






-
近年来,污水处理后作为再生水回用逐渐得到重视。2021年,国家发布《关于推进污水资源化利用的指导意见》指出,污水经无害化处理达到特定水质标准后,可作为再生水用于居民生活、生态补水、回灌地下水等。但诸多研究表明,再生水中仍能频繁检出微量抗生素等难降解有机污染物,呈现浓度低、高稳定性、高毒性和易生物富集等特性,进入水环境可诱导环境菌群产生抗药性,甚至导致超级抗药细菌的出现,对人体和生态环境产生巨大的潜在风险[1-4]。开发经济高效的污水中抗生素等微量污染物的去除技术,仍是目前污水处理领域亟需解决的重要问题之一。
高级氧化技术(advanced oxidation processes, AOPs)能够产生具有强氧化力的活性物种,可以高效降解去除上述微量高风险有机物,近年来引起了广泛的关注[5-6]。其中,基于过硫酸盐(PMS)或过二硫酸盐(PDS)活化的高级氧化技术所产生的硫酸根自由基(SO4·−)、羟基自由基(·OH)、单线态氧(1O2)等ROS,具有广谱的难降解有机污染物去除能力,且活化方法较为简单,较之芬顿体系表现出较大优势[7]。在众多活化PMS的方法中,过渡金属因其运行成本低、效率高而被广泛使用[8-9]。其中,钴(Co)被认为是最有效的PMS活化金属[10-11],然而,在PMS活化过程中,Co2+的浸出对人体健康和生态环境构成了严重威胁,因为Co2+即使在非常低的浓度下也具有高毒性和潜在致癌性[12-13]。因此,寻求绿色的过渡金属制作高效催化剂,对基于过硫酸盐活化的高级氧化技术发展尤为重要。
类水滑石(layered double hydroxide,LDHs)又称层状双金属氧化物,是一类阴离子插层的二维层状无机材料,具有层板元素和层间阴离子种类可变、比例可调、金属阳离子均匀分散等特性,在多相催化中具有广阔的应用前景。有研究[14]表明,LDHs可高效活化H2O2的过氧键(O—O),产生·OH,据此推测PMS中的O—O键亦可以被LDHs活化。然而LDHs用于活化PMS虽取得一些研究进展,但目前研究多集中于Co系二元LDHs,如ZHAO等[9]对CoMn-LDHs活化PMS降解有机染料进行了研究,发现CoMn-LDHs具有优异的催化活性。然而,Co系LDHs活化PMS仍难以避免Co2+浸出,造成严重的二次污染问题;而Mn作为Co的替代性元素对PMS的活化效能仍有待提高。有研究[15]表明,多金属掺杂可有效提高催化材料活化PMS过程中的电子传递效率,提高催化剂的催化活性,实现高效、绿色活化PMS降解抗生素等难降解有机物。然而,抗生素类微量有机物与多金属掺杂的多元LDHs表面吸附态PMS之间是否存在直接电子传递作用,并实现对这类难降解有机物的高效降解,目前仍鲜有报道。
本研究以氯四环素(chlortetracycline,CTC)为研究对象,通过水热法制备了绿色、高效的催化剂MnFeCu三元类水滑石,且将其用于活化PMS以降解CTC,分别使用扫描电镜(scanning electron microscope,SEM)、X射线粉末衍射(X ray diffractometer,XRD)及X射线光电子能谱(X-ray photoelectron spectroscopy,XPS)等分析手段对催化剂的形貌和结构进行了表征和分析;考察了CTC浓度、催化剂和PMS投加量、初始pH及温度对CTC降解效能的影响;对降解过程中产生的各类活性物种进行了淬灭实验及EPR捕获,探明了活性物种的产生情况及其在CTC降解过程中的贡献,以揭示CTC的降解机制。对活化PMS前后的催化剂理化性质进行对比分析,结合催化剂回收再利用过程中CTC的降解效能,评估了催化剂的循环稳定性。本研究结果以期为水环境中抗生素的去除提供参考。
-
氯四环素(CAS,64-72-2)、四水硝酸锰(CAS,20694-39-7)、三水硝酸铜(CAS,10031-43-3)、九水硝酸铁(CAS,7782-61-8)、对苯醌(CAS,105-51-4)、糠醇(CAS,98-00-0)购自上海Aladdin公司。盐酸(CAS,7647-01-0)、氢氧化钠(CAS,1310-73-2)、碳酸钠(CAS,497-19-8)、叔丁醇(CAS,75-65-0)、无水乙醇(CAS,64-17-5)购自上海Macklin公司。
-
采用水热法制备MnFeCu-LDHs,具体如下:将1 mmol硝酸铁、1 mmol硝酸锰和2 mmol硝酸铜溶解于100 mL去离子水中,超声分散30 min后得到锰铁铜摩尔比1:1:2的金属混合溶液,置于250 mL分液漏斗中。将0.035 mol氢氧化钠与0.015 mol碳酸钠溶解于100 mL去离子水中,超声分散30 min。在剧烈搅拌条件下,将金属混合溶液以3 mL·min−1的速度缓慢滴入制备好的碱液中,滴加过程严格控制pH至10±0.1。将所得到的悬浊液置于水热反应釜中,140 ℃反应16 h。取出后用无水乙醇及去离子水反复离心洗涤,直至上清液pH至中性。将得到的产物置于真空干燥箱中55 ℃燥24 h后取出研磨,保存备用。
-
使用SEM、XRD等对合成的三元类水滑石的表面形貌、晶型等进行表征。同时,使用XPS对反应前后三元类水滑石进行表征,比较反应前后元素价态的转化,并结合循环稳定性实验分析催化剂的稳定性。
-
在未改变反应条件的情况下,均按照以下反应条件进行实验:在25 oC、初始pH=7的条件下,投加40 mg催化剂于200 mL10 mg·L−1的CTC溶液中,使催化剂浓度为0.2 g·L−1。在黑暗条件下使用磁力搅拌器搅拌30 min,使催化剂达到吸附解吸平衡,以排除吸附对催化降解速率的影响。往体系中投加40 mg PMS,使其浓度也达到0.2 g·L−1,分别在2.5、5、10、15、20及30 min取样后在30 s内过0.22 μm微孔滤膜,使用紫外分光光度计在356 nm波长处测得吸光度,进而计算出CTC剩余浓度[16-17]。
需要说明的是,在探究各影响因素对CTC的降解影响实验中,均采用单因素控制变量法进行研究。在探究CTC初始浓度的影响实验中,分别改变CTC质量浓度至5、20、30及40 mg·L−1,其余变量保持不变;在探究催化剂投加量的影响实验中,分别改变催化剂投加量至0.05、0.1、0.3及0.4 g·L−1,其余变量保持不变;在探究PMS投加量的影响实验中,分别改变PMS投加量至0.05、0.1、0.3及0.4 g·L−1,其余变量保持不变;在探究pH的影响实验中,分别改变pH至4、6、8和10,其余变量保持不变;在探究温度的影响实验中,分别改变温度至35、45和55 ℃,其余变量保持不变。
同时,为了考察催化剂的循环稳定性,将反应后的反应液倒入离心管中,洗涤剂为去离子水及无水乙醇,使用高速离心机在8 000 r·min−1下进行离心洗涤,在去离子水及无水乙醇各洗涤两次后放入55 oC真空干燥箱中干燥24 h。
-
为了探究类水滑石的结构形貌,对所有材料采用SEM进行分析(图1(a)~(b)为SEM扫描电镜图像)。其中花瓣状是LDHs的典型形态[18],可以看到MnFeCu-LDHs具有相对平整的氢氧化物层表面和明显的叠层结构。图1(c)~(f)是SEM图像中各元素的映射图像。可见复合材料中含有O、Mn、Fe、Cu元素,进一步说明MnFeCu-LDHs具有较为均匀的层状结构。由此可以初步判断,通过水热法成功合成了MnFeCu-LDHs复合材料。
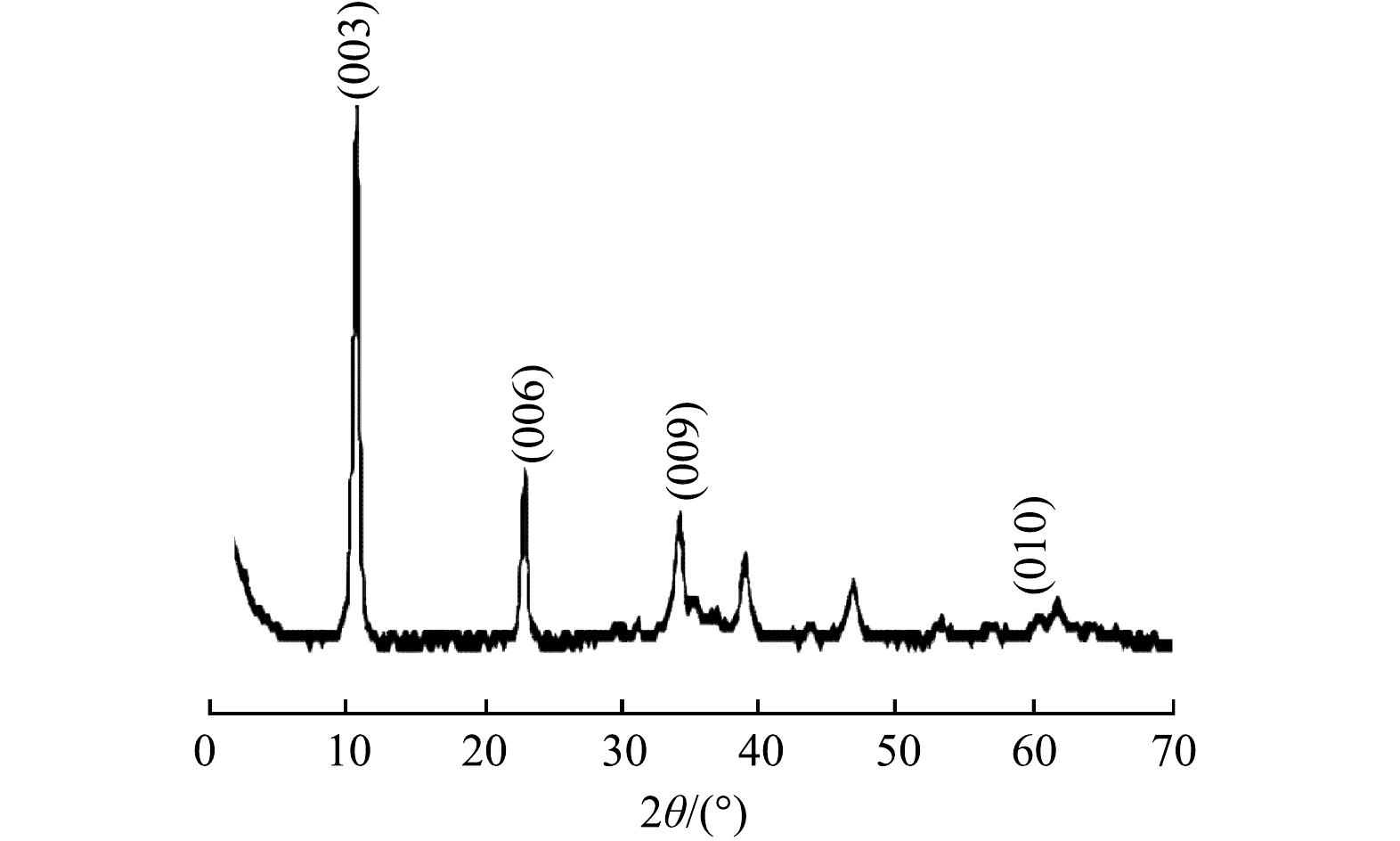
图2为所合成MnFeCu-LDHs的XRD图谱。在2θ为11.11°、22.82°、34.57°和60.30°处的特征峰,分别对应于类水滑石(003)、(006)、(009)和(110)晶面[19]。所得图像基线平稳,峰型窄且尖锐,进一步表明合成了结晶度良好的MnFeCu-LDHs。
-
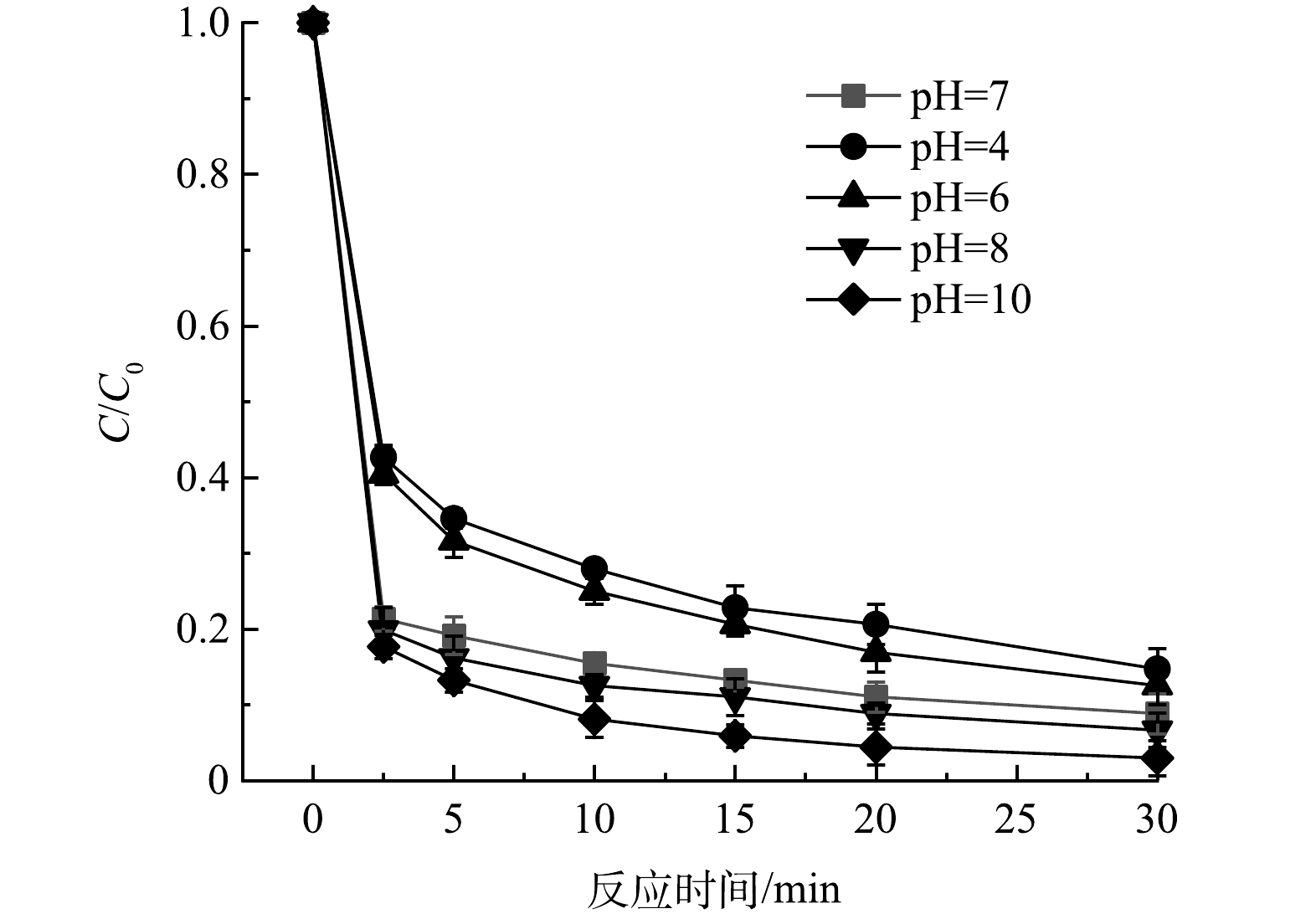
在反应温度为25 ℃,初始pH=7,催化剂投加量0.2 g·L−1,PMS投加量0.2 g·L−1和CTC质量浓度为10 mg·L−1的条件下,考察了MnFeCu-LDHs活化PMS降解CTC的效果(图3)。结果表明,PMS对CTC存在一定的降解能力,但其30 min降解率只有34.1%,这可能是由于PMS本身具有一定的氧化能力,且在水中只会产生少量的·OH[20]。而在添加MnFeCu-LDHs作为催化剂的条件下,CTC浓度在2.5 min内便迅速下降,去除率达到78.68%,30 min内去除率则达到91.18%。由此可见,MnFeCu-LDHs可有效活化PMS,产生的多种活性物种对CTC可实现高效降解。进一步改变MnFeCu-LDHs投加量至0.05、0.1、0.3、0.4 g·L−1,PMS投加量保持0.2 g·L−1不变,可以看出,当催化剂质量浓度从0.05 g·L−1增加到0.2 g·L−1时,CTC的催化降解效率得到明显提高,2.5 min时去除率达78.68%,30 min时去除率升高到91.18%。这是由于较高浓度的催化剂可为PMS活化提供更多的活性位点,从而进一步增强了CTC的降解。然而,当催化剂质量浓度继续增加至0.4 g·L−1时,降解曲线十分的接近,CTC降解的改善很小,说明该浓度下的PMS活化已达到了饱和,PMS浓度成为了CTC催化降解的限速步骤。
-
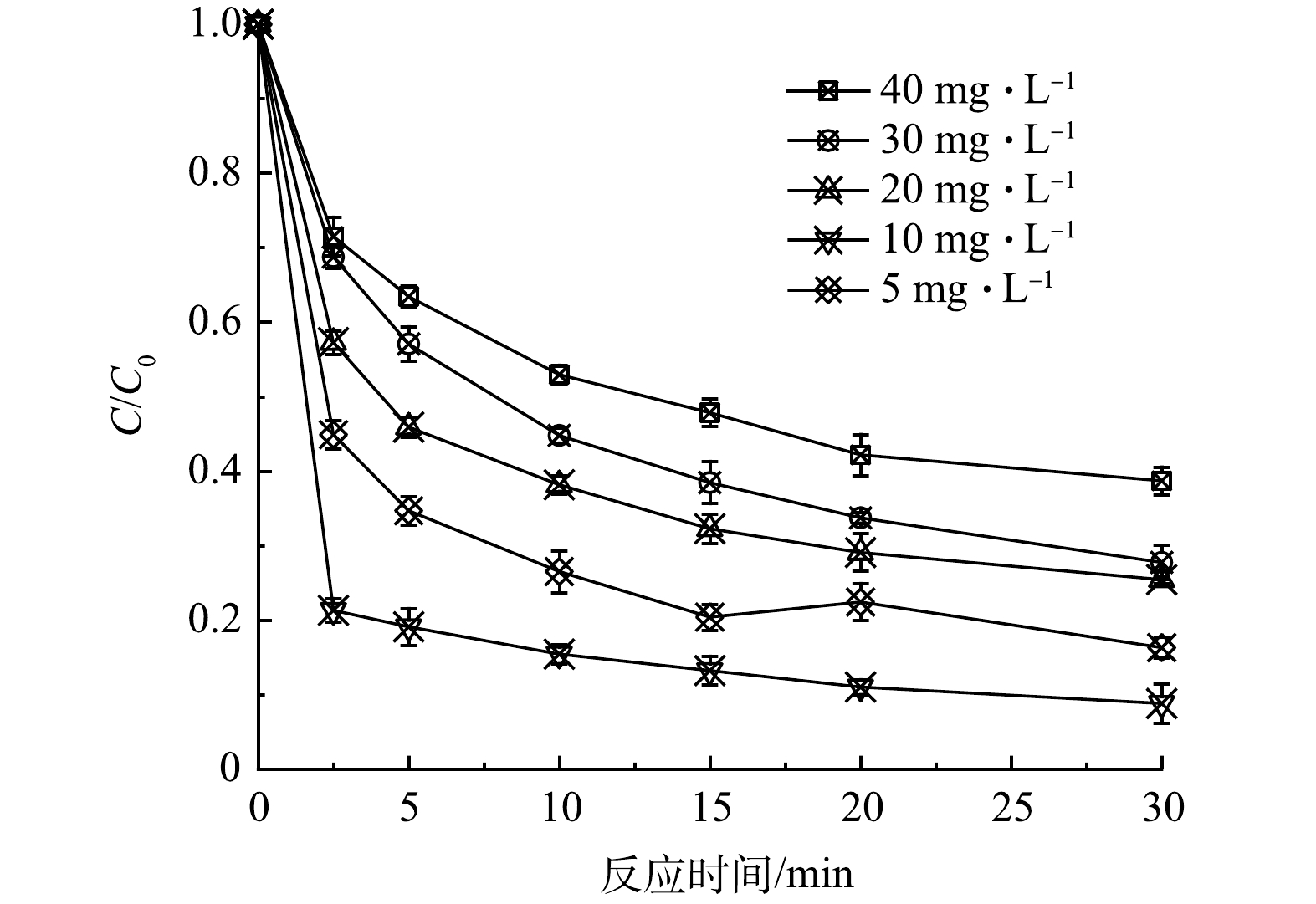
1) CTC初始浓度的影响。改变CTC质量浓度至5、20、30、40 mg·L−1,催化剂、PMS投加量保持0.2 g·L−1不变,初始pH为7,反应温度25 ℃,探究CTC初始浓度对反应体系中自身去除率的影响。如图4所示,当CTC质量浓度从5 mg·L−1上升至10 mg·L−1时,去除率达到最高,30 min去除率达到91.2%;继续升高CTC浓度时,30 min去除率则逐步下降,说明特定浓度的催化剂和PMS浓度下,MnFeCu-LHDs/PMS体系产生的活性物种有限,高浓度的CTC不足以被充分氧化降解。
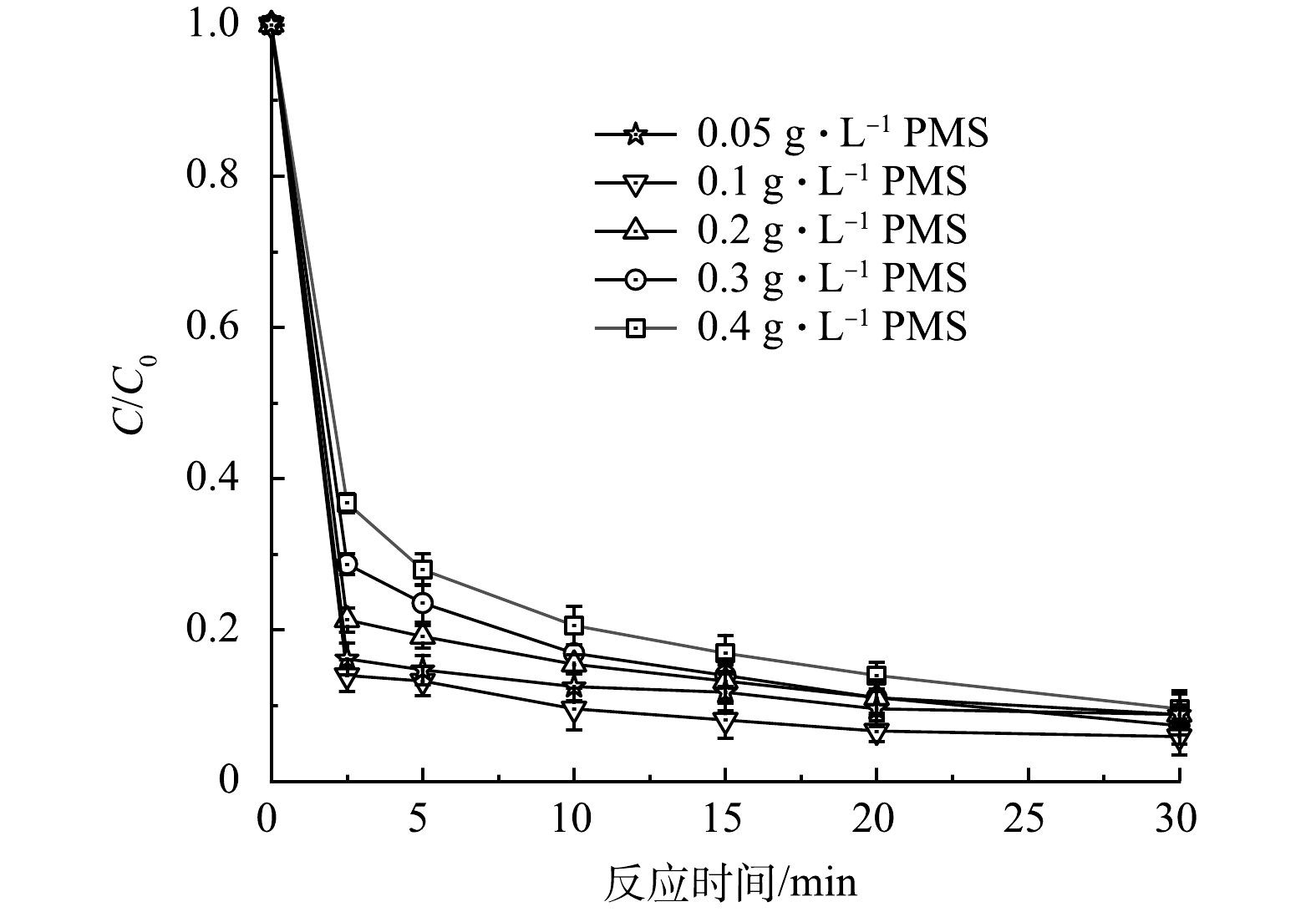
2) PMS投加量的影响。改变PMS投加量至0.05、0.1、0.3、0.4 g·L−1,其余各条件保持不变,探究PMS投加量对CTC降解的影响。如图5所示,总体来看改变PMS投加量对CTC 30 min去除率影响不大。当PMS投加量由0.05 g·L−1增加到0.1 g·L−1时,CTC降解效果有着一定程度的增加,但继续增加PMS浓度时,CTC的降解效果反而出现了削弱。这可能是由于随着PMS浓度的提高,引发了PMS自淬灭反应,多余的PMS与SO4·−和·OH生成 SO5·−(式(1)~式(2)),而这是一种较弱的氧化剂,氧化还原电位仅为1.1 V,导致CTC降解效率降低[21-22]。
3)初始pH的影响。其余各条件保持不变,改变初始pH(4~10)探究pH对反应体系中CTC降解效能的影响。如图6所示,在碱性条件下,锰铁铜水滑石对CTC的催化降解有着更好的效果。这是由于碱性条件下,OH−可以加速HSO5−的分解;此外,OH−可以与 SO4·−反应生成·OH,其在碱性条件下是一种非选择性的氧化性自由基[23]。而在酸性条件下,H+将破坏水滑石的氢氧化物层,并加速金属的溶解,同时过量的H+可以清除SO4·−和·OH(式(3)~式(5))。因此,碱性条件提高了CTC的降解效率并抑制了金属的溶解。但即使是在初始pH为4的条件下,CTC的30 min去除率仍能达到85.3%,可见该催化剂良好的催化性能,这将有效弥补芬顿体系中低pH的局限性。
4)不同温度的影响。使用控温磁力搅拌器将反应温度调整至308、318和328 K,为了更加清晰地展现温度对CTC降解的影响情况,将催化剂投加量降至0.05 g·L−1,PMS投加量保持0.2 g·L−1不变,初始pH为7,结果如图7所示。随着反应温度的增加,CTC降解速率与去除率均有所增加。对反应过程做动力学分析,可以得到不同温度下的反应速率常数k(图7(b)),根据阿伦尼乌斯方程(式(6)),通过对3个温度下的lnk与1/T进行拟合,可以得到斜率即Ea/R,进而计算出锰铁铜水滑石活化PMS降解CTC的活化能。该化学反应的活化能为50.3 kJ·mol−1。
-
为了探明反应体系中活性氧物种的产生情况,使用DMPO捕获·OH、O2·−、SO4·−,甲醇做溶剂,使用TEMP捕获1O2,去离子水做溶剂,混合均匀之后,用毛细管吸取一定量混合液,套上石英管之后,放入EPR样品腔内进行ROS信号测试。通过EPR/ESR分析各活性氧物种的信号特征峰(图8)。结果表明,反应体系中·OH、SO4·−、O2·−及1O2均有明显的信号特征峰检出。值得注意的是,在加入PMS反应1 min和10 min时,可以观测到较为明显的1O2、·OH和O2·−信号特征峰,而SO4·−则在反应1 min时信号峰特征明显,在反应10 min时,信号特征峰强度有着明显的减弱,推断SO4·−不会持续存在,在反应中不占据主导地位。
为了进一步探明反应体系中各活性物种对CTC降解的贡献,分别加入无水乙醇(EtOH)淬灭·OH和SO4·−,叔丁醇(TBA)淬灭·OH,对苯醌(PBQ)淬灭O2·−,糠醇(FFA)淬灭1O2[24-25]。如图8所示,加入4种淬灭剂后,CTC的降解率均有不同程度的下降。加入EtOH后,30 min后CTC去除率由未加入淬灭剂的91.18%下降到76.47%,而加入TBA后,30 min去除率则下降至85.29%,证明了体系中SO4·−与·OH的存在。在FFA后,30 min去除率下降至38.24%,其对降解的抑制效果远高于EtOH和TBA,证明了该体系中占据主导的是1O2;此外,可以看到,PBQ的加入对CTC的降解去除也产生了一定的抑制作用。这是由于PBQ清除 O2·−后,1O2的生成随之受到影响,进而表现出抑制作用,这些均证实了该反应体系CTC的降解过程中非自由基反应占主导。
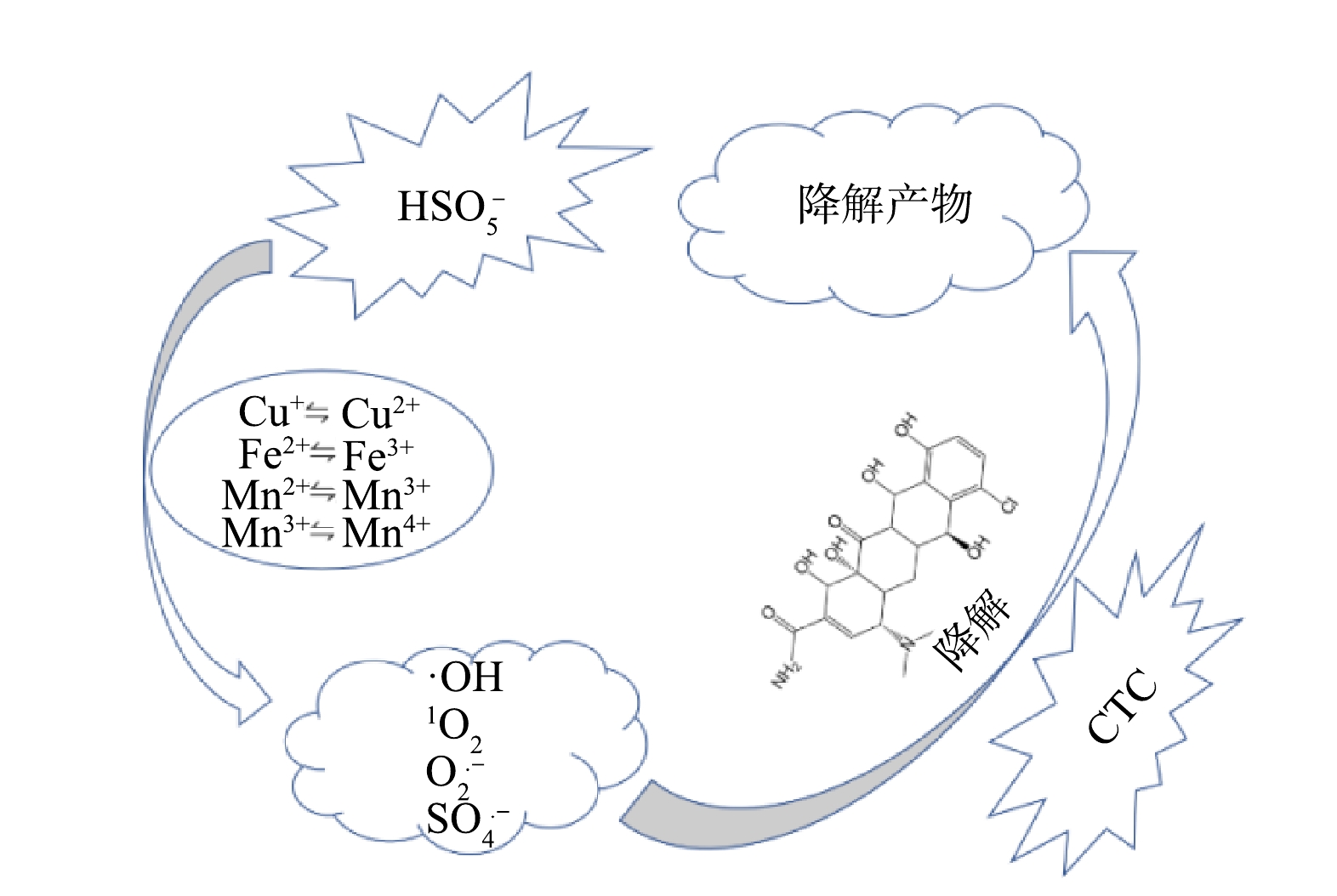
由图9中XPS图谱可见,MnFeCu-LHDs/PMS体系中存在二价和三价的锰离子和铁离子以及一价和二价的铜离子,通过氧化还原反应可以激活PMS产生强氧化性物质。可以看出,反应前后3种过渡金属不同价态的含量均发生明显变化,Mn2+从反应前的39.67%下降到反应后的35.84%,Mn3+则由44.9%增加到49.28%;Fe2+从反应前63.95%增加到反应后的64.63%,Fe3+则从36.05%下降到35.37%;Cu+从反应前的40.51%上升到反应后的44.36%,Cu2+则从59.49%下降到55.64%。此外,O1s谱图中晶格氧的含量下降了2.93%,证明催化剂表面的含氧基团可能参与了活化反应。通过以上分析,我们推理出了MnFeCu-LDHs活化PMS的过程:MnFeCu-LDHs中的Mn2+,Mn3+,Fe2+和Cu+通过电子转移活化PMS(HSO5·−)生成SO4·−(式(7)~式(9),而SO4·−可以与H2O和OH−反应生成·OH(式(10))[26-27]。反应后的高价态Mn3+、Fe3+和Cu2+可被HSO5−还原为低价态的金属离子(式(11)),同时由于Mn3+/Mn2+、Fe3+/Fe2+、Cu2+/Cu+及Mn4+/Mn3+的氧化还原电位分别是1.51V、0.77V、0.34V和0.15V,氧化还原电位间的差异将促进金属离子间的循环(式(12)~式(16)),使多金属间产生协同效应进而提高催化活性[23]。Mn2+、Fe2+和Cu+还会与催化剂表面的H2O和OH−结合,对PMS的活化有着促进作用(式(17)~式(18))[28-30]。1O2则可以通过PMS分解和O2·−与H2O反应生成(式(19)~式(20))。由此可见,CTC在SO4·−、·OH、1O2及O2·−几种自由基的共同作用下被降解。
-
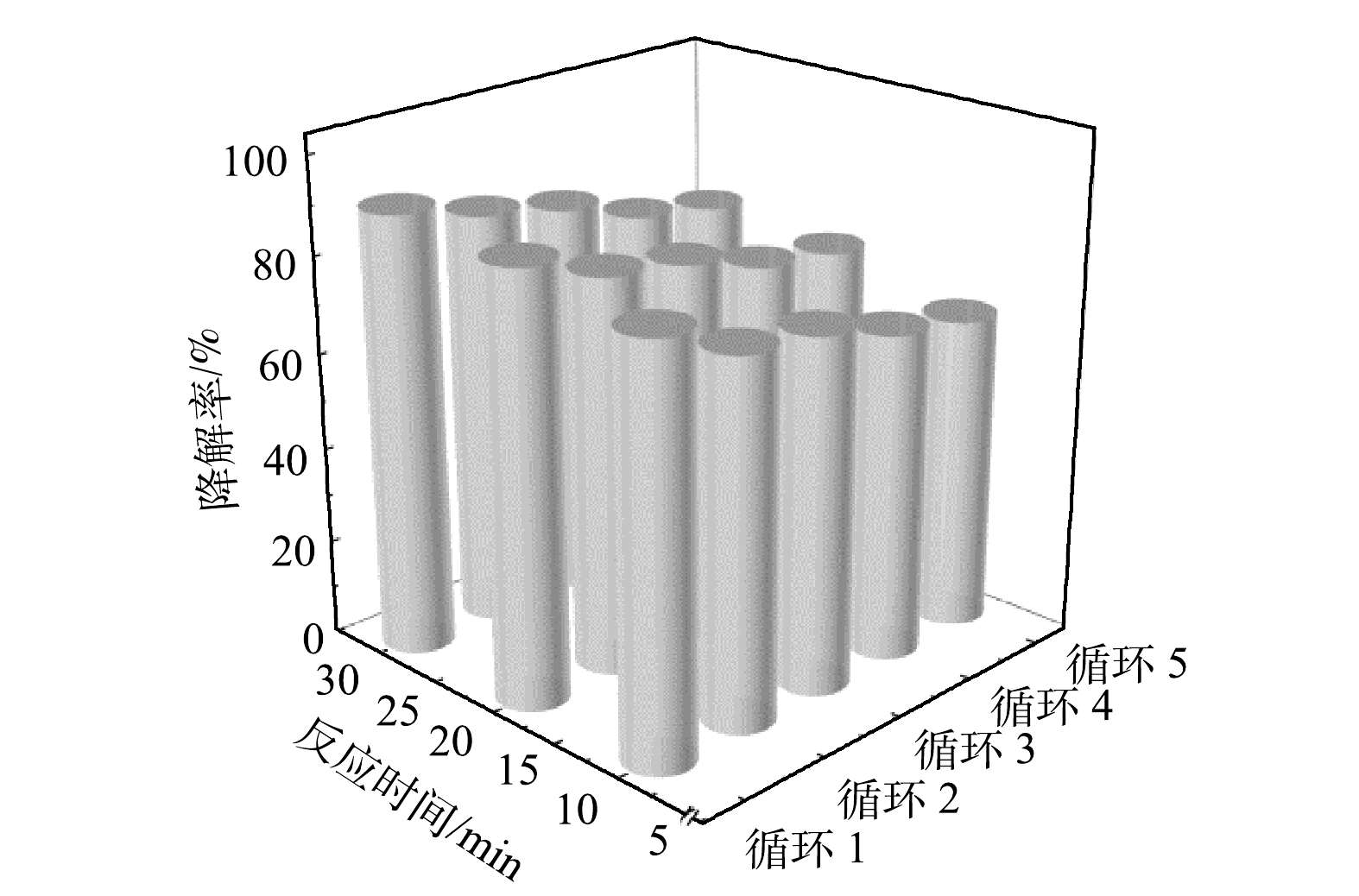
将干燥后的催化剂仔细研磨后用于活化PMS降解CTC上述过程重复5次(图10)。随着循环次数的增多,催化剂30 min催化降解效果逐次下降,从第1次的91.18%依次下降至86.11%、82.64%、76.39%及73.61%,这可能是由于金属离子的溶出和催化剂表面残留了部分CTC降解产物,导致了催化降解效率的降低。在重复使用3次后,CTC的10 min降解率仍有70%以上,30 min降解效率仍有80%以上。最终经过5次循环后,CTC 30 min催化降解率仍然有73.61%,表明锰铁铜类水滑石催化剂有着良好的循环稳定性与重复使用性。
-
结合活性氧物种分析及XPS分析结果,对MnFeCu-LDHs活化PMS机理进行总结,如图11所示,MnFeCu-LDHs表面的金属离子通过与HSO5−间电子的转移,电子转移过程如式7-式20所示,通过电子转移生成了·OH、1O2等活性氧物种,这些活性氧物种与CTC具有很好的反应活性,通过与CTC发生反应完成对CTC的降解,在此过程产生的几种活性氧物种均不同程度的参与了此过程,其降解反应以1O2的非自由基反应为主。
-
1)采用水热法成功制备了MnFeCu-LDHs,将其应用于活化PMS降解CTC,发现其具有良好的催化活性,能够有效去除水环境中的CTC:在初始pH为7,催化剂及PMS投加量为0.2 g·L−1时,CTC在30 min的去除率达到91.18%。
2)催化剂在各pH条件下都具有较好的应用性,在碱性条件下,催化剂对PMS的活化效果较好,CTC的降解效率也较高。同时增大催化剂投加量、升高温度均可强化CTC降解效能,但催化剂投加量超过0.2 g·L−1后,强化效果不再显著。此外,过多的PMS浓度将和SO4·−和 ·OH生成SO5·−,氧化能力被削弱。
3)通过淬灭实验和EPR捕获证明了在该体系中,·OH、SO4·−、O2·−、1O2均参与了CTC的降解,贡献度最高的是1O2,其次为·OH和SO4·−。
4)材料表面的不同价态金属离子因氧化还原电位间的差异将促进金属离子间的循环,使多金属间产生协同效应进而可提高催化活性;经过5次循环使用后,催化剂对CTC仍具有良好的催化降解能力,说明其具有优良的重复利用性。
MnFeCu-LDHs活化PMS降解氯四环素的效能及机制
Degradation efficiency and mechanism of chlortetracycline by activation of peroxymonosulfate via MnFeCu-LDHs
-
摘要: 针对污水处理厂尾水中抗生素等生物难降解有机物频繁检出的问题,采用相对绿色、低毒性的过渡金属元素制备了锰铁铜类水滑石(MnFeCu-LDHs),并将其用于活化过一硫酸盐(PMS)降解氯四环素(CTC)。探究了初始pH、反应温度、催化剂和PMS投量对CTC降解效能的影响规律,通过化学捕获和淬灭实验确定了活性氧物种(ROS)的种类与贡献,并对反应前后的催化剂进行理化性质表征且考察了催化剂稳定性。结果表明,在初始pH为7、反应温度为298 K、催化剂及PMS投加量均为0.2 g·L−1条件下,反应5 min后CTC去除率达到80.88%,30 min去除率达到91.18%,同时,随着初始pH和温度的提高,CTC的降解效果得到明显增强;ROS淬灭实验和EPR捕获实验结果证实了在该体系中,·OH、SO4·−、1O2均参与了CTC的降解,贡献度最高的是1O2,其次为·OH和SO4·−;基于反应前后XPS光谱对比分析,发现MnFeCu-LDHs活化PMS过程稳定性较好,此外该催化剂在重复使用5次后,CTC的30 min去除率仍达到73.61%。因此,本研究可为SR-AOPs应用于控制水环境抗生素类污染提供新思路。
-
关键词:
- MnFeCu-LDHs /
- 过一硫酸盐 /
- 高级氧化技术 /
- 氯四环素 /
- 氧化降解
Abstract: Aiming at frequent detection of biorefractory organics, such as antibiotics in the tail water of wastewater treatment plant, MnFeCu layered double hydroxide (MnFeCu-LDHs) was prepared with green and low toxic transition metals to activate persulfate (PMS) for degrading chlortetracycline (CTC). The effects of initial pH, reaction temperature, catalyst and PMS dosage on CTC degradation efficiency were investigated. The types and contributions of reactive oxygen species (ROS) were determined through chemical capture and quenching experiments. The physicochemical properties of catalysts before and after reaction were characterized to investigate the stability of catalysts. The results showed that the removal rate of CTC reached 80.88% after 5 min reaction, and 91.18% after 30 min reaction when the initial pH was 7, the reaction temperature was 298 K, and the dosages of catalyst and PMS were 0.2 g·L−1. Meanwhile, with the increase of initial pH and temperature, the degradation effect of CTC increased significantly. ROS quenching experiment and EPR capture confirmed that ·OH, SO4·− and 1O2 participated in CTC degradation in this system, of which the highest contribution was 1O2, followed by ·OH and SO4·−. Based on the comparative analysis results of XPS spectra before and after the reaction, it was found that the stability of MnFeCu-LDHs activated PMS process was reliable. In addition, after five recycles of the catalyst, the CTC degradation rate still reached 73.61% at 30 min reaction. Therefore, this study would provide a new insight for the application of SR-AOPs to control antibiotic pollution in water environment.-
Key words:
- MnFeCu-LDHs /
- peroxymonosulfate /
- advanced oxidation processes /
- chlortetracycline /
- degradation
-
电镀、化学镀等表面处理过程产生的废水中通常含有铬、锌、铜、镍等重金属及氰化物等污染物,必须对其进行严格处理[1]。铜是主要镀种之一,含铜废水主要来自镀件漂洗,其中含有大量铜和有机污染物[2]。电镀过程中广泛使用的络合剂(乙二胺四乙酸(EDTA)、柠檬酸、酒石酸)会与铜形成稳定性高且形态复杂的络合铜[3],如EDTA-Cu稳定常数比Cu(OH)2高5个数量级[4]。络合态重金属多数具有很高的水溶性,且在广泛的pH范围内能够稳定存在,使得铜难以从废水中有效去除[5]。以碱沉淀法和硫化物沉淀法为代表的化学沉淀法是目前广泛使用且经济性较好的方法[6-9],但在铜浓度较低时效果不佳,且沉淀物在酸性条件下不稳定。吸附法[10-12]、电解法[13]、离子交换法[14-15]、高级氧化还原法[16-18]等其他方法也存在成本较高、再生困难等问题。因此,废水中络合铜的有效、经济去除是一个亟待解决的问题。
重金属捕集剂能与废水中的重金属或络合态重金属迅速发生螯合作用,产生难溶的螯合沉淀,具有反应速率快、沉淀物稳定、选择性好的优点,已成为重金属污染处理领域关注的热点,具有广泛的应用前景[19-22]。在重金属捕集剂中,二硫代氨基甲酸盐(DTC)类捕集剂使用广泛。谭聪等[23]用二硫化碳、碳酰肼为原料,合成了重金属捕集剂DT-SC,对Cu2+、Cd2+、Mn2+去除率均在90%以上。张翔等[24]采用二并哌嗪、二硫化碳等在碱性条件下合成重金属捕集剂TDDP,在最佳反应条件下对Cu2+和Pb2+去除率均超过96%,对Zn2+去除率也超过91%。二乙基二硫代氨基甲酸钠(DDTC)、SDDC[25]等小分子螯合剂及二硫代羧基化羟甲基聚丙烯酰胺(DTMPAM)[26]、聚-二硫代氨基甲酸铵(PADTC)[27]等高分子螯合剂,对Cu具有良好的去除性能。其中高分子螯合剂沉降性能较好,但其较大的空间位阻使得DTC基团利用率较低,且存在成本较高的问题[28]。同时,小分子螯合剂DTC基团利用率相对较高,但含有单个螯合基团的小分子螯合剂去除能力有限。分子质量大小适当并拥有多个DTC基团的螯合剂可提升捕集效率和去除性能。因此,采用三乙烯四胺等为原料合成多硫代氨基羧基基团的重金属捕集剂,一方面可提升二硫代羧基的数量,去除能力显著增强;另一方面具有适当分子链长,可降低空间位阻,使得基团利用效率进一步提升。综上所述,本文以三乙烯四胺等为原料,在操作简单、条件温和的条件下合成重金属捕集剂N,N-双(二硫代羧基)三乙烯四胺(TDTC),对EDTA-Cu等具有优异的去除效果,以期为含铜废水的高效处理提供参考。
1. 材料与方法
1.1 实验材料
本研究所用聚丙烯酰胺(PAM,MW 300万)购于天津市大茂化学试剂厂,氢氧化钠(NaOH)、乙二醇(C2H6O2)、乙二胺四乙酸二钠(C10H14N2O8Na2·2H2O)、硫酸铜(CuSO4·5H2O)、柠檬酸(C6H8O7·H2O)、酒石酸钾钠(C4H4O6KNa·4H2O)均购于广州化学试剂厂,三乙烯四胺(TETA)、二硫化碳(CS2)均购于上海阿拉丁公司,试剂均为分析纯。
1.2 实验方法
1) TDTC的合成。向有冷凝和搅拌装置的250 mL三颈烧瓶中加入一定量的混合溶剂(V乙二醇/
VH2O 
2)模拟废水的配制。按照络合剂与Cu摩尔比为1∶1称取适量的乙二胺四乙酸二钠(EDTA-2Na)、柠檬酸(C6H8O7·H2O,CA)与酒石酸钾钠(C4H4O6KNa·4H2O,TA)与五水硫酸铜(CuSO4·5H2O)配制乙二胺四乙酸二钠络合铜(EDTA-Cu)、柠檬酸络合铜(CA-Cu)、酒石酸络合铜(TA-Cu) 3种模拟废水。
3)络合Cu去除实验。在室温条件下,在150 mL烧杯中分别加入100 mL Cu质量浓度为50 mg·L−1的EDTA-Cu、CA-Cu、TA-Cu溶液,调节pH,添加一定量TDTC固体,置于搅拌器中快速(300 r·min−1)搅拌,然后液体投加1.0 mg·L−1 PAM,慢速(70 r·min−1)搅拌一定时间,静置 10 min,取液面下2 cm处上清液,上清液中残留的Cu浓度用原子吸收分光光度计测定,Cu去除率根据式(2)进行计算。
R=C0−C1C0×100% (2) 式中:
R 1.3 分析方法
pH采用酸度计(雷磁pHS-25)测定;Cu浓度采用火焰原子吸收分光光度计(WFX-110B,检出限0.006 mg·L−1,北分瑞利)测定;TDTC及其沉淀表面形态观察使用扫描电子显微镜(Hitachi S4800,日本日立);TDTC捕集重金属前后红外光谱采用KBr压片法在傅立叶红外光谱仪(Nicolet 670,美国Thermo Fisher)上完成;XPS测试使用X射线光电子能谱仪(Thermo Scientific K-Alpha+,美国Thermo Fisher);捕集剂与螯合产物元素分析采用有机元素分析仪(Vario MACRO cube,德国Elementar)测定碳、氢、氮、硫含量。
2. 结果与分析
2.1 TDTC投加量对络合Cu去除效果的影响
在PAM添加量为1.0 mg·L−1、反应时间5 min、静置10 min、溶液pH为4.0、 Cu质量浓度为50 mg·L−1的条件下,加入不同量的重金属捕集剂,络合Cu的去除率变化如图1所示。由图1可知,当TDTC投加量<1 mmol·L−1时,3种络合Cu的去除率随TDTC投加量增加而持续增加;当捕集剂达到1 mmol·L−1时,络合Cu去除率基本稳定,达到99.6%以上,此时反应体系中的Cu质量浓度均在0.2 mg·L−1以下。此外,TDTC对3种络合态Cu均有较好的去除效果,二硫代羧基对于Cu的络合能力强于TA、CA、EDTA。在TDTC投加量相同的情况下,Cu去除率由高到低为TA-Cu>CA-Cu>EDTA-Cu。这是因为3种络合剂与Cu形成络合物的稳定常数由低到高为TA-Cu<CA-Cu<EDTA-Cu[29],TDTC在与络合剂的配位竞争中络合剂的稳定常数越小,TDTC竞争越占优势,螯合沉淀效率越高,因此,同等投加量的TDTC对稳定常数小的络合Cu去除效果更好。继续增加投加量,去除率有一定上升,但幅度不大,投加量过高造成水中COD由170 mg·L−1增加到407.5 mg·L−1,同时会导致处理成本的上升,所以选定TDTC最佳投加量为1 mmol·L−1。
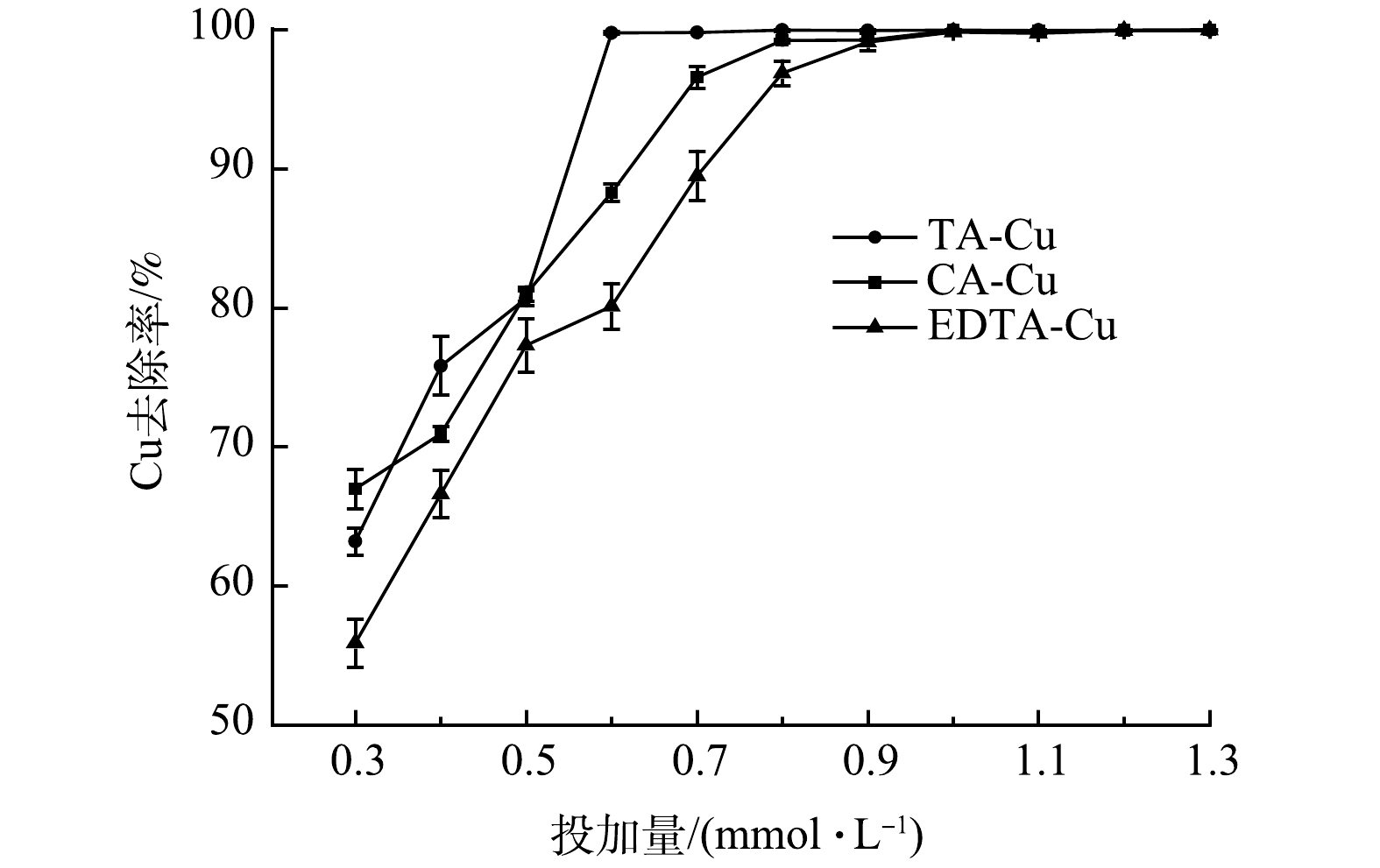 图 1 TDTC投加量对络合Cu去除的影响Figure 1. Effect of TDTC dosage on complex copper removal capacity
图 1 TDTC投加量对络合Cu去除的影响Figure 1. Effect of TDTC dosage on complex copper removal capacity2.2 初始pH对络合Cu去除效果的影响
初始pH对TDTC捕集重金属离子效果影响较大。在PAM投加量为1.0 mg·L−1、TDTC投加量为1 mmol·L−1、反应时间5 min、静置10 min、 Cu质量浓度为50 mg·L−1的条件下,采用0.1 mol·L−1的HCl/NaOH调节体系初始pH,初始pH对络合Cu去除率影响结果如图2所示。投加捕集剂之前先分别调节反应体系pH为1.0~11.0,没有Cu(OH)2沉淀物的形成,表明TA-Cu、CA-Cu、EDTA-Cu在酸性和碱性条件下处于稳定状态,碱沉淀法不能处理含络合铜废水。由图2可知,当pH为3.0 ~ 9.0时,残留Cu质量浓度均在0.2 mg·L−1以下,达到排放标准要求;当pH<3.0或pH>9.0,即强酸或强碱情况下去除效果明显下降。初始pH会影响TDTC的电离平衡,当pH>11.0时,3种络合态Cu的去除率均有下降,处于在90%以下。这是因为pH的持续上升会使式(3)中的平衡向右移动,可使更多的二硫代羧基失去质子而呈离子化状态,更容易与Cu2+螯合,但在强碱情况下,络合剂与Cu2+的配合物稳定性更强,不利于捕集。
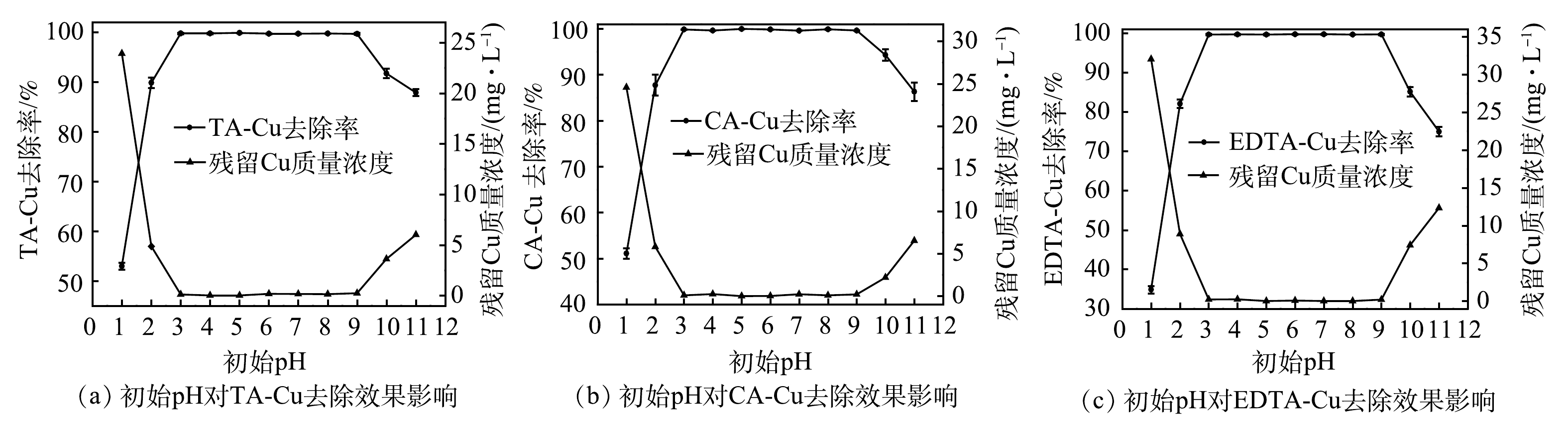 图 2 初始pH对络合Cu去除效果的影响Figure 2. Effect of system initial pH on complex copper removal capacity
图 2 初始pH对络合Cu去除效果的影响Figure 2. Effect of system initial pH on complex copper removal capacity{\rm{R}}_{1}{\rm{R}}_{2}{\rm{N}} {\text{—}} {\rm{CSSH}} \rightleftharpoons}_{2}{\rm{N}}{\text{—}}{\rm{CSS}}^- + {\rm{H}}^{+ } (3) 如柠檬酸在溶液中有C6H8O7、C6H7O7−、C6H6O72−、C6H5073− 4种形态,对应的电离常数分别为pk1=3.13、pk2=4.76、pk3=6.40。在碱性条件下,柠檬酸的酸效应减弱,与Cu2+形成的络合物更加稳定,导致去除效率下降;当pH<3.0时,式(3)中的电离平衡左移,产生的离子化二硫代羧基减少,不利于对Cu2+的捕集,且基团中的螯合位点相较于Cu2+更易与H+结合,带正电荷的—N+HR2与二硫代羧酸形成内盐,导致Cu2+去除率下降[30-31]。实际重金属废水多为酸性,TDTC处理含铜废水无需调节pH至中性或碱性,处理后的溶液也便于回用,适用范围广,比传统化学沉淀法具有明显优势。
2.3 反应时间对络合Cu去除效果的影响
采用100 mL络合Cu质量浓度为50 mg· L−1的实验废水,TDTC投加量为1 mmol·L−1,调节pH=4.0,PAM添加量为1 mg·L−1,调节搅拌速度为慢搅(70 r·min−1),搅拌0~10 min,以探究反应时间对络合Cu去除效率的影响,结果见图3。由图3可知,前30 s时间段内络合Cu的去除率为70%~90%。这可能因为该阶段捕集剂与络合剂处于对Cu2+的螯合竞争,Cu2+从络合物中释放出来并立即形成螯合物。在前3 min,Cu去除率随反应时间的延长而提高,然后去除率逐渐趋于稳定,TDTC与络合Cu的反应基本完成,Cu去除率在99.8%以上;在反应时间大于10 min后,Cu去除率稍有下降,这可能是因为长时间搅拌会导致已形成的絮体破碎,从而影响出水水质。考虑到实际应用中的成本因素,选取反应时间为3 min。
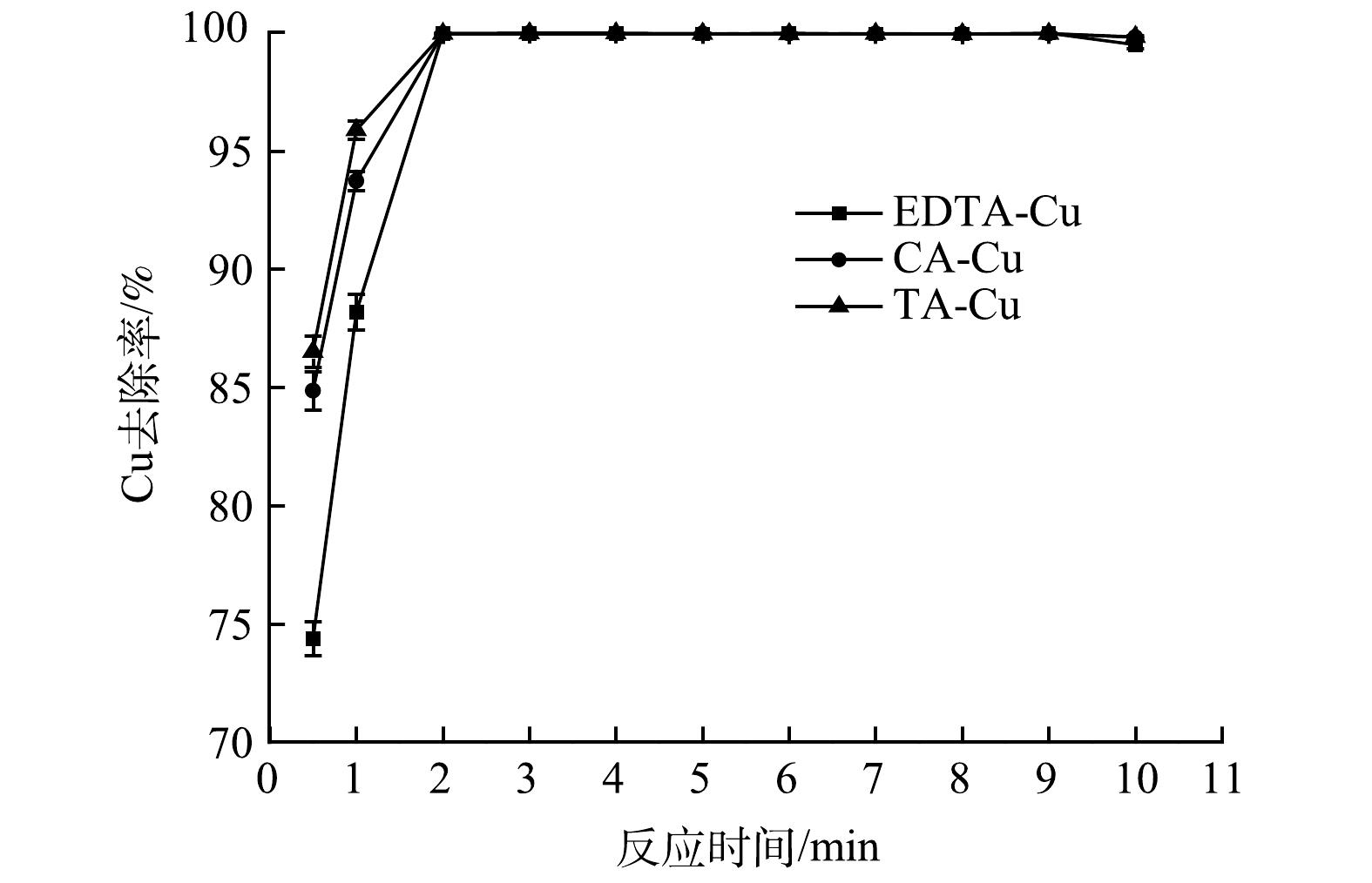 图 3 反应时间对络合Cu去除效果的影响Figure 3. Effect of reaction time on complex copper removal capacity
图 3 反应时间对络合Cu去除效果的影响Figure 3. Effect of reaction time on complex copper removal capacity2.4 絮凝剂对络合Cu去除效果的影响
取100 mL、50 mg·L−1的3种络合Cu模拟废水,在pH为4.0,TDTC投加量为1 mmol·L−1,反应时间5 min,静置10 min的条件下,加入不同量的絮凝剂PAM,以探究絮凝剂PAM在对络合Cu去除的影响。由图4可知,在不添加絮凝剂PAM的情况下,EDTA-Cu、CA-Cu与TA-Cu的去除率分别为95.63%、96.17%和96.32%,沉降性能较没有添加絮凝剂PAM时弱。随着絮凝剂用量的增加,3种络合铜的去除率随之增大,这可能是部分捕集剂在将Cu从络合剂中捕集出来后,形成的细小微粒没有与絮体一同沉降去除,而PAM与细小悬浮颗粒物通过架桥作用连接形成大絮体从而使该部分得以去除。当PAM添加量为1 mg·L−1时,3种络合Cu的去除率均在99.6%以上,且趋于稳定。综合考虑经济成本,选用PAM最佳用量为1 mg·L−1。
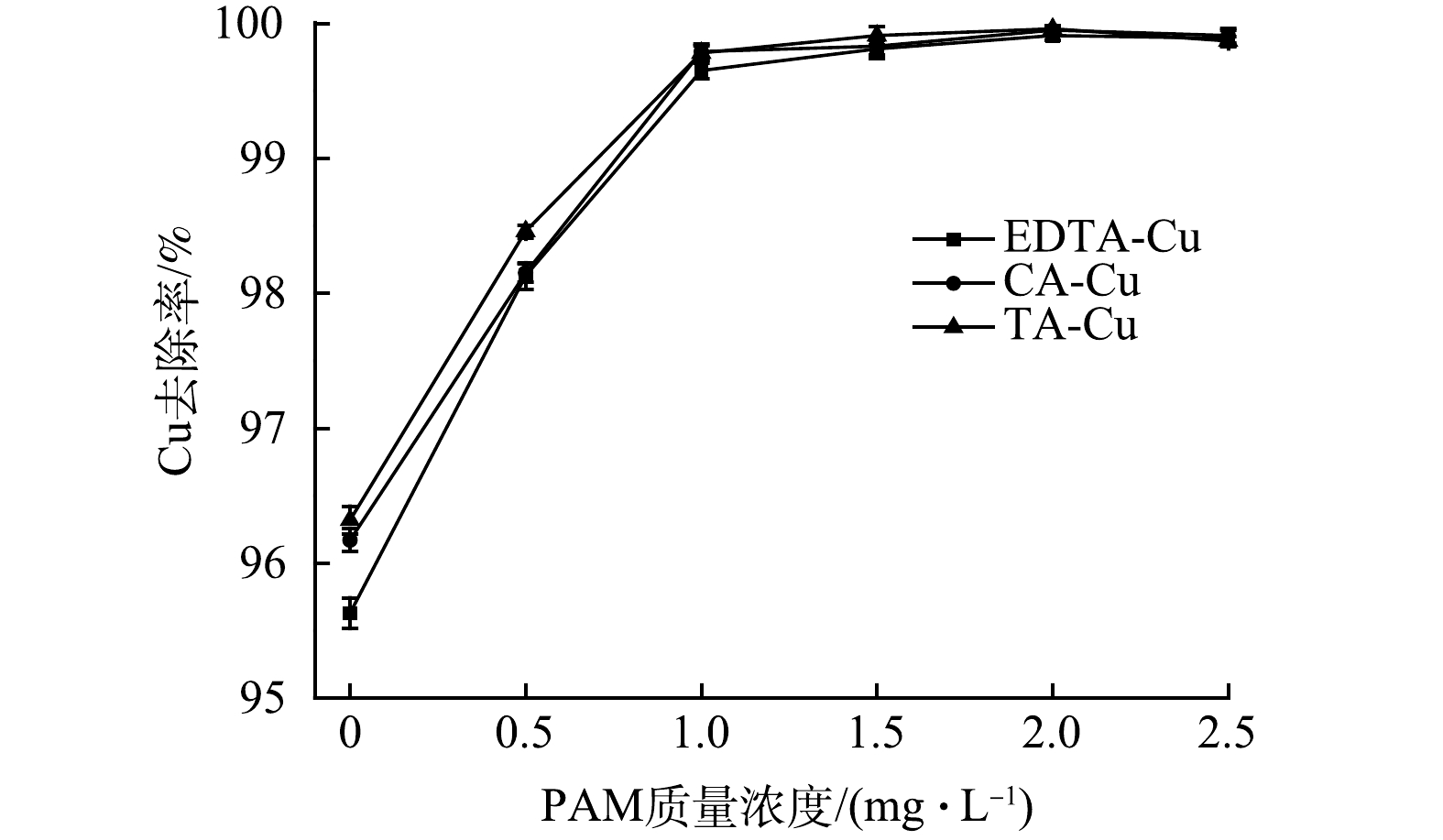 图 4 絮凝剂投加量对络合Cu去除效果的影响Figure 4. Effect of PAM dosage on complex copper removal capacity
图 4 絮凝剂投加量对络合Cu去除效果的影响Figure 4. Effect of PAM dosage on complex copper removal capacity2.5 TDTC与Cu螯合机理
1) TDTC及其捕集产物的表面形态。通过扫描电镜观察TDTC及其捕集产物的表面形态。图5(a)为TDTC固体的SEM图,图5(b)、图5(c)和图5(d)为3种螯合沉淀的SEM图。由图5(a)可看出,捕集剂形态不规则,其表面光滑,比表面积大,结构紧密,层次堆积明显。由图5(b)、图5(c)与图5(d)可看出,相较于图5(a)中的TDTC,反应后生成的螯合沉淀形态不规则,表面变得相对粗糙,颗粒之间结合更加紧密。这是由TDTC将络合Cu中的Cu有效捕集出来所致。而3种沉淀形态的差异可能是由于TDTC在与不同络合Cu反应过程中,因不同络合剂络合能力大小不一,用于争夺Cu所用的TDTC量会有差异,导致形成沉淀的形态不同。
2) TDTC元素分析。由表1中数据计算可得,TDTC中C、H、N和S的摩尔比接近于4∶9∶2∶2的理论比值。结合上述分析结果,可以认为有效合成了TDTC,化合式为C4H9N2S2。产品中C、S超过理论值,说明产品不纯,还含有一定量杂质,推测其可能是CS2等反应物。
表 1 TDTC的元素分析Table 1. Elementary analysis of TDTC元素 比例/% 物质的量相对值 C 33.97 1.91 H 3.28 3.96 N 18.64 0.9 S 40.75 1.1 | Show Table DownLoad:
CSV
DownLoad:
CSV
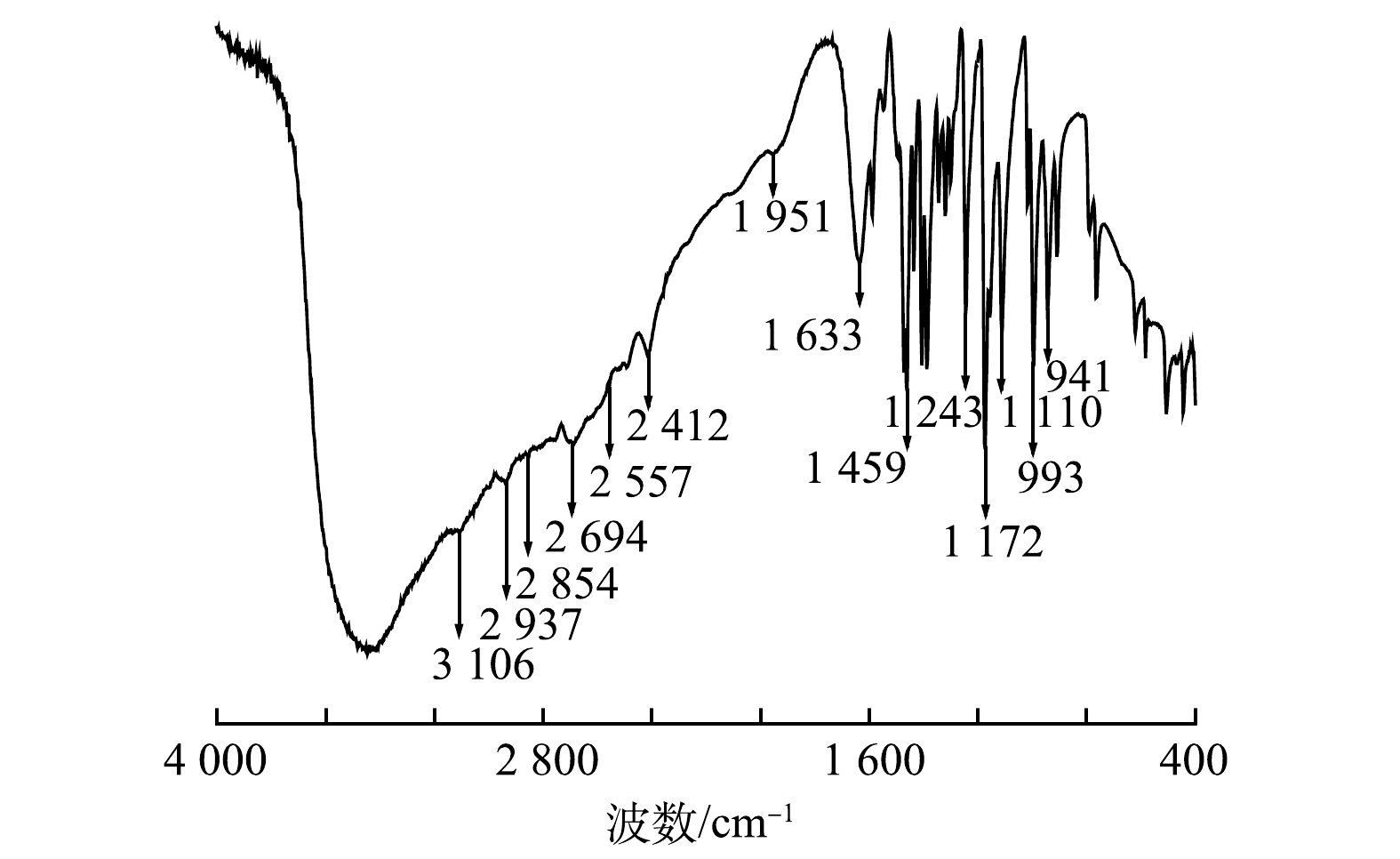
3)红外光谱分析。在400~4 000 cm−1对TDTC进行红外光谱扫描,结果如图6所示。2 937 cm−1处为C—H伸缩振动,低于3 000 cm−1,该吸收峰为C—H的饱和吸收峰[32];2 854 cm−1处为—CH2的对称伸缩振动峰[33];2 557 cm−1处为—SH的振动伸缩峰[34];1 633 cm−1处为N—H的变形振动峰[31];1 459 cm−1处为N—CS2的伸缩振动吸收峰,此峰介于C—N单键(1 300 cm−1)和C=N双键(1 600 cm−1)之间,具有部分双键性质[35];1 243 cm−1处有C=S特征吸收峰;在1 110、993、941 cm−1处有C—S的振动伸缩峰,低于C=S双键的特征吸收(1 501~1 200 cm−1),高于C—S单键的特征吸收(600~700 cm−1),且为强吸收峰,具有部分双键性质[36-37]。以上结果说明反应产物含有二硫代氨基羧基基团。
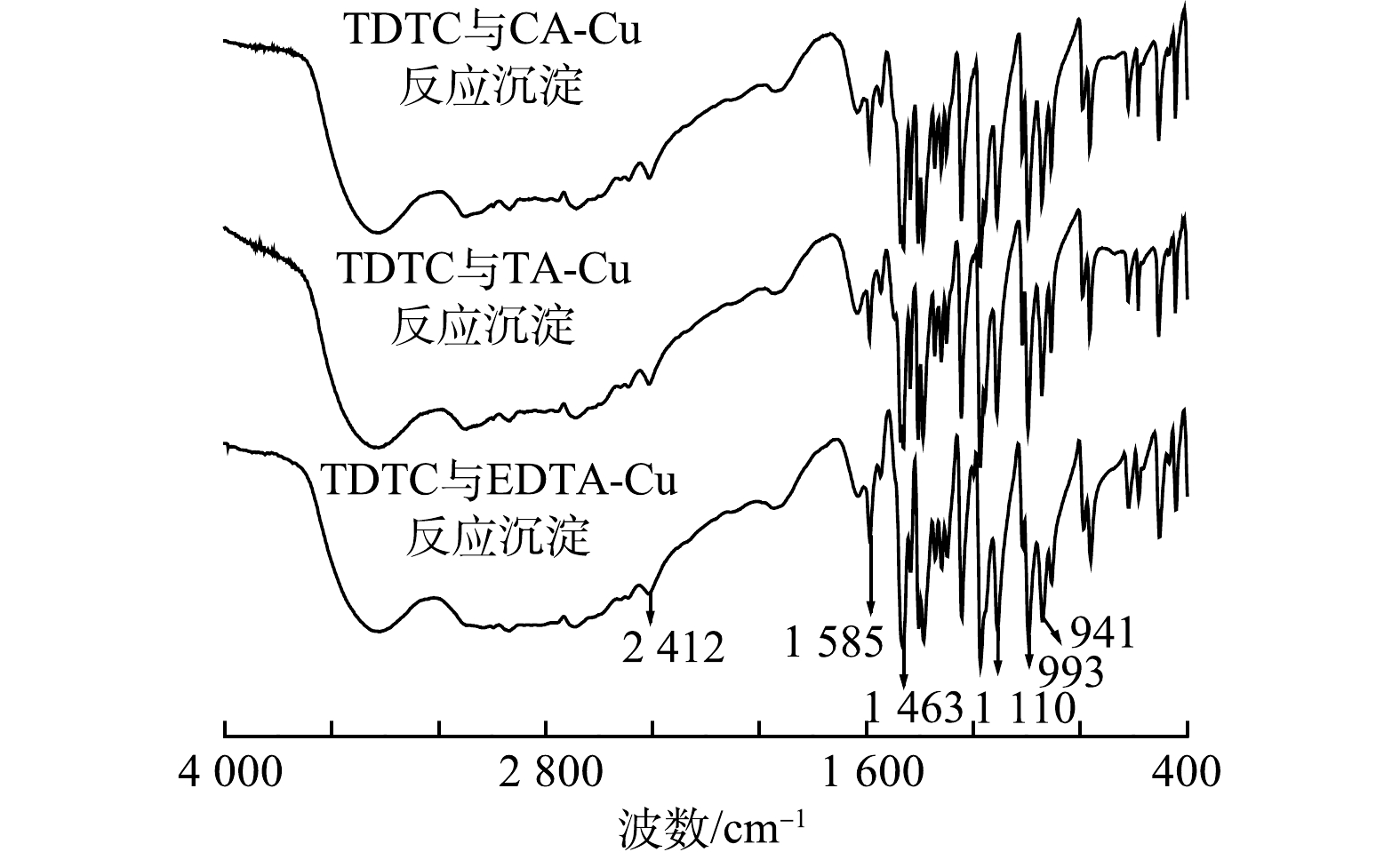
在上述最佳处理条件下,将捕集剂与3种模拟络合Cu废水反应得到棕红色沉淀,对其进行红外光谱表征,结果见图7。由图7可知,3种沉淀的红外谱图在强度及峰位上的表现差异不大,表明产生的3种沉淀在官能团类型大体上是一致的,而TA、CA、EDTA 3种络合剂官能团并不完全一致。由此可以推断,在螯合反应过程中产生的最终沉淀物不含各种络合剂,也可能是由于捕集剂和络合剂中部分官能团吸收峰重叠所致。
比较图6与图7结果可见,捕集剂在1 459 cm−1处的N—CS2的伸缩振动吸收峰发生位移至1 463 cm−1处且吸收峰强度减小,2 557 cm−1处的—SH特征吸收峰在与络合Cu反应之后消失,1 110、993、941 cm−1处的C—S振动伸缩峰与1 243 cm−1处的C=S特征吸收峰强度有所降低。以上峰变化的结果表明,反应的主要基团—CSS与络合剂上的Cu发生螯合反应,S的电负性减小,—CSS的共轭体系发生改变。同时,1 459 cm−1处的N—CS2的伸缩振动吸收峰的的位移与1 633 cm−1处的N—H峰强的变化可能是由于N与Cu形成配位键所致。
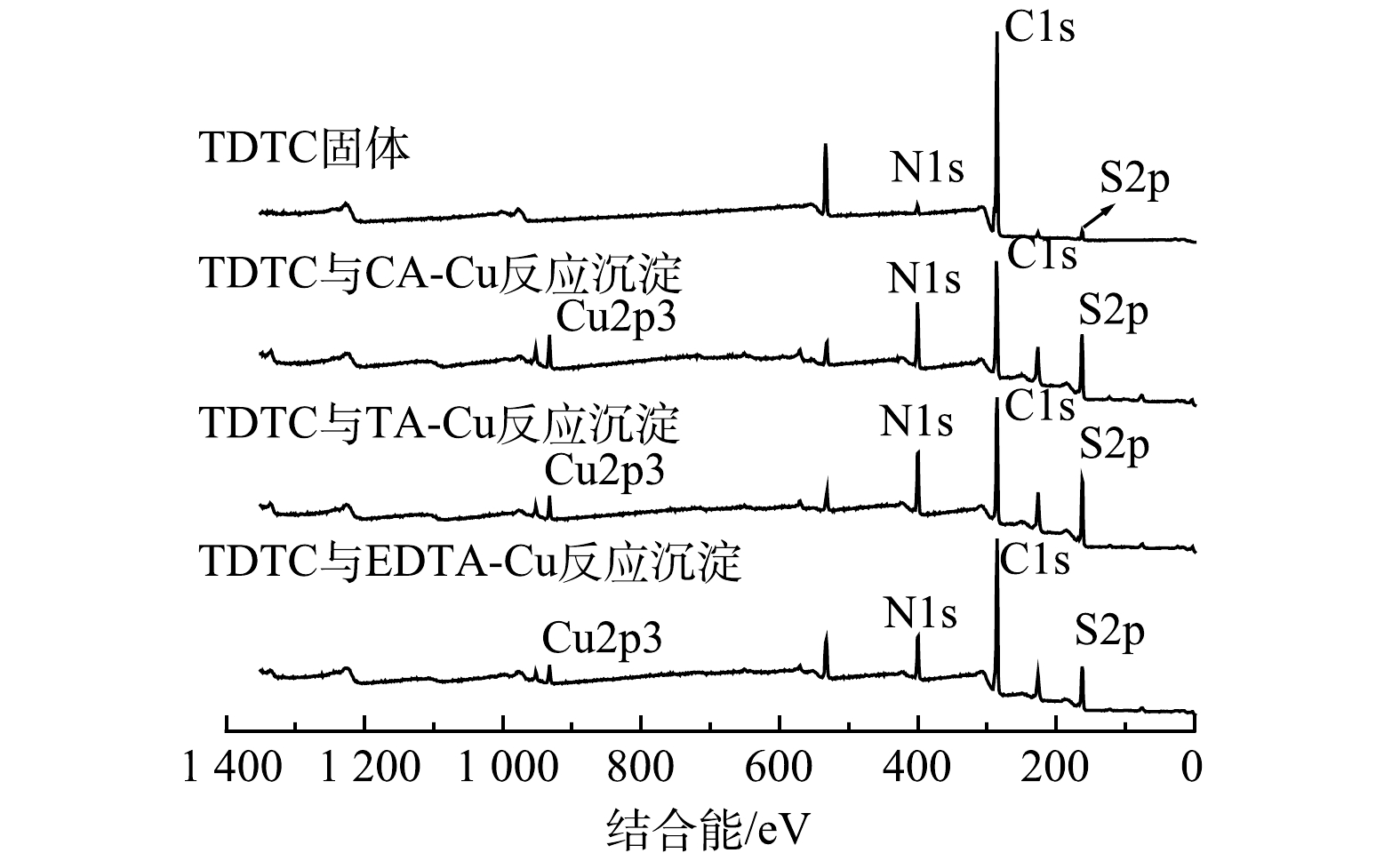
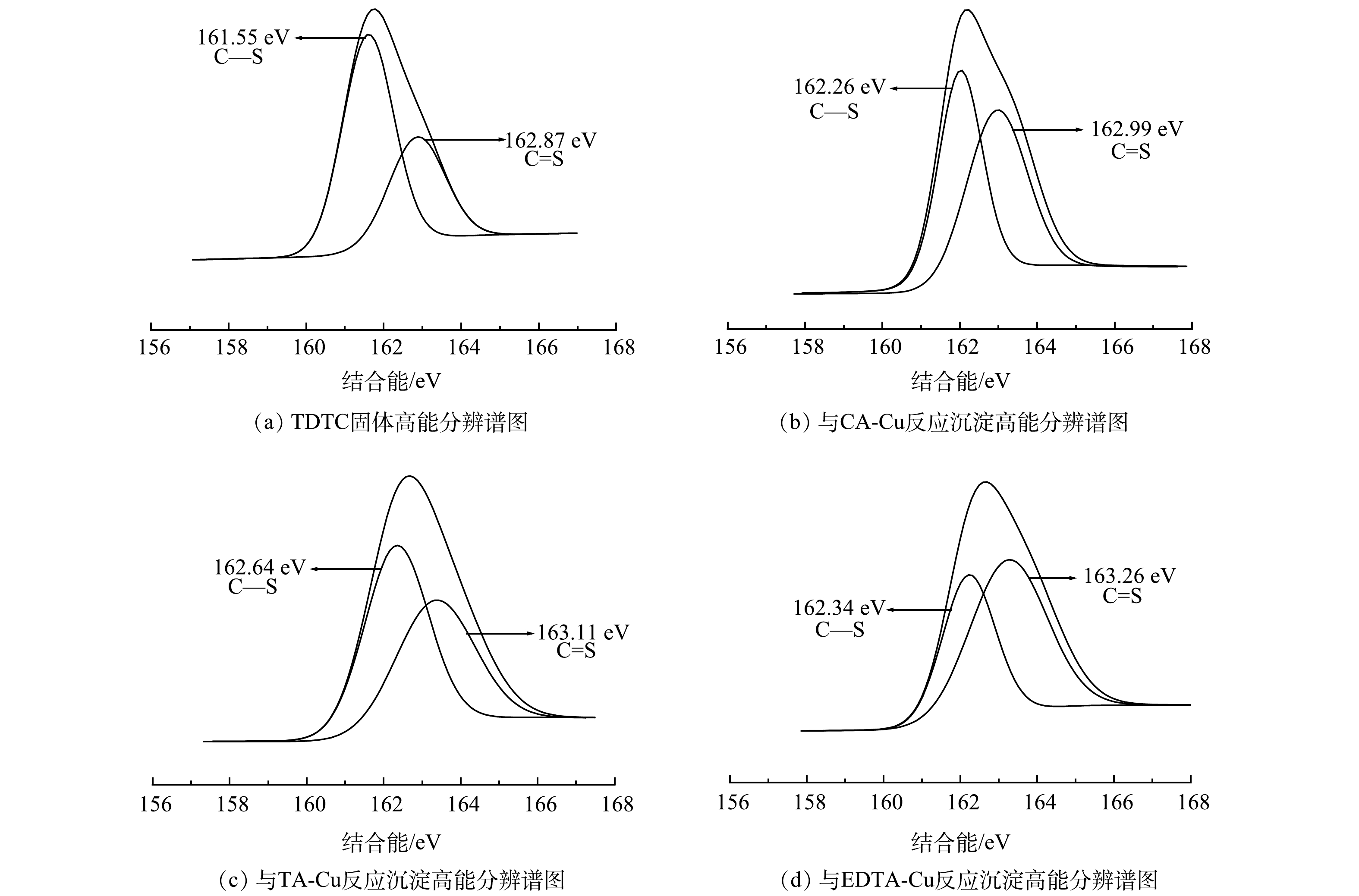
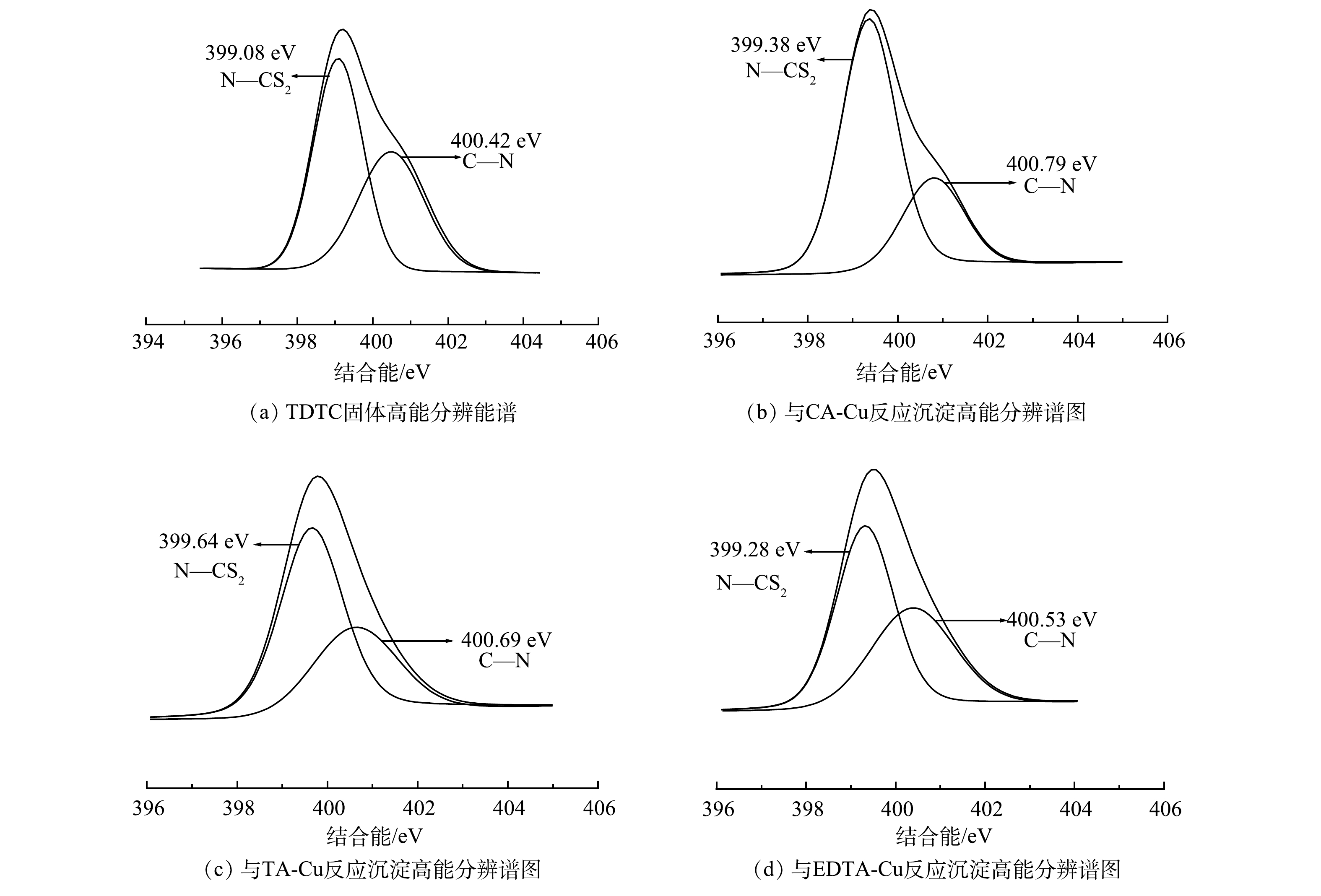
4) XPS分析。利用XPS来分析TDTC及其捕集3种不同络合Cu前后表面化学组成与结构变化及键合机理。图8为TDTC及3种螯合沉淀的XPS谱图,图9和图10为TDTC及3种螯合沉淀的S2p、N1s的XPS高分辨能谱图。由图8可知,在TDTC与CA-Cu、TA-Cu与EDTA-Cu反应形成沉淀的宽谱扫描能谱中均出现Cu2p3峰信号,而TDTC固体没有相应的峰,表明TDTC在与络合剂对Cu的竞争中成功将Cu螯合。由图9可知,TDTC与TDTC-Cu中二硫代羧基基团C—S键的S2p结合能分别为161.55 eV和162.64、162.26、162.34 eV,捕集后产生的3种沉淀S2p的结合能都大于前者。这是因为TDTC-Cu中二硫代氨基羧基中C—S的S原子螯合过程中向Cu贡献了电子,使反应后结合能升高[38]。TDTC和TDTC-Cu中二硫代羧基中C=S的S2p结合能分别为162.87 eV和163.11、162.99、163.26 eV,变化很小,说明DTC中C=S键也较微弱地参与了与Cu的螯合过程。图10中N1s高分辨率能谱图由N—CS2和C—N 2个峰组成,TDTC与TDTC-Cu中N—CS2键的结合能分别为399.08、399.64、399.38和399.28 eV,TDTC与TDTC-Cu中C—N键中N1s结合能分别为400.42、400.79、400.69与400.53 eV,后者结合能相较于前者稍有增加。这表明DTC基团中的N原子与Cu之间可能存在一定的配位作用,致使N原子上的电子向Cu移动。以上结果表示TDTC在与络合Cu反应过程中S、N与Cu存在配位作用,并主要通过二硫代羧基的螯合配位作用将Cu捕集,与红外光谱分析结果基本一致。
2.6 与其他DTC类捕集剂对铜去除效果的比较
本研究中合成的TDTC与其他研究中的重金属捕集剂的比较见表2。由表2可见,虽然本研究中初始铜浓度较高,但捕集剂与铜的质量比较低,质量比也更能说明其捕集性能。TDTC表现出更突出的捕集性能,在较宽pH范围内TDTC对络合Cu的去除率达99.6%以上,而表中其他捕集剂均在90%以下。
表 2 TDTC与其他重金属捕集剂去除效果对比Table 2. Comparison of heavy metal removal effect between TDTC and other sulfur-containing heavy metal chelation agents捕集剂种类 Cu形态 Cu质量浓度/(mg·L−1) pH 投加质量比 去除率/% 文献来源 DDTC EDTA-Cu 5 3.0 DDTC/Cu=5.4∶1 30~35 [39] DTMPAM EDTA-Cu 25 3.0 DTMPAM/Cu=9∶1 65~70 [26] DTC-S EDTA-Cu 5 9.0 DTC-S/Cu=6∶1 85~90 [40] TDTC EDTA-Cu 50 3.0~9.0 TDTC/Cu=6∶1 >99.6 本研究 | Show TableDownLoad:
CSV
二硫代羧酸盐与铜配位时,1个Cu2+需要2个二硫代羧基。DDTC与DTC-S属于小分子螯合剂,螯合基团数量为1。以DDTC为例,与铜形成C9H20N2S4Cu的类双环结构,其中C=S中的S也会微弱参与螯合,在质量比接近情况下,TDTC较DDTC二硫代羧基相对量增加,与Cu发生螯合作用的官能团变多,更利于Cu的去除。此外,拥有多个螯合官能团比只含有单个螯合团的捕集剂产生的絮体更大,提升了去除效率。DTMPAM为高分子螯合剂,聚合长链上有多个螯合基团,但在投加量大于TDTC情况下去除率只有70%左右。这可能是其部分螯合基团因空间位阻未能与金属螯合,没有充分反应,导致利用率不高。相比之下,1个TDTC分子可以提供2个二硫代羧基参与螯合成环,捕集性能得以提高。在TDTC与铜原子螯合时,推测其两端的二硫代羧基分别与2个Cu2+配位,这一点尚需进一步深入研究。
3. 结论
1)采用三乙烯四胺和CS2为原料,在混合溶剂中(V乙二醇/
VH2O 2)在pH为3.0~9.0、TDTC为1 mmol·L−1、PAM为1 mg·L−1、反应时间为3 min的条件下,3种初始质量浓度为50 mg·L−1的络合Cu溶液中,Cu去除率达到99.6%以上,络合态Cu可被TDTC有效去除,残留Cu质量浓度均小于0.2 mg·L−1,符合水污染物特别排放要求中关于Cu的排放限值要求。
3) TDTC捕集络合Cu主要通过与原有络合剂竞争及二硫代羧基与Cu螯合,将可溶性的络合Cu变为难溶的沉淀物而去除。
-

图 3 MnFeCu-LDHs活化PMS降解CTC效果图
Figure 3. Degradation efficiency of CTC by PMS activation with MnFeCu-LDHs
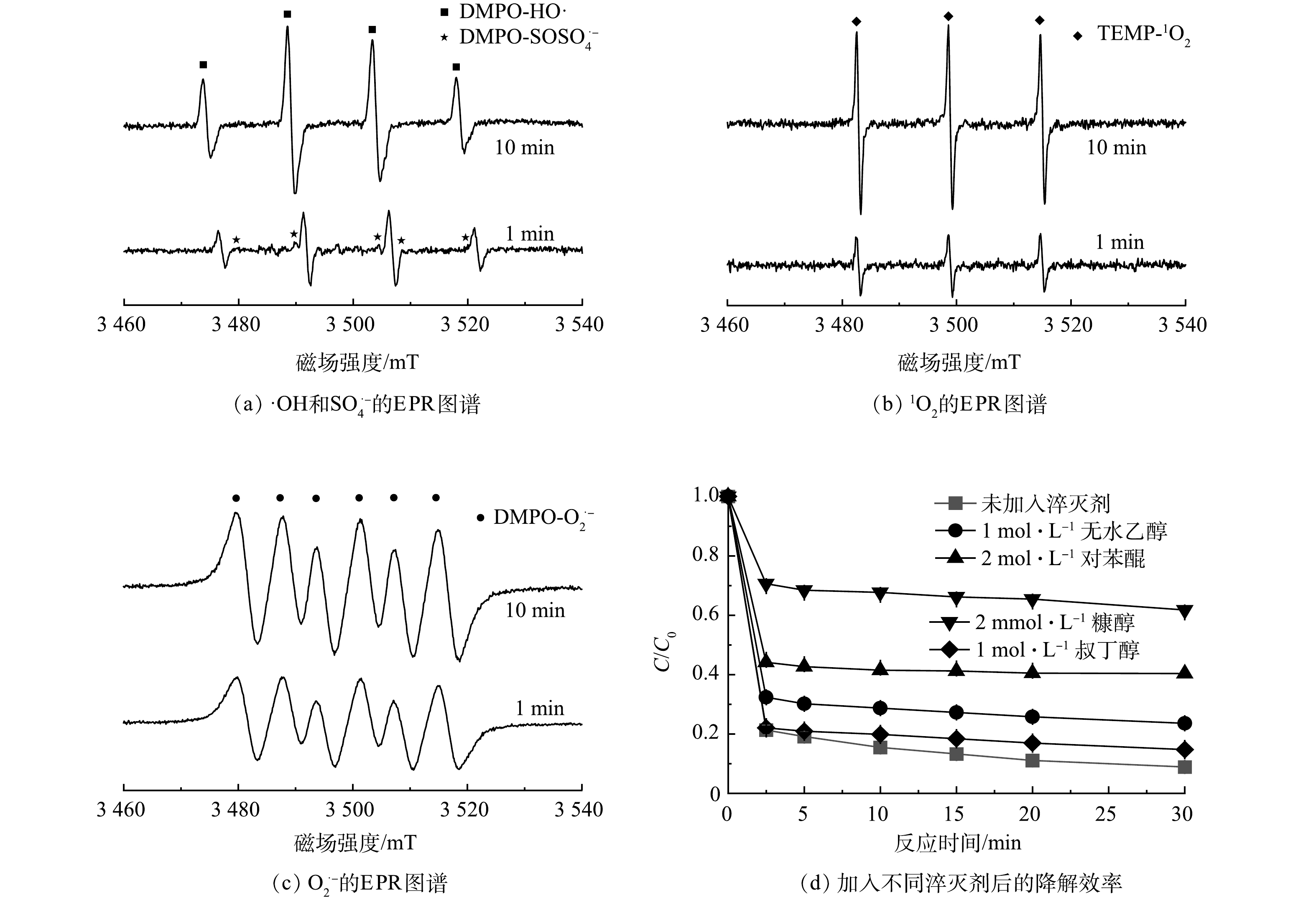
图 8 自由基淬灭对CTC降解效果的影响及MnFeCu-LDHs/PMS体系EPR图谱
Figure 8. Effect of radicals quenching on CTC degradation and EPR spectra of MnFeCu-LDHs/PMS system

图 9 反应前后锰铁铜类水滑石 XPS光谱
Figure 9. XPS spectra of Mn-Fe-Cu LDH before and after reaction.
-
[1] 曲久辉, 赵进才, 任南琪, 等. 城市污水再生与循环利用的关键基础科学问题[J]. 中国基础科学, 2017, 19: 6-12. doi: 10.3969/j.issn.1009-2412.2017.05.002 [2] 卫先宁, 季民, 李茹莹. 再生水中药品及个人护理品分布和环境风险分析[J]. 环境科学与技术, 2021, 43(12): 1-6. doi: 10.19672/j.cnki.1003-6504.2020.12.028 [3] BEN W W, ZHU B, YUAN X J, et al. Occurrence, removal and risk of organic micropollutants in wastewater treatment plants across China: Comparison of wastewater treatment processes[J]. Water Research, 2017, 130: 38-46. [4] MIKLOS D B, REMY C, JEKEL M, et al. Evaluation of advanced oxidation processes for water and wastewater treatment: A critical review[J]. Water research, 2018, 139: 118-131. doi: 10.1016/j.watres.2018.03.042 [5] LIU J, YANG Q, WANG D, et al. Enhanced dewaterability of waste activated sludge by Fe (II)-activated peroxymonosulfate oxidation[J]. Bioresource technology, 2016, 206: 134-140. doi: 10.1016/j.biortech.2016.01.088 [6] BOCZKAJ G, FERNANDES A. Wastewater treatment by means of advanced oxidation processes at basic pH conditions: A review[J]. Chemical Engineering Journal, 2017, 320: 608-633. doi: 10.1016/j.cej.2017.03.084 [7] PANG X, GUO Y, ZHANG Y, et al. LaCoO3 perovskite oxide activation of peroxymonosulfate for aqueous 2-phenyl-5-sulfobenzimidazole degradation: Effect of synthetic method and the reaction mechanism[J]. Chemical Engineering Journal, 2016, 304: 897-907. doi: 10.1016/j.cej.2016.07.027 [8] OH W D, DONG Z, LIM T T. Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal: Current development, challenges and prospects[J]. Applied Catalysis B:Environmental, 2016, 194: 169-201. doi: 10.1016/j.apcatb.2016.04.003 [9] ZHAO X, NIU C, ZHANG L, et al. Co-Mn layered double hydroxide as an effective heterogeneous catalyst for degradation of organic dyes by activation of peroxymonosulfate[J]. Chemosphere, 2018, 204: 11-21. doi: 10.1016/j.chemosphere.2018.04.023 [10] GERKEN J B, MCALPIN J G, CHEN J Y C, et al. Electrochemical water oxidation with cobalt-based electrocatalysts from pH 0–14: the thermodynamic basis for catalyst structure, stability, and activity[J]. Journal of the American Chemical Society, 2011, 133(36): 14431-14442. doi: 10.1021/ja205647m [11] GUO W, SU S, YI C, et al. Degradation of antibiotics amoxicillin by Co3O4-catalyzed peroxymonosulfate system[J]. Environmental progress & sustainable energy, 2013, 32(2): 193-197. [12] SIMONSEN L O, HARBAK H, BENNEKOU P. Cobalt metabolism and toxicology: A brief update[J]. Science of the Total Environment, 2012, 432: 210-215. doi: 10.1016/j.scitotenv.2012.06.009 [13] FENG Y, WU D, ZHOU Y, et al. A metal-free method of generating sulfate radicals through direct interaction of hydroxylamine and peroxymonosulfate: Mechanisms, kinetics, and implications[J]. Chemical Engineering Journal, 2017, 330: 906-913. doi: 10.1016/j.cej.2017.08.034 [14] FAN X, CAO Q, MENG F, et al. A Fenton-like system of biochar loading Fe–Al layered double hydroxides (FeAl-LDH@ BC)/H2O2 for phenol removal[J]. Chemosphere, 2021, 266: 128992. doi: 10.1016/j.chemosphere.2020.128992 [15] XU X, LIN R, DENG X, et al. In situ synthesis of FeOOH-coated trimanganese tetroxide composites catalyst for enhanced degradation of sulfamethoxazole by peroxymonosulfate activation[J]. Separation and Purification Technology, 2021, 275: 119184. doi: 10.1016/j.seppur.2021.119184 [16] 吴德勇, 苏积珊. Cu2O/CuO/BC复合材料活化PMS降解TC[J]. 环境科学与技术, 2022, 45(3): 188-196. [17] 林陆健, 汤帅, 孙璇, 等. 铅离子和四环素在微塑料表面的吸附机理与协同效应[J]. 环境科学学报, 2021, 41(10): 4022-4031. doi: 10.13671/j.hjkxxb.2021.0052 [18] ZENG H, ZHANG W, DENG L, et al. Degradation of dyes by peroxymonosulfate activated by ternary CoFeNi-layered double hydroxide: Catalytic performance, mechanism and kinetic modeling[J]. Journal of colloid and interface science, 2018, 515: 92-100. doi: 10.1016/j.jcis.2018.01.016 [19] WANG G, LI D, WANG S, et al. Ternary NiFeMn layered metal oxide (LDO) compounds for capacitive deionization defluoridation: The unique role of Mn[J]. Separation and Purification Technology, 2021, 254: 117667. doi: 10.1016/j.seppur.2020.117667 [20] HOU L, LI X, YANG Q, et al. Heterogeneous activation of peroxymonosulfate using Mn-Fe layered double hydroxide: Performance and mechanism for organic pollutant degradation[J]. Science of the Total Environment, 2019, 663: 453-464. doi: 10.1016/j.scitotenv.2019.01.190 [21] GONG C, CHEN F, YANG Q, et al. Heterogeneous activation of peroxymonosulfate by Fe-Co layered doubled hydroxide for efficient catalytic degradation of Rhoadmine B[J]. Chemical engineering journal, 2017, 321: 222-232. doi: 10.1016/j.cej.2017.03.117 [22] AHMADI M, GHANBARI F. Combination of UVC-LEDs and ultrasound for peroxymonosulfate activation to degrade synthetic dye: Influence of promotional and inhibitory agents and application for real wastewater[J]. Environmental Science and Pollution Research, 2018, 25(6): 6003-6014. doi: 10.1007/s11356-017-0936-8 [23] HUANG G X, WANG C Y, YANG C W, et al. Degradation of bisphenol A by peroxymonosulfate catalytically activated with Mn1.8Fe1.2O4 nanospheres: Synergism between Mn and Fe[J]. Environmental Science & Technology, 2017, 51(21): 12611-12618. [24] DENG J, WU G, YUAN S, et al. Ciprofloxacin degradation in UV/chlorine advanced oxidation process: Influencing factors, mechanisms and degradation pathways[J]. Journal of Photochemistry and Photobiology A:Chemistry, 2019, 371: 151-158. doi: 10.1016/j.jphotochem.2018.10.043 [25] TAN Y, LI C, SUN Z, et al. Natural diatomite mediated spherically monodispersed CoFe2O4 nanoparticles for efficient catalytic oxidation of bisphenol A through activating peroxymonosulfate[J]. Chemical Engineering Journal, 2020, 388: 124386. doi: 10.1016/j.cej.2020.124386 [26] LI Z, SUN Y, HUANG W, et al. Innovatively employing magnetic CuO nanosheet to activate peroxymonosulfate for the treatment of high-salinity organic wastewater[J]. Journal of Environmental Sciences, 2020, 88: 46-58. doi: 10.1016/j.jes.2019.07.011 [27] XU Y, AI J, ZHANG H. The mechanism of degradation of bisphenol A using the magnetically separable CuFe2O4/peroxymonosulfate heterogeneous oxidation process[J]. Journal of Hazardous Materials, 2016, 309: 87-96. doi: 10.1016/j.jhazmat.2016.01.023 [28] DUAN P, MA T, YUE Y, et al. Fe/Mn nanoparticles encapsulated in nitrogen-doped carbon nanotubes as a peroxymonosulfate activator for acetamiprid degradation[J]. Environmental Science:Nano, 2019, 6(6): 1799-1811. doi: 10.1039/C9EN00220K [29] YANG S, WU P, LIU J, et al. Efficient removal of bisphenol A by superoxide radical and singlet oxygen generated from peroxymonosulfate activated with Fe0-montmorillonite[J]. Chemical Engineering Journal, 2018, 350: 484-495. doi: 10.1016/j.cej.2018.04.175 [30] DONG X, REN B, SUN Z, et al. Monodispersed CuFe2O4 nanoparticles anchored on natural kaolinite as highly efficient peroxymonosulfate catalyst for bisphenol A degradation[J]. Applied Catalysis B:Environmental, 2019, 253: 206-217. doi: 10.1016/j.apcatb.2019.04.052 -

 点击查看大图
点击查看大图

计量
- 文章访问数: 7356
- HTML全文浏览数: 7356
- PDF下载数: 110
- 施引文献: 0
