



-
厌氧氨氧化(anaerobic ammonium oxidation, Anammox)可以代替传统硝化反硝化工艺去除污水中的氮,具有能耗低、产泥量少、无需外加碳源和运行成本低等优势[1],被认为是最有前途的生物脱氮工艺之一。由于厌氧氨氧化生长缓慢,倍增时间长,工程上采用生物膜[2]或颗粒污泥[3]形态持留厌氧氨氧化菌(anaerobic ammonium oxidation bacteria, AnAOB),以保证系统稳定运行。目前,厌氧氨氧化工艺已成功应用于污泥厌氧消化上清液[4]等高浓度氨氮污水处理中。由于城市污水具有氨氮浓度低、温度低等特点,限制了厌氧氨氧化的主流应用[5]。为提高AnAOB活性,通常采用投加FeS[6]、Fe(Ⅲ)[7]、纳米零价铁(nZVI)[8]、羟胺(NH2OH)[9]、肼(N2H4)[10]、石墨烯[11]以及生物炭[12]等辅助材料。其中,N2H4作为厌氧氨氧化代谢中间产物受到广泛关注。N2H4可以通过抑制其他细菌生长,降低与AnAOB对底物的竞争,同时为AnAOB的生长提供额外能量,减少NO3−-N的产生,从而提高厌氧氨氧化反应器脱氮性能[13-15]。
YAO等[16]研究表明,当加入3.99 mg·L−1的N2H4时,CANON系统中颗粒污泥的厌氧氨氧化活性增加,当N2H4质量浓度为4.86 mg·L−1时,可以缓解NO2−-N对AnAOB活性的抑制[17]。MIODOŃSKI等添加了3.7 mg·L−1的N2H4后,在高基质浓度条件下厌氧氨氧化系统在42 d内完成了快速启动,平均氮负荷率(nitrogen loading rate, NLR)比对照组高2倍[18]。蔡庆等[19]通过批式实验研究N2H4对高基质浓度下(NH4+-N约225 mg·L−1,NO2−-N约280 mg·L−1)厌氧氨氧化颗粒污泥的短期影响,结果发现,当N2H4质量浓度在1.8~9.5 mg·L−1时,厌氧氨氧化活性明显增加。XIANG等[20]研究发现当N2H4质量浓度为2~5 mg·L−1时,纯颗粒污泥和絮体-颗粒混合污泥的反应器均可保持长达4个月的稳定高效运行,但纯颗粒污泥系统具有更高效的性能,总氮去除速率(total nitrogen removal rate, TNRR)达到(0.33±0.04) g·(L·d)−1。可见,N2H4对AnAOB活性的影响不仅与N2H4质量浓度有关,还与污泥形态有关。目前大部分研究集中于N2H4对颗粒形态AnAOB的影响,而低质量浓度N2H4对生物膜形态厌氧氨氧化体系的研究尚有不足。但在城市污水低氨氮浓度条件下,厌氧氨氧化颗粒污泥粒径小,难以有效持留在系统中[21],而通过载体形成的厌氧氨氧化生物膜可被有效截留于反应器中,因此,生物膜形式的厌氧氨氧化技术具有更广泛的应用。悬浮载体上的生物膜可自发富集AnAOB,加速AnAOB的粘附与生长,从而加速主流厌氧氨氧化工艺性能的提升[2]。由于生物膜传氧限制,厌氧氨氧化生物膜可以在低溶解氧(dissolved oxygen, DO)环境下生长,对正常的北方气候温度和低氨氮底物浓度适应良好[22]。因此,探究低质量浓度N2H4对厌氧氨氧化生物膜的长期影响,可以为厌氧氨氧化的在城市污水脱氮中的应用提供技术支撑。
-
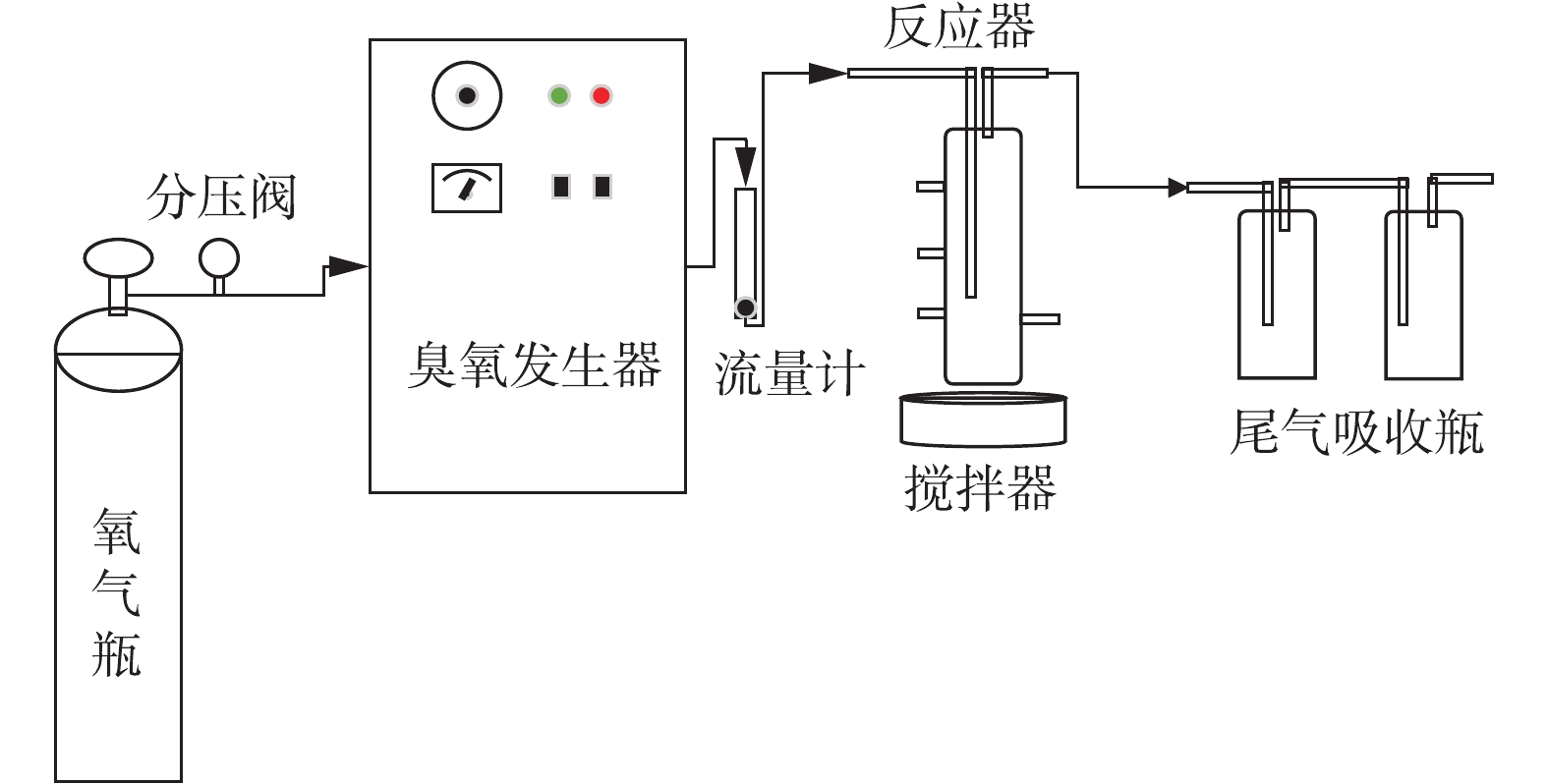
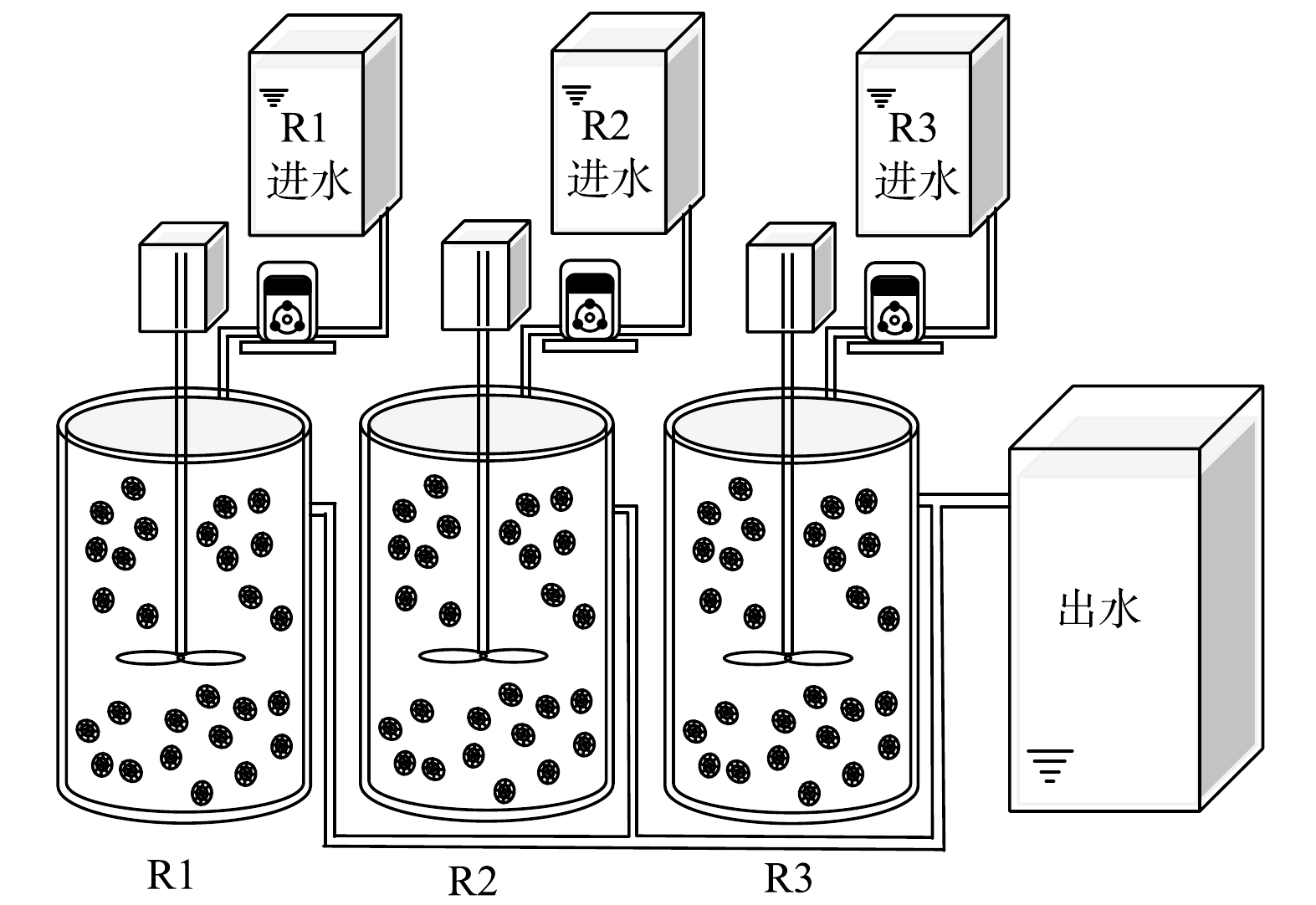
实验室规模的小试采用3个圆柱形移动床生物膜反应器,有效容积为2 L。R1为对照组,R2和R3中分别添加5 mg·L−1和10 mg·L−1的N2H4。3个反应器中均填充已挂膜的填料,填充比为35%,填料直径为25 mm,高10 mm。采用序批式活性污泥法(sequencing batch reactor activated sludge process, SBR)运行,周期为6 h (360 min),包括5 min进水,340 min搅拌,14 min沉降和1 min排水,HRT为1 d,日处理量为2 L·d−1。3个反应器均在常温条件下运行,温度在(27.6±2.4) ℃。反应器装置如图1所示。
-
实验中用到的生物膜污泥来自实验室稳定运行196 d的移动床生物膜反应器(moving bed biofilm reactor, MBBR),NLR为0.6 kg·(m3·d)−1,生物膜系统中的污泥质量浓度为(1 800±100) mg·L−1。
实验采用模拟废水,主要成分有50 mg·L−1 NH4+-N、55 mg·L−1 NO2−-N、300 mg·L−1 CaCl2·2H2O、180 mg·L−1 MgSO4·7H2O、27.2 mg·L−1 KH2PO4以及500 mg·L−1 NaHCO3。每升废水分别加入1 mL微量元素Ⅰ和Ⅱ[23]。N2H4以N2H4·H2SO4的形式投加。初始pH通过滴加1 mol·L−1的HCl或NaOH溶液调节到6.9~7.3。配水采用自来水,未对其进行脱氧处理,进水DO质量浓度为5 mg·L−1。
-
整个过程按不同运行参数分为3阶段。阶段Ⅰ(1~11 d):为获取较为稳定的实验条件,反应器在初始NLR下运行。阶段Ⅱ(12~46 d):加入N2H4运行,3个反应器N2H4质量浓度分别为0、5、10 mg·L−1。阶段Ⅲ(47~60 d):停止加入N2H4,反应器继续运行。实验设计如表1所示。
-
用比色法测定NH4+-N、NO2−-N、NO3−-N和N2H4的质量浓度。其中NH4+-N、NO2−-N和NO3−-N采用标准测试方法[24]。通过加入1 mol·L−1 HCl和0.1mol·L−1 KIO3溶液消除N2H4对NH4+-N测定的干扰[25]。N2H4依照Watt和Chrisp[26]的检测方法,通过加入0.5%的氨基磺酸溶液消除NO2−-N对N2H4测定的干扰[27]。在每个阶段结束(反应器运行第11、46和60天)留取生物样品测定生物量(suspended solids, SS)、有机物量(volatile suspended solids, VSS)、胞外聚合物(extracellular polymeric substances, EPS)、血红素和微生物群落结构。
采用水解法提取生物膜样品中的EPS(主要由蛋白质和多糖组成[28]),蛋白质(proteins, PN)用改良Folin-Lowry法测定,使用牛血清蛋白作为标准物质。多糖(polysaccharides, PS)用蒽酮-硫酸法测定,使用葡萄糖作为标准物质。EPS浓度以单位质量挥发性有机物中EPS的质量(mg·g−1)表示。
用磷酸盐缓冲液(PBS)通过细胞破碎的方式提取血红素[29],以氯化血红素作为标准物质,通过Pyridine-NaOH方法[30]测定血红素的含量。血红素浓度以单位质量挥发性有机物中血红素的物质的量(μmol·g−1)表示。
-
为直观表示出整个反应体系中的主导反应,依照亚硝化反应(式(1))、硝化反应(式(2))以及厌氧氨氧化反应(式(3))的化学计量方程计算得到含有NH4+-N、NO2−-N、NO3−-N变化量的公式。在进水无有机物的厌氧氨氧化反应器中,异养生物对NRR的贡献通常< 5%[31],可忽略其对整体反应的影响,故在计算时不考虑反硝化过程,计算得到式(4)~式(7)。
式中:
\theta 为周期时间,d;C1,C2为1个周期内NH4+-N和NO2−-N的去除量,mg·L−1;C3为NO3−-N的生成量,mg·L−1。Q1为AOB对NH4+-N的氧化速率(AOR),g·(m3·d)−1;Q2为NOB对NO2−-N的消耗速率(NOR),g·(m3·d)−1。Q3 为AnAOB对NH4+-N的消耗速率(AnAOR),g·(m3·d)−1;Q4为AnAOB对NO2−-N的消耗速率(AnANR),g·(m3·d)−1。 -
在各阶段的稳定运行期内,保留生物膜样品于-80 ℃条件下冻存。实验结束后统一进行基因组DNA的提取,之后采用341F(5’-CCTACGGGNGGCWG-CAG-3’)和785R(5’- GACTACHVGGGTATCTAATCC-3’)作为扩增引物,对细菌16S rRNA基因进行2轮PCR扩增。后续使用Illumina Novaseq 6000测序平台进行高通量并行测序,将相似水平在97%的序列归为1个OTU进行生物信息统计分析。
-
反应器各阶段运行性能如图2所示。在进水NH4+-N和NO2−-N质量浓度分别为(50.9±3.6) mg·L−1和 (55.5±3.2) mg·L−1条件下持续运行11 d后,3个反应器运行趋于平稳。阶段Ⅰ结束时NRR分别为27.7、24.7、23.2 g·(m3·d)−1,这一差异可能是由于实验前期选取的生物膜填料挂膜不均匀导致。
对照组R1内逐渐形成稳定的厌氧氨氧化环境,阶段III时NRR最高达到139.2 g·(m3·d)−1。R2和R3在加入N2H4后均表现出NRR先快速上升后下降的趋势。阶段Ⅱ运行前期(第12~15天),R2和R3中的NRR分别提升了74%和44%,此时N2H4的利用率均在74%以上。随着运行周期的增加(第16~46天),R2和R3的脱氮效能逐渐下降,其中R2内NRR下降了53%,R3内NRR下降了64%,在阶段Ⅱ结束时(第46天)R2和R3中N2H4的利用率分别从最高值(97%和79%)下降到32%。当R2和R3系统内的脱氮效能低于15 g·(m3·d)−1时停止加N2H4,开始阶段Ⅲ的运行。N2H4的抑制解除后,R2内厌氧氨氧化活性开始逐渐恢复,到阶段Ⅲ末期,NRR由阶段Ⅱ末期的11.0 g·(m3·d)−1升至到33.4 g·(m3·d)−1,R3的NRR也由10.4 g·(m3·d)−1升至22.8 g·(m3·d)−1,表明停止投加N2H4后,其抑制作用可缓慢恢复。经14 d恢复后,R2反应器的脱氮效能恢复到投加N2H4前的水平,然而,10 mg·L−1的N2H4对生物膜的抑制作用更强,生物膜恢复更加缓慢。SCHALK等[32](N2H4投加量为28.8 mg·L−1)的研究结果也证实了这一点。但GANESAN等[10](N2H4投加量为10 mg·L−1)、ZHOU等[33]( N2H4投加量为10 mg·L−1)、MIODOŃSKI等[18] (N2H4投加量为3.7 mg·L−1)和XIANG等[14](N2H4投加量为2~5 mg·L−1)的研究结果均表明,投加不同质量浓度的N2H4均能维持脱氮系统长期运行且N2H4可被快速消耗,这与本研究的结果并不一致。
本研究中出现N2H4浓度不断积累且NRR不断下降的现象,推测可能有以下2点原因。一方面,生物膜与颗粒污泥的形态结构存在差异。对比本研究中使用的生物膜,颗粒污泥结构更加密实,传质效率低,内部更容易形成较强的浓度梯度,即实际进入颗粒污泥内部的N2H4浓度会低于水中测得的浓度。溶质的浓度梯度是随着生物膜厚度的增加而形成的,生物膜厚度会影响液体的流动扩散和营养物质的传质效果,从而影响工艺整体性能[34]。本研究中生物膜厚度较薄(约550 μm),可推测扩散到生物膜内部的N2H4质量浓度较高,从而对AnAOB产生较强的毒性效应。另一方面,基质的质量浓度和N2H4质量浓度的比例不同。在前人研究N2H4对低基质(TN<100 mg·L−1)厌氧氨氧化系统的长期影响中,维持系统良好脱氮效果的基质质量浓度与N2H4质量浓度的比例在10左右。本实验中R2和R3中此比例分别为21和10.5,但显然R3受到更强的抑制作用,很可能是N2H4扩散到生物膜的速度过快,因此,需要降低N2H4的质量浓度以达到更好的TN去除效果。根据STROUS[35]提出的厌氧氨氧化分解代谢模型,可推知未反应完全的N2H4会促使反应逆向进行从而产生NH4+-N。SCHALK等[32]在加入N2H4的厌氧氨氧化批式实验中观察到,1 mol的N2H4可被转化为1.3 mol的NH4+-N,在ZEKKER等[13]的实验中,1 mol 的N2H4可形成1.63 mol NH4+-N。可见,AnAOB对N2H4有歧化作用,高质量浓度的N2H4会导致NH4+-N不断积累而升高,从而NRR也不断下降。
-
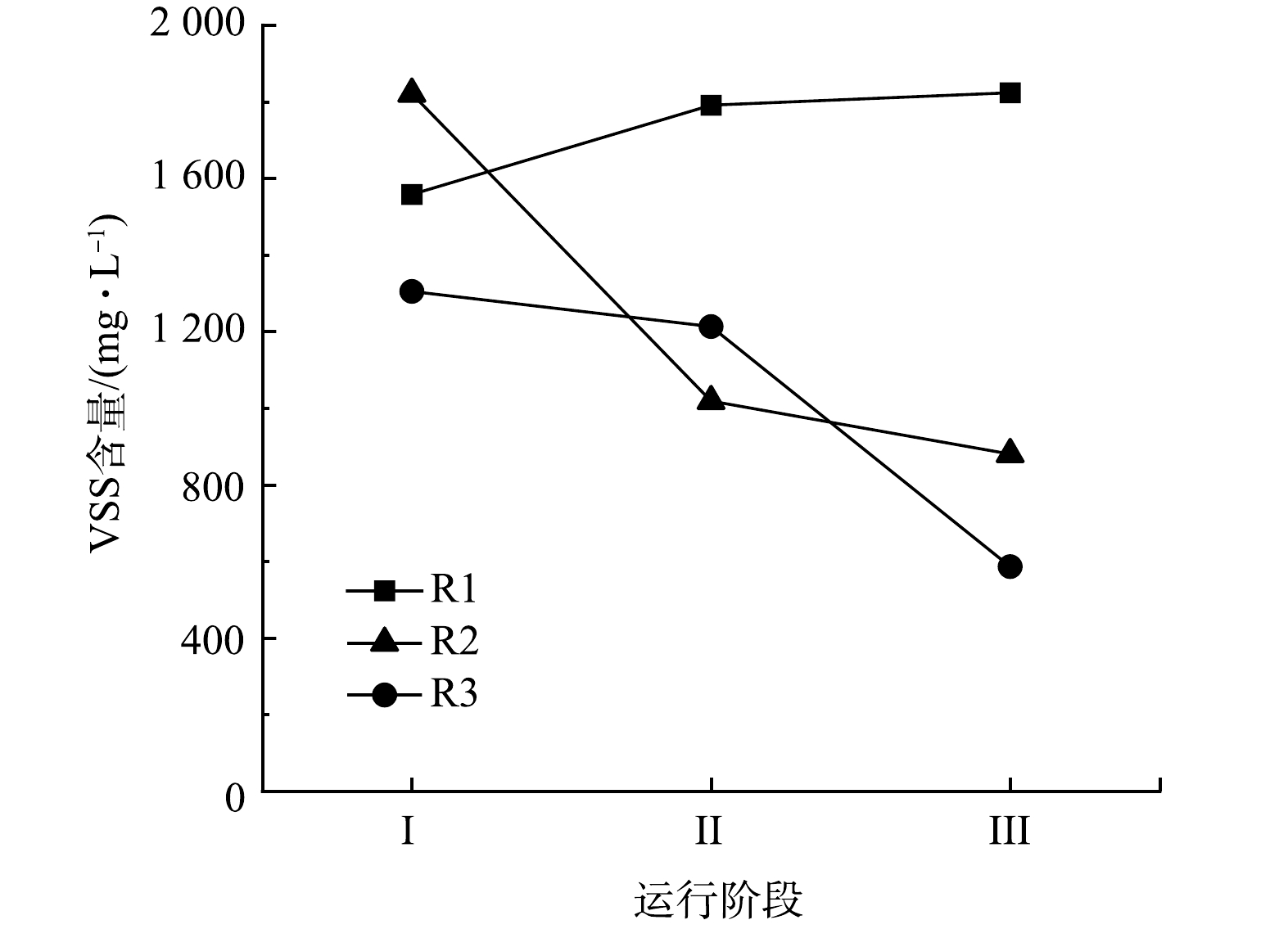
整个运行期间3个反应器内生物量变化如图3所示。对照组R1中的生物量随运行时间的增加逐渐升高,在整个实验周期结束时VSS达到1 824 mg·L−1。且R1在3个反应器中表现出最好的脱氮性能,也证实了R1内生物膜活性较高的结论。在阶段Ⅱ中,R2和R3中均出现了污泥流失情况。停止加入N2H4后,2个反应器内污泥量仍持续下降。R2在阶段Ⅱ和阶段Ⅲ结束时的VSS分别为1 019 mg·L−1和880 mg·L−1,R3在阶段Ⅱ和阶段Ⅲ内VSS分别为1 213 mg·L−1和587 mg·L−1。对比初始值,阶段Ⅲ结束时R2的VSS浓度下降了52%,R3内的VSS浓度下降了55%,可以看出,加入N2H4后反应器内部生物膜会逐渐脱落和流失,生物膜活性减弱,导致系统内部的脱氮效能下降。
-
由于进水中含有一定溶解氧,促进了脱氮系统中硝化菌的生长。为评估系统中亚硝化,硝化和厌氧氨氧化效果,对反应器中各阶段的氮素变化量进行分析,从而得到各个反应体系中氨氧化菌(ammonia-oxidizing bacteria, AOB)、亚硝酸盐氧化菌(nitrite-oxidizing bacteria, NOB)和AnAOB的变化趋势,结果如图4所示。从图4(a)可以看出,在阶段Ⅰ和阶段Ⅱ,R1中的AOB和NOB均维持在稳定状态,AnAOB活性逐渐升高,阶段Ⅱ结束时提高了约80%。但在阶段Ⅲ开始时AOB作用减弱,NOB和AnAOB作用增强,NRR最高可达139.2 g·(m3·d)−1。R2以及R3内基质消耗速率分别见图4(b)和图4(c)。加入N2H4后2个反应器内AOR、AnAOR和AnANR短暂提升,与NRR变化规律相一致,说明N2H4的投加会在短期内迅速提高厌氧氨氧化生物膜系统的脱氮效能。有研究表明,外源N2H4可以直接被AnAOB利用,促进厌氧氨氧化进程[36]。对本研究中的厌氧氨氧化生物膜来说,N2H4添加量为5 mg·L−1时的脱氮效果优于10 mg·L−1。对比对照组R1,R2和R3系统内的AnAOB和AOB的活性均只在加入N2H4的前4 d得到提升,但很快就下降到较低水平,与NRR的变化规律一致。这可能是由于N2H4的长期抑制作用导致反应器内生物量随着生物膜不断脱落而流失 (SS分别为6 194 mg·L−1和4 322 mg·L−1),因此,反应器内功能菌的数量进一步降低。阶段Ⅱ运行后期(第31~46天)R2和R3出水N2H4质量浓度分别稳定在1~3 mg·L−1和 3~5 mg·L−1,推测在此阶段N2H4的投加量超过AnAOB的利用量,AnAOB不能有效降解N2H4,继续添加会导致N2H4逐渐积累从而抑制AnAOB的活性。由于N2H4对AOB和NOB均有毒性作用[16],因此,整个体系中AOB和NOB作用均较弱,而NOB比AOB更敏感,会导致NOB的占比更低。在阶段Ⅲ中,停止加入N2H4后,R2和R3中AOB和AnAOB的占比逐渐上升,体系中的脱氮效果也在缓慢提升,可见N2H4的抑制效果是可逆的,但N2H4为5 mg·L−1的R2能逐渐恢复到抑制前的脱氮水平,而投加10 mg·L−1 N2H4的R3较难恢复。
-
EPS对生物膜的形成和稳定有重要作用[37],因此,有必要探讨N2H4对生物膜EPS的影响。3个反应器中EPS变化量及蛋白质和多糖的比值(PN/PS)变化情况如图5所示。对照组R1的EPS处于稳定增长状态,各阶段EPS含量分别为6.76、10.03和11.22 mg·g−1,PN/PS稳定在2.19~2.59,与反应器中NRR的变化趋势一致。阶段Ⅱ结束时(第46天),R2中EPS由阶段I的6.60 mg·g−1增加到17.68 mg·g−1,同时PN和PS的含量也发生变化,PN/PS由3.49增加到8.23。由于外部环境的改变会刺激细菌分泌较多的EPS,这种自我保护行为会适当减轻不良环境造成的影响[38],N2H4的毒性作用导致R2内的细菌分泌大量EPS。EPS中的PN/PS通常用于定义生物膜的状态[34],活性污泥的PN/PS维持在1~4是一个适宜的水平[39]。由于PN会比PS优先响应外部环境变化[40],且较高质量浓度N2H4可通过分泌大量结合蛋白(bound protein, B-PN)触发厌氧氨氧化污泥的自我保护机制[36],导致R2中的PN增长了2倍,生物膜变得蓬松不稳定,促进了生物膜的脱落和流失,引起反应器运行效能的下降。这与阶段Ⅱ观察到的反应器出水浊度增大以及NRR的下降一致。与R2相反,R3内的EPS由9.23 mg·g−1下降到4.38 mg·g−1,PN/PS的值由2.33增加到6.88。这可能是由于R3中N2H4质量浓度过高,对生物膜产生更强的毒性致使微生物死亡,导致EPS含量下降。停止加入N2H4后,R2的EPS含量下降了42%,PN/PS为2.20,与对照组R1近乎相平。R3的EPS含量逐渐升高至8.84 mg·g−1,PN/PS为2.25。PN/PS的值越低则系统稳定性越高[41],2个反应器内PN/PS均降到适宜范围(1~4)内,可证明污泥结构已经趋于稳定,受N2H4刺激后生物膜缓慢恢复。
-
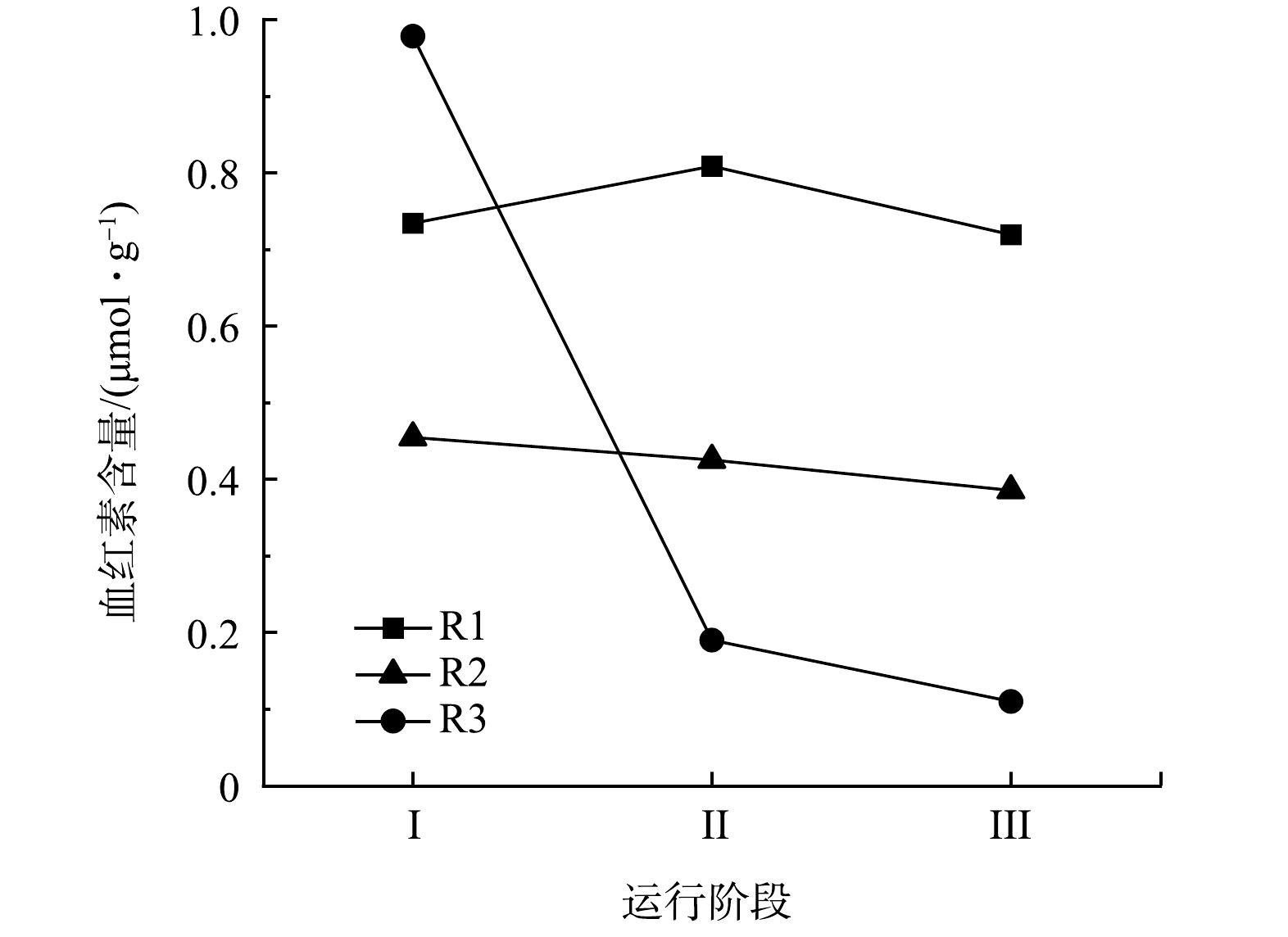
血红素参与AnAOB的主要代谢反应,具有催化和电子转移潜力,可以作为评估厌氧氨氧化性能的指标[29]。不同质量浓度N2H4对AnAOB中血红素的影响如图6所示。对照组R1各阶段的血红素含量分别为0.73、0.81、0.72 μmol·g−1,整体维持在平稳状态。R2和R3中血红素含量均呈现下降的趋势。R2从0.45 μmol·g−1 降至0.39 μmol·g−1,R3则从0.78 μmol·g−1降至0.11 μmol·g−1。在厌氧氨氧化的代谢过程中,N2H4作为厌氧氨氧化过程的中间产物,会被脱氢酶(hydrazine dehydrogenase, HDH)氧化成N2,从而完成脱氮过程[10]。细胞色素c的含量与HDH活性存在正相关,HDH酶的活性越高,处于还原状态的细胞色素c的就越多,而血红素是细胞色素c的关键组分[42]。在本研究中,加入N2H4后的生物膜中的血红素含量一直下降,即HDH的活性一直下降,阻碍了厌氧氨氧化过程中催化N2H4氧化生成N2这一反应的正向进行,从而导致N2H4积累。由于N2H4具有强毒性,HDH受到高质量浓度N2H4的抑制,血红素的还原能力下降,AnAOB的活性受抑制,最终导致系统中NRR下降。此外,通过对比R2和R3在整个反应阶段血红素的变化量,可看出N2H4的加入量越高,抑制作用越明显,厌氧氨氧化活性越难以恢复。
-
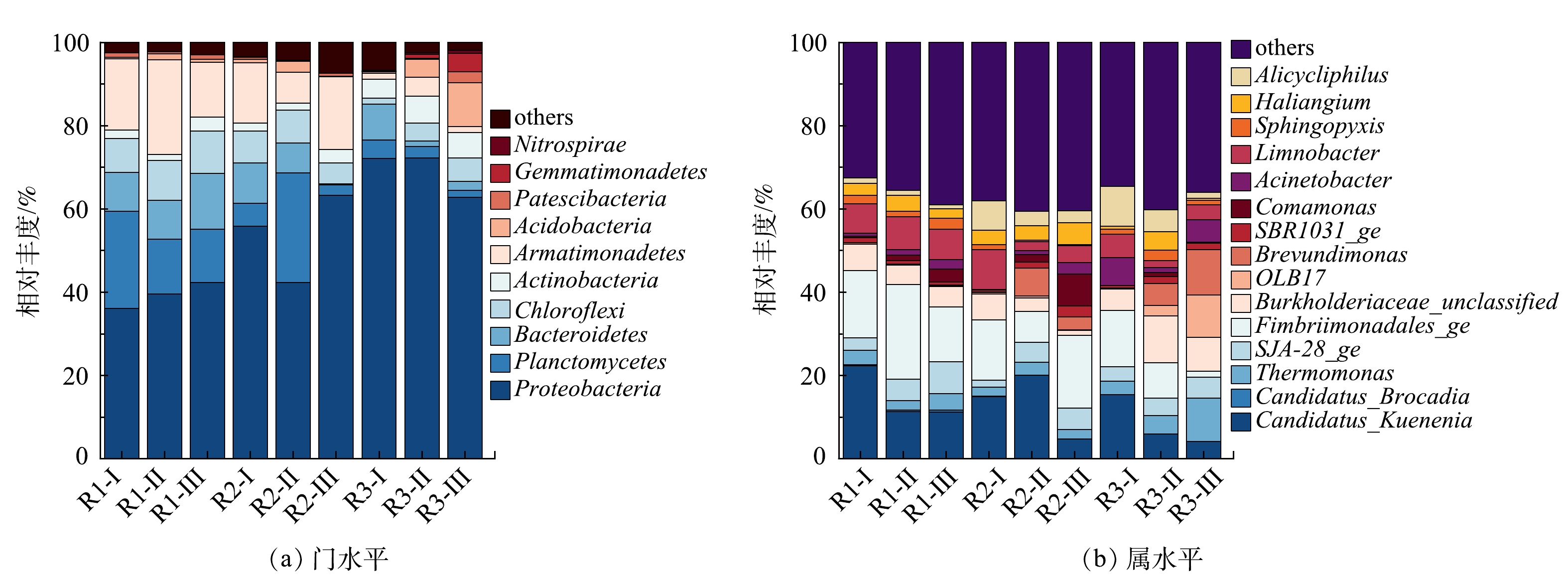
反应器在不同运行周期门水平和属水平的微生物群落结构如图7所示。可以看出,细菌的相对丰度随运行条件的改变有明显差异,表明N2H4对微生物群落的影响逐渐显现。由图7(a)中可观察到所有样本中的主要优势菌门有变形菌门(Proteobacteria)、浮霉菌门(Planctomycetes)、拟杆菌门(Bacteroidetes)、绿弯菌门(Chloroflexi)和装甲菌门(Armatimonadetes),这些都是脱氮系统中常见的典型细菌[43]。有研究[44]表明AnAOB隶属于浮霉菌门,对照组R1在整个运行周期内Planctomycetes一直维持在较高水平(12.86%~23.36%),实现了MBBR脱氮系统的长期稳定运行。加入N2H4后,R2中Planctomycetes呈现先上升至26.33%,而后下降至2.50%的现象,丰度的上升与NRR的短暂增加的结果相一致。R3内几乎不存在Planctomycetes,其相对丰度从4.44%下降到2.86%,同时可观察到放线菌门(Actinobacteria)和酸杆菌门(Acidobacteria)占明显优势,其相对丰度分别由0.55%和0.31%增长至7.17%和10.46%,说明N2H4会促进其它菌门的生长,Planctomycetes与Actinobacteria等菌门之间对底物的竞争增大,降低了AnAOB的占比,破坏脱氮系统的稳定性。
图7(b)反映了了各反应器在不同运行阶段,相对丰度占比在前15的主要菌属。样品中均检测出2种属水平AnAOB,Candidatus_Kuenenia和Candidatus_Brocadia,其中以Candidatus_Kuenenia为主。对照组R1在整个运行周期内Candidatus_Kuenenia一直维持在较高水平(11.18%~22.37%),即使在阶段Ⅱ略有下降但仍占主导地位。对比阶段Ⅲ和阶段Ⅰ,R2和R3中Candidatus_Kuenenia所占比例分别下降了64%和61%,表明长期添加微量N2H4会使AnAOB相对丰度降低,这与反应器运行过程中系统脱氮效率降低的现象一致。Fimbriimonadales在对照组R1中的相对丰度较高,为13.18%~22.68%。有研究[45]表明,Fimbriimonadales是一种异养菌,也可以利用NH4+-N和NO2−-N生成N2。本研究中Fimbriimonadales可能与AnAOB共存于反应器中协同脱氮。对照组R1中Burkholderiaceae一直维持在4.89%~6.37%,R3中Burkholderiaceae的相对丰度从阶段Ⅰ的5.16%上升到阶段Ⅱ的11.17%。Burkholderiaceae属于反硝化细菌,具有还原NO3−-N或NO2−-N的能力[31]。可见,N2H4的长期加入促进了Burkholderiaceae的生长,加大了对底物NO2−-N的竞争,抑制了AnAOB生长。有研究[46]表明,Limnobacter可与AnAOB共生,这可以保护AnAOB免受不利环境的影响。对照组中Limnobacter的丰度稳定在7%左右,但该菌属在R2中的丰度由9.55%降至2.12%,R3中由5.64%降至1.62%。N2H4会破环这种保护平衡,令AnAOB暴露在不利环境中,从而降低其活性。终上所述,N2H4不仅直接降低Candidatus_Kuenenia的丰度,还对与AnAOB菌属协同脱氮的其它菌属起到抑制作用,从而降低反应器整体功能菌属的活性,进而影响脱氮效果。
-
1)在进水NH4+-N质量浓度在(50.9±3.6) mg·L−1,NO2−-N质量浓度在(55.5±3.2) mg·L−1的条件下,加入5 mg·L−1和10 mg·L−1的N2H4可以使NRR短暂增长,但长期运行后NRR分别下降了53%和 64%。N2H4的长期加入会对生物膜产生生物毒性,抑制脱氮过程。
2) 5 mg·L−1的N2H4使生物膜的EPS分泌量提高,触发生物膜保护机制,解除N2H4抑制后,EPS浓度恢复,但生物膜变得松散,易脱落,导致污泥流失。10 mg·L−1的N2H4抑制了生物膜中AnAOB的活性,EPS和血红素含量均明显下降,解除N2H4抑制后,生物膜脱氮活性也难以恢复。
3)长期添加微量N2H4会降低厌氧氨氧化生物膜脱氮系统中Planctomycetes和Candidatus_Kuenenia的丰度。
微量肼对厌氧氨氧化生物膜长期运行效果的影响
Effects of trace hydrazine on long-term operation of anammox biofilm
-
摘要: 为探究微量肼(N2H4)对厌氧氨氧化生物膜的长期影响,采用3个移动床生物膜反应器(moving bed biofilm reactor, MBBR)处理低浓度氨氮(50.9±3.6) mg·L−1废水,分别加入0 mg·L−1 (对照组,R1)、5 mg·L−1 (R2)和10 mg·L−1(R3)的微量N2H4后连续运行35 d,考察N2H4对MBBR系统中总氮去除速率(total nitrogen removal rate, TNRR)、生物量、胞外聚合物(extracellular polymeric substances, EPS)、血红素和微生物群落的影响。结果显示,运行末期相对于R1,R2和R3的NRR分别下降了53%和 64%。N2H4 质量浓度为5 mg·L−1时,生物膜的EPS分泌量提高,触发了生物膜保护机制;当N2H4 质量浓度为10 mg·L−1时,生物膜的EPS和血红素含量均明显下降,N2H4对生物膜产生抑制作用。长期添加微量N2H4导致门水平中Planctomycetes和Candidatus_Kuenenia的相对丰度降低,可见,N2H4的加入可以使NRR短暂增长,但长期加入会对厌氧氨氧化生物膜产生生物毒性,抑制厌氧氨氧化菌(AnAOB)的活性。整体而言,5 mg·L−1和10 mg·L−1的N2H4的加入都难以维持MBBR长期稳定高效运行,其对厌氧氨氧化生物膜的负面影响更为明显。Abstract: The long-term effect of trace N2H4 on the anammox biofilm was investigated in moving bed biofilm reactors (MBBR) treating wastewater with low ammonia nitrogen concentration of (50.9±3.6) mg·L−1. After adding 0 mg·L−1 (control group, R1), 5 mg·L−1 (R2) and 10 mg·L−1 (R3) of N2H4 for 35 days, the effect of N2H4 on the total nitrogen removal rate (TNRR), biomass, extracellular polymeric substances (EPS), and heme in the MBBR systems were evaluated. The results showed that compared with R1, the NRR decreased by 53% and 64% at the end of the operation period in R2 and R3, respectively. 5 mg·L−1 N2H4 could trigger the protective mechanism of the biofilm and increased EPS secretion from it. 10 mg·L−1 N2H4 resulted in the obvious decrease of EPS and heme content, and N2H4 inhibited the activity of anammox in biofilms. Meanwhile, long-term addition of trace N2H4 reduced the abundance of Planctomycetes at phylum level and reduced that of Candidatus_Kuenenia at genus level. It can be seen that the addition of N2H4 initially increased the NRR, but the long-term addition produced biological toxicity and inhibit the activity of AnAOB. Overall, the addition of 5 mg·L−1 and 10 mg·L−1 N2H4 could not lead to a stably and efficiently long-term operation of MBBR, but result in a more significant negative effect on the anammox biofilm.
-
Key words:
- exogenous N2H4 /
- Anammox /
- MBBR /
- EPS /
- heme
-
近年来,由于高级氧化技术可以产生大量活性氧组分(ROS,如•OH、
SO⋅−4 、1O2等),从而有效促进痕量微污染物的降解,故而该技术成为降解高风险微量有机物的重要手段,而且逐渐成为研究热点[1-2]。虽然单一的臭氧氧化法能有效去除不饱和芳香族和脂肪族化合物,但其对饱和有机化合物的去除率很低[3]。为了高效去除难降解的饱和的持久性有机污染物,本文通过选用一种高性能的催化剂催化臭氧产生更多的活性物质[4-6],以达到彻底去除污染物的目的。锰酸铜(CuMn2O4)尖晶石是一种密度较大的空心六面体,其晶体结构主要是由Mn4+与Cu2+搭建的,还有较少的Mn3+与Cu+增加了结构的缺陷程度。CuMn2O4可以催化臭氧产生具有氧化能力强、无选择性的·OH,从而有效提高臭氧对水体中难降解污染物的去除效果。有研究[7]指出,臭氧与CuMn2O4的结合对二苯甲酮-3的降解有明显的协同作用。然而,在实际使用过程中,CuMn2O4的密度大、容易团聚、不易分散的特性使其利用率很低。为了提高其利用率,需要选用另外一种催化剂进行耦合,弥补其在使用过程中的缺陷。研究表明,二维层状碳材料在催化臭氧氧化领域中有很好的效果[8]。因为二维层状碳材料不仅在平面内的热运输和电荷运输过程中具有突出的物理化学特性,而且与其他材料复合后可以产生良好的耦合效应[9-10]。石墨烯/还原氧化石墨烯(rGO)是其中一种极具吸引力的二维材料,具有卓越的化学稳定性、导电性和表面体积比[11-12]。此外,石墨相氮化碳(g-C3N4)是一种具有2.7 eV带隙的二维非金属聚合物半导体[13],在化学、热和光照射过程中具有较好的稳定性。同时,g-C3N4还是一种有效的催化剂载体[14-15],在其中掺杂选定的杂原子,通过电荷转移可以形成络合的复合材料[16-17]。因此,可以考虑将rGO和g-C3N4与CuMn2O4进行复合,应用于催化臭氧氧化过程中。与大多数亲脂性的有机防晒剂不同的是,二苯甲酮-4(BP-4)是一种亲水性的紫外线吸收剂,因其质地更轻、油性更少,被广泛应用于洗发水、剃须凝胶、止汗剂、化妆品和牙膏等日用品中[18]。但是,由于BP-4化学稳定性好、不易降解,因而被认为是一种伪持久性有机污染物,越来越受到人们的关注[19]。在目前的废水处理领域中常见的水处理方法并不能将其完全去除[20]。此外,由于rGO和g-C3N4在催化臭氧氧化过程中对臭氧氧化副产物溴酸盐有很好的抑制效果[8],因此本研究将rGO和g-C3N4与CuMn2O4复合,来探究他们在催化臭氧氧化过程中对BP-4的降解效果以及溴酸盐生成的影响。
1. 材料与方法
1.1 实验原料及仪器
一水合硫酸锰(MnSO4·H2O,≥99.0%)、三水合硝酸铜(Cu(NO3)2·3H2O,≥99.0%)、碳酸钠(Na2CO3,≥99.8%)、尿素(CO(NH2)2,99%)、硝酸钠(NaNO3,≥99.0%)、高锰酸钾(KMnO4,≥99.0%)、二苯甲酮-4(C14H12O6S,98%)、硫酸(H2SO4,≥70%)、石墨粉、过氧化氢(H2O2,≥27.5%)和盐酸(HCl,38%)。
X射线衍射仪(XRD-7000,日本岛津公司);比表面积测试仪(SSA-7000,北京博德电子科技公司);X射线光电子能谱仪(AXIS Ultra,日本岛津公司);高效液相色谱仪(Waters 2695,沃特世科技(上海)有限公司);智能箱式高温炉(DC-B06/02,北京独创科技有限公司);恒温水浴振荡器(SHA-B,常州市金坛友联仪器研究所);鼓风干燥箱(DGA-9073 B-2,上海福玛实验设备有限公司);电子天平(AL 104,梅特勒-托利多仪器有限公司);pH计(S210 Seven Compact,梅特勒-托利多仪器有限公司);离子色谱仪(ICS-3000,上海普迪生物技术有限公司);电化学工作站(CHI660E,上海辰华仪器有限公司);磁力搅拌器(RH Digita,德国艾卡IKA公司);电子扫描显微镜(S4800,日本Hitachi公司)。
1.2 催化剂的制备
采用共沉淀法制备CuMn2O4。具体操作步骤如下:将3.561 7 g Cu(NO3)2·3H2O溶于120 mL去离子水中,得到溶液1;将4.969 2 g MnSO4·H2O溶于120 mL去离子水中,得到溶液2;将6.359 4 g Na2CO3溶于60 mL去离子水中,得到1 mol·L−1的Na2CO3溶液。磁力搅拌溶液2,使用塑料滴管将溶液1逐滴加入溶液2中(用时20 min)。溶液1加完后,使用pH计测定混合溶液的初始pH,此时的溶液显酸性。将溶液2使用水浴锅加热至80 ℃,磁力搅拌。将Na2CO3溶液逐滴加入溶液2中,每滴加一滴,停止滴加,搅拌均匀后,继续滴加,使用pH计测定溶液的pH,在pH=10时停止滴加(用时20 min),在水浴条件下继续搅拌20 min后停止搅拌,停止水浴加热,静置1 h。使用砂芯漏斗对沉淀进行真空抽滤,并使用去离子水洗涤,取滤液测定其pH,直到pH基本不变,再用无水乙醇洗涤5次。将沉淀转移到表面皿,将有残留样品的滤纸和表面皿在烘箱干燥,温度为120 ℃,时间为15 h。将干燥后的样品收集,置于马弗炉中900 ℃煅烧,升温速率为1 ℃·min−1,保温时间为6 h。
热缩聚的方法制备g-C3N4。称量10 g尿素,研磨成粉末,置于马弗炉中350 ℃煅烧,升温速率为5 ℃·min−1,保温时间为1 h。使用Hummers法[21]制备氧化石墨烯(GO)。
两步煅烧法制备CuMn2O4/rGO和CuMn2O4/g-C3N4。将共沉淀法制备的CuMn2O4和GO按照质量比例1∶1进行混合,研磨均匀,将粉末转移到坩埚中,置于马弗炉中煅烧,温度为350 ℃,升温速率为5 ℃·min−1,保温时间为1 h。在反应结束后,冷却至室温,得到复合的CuMn2O4/rGO粉末。将10 g尿素与0.2 g的CuMn2O4研磨均匀后,每次取定量研磨好的粉末置于马弗炉中,加热到350 ℃,加热时间为120 min,加热速率为10 ℃·min−1。在反应结束后,冷却至室温,得到复合的CuMn2O4/g-C3N4粉末。
1.3 实验装置
催化臭氧氧化BP-4的效能实验采用间歇反应模式进行。反应器为圆柱形的玻璃容器,直径为6.2 cm,高为26.5 cm,有效容积为300 mL。实验使用北京同林高科技有限责任公司生产的3S-A5型臭氧发生器(臭氧产量为0~1 g·h−1),以高纯氧气为气源,本实验所用的实验装置如图1所示。实验中每次处理的水样为300 mL,所有的溶液均用去离子水配制。臭氧进气浓度通过臭氧发生器的放电功率来调节。在每次实验开始之前,用纯氧进行吹扫,并预臭氧化处理。打开臭氧发生器,调节气流量为400 mL·min−1,臭氧发生器电流为0.025 A,预热时间为60 min,预臭氧时间为30 min。
1.4 实验方法
在反应器中加入290 mL超纯水,O3曝气30 min,搅拌器的转速为800 r·min−1。加入10 mL BP-4母液(反应器中的浓度是0.084 mmol·L−1(25.91 mg·L−1));100 μL Br−母液(母液浓度为300 mg·L−1,反应器中的浓度是100 μg·L−1),开始反应并计时。在反应时间为0、1、2、5、7、10、15、30 min时分别取样,并使用浓度为10 mmol·L−1的亚硫酸钠溶液还原残留臭氧;使用0.22 μm的水系滤膜过滤粉体催化剂后待分析。
1.5 分析方法
使用X-射线衍射仪(XRD)对制得的粉体催化剂的矿物组成与结晶结构进行分析;使用比表面积分析仪对制得的粉末催化剂的比表面积及表面孔结构进行表征;使用X射线光电子能谱仪(XPS)表征粉末状催化剂中各元素的价态;使用电化学工作站分析粉末催化剂的阻抗。使用扫描电镜(SEM)对制得的粉末催化剂进行表观形貌的分析。
BP-4的浓度由高效液相色谱仪Waters 2695测定。使用的色谱柱为Symmetry C18,柱温为30 ℃。流动相为0.3%甲酸缓冲液和甲醇的混合溶液,其中0.3%甲酸缓冲液的体积分数为35%,流速为1 mL·min−1,检测波长为286 nm,进样量为10 μL,保留时间为8.5 min。
Br−和
BrO−3 的浓度由离子色谱仪ICS-3 000测定。样品先经过AG19保护柱,然后进入分析柱AS19进行分析。取样量为500 μL,流速为1 mL·min−1,柱温为35 ℃。淋洗条件为梯度淋洗(0~18.0 min:10.0 mmol·L−1 NaOH;18.1~26.0 min:35.0 mmol·L−1 NaOH;26.1~40.0 min:10.0 mmol·L−1 NaOH)。催化臭氧氧化过程中溴酸盐的生成速率可根据式(1)进行计算。v=(CBrO3−)max (1) 式中:v为生成速率,min−1;(
C_{{\rm{BrO}}_3^ -} )max为整个催化臭氧氧化过程中的{\rm{BrO}}_3^ - 的最大浓度,mol·L−1;(CBr-)0为溴离子的初始浓度,mol·L−1,本研究中该值为1.25 μmol·L−1;t为{\rm{BrO}}_3^ - 达到最大浓度时的反应时间,min。2. 结果与讨论
2.1 CuMn2O4、CuMn2O4/rGO与CuMn2O4/g-C3N4的结构与表面特性分析
1)矿物组成与晶体结构。如图2所示,CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4具有相似的XRD谱图。分别在2θ=18.4°、30.2°、35.6°、43.5°、54.1°、57.5°、63.3°和77.5°处出现的衍射峰与CuMn2O4的标准卡片(JCPDS#84-0543)中特征衍射峰的位置是一致的。这些衍射峰分别对应着CuMn2O4的(111)、(220)、(311)、(222)、(400)、(422)、(511)、(440)和(533)晶格面。此外,CuMn2O4/rGO的谱图中在2θ=25.4°处对应的是rGO的特征衍射峰;CuMn2O4/g-C3N4的谱图中在2θ=27.3°处对应的是g-C3N4的特征衍射峰。可见,通过两步煅烧的方法已经将CuMn2O4与g-C3N4和rGO成功复合在一起。值得注意的是,与CuMn2O4/rGO相比,CuMn2O4/g-C3N4中CuMn2O4的衍射峰强变弱。ZHU等[22]将NiFe2O4与g-C3N4复合后,通过XRD分析,同样发现NiFe2O4的峰强减弱。可见,rGO的掺入不会影响CuMn2O4的物相结构和结晶度;g-C3N4的掺入虽然不会影响CuMn2O4的物相结构,但会降低CuMn2O4的结晶度。
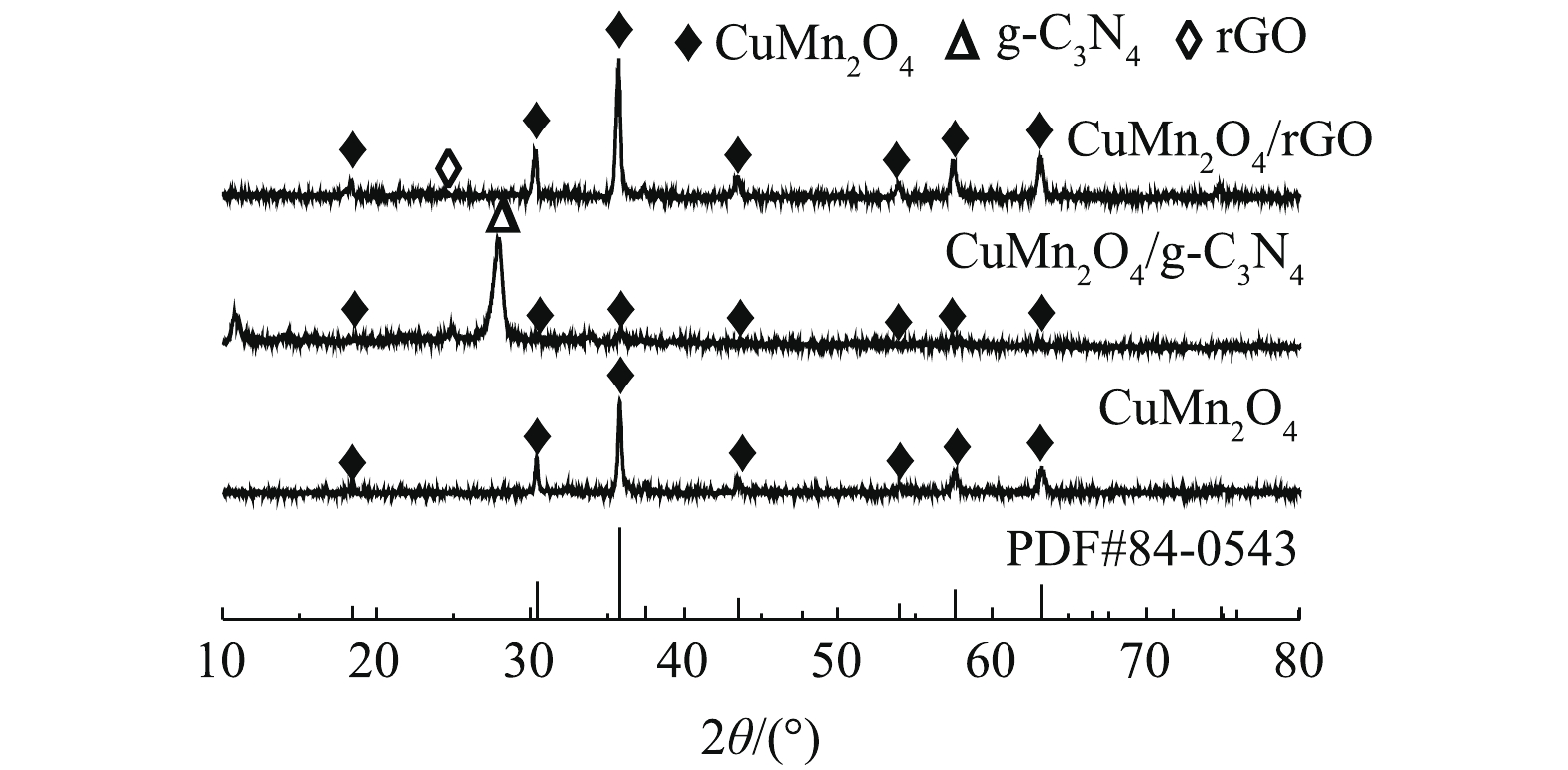 图 2 CuMn2O4,CuMn2O4/rGO与CuMn2O4/g-C3N4的矿物组成与结晶情况Figure 2. Mineral composition and crystal state of CuMn2O4, CuMn2O4/rGO and CuMn2O4/g-C3N4
图 2 CuMn2O4,CuMn2O4/rGO与CuMn2O4/g-C3N4的矿物组成与结晶情况Figure 2. Mineral composition and crystal state of CuMn2O4, CuMn2O4/rGO and CuMn2O4/g-C3N42)催化剂形貌分析。通过SEM对CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4的形貌进行分析,结果如图3所示。如图3(a)所示,CuMn2O4是表面光滑的不规则立方体,平均粒径为2~3 μm。将其与rGO复合后,SEM表征结果如图3(b)所示,与CuMn2O4相比,催化剂颗粒的体积变小,这可能是因为rGO的存在阻碍了CuMn2O4的团聚。还可以发现,层状rGO基本全部包覆在CuMn2O4的外表面。将CuMn2O4与g-C3N4复合后,形貌如图3(c)所示。与CuMn2O4相比,催化剂颗粒的体积减小,这可能也是因为g-C3N4阻碍了CuMn2O4的团聚。此外,还可以发现,与CuMn2O4/rGO不同的是,在CuMn2O4/g-C3N4样品中,碎片状的g-C3N4一部分覆盖在CuMn2O4的外表面,另一部分独立分散在CuMn2O4颗粒之间。据此可以初步推测,与g-C3N4相比,rGO可能更容易与CuMn2O4结合,CuMn2O4/rGO的结构更稳定。
 图 3 CuMn2O4,CuMn2O4/rGO和CuMn2O4/g-C3N4的SEM表征结果Figure 3. SEM images of CuMn2O4, CuMn2O4/rGO and CuMn2O4/g-C3N4
图 3 CuMn2O4,CuMn2O4/rGO和CuMn2O4/g-C3N4的SEM表征结果Figure 3. SEM images of CuMn2O4, CuMn2O4/rGO and CuMn2O4/g-C3N43)比表面积分析。使用比表面积分析仪分析催化剂CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4的孔隙结构特征,结果如图4所示。与CuMn2O4相比,CuMn2O4/rGO和CuMn2O4/g-C3N4的比表面积、总孔容积和平均孔半径均有所增加。与rGO和g-C3N4分别复合后,CuMn2O4的比表面积从2.058 m2·g−1分别增加到38.438 m2·g−1和12.553 m2·g−1;总孔容积从0.011 cm3·g−1分别提升到0.264 cm3·g−1和0.09 cm3·g−1;平均孔半径从10.47 nm分别增至13.74 nm和14.35 nm。DEVI等[23]将rGO与CoFe2O4复合到一起后,也得到了相似的结果:在掺入rGO后,催化剂的比表面积从6.202 m2·g−1增加到了25.18 m2·g−1,总孔容积从0.003 1 cm3·g−1增至0.022 cm3·g−1。他们认为,比表面积和总孔容积的增加是由于CoFe2O4在rGO片层中随意堆积导致的,这也极有可能是CuMn2O4/rGO和CuMn2O4/g-C3N4的比表面积和总孔容积的增加的原因。此外,CuMn2O4/rGO的比表面积是CuMn2O4/g-C3N4的3.06倍,总孔容积为2.93倍,平均孔径为0.98倍。由此可见,rGO的掺入对增大催化剂比表面积和总孔容积的效果更好。
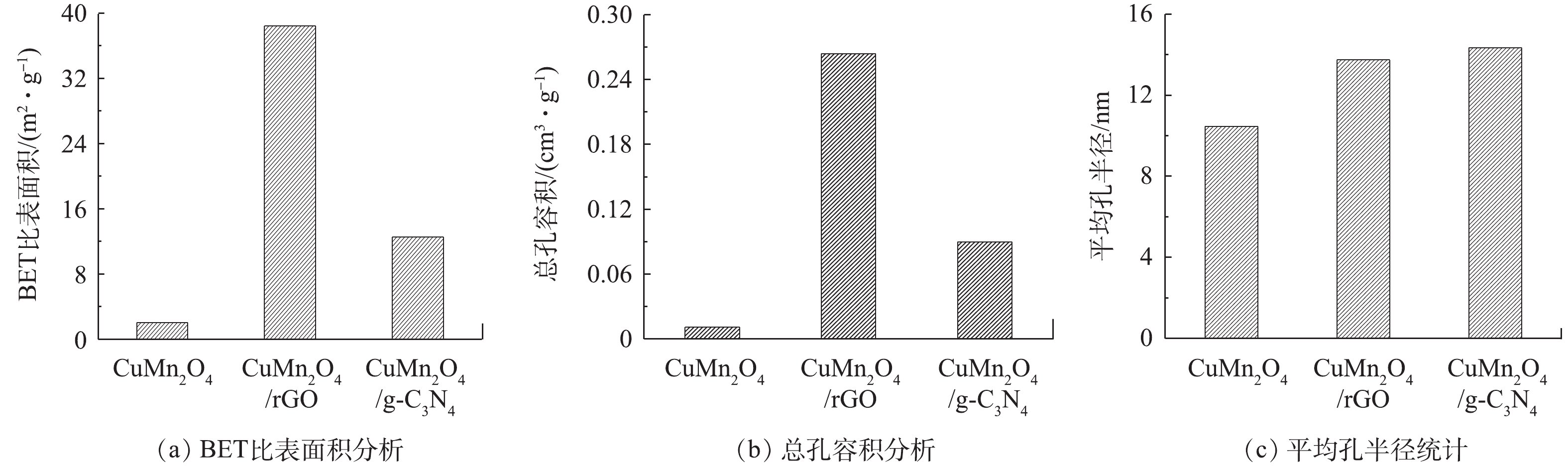 图 4 CuMn2O4,CuMn2O4/rGO与CuMn2O4/g-C3N4的BET表征结果Figure 4. BET characterization results of CuMn2O4, CuMn2O4/rGO and CuMn2O4/g-C3N4
图 4 CuMn2O4,CuMn2O4/rGO与CuMn2O4/g-C3N4的BET表征结果Figure 4. BET characterization results of CuMn2O4, CuMn2O4/rGO and CuMn2O4/g-C3N44)表面元素价态分析。为了进一步确定CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4中Cu、Mn与O元素的价态,对3种催化剂分别进行XPS表征,并对测试结果进行解卷积分析,结果如表1所示。Cu、Mn这两种元素在复合前后的价态分布有明显的变化。CuMn2O4中Cu2+的含量为12.89%,当其与层状碳材料复合后,Cu2+的含量分别增加到了70.92%(g-C3N4)和53.96%(rGO);CuMn2O4中Mn4+的含量为28.41%,当其与层状碳材料复合后,Mn4+的含量分别增至34.30%(rGO)和34.30%(g-C3N4)。这说明层状碳材料rGO和g-C3N4都可以氧化CuMn2O4中的Cu+与Mn3+,使他们失去电子导致其化合价升高,失去的电子通过层状碳材料表面的官能团(如羟基)进行转移。与rGO相比,当g-C3N4掺入CuMn2O4后催化剂中Cu2+的含量更高,可见g-C3N4对CuMn2O4中的Cu+有更强的氧化能力,这也意味着CuMn2O4/g-C3N4的表面会有更多的转移电子。
表 1 CuMn2O4,CuMn2O4/rGO与CuMn2O4/g-C3N4的XPS表征结果Table 1. XPS characteristics of CuMn2O4, CuMn2O4/rGO and CuMn2O4/g-C3N4% 催化剂 Cu元素 Mn元素 O元素 Cu+/Cu Cu2+/Cu Mn2+/Mn Mn3+/Mn Mn4+/Mn OV/O OOH/O Olatt/O CuMn2O4 87.11 12.89 42.6 50 7.4 13.6 61.4 25 CuMn2O4/g-C3N4 29.08 70.92 28.8 36.9 34.3 50.8 47.4 1.8 CuMn2O4/rGO 46.24 53.76 23.1 42.6 34.3 47.7 48.2 4.1 | Show Table DownLoad:
CSV
DownLoad:
CSV
此外,在3种催化剂中均可以观察到晶格氧(Olatt)、表面羟基氧(OOH)和表面氧空位(OV)[24]。粉末催化剂中的氧空位是催化臭氧氧化重要的潜在催化位点[25],CuMn2O4中OV的含量为13.6%,CuMn2O4/rGO与CuMn2O4/g-C3N4中OV的含量分别增加至50.8%和47.7%。这一结果归因于层状碳材料rGO和g-C3N4与CuMn2O4晶格结合后,导致其晶格膨胀或畸变,产生了更多的缺陷位,从而形成更多氧空位[25]。与CuMn2O4/rGO相比,CuMn2O4/g-C3N4中OV的含量更多。
5)阻抗分析。CuMn2O4,CuMn2O4/rGO和CuMn2O4/g-C3N4的Nyquist曲线如图5所示。与CuMn2O4相比,CuMn2O4/rGO和CuMn2O4/g-C3N4的半圆弧形半径增大。这说明,复合催化剂CuMn2O4/rGO和CuMn2O4/g-C3N4的电荷转移电阻变大。通过进一步使用模拟电路进行拟合计算,CuMn2O4、CuMn2O4/rGO和CuMn2O4/g-C3N4的阻抗值分别为105.9、125.7和155.6 Ω。这与LI等[26]的实验结果不一致,他们将石墨烯纳米片与CuMn2O4复合后,CuMn2O4/rGO的阻抗小于CuMn2O4。CHEN等[27]将LiMn2O4与rGO通过水热法和直接将2种物质混合的方法分别进行复合后,也得到了相似的结果:2种方法得到的LiMn2O4@rGO的阻抗分别为56.79 Ω和178.10 Ω,而复合前LiMn2O4的阻抗为287.0 Ω。CHEN等[27]认为,与LiMn2O4相比,复合催化剂阻抗的减小主要归因于rGO自身优越的导电性;与直接将2种材料混合的方法相比,水热法制备的复合催化剂阻抗更小则是因为借由水热反应,rGO与LiMn2O4之间的结合方式更有利于电荷转移。据此可以推测,本研究中复合材料CuMn2O4/rGO和CuMn2O4/g-C3N4的阻抗比CuMn2O4大的原因极有可能是,因为通过两步煅烧的方法制备的复合催化剂中CuMn2O4与rGO和g-C3N4之间的结合方式不利于电荷转移。此外,与CuMn2O4/rGO相比,CuMn2O4/g-C3N4的阻抗更大。可见,rGO掺入CuMn2O4后催化剂的导电能力增强。
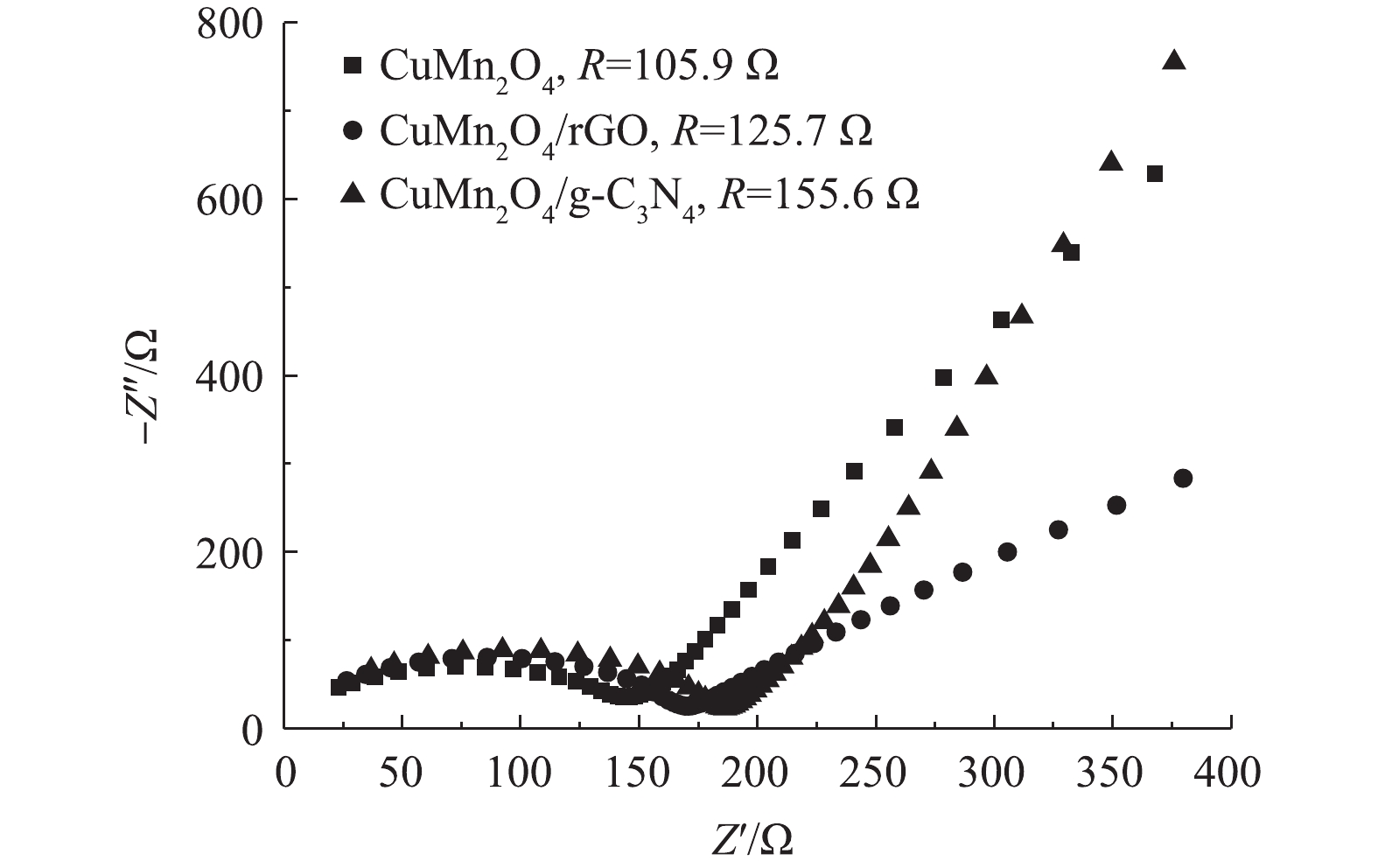 图 5 CuMn2O4,CuMn2O4/rGO与CuMn2O4/g-C3N4的EIS谱线Figure 5. EIS characteristics CuMn2O4,CuMn2O4/rGO and CuMn2O4/g-C3N4
图 5 CuMn2O4,CuMn2O4/rGO与CuMn2O4/g-C3N4的EIS谱线Figure 5. EIS characteristics CuMn2O4,CuMn2O4/rGO and CuMn2O4/g-C3N42.2 rGO和g-C3N4改性的CuMn2O4催化臭氧氧化性能研究
1)rGO 和g-C3N4改性的CuMn2O4催化臭氧降解BP-4的速率对比。单独臭氧氧化、CuMn2O4、rGO、g-C3N4、CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧氧化对BP-4的降解效果如图6所示。如图6(a)所示,单独臭氧氧化和CuMn2O4催化臭氧氧化降解BP-4时,反应30 min后BP-4才能被完全降解;g-C3N4、rGO、CuMn2O4/g-C3N4和CuMn2O4/rGO催化臭氧氧化则可以在10 min后将BP-4完全降解。对BP-4的降解进行拟一级动力学分析,结果如图6(b)所示,单独臭氧氧化、CuMn2O4、g-C3N4、rGO、CuMn2O4/g-C3N4和CuMn2O4/rGO催化臭氧氧化降解BP-4的动力学常数k分别为0.114、0.440、0.441、0.462、0.613和0.796 min−1,可以发现,与g-C3N4和rGO相比,CuMn2O4/g-C3N4和CuMn2O4/rGO催化臭氧氧化降解BP-4的速率得到进一步提升。AKHUNDI等[28]将g-C3N4纳米片(g-C3N4-NS)与CuCr2O4复合后,将其应用于光催化降解RhB、MB染料和苯酚,也得到了相似的结果,他们发现,复合催化剂g-C3N4-NS/CuCr2O4降解RhB的速率分别是块状g-C3N4和g-C3N4-NS单独光催化的11.8倍和4.8倍。此外,进一步对比可以发现,与CuMn2O4/g-C3N4相比,CuMn2O4/rGO催化臭氧氧化降解BP-4的速率更快。结合之前对于2种复合材料结构的分析与比较,可以推测导致以上结果的原因归为以下3点:其一,尽管CuMn2O4/g-C3N4表面有较多的转移电子和氧空位,但是CuMn2O4/rGO的结晶度更高、更稳定;其二,CuMn2O4/rGO的比表面积和总孔容积更大,与O3分子的接触面积更大;其三,CuMn2O4/rGO的导电性更好,更有利于反应过程中的电荷转移,故而催化臭氧氧化降解BP-4的速度更快。
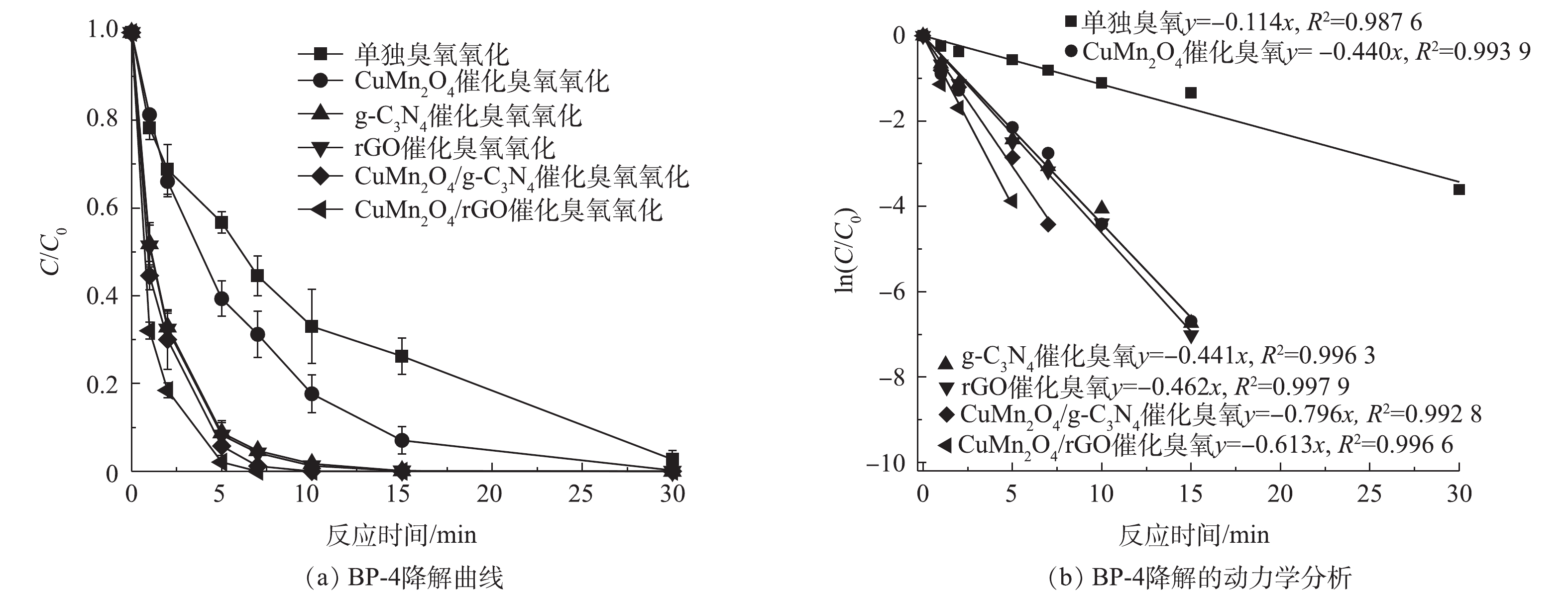 图 6 单独臭氧、CuMn2O4、rGO、g-C3N4、CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧氧化对BP-4的降解效果及对BP-4降解的拟一级动力学分析Figure 6. Degradation efficiency of BP-4 and the pseudo first order reaction kinetics of BP-4 degradation by the sole ozonation and CuMn2O4, rGO, g-C3N4, CuMn2O4/rGO and CuMn2O4/g-C3N4 catalytic ozonation
图 6 单独臭氧、CuMn2O4、rGO、g-C3N4、CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧氧化对BP-4的降解效果及对BP-4降解的拟一级动力学分析Figure 6. Degradation efficiency of BP-4 and the pseudo first order reaction kinetics of BP-4 degradation by the sole ozonation and CuMn2O4, rGO, g-C3N4, CuMn2O4/rGO and CuMn2O4/g-C3N4 catalytic ozonation2) rGO 和g-C3N4改性的CuMn2O4催化臭氧对溴酸盐的抑制效果的比较。单独臭氧氧化、CuMn2O4、rGO、g-C3N4、CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧氧化过程中溴酸盐的生成情况如图7所示。如图7(a)所示,在单独臭氧氧化过程中,溴酸盐的含量随着反应时间延长显著增多;而在催化臭氧氧化过程中,溴酸盐的生成量得到有效控制。值得注意的是,不同催化臭氧氧化体系中溴酸盐含量的变化趋势存在差异:在CuMn2O4和CuMn2O4/g-C3N4这2种催化臭氧氧化体系中,溴酸盐的生成量随着反应的进行缓慢增加,与单独臭氧氧化相比,溴酸盐的生成量分别减少了86.30%和84.23%;而在g-C3N4、rGO和CuMn2O4/rGO这3种催化臭氧氧化体系中,溴酸盐的生成量迅速增加后又逐渐减少,具体变化过程如下:在反应前20 min溴酸盐的生成量迅速增加,在20 min时溴酸盐的含量最高;反应20 min后,溴酸盐的含量开始减少,反应120 min后溴酸盐的含量接近于0,与单独臭氧氧化相比,溴酸盐的生成量减少了100%。溴酸盐含量的第一种变化过程与之前大部分[29-31]的研究结果是一致的,在他们的实验过程中,溴酸盐的含量也是在不断增加并最终保持不变。这些研究中对于溴酸盐的消除机理有两种不同的解释,其中一种观点认为溴酸盐的减少得益于β-FeOOH/Al2O3和Fe-Al-LDH/Al2O3[29-30]表面上的Fe2+对溴酸盐的还原,另一种观点则认为催化剂加速了O3的分解,生成更多的•OH,减少了可用于氧化Br−的O3[31]。溴酸盐生成量的第2种变化过程与ZHANG等[32-33]先后使用LaFeO3和LaCoO3与LaCoO3/g-C3N4催化臭氧氧化降解BZA过程中溴酸盐的变化趋势是一致的,溴酸盐的含量均是先增加后减少,他们认为溴酸盐的消除主要归因于H2O2对溴酸盐的还原。此前,SONG等[8]在rGO和g-C3N4催化臭氧氧化降解BZA及去除溴酸盐的研究中指出,rGO表面的含氧官能团和碳层结构中的π电子可以促进臭氧分解形成H2O2;g-C3N4表面的含氧官能团(如-C=O)也会促进H2O2的产生。结合本研究中的实验结果可以推测,在CuMn2O4和CuMn2O4/g-C3N4这2种催化臭氧氧化体系中,溴酸盐的消除有2个方面的原因:其一,催化剂加速了O3的分解,生成更多的·OH,减少了可用于氧化Br−的O3;其二,催化剂可以催化臭氧产生H2O2来还原溴酸盐。在g-C3N4、rGO和CuMn2O4/rGO这3种催化臭氧氧化体系中,溴酸盐的减少极有可能是因为生成的H2O2促进了溴酸盐的还原。对实验结果进一步分析可以发现,与CuMn2O4/g-C3N4相比,CuMn2O4/rGO催化臭氧氧化过程对于溴酸盐的控制效果更好。
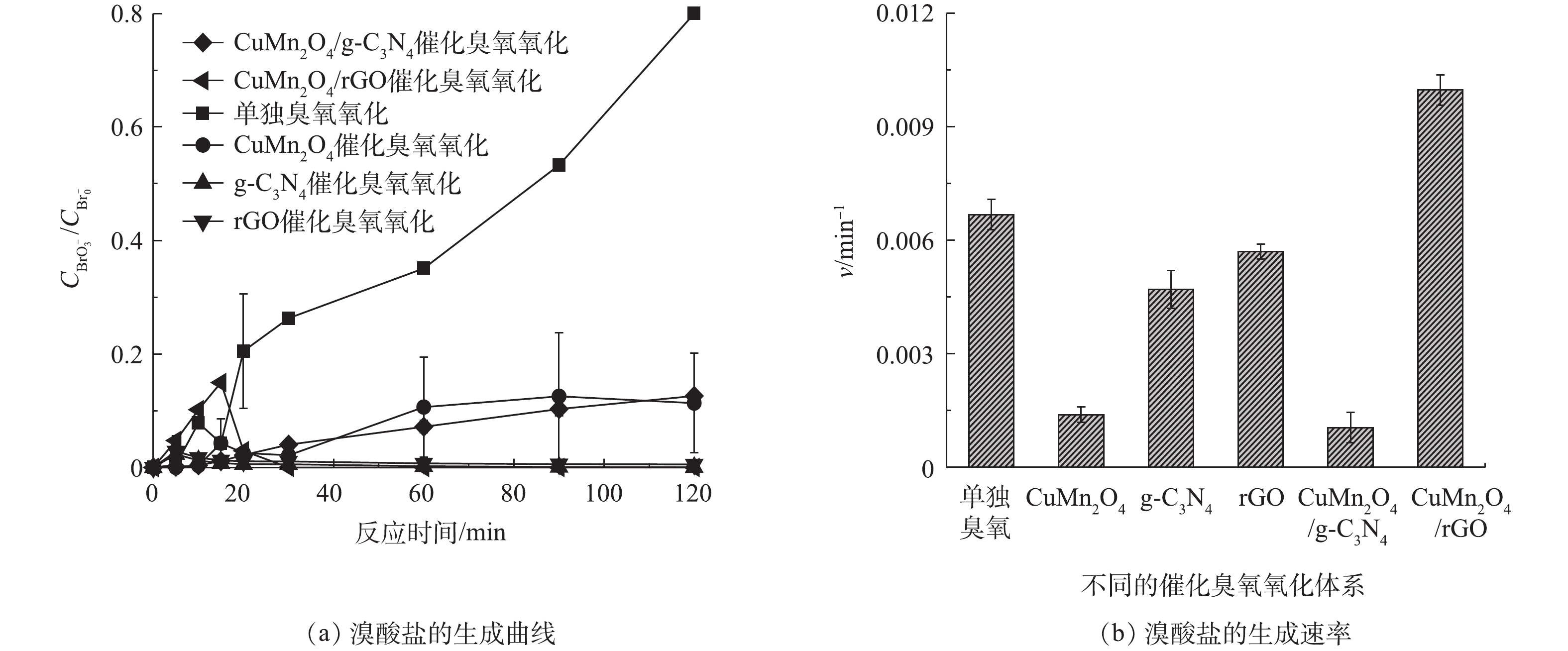 图 7 单独臭氧氧化及CuMn2O4,g-C3N4,rGO,CuMn2O4/rGO与CuMn2O4/g-C3N4催化臭氧氧化对溴酸盐生成量的影响Figure 7.
图 7 单独臭氧氧化及CuMn2O4,g-C3N4,rGO,CuMn2O4/rGO与CuMn2O4/g-C3N4催化臭氧氧化对溴酸盐生成量的影响Figure 7.{\rm{BrO}}_3^ - yield during the sole ozonation and CuMn2O4, g-C3N4, rGO, CuMn2O4/rGO and CuMn2O4/g-C3N4 catalytic ozonation为了进一步分析反应过程中溴酸盐的生成规律,通过式(1)计算了不同反应体系中溴酸盐的生成速率v,结果如图7(b)所示,在不同催化臭氧氧化体系中,溴酸盐的生成速率依次是CuMn2O4/rGO>rGO>g-C3N4>CuMn2O4>CuMn2O4/g-C3N4,可见CuMn2O4/rGO催化臭氧体系中溴酸盐的生成速率要明显快于CuMn2O4/g-C3N4,这与二者对BP-4的降解速率的快慢是一致的。据此可以推测,CuMn2O4/rGO催化臭氧生成溴酸盐的速率更快的原因主要是因为CuMn2O4/rGO的结晶度更高、更稳定、比表面积和总孔容积更大、导电性更好。
2.3 结构与性能之间关系分析
为了进一步确定CuMn2O4/rGO和CuMn2O4/g-C3N4的结构与催化性能之间的关系,使用雷达图对2种催化剂的结构和性能进行了比较,结果如图8所示。图8中7个指标的初始值均为CuMn2O4的相关参数,结果表示的分别是CuMn2O4/rGO和CuMn2O4/g-C3N4中相应指标的相对值。从结构上看,rGO和g-C3N4的掺入对CuMn2O4的比表面的影响程度最大。与CuMn2O4相比,CuMn2O4/rGO的比表面积增大了17.68倍,CuMn2O4/g-C3N4的比表面积增大了5.09倍。rGO和g-C3N4的掺入对Cu2+、Mn4+和OV的相对含量的影响次之,CuMn2O4/rGO中Cu2+和Mn4+的相对含量分别增加了3.17倍和3.64倍,CuMn2O4/g-C3N4中Cu2+和Mn4+的相对含量分别增加了4.50倍和3.64倍;CuMn2O4/rGO中OV的相对含量增加了2.51倍,CuMn2O4/g-C3N4中OV的相对含量增加了2.74倍。rGO和g-C3N4的掺入对阻抗R的影响最小,CuMn2O4/rGO的阻抗增大了18.70%,CuMn2O4/g-C3N4的阻抗增大了46.93%。rGO和g-C3N4掺入CuMn2O4后带来的结构上的显著变化可能是因为CuMn2O4中的金属离子化学掺杂到rGO和g-C3N4的骨架中形成复合物,其作为催化中心改变了rGO和g-C3N4的表面性质和电子分布。
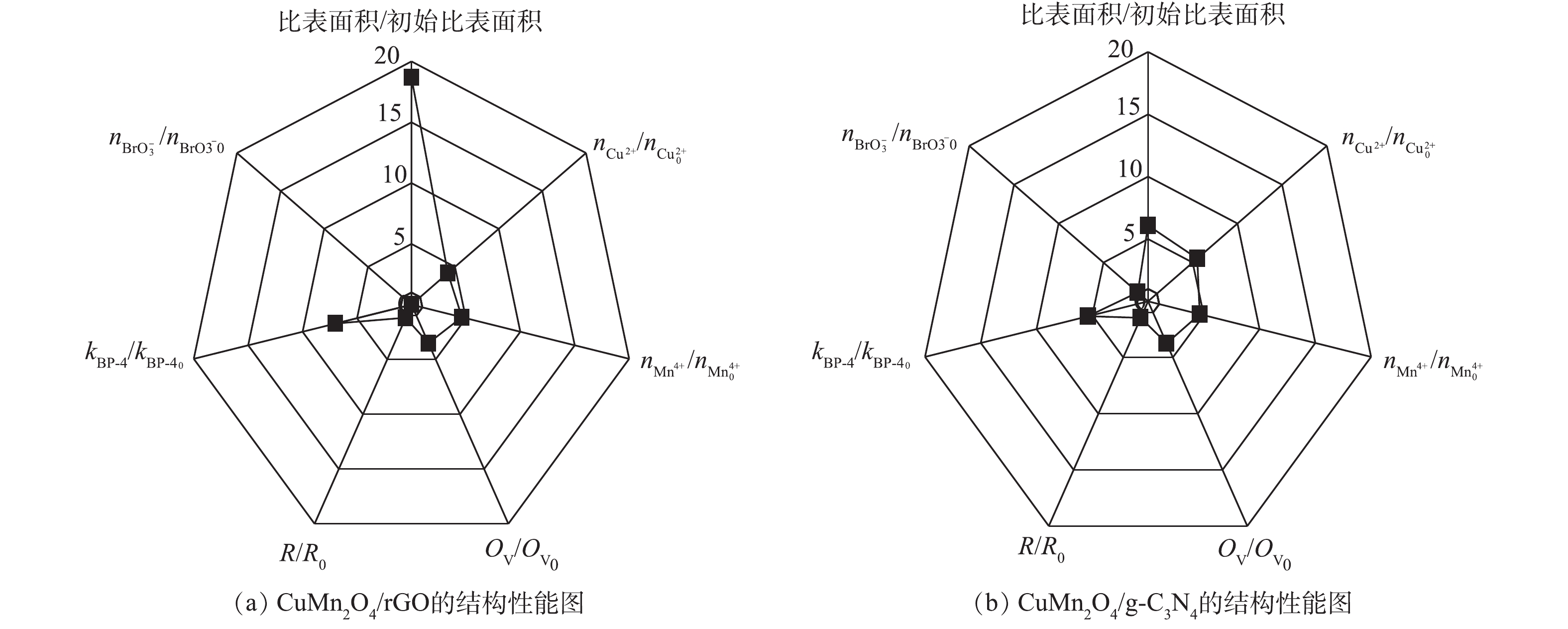 图 8 CuMn2O4/rGO、CuMn2O4/g-C3N4的结构和性能分析图Figure 8. Analysis of structure and performance of CuMn2O4/rGO and CuMn2O4/g-C3N4
图 8 CuMn2O4/rGO、CuMn2O4/g-C3N4的结构和性能分析图Figure 8. Analysis of structure and performance of CuMn2O4/rGO and CuMn2O4/g-C3N4从降解效能上看,rGO和g-C3N4的掺入均能显著加快BP-4的降解,与CuMn2O4相比,CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧降解BP-4的速率分别提高了5.98倍和5.37倍。但是,rGO和g-C3N4的掺入对溴酸盐生成量的抑制效果有显著差异。与CuMn2O4相比,CuMn2O4/rGO催化臭氧使
{\rm{BrO}}_3^ - 的含量降低了100%,而CuMn2O4/g-C3N4催化臭氧生成{\rm{BrO}}_3^ - 的含量没有进一步减少。结合rGO和g-C3N4掺入CuMn2O4后结构上发生的变化,可以推测,反应速率的增强主要得益于3个方面:其一,具有平面片状结构的rGO和g-C3N4阻碍了CuMn2O4的团聚,同时可以作为一个比表面积大、导电性高的框架来维持CuMn2O4纳米颗粒在催化臭氧氧化过程中电子的转移,产生更多的活性物种;其二,rGO和g-C3N4的掺入,可以促进金属离子的电荷转移,使催化剂表面有更多的转移电子,加速反应的进行;其三,rGO和g-C3N4掺入CuMn2O4后,催化剂中含有更多的氧空位,这为催化臭氧氧化提供了更多潜在的反应位点。综合比较CuMn2O4/rGO和CuMn2O4/g-C3N4催化臭氧氧化降解BP-4的效果和对溴酸盐的控制效果,CuMn2O4/rGO更适合应用于催化臭氧氧化降解微污染物及控制溴酸盐生成的反应过程中。3. 结论
1)使用两步煅烧法成功制备出了CuMn2O4/rGO与CuMn2O4/g-C3N4。通过XRD表征、BET比表面积分析、XPS分析以及电化学交流阻抗测试分析发现,尽管CuMn2O4/g-C3N4比CuMn2O4/rGO电子转移速率更快、氧空位更多,但是CuMn2O4/rGO比CuMn2O4/g-C3N4的结晶度更高、比表面积更大、导电性更好。
2)通过催化臭氧氧化降解BP-4的实验结果表明,rGO和g-C3N4的掺入均能有效提升CuMn2O4催化臭氧氧化降解BP-4的速率。但是,二者的掺入对于溴酸盐生成的控制效果有显著差异。在掺入rGO后,溴酸盐的生成量能进一步减少;而g-C3N4的掺入对溴酸盐生成的控制效果没有提升。
3)进一步比较CuMn2O4/rGO与CuMn2O4/g-C3N4的结构和性能发现,rGO和g-C3N4掺入CuMn2O4后可以阻碍了CuMn2O4的团聚的同时,还可以作为一个高导电性的框架,促进CuMn2O4在催化臭氧氧化过程中电子的转移,此外,由于其具有高导电性和大表面积而提高了催化效率。综合考虑2种复合催化剂对BP-4的降解效果与对溴酸盐的控制效果,CuMn2O4/rGO更适用于催化臭氧氧化。
-
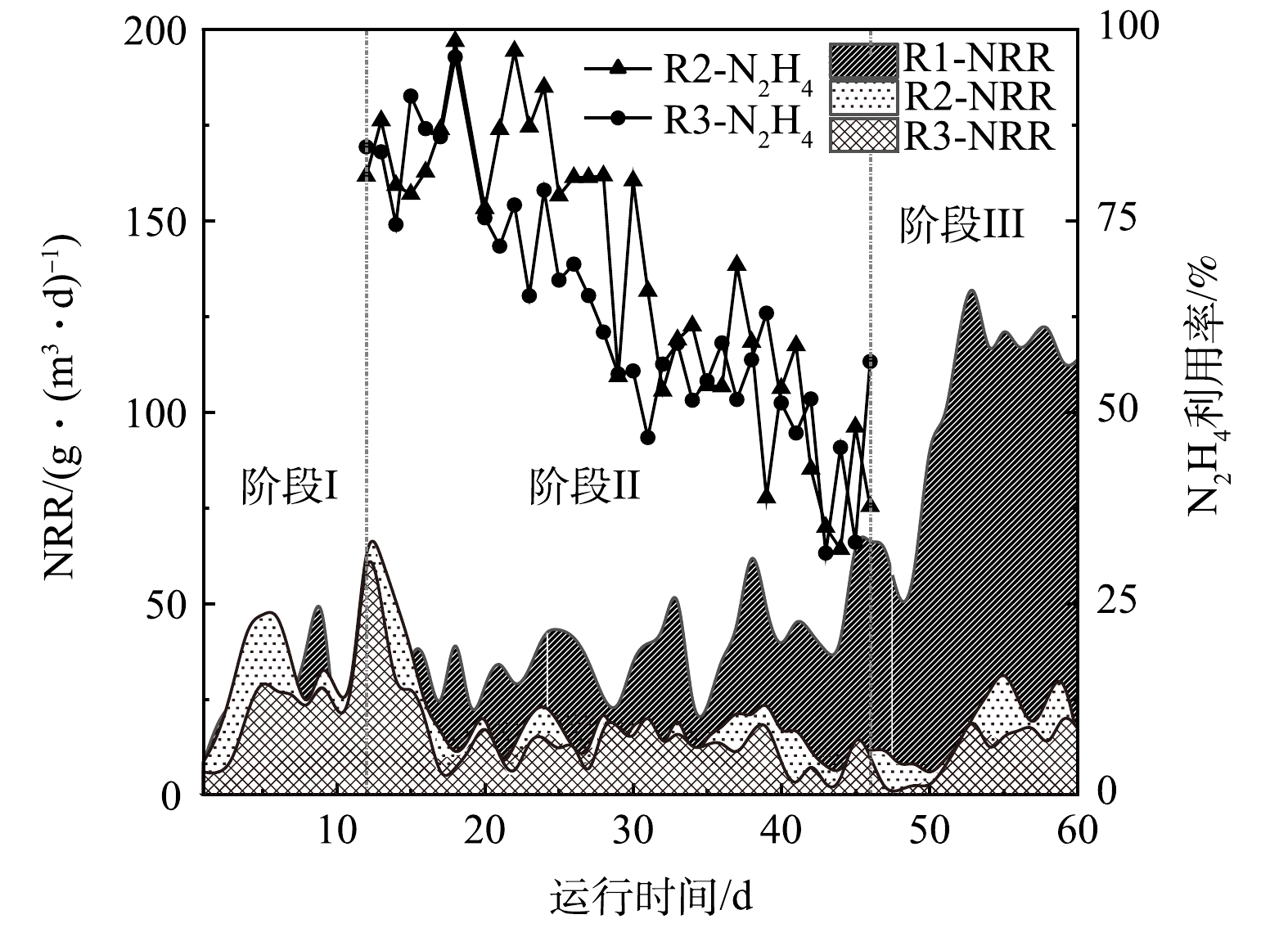
图 2 运行期间NRR以及N2H4利用率的变化
Figure 2. Variations of NRR and N2H4 utilization during operation
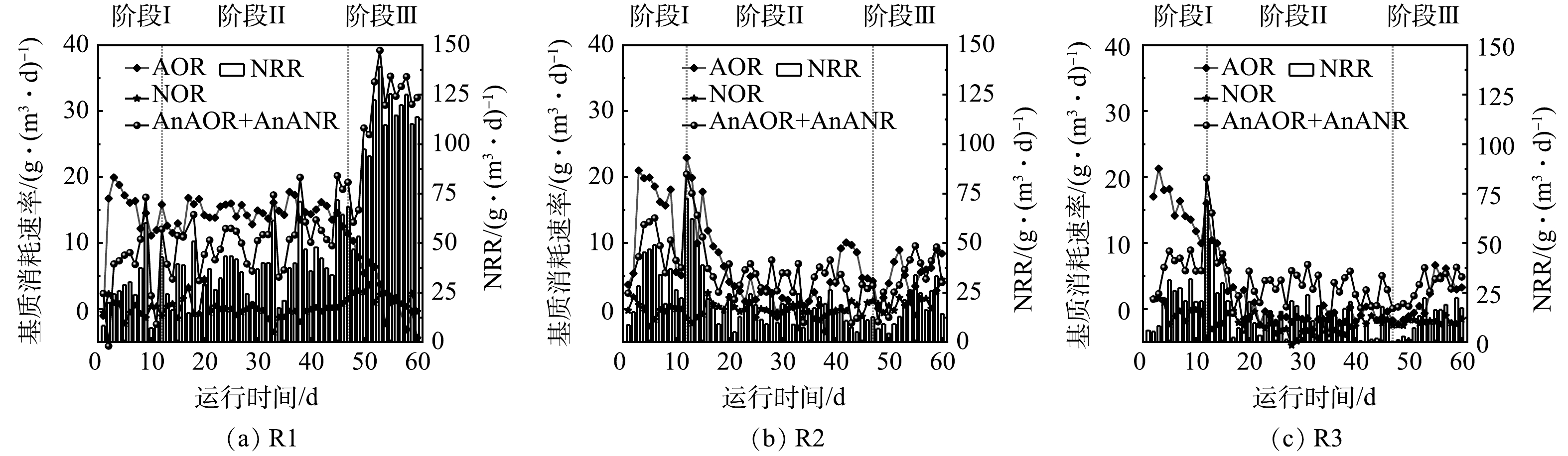
图 4 运行期间亚硝化、硝化和厌氧氨氧化作用的变化
Figure 4. Variations of nitrosation, nitrification and Anammox during operation
表 1 实验设计表
Table 1. The experimental design table
反应器 阶段Ⅰ(1~11 d) 阶段Ⅱ(12~46 d) 阶段Ⅲ(47~60 d) R1 未添加N2H4 未添加N2H4 未添加N2H4 R2 添加5 mg·L-1 N2H4 R3 添加10 mg·L-1 N2H4
下载: 导出CSV
-
[1] JIA Z, YUAN Q, ROOTS P, et al. Partial Nitritation/Anammox and biological phosphorus removal integration in a single bioreactor under mainstream conditions[J]. Bioresource Technology, 2023, 373: 128714. doi: 10.1016/j.biortech.2023.128714 [2] YUAN Q, JIA Z, ROOTS P, et al. A strategy for fast anammox biofilm formation under mainstream conditions[J]. Chemosphere, 2023, 318: 137955. doi: 10.1016/j.chemosphere.2023.137955 [3] 卢帅宇, 由昆, 周伟伟, 等. MBBR厌氧氨氧化工艺污水脱氮的研究进展[J]. 能源环境保护, 2022, 36(6): 22-31. doi: 10.3969/j.issn.1006-8759.2022.06.003 [4] XU Y, XU Y, LI T, et al. Two-step partial nitrification-anammox process for treating thermal-hydrolysis anaerobic digester effluent: Start-up and microbial characterisation[J]. Journal of Cleaner Production, 2020, 252: 119784. doi: 10.1016/j.jclepro.2019.119784 [5] TRINH H P, LEE S H, JEONG G, et al. Recent developments of the mainstream anammox processes: Challenges and opportunities[J]. Journal of Environmental Chemical Engineering, 2021, 9(4): 105583. doi: 10.1016/j.jece.2021.105583 [6] MA J, WEI J, KONG Q, et al. Synergy between autotrophic denitrification and Anammox driven by FeS in a fluidized bed bioreactor for advanced nitrogen removal[J]. Chemosphere, 2021, 280: 130726. doi: 10.1016/j.chemosphere.2021.130726 [7] ZHANG S, ZHANG L, YAO H, et al. Responses of anammox process to elevated Fe(III) stress: Reactor performance, microbial community and functional genes[J]. Journal of Hazardous Materials, 2021, 414: 125051. doi: 10.1016/j.jhazmat.2021.125051 [8] WANG Z, LIU X, NI S Q, et al. Nano zero-valent iron improves anammox activity by promoting the activity of quorum sensing system[J]. Water Research, 2021, 202: 117491. doi: 10.1016/j.watres.2021.117491 [9] LIU W, SHEN C, LIU C, et al. Achieving stable mainstream nitrogen and phosphorus removal assisted by hydroxylamine addition in a continuous partial nitritation/anammox process from real sewage[J]. Science of the Total Environment, 2021, 794: 148478. doi: 10.1016/j.scitotenv.2021.148478 [10] GANESAN S, VADIVELU V M. Effect of external hydrazine addition on anammox reactor start-up time[J]. Chemosphere, 2019, 223: 668-674. doi: 10.1016/j.chemosphere.2019.02.104 [11] YIN X, QIAO S, ZHOU J, et al. Fast start-up of the anammox process with addition of reduced graphene oxides[J]. Chemical Engineering Journal, 2016, 283: 160-166. doi: 10.1016/j.cej.2015.07.059 [12] ADAMS M, XIE J, CHANG Y, et al. Start-up of Anammox systems with different biochar amendment: Process characteristics and microbial community[J]. Science of The Total Environment, 2021, 790: 148242. doi: 10.1016/j.scitotenv.2021.148242 [13] ZEKKER I, KROON K, RIKMANN E, et al. Accelerating effect of hydroxylamine and hydrazine on nitrogen removal rate in moving bed biofilm reactor[J]. Biodegradation, 2012, 23(5): 739-749. doi: 10.1007/s10532-012-9549-6 [14] XIANG T, GAO D. Comparing two hydrazine addition strategies to stabilize mainstream deammonification: Performance and microbial community analysis[J]. Bioresource Technology, 2019, 289: 121710. doi: 10.1016/j.biortech.2019.121710 [15] WEN R, WEI Y, ZHANG W. Recovery of nitrogen removal by N2H4 after nitrite inhibited anammox reaction[J]. Global NEST Journal, 2021, 23: 249-256. [16] YAO Z B, CAI Q, ZHANG D J, et al. The enhancement of completely autotrophic nitrogen removal over nitrite (CANON) by N2H4 addition[J]. Bioresource Technology, 2013, 146: 591-596. doi: 10.1016/j.biortech.2013.07.121 [17] YAO Z, ZHANG D, XIAO P, et al. Long-term addition of micro-amounts of hydrazine enhances nitrogen removal and reduces NO and NO3− production in a SBR performing Anammox: Long-term addition of micro-amount hydrazine enhanced Anammox[J]. Journal of Chemical Technology & Biotechnology, 2016, 91(2): 514-521. [18] MIODOŃSKI S, MUSZYŃSKI-HUHAJŁO M, ZIĘBA B, et al. Fast start-up of anammox process with hydrazine addition[J]. SN Applied Sciences, 2019, 1(6): 523. doi: 10.1007/s42452-019-0514-4 [19] 蔡庆, 黄阳全, 罗乐, 等. 厌氧氨氧化颗粒污泥的培养及影响因素[J]. 工业安全与环保, 2016, 42(11): 68-71. doi: 10.3969/j.issn.1001-425X.2016.11.019 [20] XIANG T, GAO D, WANG X. Performance and microbial community analysis of two sludge type reactors in achieving mainstream deammonification with hydrazine addition[J]. Science of the Total Environment, 2020, 715: 136377. doi: 10.1016/j.scitotenv.2019.136377 [21] YUAN Q, ZHANG Y, XUE X, et al. Morphological, kinetic, and microbial community characterization of anammox bacteria with different inoculations and biofilm types for low-ammonium wastewater treatment[J]. Journal of Water Process Engineering, 2022, 47: 102748. doi: 10.1016/j.jwpe.2022.102748 [22] YUAN Q, HE B, QIAN L, et al. Role of air scouring in anaerobic/anoxic tanks providing nitrogen removal by mainstream anammox conversion in a hybrid biofilm/suspended growth full-scale WWTP in China[J]. Water Environment Research, 2021, 93(10): 2198-2209. doi: 10.1002/wer.1592 [23] VAN DE GRAAF A, DE BRUIJN P, ROBERTSON L A, et al. Autotrophic growth of anaerobic ammonium-oxidizing micro-organisms in a fluidized bed reactor[J]. Microbiology, 1996, 142(8): 2187-2196. doi: 10.1099/13500872-142-8-2187 [24] 国家环境保护总局. 水和废水监测分析方法[M]. 4版. 北京: 中国环境科学出版社, 2002. [25] CROSBY N T. Determination of ammonia by the Nessler method in waters containing hydrazine[J]. The Analyst, 1968, 93(1107): 406-408. doi: 10.1039/an9689300406 [26] WATT G W, CHRISP J D. Spectrophotometric method for determination of hydrazine[J]. Analytical Chemistry, 1952, 24(12): 2006-2008. doi: 10.1021/ac60072a044 [27] GEORGE M, NAGARAJA K S, BALASUBRAMANIAN N. Spectrophotometric determination of hydrazine[J]. Talanta, 2008, 75(1): 27-31. doi: 10.1016/j.talanta.2007.09.002 [28] YU G H, HE P J, SHAO L M. Characteristics of extracellular polymeric substances (EPS) fractions from excess sludges and their effects on bioflocculability[J]. Bioresource Technology, 2009, 100(13): 3193-3198. doi: 10.1016/j.biortech.2009.02.009 [29] MA H, ZHANG Y, XUE Y, et al. Relationship of heme c, nitrogen loading capacity and temperature in anammox reactor[J]. Science of the Total Environment, 2019, 659: 568-577. doi: 10.1016/j.scitotenv.2018.12.377 [30] BERRY E A, TRUMPOWER B L. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra[J]. Analytical Biochemistry, 1987, 161(1): 1-15. doi: 10.1016/0003-2697(87)90643-9 [31] XIAO R, ZHU W, ZHENG Y, et al. Active assimilators of soluble microbial products produced by wastewater anammox bacteria and their roles revealed by DNA-SIP coupled to metagenomics[J]. Environment International, 2022, 164: 107265. doi: 10.1016/j.envint.2022.107265 [32] SCHALK J, OUSTAD H, KUENEN J G, et al. The anaerobic oxidation of hydrazine: A novel reaction in microbial nitrogen metabolism[J]. FEMS Microbiology Letters, 1998, 158(1): 61-67. doi: 10.1111/j.1574-6968.1998.tb12801.x [33] ZHOU S, CHEN Z, WANG J, et al. Recovery of anaerobic ammonium oxidation via hydrazine following sulfate inhibition[J]. Environmental Science:Water Research & Technology, 2022, 8(7): 1458-1465. [34] DI BIASE A, KOWALSKI M S, DEVLIN T R, et al. Moving bed biofilm reactor technology in municipal wastewater treatment: A review[J]. Journal of Environmental Management, 2019, 247: 849-866. [35] STROUS M, HEIJNEN J J, KUENEN J G, et al. The sequencing batch reactor as a powerful tool for the study of slowly growing anaerobic ammonium-oxidizing microorganisms[J]. Applied Microbiology and Biotechnology, 1998, 50(5): 589-596. doi: 10.1007/s002530051340 [36] XIANG T, LIANG H, GAO D. Effect of exogenous hydrazine on metabolic process of anammox bacteria[J]. Journal of Environmental Management, 2022, 317: 115398. doi: 10.1016/j.jenvman.2022.115398 [37] TANG B, YU C, BIN L, et al. Essential factors of an integrated moving bed biofilm reactor–membrane bioreactor: Adhesion characteristics and microbial community of the biofilm[J]. Bioresource Technology, 2016, 211: 574-583. doi: 10.1016/j.biortech.2016.03.136 [38] SHENG G P, YU H Q, LI X Y. Extracellular polymeric substances (EPS) of microbial aggregates in biological wastewater treatment systems: A review[J]. Biotechnology Advances, 2010, 28(6): 882-894. doi: 10.1016/j.biotechadv.2010.08.001 [39] ZHANG M, GAO J, FAN Y, et al. Combined effects of volume ratio and nitrate recycling ratio on nutrient removal, sludge characteristic and microbial evolution for DPR optimization[J]. Journal of Environmental Sciences, 2021, 104: 69-83. doi: 10.1016/j.jes.2020.12.003 [40] 杨明明, 党超军, 张爱余, 等. 厌氧氨氧化颗粒污泥胞外聚合物金属元素特性[J]. 中国环境科学, 2020, 40(11): 4728-4734. doi: 10.3969/j.issn.1000-6923.2020.11.011 [41] CHEN J, HAI Y, ZHANG W, et al. Insights into deterioration and reactivation of a mainstream anammox biofilm reactor response to C/N ratio[J]. Journal of Environmental Management, 2022, 320: 115780. doi: 10.1016/j.jenvman.2022.115780 [42] KANG D, LI Y, XU D, et al. Deciphering correlation between chromaticity and activity of anammox sludge[J]. Water Research, 2020, 185: 116184. doi: 10.1016/j.watres.2020.116184 [43] LIU W, YANG D, CHEN W, et al. High-throughput sequencing-based microbial characterization of size fractionated biomass in an anoxic anammox reactor for low-strength wastewater at low temperatures[J]. Bioresource Technology, 2017, 231: 45-52. doi: 10.1016/j.biortech.2017.01.050 [44] 杨瑞丽, 王晓君, 吴俊斌, 等. 厌氧氨氧化工艺快速启动策略及其微生物特性[J]. 环境工程学报, 2018, 12(12): 3341-3350. [45] HUANG D Q, WANG Y, WU Q, et al. Anammox sludge preservation: Preservative agents, temperature and substrate[J]. Journal of Environmental Management, 2022, 311: 114860. doi: 10.1016/j.jenvman.2022.114860 [46] WANG C, LIU S, XU X, et al. Achieving mainstream nitrogen removal through simultaneous partial nitrification, anammox and denitrification process in an integrated fixed film activated sludge reactor[J]. Chemosphere, 2018, 203: 457-466. doi: 10.1016/j.chemosphere.2018.04.016 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2830
- HTML全文浏览数: 2830
- PDF下载数: 108
- 施引文献: 0

