

 下载:
下载:




-
抗菌类药物抗生素的广泛应用,给人类带来了便利,也对环境造成了危害[1]。四环素类抗生素很难被动物吸收,大部分通过排泄残留在养殖废水里[2-3],不仅对水生动植物的生长和繁衍造成不利影响[4-5],还危害到人体健康,破坏人体免疫系统[6]。抗生素废水是一种有机废水,毒性大,难降解[7]。以现有的研究水平很难完全去除水环境中所有抗生素[8-9]。因此,四环素废水的高效处理方法已成为研究热点[10]。高压脉冲放电等离子体技术是一种处理难降解有机物的高级氧化技术,因其处理效率高,无二次污染,而受到广泛关注[11-12]。仇聪颖等[13]采用多针-网式反应器,通过电晕放电等离子体技术循环处理酸性红73(AR73)的模拟废水,放电30 min后AR73降解率可以达到83.20%。Hao等[14]研究了脉冲放电处理水溶液中的四环素,结果表明,初始浓度为50 mg·L−1的四环素降解效率可达到92.3%。然而,单纯的等离子体技术存在能量利用率低,选择性差等问题。催化剂有较好的产物选择性,且可降低反应活化能,达到节能要求[15]。因此,将催化剂与脉冲放电等离子体技术相结合,能有效提高废水降解效率,引起了科研工作者的关注[16-17]。Yan等[18]采用介质阻挡放电(DBD)与纳米氧化锌协同降解双酚A,结果表明,在添加纳米氧化锌50 mg·L−1时降解效果最好,降解效率为85.4%,比单一DBD体系提高了17%。
本课题组已对催化剂与高压脉冲放电等离子体协同处理废水技术进行了相关研究,董冰岩等[19]确定了本次实验最佳的基础参数(放电电压、脉冲频率、气体流量、电极间距、溶液的初始浓度、溶液电导率)。Ag/BiVO4复合型催化剂具有较窄的带隙、无毒和高稳定性等优点,可与放电产生的H2O2、O3作用,产生大量·O2-、·OH等强氧化性活性粒子,促进等离子通道的形成,加强反应速率[20]。为进一步提高TCH的去除效率,本实验通过高压脉冲放电技术协同Ag 改性的BiVO4催化剂对TCH去除率进行研究,并通过XRD、SEM、BET 对所有催化剂进行表征分析[21],为脉冲放电协同催化剂去除TCH提供一定的科学指导意义。
-
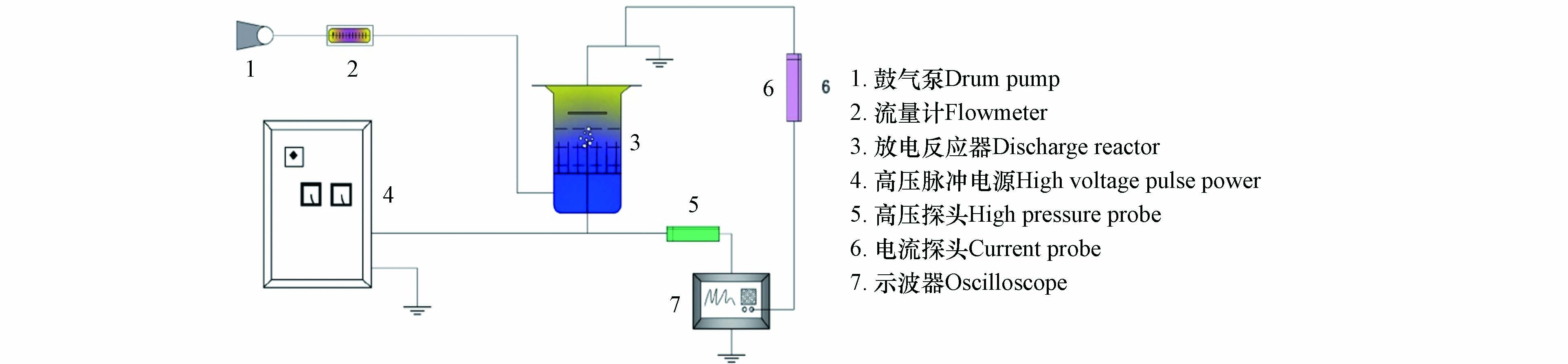
实验装置系统图如图1所示。主要由高压脉冲电源(0—60 kV,0—200 Hz)、流量控制装置、等离子体反应器和脉冲测量装置4个部分构成。高压脉冲电源由直流电源,两个火花间隙,储能电容和脉冲电容组成。气流量控制装置采用IPX4 ACO-9610型鼓气泵和D07-19CM型流量计。脉冲测量装置包括高压探头、流量探头、示波器和高效液相色谱仪(TeK P6015A 、DSO-X-3054A、Agilent1260 LC)。
-
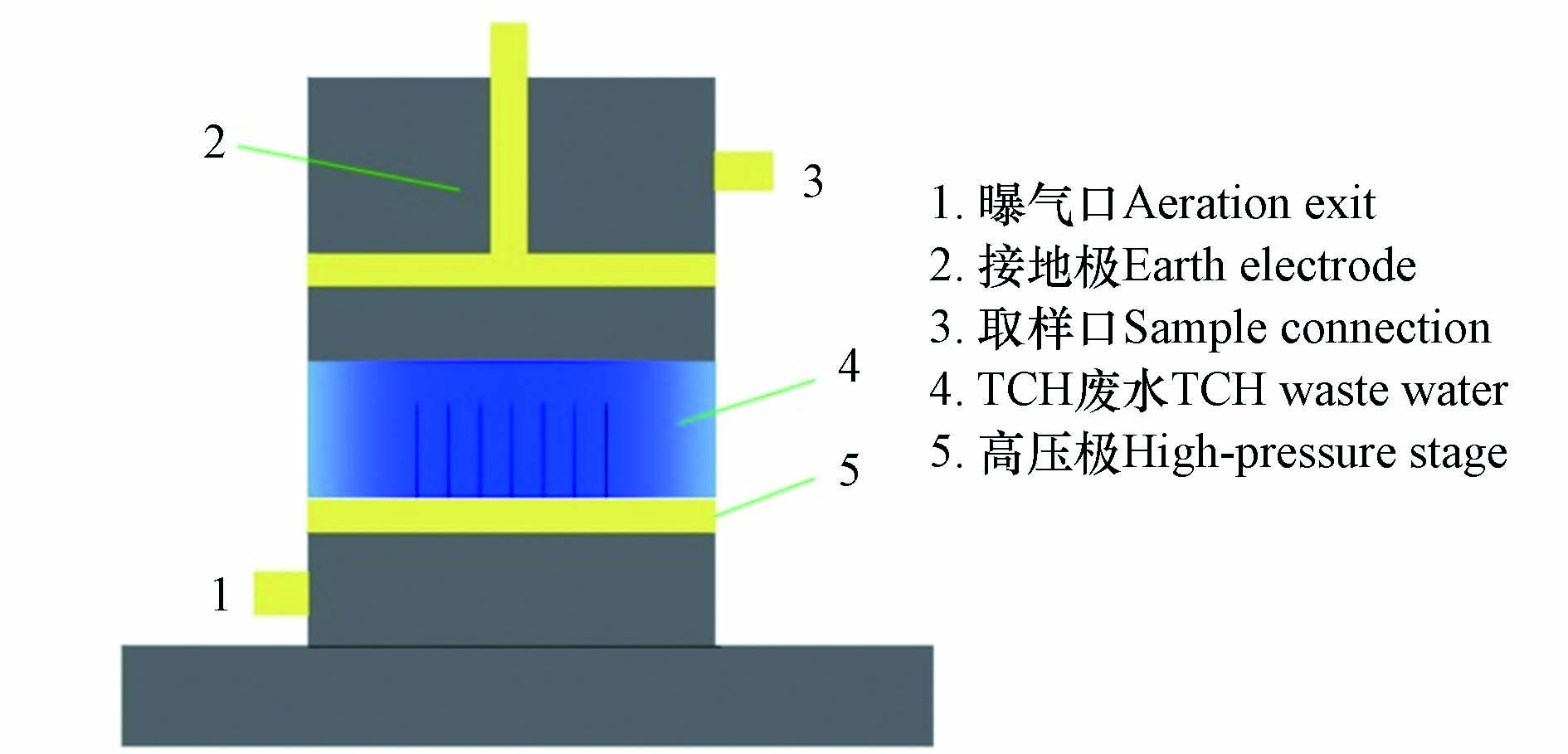
实验反应器结构如图2所示。实验反应器利用有机玻璃制成,内径为80 mm,总高度为140 mm。中间有7根不锈空心钢针构成了反应器的高压极,并利用鼓气泵不断向溶液中鼓气。不锈钢板作为接地极,上下移动不锈钢板来调节两个电极之间的距离。
-
Ag/BiVO4催化剂的制备方法为:首先将五水硝酸铋溶解于10%稀硝酸溶液中,滴加柠檬酸搅拌,形成溶液A;之后将0.01 mol偏钒酸铵加入80 °C超纯水中,滴加柠檬酸搅拌,形成溶液B。将B溶液缓慢倒入A溶液形成C溶液,不断加热搅拌,并滴加氨水,使溶液pH在6.5—7之间,保持温度为80 °C,匀速搅拌至形成深蓝色凝胶为止。将深蓝色凝胶在80 °C鼓风烘干箱中烘干10 h,冷却后在马弗炉中焙烧5 h,用研钵研磨成粉末,得到纯BiVO4催化剂。分别称取一定量BiVO4,Ag粉,倒入蒸馏水,40 °C恒温水浴,并用多头恒温磁力搅拌器持续搅拌 2 h,超声分散15 min,10 °C以下烘干。研碎后在马弗炉中,以 2 °C·min−1的速度升温至 500 °C,持续处理3 h,得到有活化性能的Ag/BiVO4,研碎至200目过筛状态待用。
-
(1)扫描电镜分析(SEM)
取不同比例Ag/BiVO4复合催化剂粉末,在金属托盘上用导电胶带粘贴样品粉末并依次编号。随后将样品放入电镜试样镀膜仪进行喷金处理,再使用MLA650F场发射扫描电子显微镜观察不同比例的Ag/BiVO4样品的微观结构。
(2)X射线衍射(XRD)
采用XRD(XRD-2700射线衍射仪,丹东方圆公司)对催化剂表征分析,绘制相应衍射图谱,进一步判断催化剂的物相结构。
(3)比表面积及孔隙度分析(BET)
采用康塔全自动比表面积及孔隙度分析仪对所制备催化剂表面积及孔径进行测试分析。运用BET多点测试法计算比表面积,BJH法计算孔径分布。
-
本实验的最佳放电条件依据本课题前期实验测试确定为:放电电压24 kV,频率60 Hz,气体流量2.5 L·min−1,电极间距7 mm,溶液初始浓度100 mg·L−1,溶液电导率200 μs·cm−1。
-
图3分别为BiVO4、1%Ag/BiVO4和3%Ag/BiVO4等3种催化剂的SEM表征,展示了3种催化剂颗粒在放大5 μm时微观形貌。由图3可见,纯BiVO4催化剂颗粒分布松散,形状不规则且有少量棒状结构(如图3a);当掺杂1%Ag后,催化剂颗粒变为大小均匀的蚕茧状,且颗粒之间存在许多微孔,孔隙的存在有利于放电产生的活性粒子进入与TCH分子的吸附。催化剂表面连结着许多小颗粒,增加了活性位点,有利于提高催化剂的比表面积 (如图3b);当掺杂 3% Ag时,催化剂形貌有团聚、结板的现象,且出现大块长形晶型(如图3c),减小了催化剂的比表面积,使TCH分子与活性粒子的碰撞率降低,不利于TCH的去除。这表明过量Ag掺杂会抑制颗粒的生长,这一种现象与 Wang 课题组[22]中的发现相似。通过SEM表征结果表明,Ag的最佳掺杂量为1%。
-
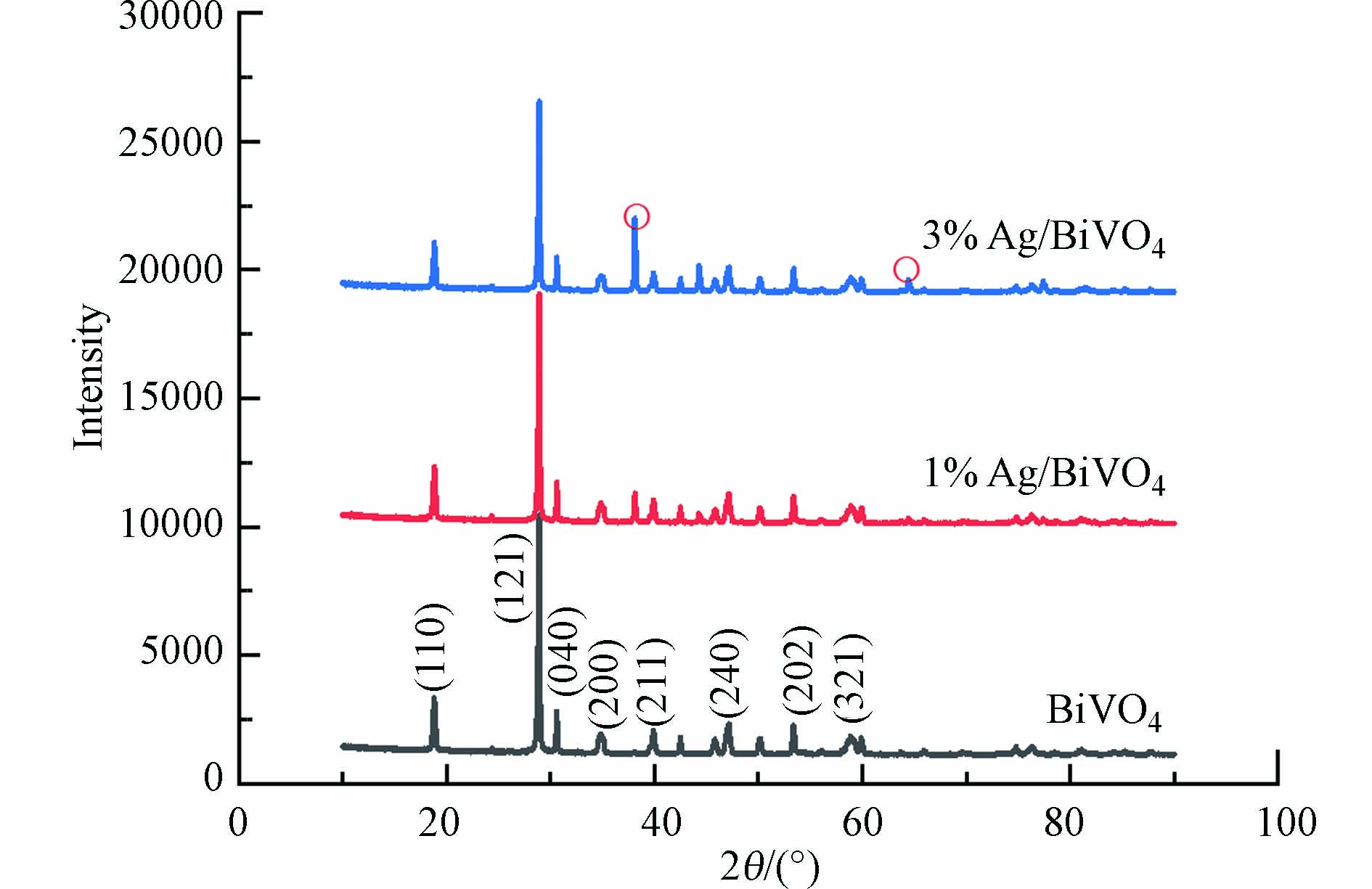
如图4所示,从下往上依次是BiVO4、1%Ag/BiVO4、3%Ag/BiVO4的3种催化剂XRD图谱。由XRD图可见,3种催化剂均出现尖锐的衍射峰。在X射线衍射图谱中,峰出现在2θ值18.8°、28.8°、30.5°、34.8°、39.9°、47.2°、53.3°、58.8°处,分别对应了单斜晶型BiVO4 标准卡(JCPDS 14-0688)的(110)、(121)、(040)、(200)、(211)、(240)、(202)、(321)[23]。当Ag掺杂量为3%时,XRD图中可清晰看到,在2θ值38.1°、64.4°处有特征峰出现,参照Ag的标准卡(JCPDS 04-0783),可以确定其为Ag单质[24],没有发现其他相或杂质,可见所制复合型催化剂Ag/BiVO4纯度较高。
由图4可知,虽Ag的掺杂量不同,但衍射峰的位置大致相似,强度相对减弱,说明负载Ag没有改变BiVO4的晶型结构,仍然保持完好的孔道结构。由于Ag3+与Bi3+的半径不同,使得与纯BiVO4相比,掺杂1%和3%Ag后的衍射峰有较小角度的偏移。此外,掺杂Ag越多,所对应最高峰的强度大大减弱,这意味着掺杂过量的Ag阻碍了BiVO4的结晶[25]。
-
图5为所制复合型催化剂1%Ag/BiVO4的N2吸附-脱附等温线(a)和孔径分布(b)。在图5(a)中,样品的氮气吸附-脱附等温线呈现Ⅳ型等温线,且明显为H3型滞后现象,表明催化剂表面存在中孔结构。由图5(b)可知,样品的孔径分布大多在0—10 nm范围内,且存在小于5 nm的介孔,有利于吸附和解吸,进一步增强催化剂表面的活性位点。另外,根据实验结果,运用BET方程和BJH法计算出样品的比表面积为26.650 m2 ·g−1、孔容为0.168 cm3·g−1、孔径为3.986 nm。
-
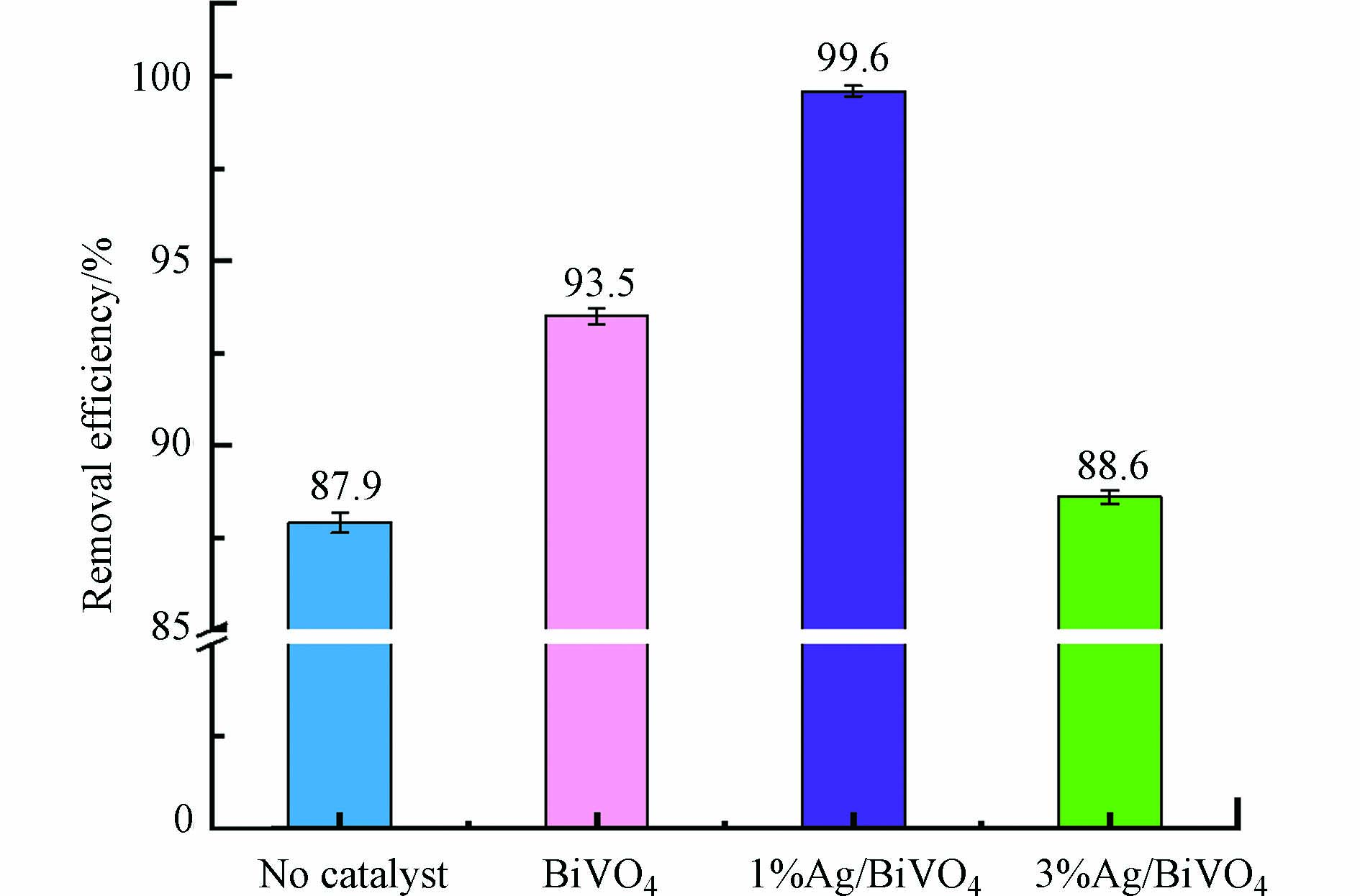
为研究不同Ag负载量对TCH去除效果的影响,在最佳放电条件(放电电压24 kV,频率60 Hz,气体流量2.5 L·min−1,电极间距7 mm,溶液初始浓度100 mg·L−1,溶液电导率200 μs·cm−1)基础上,分别加入BiVO4,1%Ag/BiVO4,3%Ag/BiVO4的3种催化剂,放电60 min,并与无催化剂情况下单独脉冲放电(HVPD)实验进行对比,结果如图6所示。
由图6可见,加入催化剂后,盐酸四环素的降解效率均有所提升,表明高压脉冲放电等离子体技术与催化剂技术存在明显的协同作用。可能是因为催化剂表面积大,能与TCH分子充分接触,且催化剂结构存在许多孔隙,明显有利于TCH分子的吸附,增加了TCH分子在放电区的停留时间,使放电产生的活性粒子能与TCH分子充分反应,提高TCH降解效率。并且可以看出,增加Ag负载量,TCH去除率先增大再减小。纯BiVO4对TCH的去除率为93.5%;在加入1%Ag/BiVO4催化剂,TCH去除率最高,为99.6%;而加入3%Ag/BiVO4,TCH的去除率却只有88.6%。由于掺杂过量的Ag,会把催化剂表面孔道堵塞,覆盖活性位点,使催化效率降低,这种现象与先前报道[26]的解释是一致的。所以该实验Ag负载量的最佳值为1%,与之前所做SEM表征结果相符。
-
为研究催化剂投加量对TCH去除效果的影响,改变1%Ag/BiVO4催化剂投加量,分别为0.05、0.1、0.15、0.2、0.25 g,在最佳放电条件基础上,放电60 min,结果如图7所示。
由图7可见,随着1%Ag/BiVO4催化剂投加量的增大,TCH的去除率先增大再减小。当向反应器内投加0.1 g和0.15 g催化剂时,TCH去除率达100%,但投加催化剂0.2 g时,TCH去除率却下降到94.3%。可以看出,催化剂投加量对有机废水的降解效率有较大影响。可能是由于在放电区,低温等离子体会放出波长250—450 nm,能量3—4 eV的紫外辐射,可作为光能利用,向反应体系中添加少量催化剂,催化剂吸收光能,产生电子空穴对,会与吸附在其表面的水和氧等物质作用,产生强氧化性自由基·OH、·O2;且催化剂还能与放电产生的H2O2、O3等活性粒子作用,形成多效催化,使体系中有更多·OH生成,充分降解稳定吸附在催化剂表面的TCH分子,提高TCH降解效率,同时提升脉冲放电体系中能量利用率。而投加过量催化剂,由于催化剂的不透明,使反应器内溶液变浑浊,抑制了紫外光的透射,阻碍等离子体通道的形成,使产生的活性物质减少。反应体系吸收的紫外光变少,同时聚集的催化剂使表面活性位点数量减少[27],因此TCH的去除率降低。从经济角度出发,本次实验最佳投加量为0.1 g。
-
为研究不同pH值对TCH去除效果的影响,在最佳放电条件基础上,投加0.1 g 1%Ag/BiVO4催化剂,改变反应体系的pH值,分别为2.3、5.3、8.3、11.3,放电60 min。结果如图8所示。
由图8可见,随着pH增大,TCH去除率先增大再减小。在pH为5.3时TCH的去除率最高,为99.9%,而pH为2.3、8.3和11.3时,TCH的去除率仅为92%、96.2%、92.2%。说明反应体系过酸或过碱都会影响TCH的去除效率。可能由于在pH过低时,氢离子会与放电产生的活性自由基·OH反应,使体系中产生的强氧化性活性粒子变少,降低TCH去除率;pH过高会加快O3分解速度,使与TCH反应的O3量减少,TCH去除率降低。所以该实验最佳溶液原始pH值为5.3。
-
为研究所制复合型催化剂Ag/BiVO4的稳定性,通过使用1%Ag/BiVO4多次催化降解TCH溶液。每次实验完成后,记录降解率,并将催化剂用水洗涤再在60 °C下干燥1 h,对其进行活化处理。在相同的条件下继续重复使用4个循环,测试TCH 降解效果。结果如图9所示。
由图9可见,在高压脉冲放电协同1%Ag/BiVO4催化剂去除TCH的实验中,催化剂连续使用4个循环得出的4次TCH的去除率分别为99.55%、98.6%、97.2%、96.8%,虽有所降低,但总体相差不大,第一次与第四次相比降解率下降了2.75%。可能由于在回收利用过程中,催化剂的质量会有些损失,且表面可能有反应过程中产生的中间产物吸附,堵塞孔隙,导致TCH去除率有稍许差别,但都在95%以上。由此可见,该催化剂稳定性非常高,可重复利用[28]。
-
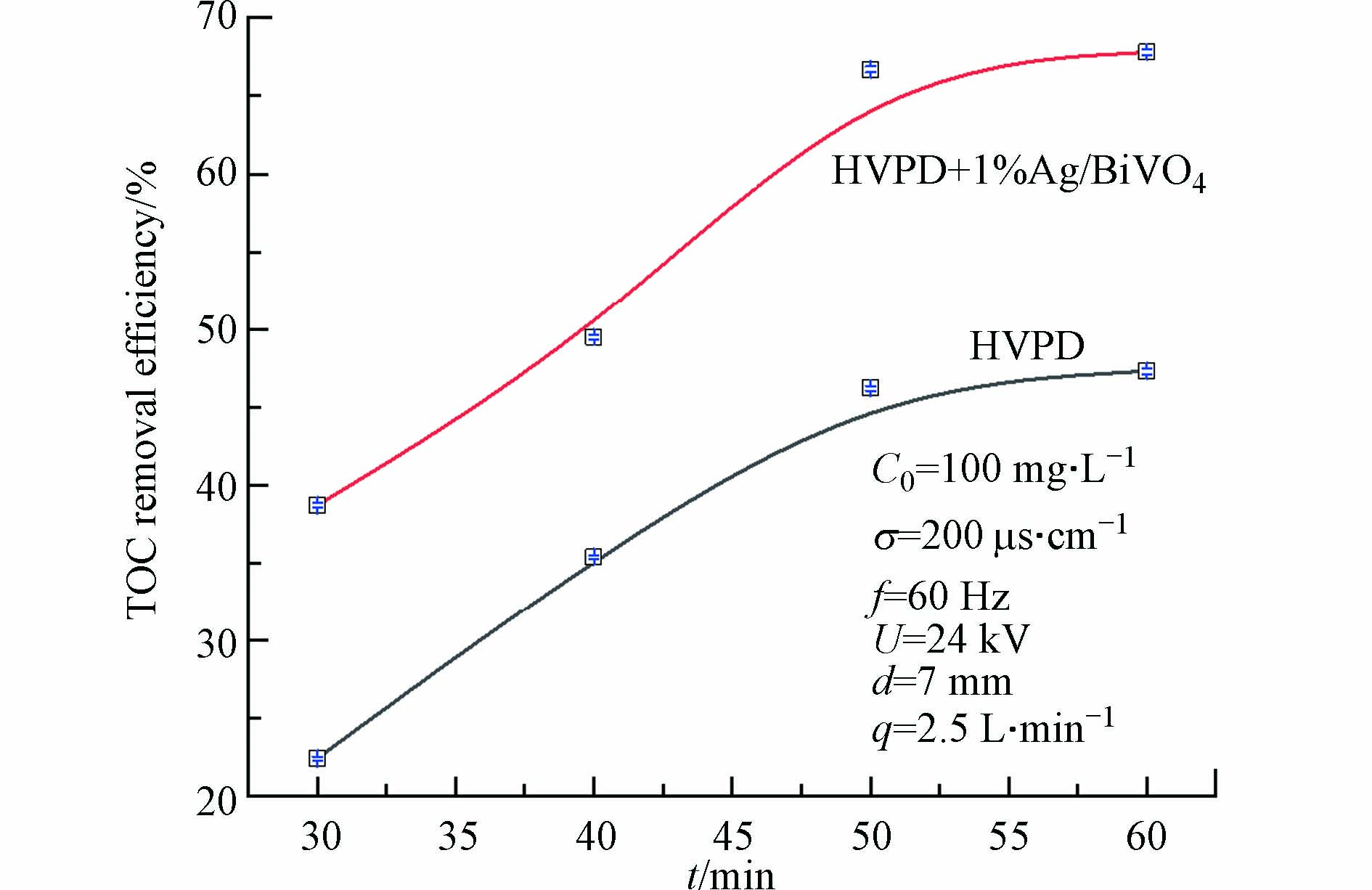
为研究TCH溶液中TOC的变化,在最佳放电条件基础上,投加0.1 g 1%Ag/BiVO4催化剂,放电60 min,分析TOC去除率随放电时间的变化规律。并与无催化剂情况下单独脉冲放电(HVPD)实验进行对比,结果如图10所示。由图10可见,放电时间延长,盐酸四环素溶液中TOC的去除率均逐渐增大。在单独脉冲放电过程中,放电60 min后,矿化率最高为47.3%,矿化程度偏低。而加入1%Ag/BiVO4催化剂与高压脉冲放电协同处理盐酸四环素废水,在放电60 min时,矿化率最高可达67.8%,与单独脉冲放电相比,加入催化剂后矿化率有了明显提升。与前文实验相比,在相同条件下,TCH的去除率高于矿化率。由此可知在高压脉冲放电协同1%Ag/BiVO4处理盐酸四环素过程中,只有一部分TCH分子被矿化为CO2和H2O,另一部分被转化为小分子物质或很难被矿化的中间降解产物。
-
(1)催化剂与脉冲放电等离子体存在协同作用,能有效提高TCH的降解效率,随着催化剂投加量的增多,TCH去除率先增大后减小,在投加0.1 g时,去除效果最好。
(2)负载Ag的BiVO4催化剂结合高压脉冲放电去除TCH效果更好。放电电压24 kV,频率60 Hz,气体流量2.5 L· min−1,电极间距7 mm,溶液的初始浓度100 mg·L−1,溶液电导率200 μs·cm−1时,投加掺杂1%Ag的BiVO4催化剂,TCH去除率可达到99.8%。矿化率最高达67.8%。
(3)pH值为高压脉冲放电结合Ag/BiVO4催化剂去除实验TCH实验的显著影响因素,测试结果显示随着pH的增大,TCH去除效率先增大后减小,在pH为5.3时,TCH的去除效果最佳为99.9%。
(4)Ag/BiVO4催化剂的表征技术显示,脉冲放电能改变催化剂的形貌和结构,掺杂一定量Ag使BiVO4催化剂表面更加密集,颗粒均匀,比表面积增大,反应所需活性位点增多,促进反应进行。
高压针-板脉冲火花放电协同Ag改性的复合型催化材料处理盐酸盐四环素
Treatment of tetracycline hydrochloride by high voltage needle-plate pulsed spark discharge combined with Ag modified composite catalytic materials
-
摘要: 采用高压针-板脉冲火花放电协同负载型光催化剂Ag/BiVO4对盐酸盐四环素(TCH)抗生素模拟废水进行去除实验。通过溶胶-凝胶与掺杂法合成复合型Ag/BiVO4催化剂,用X 射线衍射(XRD)、扫描电子显微镜(SEM)及BET表征技术分析Ag/BiVO4催化剂的基本特性,考察了不同Ag 负载量的催化剂、不同投加量和不同反应体系pH值对TCH去除率的影响。结果显示,在放电电压为24 kV,频率为 60 Hz,气体流量为2.5 L·min−1,电极间距为7 mm,溶液的初始浓度为100 mg·L-1,溶液电导率为200 μs·cm−1,同时添加含量为0.1 g 1%Ag/BiVO4催化剂与脉冲放电等离子体协同去除TCH的效果最好,盐酸四环素的去除率可达到99.8%,矿化率最高为67.8%。高压脉冲放电可以改变催化剂的晶形和结构,并且在掺杂一定量的Ag后BiVO4催化剂表面变得更加密集,颗粒均匀,比表面积增大,活性位点增多,促进了催化反应,进一步加强了对TCH的去除。Abstract: A high-voltage needle-plate pulsed spark discharge synergistically supported photocatalyst Ag/BiVO4 was employed for the removal experiment of tetracycline hydrochloride (TCH) antibiotic simulated wastewater. The composite Ag/BiVO4 catalyst was synthesized by sol-gel and doping method. X-ray diffraction (XRD), scanning electron microscope (SEM) and BET characterization techniques were used to analyze the basic characteristics of Ag/BiVO4 catalyst. The effects of catalysts with different Ag loads, dosage and pH values of reaction systems on TCH removal rate were investigated. When the concentration of TCH was 100 mg·L−1, under the conditions of discharge voltage 24 kV, pulse frequency 60 Hz, aeration 2.5 L·min−1, electrode spacing 7 mm, the discharge plasma with the 1%Ag/BiVO4 catalyst of 0.1 g gave the best degradation effect. The removal rate of tetracycline hydrochloride can reach 99.8%, and highest mineralization rate can be 67.8%. The high voltage pulse discharge can change the crystal shape and structure of catalyst. In addition, after doping a certain amount of Ag, the surface of BiVO4 catalyst becomes denser, the particles are uniform, the specific surface area is enlarged, and the active sites are increased, which promotes the catalytic reaction and further strengthens the removal of TCH.
-
Key words:
- pulse discharge /
- catalyst /
- wastewater /
- degradation /
- environment
-
药品和个人护理品类污染物(如内分泌干扰物、抗生素等)导致的水体污染问题是生态安全和人类身体健康的一大威胁[1-3]。基于过硫酸盐的高级氧化技术可通过产生羟基自由基(OH•)、硫酸根自由基(SO4•−)、单线态氧(1O2)等活性氧物种降解水中多种类型的有机污染物,在药、护品类污染物处理方面受到了广泛关注[1]。与过一硫酸盐(peroxymonosulfate,PMS)相比,过二硫酸盐(peroxydisulfate,PDS)价格低廉、水溶性好、稳定性高便于储存和运输,已被广泛用于催化降解有机污染物[1]。PDS可通过热活化、碱活化、金属离子催化、金属氧化物催化等多种方法活化,其中基于纳米金属氧化物(如Co3O4等)的异相催化由于催化剂价格低廉、反应条件温和、催化活性高等优点,是目前研究的一个重要方向[2]。然而,纳米尺寸的金属氧化物分散于水中易团聚,导致催化剂活性位点暴露有限,并且分散于水中的纳米催化剂还存在难以分离和回收的问题,极大地限制了其应用。
将金属氧化物纳米催化剂负载于活性炭、泡沫金属、矿物颗粒以及陶瓷膜等三维载体的表面,不仅可以有效缓解氧化物纳米颗粒的团聚问题,而且便于分离和回收,对纳米金属氧化物活化过硫酸盐降解有机物具有重要意义[4]。现阶段,金属氧化物主要通过浆料涂覆的方式负载于载体表面,合成方法简单但稳定性较差[5]。添加高分子粘结剂可增强其稳定性,但催化体系中引入导电性较差的高分子不利于过硫酸活化过程中电子的转移,同时添加粘结剂也可能会带来二次污染[6]。因此,亟需发展一种可将金属氧化物原位负载于三维载体表面的绿色合成方法。植物多酚如单宁酸(tannic acid,TA)普遍存在于植物的根、茎、叶及果实中,是一种价格低廉的生物质[7]。TA分子富含邻二酚羟基,具有较强的金属离子络合能力和优异的表面粘附性能,可与多种金属离子(Co2+、Fe2+、Cu2+等)在聚乙烯球、陶瓷、玻璃等不同载体表面配位形成金属-多酚配合物[7]。作为由金属离子和有机配体组成的复合物,金属-多酚配合物可进一步热解制成不同种类的金属氧化物,并且研究表明有机配体TA分解还会产生多孔结构而形成多孔结构的金属氧化物[8]。
基于TA分子的金属离子络合能力和表面粘附性能,本研究以具有稳定物理化学性质且价格低廉的气泡石为载体,通过在其表面包覆Co2+与TA的配合物,进一步热解将纳米尺寸的Co3O4原位负载于气泡石表面,制备易分离回收的负载型Co3O4催化剂,通过SEM、XRD、ICP等详细分析了负载型Co3O4的结构和组成。利用负载型Co3O4催化剂活化PDS降解水中的双酚A(bis-phenol A,BPA)、磺胺甲恶唑(sulfamethoxazole,SMX)等药、护品类有机污染物,分析了溶液初始pH、PDS浓度、催化剂投加量、共存化学组分等对有机物降解性能的影响,并通过电子顺磁共振分析、化学淬灭实验、光谱分析等手段深入分析了PDS的活化机制和有机物的降解机理。
1. 材料与方法
1.1 实验试剂
主要试剂包括六水合硝酸钴(Co(NO3)2·6H2O)、单宁酸(tannic acid)、双酚A(bisphenol A)、盐酸(HCl,1 mol·L−1)、过硫酸钠(sodium persulfate, Na2S2O8)、氢氧化钠(NaOH,1 mol·L−1)、叔丁醇(tert-butyl alcohol, TBA)、对苯醌(p-benzoquinone, BQ)、碳酸钠(Na2CO3)、氯化钠(NaCl)、硝酸钠(NaNO3)、硫酸钠(Na2SO4)、腐殖酸(HA)、甲醇、乙腈、无水乙醇等,所有试剂均为分析纯,实验用水为超纯水。
1.2 实验方法
1)负载型Co3O4的制备:将尺寸0.5 cm×0.5 cm×0.5 cm的气泡石颗粒置于Co(NO3)2与TA的混合溶液中,利用TA分子的表面粘附特性及其与Co2+的配位反应,在气泡石颗粒表面形成Co-TA包覆层,进一步在空气气氛下焙烧制得负载于气泡石表面的Co3O4催化剂。具体步骤如下:将5 g Co(NO3)2和10 g TA依次溶解于250 mL纯水,然后加入30 g气泡石载体搅拌30 min,加入5 mL氨水后继续搅拌1 h,将修饰后的气泡石用纯水反复清洗。重复上述负载步骤4次后,制得负载有Co-TA配合物的气泡石颗粒,充分干燥后将其置于马弗炉中在400 °C下焙烧2 h (升温速率设置为2 ℃·min−1),即制得负载型Co3O4催化剂。
2)材料表征:通过场发射扫描电子显微镜(Hitachi, S-4800)分析负载型Co3O4的形貌与元素组成,利用Rigaku Ultimate IV型X射线衍射(XRD)表征其晶体结构,借助X射线光电子能谱(XPS)研究催化剂反应前后各元素的化学价态,采用电感耦合等离子体发射光谱仪(ICP, NexION 350D)测定浸出钴离子的浓度以及气泡石颗粒表面Co3O4的负载量。ATR-FTIR测试以超纯水为背景,将PDS与Co3O4纳米催化剂的混合物滴至ATR附件晶体表面,在400~3000 cm−1内至少扫描3次,扫描分辨率为4 cm−1。
3)BPA降解实验:本研究通过催化降解BPA评价负载型Co3O4活化PDS的性能。室温条件下,配制50 mL 0.04 mmol·L−1的BPA溶液,加入3.0 g负载型Co3O4催化剂和0.5 mL 40 mmol·L−1的PDS溶液,然后于特定时间取1 mL水样与0.5 mL甲醇混合以终止反应,进一步用0.22 μm微孔滤膜过滤后,通过高效液相色谱仪测定BPA的浓度,流动相为水和乙腈的混合溶液,两者的体积比为1:1,流速为1 mL·min−1。
2. 结果与讨论
2.1 负载型Co3O4催化剂的表征
图1为气泡石载体负载Co3O4前后的照片及其表面Co3O4纳米催化剂的微观结构。如图1(a)所示,气泡石表面粗糙呈白色,负载Co3O4后变为灰褐色(图1(b)),表明Co3O4纳米催化剂成功负载于其表面。通过SEM表征了气泡石表面Co3O4纳米催化剂的微观结构,如图1(c)所示,气泡石光滑的表面存在大量的纳米颗粒团簇,进一步表明Co3O4纳米催化剂的成功负载。纳米团簇的高倍SEM图表明Co3O4纳米催化剂呈多孔结构(图1(d)),这可能是Co-TA配合物中有机组分TA分解导致的。多孔结构利于催化位点的充分暴露,利于PDS的活化和有机污染物的降解[9]。
 图 1 气泡石与气泡石负载Co3O4的电子照片与SEM分析Figure 1. Digital images of airstone with and without Co3O4 and their SEM analysis
图 1 气泡石与气泡石负载Co3O4的电子照片与SEM分析Figure 1. Digital images of airstone with and without Co3O4 and their SEM analysis图2(a)为Co-TA焙烧衍生粉体Co3O4及气泡石负载Co3O4的XRD谱图。粉体催化剂在2θ为31.27°、36.85°、44.8°、59.35°、65.23°的衍射峰分别对应于立方相Co3O4的(220)、(311)、(400)、(511)和(440)晶面(JCPDS-no.42-1467)。气泡石的XRD图谱表明其主要成分为SiO2 (JCPDS-no.78-2315);SiO2化学性质稳定,便于在复杂环境中使用。气泡石表面负载催化剂后,在2θ为31.27°、44.81°检测到了属于Co3O4的衍射峰,分别对应于立方相Co3O4的(220)和(400)晶面,进一步表明Co3O4成功负载于气泡石表面。通过热重分析研究了Co-TA配合物的热分解过程。如图2(b)所示,Co-TA配合物的残余质量在20~400 °C内持续降低,这主要归咎配合物表面水分子的脱附以及有机配体TA的氧化分解[10]。DTA分析表明Co-TA分解过程中在380 °C出现了明显的吸热峰,表明Co-TA热解生成了新的固相。400 °C后配合物的质量损失保持不变,表明Co-TA中的有机配体在400 °C下完全分解,因此本研究将负载型Co3O4催化剂的合成温度设定为400 °C。通过ICP-MS分析了气泡石表面Co3O4的负载量,结果表明每克气泡石表面负载有1.25 mg的Co3O4纳米颗粒。
 图 2 负载型Co3O4和粉体Co3O4的XRD以及Co-TA配合物的热解分析Figure 2. XRD patterns for powdery Co3O4 and supported Co3O4 and TG curve for Co-TA polymer
图 2 负载型Co3O4和粉体Co3O4的XRD以及Co-TA配合物的热解分析Figure 2. XRD patterns for powdery Co3O4 and supported Co3O4 and TG curve for Co-TA polymer2.2 负载型Co3O4的催化性能研究
在反应温度为(25±1) °C、负载型Co3O4投加量60 g·L−1(即0.075 g·L−1 Co3O4纳米颗粒)、BPA初始浓度为0.04 mmol·L−1、PDS初始浓度为0.4 mmol·L−1和pH为7的条件下测试了负载型Co3O4活化PDS降解BPA的性能。如图3(a)所示,PDS氧化降解BPA的能力有限,120 min内BPA的去除率仅为9%,同时负载型Co3O4仅吸附了4.8%的BPA。负载型Co3O4活化PDS可有效降解BPA,120 min内BPA的去除率达到了100%。BPA的降解遵循拟一级反应动力学,反应动力学常数为0.04 min−1 (图3(b))。ICP分析表明催化反应结束后溶液中Co2+的质量浓度为0.55 mg·L−1,低于地表水环境质量标准(GB 3838-2002D)规定的标准(1.0 mg·L−1),表明负载型Co3O4具有良好的稳定性。溶出的Co2+在相同实验条件下催化PDS仅降解了11%的BPA,表明负载型Co3O4主要通过异相催化反应降解有机物[10]。图3(b)反映了负载型Co3O4活化PDS降解不同类型有机物的性能,苯酚(Phenol)、对乙酰氨基酚(AAP)、磺胺甲恶唑(SMX)均可被有效去除,相同反应条件下120 min内的去除率分别为78.2%、93.6%、67.3%,对应的反应动力学常数分别为0.01、0.02、0.01 min−1(图3(d))。以上结果表明负载型Co3O4活化PDS可催化降解不同类型的药、护品类有机污染物。
 图 3 负载型Co3O4活化PDS降解不同有机物的性能Figure 3. Degradation of different organic pollutants by PDS activated over supported Co3O4
图 3 负载型Co3O4活化PDS降解不同有机物的性能Figure 3. Degradation of different organic pollutants by PDS activated over supported Co3O42.3 实验因素的影响
图4(a)反映了溶液初始pH分别为3、5、7、9、11时,负载型Co3O4活化PDS降解BPA的性能。在pH=3~9内,BPA均可得到有效去除,120 min内BPA的去除率均达到100%,同时反应速率也并未受到显著影响;然而,当pH提升至11时,BPA的降解受到抑制,120 min内BPA的去除率降低了24.4%,反应速率也由中性时的0.04 min−1降至0.01 min−1,这可能是由于碱性条件下OH•的氧化还原电位降低所致[11]。图4(b)为不同PDS投加量下,负载型Co3O4对BPA的催化降解性能。当PDS浓度由0.1 mmol·L−1增加到0.4 mmol·L−1时,BPA的去除率从20.9%增加到98.2%,反应速率也由0.002 min−1提升至0.03 min−1。增加PDS的浓度可增加活性氧物种的生成量,进而促进有机物的催化降解[12]。然而,进一步增加PDS的浓度至0.8 mmol·L−1和1.6 mmol·L−1时,BPA的降解性能并未得到进一步提升。这可能是由于负载型Co3O4的催化位点有限,不能完全活化反应体系中的PDS[13]。增加催化剂的投加量可提供更多的催化位点而充分活化PDS,进而产生更多的活性氧物种而显著提升有机物的降解效率[14]。如图4(c)所示,在BPA溶液体积为50 mL的情况下,当负载型Co3O4催化剂的投加量由1.5 g增加至3.0 g时,BPA的降解速率由0.02 min−1提高至0.037 min−1,当催化剂的投加量进一步增加至6.0 g时,BPA在60 min即可被完全去除,降解速率达0.039 min−1。这表明增加负载型Co3O4的剂量可显著提升PDS的有效利用率。
 图 4 负载型Co3O4活化PDS降解BPA的影响因素Figure 4. Effect of operation parameters on BPA removal in the supported Co3O4/PDS system
图 4 负载型Co3O4活化PDS降解BPA的影响因素Figure 4. Effect of operation parameters on BPA removal in the supported Co3O4/PDS system为考察水中常见无机阴离子对负载型Co3O4活化PDS降解有机物性能的影响,研究了Cl−、NO3−、CO32−对BPA去除效率和降解速率的影响。如图4(d)所示,在负载型Co3O4/PDS体系中加入10 mmol·L−1 Cl−并未降低BPA的去除效率,同样加入10 mmol·L−1的NO3−也未影响BPA的去除。Co3O4活化PDS降解有机物主要有2种途径:一种是基于SO4•−和OH•的自由基氧化;另一种是基于单线态氧(1O2)的非自由基氧化[15]。在自由基途径中,Cl−、NO3−等无机阴离子会竞争消耗强氧化性的SO4•−和OH•,进而抑制有机污染物的降解[16-18]。在本研究中,Cl−和NO3−对BPA的去除影响较弱,表明自由基氧化不是Co3O4活化PDS降解有机物的主要途径。然而,向反应体系中添加10 mmol·L−1的CO32−可显著抑制BPA的去除,同样反应条件下BPA仅去除了16.64%,同时反应速率由0.04 min−1降低至了0.001 min−1。CO32−是一种常用的1O2淬灭剂,其对BPA降解的显著抑制表明负载型Co3O4活化PDS降解有机物是1O2主导的非自由途径[19]。除了无机阴离子,天然有机物如腐殖酸(HA)也是一种常见的水体背景成分[6]。在10 mg·L−1 HA存在的情况下,负载型Co3O4催化降解有机物的效率也未受到显著影响,进一步说明本反应体系中有机物的降解为非自由基途径[20]。综上所述,负载型Co3O4/PDS体系不仅能够在较宽的pH范围内降解有机物,而且对无机阴离子和腐殖酸等常见的水体背景成分具有较强的抗干扰能力,因此,有着较强的应用潜力。
2.4 活性氧物种鉴定
为明确负载型Co3O4活化PDS降解有机物的机理,通过EPR鉴定分析了该体系产生的活性氧的种类。如图5(a)所示,当仅有PDS时,以DMPO为自旋捕获剂未检测到任何信号,而当加入负载型Co3O4催化剂后,EPR谱图观测到了峰强度为1: 2 : 2 : 1的特征峰,这是催化产生的OH•被DMPO捕获所致,表明负载型Co3O4活化PDS产生了OH•[21]。此外,以TEMP为1O2捕获剂还检测到了TEMP-1O2的特征峰,因此,负载型Co3O4活化PDS也产生了1O2[22]。进一步通过化学淬灭实验判别了反应体系中OH•和1O2对BPA降解的贡献。图5(b)反映了不同浓度MeOH的淬灭效果,可以看出MeOH对BPA的降解影响有限,在1 000 mmol·L−1 MeOH存在的情况下,BPA的去除率仍高达94.7%。MeOH可与SO4•−和OH•快速反应,反应速率常数分别为k 分别为 2.5×107 (mol·s)−1和9.7×108 (mol·s)−1,是常用的自由基淬灭剂;MeOH对负载型Co3O4/PDS有限的淬灭能力表明该体系降解有机物的过程中,自由基的氧化作用有限[7]。然而,L-组氨酸可显著抑制BPA的降解,如图5(c)所示,1 mmol·L−1的组氨酸使得BPA的降解率由100%降低至89%;当L-组氨酸的浓度增加至5 mmol·L−1时,BPA的去除率进一步降低至21.98%,反应速率也由0.04 min−1降低至0.002 min−1。L-组氨酸具有一定的还原性,可能会消耗PDS而降低负载型Co3O4催化降解BPA的能力[23]。为排除L-组氨酸消耗PDS对淬灭实验的干扰,研究了不同浓度L-组氨酸对PDS的降解效率。如图5(d)所示,L-组氨酸分解PDS的能力有限,例如5 mmol·L−1的L-组氨酸在120 min内仅分解了5.8%的PDS。因此,L-组氨酸对负载型Co3O4/PDS降解BPA的抑制作用主要归咎于其对活性氧物种的淬灭,而L-组氨酸是常用的1O2淬灭剂,这表明负载型Co3O4活化PDS降解有机物是以1O2为主导的非自由基氧化途径。过渡金属氧化物活化PDS产生1O2已被广泛报道,有研究表明,O2•−是1O2生成的重要中间体(
2O2·−+2H2O→21O2+2OH−+H2O2 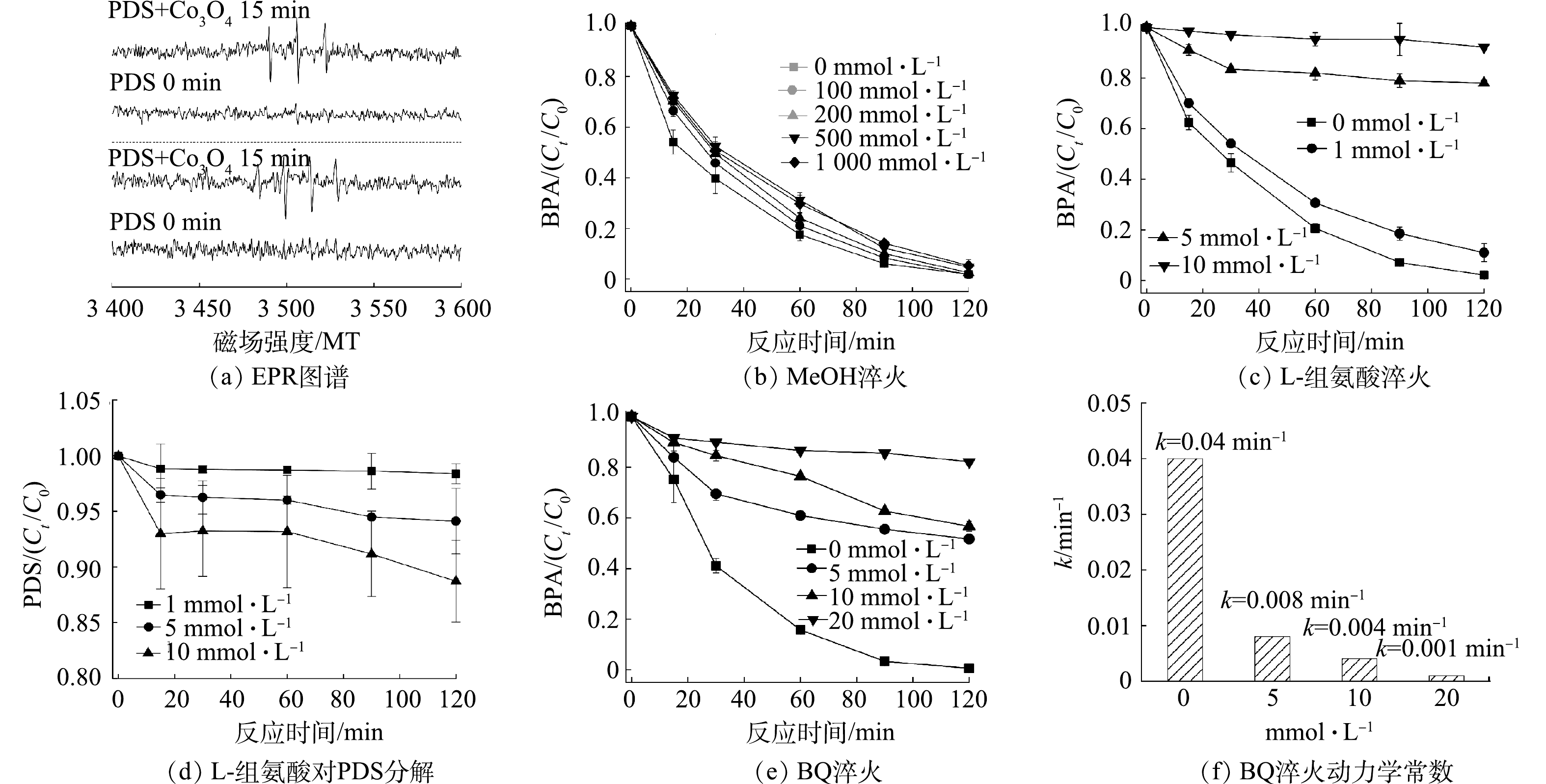 图 5 负载型Co3O4活化PDS的电子自旋共振测试和淬灭实验Figure 5. EPR spectra and quenching experiments on BPA removal
图 5 负载型Co3O4活化PDS的电子自旋共振测试和淬灭实验Figure 5. EPR spectra and quenching experiments on BPA removal2.5 PDS活化机理研究
过渡金属氧化物主要通过金属离子的价态循环活化过硫酸盐[26]。为解析负载型多孔Co3O4活化PDS的机理,通过XPS分析了Co3O4中Co元素的价态及其反应前后含量的变化。如图6(a)所示,Co2p在794.9 eV和779.7 eV的特征峰分别对应于Co2p1/2和Co2p3/2,而Co2p3/2的高分辨XPS谱图拟合结果表明,781.2 eV和779.7 eV处的峰分别对应于Co2+和Co3+,所占比例分别为34.2%和54.5%。反应后,Co3O4表面Co2+、Co3+比例变化表明Co3+/Co2+的氧化还原参与了PDS的活化[27]。进一步通过原位ATR-FTIR分析研究了PDS分子在负载型Co3O4表面的反应机理。如图6(b)所示,在1 050 cm−1和1 272 cm−1处检测到PDS振动峰,加入Co3O4催化剂后在其表面观测到了PDS的特征峰的位置并且未发生偏移,表明PDS吸附于Co3O4催化剂的表面。此外,在Co3O4/PDS体系中还观测到了SO42‒的特征峰(1 107 cm−1),表明Co3O4活化PDS产生了SO42‒;随着反应时间的增加PDS特征峰的强度逐渐降低,而SO42‒特征峰的强度逐渐增加,表明PDS在Co3O4催化剂表面持续分解[1]。
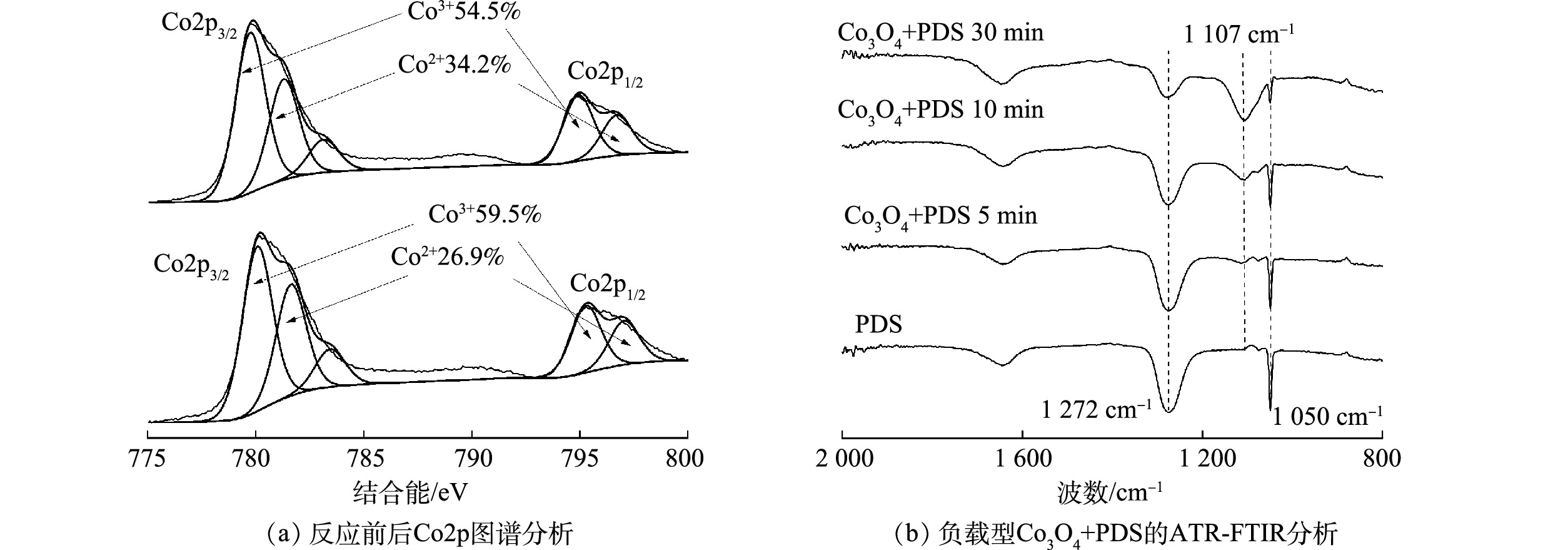 图 6 负载型Co3O4活化PDS的机理研究Figure 6. Mechanism of PDS activated over the supported Co3O4 catalyst
图 6 负载型Co3O4活化PDS的机理研究Figure 6. Mechanism of PDS activated over the supported Co3O4 catalyst金属氧化物(MOx)活化过硫酸盐时,吸附于其表面的水分子会解离形成MOx-OH[19]。因此,结合XPS和ATR-FTIR分析,提出了负载型Co3O4活化PDS降解BPA的可能反应机理(图7)。首先,PDS分子通过氢键与[≡Co3+—OH]2+结合形成[≡Co3+—O—O—SO3]+络合物(式(1)),该络合物具有较高的氧化电势,还原PDS分子生成O2•−,同时Co3+被还原为Co2+形成中间态[≡Co2+—OH] (式(2)),随之生成的O2•−进一步反应生成1O2 (式(3))。中间态[≡Co2+—O—O—SO3]通过内电子转移被氧化为≡Co3+—OH,同时生成少量的自由基(式(4))。如上所述,负载型Co3O4/PDS体系中自由基对有机物的降解贡献有限,因此1O2进一步氧化有机物将其分解为中间产物或者H2O和CO2 (式(5))。本研究中,负载型Co3O4有效活化PDS主要有2点原因,一是负载型Co3O4避免了纳米催化剂的团聚,可暴露更多的活性位点而提升催化效率;另一方面,Co-TA配合物热解会产生富含氧缺陷的Co3O4[28],而氧缺陷可有效提升PDS分子在Co3O4表面的吸附,也会提升内电子催化PDS的效能。
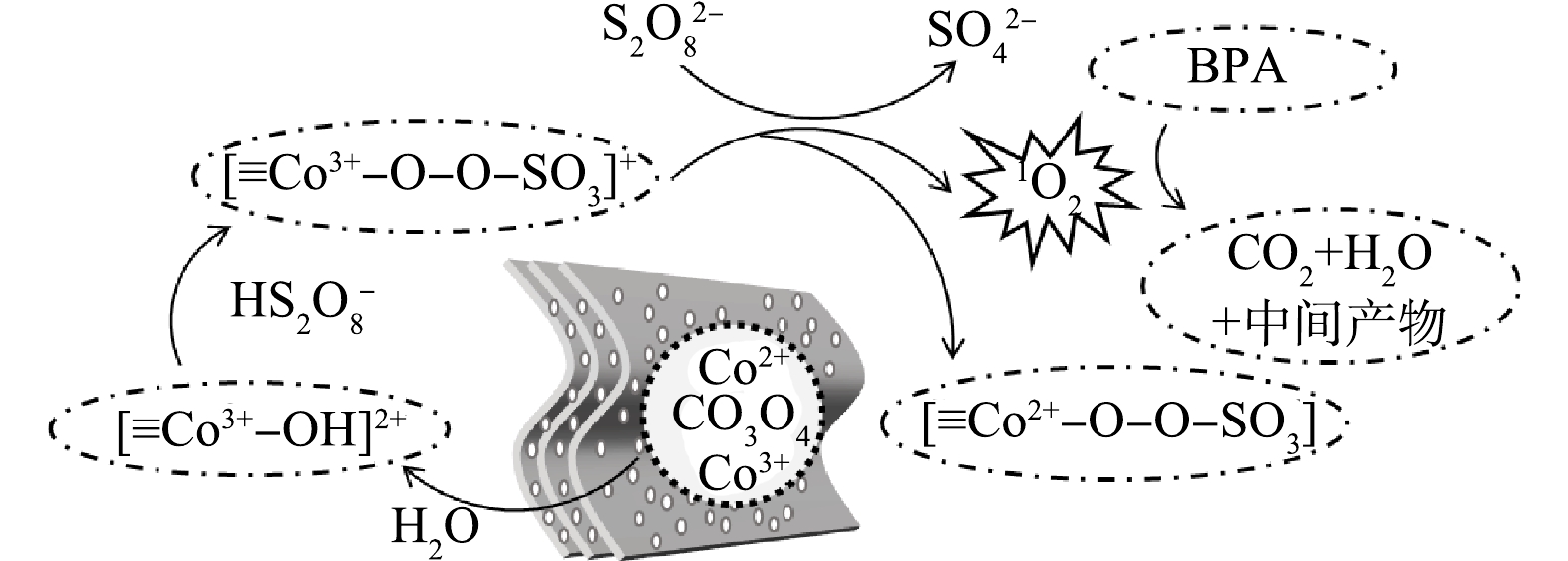 图 7 负载型Co3O4活化PDS降解BPA的机理示意图Figure 7. Schema for BPA degradation by PDS activated over the supported Co3O4 catalyst
图 7 负载型Co3O4活化PDS降解BPA的机理示意图Figure 7. Schema for BPA degradation by PDS activated over the supported Co3O4 catalyst2[≡Co3+−OH]2++HS2O8−→2[≡Co3+−O−O−SO3]++3H+ (1) 2[≡Co3+−O−O−SO3]++4H2O+S2O82−→2[≡Co2+−OH]+4SO42−+2O2·−+6H+ (2) 2O2·−+2H2O→21O2+2OH−+H2O2 (3) [≡Co2+−O−O−SO3]+H2O→≡Co3+−OH+SO4·−+OH· (4) 1O2+有机物→H2O+CO2+中间产物 (5) 催化剂的稳定性是衡量其实际应用潜力的一个重要因素,本研究通过循环降解实验研究了负载型Co3O4的稳定性。如图8(a)所示,3次循环后负载型Co3O4对BPA的去除率仍保持在89%,表明负载型Co3O4具有良好的稳定性。循环降解过程中催化活性的轻微降低可能是BPA的降解产物堵塞活性位点导致的[10]。此外,XRD分析表明循环使用后负载型Co3O4的晶相没有明显变化(图2(a)),进一步说明负载型Co3O4具有良好的稳定性。负载型Co3O4可有效矿化BPA(图8(b)),TOC去除率高达73%,而且3次循环后TOC的去除率仍高达63%,表明负载型Co3O4可持续稳定地处理水中酚类有机污染物。
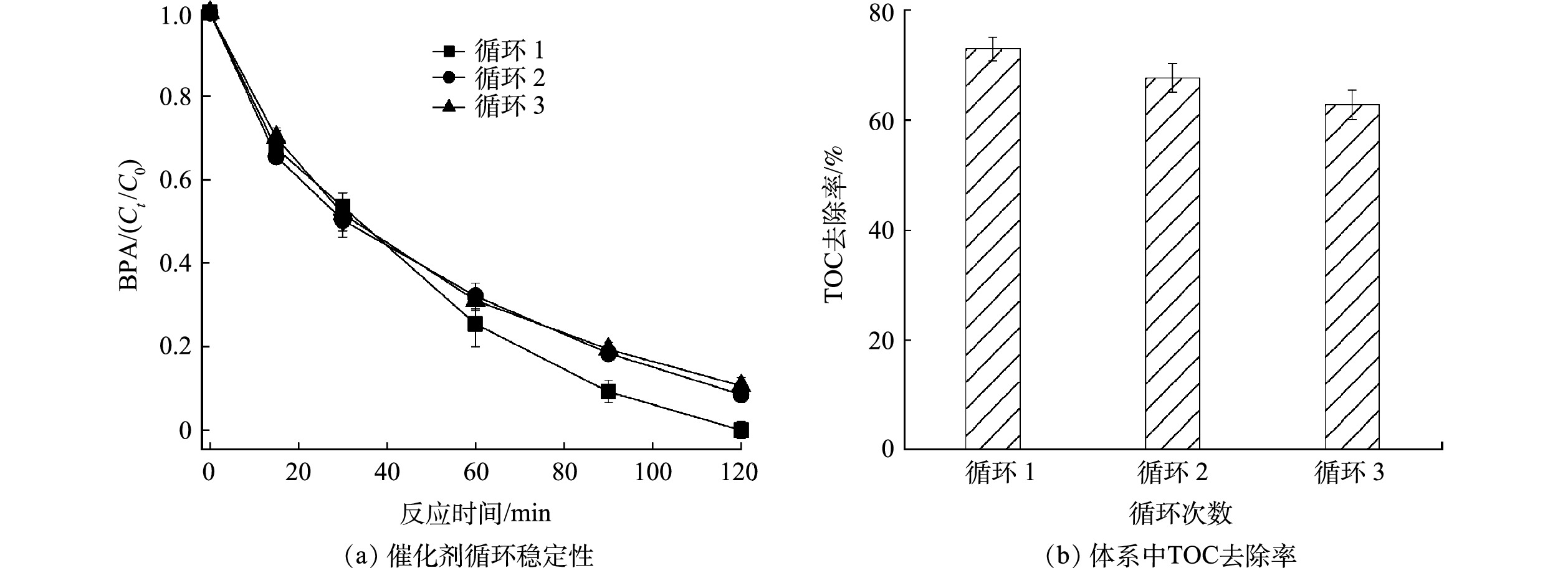 图 8 循环降解过程中BPA的去除性能和TOC的去除效率Figure 8. Removal of BPA and corresponding TOC during the consecutive runs over the supported Co3O4
图 8 循环降解过程中BPA的去除性能和TOC的去除效率Figure 8. Removal of BPA and corresponding TOC during the consecutive runs over the supported Co3O43. 结论
1)基于植物多酚的界面配位原理,通过在三维矿物载体表面原位负载Co3O4纳米颗粒制得了易分离回收的负载型Co3O4催化剂,比Co3O4粉体催化剂展现出更优异的催化活性和良好的稳定性。
2)负载型Co3O4催化剂活化PDS可高效降解BPA、SMX、AAP等多种类型的药、护品类有机污染物,中性条件下,负载型Co3O4投加量为60 g·L−1(即0.075 g·L−1 Co3O4纳米颗粒),在BPA/PDS摩尔比为1:10的情况下可在120 min内将BPA完全降解。
3)负载型Co3O4主要通过内电子转移活化PDS,降解有机物时自由基氧化的作用有限,主要是1O2主导的非自由基氧化途径,对无机阴离子和腐殖酸等水体背景成分有着较强的抗干扰能力,在实际水体处理方面展现出广阔的应用前景。
-
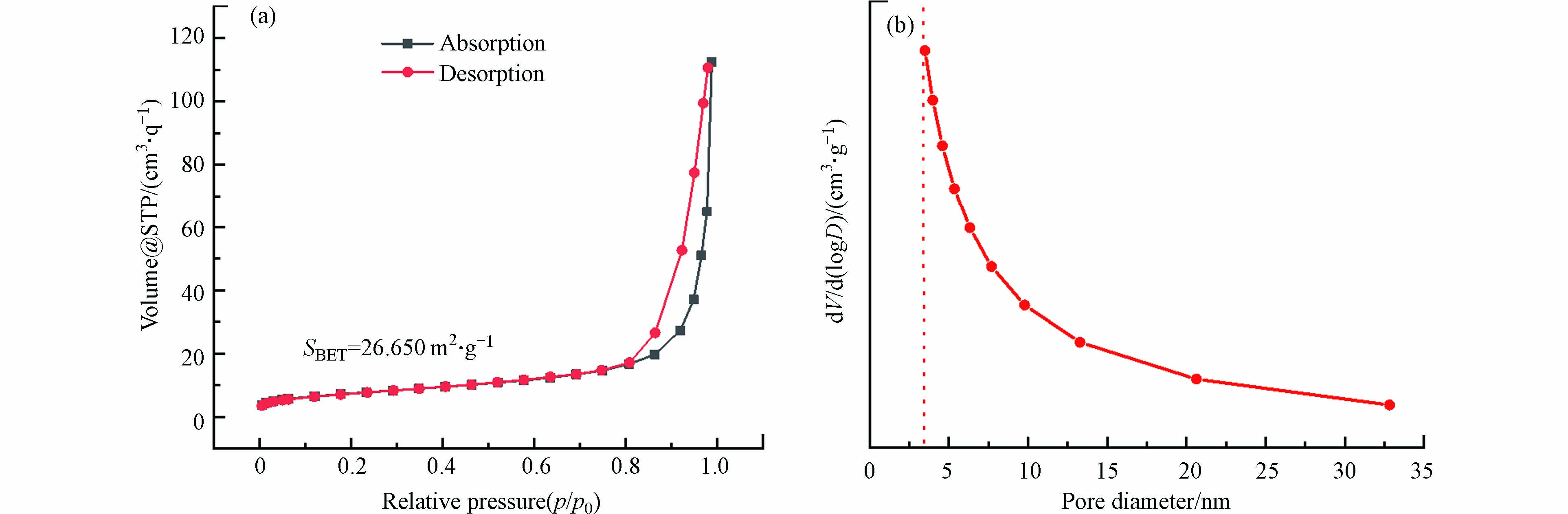
图 5 1%Ag/BiVO4的氮气吸附-脱附等温线(a)和孔径分布(b)
Figure 5. Nitrogen adsorption-desorption isotherm (a) and pore size distribution (b) of 1%Ag/BiVO4
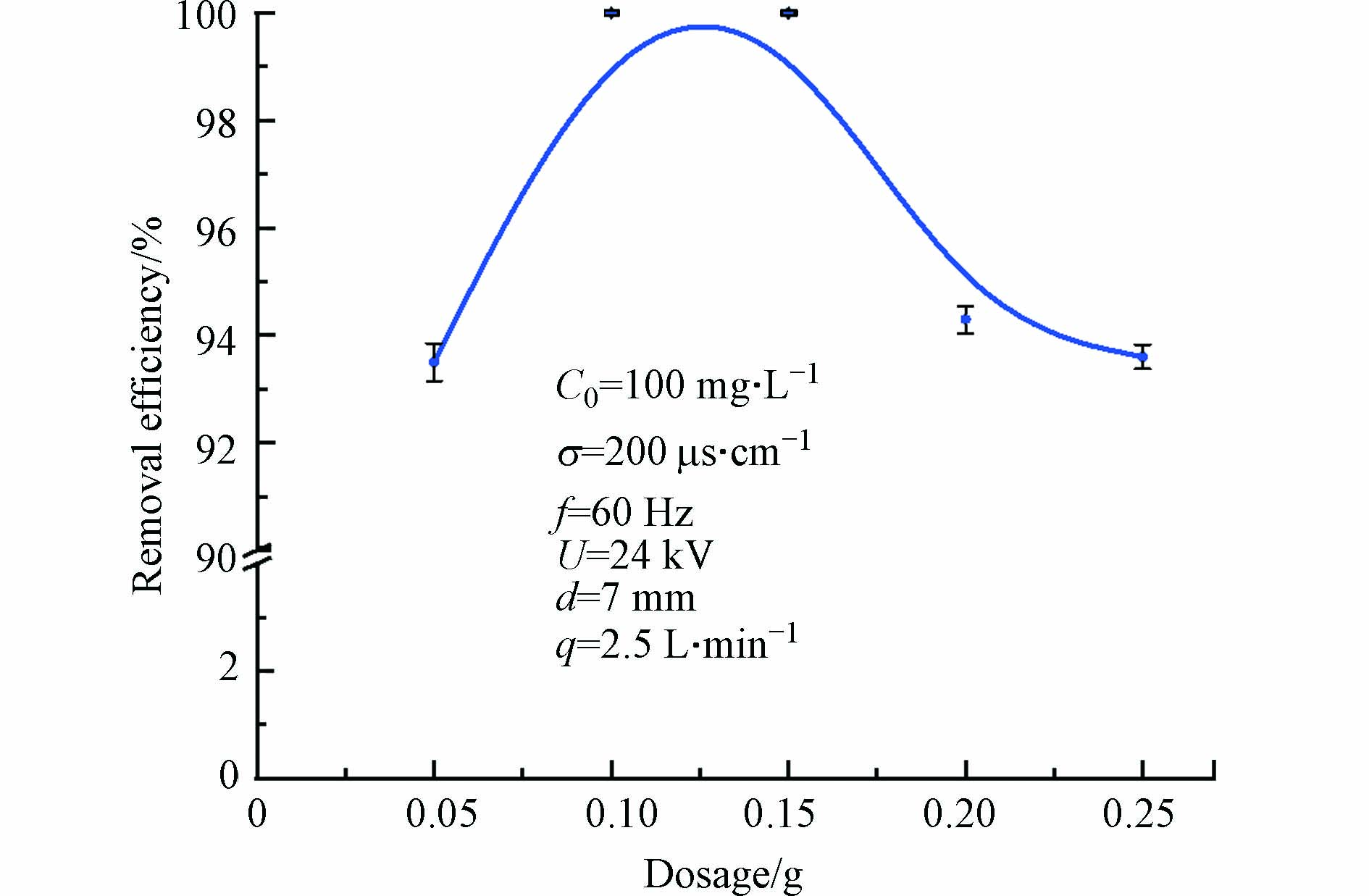
图 7 1%Ag/BiVO4催化剂投加量对TCH去除率的影响
Figure 7. Effect of 1%Ag/BiVO4 catalyst dosage on TCH removal rate
-
[1] 张国栋, 董文平, 刘晓晖, 等. 我国水环境中抗生素赋存、归趋及风险评估研究进展 [J]. 环境化学, 2018, 37(7): 1491-1500. doi: 10.7524/j.issn.0254-6108.2017112003 ZHANG G D, DONG W P, LIU X H, et al. Occurrence, fate and risk assessment of antibiotics in water environment of China [J]. Environmental Chemistry, 2018, 37(7): 1491-1500(in Chinese). doi: 10.7524/j.issn.0254-6108.2017112003
[2] 陈小桐, 安静, 杨合, 等. 辽宁省畜禽养殖业抗生素排放特征及减排路径分析 [J]. 环境科学学报, 2021, 41(7): 2896-2904. doi: 10.13671/j.hjkxxb.2020.0468 CHEN X T, AN J, YANG H, et al. Analysis of antibiotics emission characteristics and reduction path of livestock and poultry industry in Liaoning Province [J]. Acta Scientiae Circumstantiae, 2021, 41(7): 2896-2904(in Chinese). doi: 10.13671/j.hjkxxb.2020.0468
[3] 朱婉婷, 于晓彩, 田思瑶, 等. CuO/ZnO复合光催化剂降解海水养殖废水中盐酸四环素 [J]. 环境污染与防治, 2020, 42(3): 305-309,316. doi: 10.15985/j.cnki.1001-3865.2020.03.009 ZHU W T, YU X C, TIAN S Y, et al. Photocatalytic degradation of tetracycline hydrochloride in mariculture wastewater by CuO/ZnO composite photocatalyst [J]. Environmental Pollution & Control, 2020, 42(3): 305-309,316(in Chinese). doi: 10.15985/j.cnki.1001-3865.2020.03.009
[4] WANG L, CHEN Y, ZHAO Y, et al. Toxicity of two tetracycline antibiotics on Stentor coeruleus and Stylonychia lemnae: Potential use as toxicity indicator [J]. Chemosphere, 2020, 255: 127011. doi: 10.1016/j.chemosphere.2020.127011 [5] 苗淑彦, 韩蓓, 胡俊涛, 等. 饲料中添加不同浓度四环素对乌鳢生长性能、肠道菌群组成和组织形态的影响 [J]. 动物营养学报, 2019, 31(12): 5813-5822. doi: 10.3969/j.issn.1006-267x.2019.12.047 MIAO S Y, HAN B, HU J T, et al. Effects of dietary different concentrations of tetracycline on growth performance, intestinal microbiota composition and morphology of Channa argus [J]. Chinese Journal of Animal Nutrition, 2019, 31(12): 5813-5822(in Chinese). doi: 10.3969/j.issn.1006-267x.2019.12.047
[6] 秦丽婷, 童蕾, 刘慧, 等. 环境中磺胺类抗生素的生物降解及其抗性基因污染现状 [J]. 环境化学, 2016, 35(5): 875-883. doi: 10.7524/j.issn.0254-6108.2016.05.2015113004 QIN L T, TONG L, LIU H, et al. Biodegradation of sulfonamides and the pollution characteristics of sulfonamide resistance genes in the environment [J]. Environmental Chemistry, 2016, 35(5): 875-883(in Chinese). doi: 10.7524/j.issn.0254-6108.2016.05.2015113004
[7] 印献栋, 段锋. O3/H2O2氧化预处理高浓度抗生素制药废水研究[J]. 现代化工, 2020, 40(S1): 121-123, 127. YIN X D, DUAN F. Study on oxidation pretreatment of high-concentration wastewater from antibiotics production by ozone/hydrogen peroxide[J]. Modern Chemical Industry, 2020, 40(Sup 1): 121-123, 127(in Chinese).
[8] 汪春霞. 医药废水处理技术研究进展 [J]. 四川化工, 2018, 21(5): 28-30. doi: 10.3969/j.issn.1672-4887.2018.05.008 WANG C X. Research progress in pharmaceutical wastewater treatment technology [J]. Sichuan Chemical Industry, 2018, 21(5): 28-30(in Chinese). doi: 10.3969/j.issn.1672-4887.2018.05.008
[9] 张婷, 郎朗. 环境水体中抗生素残留及处理方法现状分析 [J]. 黑龙江科技信息, 2017(9): 84-85. ZHANG T, LANG L. Analysis of antibiotic residues and treatment methods in environmental water [J]. Heilongjiang Science and Technology Information, 2017(9): 84-85(in Chinese).
[10] 林爱秋, 程和发. 芬顿及光芬顿法降解氟喹诺酮类抗生素研究进展 [J]. 环境化学, 2021, 40(5): 1305-1318. doi: 10.7524/j.issn.0254-6108.2021011401 LIN A Q, CHENG H F. Recent development in the degradation of fluoroquinolones by Fenton and photo-Fenton processes [J]. Environmental Chemistry, 2021, 40(5): 1305-1318(in Chinese). doi: 10.7524/j.issn.0254-6108.2021011401
[11] 朱鋆珊, 郭丽, 李平. 高级氧化技术在四环素废水处理中的应用进展 [J]. 化工科技, 2019, 27(4): 65-68. doi: 10.3969/j.issn.1008-0511.2019.04.013 ZHU Y S, GUO L, LI P. Application progress of advanced oxidation technology in tetracycline wastewater treatment [J]. Science & Technology in Chemical Industry, 2019, 27(4): 65-68(in Chinese). doi: 10.3969/j.issn.1008-0511.2019.04.013
[12] 屈广周, 李杰, 梁东丽, 等. 低温等离子体技术处理难降解有机废水的研究进展 [J]. 化工进展, 2012, 31(3): 662-670. doi: 10.16085/j.issn.1000-6613.2012.03.039 QU G Z, LI J, LIANG D L, et al. Research progress in organic wastewater treatment by low-temperature plasma discharge technology [J]. Chemical Industry and Engineering Progress, 2012, 31(3): 662-670(in Chinese). doi: 10.16085/j.issn.1000-6613.2012.03.039
[13] 仇聪颖, 管显涛, 刘振, 等. 纳秒脉冲放电处理有机染料废水的实验研究 [J]. 强激光与粒子束, 2020, 32(2): 56-62. doi: 10.11884/HPLPB202032.190390 QIU C Y, GUAN X T, LIU Z, et al. Degradation of organic dyes by nanosecond pulsed discharge plasma [J]. High Power Laser and Particle Beams, 2020, 32(2): 56-62(in Chinese). doi: 10.11884/HPLPB202032.190390
[14] HAO C J, YAN Z Y, LIU K F, et al. Degradation of pharmaceutical contaminant tetracycline in aqueous solution by coaxial-type DBD plasma reactor [J]. IEEE Transactions on Plasma Science, 2020, 48(2): 471-481. doi: 10.1109/TPS.2020.2964612 [15] 鲁美娟, 汪怀建, 黄荣, 等. 催化剂对等离子体协同催化降解挥发性有机物影响的研究进展 [J]. 环境污染与防治, 2018, 40(1): 88-94. doi: 10.15985/j.cnki.1001-3865.2018.01.019 LU M J, WANG H J, HUANG R, et al. Research of effect of catalyst on the VOCs degradation using plasma-assisted catalysis technology [J]. Environmental Pollution & Control, 2018, 40(1): 88-94(in Chinese). doi: 10.15985/j.cnki.1001-3865.2018.01.019
[16] 马瑾, 王新海. 金属掺杂改性纳米二氧化钛对废水中有机物催化降解的研究进展 [J]. 能源化工, 2020, 41(4): 7-13. doi: 10.3969/j.issn.1006-7906.2020.04.003 MA J, WANG X H. Research progress of metal-doped modified titanium dioxide nanoparticles for catalytic degradation of organic compounds in waste water [J]. Energy Chemical Industry, 2020, 41(4): 7-13(in Chinese). doi: 10.3969/j.issn.1006-7906.2020.04.003
[17] 汪超, 王铁成, 屈广周, 等. 脉冲电晕放电等离子体耦合土壤颗粒去除废水中盐酸四环素 [J]. 高电压技术, 2018, 44(9): 3076-3082. doi: 10.13336/j.1003-6520.hve.20171101001 WANG C, WANG T C, QU G Z, et al. Removal of tetracycline HCl in wastewater by pulsed Corona discharge plasma coupled with soil particles [J]. High Voltage Engineering, 2018, 44(9): 3076-3082(in Chinese). doi: 10.13336/j.1003-6520.hve.20171101001
[18] YAN X, YI C W, WANG Y H, et al. Multi-catalysis of nano-zinc oxide for bisphenol A degradation in a dielectric barrier discharge plasma system: Effect and mechanism [J]. Separation and Purification Technology, 2020, 231: 115897. doi: 10.1016/j.seppur.2019.115897 [19] 董冰岩, 李琼, 王慧, 等. 高压脉冲放电降解选矿废水中残余黄药的研究 [J]. 现代化工, 2019, 39(9): 87-91. doi: 10.16606/j.cnki.issn0253-4320.2019.09.019 DONG B Y, LI Q, WANG H, et al. Study on degradation of residual xanthate in mineral separation wastewater by high voltage pulse discharge [J]. Modern Chemical Industry, 2019, 39(9): 87-91(in Chinese). doi: 10.16606/j.cnki.issn0253-4320.2019.09.019
[20] DONG Q, YANG F L, LIANG F, et al. Silver particle on BiVO4 nanosheet plasmonic photocatalyst with enhanced photocatalytic oxidation activity of sulfadiazine [J]. Journal of Molecular Liquids, 2021, 331: 115751. doi: 10.1016/j.molliq.2021.115751 [21] PANDIYARAJ K N, VASU D, GHOBEIRA R, et al. Dye wastewater degradation by the synergetic effect of an atmospheric pressure plasma treatment and the photocatalytic activity of plasma-functionalized Cu-TiO2 nanoparticles [J]. Journal of Hazardous Materials, 2021, 405: 124264. doi: 10.1016/j.jhazmat.2020.124264 [22] WANG M, HAN J, LV C M, et al. Ag, B, and Eu tri-modified BiVO4 photocatalysts with enhanced photocatalytic performance under visible-light irradiation [J]. Journal of Alloys and Compounds, 2018, 753: 465-474. doi: 10.1016/j.jallcom.2018.04.068 [23] SHAFIQ I, HUSSAIN M, SHEHZAD N, et al. The effect of crystal facets and induced porosity on the performance of monoclinic BiVO4 for the enhanced visible-light driven photocatalytic abatement of methylene blue [J]. Journal of Environmental Chemical Engineering, 2019, 7(4): 103265. doi: 10.1016/j.jece.2019.103265 [24] 王晓亮, 王振华, 何瑾馨. Ag掺杂BiVO4光催化剂的制备及其性能 [J]. 印染, 2013, 39(19): 5-8,24. WANG X L, WANG Z H, HE J X. Preparation and photocatalytic activity of Ag doped BiVO4 photocatalyst [J]. Dyeing & Finishing, 2013, 39(19): 5-8,24(in Chinese).
[25] 贾恒. 铋基可见光催化剂的制备及降解四环素废水的研究[D]. 邯郸: 河北工程大学, 2020. JIA H. Preparation of Bi based-photocatalyst and photocatalytic degradation of tetracycline wastewater under visible light illumination[D]. Handan: Hebei University of Engineering, 2020(in Chinese).
[26] ZHAO W, TU X Y, WANG X M, et al. Novel p-n heterojunction photocatalyst fabricated by flower-like BiVO4 and Ag2S nanoparticles: Simple synthesis and excellent photocatalytic performance [J]. Chemical Engineering Journal, 2019, 361: 1173-1181. doi: 10.1016/j.cej.2018.12.120 [27] KONSTANTINOU I K, ALBANIS T A. TiO2-assisted photocatalytic degradation of azo dyes in aqueous solution: Kinetic and mechanistic investigations: A review [J]. Applied Catalysis B:Environmental, 2004, 49(1): 1-14. doi: 10.1016/j.apcatb.2003.11.010 [28] 陈宇溪, 罗力莎, 时峥, 等. Ag掺杂型TiO2粉末光催化降解四环素类抗生素废水 [J]. 科技创新与应用, 2018(13): 30-32. CHEN Y X, LUO L S, SHI Z, et al. Photocatalytic degradation of tetracycline antibiotic wastewater by Ag-doped TiO2 powder [J]. Technology Innovation and Application, 2018(13): 30-32(in Chinese).
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2593
- HTML全文浏览数: 2593
- PDF下载数: 84
- 施引文献: 0
