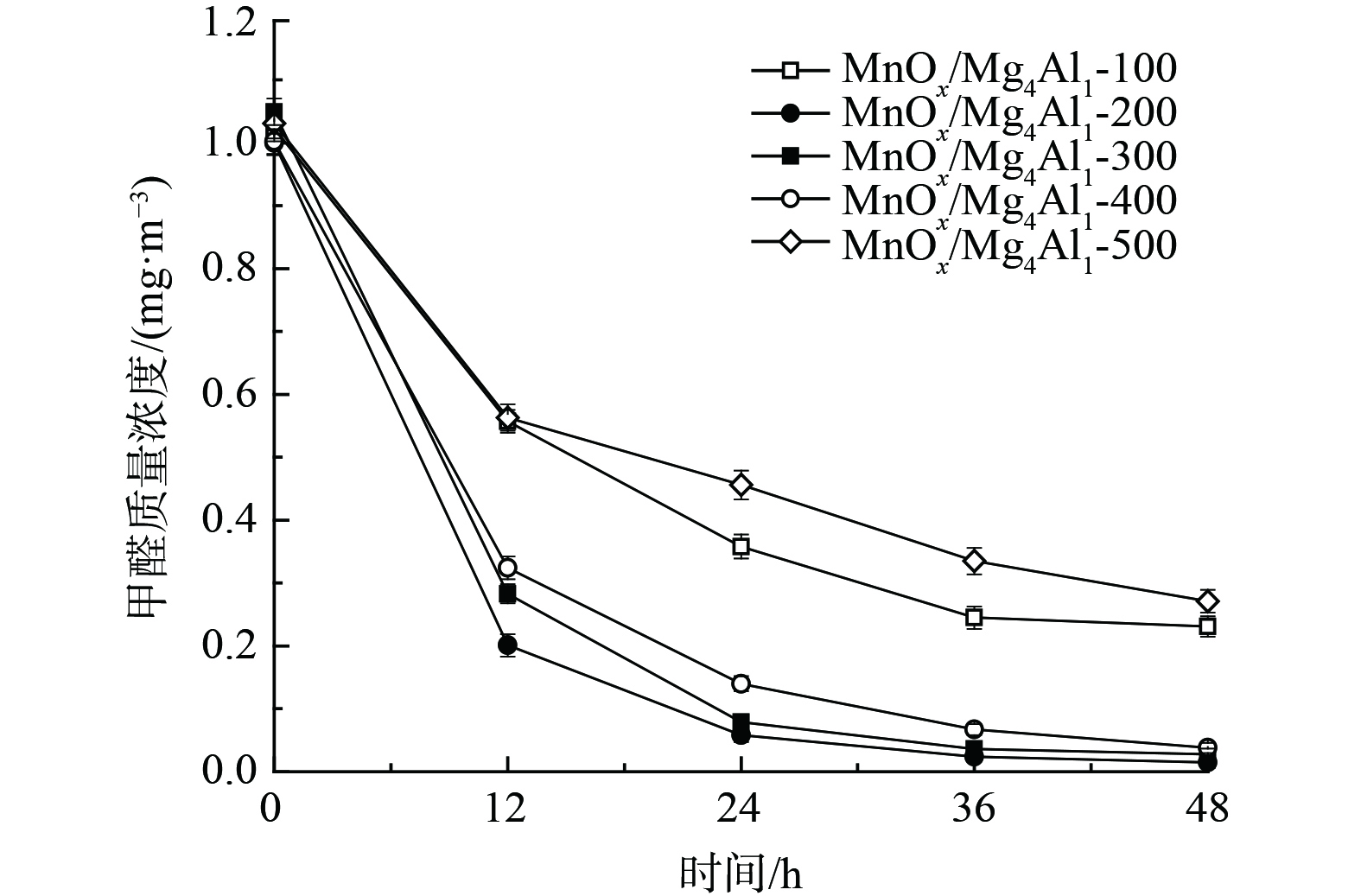
图 3 煅烧温度对催化剂氧化性能的影响
Figure 3. The effect of calcination temperature on the oxidation performance of catalysts
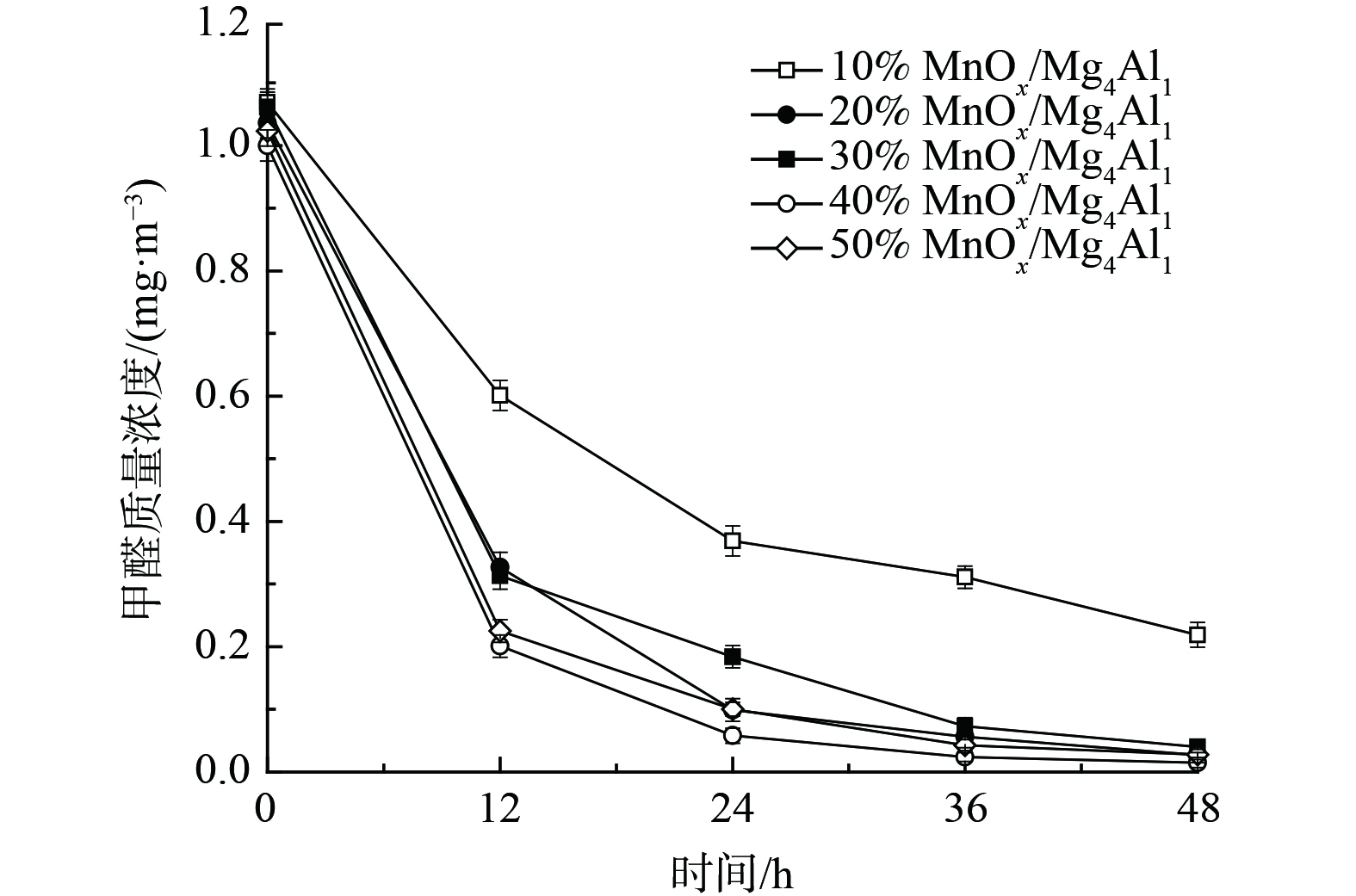
图 4 负载量对催化剂氧化性能的影响
Figure 4. The effect of loading on the oxidation performance of catalysts
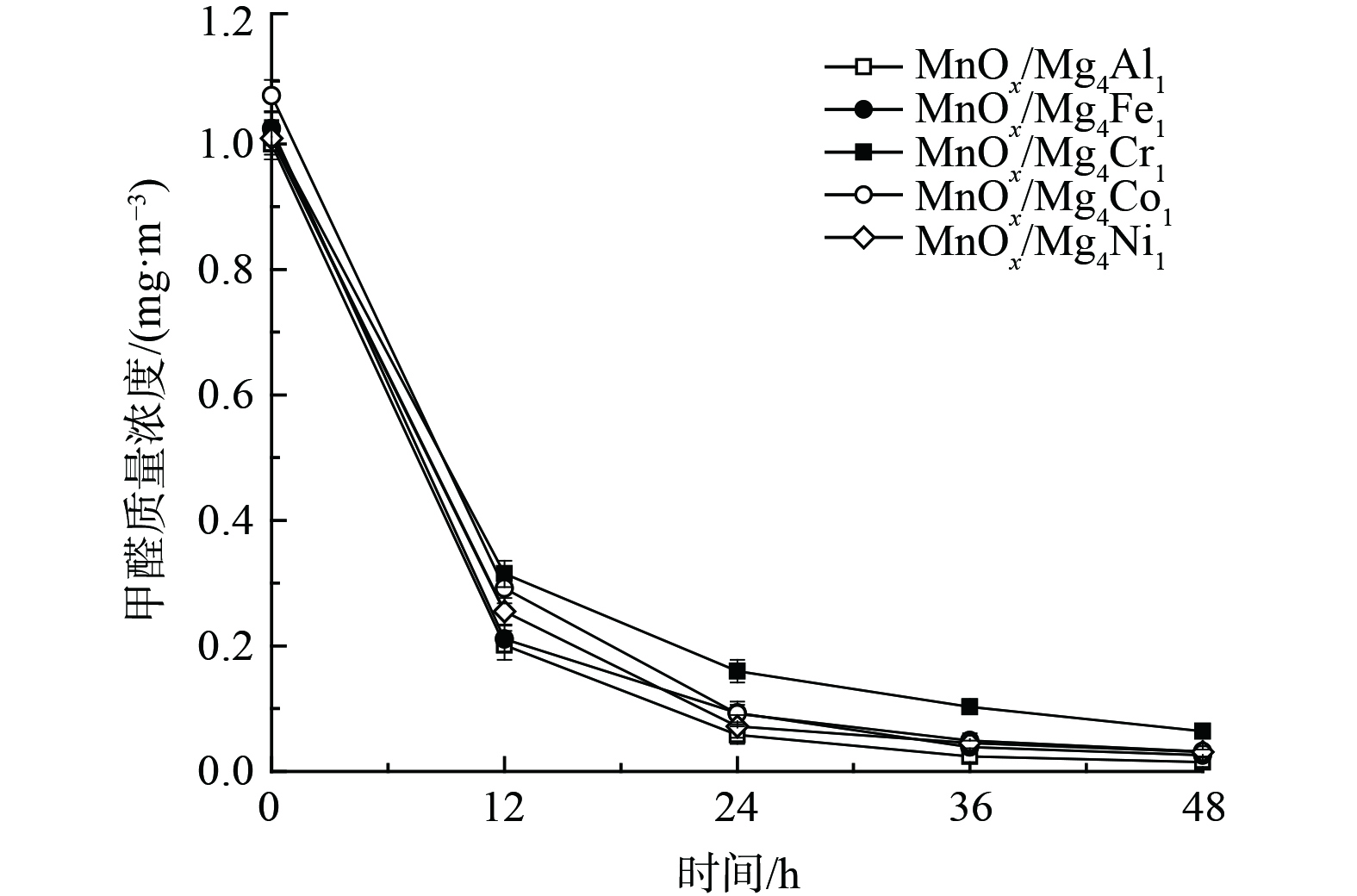
图 5 类水滑石组分对催化剂氧化性能的影响
Figure 5. The effect of hydrotalcite-like component on the oxidation performance of catalysts
表 1 催化剂的比表面积、孔体积和平均孔径
Table 1. Specific surface area, pore volume and average pore diameter of catalysts
样品 比表面积(BET)/(m2·g−1) 孔体积/(cm3·g−1) 平均孔径/nm MnOx-200 22.6 0.222 28.4 Mg4Al1-200 58.2 0.109 5.6 MnOx/Mg4Al1-100 62.0 0.231 13.0 MnOx/Mg4Al1-200 78.8 0.437 14.2 MnOx/Mg4Al1-300 81.4 0.497 19.1 MnOx/Mg4Al1-500 66.3 0.566 16.4 MnOx/Mg1Al1-300 79.0 0.187 6.5 MnOx/Mg1Al4-300 156.7 0.438 8.7  下载: 导出CSV
下载: 导出CSV
-
[1] HU Q Q, LIU K J, YE J W, et al. Hierarchical Pt/NiCo2O4 nanosheets decorated carbon nanofibers for room-temperature catalytic formaldehyde oxidation[J]. Applied Surface Science, 2023, 623: 157012. doi: 10.1016/j.apsusc.2023.157012 [2] MIAO L, WANG J L, ZHANG, P Y. Review on manganese dioxide for catalytic oxidation of airborne formaldehyde[J]. Applied Surface Science, 2019, 466: 441-453. doi: 10.1016/j.apsusc.2018.10.031 [3] FANG R M, HUANG, X Y, SUN Y J, et al. Excellent stability for catalytic oxidation formaldehyde over defective δ-MnO2 nanoparticles at room temperature[J]. Journal of Environmental Chemical Engineering, 2023, 11(1): 109064. doi: 10.1016/j.jece.2022.109064 [4] 武强, 潘奕君, 裴晶晶. 黑色TiO2/活性炭复合材料的制备及其光催化净化甲醛性能[J]. 环境工程学报, 2023, 17(3): 841-849. [5] YE J, ZHU X, CHENG B, et al. Few-layered graphene-like boron nitride: a highly efficient adsorbent for indoor formaldehyde removal[J]. Environmental Science & Technology Letters, 2017, 4: 20-25. [6] LI Q, ODOOM-WUBAH T, ZHOU Y P, et al. Coral-like CoMnOx as a highly active catalyst for benzene catalytic oxidation[J]. Industrial & Engineering Chemistry Research, 2019, 58(8): 2882-2890. [7] 潘菊霜, 麻春艳, 宋茂勇. 环境水对FeMnO3和NiMnO3室温催化氧化甲醛的影响[J]. 环境科学学报, 2022, 42(6): 346-356. [8] 蒋昕楠, 孔振凯, 王际童, 等. 高锰酸钾改性球形中孔炭的甲醛吸附性能[J]. 环境工程学报, 2018, 12(6): 1676-1 682. [9] 孙婷, 刘凤娟. 室内甲醛污染治理方法研究进展[J]. 环境生态学, 2021, 3(8): 91-95. [10] 雷春生, 朱晓峰. 胺化凹凸棒黏土的制备及甲醛吸附性能研究[J]. 环境工程, 2016, 34(6): 95-100. [11] BOYRAZ E, YALCINKAYA F. Hydrophilic surface-modified PAN nanofibrous membranes for efficient oil-water emulsion separation[J]. Polymers, 2021, 13(2): 197. doi: 10.3390/polym13020197 [12] LI J Y, CUI W, CHEN P, et al. Unraveling the mechanism of binary channel reactions in photocatalytic formaldehyde decomposition for promoted mineralization[J]. Applied Catalysis B:Environmental, 2020, 260: 118130. doi: 10.1016/j.apcatb.2019.118130 [13] BERRICHI A, BACHIR R, BEDRANE S, et al. Heterogeneous bimetallic Au-Co nanoparticles as new efficient catalysts for the three-component coupling reactions of amines, alkynes and CH2Cl2[J]. Research on Chemical Intermediates, 2019, 45(6): 3481-3495. doi: 10.1007/s11164-019-03803-6 [14] 张珍珍, 李鑫恒. 基于催化氧化技术去除甲醛的研究进展[J]. 分子催化, 2019, 33(4): 381-389. doi: 10.16084/j.cnki.issn1001-3555.2019.04.005 [15] 朱杰, 孙月吟, 顾名扬, 等. TiO2负载MnFeOx催化剂的制备及常温催化氧化甲醛性能研究[J]. 功能材料, 2022, 53(4): 4011-4019. [16] CHEN X Y, WANG H H, CHEN M, et al. Co-function mechanism of multiple active sites over Ag/TiO2 for formaldehyde oxidation[J]. Applied Catalysis B:Environmental, 2021, 282: 119543. doi: 10.1016/j.apcatb.2020.119543 [17] TIAN M Z, LIU S J, WANG L L, et al. Complete degradation of gaseous methanol over Pt/FeOx catalysts by normal temperature catalytic ozonation[J]. Environmental Science & Technology, 2020, 54(3): 1938-1945. [18] TIAN Z Y, NGAMOU P H T, VANNIER V, et al. Catalytic oxidation of VOCs over mixed Co-Mn oxides[J]. Applied Catalysis B:Environmental, 2012, 117/118: 125-134. doi: 10.1016/j.apcatb.2012.01.013 [19] YAO X, GAO M X, WEI Z D, et al. Removal of hexanal in cooking fume by combination of storage and plasma-catalytic oxidation on alkali-modified Co-Mn solid solution[J]. Chemosphere, 2019, 220: 738-747. doi: 10.1016/j.chemosphere.2018.12.201 [20] FANG R M, HUANG H B, JI J, et al. Efficient MnOx supported on coconut shell activated carbon for catalytic oxidation of indoor formaldehyde at room temperature[J]. Chemical Engineering Journal, 2018, 334: 2050-2 057. [21] 薛花, 崔家浩, 邢中鹏, 等. MnOx/ACF负载型催化剂常温催化氧化二氯甲烷[J]. 化工环保, 2021, 41(1): 77-82. doi: 10.3969/j.issn.1006-1878.2021.01.013 [22] LI R, SHI, X, HUANG Y, et al. Catalytic oxidation of formaldehyde on ultrathin Co3O4 nanosheets at room temperature: Effect of enhanced active sites exposure on reaction path[J]. Applied Catalysis B:Environmental, 2022, 319: 121902. doi: 10.1016/j.apcatb.2022.121902 [23] 赵海楠, 王健, 徐文青, 等. 锰氧化物催化氧化挥发性有机物(VOCs)研究进展[J]. 环境工程, 2019, 37(10): 157-167. doi: 10.13205/j.hjgc.201910027 [24] LU L, TIAN H, HE J H, et al. Graphene-MnO2 hybrid nanostructure as a new catalyst for formaldehyde oxidation[J]. The Journal of Physical Chemistry C, 2016, 120(41): 23660-23668. doi: 10.1021/acs.jpcc.6b08312 [25] ZHU L, WANG J L, RONG S P, et al. Cerium modified birnessite-type MnO2 for gaseous formaldehyde oxidation at low temperature[J]. Applied Catalysis B:Environmental, 2017, 211: 212-221. doi: 10.1016/j.apcatb.2017.04.025 [26] LI J W, PAN K L, YU S J, et al. Removal of formaldehyde over MnxCe1-xO2 catalysts: thermal catalytic oxidation versus ozone catalytic oxidation[J]. Journal of Environmental Sciences, 2014, 26(12): 2546-2553. doi: 10.1016/j.jes.2014.05.030 [27] SZABÓ V, MESZAROS R, KÓNYA Z, et al. Preparation and characterization of MnIn-layered double hydroxides (LDHs), extension of the synthesis to fabricate MnM (III)-LDHs (M=Al, Sc, Cr, Fe, Ga), and the comparison of their photocatalytic and catalytic activities in the oxidation of hydroquinone[J]. Journal of Molecular Structure, 2022, 1261: 132966. doi: 10.1016/j.molstruc.2022.132966 [28] YAN Q H, HOU X T, LIU G C, et al. Recent advances in layered double hydroxides (LDHs) derived catalysts for selective catalytic reduction of NOx with NH3[J]. Journal of Hazardous Materials, 2020, 400: 123260. doi: 10.1016/j.jhazmat.2020.123260 [29] DANIEL S, THOMAS S. Layered double hydroxides: fundamentals to applications[M]. Cambridge: Woodhead Publishing, 2020: 1-76. [30] LI, D D, YANG G L, LI P L, et al. Promotion of formaldehyde oxidation over Ag catalyst by Fe doped MnOx support at room temperature[J]. Catalysis Today, 2016, 277: 257-265. doi: 10.1016/j.cattod.2016.02.040 [31] YU H R, LIU D X, WANG H Y, et al. Singlet oxygen synergistic surface-adsorbed hydroxyl radicals for phenol degradation in CoP catalytic photo-Fenton[J]. Chinese Journal of Catalysis, 2022, 43(10): 2678-2689. doi: 10.1016/S1872-2067(22)64117-2 [32] WU Z B, JIN R B, WANG H Q, et al. Effect of ceria doping on SO2 resistance of Mn/TiO2 for selective catalytic reduction of NO with NH3 at low temperature[J]. Catalysis Communications, 2009, 10(6): 935-939. doi: 10.1016/j.catcom.2008.12.032 [33] 张先龙, 彭真, 刘鹏等. 基于PPS的锰基催化脱硝-除尘功能一体化滤料的制备及其低温SCR脱硝[J]. 功能材料, 2015, 46(S2): 160-164. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2982
- HTML全文浏览数: 2982
- PDF下载数: 80
- 施引文献: 0
