
























-
大气颗粒物PM2.5与气候变化、人体健康、生态环境等密切相关[1]. 三亚市(18°18′N,109°31′E)位于海南岛的最南端,地处低纬度,属热带海洋性季风气候区. 随着海南自由贸易港建设进展加快、国际旅游岛对外开放和海南热带气候等区位优势,海南人口激增与经济快速发展,空气质量的压力越来越大[2],三亚市政府和国内外游客也日益重视PM2.5的健康效应.
三亚PM2.5研究报道较少,与之关联的是南海大气化学研究. 前人研究如下:三亚PM2.5中OC和EC [3-4];PM2.5光学特征[5-6];PM2.5的潜在源定性分析[4,7];PMF模型定量源解析[8]. 海口CMB模型源解析[9-10];南海PMF模型源解析[11-12];南海有机气溶胶分子标识物和同位素示踪来源[13-15];上述源解析很少单用微量元素. 受体模型PMF源解析优点在于灵活性(不需源谱)和操作性(模型软件化和源个数“眼球法”),缺点在于共线源问题和源解析结果时空差异大[16]. 基于PMF的优缺点,在实际运用中,呈现两种局面:一方面是接受PMF优点,广泛应用,同时,单用微量元素组分进行源解析,多与PM2.5中重金属健康效应结合 [17-18];另一方面是将受体模型、源清单法和源过程法等方法结合,进行多种源解析,相互验证解决PMF不足 [19]. 本文基于微量元素数据及其健康评价,采用PMF源解析第一情况,其不足在于微量元素标识物的共线源问题,优势在于微量元素相对于有机组分来说,其变化程度小. 三亚市PM2.5微量元素的源解析尚鲜见报道,研究PM2.5中微量元素污染特征及健康评价具有十分重要的意义.
本文通过离线滤膜样品采集得到三亚市PM2.5微量元素数据,结合富集因子分析,表征三亚市PM2.5微量元素的化学特征;使用受体PMF模型进行源解析,定量估算微量元素排放源对三亚市PM2.5的贡献比例;利用暴露评估模型,评价PM2.5中微量元素的健康效应. 此论文为三亚市PM2.5的空气质量监测及防治提供思路及措施.
-
本研究样品采集地点位于三亚市吉阳区热带海洋学院实验楼8栋楼顶大气颗粒物采样室. 采样时间选用2019年6月(代表夏季)和10月(秋季)、12月(冬季)作为季节性代表进行采样. 每天1个样,每个滤膜样品每天收集大气颗粒物至少20 h,每月30个样品,共计90个样品. 样品采集使用仪器为mini-volume便携式大气颗粒物PM2.5采样器,流量是5 L·min−1,该仪器已得USEPA(美国环保局)认可,选择特氟龙滤膜收集元素组分.
-
本实验样品分析地点是在中科院地球与环境研究所化学与物理重点实验室,使用X射线荧光光谱仪对特氟龙膜上气溶胶所含的26种元素(Na、Mg、Al、Si、P、S、Cl、K、Ca、Sc、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn、Ga、As、Se、Br、Sr、Ba、Pb)进行了分析测定. 仪器采用美国Micro Matter公司的薄膜滤纸作为标准物质建立工作曲线;采用NIST SRM 2783号标准物质进行质量控制,每8个样品任意挑选1个做复检. 检查各个元素的浓度测量值,并给出最终结果. 结合采样面积和采样流量,将初始浓度转化为最终的大气质量浓度单位. 样品分析过程中的质量保证/质量控制详见文献 [20].
-
元素富集因子法简称“富集因子法”(Enrichment Factor,EF). 用以表示大气颗粒物中元素的富集程度,并判断和评价颗粒物中元素来源(自然来源和人为来源)的因子. 根据前人研究,元素的富集因子<10,主要来源为自然源;元素的富集因子>10,则认为其受到人为污染源的影响,富集因子越高,其受人为活动影响越大[1]. 富集因子EF的定义如下:
式中,Cx是指研究中感兴趣的元素浓度,Cr为参考元素的浓度,分子的含义是样品中待测元素与参考元素的浓度比值,分母的含义为参考质中待元素与参考元素的浓度比值.
-
PMF法(Positive Matrix Factorization)是美国环保署EPA确定的区域环境污染评价的重要方法之一[21]. PMF法不依赖于排放源的排放条件、气象、地形等数据,能够定量确定污染源的类型和贡献比例 [19]. 受体模型PMF法的原理为:假设有p种来源、来源类型或来源区域影响一个受体,对观测得到的大气颗粒物中各种组分的浓度按照各因素的影响进行线性拟合. 数学表达式如下:
式中,Xij是受体第i天第j种组分的浓度;gik是第k种因素在第i天对受体的贡献;fkj是组分j在第k种因素中所占的部分;eij是第i天第j种组分的残余部分. EPA PMF的任务就是最小化总方差,即Q值最小.
式中,Sij是第i天第j种组分的不确定性,用户可以根据公式(4)和(5)确定每个Xij的不确定性,当化学组分浓度小于最低检测限(MDL)时,不确定度用公式(4)计算;当化学组分浓度大于最低检测限(MDL)时,不确定度用公式(5)计算,式中Error Fraction 的计算基于重复样测试结果计算,MDL 组分分析设备都会有该数据.
本研究采用美国环保署认可的 EPA PMF 5.0 模型软件进行分析运算,以采样点位共计90个 PM2.5 样品中的26个微量组分浓度及其不确定性作为输入文件. 根据各因子的特征来解释污染源类型和贡献大小 (下载地址:http://www.epa.gov/air-research/positive-matrix-factorization-model-environmental-data-analyses).
-
本文选取9种重金属元素(As、Cr、Ni、Al、Cu、Mn、Pb、Se、Zn)进行健康评价. 依据美国环保署综合风险信息数据库和国际癌症研究机构的研究结论,大气细颗粒物重金属中的As、Cr、Ni属于致癌性物质,Al、Cu、Mn、Pb、Se、Zn属于非致癌性物质 [22-23]. 经呼吸途径暴露的健康风险评价公式如下:
其中,R为人群超额危险度,无量纲;LADD(life average daily dose)为致癌物质终身日均暴露剂量[μg·(kg·d)−1];ADD(average daily dose)为非致癌物质日均暴露剂量[μg·(kg·d)−1];AL(average life)为人均寿命参数,取常量70;SF(slope factor)为致癌化学物质的致癌斜率因子[(kg·d)·μg−1;RfD(reference dose)为参考剂量[μg·(kg·d)−1];SF与RfD获得自美国环保署颁布的化学物质人体健康效应评价文件(HEA)(https://www.epa.gov/),见表1.
暴露剂量率计算方法为:
其中,C(concentration)为污染物浓度(μg·m−3);IR(inhalation rate)为呼吸速率参数(m3·d−1);ED(exposure duration)为暴露持续时间(d);BW(body weight)为体重(kg);AT(averaging time)为平均暴露时间(d);经呼吸途径进入人体的暴露剂量参数来自美国环保署( https://www.epa.gov/),其中成年男性与女性的IR与BW来自生态环境部(https://www.mee.gov.cn/),见表2.
-
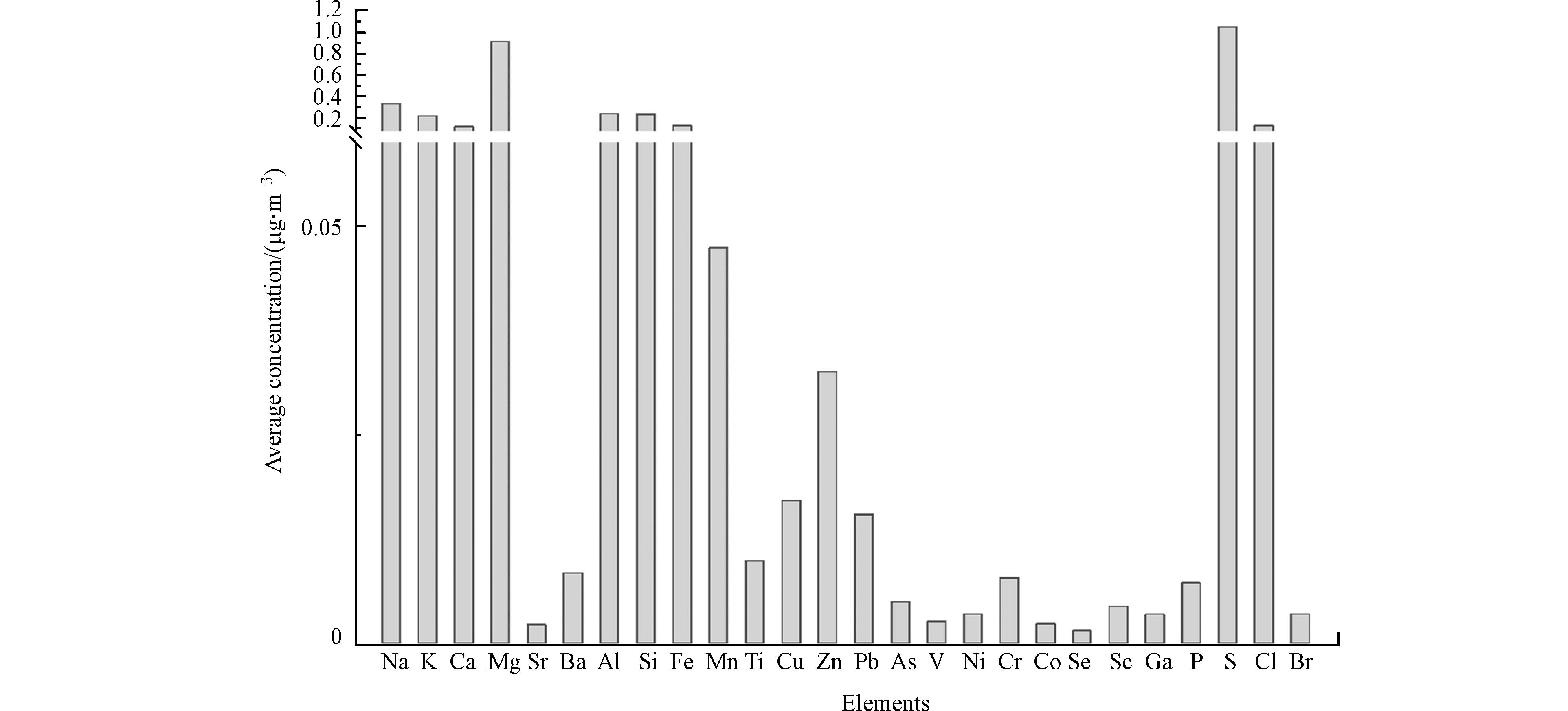
26种微量元素平均浓度见图1. 从图1可知,S(1.040 μg·m−3)、Mg(0.906 μg·m−3)、Na(0.325 μg·m−3)、Al(0.235 μg·m−3)、Si(0.228 μg·m−3)、K(0.214 μg·m−3)、Fe(0.126 μg·m−3)、Ca(0.112 μg·m−3)等元素含量较高. Na和K是碱金属元素,Ca和Mg是碱土金属元素,Al、Si和Fe是造岩元素或土壤中主量元素,Mg含量排序第二,表明样品受土壤排放源影响大. S是非金属元素,来源于含硫矿物燃烧,含量排序第一,表明三亚市大气颗粒物PM2.5已受人为源的影响[3-4,7-8].
表3 是PM2.5中7种重金属元素浓度均值. 由表3可知,三亚市重金属元素的年均质量浓度排序为:Zn>Cu>Pb>Cr>As>Ni>Se. Zn的质量浓度最高,其中致癌性重金属Cr(7.70×10−3 μg·m−3)已超过《环境空气质量标准》(GB 3095—2012)中规定标准限值2.50×10−5 μg·m−3. 大气Zn排放源较多,除受电镀、冶金、化工等工业影响外,还与机动车辆橡胶轮胎磨损、垃圾焚烧、含锌矿石的开采等多种因素有关[24];Cr广泛应用于汽车零件,铝合金和钛合金等制品、轮胎摩擦产生的粉尘也含有Cr元素[25],研究表明,受海洋气候影响,滨海城市Cr高于内陆城市. 三亚是国际旅游城市,高速公路和景区常见大陆各省车辆,加之行车道路环境的不熟悉,高浓度的Zn和致癌性重金属Cr可能与车辆橡胶轮胎磨损有关;同时,海南岛处于季风交互地带,受大陆污染影响严重[4,7-8],这也是三亚重金属Cr超标原因之一.
-
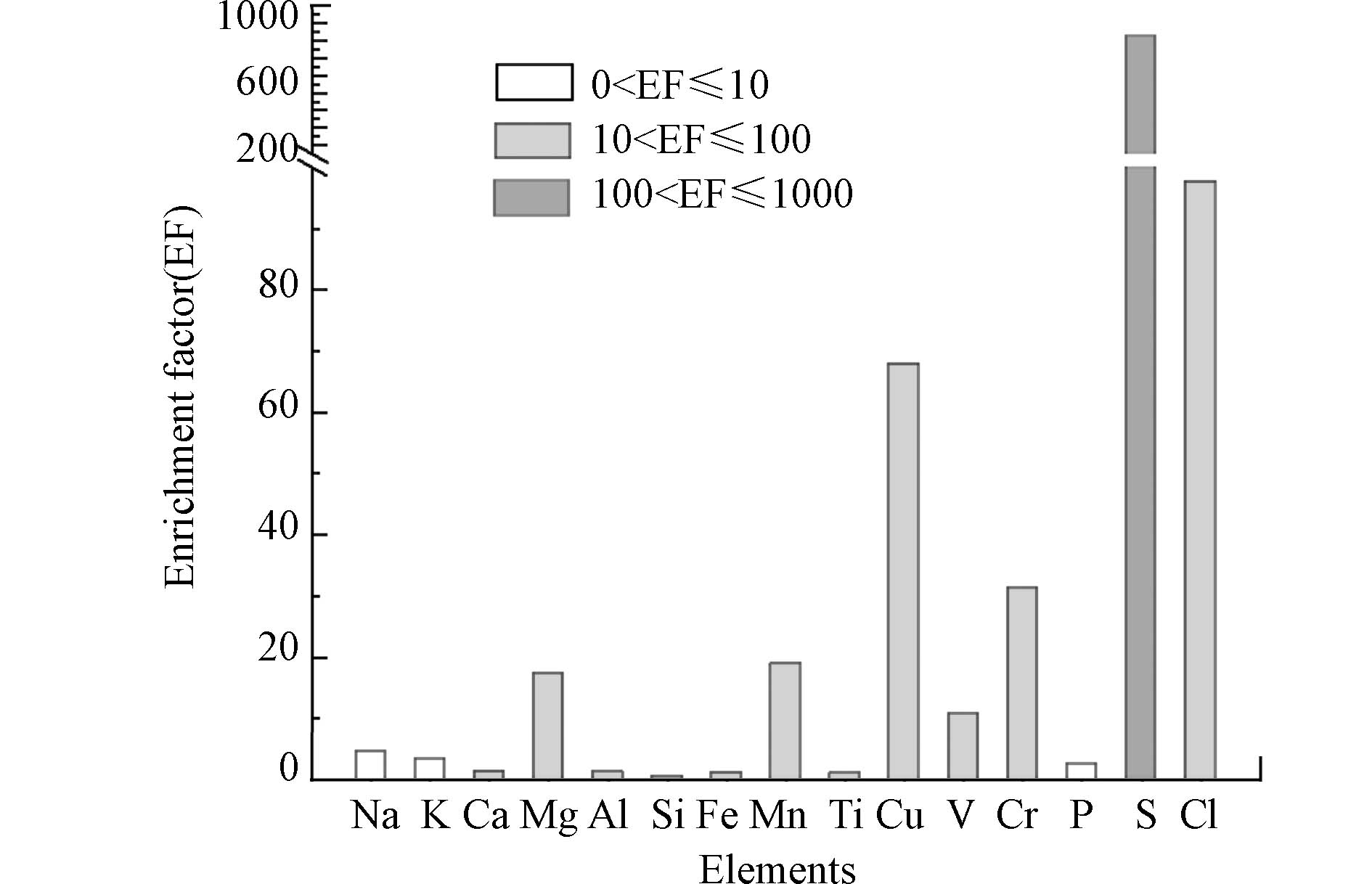
以Fe为参考物质,对Na、Mg、Al、Si、P、S、Cl、K、Ca、Ti、V、Cr、Mn、Fe、Cu等15种元素进行富集因子分析,分析结果如图2. 由图2可知,S(825.46)、Cl(97.67)、Cu(67.87)、Cr(31.21)、Mn(18.80)、Mg(17.20)、V(10.57)均出现较高的EF值,其中S的富集因子高达825.46,表明这些元素已经明显受到人为活动的影响,与前人研究三亚PM2.5空气质量结果一致[4,7-8]. 三亚市PM2.5主要来源是海洋源、工业源和交通源等,故S、Cl、Cu等元素富集因子较高.
-
为了确定因子(源)数量,分别对模型进行了迭代运算,通过比较分析,最终确定5个因子最为合理(“眼球法”) [21]. 5个因子的贡献比例如图3所示.
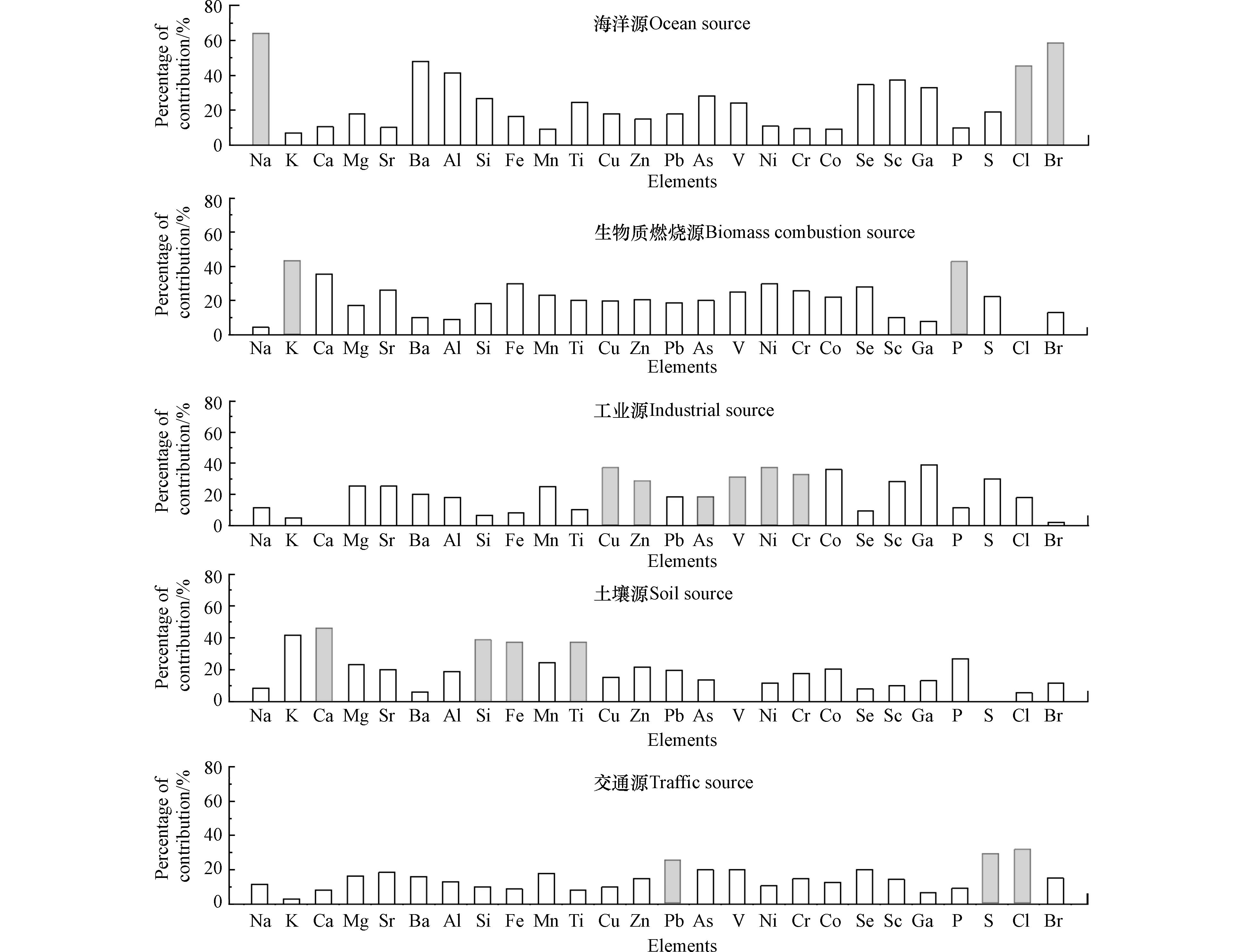
本研究根据前人文献及研究,交通源用Pb、Zn等来指示,K来指示生物质燃烧源,Na来指示海洋源[1], Mn、Si、Ca、Fe来指示土壤源,V、Cu、Zn来指示工业源[16]. 对照相关因子浓度贡献占比及其时间序列,第1个因子是海洋源:标识物为Na、Cl和Br,Na是碱金属元素,Cl和Br是海洋卤族元素;第2个因子是生物质燃烧源:标识物为K、P,二者是植物生长的营养元素;第3个因子是工业源,标识物为Cu、Zn、V、Ni、Cr,这些都是重金属元素;第4个因子为土壤源,标识物为Ca、Si、Fe、Ti,其中,Ca是碱土金属,剩余的都是造岩元素,也是土壤的主量元素;第5个因子是交通源,标识物为S、Pb、Cl,其中,S和Pb是交通燃烧源的指纹,Cl是海洋源和燃烧源(如生物质燃烧、机动车燃烧和燃煤燃烧)的标识物 [26-28]. 虽然Cl是作物生长发育不可或缺的一种元素,是构成作物生长所必须的16种元素之一,可归属于生物质燃烧源,但限于Cl元素的共线源问题和PMF模型“眼球法”识别源个数的主观性,综合考虑,本文将Cl归属于交通源排放.
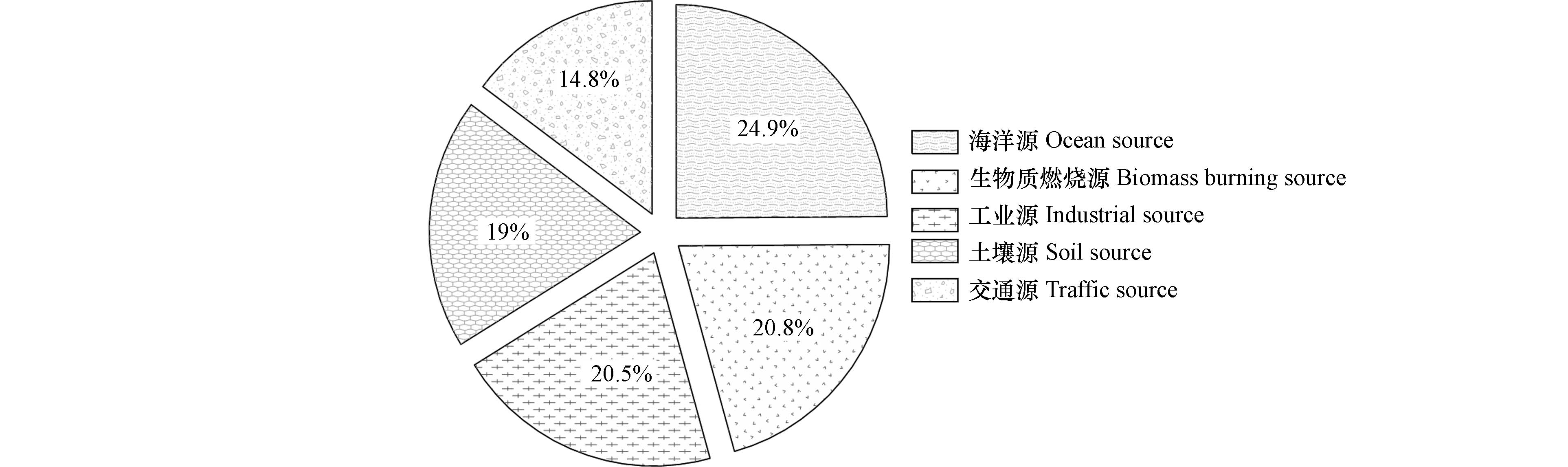
图4是三亚市PM2.5源解析结果,5种排放源大小比例分别是:海洋源(24.9%)>生物质燃烧源(20.8%)>工业源(20.5%)>土壤源(19%)>交通源(14.8%). 结果表明,三亚比邻南海,海洋表层微生物或植物是第一自然排放源,与前人研究结果一致[6-7];海南岛属热带气候,森林覆盖率达80%,生物质燃烧多以谷物秸秆、烤槟榔、枯枝落叶等燃烧排放为主 [29],生物质燃烧是第二排放源;工业源为第三排放源;土壤源为第四贡献源;交通源是第五贡献源.
表4是南海周边国家利用受体模型对PM(TSP、PM10、PM2.5)进行源解析的结果比较. 结果表明如下:1) 滨海城市中海盐贡献占比<10%, 如马来西亚八打灵海盐(7.4%),在贫营养化的三沙永兴岛,海盐占比高达46.6%;在离大陆最近的城市海口,海盐占比低达3%,这一现象说明海盐贡献比例与采样点离岸距离有梯度变化特征 [30]. 2) 滨海城市的主要人为排放源受制于城市经济发展程度,如三亚旅游国际岛,交通源占比高达37.9%;海口是海南省会城市,经济综合发展,交通源占比17.5%;中国台湾省高雄,港口贸易发达,船舶排放占比15.6%;马来西亚八打灵市工业发达,制造业(12%)+混合冶炼工业和道路扬尘(5.6)+冶金工业(5.1%)+ 矿物尘(7.2%),工业源的贡献比例高达30%;菲律宾马尼拉市是工业欠发达,以基础工业为主,如燃煤(17.4%),工业(17.7%),机动车(12.6%)等,工业源的贡献比例更高达50% [30-31]. 3) 二次无机气溶胶由于是二次生成,在滨海城市中,随着工业污染占比高,其贡献比呈现下降趋势,比例范围在20%—30%. 如海南省三沙市二次无机气溶胶(30.1%);海南省海口SO42-(9.5%);马来西亚八打灵二次无机气溶胶(28.5%);中国台湾省高雄港二次无机气溶胶(24%);菲律宾马尼拉二次气溶胶(21.3%)等 [30-31]. 4) 二次无机气溶胶是有SO42-、NO3-和NH4+组成(简称SNA),在PMF模型源解析中,中国大陆学者将其归为二次污染物,但溯源对应不了经济产业部门,而东南亚学者将标识物SO42-基本归为工业燃煤源;NO3-归为交通源;NH4+归为农业源,这是PMF源解析结果差异之一 [21]. 5) 具体确定生物质燃烧源的贡献比,如通用K+标识物外,应加上左旋葡萄糖等有机分子标识物,因为K+还来源于土壤等[16].
-
根据经呼吸途径暴露的健康风险评价模型[18],分别计算出的PM2.5重金属元素对儿童、成年女性和成年男性的平均超额危险度见表5. 9种重金属元素的年均超额危险度在5.40×10-12—7.63×10−7之间,均低于人群可接受危险水平标准1.00×10-6 [32];9种重金属元素对3类人群经呼吸途径暴露的健康风险均为Cr > As > Ni > Al > Mn > Pb > Cu > Zn > Se,致癌性物质As、Cr、Ni年均超额危险度数量级为10−7—10−9,远高于非致癌性物质Al、Cu、Mn、Pb、Se、Zn在量级为10−9—10-12的年均超额危险度;结合表3可看出,Cr元素质量浓度超标,造成Cr的年均超额危险度接近人群可接受危险水平标准. 得出三亚市大气颗粒物PM2.5重金属元素对人群基本无健康风险,但致癌性元素Cr浓度超过国家标准,并且对人群具有潜在的危害. 较之前人研究结果[17],三亚市年均超额危险度量级更低,三亚人口较少,且无采暖季节,海陆风盛行导致扩散条件好,污染物不容易累积[3].
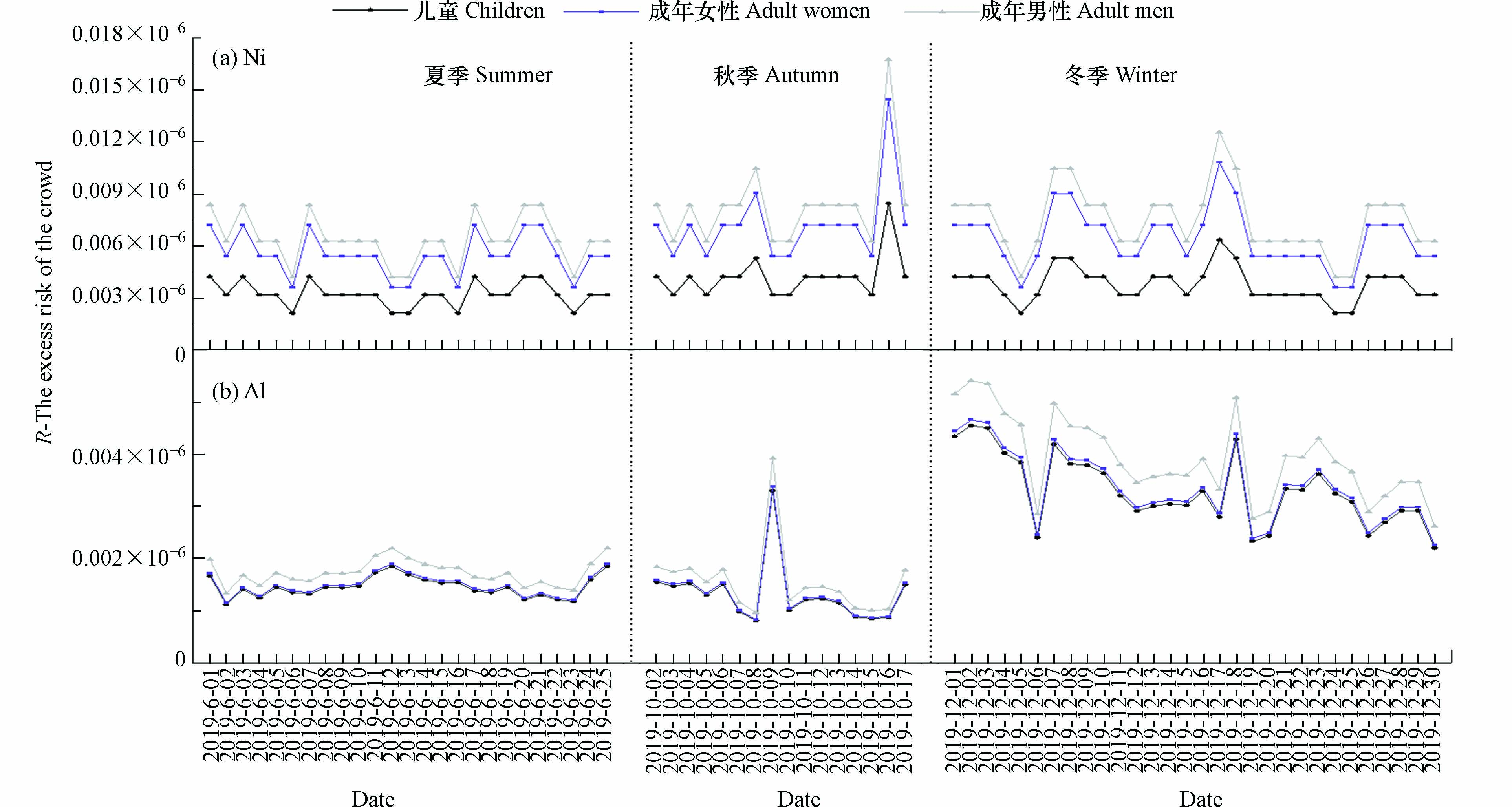
各选取一种代表性致癌性重金属元素和非致癌性重金属元素代入健康风险评价公式,得出每日人群超额危险度见图5. 致癌性重金属代表元素Ni对儿童、成年女性、成年男性人群超额危险度分别为3.68×10−9、6.27×10−9、7.28×10−9;非致癌性重金属Al对儿童、成年女性、成年男性人群超额危险度分别为2.21×10−9、2.26×10−9、2.62×10−9,结合图5得出不同人群的健康风险评价结果为:成年男性>成年女性>儿童,与山东省枣庄市研究结果一致[17].
-
(1)26种微量元素中S、Mg、Na、Al、Si、K、Fe、Ca元素平均质量浓度较高,其中,重金属元素Zn的质量浓度最高,致癌性重金属Cr已经超过标准限值,S的富集因子高达825.46.
(2)PMF源解析结果为海洋源(24.9%)>生物质燃烧源(20.8%)>工业源(20.5%)>土壤源(19%)>交通源(14.8%);对比南海周边滨海城市PMF,结果表明,海盐贡献比例与采样点离岸距离有梯度变化特征;滨海城市的主要排放源受制于城市主导经济产业发展;二次无机气溶胶的贡献与工业源的比例呈反比,取决于SNA标识物的分配;除K+作为生物质标识物外,应加上左旋葡萄糖等有机分子标识物.
(3)三亚市大气颗粒物PM2.5重金属污染对不同人群的影响为:成年男子>成年女子>儿童;不同季节影响为冬季>秋季>夏季:9种重金属元素对3类人群经呼吸途径暴露的健康风险均为Cr > As > Ni > Al > Mn > Pb > Cu > Zn > Se,其年均超额危险度均低于可接受危险水平标准.
2019年三亚市PM2.5微量元素的源解析和健康评价
Source analysis and health assessment of PM2.5 trace elements in Sanya City in 2019
-
摘要: 本研究通过滤膜采样分析得到三亚市PM2.5微量元素数据,结合富集因子,表征化学特征;使用PMF模型进行源解析,定量估算各排放源的贡献比例,并与南海周边城市源解析比较;根据暴露评估模型评估健康效应. 结果表明,三亚市致癌性重金属Cr(7.70×10-3 μg·m−3)已经超过标准限值(2.50×10−5 μg·m−3),S的富集因子高达825.46,表明三亚PM2.5受S元素污染严重;源排放贡献大小比例分别为:海洋源(24.9%)>生物质燃烧源(20.8%)>工业源(20.5%)>土壤源(19%)>交通源(14.8%),源解析结果比较得知,海盐贡献比例与采样点离岸距离有梯度变化特征;滨海城市的主要人为排放源受制于城市经济发展程度;二次无机气溶胶的贡献与工业源的比例呈反比,取决于SAN SNA标识物的配分;重金属污染对三亚不同人群的影响大小顺序为:成年男子>成年女子>儿童;9种重金属元素对 3 类人群经呼吸途径暴露的健康风险均为Cr>As>Ni>Al>Mn>Pb>Cu>Zn>Se.Abstract: This study used membrane filter sampling to collect PM2.5 trace element data of Sanya, China. Enrichment factor analysis and chemical characterization were used with a positive matrix factorization model to conduct source apportionment. The contribution proportions of each emission source were quantified and compared with the source apportionment results of coastal cities of South China Sea. According to an exposure assessment model, the effects of the elements on health was evaluated. The results revealed that the carcinogenic heavy metal Cr (7.70×10−3 μg·m−3)at Sanya exceeded the threshold of 2.50×10−5 μg·m−3. The enrichment factor of S reached as high as 825.46, implying that the PM2.5 in Sanya was severely polluted by S. The contribution proportions of emission sources ranking from high to low were the ocean (24.9%) > biomass combustion (20.8%) > industry (20.5%) > soil (19%) > traffic (14.8%). The source apportionment results revealed that the contribution proportion of sea salt exhibited gradient changes in relation to the distance of the sampling location to the coast. The main anthropogenic emissions of coastal cities were subject to the level of economic development of individual cities. The contribution proportions of secondary inorganic aerosols were inversely proportional to that of industrial emission sources, depending on the ratios of SNA markers. The level of influence of heavy metal pollution on different populations in Sanya was highest on adult man, followed by adult women and children. The risks of the 3 populations exposed to 9 heavy metals through respiratory tracts were Cr > As > Ni > Al > Mn > Pb > Cu > Zn > Se.
-
Key words:
- Sanya /
- PM2.5 /
- trace elements /
- source apportionment /
- health assessment.
-
纳米银(silver nanoparticles, AgNPs)是三维空间中至少有一维处于1—100 nm的单质银颗粒[1],其拥有高效、广谱的杀菌性能,因此广泛应用于医药、食品、化妆品、纺织品等领域[2]. 随着近年来AgNPs技术的不断发展[3],越来越多的AgNPs产品在生产、使用和废弃过程中释放进入水环境[4-5],并对水生生物产生毒害[6],因此有必要深入了解其环境归宿及潜在危害.
AgNPs化学性质活泼,进入水环境后很容易在氧气(O2)和质子(H+)的作用下发生氧化溶解,释放出银离子(silver ions, Ag+)[7]. 由于银的氧化还原电位适中(Ψ(Ag+/Ag0=0.80 V)),自然界中Ag+也可被环境中普遍存在的天然有机质以及一些动物、植物和微生物等还原成零价的AgNPs[8-10]. 因此,水环境中AgNPs与Ag+会相互转化,呈高度动态性. 而由于形态不同,AgNPs和Ag+的毒性效应存在较大差异[11-12]. 例如,虽然AgNPs和Ag+都会对蚯蚓产生细胞毒性,但Ag+主要积聚在含胞质溶胶的部分,而AgNPs主要破坏细胞膜隔室[13]. 此外,AgNPs和Ag+的生物利用度及在生物体的富集过程也存在差异[14-15]. 因此,研究水环境中的AgNPs与Ag+的转化过程对评估AgNPs的生态风险具有重要意义.
溶解性有机物(dissolved organic matter, DOM) 是一类广泛存在于自然水体,由各种活性有机物(如腐殖酸(humic acid, HA)和富里酸(fluvic acid, FA)、蛋白质、多糖和胞外聚合物(extracellular polymeric substances, EPS))组成的非均质复合物[16]. DOM具有多种活性官能团,如硫醇(—SH)、醇/酚羟基(—OH)、醛、羰基、酮、醚基、羧基(—COOH)、胺和甲氧基等,因此其具有较强的氧化还原性,能够介导水体中重金属的迁移转化、毒性和生物利用度的改变[17-18].
现有研究表明,DOM是影响AgNPs和Ag+相互转化的重要因素之一[19-21]. 然而DOM对AgNPs/Ag+的氧化还原存在双面性[22-24],既可氧化AgNPs释放Ag+,又可还原Ag+生成AgNPs,因此,在含有DOM的水环境中AgNPs/Ag+如何转化,环境风险会有多大,目前仍难以预测.
本文首先介绍了DOM促进/抑制AgNPs氧化溶解的机理,然后阐述了DOM还原Ag+形成AgNPs的机理,在此基础上总结了环境因素对DOM介导AgNPs与Ag+相互转化的影响. 最后提出了目前研究存在的不足,并为未来研究方向提供一定的建议.
1. DOM对纳米银转化为银离子的影响(The effect of DOM on the conversion of silver nanoparticles into silver ions)
1.1 DOM抑制AgNPs氧化释放Ag+
DOM可通过氢键、静电引力、疏水性作用、配体交换和离子架桥等方式吸附在无涂层AgNPs、聚乙烯吡咯烷酮包埋的AgNPs(PVP-AgNPs)或柠檬酸盐包埋的AgNPs(Cit-AgNPs)表面,改变其界面特性,进而影响AgNPs的溶解速率及溶解平衡[25-26]). 多数研究表明,DOM在AgNPs表面的吸附抑制了AgNPs的氧化溶解和Ag+的释放,其机理可总结为以下3点:
(a)吸附在AgNPs表面的DOM会屏蔽AgNPs对光子的吸收,进而抑制AgNPs的氧化蚀刻及Ag+释放[27-28]. Zhang等[29]研究发现,由于光屏蔽效应,AgNPs在含有聚苯乙烯微塑料溶液中光氧化释放的Ag+浓度显著低于纯水环境.
(b)阻塞AgNPs表面活性位点并降低其与水体氧化剂(如O2、H2O2和·OH)及H+的反应性[30],这是DOM抑制AgNPs氧化溶解的主要机理. Li等[31]发现,AgNPs的氧化与DOM在其表面的覆盖率呈反比,当全氟羧酸在Cit-AgNPs表面覆盖率为0、20%和50%时,Ag+释放量分别为35.5、31.4、18.8 µg·L−1.
(c)形成物理屏障限制AgNPs表面的Ag+扩散到溶液中,并将氧化释放的Ag+还原为新的AgNPs[32-33]. Fernando等[34]研究发现,HA介导下AgNPs在短时间内释放大量的Ag+,然而在较长时间后,溶液中Ag+会被还原成AgNPs,导致溶液中Ag+浓度降低.
1.2 DOM促进AgNPs氧化释放Ag+
DOM也可促进AgNPs氧化释放Ag+. 如Ostermeyer、Zhang和Yang [35-37]等研究发现,添加600 mg·L−1的牛血清白蛋白、10 mg·L−1的HA和总有机碳含量(Total Organic Carbon, TOC)为10 mg·L−1 C的EPS后,溶液中Ag+含量分别是未加DOM时的2倍、2.5倍和3倍. DOM促进AgNPs氧化溶解的机理可总结为以下3点:
(a)DOM可通过官能团(如—COOH、—OH和—SH等)与AgNPs、Ag2O相互作用形成复合物,削弱Ag—Ag键和Ag—O键,从而促进Ag+的释放[38]. 而且,DOM还可以通过与吸附在AgNPs表面的Ag+络合,使反应(1)平衡右移,促进AgNPs氧化溶解[39].
4Ag0+O2+4H+⇌Ag++2H2O (1) Gondikas等[40]发现,半胱氨酸(cysteine, Cys)能通过—SH与AgNPs释放的Ag+配位结合,促进溶液中AgNPs的氧化溶解.
(b)DOM中含量较多的酸性官能团(如羧基和酚羟基等)在水环境中会电离释放H+,较高的H+浓度会促进AgNPs表面氧化层的溶解,释放Ag+[41]. Zhang等[36]研究发现,HA在溶液中的酸释放促进了AgNPs的氧化溶解. 虽然吸附在AgNPs表面的DOM一定程度上阻碍了AgNPs与O2和H+的相互作用,但吸附层是可渗透的,AgNPs依然可与O2和H+反应[31].
(c)DOM具有很强的光化学活性,其在光照下可生成H2O2、1O2和·OH等强氧化性的活性氧物质(reactive oxygen species, ROS),氧化AgNPs[42]. Tong等[43]证实了光照下聚苯乙烯微塑料产生的1O2和·OH可诱导AgNPs氧化溶解.
DOM介导下促进/抑制AgNPs氧化释放Ag+的机理可总结为图1.
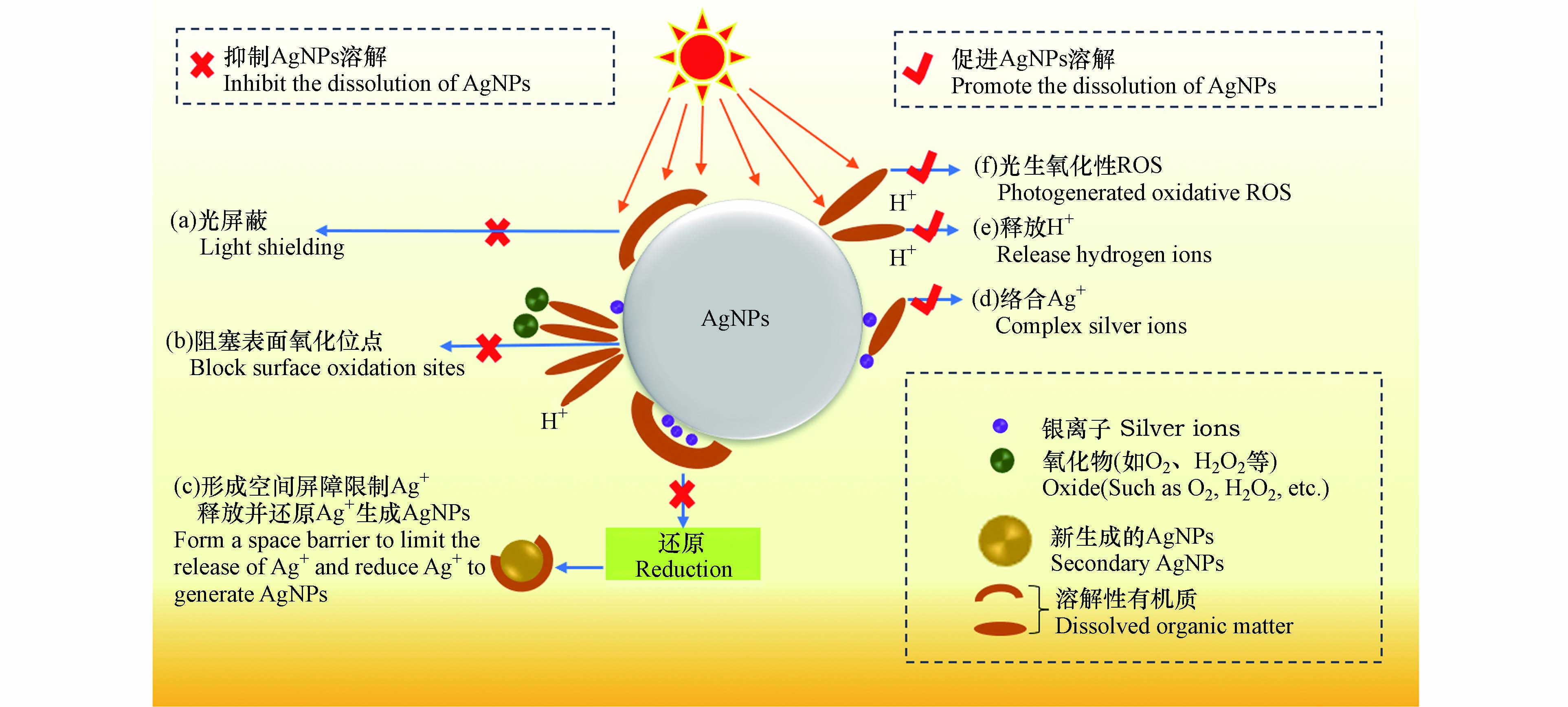 图 1 DOM介导AgNPs转化为Ag+的机理Figure 1. Mechanism of DOM mediated AgNPs transformation to Ag+
图 1 DOM介导AgNPs转化为Ag+的机理Figure 1. Mechanism of DOM mediated AgNPs transformation to Ag+1.3 环境条件对DOM介导AgNPs氧化释放Ag+的影响
1.3.1 DOM/Ag浓度(物质的量)比
DOM/Ag浓度(物质的量)比会影响DOM对AgNPs表面活性位点的占用以及DOM-Ag配体的形成,进而影响AgNPs的氧化溶解. 一般来说,当DOM/Ag较高时,DOM会占据AgNPs更多反应活性位点,抑制AgNPs与O2和H+作用,Ag+释放显著减少[44]. 当系统中萨旺尼河腐殖酸(Suwannee River humic acid, SRHA)的TOC浓度由0增加至6.6 mg·L−1C时,2.78 mg·L−1的AgNPs释放Ag+浓度由1383 µg·L−1降低至339 µg·L−1[45]. 同样,Ag+的释放也随其他组分DOM(如多糖、蛋白质和胞外聚合物等)浓度的增加而显著降低[46-48].
而当DOM/Ag浓度比较低时,DOM占据AgNPs表面活性位点较少,其还可以通过鳌合Ag+而促进AgNPs氧化溶解[35]. Cáceres-Vélez等[49]研究发现,20 mg·L−1的HA促进了10 mg·L−1 AgNPs的溶解(DOM/Ag=2),而抑制了0.5、1、3 mg·L−1 AgNPs (DOM/Ag > 6)的溶解. Boehmler等[50]同样发现,当牛血清白蛋白浓度由0增加至2 nmol·L−1时,其可通过—SH鳌合Ag+使得粒径为10 nm的 Cit-AgNPs的溶解速率增加1.5倍.
1.3.2 DOM种类
DOM是非均质的混合物,其种类复杂,性质多变,如元素含量、官能团和芳香性会存在差异. 因此不同DOM种类作用下,AgNPs的氧化溶解差异明显. Gunsolus等[51]研究发现,小马湖富里酸(Pony Lake fluvic acid, PLFA)对Cit-AgNPs氧化溶解的抑制作用强于同样浓度的SRHA和萨旺尼河富里酸(Suwannee River fluvic acid, SRFA),进一步探究发现DOM介导下Ag+释放量与DOM的S、N含量呈负相关. 高S、N元素的DOM对AgNPs/Ag+有很强的亲和性,因此会占据更多AgNPs表面活性位点进而抑制AgNPs氧化溶解[52]. 而对Ag+有强亲和力的官能团也可促进AgNPs的氧化溶解. Liu[39]和Gondikas[40]均研究发现,DOM可通过—SH络合Ag+从而促进AgNPs的氧化溶解. 芳香性较强的DOM不会占据太多AgNPs的活性位点,进而增强AgNPs的反应性. Pokhrel等[53]研究发现,溶液中较高芳香性的风化褐煤腐殖酸(Leonardite humic acid, LHA)作用下Ag+释放量是无LHA条件下的4—5倍.
1.3.3 共存离子及浓度
离子浓度会影响DOM的分子结构,改变DOM在AgNPs表面的吸附[54],进而影响DOM介导的AgNPs氧化释放Ag+. 较低离子浓度下,DOM分子结构较膨胀,DOM在AgNPs表面的吸附会占据更多氧化位点,且许多环境阴离子(
CO2−3 SO2−4 PO3−4 在较高离子浓度环境中,DOM分子结构紧凑,吸附在AgNPs表面的DOM占据表面氧化位点较少,并且与释放的Ag+络合促进AgNPs氧化溶解[49, 57]. 高浓度的Cl−对AgNPs氧化溶解的影响最为显著[58]. 当Cl−/Ag <26750时,反应主要生成AgCl(s)沉淀;当Cl−/Ag ≥26750时,主要生成溶解性的配合物
AgCl1−xx AgCl2−3 AgCl3−4 AgCl1−x 阳离子(如Na+、Ca2+、Mg2+)会促使AgNPs聚集,减小比表面积,从而抑制AgNPs氧化释放Ag+[60]. 虽然有研究证明DOM抑制了AgNPs的聚集,但在二价阳离子(如Ca2+、Mg2+)作用下,吸附在AgNPs表面的DOM会通过络合Ca2+、Mg2+而桥连,发生更强烈的聚集,使Ag+释放量显著降低[61]. Huang等[62]研究发现, AgNPs在HA与Ca2+共同作用下氧化释放Ag+的浓度依次低于其在HA作用下、Ca2+作用下和纯水中的Ag+释放量.
1.3.4 pH
pH会影响AgNPs向Ag+的转化过程,且会改变DOM在水体中的分子结构,影响DOM的光化学反应,因此pH显然会影响DOM介导下AgNPs的氧化溶解. 在低pH条件下溶液中H+含量较高,会促进AgNPs的氧化溶解平衡(反应式1)右移[63];并且低pH时DOM的分子结构较紧凑,占据AgNPs氧化位点较少,削弱了其对Ag+释放的抑制作用[64];此外,相比碱性条件下,酸性溶液中DOM光生氧化性自由基显著增加[65]. 因此,低pH条件下促进了AgNPs的氧化溶解;反之,高pH条件下会抑制Ag+的释放.
1.3.5 光照
AgNPs具有很强的光吸收能力,短时间光照会促进AgNPs的光裂解及氧化蚀刻,迅速释放Ag+. Shi等[28]研究发现,光照下三磷酸腺苷包埋的AgNPs会在短时间内(≤1 h)迅速氧化,其Ag+释放量显著高于黑暗条件. 但长时间光照会破坏AgNPs表面涂层,使AgNPs失稳聚集,表面活性位点减少,进而氧化速率降低. Yu等[32]研究发现在短时间光照(≤ 12 h)下,Cit-AgNPs的平均溶解速率常数是长时间光照(70 h)下的4.5倍,且AgNPs发生明显聚集.
光照会促进DOM介导的AgNPs氧化或降低DOM对AgNPs氧化溶解的抑制作用. Rong等[66]发现,当SRFA的浓度为0、5、10 mg·L−1时,10 min光照可使Cit-AgNPs氧化率分别比黑暗条件下提高了3.4%、4.6%和6.0%. Yu等[67]研究发现,黑暗条件下,TOC浓度为5 mg·L−1 C的SRHA使1.02 mg·L−1的AgNPs在20 h后的Ag+释放量由830 µg·L−1降至100 µg·L−1;而20 h的光照后,Ag+释放量由738 µg·L−1降至225 µg·L−1.
2. DOM对银离子转化为纳米银的影响(The effect of DOM on the conversion of silver ions into silver nanoparticles)
2.1 DOM介导Ag+还原生成AgNPs
在黑暗/光照条件下,DOM均可介导Ag+还原成AgNPs,其还原机理可概括为以下3点,见图2
 图 2 DOM 介导下Ag+转化为AgNPs的机理Figure 2. Mechanism of DOM mediated Ag+ transformation into AgNPs
图 2 DOM 介导下Ag+转化为AgNPs的机理Figure 2. Mechanism of DOM mediated Ag+ transformation into AgNPs(1)黑暗条件下的自催化. 溶液中游离的Ag+通过沉积在Ag2O/AgNPs簇表面进而提高其氧化还原电位(游离状态:Ψ(Ag+/Ag0 = −1.8 V);吸附在固态表面的Ag+:Ψ(Ag+/Ag0 = 0.7996 V),进而使DOM还原Ag+的反应在热力学上是可行的,如式(2,3)所示[68].
Ag+−Solidsurface+HA(red)→Ag0+HA(ox)E0=∼0.1Vvs.NHE (2) Ag+−Solidsurface+FA(red)→Ag0+FA(ox)E0=∼0.3Vvs.NHE (3) 自催化过程可以被描述为以下几个步骤[69]:
2Ag++2OH−→Ag2O+H2Ofast (4) Ag2O+(Ag+)n→Ag2O−(Ag+)nfast (5) Ag2O−(Ag+)n+DOM⇌Ag2O−(Ag+)n−DOMKad (6) Ag2O−(Ag+)n−DOM⇌Ag2O−(Ag+)n−1+Ag0+DOM−(ox)Kred (7) DOM首先发生脱质子化,然后通过静电作用/络合作用与Ag+结合[70-71],DOM-Ag复合物通过还原性官能团(如—COOH、—OH、—SH、醛基和酮基等)将e−转移给Ag+,生成AgNPs[39, 72].
(2)光照生成还原性自由基. 在光照条件下,DOM充当光吸收体,产生强还原性自由基(如
e−aq O•−2 O•−2 O•−2 O•−2 (3)光照条件下配体-金属电荷转移(ligand-to-metal charge transfer, LMCT). 当Ag+吸附到DOM表面后,光照促进DOM配体将e−转移至Ag+,进而生成AgNPs[75]. Hou等[76]研究发现,HA光还原Ag+生成AgNPs的速率随着溶液中Na+浓度的增加而显著降低,证实了Na+通过竞争HA表面金属离子结合位点进而抑制了HA通过LMCT还原Ag+.
2.2 环境条件对DOM介导Ag+还原生成AgNPs的影响
2.2.1 DOM浓度
DOM介导的AgNPs形成速率可由以下式子表示[74]:
r=k[Ag+][DOM] (8) r=(dA/dt)t→0 (9) 其中,r为AgNPs形成速率,单位h−1;[Ag+]为Ag+浓度,单位mg·L−1;[DOM]为DOM浓度,单位mg·L−1;A为AgNPs表面等离子体共振(Surface Plasmon Resonance, SPR)峰吸光度.
由于DOM介导的AgNPs/Ag+氧化还原是同时进行的,当DOM浓度一定时,需要足够的Ag+浓度才能实现AgNPs团簇的快速生长,这个浓度称为临界诱导浓度. 低于临界诱导浓度时,Ag+还原生成AgNPs不稳定,会马上被氧化,无法实现AgNPs团簇的生长[77];而高于该浓度时,AgNPs生成速率与DOM浓度呈正相关. Xiong等[78]研究发现,初始浓度为30 µg·L−1的Ag+无法被EPS被还原成AgNPs;而Yin等[79]研究发现,初始浓度为0.2 mmol·L−1的Ag+会随DOM浓度的升高而加速转化为AgNPs.
2.2.2 DOM组分
不同组分DOM的性质差异(比如对Ag+的吸附能力、芳香性、分子量和官能团)会影响DOM介导的Ag+还原成AgNPs. 高Ag+吸附性的DOM能更好地通过LMCT还原Ag+. Liu等[75]比较了吸附性很强的溶解性黑炭和吸附性较弱的SRHA对Ag+的光还原能力,发现溶解性黑炭介导的AgNPs生成速率显著高于SRHA. Nie等[80]研究发现,DOM的芳香成分会限制Ag+与DOM的还原性官能团结合. 低芳香性的泥炭HA 和泥炭FA可在24 h内介导Ag+还原成AgNPs;而同等浓度的高芳香性商用HA需要120 h才能介导Ag+还原成AgNPs. 由于低分子量的DOM光屏蔽能力较弱,Guo等[81]研究发现,分子量<3 kDa的DOM作用下Ag+光还原效率远高于>3 kDa DOM. 此外,DOM官能团的还原性会影响Ag+的还原. Nie等[80]研究发现,酚基比羰基具有更强的还原性,其利用NaBH4将羰基转化为酚基后,发现DOM介导的AgNPs生成速率和浓度均显著增加.
2.2.3 共存离子及浓度
环境共存离子会影响Ag+还原为AgNPs. 其中环境中常见的阴离子如
CO2−3 SO2−4 PO3−4 SO2−4 CO2−3 PO3−4 此外,一些具有光敏性的银盐(如AgCl)被认为是AgNPs的前体物质,在光照下其会激发出从价带跃迁到导带的电子e−,e−通过界面电子转移至AgCl表面的Ag+,从而生成AgNPs[83]. Cl−介导下Ag+还原可以由以下式子表示[84]:
AgCl(aq)+hѵ→h++e− (10) Ag++e−→Ag0 (11) 离子浓度对Ag+转化成AgNPs影响很大. 当离子浓度较低时(如Cl−/Ag+ ≤5000),Cl−与Ag+结合主要生成AgCl,在光照下可被还原成AgNPs;而在高离子浓度下(Cl−/Ag+ > 1.8×105),则AgCl会转化为光活性较弱的
AgCl1−xx 环境中常见的阳离子(如Ca2+、Mg2+、Na+、Fe2+和Fe3+)也会影响环境中Ag+转化为AgNPs. 如在光照下DOM会与Fe2+/Fe3+形成氧化还原循环,催化DOM对Ag+的还原,如化学式(12—14)所示[85]. Yin等[86]研究表明,添加10 µmol·L−1的Fe2+/Fe3+可使DOM-Ag+溶液中还原生成的AgNPs浓度显著增加.
DOM+O2hѵ→DOM·++O·−2 (12) Fe3++O·−2→Fe2++O2 (13) Fe2++Ag+→Fe3++Ag0 (14) Ca2+、Mg2+和Na+等阳离子的存在会竞争DOM表面的吸附位点,抑制DOM对Ag+的吸附及还原,并且这些阳离子会压缩AgNPs表面的双电层,使其失稳聚集,导致溶液中AgNPs浓度降低[87]. Yin等[79]研究发现,溶液中 Ca2+浓度越高则DOM还原Ag+生成的AgNPs浓度越低,且AgNPs的粒径显著增大. 而其他贵重金属离子如(Au3+)对应的纳米粒子具有较高的内聚能,成核速率较快,因此这类金属会优先与DOM作用并生成相应的纳米粒子,吸附Ag+在其表面并提供成核位点,促进了Ag+转化为AgNPs[88].
2.2.4 pH
DOM还原Ag+受溶液pH影响显著. 随着pH的升高,DOM的氧化还原电位逐渐降低,这促进了Ag+转化为AgNPs[74];而且高pH条件下DOM的酸性官能团去质子化,其表面电性更负,带正电的Ag+与带负电的DOM之间的强静电引力增强了Ag+与DOM络合,促进了光照条件DOM通过LMCT途径还原Ag+或黑暗条件下DOM通过给电子官能团还原Ag+[76].
2.2.5 光照
黑暗条件下DOM还原Ag+需要很高的活化能,而光照可以加快DOM介导的Ag+还原生成AgNPs速率[89]. 光照可作为AgNPs生成的催化剂,既会诱导DOM产生
O•−2 e−aq 鉴于研究DOM介导AgNPs/Ag+转化会得到DOM促进/抑制AgNPs氧化释放Ag+以及还原Ag+生成AgNPs 3种不同的结论,可能源于这些研究中设计的特定实验条件影响了DOM对
Ag0⇄Ag+ 表 1 DOM介导下的AgNPs氧化或Ag+还原条件汇总Table 1. Summary of AgNPs oxidation or Ag+ reduction conditions mediated by DOM离子Ion Ag初始浓度Initial concentration of Ag species DOM种类及初始浓度DOM species and initial concentration 光照Illumination state pH 时间Time 结果Result 参考文献Reference 0.08—16.67 mg·L−1 盐度天然水 5 mg·L−1 AgNPs 3.8—7.2 mg·L−1 C 天然水体DOM 黑暗 — 7 d 高盐度↑低盐度↓ Li et al., 2020[57] 0、0.01、0.1、0.3 mol·L−1 NaCl 1 mg·L−1 AgNPs 1.5—2 mg·L−1 C 天然水体DOM — 5.5 168 h ↑/↓ Zhao et al., 2021[56] — 20 mg·L−1 Ag+ 5—20 mg·L−1 C HA 光暗交替 7 96 h 光: ←;暗:↑ Liu et al., 2020[92] 7 mmol·L−1 NaHCO310 mmol·L−1 NaNO3 8 µmol·L−1 AgNPs 400 µmol·L−1 Cys — 7.5— 8.1 50 h ↑ Gondikas et al., 2012[40] 280 mg·L−1 CaCO3 10 mg·L−1 AgNPs 2—20 mg·L−1 LHA — 7 4 h ↑ Pokhrel et al., 2014[53] — 1.08 mg·L−1 AgNPs 0—10 mg·L−1 C EPS 132 W·m−2 Xe灯 7.1 72 h ↑ Yang et al., 2021[37] 柠檬酸盐缓冲液 1 mg·L−1 AgNPs 0—2 nmol·L−1 BSA — 6.5 4 h ↑ Boehmler et al., 2020[50] 硼酸盐、硝酸根 20 mg·L−1 AgNPs 20 mg·L−1 PS MPs 550 W·m−2 Xe灯/黑暗 5.5 72 h ↑ Tong et al.,2022[43] 硼酸盐、硝酸根 20 mg·L−1 AgNPs 20 mg·L−1 PS MPs 550 W·m−2 Xe灯/黑暗 8.5 24 h ↓ Zhang et al.,2021[29] 0.1 mmol·L−1 KH2PO4 500 µg·L−1 AgNPs 0.1—10 µmol·L−1 Cys — 7 72 h ↓ Afshinnia et al., 2018[52] 人工介质ASW38 1 µg·L−1 AgNPs 0—50 µmol·L−1 BSA 自然光 8 15 h ↓ Levak et al., 2017[47] — 1.02 mg·L−1 AgNPs 5 mg·L−1 C SRHA 550 W·m−2 Xe灯/黑暗 5—8.3 48 h ↓ Yu et al., 2014[67] 0.1 mol·L−1 KH2PO4 5 mg·L−1 AgNPs 10 mg·L−1 PLFA、SRHA、 SRFA — 7 5 h ↓ Gunsolus et al., 2015[51] 硼酸盐缓冲液 1—1000 µg·L−1 Ag+ 25 mg·L−1 SRHA 黑暗 6—9 2 d ← Dong et al., 2019[82] 0—150 mg·L−1 NaCl 5 mg·L−1 Ag+ 20、40 mg·L−1 EPS 荧光灯/黑暗 8 36 h ← Xiong et al., 2021[78] 0—10 µmol·L−1 Fe2+/Fe3+ 1 mmol·L−1 Ag+ 30 mg·L−1 DOM 550 W·m−2Xe灯 6.3 8 h ← Yin et al., 2017[86] 12.7 mg·L−1 NaCl 10 mg·L−1 Ag+ 50 mg·L−1 DBC/SRHA 50 W Xe灯 7.3 2 h ← Liu et al., 2021[75] 磷酸盐–硼酸盐缓冲液 1 mmol·L−1 Ag+ 15—100 mg·L−1 HA 黑暗 8 5 d ← Nie et al., 2020[80] — 0.2 mmol·L−1 Ag+ 20 mg·L−1 C EPS Xe灯/黑暗 7.6 16 h ← Zhang et al., 2016[90] 注:↑,↓分别表示促进和抑制Ag+释放;←表示促进Ag+还原生成AgNPs;-为文献未提及该因素;PS MPs代表聚苯乙烯微塑料;DBC代表溶解性黑炭;BSA代表牛血清白蛋白. Note:↑ and ↓ respectively promote and inhibit the release of Ag+;← means promoting Ag+ reduction to generate AgNPs; - is not mentioned in the literature; PS MPs stands for polystyrene microplastics; DBC stands for dissolved black carbon; BSA stands for bovine serum albumin. | Show Table DownLoad:
CSV
DownLoad:
CSV
3. 结语(Conclusions and recommendations for further work)
目前研究表明DOM可通过占据氧化位点、还原Ag+、光衰弱来抑制AgNPs氧化,或释放H+、络合Ag+和光生氧化性ROS来促进Ag+释放,还可以通过自催化、光生还原性ROS和LMCT等途径将Ag+还原成AgNPs. 而环境因素(如DOM组分及浓度、AgNPs/Ag+浓度、离子、光照和pH等)会影响AgNPs/Ag+的转化过程,因此判断真实环境中DOM介导下AgNPs/Ag+的转化方向是比较困难的. 鉴于目前银基抗菌产品的市场在世界范围内进一步扩大,这将导致AgNPs与Ag+释放到生态环境中,并对生态系统和人体健康造成潜在危害,因此迫切需要更科学的理论基础去预测和评估其环境及健康风险.
目前DOM存在下AgNPs氧化/Ag+还原是AgNPs研究领域的热点之一,研究仍存在一些问题值得今后进一步研究:
(1)从表1可以看出,反应条件会显著影响DOM介导的AgNPs氧化及Ag+还原过程,区分和量化各种反应条件对这些过程的影响[26],将有利于判定AgNPs与Ag+转化的进行.
(2)光照对DOM介导的纳米银/银离子转化过程具有重要的影响,光照既可促进纳米银转化为银离子,也可促进Ag+还原生成纳米银,因此难以预测在现实环境中光暗交替下纳米银/银离子转化,未来可加强这方面的研究.
(3)目前关于DOM与AgNPs相互作用的研究存在使用的DOM模型简单(常将HA/FA作为DOM模型)的问题[93]. 未来研究中应考虑研究其他DOM组分(如溶解性黑炭[94]和人工合成类DOM)与AgNPs/Ag+相互作用的效应及作用机理.
(4)AgNPs粒径、形貌和表面包被等对AgNPs氧化溶解的影响已研究得较为透彻,但目前尚不清楚在DOM存在的条件下AgNPs粒径、形貌和表面包被会怎样影响DOM与AgNPs的相互作用及AgNPs的氧化溶解. 今后可研究上述因素与DOM耦合作用下的AgNPs氧化溶解,并深入探究其机理.
-
表 1 经呼吸进入人体的暴露参数
Table 1. Exposure parameters of breathing into human body
重金属元素Heavy metal element 性质Characteristic 致癌因子/((kg·d)· μg−1)SF 参考剂量/(μg·(kg·d) −1)RfD As 致癌 2.01×10−2 — Cd 致癌 8.40×10−3 — Ni 致癌 1.19×10−3 — Al 非致癌 — 4.00×10−1 Cu 非致癌 — 2.00 Mn 非致癌 — 3.00×10−1 Pb 非致癌 — 4.30×10−1 Se 非致癌 — 1.00 Zn 非致癌 — 1.00×10−2
下载: 导出CSV
表 2 重金属经呼吸进人体的剂量-反应参数
Table 2. Dose-response parameters of heavy metals breathing into human body
人群Crowd 呼吸速率/(m3·d−1)IR 体重/kgBW 暴露持续时间/dED 致癌暴露时间/dAT-Carcinogenesis 非致癌暴露时间/dAT-Non-carcinogens 儿童 8.70 36.00 18.00 70.00 18.00 成年女性 14.17 57.30 30.00 70.00 30.00 成年男性 19.02 66.20 30.00 70.00 30.00
下载: 导出CSV
表 3 三亚市PM2.5重金属元素的质量浓度
Table 3. Mass concentration of PM2.5 heavy metal elements in Sanya
元素Element 2019年均值/(μg·m−3)2019 mean 6月均值/(μg·m−3)June mean 10月均值/(μg·m−3)October mean 12月均值/(μg·m−3)December mean 标准限值/(μg·m−3)Standard limit As 4.30×10−3 — 4.00×10−4 4.90×10−3 6.00×10−3 Cr 7.70×10−3 7.60×10−3 8.50×10−3 7.40×10−3 2.50×10−5 Ni 3.50×10−3 3.00×10−3 4.00×10−3 3.60×10−3 — Cu 1.74×10−2 1.44×10−2 1.64×10−2 2.04×10−2 — Pb 1.53×10−2 1.19×10−2 1.75×10−2 1.68×10−3 5.00×10−1 Se 1.60×10−3 1.00×10−3 1.80×10−3 2.00×10−3 — Zn 3.23×10−2 2.04×10−2 3.79×10−2 3.92×10−2 —
下载: 导出CSV
表 4 南海周边大气颗粒物PM受体模型源解析比较表
Table 4. Comparison of source analysis of PM receptor models around the South China Sea
采样点Sampling site 粒径Size 采样时间Date 模型Model 源解析结果Source analysis results 参考文献References 中国海南省三亚市 PM2.5(n=34) 2012, 1.6 — 2.8; 2013.6.6—7.25 PMF 1.生物质燃烧(23.2%);2.机动车(37.9%); 3.燃煤(22.6%); 4.其他(16.3%); [8] PM2.5(n=90) 2019, 6 — 2019,12 PMF 1. 海洋源(24.9%); 2. 生物质燃烧源(20.8%); 3. 工业源(20.5%); 4. 土壤源(19%); 5. 交通源(14.8%) 本研究 中国海南省三沙市 TSP(n=73) 2014.3—2015.2 PMF 1. 海盐(46.6%); 2. 土壤尘(11.9%); 3. 二次无机气溶胶(30.1%); 4. 海洋排放(11%) [11] 中国海南省海口市 PM2.5(n=38) 2011.12.26 — 2012.6.3; 2012.4.17—26 CMB 1. 扬尘(14.9%); 2. 机动车尾气(17.5%); 3. SO42-(9.5%); 4. 海盐(3%); [9] PM10(n=76) 1. 扬尘(23.6%); 2. 机动车尾气(35%); 3. SO42-(15.7%); 4. 海盐(8%); 中国台湾省高雄港 PM2.5(n=28) 2018.5— 2019.1 PMF 1. 船舶排放(15.6%);2. 二次无机气溶胶(24%);3. 机动车尾气(12.2%);4. 海盐(20.7%);5. 海盐和生物质燃烧(14.4%);6. 重油, 生物质燃烧(13.2%) [30] 菲律宾马尼拉 PM2.5(n=28) 2018.5— 2019.1 PMF 1. 道路扬尘, 燃煤(17.4%);2. 船舶排放, 机动车(19.1%);3. 工业(17.7%);4. 机动车(12.6%);5. 二次气溶胶,土壤, 生物质燃烧(21.3%);6. 海盐, 生物质燃烧(11.8%) [30] 马来西亚八打灵 PM2.5(n=247) 2017.1.11—2018.2.19 PMF 1. 混合冶炼工业和道路扬尘(5.6);2. 矿物尘(7.2%);3. 海盐(7.4%);4. 冶金工业(5.1%);5. 农业(19.2%);6. 制造业 (12%);7. 二次无机气溶胶,交通 (28.5%);8.生物质燃烧(15.2%) [31]
下载: 导出CSV
表 5 三亚市大气PM2.5中重金属元素经呼吸途径对人群的年均超额危险度
Table 5. Average annual excess risk of heavy metal elements in atmospheric PM2.5 in Sanya City to the population through respiratory pathway
元素Elements 年均超额危险度R(无量纲) Annual excess risk R(No unit) 儿童Child 成年女性Adult women 成年男性Adult men As 7.64×10−8 1.30×10−7 1.51×10−7 Cr 3.85×10−7 6.57×10−7 7.63×10−7 Ni 3.68×10−9 6.27×10−9 7.28×10−9 Al 2.21×10−9 2.26×10−9 2.62×10−9 Cu 3.01×10-11 3.08×10-11 3.58×10-11 Mn 5.46×10-10 5.58×10-10 6.49×10-10 Pb 1.23×10-10 1.26×10-10 1.46×10-10 Se 5.40×10-12 5.52×10-12 6.42×10-12 Zn 1.12×10-11 1.14×10-11 1.33×10-11
下载: 导出CSV
-
[1] 曹军骥. PM2.5与环境[M]. 北京: 科学出版社, 2014. CAO J J. PM2.5 and environment[M]. Beijing: Science Press, 2014 (in Chinese) .
[2] 周元. 努力建设以人民为中心的海南自贸港[N]. 海南日报, 2021-07-27(A02). ZHOU Y. Strive to build people-centered Hainan Free Trade Port[N]. Hainan Daily, 2021 /7 /27 / A02 edition (in Chinese).
[3] 周家茂, 赵由之, 刘随心, 等. 三亚冬季大气PM2.5及碳气溶胶特征与来源分析 [J]. 地球环境学报, 2012, 3(5): 1060-1065. ZHOU J M, ZHAO Y Z, LIU S X, et al. Characteristic and source identifications of PM2.5 and carbonaceous aerosol at Sanya durning 2011 winter [J]. Journal of Earth Environment, 2012, 3(5): 1060-1065(in Chinese).
[4] ZHOU J M, HO S, CAO J J, et al. Chemical characterization of PM2.5 from a southern coastal city of China: Applications of modeling and chemical tracers in demonstration of regional transport [J]. Environmental Science and Pollution Research International, 2018, 25(21): 20591-20605. doi: 10.1007/s11356-018-2238-1 [5] TIAN J, WANG Q Y, HAN Y M, et al. Contributions of aerosol composition and sources to particulate optical properties in a southern coastal city of China [J]. Atmospheric Research, 2020, 235: 104744. doi: 10.1016/j.atmosres.2019.104744 [6] WANG Q Y, LIU H K, WANG P, et al. Optical source apportionment and radiative effect of light-absorbing carbonaceous aerosols in a tropical marine monsoon climate zone: The importance of ship emissions [J]. Atmospheric Chemistry and Physics, 2020, 20(24): 15537-15549. doi: 10.5194/acp-20-15537-2020 [7] WANG P, HAN C, ZHAO Y Z, et al. Characterization of PM2.5 mass concentration in the onshore of Sanya, China [J]. Journal of Atmospheric Science Research, 2020, 3(2): 32-38. [8] WANG J Z, HO S, CAO J J, et al. Characteristics and major sources of carbonaceous aerosols in PM2.5 from Sanya, China [J]. The Science of the Total Environment, 2015, 530/531: 110-119. doi: 10.1016/j.scitotenv.2015.05.005 [9] FANG X Z, BI X H, XU H, et al. Source apportionment of ambient PM10 and PM2.5 in Haikou, China [J]. Atmospheric Research, 2017, 190: 1-9. doi: 10.1016/j.atmosres.2017.01.021 [10] LIU B S, ZHANG J Y, WANG L, et al. Characteristics and sources of the fine carbonaceous aerosols in Haikou, China [J]. Atmospheric Research, 2018, 199: 103-112. doi: 10.1016/j.atmosres.2017.08.022 [11] XIAO H W, XIAO H Y, LUO L, et al. Atmospheric aerosol compositions over the South China Sea: Temporal variability and source apportionment [J]. Atmospheric Chemistry and Physics, 2017, 17(4): 3199-3214. doi: 10.5194/acp-17-3199-2017 [12] GENG X F, MO Y Z, LI J, et al. Source apportionment of water-soluble brown carbon in aerosols over the northern South China Sea: Influence from land outflow, SOA formation and marine emission [J]. Atmospheric Environment, 2020, 229: 117484. doi: 10.1016/j.atmosenv.2020.117484 [13] SONG J W, ZHANG Y Y, ZHANG Y L, et al. A case study on the characterization of non-methane hydrocarbons over the South China Sea: Implication of land-sea air exchange [J]. The Science of the Total Environment, 2020, 717: 134754. doi: 10.1016/j.scitotenv.2019.134754 [14] ZHANG Y L, LI J, ZHANG G, et al. Radiocarbon-based source apportionment of carbonaceous aerosols at a regional background site on Hainan Island, South China [J]. Environmental Science & Technology, 2014, 48(5): 2651-2659. [15] XIAO H W, XIE L H, LONG A M, et al. Use of isotopic compositions of nitrate in TSP to identify sources and chemistry in South China Sea [J]. Atmospheric Environment, 2015, 109: 70-78. doi: 10.1016/j.atmosenv.2015.03.006 [16] 郑玫, 张延君, 闫才青, 等. 中国PM2.5来源解析方法综述 [J]. 北京大学学报(自然科学版), 2014, 50(6): 1141-1154. ZHENG M, ZHANG Y J, YAN C Q, et al. Review of PM2.5 source apportionment methods in China [J]. Acta Scientiarum Naturalium Universitatis Pekinensis, 2014, 50(6): 1141-1154(in Chinese).
[17] 魏青, 陈文怡, 金麟先. 枣庄市大气PM2.5重金属元素健康风险评价及污染来源解析 [J]. 中国粉体技术, 2020, 26(6): 69-78. WEI Q, CHEN W Y, JIN L X. Health risk assessment and source analysis of heavy metal elements in PM2.5 in Zaozhuang City [J]. China Powder Science and Technology, 2020, 26(6): 69-78(in Chinese).
[18] 单慧, 欧阳钏, 柯鸿阳, 等. 西北地区典型城市PM2.5中重金属污染特征及健康风险评价 [J]. 中国公共卫生, 2022, 38(4): 476-480. doi: 10.11847/zgggws1127718 SHAN H, OUYANG C, KE H Y, et al. Heavy metals in PM2.5 in four metropolitan cities in Northwest China: Pollution characteristics and health risk assessment [J]. Chinese Journal of Public Health, 2022, 38(4): 476-480(in Chinese). doi: 10.11847/zgggws1127718
[19] CAO J J, WU F, CHOW J C, et al. Characterization and source apportionment of atmospheric organic and elemental carbon during fall and winter of 2003 in Xi'an, China [J]. Atmospheric Chemistry and Physics, 2005, 5(11): 3127-3137. doi: 10.5194/acp-5-3127-2005 [20] WU F, ZHANG D Z, CAO J J, et al. Soil-derived sulfate in atmospheric dust particles at Taklimakan desert [J]. Geophysical Research Letters, 2012, 39(24): L24803. [21] ANTONY CHEN L W, CAO J J. PM2.5 source apportionment using a hybrid environmental receptor model [J]. Environmental Science & Technology, 2018, 52(11): 6357-6369. [22] USA EPA. Integrated risk information system [DB/OL] [2019-02-03]https://www.epa.gov/research-states/integrated-risk-information-system-iris-webinar-archive. [23] ATSDR. Agency for Toxic Substances and Disease Registry[DB/OL]. [2019-02-03] . [24] 范逸飞, 陈秀玲, 方滋婧, 等. 漳州市城市公园灰尘重金属来源及健康风险评价 [J]. 地球环境学报, 2021, 12(1): 104-120. FAN Y F, CHEN X L, FANG Z J, et al. Heavy metal sources and health risk assessment of dust in Zhangzhou urban parks [J]. Journal of Earth Environment, 2021, 12(1): 104-120(in Chinese).
[25] 郑乃嘉, 谭吉华, 段菁春, 等. 大气颗粒物水溶性重金属元素研究进展 [J]. 环境化学, 2014, 33(12): 2109-2116. doi: 10.7524/j.issn.0254-6108.2014.12.005 ZHENG N J, TAN J H, DUAN J C, et al. Research progress on water-soluble heavy metal in atmospheric particulate mattters [J]. Environmental Chemistry, 2014, 33(12): 2109-2116(in Chinese). doi: 10.7524/j.issn.0254-6108.2014.12.005
[26] YAO X H, CHAN C K, FANG M, et al. The water-soluble ionic composition of PM2.5 in Shanghai and Beijing, China [J]. Atmospheric Environment, 2002, 36(26): 4223-4234. doi: 10.1016/S1352-2310(02)00342-4 [27] LEE Y N, WEBER R, MA Y, et al. Airborne measurement of inorganic ionic components of fine aerosol particles using the particle-into-liquid sampler coupled to ion chromatography technique during ACE-Asia and TRACE-P [J]. Journal of Geophysical Research:Atmospheres, 2003, 108(D23): 8646. doi: 10.1029/2002JD003265 [28] MA Y, WEBER R J, LEE Y N, et al. Characteristics and influence of biosmoke on the fine-particle ionic composition measured in Asian outflow during the Transport and Chemical Evolution Over the Pacific (TRACE-P) experiment[J]. Journal of Geophysical Research: Atmospheres, 2003, 108(D21),doi:10.1029/2002JD003128. [29] 海南日报. 2020 年海南省生态环境状况公报[N]. 2021 年/6 月/5 日/第A06 版. Hainan Daily. Ecological and Environmental Status Report of Hainan Province in 2020 [N] . 2021/26/25/A06 edition (in Chinese).
[30] TSENG Y L, WU C H, YUAN C S, et al. Inter-comparison of chemical characteristics and source apportionment of PM2.5 at two harbors in the Philippines and Taiwan [J]. Science of the Total Environment, 2021, 793: 148574. doi: 10.1016/j.scitotenv.2021.148574 [31] HASSAN H, LATIF M T, JUNENG L, et al. Chemical characterization and sources identification of PM2.5 in a tropical urban city during non-hazy conditions [J]. Urban Climate, 2021, 39: 100953. doi: 10.1016/j.uclim.2021.100953 [32] ROVIRA J, ROIG N, NADAL M, et al. Human health risks of formaldehyde indoor levels: An issue of concern [J]. Journal of Environmental Science and Health. Part A, Toxic/Hazardous Substances & Environmental Engineering, 2016, 51(4): 357-363. 期刊类型引用(3)
1. 彭晓,杨萌,王笑欢,娄英斌,张玉凤,曹姗姗. 大连市PM_(2.5)中重金属的污染特征、来源及健康风险评价. 生态毒理学报. 2024(04): 294-310 .  百度学术
百度学术
2. 郭昭伟,王平,路放,赵由之,丁文慈,黄鼎,杜嘉欣. 海南生物质燃烧源排放清单的时空配分. 生态学杂志. 2024(11): 3470-3477 . 百度学术
3. 吴凯章,刘明,罗中华,陈来国,蔡立梅,王安侯,郑昱,陆海涛. 大宝山矿区周边大气重金属来源与风险评估. 中国环境科学. 2023(12): 6270-6280 . 百度学术
其他类型引用(2)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 3175
- HTML全文浏览数: 3175
- PDF下载数: 46
- 施引文献: 5
