























-
近年来,药品和个人护理产品(PPCPs)的消费量逐年增加[1],引起的环境污染越来越受到关注[2-3]。大多数PPCPs在污水处理厂无法完全去除,其会随着污水厂出水进入受纳水体,导致其在自然水环境中的暴露。例如,被广泛用作止疼和抗风湿的双氯芬酸(DCF),在地表水甚至自来水中都被检测出[4],DCF的基本理化性质见表1[5]。此外,低剂量的PPCPs即可对生物产生生理效应[1, 6],这使得处理含有PPCPs的工业废水和生活污水尤为重要。
高级氧化工艺(AOPs)被认为是降解PPCPs的有效方法之一,常用的氧化剂包括过氧化氢(H2O2)、过一硫酸盐(PMS)和过硫酸盐(PS)等[7-9]。过氧乙酸(PAA, CH3C(=O)OOH)是一种类似H2O2的新型氧化剂,近年来在AOPs中得到广泛研究[10-12]。此前,PAA因其良好的消毒性能和副产物的低毒性而被广泛用于废水的消毒[13-15]。PAA的过氧键键能仅为159 kJ·mol−1,低于H2O2的213 kJ·mol−1[16],这意味着PAA更容易被活化[17]。迄今为止,已报道活化PAA的方法有紫外(UV)、过渡金属、热等[15, 18-22]。UV能激活PAA产生活性物质进而降解污染物[18],然而UV在水中的穿透力有限,如果没有足够的UV强度,这一过程很难实现。过渡金属活化PAA的反应条件简单,能够活化PAA的过渡金属离子包括Co2+、Mn2+、Fe2+、Cu2+等[15, 19-21],然而Co2+和Mn2+具有一定毒性,进而可能造成二次污染;Fe2+虽然无毒性,但在非酸性条件下易被氧化而降低活化效率;Cu2+的毒性较低,但低剂量的Cu2+活化PAA效果不佳。热能激活PAA产生活性物质,从而快速降解磺胺甲恶唑(SMX)[22],如公式(1)所示,但断裂过氧键需要消耗大量能量。
根据预实验发现,Cu(Ⅱ)和热联用活化PAA (Cu(Ⅱ)-heat/PAA)只需要较低浓度的Cu(Ⅱ)(低至μmol·L−1级别)和消耗较少的能量(与单独热激活PAA比较);相比于单独Cu(Ⅱ)活化PAA(Cu(Ⅱ)/PAA)和热激活PAA(heat/PAA)体系,Cu(Ⅱ)-heat/PAA体系显著加速了DCF的去除。因此,本文拟对该现象进行深入探讨研究,考察该协同效应的作用机理;调查常见水基质(Cl−、SO42−、NO3−、HCO3−、天然有机质(NOM))对Cu(Ⅱ)-heat/PAA体系降解DCF的影响;评估该技术对实际水体中DCF的去除效果。
-
双氯芬酸钠(≥ 99%)、富里酸(FA)和叔丁醇(TBA,≥ 99.5%)购买于上海阿拉丁生化科技有限公司(中国);过氧乙酸(PAA,质量分数15%,H2O2/PAA的摩尔比 = 1.4)、五水合硫代硫酸钠(Na2SO3·5H2O)、五水合硫酸铜(CuSO4·5H2O)、硝酸钾(KNO3)、氢氧化钠(NaOH)、硫酸钠(Na2SO4)、氯化钠(NaCl)和硫酸(H2SO4)购买于成都科隆化学试剂有限公司。甲醇(MeOH,LC/MS级)购买于Fisher公司。除了实际水体,实验用水均使用超纯水(18.2 MΩ·cm)。
-
实验均在烧杯(250 mL)中进行,通过恒温水浴锅控制反应温度,转速定为350 r·min−1。具体步骤如下:首先配制5 μmol·L−1 DCF反应液100 mL,然后加入指定量的Cu(Ⅱ),再加入一定量的5% H2SO4或10 g·L−1 NaOH溶液调节反应体系初始pH值;当溶液达到指定温度后,加入PAA启动反应。在给定时间点,取样1 mL加入到含有0.1 mL 0.2 mol·L−1 Na2SO3溶液的液相小瓶中进行DCF定量分析。在自由基清除实验和水基质实验中,先向反应液加入清除剂(TBA或MeOH)或者常见水基质(Cl−、SO42−、NO3−和NOM),再加入DCF。所有实验至少进行3次重复,图中的数据为带有误差棒的平均值。
-
使用配备光电二极管阵列检测器(Waters 2998)的Waters ACQITY UPLC I-Class测定DCF浓度。固定相为ACQUITY-UPLC-BEH-C18柱(2.1 mm× 50 mm,1.7 μm);流动相为甲醇和0.1 %醋酸的混合物(体积比 61:39);进样量设为10 μL;波长设置为276 nm;柱温30 ℃。通过滴定法[23]定期测量PAA储备液的浓度。使用pH计(PHS-3C,上海雷磁)测量溶液的pH值。
-
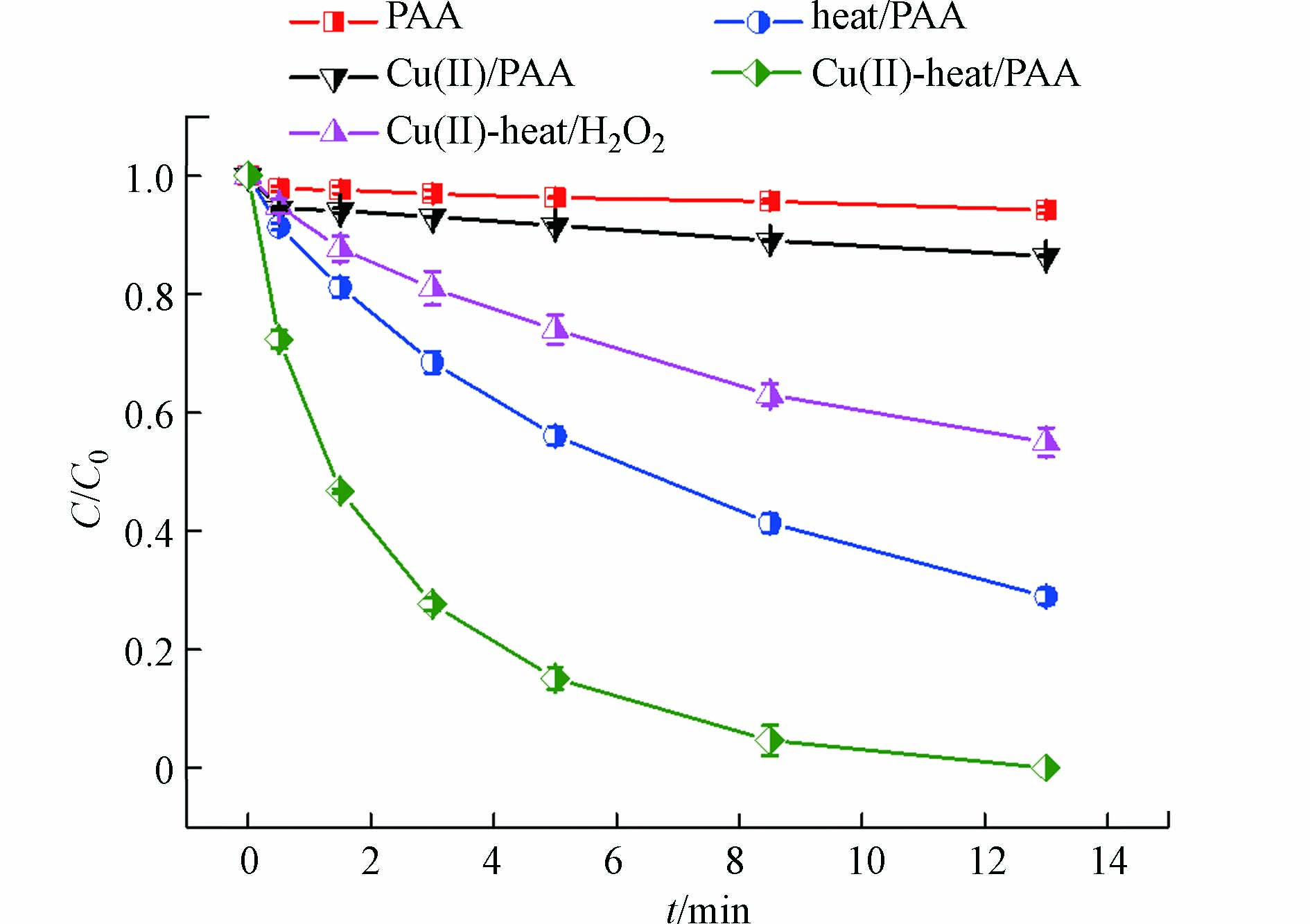
如图1所示,反应13 min后,在单独PAA体系中仅有6% DCF被降解;而在Cu(Ⅱ)/PAA体系中,DCF去除率增加至13%,这说明Cu(Ⅱ)对PAA有一定的活化效果。PAA能被热激活产生活性物质[22],故在60 ℃时,heat/PAA体系对DCF的降解率为72%。而在相同的温度下,Cu(Ⅱ)-heat/PAA体系能够完全消除DCF,远大于heat/PAA体系和Cu(Ⅱ)/PAA体系单独作用,这表明Cu(Ⅱ)和热联用活化PAA存在协同效应。
据报道,在Cu(Ⅱ) 协同热活化H2O2(Cu(Ⅱ)-heat/H2O2)过程中也有类似现象[24-25]。在温度高于35 ℃后,Cu(Ⅱ)-heat/H2O2体系对硝基苯的降解速率快速增加,表观活化能由12.6 kcal·mol−1(20—35 ℃)增加到27.3 kcal·mol−1(35—50 ℃),由此作者认为Cu(Ⅱ)被还原成Cu(I)是Cu(Ⅱ)-heat/H2O2体系降解硝基苯的限速步骤之一。因此,在Cu(Ⅱ)-heat/PAA体系中,Cu(Ⅱ)被PAA还原为Cu(I)也可能是限速步骤之一,体系温度升高能够明显提高该限速反应,从而加速了Cu(Ⅱ)的还原和Cu(I)的生成。Cu(I)能够有效激活PAA产生CH3C(=O)O·和·OH,如式(2)—(4)所示,进而加速了DCF的去除。由于实验使用的PAA溶液是PAA和H2O2的混合物,因此有必要考察Cu(Ⅱ)-heat/PAA体系中H2O2对DCF降解的贡献。由图1可知,在相同条件下Cu(Ⅱ)-heat/H2O2体系对DCF的去除率仅为45%,故H2O2在Cu(Ⅱ)-heat/PAA体系中的贡献较小,主要活性氧化剂仍为PAA。DCF在各个体系中降解符合伪一级反应动力学模型,其表观降解速率常数(kobs)可以通过式(5)计算得出,结果如表2所示。
式中,kobs为表观降解速率常数(min−1);C和C0分别为指定时间和初始时间时DCF的摩尔浓度(μmol·L−1);t为反应时间(min)。
-
在不同pH或温度下,物质存在的形态可能不同,利用环境水化学平衡软件(Visual MINTEQ 3.0)[26]模拟了25 ℃和60 ℃时不同pH条件下Cu(Ⅱ) 的存在形态,所用参数均为数据库默认值。如图2所示,在pH 8时,从25 ℃升高至60 ℃过程中,虽然Cu(OH)+浓度有所降低,但其仍在Cu(Ⅱ)存在形态中占有较高比例,故Cu(OH)+可能是被PAA还原的Cu(Ⅱ)形态之一。此外,Cu(OH)2 (aq)浓度随温度升高而增大,在60 ℃时已成为Cu(Ⅱ)的主要存在形态,因此Cu(OH)2 (aq)也可能是被PAA还原的Cu(Ⅱ)形态。Millero等[27]分别在pH 6—9和温度5—45 ℃条件下测量了海水中H2O2对Cu(Ⅱ)的还原率,认为Cu(OH)2 (aq)是被H2O2还原的活性物质。这与模拟Cu(OH)2 (aq)是被PAA还原的Cu(Ⅱ)形态结论类似。综上所述,Cu(OH)2 (aq)和Cu(OH)+可能是被PAA还原的活性Cu(Ⅱ)形态。
如上所述,热能激活PAA产生CH3C(=O)O·和·OH。此外,温度升高能加速Cu(Ⅱ)/PAA体系中Cu(Ⅱ)的还原和Cu(I)的生成,Cu(I)也能够有效活化PAA产生CH3C(=O)O·和·OH。产生的CH3C(=O)O·和·OH均可与PAA反应生成CH3C(=O)OO·,如式(6)和(7)所示[20]。而CH3C(=O)O·可进一步分解为·CH3和CO2,形成的·CH3可与O2反应生成CH3OO·,如式(8)和(9)所示[18]。因此,在Cu(Ⅱ)-heat/PAA体系中,可能产生·OH和有机自由基R-O·(CH3C(=O)O·、CH3C(=O)OO·、·CH3和CH3OO·)。为了识别该体系中负责DCF降解的活性自由基,分别将TBA和MeOH添加到反应体系中。TBA是一种常用的·OH淬灭剂(7.6×108 L·mol−1·s−1)[28],但不能有效淬灭R-O·[29]。而MeOH不仅可以清除·OH(9.7×108 L·mol−1·s−1)[30],也可以抑制R-O·[31]。如图3所示,当TBA投加到Cu(Ⅱ)-heat/PAA体系中,DCF的降解部分被抑制;然而加入MeOH后,抑制作用明显增强,这表明·OH和R-O·都是Cu(Ⅱ)-heat/PAA体系中的活性物质,且R-O·对DCF去除的作用更大。由于CH3COO·和·CH3氧化性较弱[22],它们对DCF降解的作用可能较小,因此CH3C(=O)O·和CH3C(=O)OO·可能是主要活性的有机自由基。类似的结论在Co2+/PAA体系降解SMX中也有报道[31]。综上,在Cu(Ⅱ)-heat/PAA体系中,·OH和R-O·(CH3C(=O)O·和CH3C(=O)OO·)是主要的活性物质,且R-O·对DCF降解的贡献更大。
-
如图4(a)所示,1—5 mmol·L−1 Cl−会微弱促进Cu(Ⅱ)-heat/PAA体系降解DCF。
由于Cl−和Cu(Ⅱ)/Cu(I)能够形成氧化还原电位较高的Cu(Ⅱ)/Cu(I)-Cl络合物,故Cu(Ⅱ)-heat/PAA体系中Cl−的存在可能会促进Cu(Ⅱ)还原为Cu(I)[32]。此外,虽然Cl−能与CH3C(=O)OO·和·OH反应生成氧化性较弱的含氯自由基[18, 33],如式(10)—(13)所示,但是这些含氯自由基(Cl·和Cl2·−)也可能氧化降解DCF [18]。如图4(b)和4(c)所示,SO42− 和NO3−对Cu(Ⅱ)-heat/PAA体系降解DCF几乎没有影响,这可能是因为它们不与体系中的活性物质发生反应[33, 34]。HCO3−是常见的·OH淬灭剂,其可能会抑制·OH氧化降解污染物。如图4(d)所示,HCO3−加入后虽然会减缓DCF的降解速率,但是反应结束后其去除率基本不受影响。这可能是因为HCO3−能够与Cu(Ⅱ)形成配合物,从而提高了Cu(Ⅱ)催化PAA的能力[35],其促进作用可能刚好与HCO3−对·OH的抑制作用相抵消,结果表现出HCO3−不影响DCF的降解率。本研究使用富里酸(FA)代表NOM,考察其对DCF降解的影响,结果如图4(e)所示。NOM的存在抑制了Cu(Ⅱ)-heat/PAA体系对DCF的降解,且NOM浓度越高,抑制作用越显著。NOM能够与DCF竞争反应体系中的活性自由基(·OH和R-O·)[11],从而导致DCF去除率的下降.
-
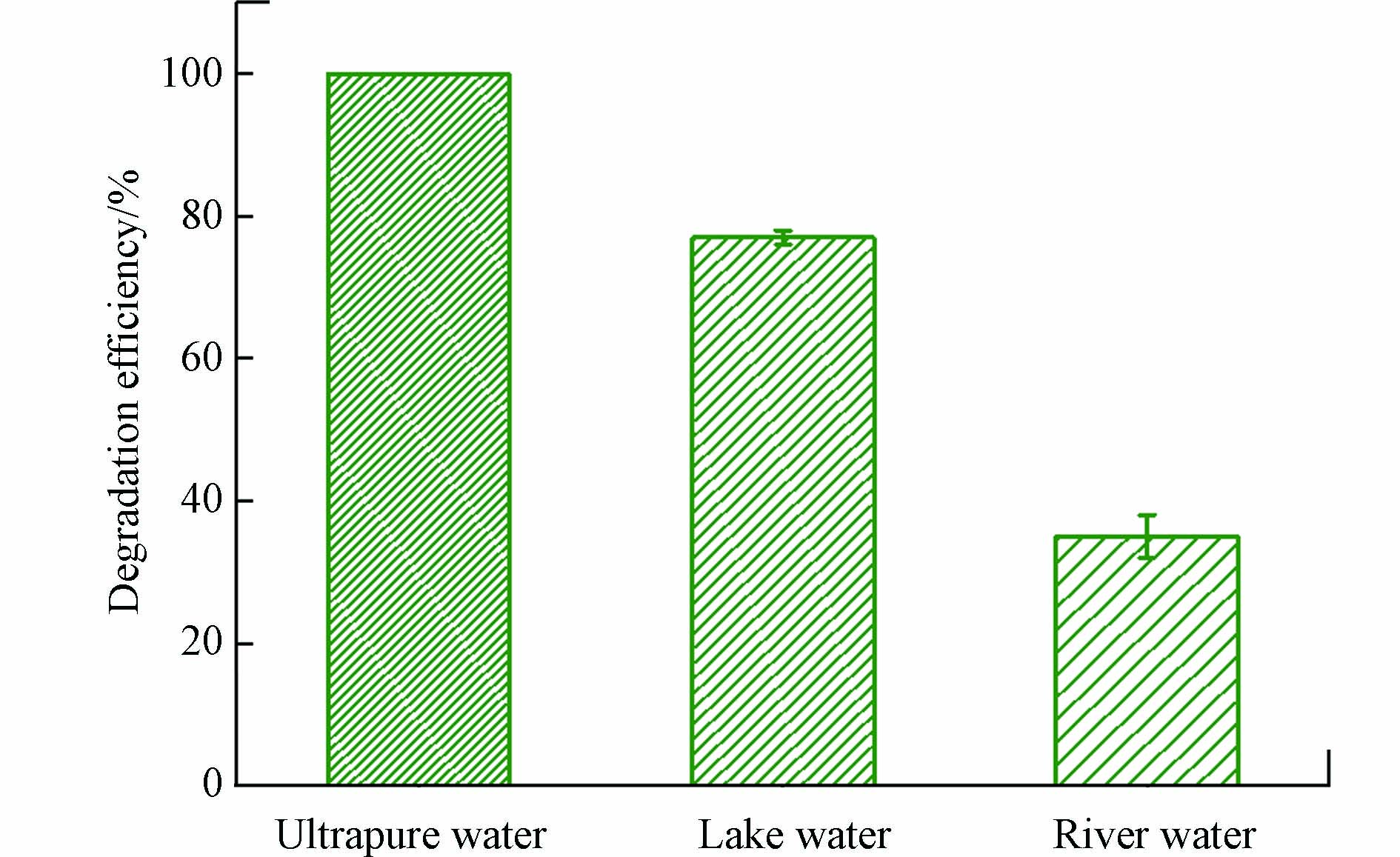
为了评估Cu(Ⅱ)-heat/PAA体系在实际运用中对有机污染物的去除能力,考察了其对实际水体中DCF的降解,结果如图5所示。与超纯水相比,DCF在湖水和河水中均受到抑制。两种实际水体的水质参数见先前的报道[36]。湖水和河水的CODcr、UV254分别为20.8 mg·L−1、0.0365和22.4 mg·L−1、0.0672,表明两种实际水体中都含有一定量的NOM,而NOM会抑制Cu(Ⅱ)-heat/PAA体系降解DCF,造成DCF在两种实际水体中的降解均受到抑制。由于河水中的NOM浓度高于湖水,故DCF在河水中的降解效率低于其在湖水中的去除。
-
(1) Cu(Ⅱ)-heat/PAA体系降解DCF明显高于heat/PAA和Cu(Ⅱ)/PAA体系,表明Cu(Ⅱ)和热联用活化PAA存在协同效应,这主要是由于温度升高能够加快体系中Cu(Ⅱ)向Cu(I)的还原。
(2) 在pH 8时,通过自由基清除实验证明·OH和R-O·(CH3C(=O)O·和CH3C(=O)OO·)是Cu(Ⅱ)-heat/PAA体系降解DCF的主要活性物质,且R-O·对DCF去除的贡献更大。
(3) 由于Cl−与Cu(Ⅱ)/Cu(I)可能形成氧化还原电位较高的Cu(Ⅱ)/Cu(I)-Cl络合物,从而微弱促进了DCF的降解;SO42−、NO3−和HCO3−对DCF的去除几乎没有影响;而NOM抑制DCF降解,导致DCF在河水和湖水中的降解受到抑制。
Cu(II)协同热活化过氧乙酸降解水中双氯芬酸
Degradation of diclofenac in water by Cu(II)-combined with heat activation of peracetic acid
-
摘要: 采用Cu(Ⅱ)协同热活化过氧乙酸(Cu(Ⅱ)-heat/PAA)降解水中双氯芬酸(DCF),识别了Cu(Ⅱ)-heat/PAA体系中的主要活性物质;考察了常见水基质(Cl−、SO42−、NO3−、HCO3−、天然有机质(NOM))对DCF去除的影响;探讨了该体系对天然水体中DCF的去除。结果表明,Cu(Ⅱ)-heat/PAA体系去除DCF的效率明显高于Cu(Ⅱ)/PAA和heat/PAA体系,表明热与Cu(II)两者结合对PAA的活化具有协同作用。在pH 8时,羟基自由基(·OH)和有机自由基R-O·(CH3C(=O)O·和CH3C(=O)OO·)是Cu(Ⅱ)-heat/PAA体系去除DCF的主要活性物质,且R-O·对DCF去除的作用更大。在Cu(Ⅱ)-heat/PAA 体系中,SO42−、NO3−和HCO3−对DCF的去除几乎没有影响;Cl−对DCF去除有微弱的促进效果;而NOM抑制DCF的降解,导致DCF在湖水和河水中的去除受到抑制。Abstract: Removal of diclofenac (DCF) in water by Cu(Ⅱ)-combined with heat activation peracetic acid (Cu(Ⅱ)-heat/PAA) was investigated, and the main active species in Cu(Ⅱ)-heat/PAA system were identified. The effects of common water matrix (Cl−, SO42−, NO3−, HCO3− and natural organic matter (NOM)) on the removal of DCF were explored and the degradation of DCF in natural water was studied. As a result, the removal efficiency of DCF in Cu(Ⅱ)-heat/PAA process presented a relatively faster rate than those in Cu(Ⅱ)/PAA and heat/PAA systems, indicating that the combination of heat and Cu(Ⅱ) had a synergistic effect on the activation of PAA. At pH 8, hydroxyl radical (·OH) and organic radicals (R-O·, CH3C(=O)O· and CH3C(=O)OO·) were the main active substances for DCF removal in Cu(Ⅱ)-heat/PAA system and R-O· played a major role in DCF removal. Presence of SO42−, NO3− and HCO3− had little effect on DCF degradation; Cl− could weakly promote the degradation of DCF; NOM inhibited DCF removal, which resulted in the inhibition on DCF removal in lake water and river water.
-
Key words:
- diclofenac /
- peracetic acid /
- Cu(II) /
- heat /
- organic radicals /
- advanced oxidation process
-
有机磷酸酯(organophosphorus esters, OPEs)是一类重要的有机磷阻燃剂(organophosphate flame retardants, OPFRs),主要以磷作为支架,用链烃、芳香烃或卤代烃取代磷酸上的氢原子组成。按性质的差异,OPEs可划分为卤代和非卤代有机磷阻燃剂[1],前者主要是氯代有机磷酸酯(Chlorinated organophosphates, Cl-OPEs),主要包括磷酸三(2-氯乙基)酯(TCEP),磷酸三(2-氯丙基)酯(TCPP)和磷酸三(1,3-二氯-2-丙基)酯(TDCPP)等。20世纪80年代之后,随着溴代阻燃剂在世界范围内的禁用,OPFRs因具有良好的阻燃效果及其低烟、低毒、低卤等特点,得到了广泛应用[2],2015年全球OPFRs消耗量达到68万t[3]。鉴于OPEs是以简单物理添加而非键合的方式添加到材料中,可以通过挥发和表面磨损进入环境,近年来,OPEs在室内灰尘[4-5]、大气[6-7]、水体[8-13] 、土壤[14]、沉积物[11,15]、生物体[12,16]、污泥[11,17]和垃圾填埋场[18]等多种环境介质中频繁检出。尽管确切的人体暴露数据还比较有限,大量利用斑马鱼、大型溞、小鼠等模式生物或借助体外细胞进行的试验表明,OPEs具有多种毒性效应或指向了明显的致毒趋势,如急性毒性[19-22]、生殖与发育毒性[22-30]、神经毒性[24,31-34]、脏器毒性[35]、基因毒性和致突变性[19,36-38]、内分泌干扰性[39-43]和致癌性[20,44]。表1列出了主要OPEs的基本信息。
表 1 主要OPEs名称和理化性质Table 1. Names and Physicochemical properties of the major OPEs化合物名称及简写Compounds and Abbreviation CAS 分子式Molecular formula 取代基Substituent lgKow Vp(Torr) 磷酸三乙酯(Triethyl phosphate, TEP) 78-40-0 C6H15O4P 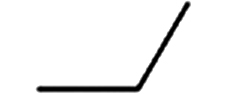
0.80 3.93×10−1 磷酸三正丁酯(Tri-n-butyl phosphate, TnBP) 126-73-8 C13H27O5P 
4.00 1.13×10−3 磷酸三异丁酯(Tri-iso-butyl phosphate, TiBP) 126-71-6 C12H27O4P 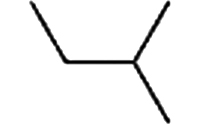
3.60 1.28×10−3 磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP) 78-51-3 C18H39O7P 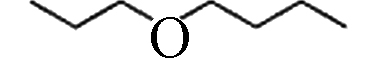
3.75 2.50×10−8 磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP) 115-96-8 C6H12Cl3O4P 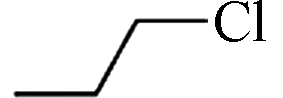
1.44 6.13×10−2 磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP) 13674-84-5 C9H18Cl3O4P 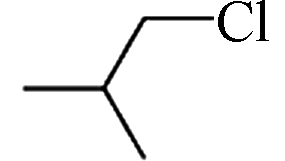
2.59 2.02×10−5 磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP) 13674-87-8 C9H15Cl6O4P 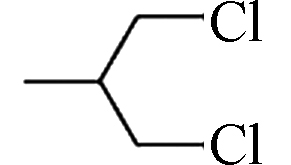
3.65 7.36×10−8 磷酸三苯酯(Triphenyl phosphate, TPhP) 115-86-6 C18H15O4P 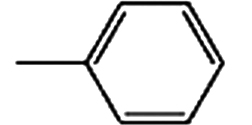
4.59 6.28×10−6 磷酸三甲苯酯(Tricresyl phosphate, TCrP) 1330-78-5 C21H21O4P 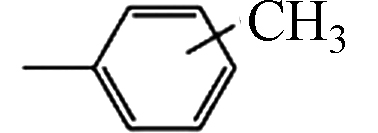
5.11 6.00×10−7 三苯基氧化膦(Triphenylphosphine oxide, TPPO) 791-28-6 C18H15OP 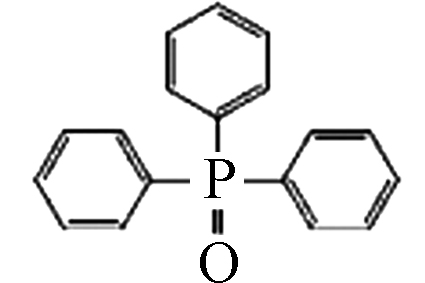
2.83 2.62×10−8 注:Kow为正辛醇-水分配系数,Vp为蒸汽压。Note: Kow is the n-octyl alcohol-water distribution coefficient, Vp is the vapor pressure. | Show Table DownLoad:
CSV
DownLoad:
CSV
随着研究的深入,Cl-OPEs作为特殊的一类OPEs越来越受到学界关注。首先,Cl-OPEs是环境赋存最广的一类OPEs,在各介质的检出组成中占据主导地位,其浓度水平为ng·L−1—μg·L−1(水体和垃圾渗滤液),pg·m−3(空气),ng·g−1(土壤、室内灰尘、生物体和沉积物)。例如,TCPP和TCEP被证实是水环境中丰度最高的两种OPEs[8-10],浓度分别可达921 ng·L−1 (TCPP)和268 ng·L−1 (TCEP)[10];以TCPP为主的Cl-OPEs在我国土壤OPEs中占74%以上[14];TCEP在我国城市家庭和大学宿舍室内灰尘中的含量高达208 μg·g−1[5];TCPP在德国北海大气OPEs中的占比高达60%±16%[7],TCPP和TDCPP是韩国石洼人工湖沉积物中的主导OPEs[45],其中TCPP浓度达到2500 ng·g−1。其次,Cl-OPEs具有显著、广泛的生物毒性效应。一方面,在所有OPEs表现出的生物毒性种类中均观察到Cl-OPEs,且只有Cl-OPEs被报道具有致癌性[44];另一方面,研究也证实Cl-OPEs中的TDCPP生物毒性极强,例如TDCPP对虹鳟鱼的96 h-LC50仅为1.1 mg·L−1[20],对斑马鱼仔鱼和胚胎的116 h-LC50仅为7 mg·L−1[19],室内灰尘中的TDCPP可能引起男性激素水平改变和精液质量显著下降[29],其代谢物双(1,3-二氯-2-丙基)磷酸盐(BDCPP)对斑马鱼胚胎是比本体强4个数量级的致畸剂[31]等。另外,Cl-OPEs是最难降解的一类OPEs,由于其对生物和化学降解的抵制倾向,以活性污泥法为基础的常规污水处理技术收效甚微[46],改性沸石[47]和碳纳米管[48]等吸附剂被报道可以有效地吸附水中的Cl-OPEs,但后续处理较为棘手,在此背景下,高级氧化技术(AOPs)在Cl-OPEs降解上得到应用。然而,Cristale等的研究表明TCEP、TDCPP和TCPP在AOPs的应用中也是最顽固的OPFRs[49],例如,UV/H2O2过程中,TCPP、TCEP和TDCPP烷基链上的Cl的存在显著降低了·OH对烷基磷酸酯的反应活性,有机物如腐殖质对UV的辐射吸收抑制了H2O2的消耗,增大了污水处理厂(WWTP)废水中Cl-OPEs的去除难度。
由于Cl-OPEs在环境中检出浓度较高、毒性效应显著且难以生化降解,亟需研究控制Cl-OPEs的替代处理技术。目前,在Cl-OPEs降解领域三种具有代表性的探索方向是水解、微生物降解和光降解。近来,以复合金属/半导体基光催化剂、强自由基发生基质(例如过硫酸盐的活化溶液)和可持续光能体系为优化方向的光催化氧化技术日渐流行。本文综述了Cl-OPEs的3种主要降解途径和机理,其中重点介绍了光降解Cl-OPEs的方法、特点和效果,通过分析比较不同降解途径的优势与不足,对未来Cl-OPEs降解的研究方向和前景提出展望。
1. 氯代有机磷酸酯的水解(Hydrolysis of chlorinated organophosphate)
1.1 Cl-OPEs的水解途径
有机磷物质的水解可以通过两条途径进行:(1) OH−和H2O攻击P原子,该亲电位点较“硬”,反应条件较为苛刻;(2) H2O攻击烷氧基上的α-碳,该亲电位点较“软”,反应较易进行[50]。这两条途径的断键机理如图1所示[51]。

1.2 Cl-OPEs水解的pH依赖性
鉴于OPEs的基本分子结构(即酯键),磷酸酯与磷酸的非生物水解与它们在水环境中的稳定性密切相关,已有研究证实OPEs的水解具有显著的pH依赖性,而对于含有特征官能团Cl原子的Cl-OPEs,这种依赖性表现为碱性pH有利于水解。早年间,曾有报道称TCPP在海洋固体废物处理场的渗滤液中不会发生非生物降解[52]。近来,Su等将16种OPEs划分为烷基侧链(通式CnH2(n + 1))、氯化烷烃侧链(通式CnHxCly,x+y = 2(n+1))、芳基侧链等类型,分别在pH=7、9、11、13时水解[53],降解动力学表明不同支链类型OPEs的稳定性排序:烷基>含氯烷烃>芳基,3种主要Cl-OPEs即TCEP、TCPP、TDCPP半衰期分别为0.083、11、0.044 d,降解速率常数分别为8.36、0.0621、15.6 d−1,三者均在35 d内被广泛降解。在pH=13的溶液中观察到Cl-OPEs降解速度比烷基OPEs快得多,例如,尽管每个取代基只有一个原子(Cl原子或H原子)的差异,TEP的t1/2,pH=13为16 d,而TCEP的t1/2,pH=13仅为0.083 d。水解产物筛选和质量平衡研究表明Cl-OPEs碱基催化水解的最终产物是二酯而非推测中的单酯,可能是二酯磷酸在碱性条件下被脱质子到相应的共轭碱后性质相当稳定导致的。近来,Wu等通过化合物特异性同位素分析方法(CSIA)表征了OPEs在水生环境中依赖于水解过程的同位素分馏模式[54],发现在较低的pH (pH=2−7)下有显著的碳分馏,但在较高的pH (pH=12)下无明显的碳分馏,在任何水解实验中都没有观察到氢的分馏,表明酸性或中性条件下造成C—O键的断裂,碱性水解则造成P—O键的断裂,进一步区分了两种不同的pH依赖的水解途径。
1.3 矿物催化水解Cl-OPEs
近两年的研究发现矿物催化水解是实际土壤或水生环境Cl-OPEs控制中一个重要的潜在途径。Fang等报道了TDCPP、TCPP、TCEP等8种OPEs在没有和存在金属氧化物矿物情况下的水解[55]。通常Cl-OPEs在中性pH条件的均相溶液中水解缓慢,半衰期t1/2>2年,但在矿物悬浮液中(pH=6,25℃,矿物表面积负载为15 m2·L−1),t1/2可缩短至不到10 d,其中三价铁系氢氧化物和氧化物(α-FeOOH、γ-FeOOH和α-Fe2O3)催化效果最好,铝和硅氧化物(Al2O3和SiO2)催化作用不明显。以代表性的铁系矿物针铁矿为例证实了表面相互作用是促进水解反应的关键,这种相互作用取决于矿物表面和OPFR的结构,进一步阐明了溶解金属离子和矿物表面催化有机磷酸酯水解反应的3种机理,包括表面金属原子与磷酸盐中心直接配位使SN2@P反应更易进行,表面金属原子与酯基直接配位削弱其与磷中心的键合从而促进裂解,以及形成表面Fe(III)羟基物种来提高矿物−水界面上的亲核剂浓度从而促进水解反应。另外,转化产物的高效液相色谱-四极杆飞行时间串联质谱分析(HPLC-Q-TOF-MS)证实酯水解为活性降解途径,单个OPE的kB值是高度可变的,并且与相应的醇离去基的酸离解常数(pKa)密切相关,矿物与OPEs的相互作用是速率控制的,可以通过调控反应速率促进Cl-OPEs的矿物水解。
2. 氯代有机磷酸酯的微生物降解(Microbial degradation of chlorinated organophosphates)
2.1 Cl-OPEs生物降解性能
微生物降解方法因能有效避免由化学降解方法产生的二次污染而受到关注。WWTPs中使用的活性污泥法对芳基和非卤代烷基OPFRs有一定的降解效果,但无法降解Cl-OPEs,因为活性污泥菌胶团中的微生物没有能使Cl-OPEs磷酯键断裂的特异性酶。微生物降解法的突出特点是选择性好、降解率高、成本低廉和环境友好,研究的重难点在于筛选出能降解目标化合物的菌种。
近十年来,被报道的可生物降解的OPEs主要是非卤代烷基磷酸酯中的TBP[56-62]和芳香族磷酸酯中的TPhP[63-66],针对Cl-OPEs的生物降解展开研究的主要是日本学者Takahashi领导的课题组,主要目标化合物为TCEP和TDCPP[67-72]。虽然研究规模较小,但该团队在筛选、培养、分离有效菌种,优化降解条件,细胞系统设计,解毒技术开发,基因工程,产物鉴定等方面做了一系列细致而深入的工作。具有降解Cl-OPEs能力的微生物及其降解性能总结见表2。由于TCEP和TDCPP各自的代谢产物2-氯乙醇(2-CE)和1,3-二氯-2-丙醇(1,3-DCP)也具有毒性,仅用目标化合物的去除率不足以完整评估其微生物降解效果,相关研究中特别引入“解毒率”这一概念来量化用微生物方法去除目标化合物以及各自的致毒代谢产物的程度。
表 2 具有降解Cl-OPEs性能的微生物及其降解性能总结Table 2. Summary of (Cl-OPEs)-degrading bacterial species and their performance菌种Bacterial species Cl-OPEs及代谢物Cl-OPEs and metabolites 降解/解毒率Degradation/detoxification rate 参考文献Reference (TCEP) Acidovorax spp.,Sphingomonas spp.(TDCPP) Acidovorax spp.,Aquabacterium spp.,Sphingomonas spp. TCEP TDCPP 100% (6 h, [TCEP]0=20 µmol·L−1)100% (3 h, [TDCPP]0=20 µmol·L−1) [67] (TCEP) Sphingobium sp.strain TCM1,(2-CE) Xanthobacter autotrophicus strain GJ10 TCEP2-CE 最佳条件测试:降解100% (30 ℃, pH=8.5, [Co2+]=50 μmol·L−1, OD660=0.8, [TCEP]0=10 µmol·L−1) 解毒100%(24 h, [2-CE]0=180 µmol·L−1)分步解毒:降解/解毒100%(①4 h,[TCEP]0=10 µmol·L−1; ②144 h,[2-CE]0=29 µmol·L−1) [68] (TDCPP)Sphingobium sp.strain TCM1,(1,3-DCP)Arthrobacter sp.strain PY1 TDCPP1,3-DCP 最佳条件测试:降解100% (30 ℃, pH=8.5, [MOPS]=50 mmol·L−1, [TDCPP]0=50 µmol·L−1)解毒100% (35 ℃, pH=9.5,[Tris-H2SO4]=50 mmol·L−1, [1,3-DCP]0=5 mmol·L−1)同时解毒:降解/解毒率100% (12 h, 30 ℃, pH=9, [Tris-H2SO4]=50 mmol·L−1,[TDCPP]0=53.2 µmol·L−1,) [69] Pseudomonas spp.Sphingobium spp. TCEP TCPP (48.37±9.52)%—(82.28±7.48)% [73] | Show TableDownLoad:
CSV
2008年,Takahashi等以TCEP或TDCPP为唯一磷源,尝试富集具有降解Cl-OPEs性能的菌种,首次报道了能降解TCEP和TDCPP的微生物[67],降解TCEP和TDCPP的优势菌种Acidovorax-和Sphingomonas-属可分别催化氯醇脱卤和OPEs的磷酸酯键裂解。接着,该团队就TCEP和TDCPP及各自代谢产生的氯醇2-CE和1,3-DCP分别设计了完全解毒的细胞系统[68-69]。一方面,利用TCM1菌株和2-CE降解菌(Xanthobacter autotrophicus菌株GJ10)开发了TCEP的两步反应解毒技术[68],包括TCEP在静息细胞TCM1的作用下24 h内水解成2-CE,然后在生长细胞GJ10的作用下经过144 h降解所有生成的2-CE,联合反应完全解毒9.6 μmol·L−1 TCEP。另一方面,开发了使用菌株TCM1和Arthrobacter sp.株PY1同时降解1,3-DCP而使TDCPP完全解毒的技术[69]。由于TCM1静息细胞对TDCPP脱磷和菌株PY1静息细胞对1,3-DCP脱卤的最佳条件相似,采用两种菌株同时对TDCPP及其代谢物进行降解以缩短反应时间,在30 ℃、pH=9.0、[Tris–H2SO4]=50 mmol·L−1菌株PY1细胞OD660=4.0的情况下,12 h内实现了TDCPP的完全解毒。这两个系统的设计为微生物解毒潜在的有毒持久性有机磷化合物开辟了一条道路。
此外,该团队在基因工程和降解路径推导方面也取得了一些研究成果,降解路径的推导依赖于产物鉴定,主要手段有离子色谱法[57]或衍生化气相色谱法[60]。Abe等从能够降解TCEP和TDCPP的两种菌株Sphingobium sp.TCM1和Sphingobium sp.TDK1中纯化克隆出两种卤代烷基磷水解酶,并分别命名为TDK-HAD和TCM-HAD,这两种磷酸三酯酶成功降解了Cl-OPEs,同时也能广泛降解溴化有机磷(如TDBPP)和三芳基磷酸盐(如TCP和TPhP)[70]。有机磷化合物对微生物细胞生长的利用被认为是由三种类型的磷酸酯酶介导的:磷酸三酯酶(PTE),磷酸二酯酶(PDE)和磷酸单酯酶(PME)。Takahashi等利用TCEP对Sphingobium sp.菌株TCM1进行碱性磷酸酶基因鉴定并推导了TCEP分步降解的路径[72],如图2所示,TCEP在菌株TCM1中的代谢包括全部3种磷酸酯酶,在每一步中,磷酸酯键中的一个被水解生成2-CE,最后一步生成Pi用于细胞生长。

2.2 Cl-OPEs生物修复案例
Cl-OPEs生物降解的最新进展源于针对混合OPFRs废水的实地生物修复案例。Liu等建立了综合垂直流人工湿地(IVCWs)以研究短期暴露于3种OPFRs(TCrP,TCEP,TCPP)后的环境行为和根际微生物反应[73],揭示了利用IVCWs处理以Cl-OPEs为主的OPFRs混合污染的潜力。研究发现,在处理低碳/氮比废水时,IVCWs对OPFRs废水既表现出良好的TN去除效果((628.13±110.63) mg·m−2·d−1),又具有相当的OPFRs去除效率((48.37%±9.52%)%—(82.28%±7.48%));对比高疏水化合物磷酸三甲苯(TCrP)在土壤、火成岩、沸石和植物组织中的吸附,Cl-OPEs则更容易被吸收并转移到植物体内;OPFRs与根际微生物的丰度、多样性和氮相关的微生物之间存在显著的相互作用;Pseudomonas属和Sphingobium属可能对混合OPFRs具有生物降解功能。
3. 氯代有机磷酸酯的光降解(Photodegradation of chlorinated organophosphate)
高级氧化技术(AOPs)是去除水溶液中难降解有机污染物的有效技术,包括臭氧化、光催化氧化、芬顿氧化、硫酸根自由基氧化等[74],其本质是通过各种途径生成羟基自由基(·OH)和硫酸盐自由基(
⋅SO−4 ⋅SO−4 按光源的不同,光氧化降解Cl-OPEs可分为紫外光(UV)降解和可见光(Vis)降解。鉴于Cl-OPEs的降解难度和高能量UV降解的稳定、高效性能,UV是学者开展Cl-OPEs降解研究的首选光源。然而,由于Cl-OPEs侧链烷基缺乏发色团,没有吸收波峰,很难直接光解,通常需要采用基于UV的复合光降解技术。按研究历程有三种复合思路:UV结合生成·OH的氧化剂如H2O2、O3[49,54,75,77-84],UV结合催化剂如TiO2和MOF材料[85-90],UV结合生成
⋅SO−4 ⋅SO−4 3.1 紫外光降解
3.1.1 UV/H2O2
在含有氧化剂H2O2的UV照射水中能形成大量的·OH,引发了UV/H2O2在全规模的水处理厂降解微污染有机物中的设计和实施[96]。Cl-OPEs在UV-C范围内是较差的光吸收剂,空白实验也表明H2O2不能单独促进化合物的降解,故UV/H2O2是UV和H2O2光解生成·OH氧化降解污染物的协同作用[80]。
多项研究表明,UV/H2O2是降解Cl-OPEs有效、可靠的技术,其光降解性能参数和效率见表3。目前,使用UV/H2O2降解OPEs的研究已经比较体系化,内容包括探究支链结构、光强、pH、底物浓度、氧化剂浓度、天然有机物的竞争性等因素对降解效率的影响,用C/C0、Cl−和
PO3−4 表 3 光降解Cl-OPEs的方法和效率总结Table 3. Summary of methods, efficiency and kinetics of photodegradation of Cl-OPEs方法Methods Cl-OPEs浓度Cl-OPEs concentration 关键试剂Key reagents 照射波长Irradiation wavelength 光降解效率Photodegradation efficiency 参考文献Reference UV/H2O2 5 mg·L−1 TCEP 50 mg·L−1,30% H2O2 254 nm >95% [77] UV/H2O2 143 mg·L−1 TCEP 5 mmol·L−1,30% H2O2 200—420 nm (1 h)>85% [80] UV/H2O2 50 μg·L−1 TCPP50 μg·L−1 TCEP50 μg·L−1 TDCPP 30% H2O2 254 nm (1 h)97%(1 h)91%(1 h)84% [81] UV/H2O2 500 μg·L−1 TCEP 1.5 mg·L−1,30% H2O2 200—400 nm (6 h)100% [82] UV/H2O2 500 mg·L−1 TCEP 30% H2O2,50 mg·L−1 185—400 nm (13 h)97% [54] UV/H2O2 5 mg·L−1 TCPP 30% H2O2,0.1mmol·L−1 200—400 nm (15 h)96% [75] UV/H2O2 4 mg·L−1 TCPP 30% H2O2 200—400 nm (25 min)96.1% [84] UV/TiO2(P25) 4 mg·L−1 TCPP 1000 mg·L−1 TiO2-P25 365 nm (12 h)80% [85] UV/TiO2-001 4 mg·L−1 TCPP 1000 mg·L−1 TiO2-001 365 nm (6 h)100% [85] UV/TiO2(P25) 1 mg·L−1 TCEP 70 mg·L−1 TiO2 254 nm (10 min)99% [86] UV/TiO2(P25) 1 mg·L−1 TCPP 100 mg·L−1 TiO2 254 nm (25 min)100% [90] UV/MIL-101(Fe)/PS 1 mg·L−1 TCEP 500 mg·L−1 MIL-101(Fe)500 mg·L−1 PS 420 nm (3 h)>80% [89] UV/PS 1 mg·L−1 TCEP 500 mg·L−1 PS 280 nm (3 h)>95% [89] UV/PS 1 mg·L−1 TCEP 175 μmol·L−1 PS 254 nm (30 min)99% [91] UV/PMS 1 mg·L−1 TCEP 20 mg·L−1 PMS 365 nm (30 min)94.6% [92] UV/PS 1 mg·L−1TCPP 75 mg·L−1 PS 254 nm (25 min)98.2% [93] 模拟太阳光/TiO2(P25) 250 μg·L−1 TCPP 50 mg·L−1 TiO2 290—800 nm (2 h)95% [97] Vis/N、-TiO2 100 μg·L−1 TCPP 250 mg·L−1 N、S-TiO2 400—800 nm (2 h)65%—70% [94] Vis/GO@MIL-101(Fe)/H2O2 1 mg·L−1 TCEP 500 mg·L−1 GO@MIL-101(Fe)165 mmol·L−1 30% H2O2 420 nm (30 min)95% [95] | Show TableDownLoad:
CSV
OPEs支链的分子结构显著影响UV/H2O2条件下·OH对目标物的降解速率。Watts等研究了TCEP、TCPP、TBP和TBEP等4种磷酸三酯的二阶反应速率,kOH, TCEP=5.6×108 mol·L−1·s−1,kOH, TCPP=1.98×108 mol·L−1·s−1,kOH, TBP=6.4×109 mol·L−1·s−1,kOH, TBEP=1.03×1010 mol·L−1·s−1,排序为TBEP>TBP>TCEP>TCPP[78],即烷基OPEs的降解速率高于Cl-OPEs,与水解时的规律一致。Cl-OPEs支链上的Cl原子显著降低了H原子的可用性,导致降解速率较慢。另外,随着烷基链的卤化,碳链数和加成物α链上的杂原子(O)也影响了·OH攻击的速率。理论上,TCPP在每个碳链上比TCEP多一个碳,降解速率应高于TCEP,然而实验结果正相反,很可能是受TCPP的α-碳处附加的—CH3的影响。此后,He等在超纯水中,用250 W紫外光照射50 mg·L−1 H2O2降解TCPP[75],动力学计算表明速率常数为0.0035 min−1(R2=0.9871),降解率达到96%,但TOC去除率仅有43.02%,证实了TCPP的矿化难度较大。
UV/H2O2体系中TCEP的去除效果与H2O2的投加量及其氧化特性密切相关。一方面,调控初始H2O2浓度对TCEP的去除非常关键。在5 mg·L−1 TCEP,pH=7碳酸盐缓冲溶液中外加50 mg·L−1 30% H2O2,用低压汞灯(λ=254 nm)照射,降解率达95%以上[77]。但31P核磁共振波谱法对UV/H2O2体系的监测表明,使用高浓度的氧化剂来改善混合OPEs中TCEP的去除可能会导致·OH氧化的抑制,使得TCEP无法完全降解[82]。以反应时间1 h为基本条件考察H2O2用量对UV/H2O2降解143 mg·L−1TCEP的影响[80],发现增加H2O2的用量提高了Cl−和
PO3−4 PO3−4 3.1.2 UV/M(光催化剂)
光催化技术及光催化材料的研究是目前最为活跃的研究方向之一。使用光催化技术降解Cl-OPEs的优势在于不需使用昂贵的氧化剂,也不易产生毒性更高的中间产物。
TiO2及其改性产品是最常用和最早实现商品化的光催化剂,具有催化活性高、应用范围广、清洁无二次污染、价格低廉等优点。TiO2的禁带宽度大(3.2 eV),产生的光生电子和空穴氧化还原电势高,且稳定性强,因而在光催化剂向有机半导体基/金属基高分子纵深发展的今天,TiO2仍然被广泛应用于实际工程领域中污染物的去除。UV/TiO2在降解Cl-OPEs上的有效性很早就得到了证实。刘青等采用商品化的TiO2-P25在UV下降解TCPP[85],UV和P25联合作用下12 h降解率达到80%,用高温醇热法进一步合成暴露(001)活性晶面TiO2,与其特征波长365 nm的紫外光联合作用,90 min TCPP降解率超过50%,360 min内完全降解,最优催化加投加量为1 g·L−1。中性和酸性时的降解率要略微大于碱性条件,但不同pH下TCPP在6 h内降解率都达到90%,表明改性催化剂TiO2-001的pH适应性更强,有潜力应用于不同酸碱度TCPP废液的处理。
天然有机物、无机离子和腐殖酸能够显著影响UV/TiO2对Cl-OPEs的降解。有报道称光催化降解TCEP的效率受pH、天然有机物和阴离子影响很大,在实际的水处理过程中,TCEP很难完全矿化[86]。Tang等在使用UV/TiO2光催化降解TCEP、TCPP和TDCPP时重点考察了无机离子和腐殖酸的作用[87],结果表明无机离子如
PO3−4 SO2−4 NO−3 PO3−4 SO2−4 NO−3 金属-有机骨架(MOFs)是一类具有超高的比表面积、较高且可调的孔隙率和开放的金属位点的新型多孔材料,作催化剂时具有多相催化后易回收分离、可循环使用的优点。光和MOFs的结合是降解Cl-OPEs的新方向。Hu等制备了禁带宽度为2.41 eV的高纯度规整晶体MIL-101(Fe),结合UV在过硫酸盐体系中降解TCEP[89],合成的MIL-101(Fe)可利用短波可见光(400—520 nm)和紫外光(200—400 nm),被光激活的MIL-101(Fe)使Fe(Ⅲ)转化为Fe(Ⅱ),进而使
S2O2−8 SO−4 S2O2−8 3.1.3 UV/PS和UV/PMS(过硫酸盐)
活化过硫酸盐(包括过二硫酸盐PS和过一硫酸盐PMS)由于操作简单、氧化性高、应用广泛,被认为是处理OPEs的有前景的技术,其原理主要是活化产生硫酸根自由基(
⋅SO−4 ⋅SO−4 ⋅SO−4 3.2 可见光降解
在光催化降解污染物的过程中,材料的选择与制备对于降解性能至关重要。尽管无毒、低成本的TiO2具有良好的化学稳定性,但其禁带宽度较大(−3.2 eV)只能吸收紫外光,造成太阳光的利用率严重受限(小于5%)。尽管利用可见光降解Cl-OPEs的研究非常有限,但其体现了光催化发展从紫外光向可见光转变的趋势。在光源选择发生转变的同时,学者也在积极探索光催化剂和光催化体系的优化,催化剂的优化表现为从以TiO2为典型的无机催化剂转向表面掺杂高聚合TiO2或有机高分子MOF材料,体系优化表现为从较为简单的UV/TiO2、UV/H2O2体系转向光/复合金属或半导体基光催化材料/过硫酸盐的多相光催化体系。例如,Antonopoulou等通过模拟太阳光,使用TiO2作为催化剂降解溶解在纯水中的TCPP,降解速率在0.0141—0.142 min−1(C0=25—500 μg·L−1)之间,处理后毒性降低[97]。通过掺杂改性等手段可以拓宽TiO2的光响应至可见波段,采用N、S掺杂改性TiO2对TCPP也进行了催化降解[94],在UV-Vis照射下,N、S共掺杂的TiO2是去除TCPP的最具光活性的催化剂,但6 h内,TCPP的去除率也不足65%。Lin等则在MIL-101(Fe)的基础上附载了氧化石墨烯,制备MOF材料GO@MIL-101(Fe),并在此基础上开发了可见光/MOF/H2O2的光催化体系增强可见光条件下对TCEP的降解能力,附载了氧化石墨烯后,MIL-101(Fe)材料电位从2.41eV降低到2.71 eV,从而对光的吸收范围从520 nm扩展到570 nm。GO的高电导率实现了快速活化和电子转移,30 min TCEP降解率达95%,且GO@MIL-101(Fe)在水中稳定[95]。
4. 氯代有机磷酸酯降解的安全风险评估(Safety risk assessment of chlorinated organophosphate degradation)
随着研究的深入,Cl-OPEs降解的安全风险越来越受到关注,这种风险主要源于降解过程中产生的高毒性含氯中间产物。由于pH依赖和矿物催化的水解方法研究尚不充分,微生物降解方法虽然选育菌种较受限但本身可以完全解毒代谢产物,对Cl-OPEs降解的安全风险探究主要聚焦在UV/H2O2、UV/TiO2、UV/PS等光降解方法研究方面。
UV/H2O2降解Cl-OPEs前后生物毒性的变化最早引发注意,但毒性结论并不统一,其升高或降低可能取决于受试生物。例如,用盐湖卤虫卵进行UV/H2O2降解TCEP前后的急性毒性测试,不论是单一TCEP还是混合OPEs降解,LC50都升高,表明急性毒性降低[82]。使用大肠杆菌评估TCPP在UV/H2O2降解前后的毒性,显示活性氧(ROS)和细胞凋亡减少,大肠杆菌的膜电位(MP)升高,生物分子功能得到修正,说明TCPP降解后毒性降低[84]。然而,根据发光菌实验数据,随着反应的进行,TCPP产物的毒性明显增加[75]。上述结果表明,需要更多降解前后的毒性变化案例佐证和完善现有的研究结果。
UV/TiO2降解Cl-OPEs的安全风险评估的研究热点是中间产物的蛋白质组学分析,因为蛋白质相互作用是揭示Cl-OPEs及其中间体在蛋白质组水平上生物分子影响的重要视角。Ye等研究了UV/TiO2非均相光催化降解水中TCEP及其对细菌蛋白质组的影响[86],自组装紫外辐射装置在投加70 mg·L−1 TiO2的情况下,对1 mg·L−1 TCEP 10 min降解率达到99%,·OH是主要的反应物种,降解过程符合准一级动力学模型,kobs=0.3167 min−1;对代谢反应、途径和网络的蛋白质组学分析发现初步降解产物能被大肠杆菌通过细胞代谢进行转运和利用;观察到抗逆性下降趋势,处理后产物毒性明显减弱,表明UV/TiO2体系下矿化不完全的TCEP的羟基化和脱氯作用对其解毒同样有效。随后Yu等将目标转向TCPP,发现Cl−和
PO3−4 UV/PS降解Cl-OPEs不但高效,且毒理学评价证实其安全风险较小。例如,Ou等观察到TCEP在254 nm UV/PS体系下降解中间产物的毒性降低[91]。Xu等使用流式细胞术(FCM)分析对降解中间混合物进行的毒理学评价显示,细胞内活性氧(ROS)和细胞凋亡率明显下降,细胞膜电位(MP)较原TCPP升高,细胞周期分析表明这些降解产物对大肠杆菌DNA生物合成的负面影响也有所减弱,说明经过UV/PS处理后降解产物的毒性明显降低[92]。UV/过硫酸盐能在部分矿化Cl-OPEs的同时进行有效解毒,提示其能控制废/污水中Cl-OPEs的污染。
另外,对于MOFs材料或可见光催化降解Cl-OPEs,因为降解效果不够理想,降解机制尚不清楚,所以研究还未拓展到安全风险评估方面。
5. 结论与展望(Conclusion and prospects)
Cl-OPEs在环境中的高丰度、高毒性和难降解性使其去除技术成为研究热点。本文主要介绍了3种代表性的去除技术,包括pH依赖的水解、特异性磷酸水解酶促进的微生物降解和光驱动的高级氧化降解,另外还讨论了各种降解方法的安全风险。总的来说,碱性条件和矿物催化有利于Cl-OPEs水解,但这方面研究仅限于实验室领域;微生物降解具有彻底矿化和完全解毒Cl-OPEs的潜力,不足在于前期选育菌种和反应用时过长,且需要依靠两种菌株共同解毒,在实际水生或土壤环境中还可能面临低温制约;光驱动的高级氧化技术通过激发催化剂或氧化剂生成具有高度活性的·OH和
⋅SO−4 因此,未来Cl-OPEs的降解研究应集中开发和优化高效、绿色、无二次污染的技术。水解方面要补充基于实际水生环境和土壤基质的实验数据,尤其关注天然pH条件下降解稳态Cl-OPEs的新方法、新材料研发。微生物降解方面应注意选育能在降解目标物的同时降低其毒性的高效菌株。当然,研究着力点仍应放在光催化和高级氧化技术的联用方面,侧重点包括:(1)光催化剂性能的提升,通过改性和引入适当的有机高分子材料优化催化效果;(2)反应体系的设计优化;(3)含氯中间产物的毒性评价方法优化;(4)开发新型可见光驱动光催化剂以拓展光响应范围,并促进电荷有效利用。
-
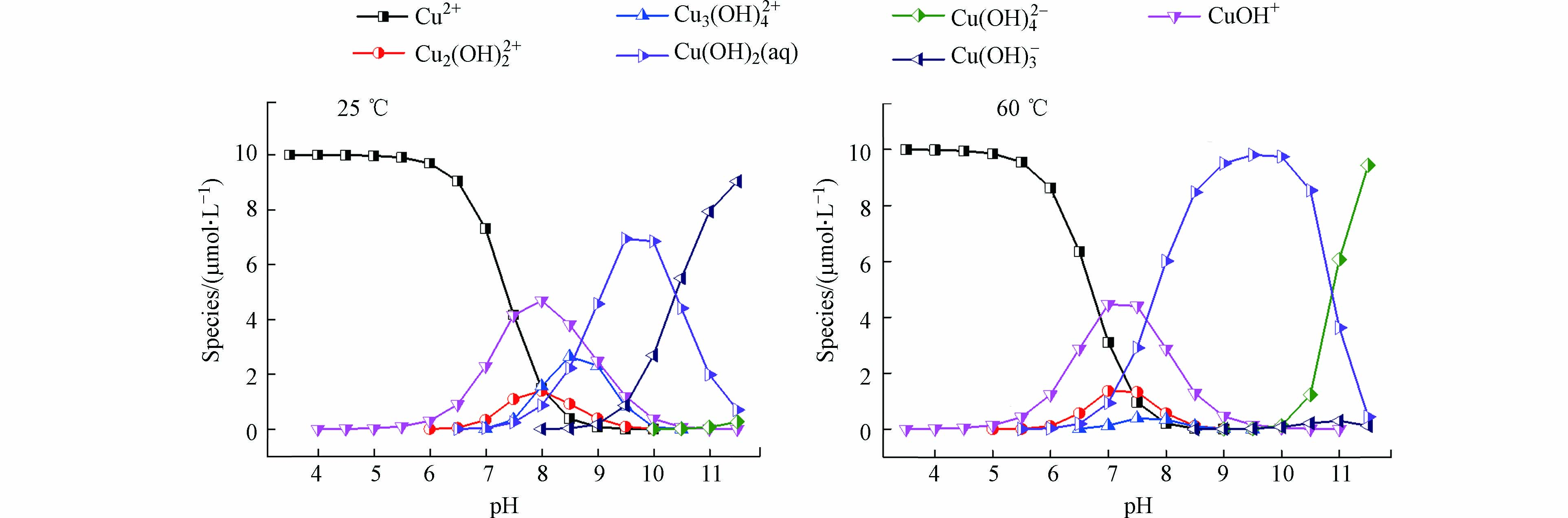
图 2 不同温度和pH条件下Cu(Ⅱ)存在的形态
Figure 2. Existing form of Cu(Ⅱ) at different temperature and pH
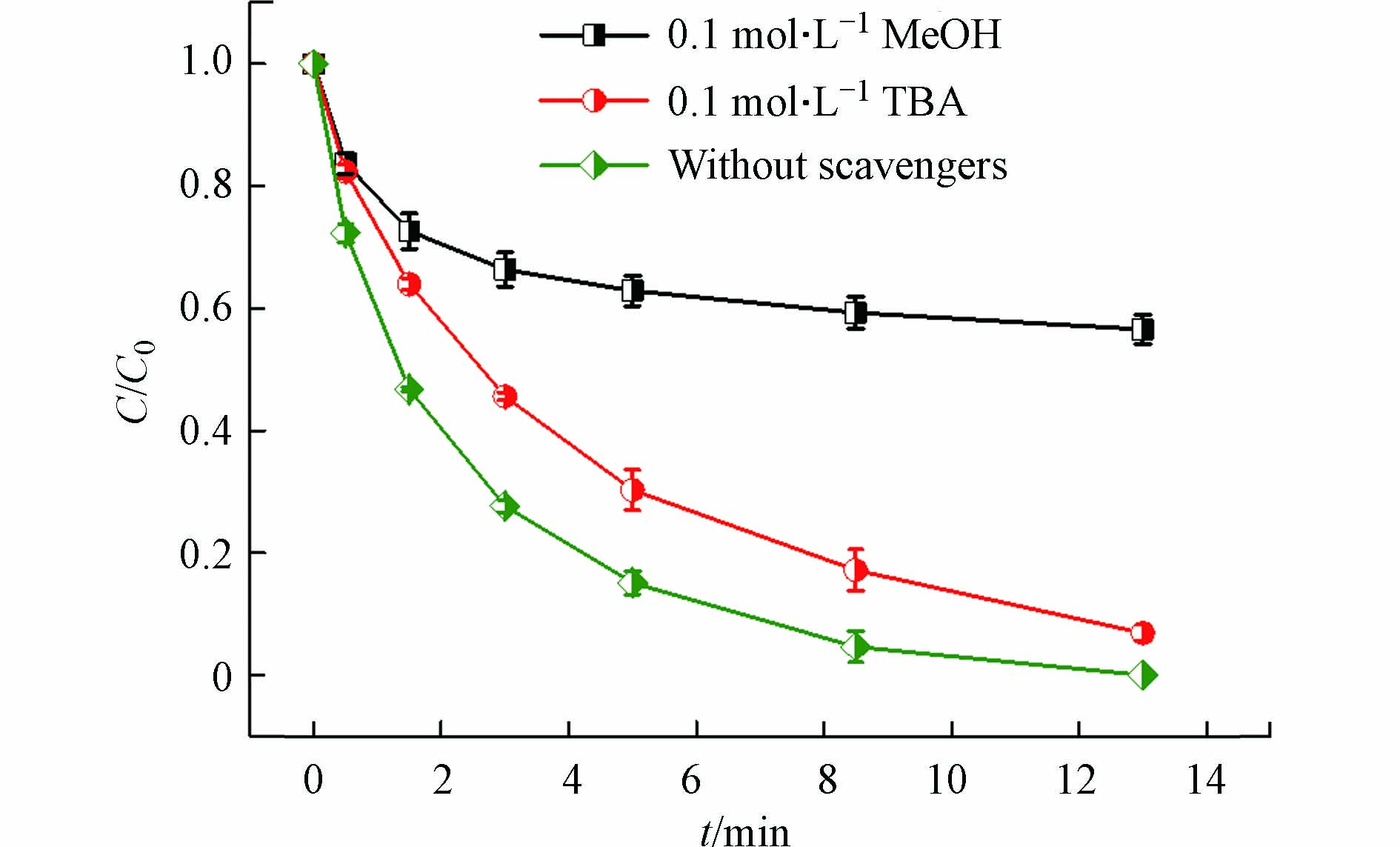
图 3 TBA和MeOH对Cu(Ⅱ)-heat/PAA体系中DCF去除的影响
Figure 3. Effect of TBA and MeOH on DCF removal in Cu(Ⅱ)-heat/PAA system
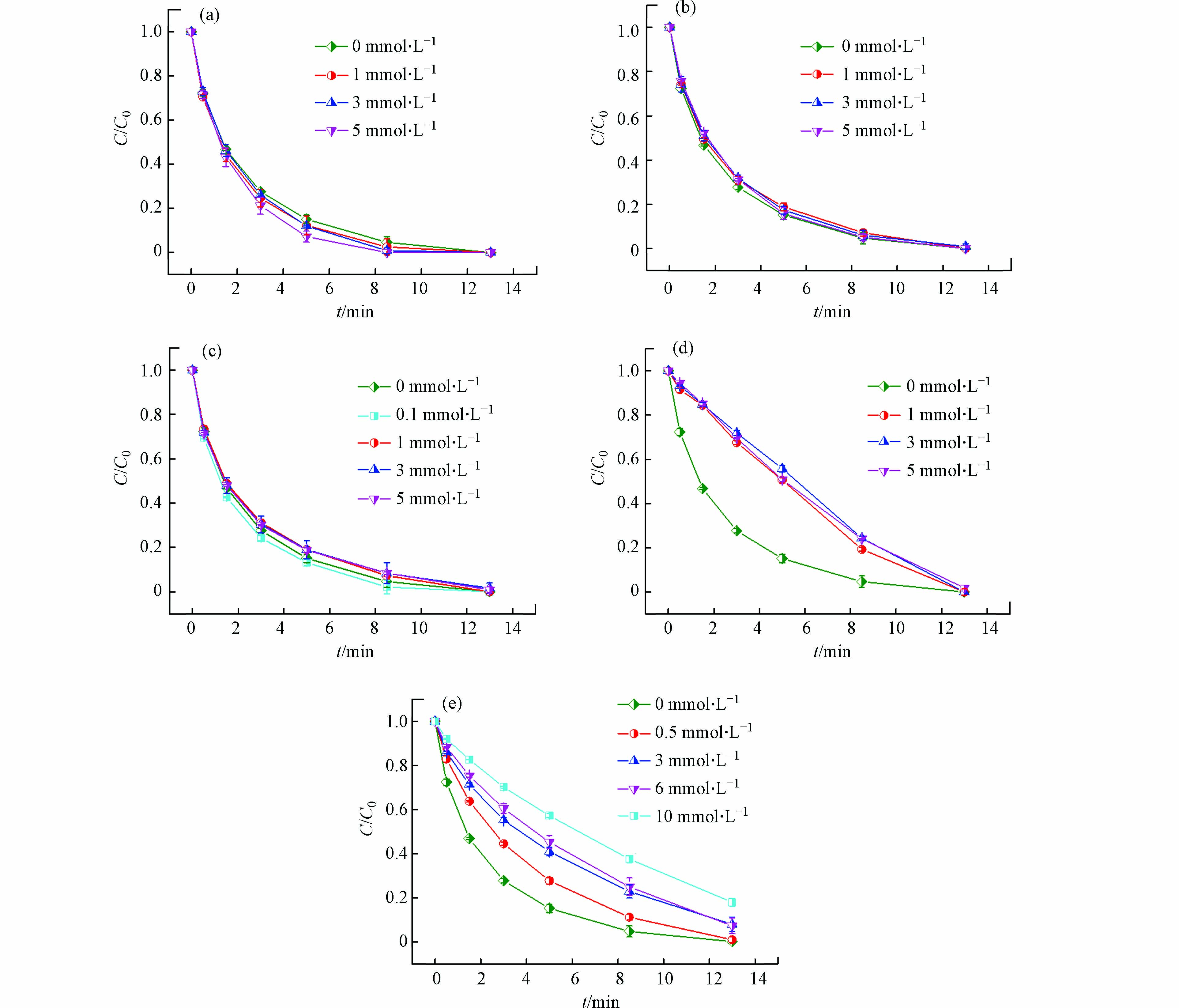
图 4 Cu(Ⅱ)-heat/PAA体系中水体基质对DCF去除的影响(a) Cl−; (b) SO42−; (c) NO3−; (d) HCO3−; (e) NOM
Figure 4. Effect of water matrix on DCF removal in Cu(Ⅱ)-heat/PAA system
表 1 DCF的理化性质
Table 1. Physicochemical properties of DCF
分子式Formula 溶解度Solubility 分子量Molecular weight 结构式Structure C14H11Cl2NO2 2.37 mg·L−1(25 ℃) 296.16 </td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">296.16</td><td class="table_top_border2 table_bottom_border" style="padding-bottom:2pt;" align="center" valign="middle"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">分子式Formula</td><td class="table_top_border" align="center" valign="middle">溶解度Solubility</td><td class="table_top_border" align="center" valign="middle">分子量Molecular weight</td><td class="table_top_border" align="center" valign="middle">结构式Structure</td></tr></thead>
<tbody><tr><td class="table_top_border2 table_bottom_border" align="center" valign="middle">C<sub>14</sub>H<sub>11</sub>Cl<sub>2</sub>NO<sub>2</sub></td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">2.37 mg·L<sup>−1</sup>(25 ℃)</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">296.16</td><td class="table_top_border2 table_bottom_border" style="padding-bottom:2pt;" align="center" valign="middle"><img class="graphic" src="2021111602-b1-1.jpg"></td></tr></tbody>
</table></div></foreignObject></svg>"></td></tr></tbody>
</table></div></foreignObject></svg>) 下载: 导出CSV
下载: 导出CSV
表 2 不同体系的kobs和R2值
Table 2. kobs and R2 in different systems
体系 PAA Cu(Ⅱ)/PAA Cu(Ⅱ)-heat/H2O2 heat/PAA Cu(Ⅱ)-heat/PAA kobs /min−1 0.004 0.008 0.054 0.094 0.394 R2 0.910 0.945 0.990 0.990 0.996
下载: 导出CSV
-
[1] KOSTICH M S, BATT A L, LAZORCHAK J M. Concentrations of prioritized pharmaceuticals in effluents from 50 large wastewater treatment plants in the US and implications for risk estimation [J]. Environmental Pollution, 2014, 184: 354-359. doi: 10.1016/j.envpol.2013.09.013 [2] EBELE A J, ABOU-ELWAFA ABDALLAH M, HARRAD S. Pharmaceuticals and personal care products (PPCPs) in the freshwater aquatic environment [J]. Emerging Contaminants, 2017, 3(1): 1-16. doi: 10.1016/j.emcon.2016.12.004 [3] YANG Y, OK Y S, KIM K H, et al. Occurrences and removal of pharmaceuticals and personal care products (PPCPs) in drinking water and water/sewage treatment plants: A review [J]. Science of the Total Environment, 2017, 596/597: 303-320. doi: 10.1016/j.scitotenv.2017.04.102 [4] 王鸿斌, 王群, 刘义青, 等. 亚铁活化过硫酸盐降解水中双氯芬酸钠 [J]. 环境化学, 2020, 39(4): 869-875. doi: 10.7524/j.issn.0254-6108.2019040806 WANG H B, WANG Q, LIU Y Q, et al. Degradation of diclofenac by ferrous activated persulfate [J]. Environmental Chemistry, 2020, 39(4): 869-875(in Chinese). doi: 10.7524/j.issn.0254-6108.2019040806
[5] LONAPPAN L, BRAR S K, DAS R K, et al. Diclofenac and its transformation products: Environmental occurrence and toxicity - A review [J]. Environment International, 2016, 96: 127-138. doi: 10.1016/j.envint.2016.09.014 [6] CHEN W P, XU J, LU S D, et al. Fates and transport of PPCPs in soil receiving reclaimed water irrigation [J]. Chemosphere, 2013, 93(10): 2621-2630. doi: 10.1016/j.chemosphere.2013.09.088 [7] 张楠, 刘国光, 刘海津, 等. 双氯芬酸在水环境中光降解的初步研究 [J]. 环境化学, 2013, 32(1): 42-47. doi: 10.7524/j.issn.0254-6108.2013.01.007 ZHANG N, LIU G G, LIU H J, et al. Photodegration mechanism of diclofenac in aqueous environment [J]. Environmental Chemistry, 2013, 32(1): 42-47(in Chinese). doi: 10.7524/j.issn.0254-6108.2013.01.007
[8] 谷得明, 郭昌胜, 冯启言, 等. 基于硫酸根自由基的高级氧化技术及其在环境治理中的应用 [J]. 环境化学, 2018, 37(11): 2489-2508. doi: 10.7524/j.issn.0254-6108.2018012102 GU D M, GUO C S, FENG Q Y, et al. Sulfate radical-based advanced oxidation processes and its application in environmental remediation [J]. Environmental Chemistry, 2018, 37(11): 2489-2508(in Chinese). doi: 10.7524/j.issn.0254-6108.2018012102
[9] 刘萌, 胡莉敏, 张广山, 等. Co/Zn双金属氧化物活化过一硫酸盐降解双酚A的性能研究 [J]. 环境化学, 2018, 37(4): 753-760. doi: 10.7524/j.issn.0254-6108.2017081605 LIU M, HU L M, ZHANG G S, et al. Activation of peroxymonosulfate by the Co/Zn bimetallic oxide for the degradation of bisphenol A [J]. Environmental Chemistry, 2018, 37(4): 753-760(in Chinese). doi: 10.7524/j.issn.0254-6108.2017081605
[10] SUN P Z, ZHANG T Q, MEJIA-TICKNER B, et al. Rapid disinfection by peracetic acid combined with UV irradiation [J]. Environmental Science & Technology Letters, 2018, 5(6): 400-404. [11] WANG Z R, SHI H L, WANG S X, et al. Degradation of diclofenac by Fe(II)-activated peracetic acid [J]. Environmental Technology, 2021, 42(27): 4333-4341. doi: 10.1080/09593330.2020.1756926 [12] ZHANG L, FU Y S, WANG Z R, et al. Removal of diclofenac in water using peracetic acid activated by zero valent copper [J]. Separation and Purification Technology, 2021, 276: 119319. doi: 10.1016/j.seppur.2021.119319 [13] KITIS M. Disinfection of wastewater with peracetic acid: A review [J]. Environment International, 2004, 30(1): 47-55. doi: 10.1016/S0160-4120(03)00147-8 [14] KOIVUNEN J, HEINONEN-TANSKI H. Peracetic acid (PAA) disinfection of primary, secondary and tertiary treated municipal wastewaters [J]. Water Research, 2005, 39(18): 4445-4453. doi: 10.1016/j.watres.2005.08.016 [15] LUUKKONEN T, HEYNINCK T, RÄMÖ J, et al. Comparison of organic peracids in wastewater treatment: Disinfection, oxidation and corrosion [J]. Water Research, 2015, 85: 275-285. doi: 10.1016/j.watres.2015.08.037 [16] BIANCHINI R, CALUCCI L, LUBELLO C, et al. Intermediate free radicals in the oxidation of wastewaters [J]. Research on Chemical Intermediates, 2002, 28(2/3): 247-256. [17] da SILVA W P, CARLOS T D, CAVALLINI G S, et al. Peracetic acid: Structural elucidation for applications in wastewater treatment [J]. Water Research, 2020, 168: 115143. doi: 10.1016/j.watres.2019.115143 [18] CHEN S A, CAI M Q, LIU Y Z, et al. Effects of water matrices on the degradation of naproxen by reactive radicals in the UV/peracetic acid process [J]. Water Research, 2019, 150: 153-161. doi: 10.1016/j.watres.2018.11.044 [19] ROTHBART S, EMBER E E, van ELDIK R. Mechanistic studies on the oxidative degradation of Orange II by peracetic acid catalyzed by simple manganese(ii) salts. Tuning the lifetime of the catalyst [J]. New J Chem, 2012, 36(3): 732-748. doi: 10.1039/C2NJ20852K [20] KIM J, ZHANG T Q, LIU W, et al. Advanced oxidation process with peracetic acid and Fe(II) for contaminant degradation [J]. Environmental Science & Technology, 2019, 53(22): 13312-13322. [21] KIM J, DU P H, LIU W, et al. Cobalt/peracetic acid: Advanced oxidation of aromatic organic compounds by acetylperoxyl radicals [J]. Environmental Science & Technology, 2020, 54(8): 5268-5278. [22] WANG J W, WAN Y, DING J Q, et al. Thermal activation of peracetic acid in aquatic solution: The mechanism and application to degrade sulfamethoxazole [J]. Environmental Science & Technology, 2020, 54(22): 14635-14645. [23] ZHANG K J, ZHOU X Y, DU P H, et al. Oxidation of β-lactam antibiotics by peracetic acid: Reaction kinetics, product and pathway evaluation [J]. Water Research, 2017, 123: 153-161. doi: 10.1016/j.watres.2017.06.057 [24] IBOUKHOULEF H, AMRANE A, KADI H. Removal of phenolic compounds from olive mill wastewater by a Fenton-like system H2O2/Cu(II)—thermodynamic and kinetic modeling [J]. Desalination and Water Treatment, 2016, 57(4): 1874-1879. doi: 10.1080/19443994.2014.978385 [25] NICHELA D A, BERKOVIC A M, COSTANTE M R, et al. Nitrobenzene degradation in Fenton-like systems using Cu(Ⅱ) as catalyst. Comparison between Cu(Ⅱ)- and Fe(Ⅲ)-based systems [J]. Chemical Engineering Journal, 2013, 228: 1148-1157. doi: 10.1016/j.cej.2013.05.002 [26] WANG H B, DENG J W, LU X H, et al. Rapid and continuous degradation of diclofenac by Fe(Ⅱ)-activated persulfate combined with bisulfite [J]. Separation and Purification Technology, 2021, 262: 118335. doi: 10.1016/j.seppur.2021.118335 [27] MILLERO F J, SHARMA V K, KARN B. The rate of reduction of copper(Ⅱ) with hydrogen peroxide in seawater [J]. Marine Chemistry, 1991, 36(1/2/3/4): 71-83. [28] LIU Y Q, HE X X, DUAN X D, et al. Significant role of UV and carbonate radical on the degradation of oxytetracycline in UV-AOPs: Kinetics and mechanism [J]. Water Research, 2016, 95: 195-204. doi: 10.1016/j.watres.2016.03.011 [29] CAI M Q, SUN P Z, ZHANG L Q, et al. UV/peracetic acid for degradation of pharmaceuticals and reactive species evaluation [J]. Environmental Science & Technology, 2017, 51(24): 14217-14224. [30] WANG H B, WANG S X, LIU Y Q, et al. Degradation of diclofenac by Fe(Ⅱ)-activated bisulfite: Kinetics, mechanism and transformation products [J]. Chemosphere, 2019, 237: 124518. doi: 10.1016/j.chemosphere.2019.124518 [31] WANG Z P, WANG J W, XIONG B, et al. Application of cobalt/peracetic acid to degrade sulfamethoxazole at neutral condition: Efficiency and mechanisms [J]. Environmental Science & Technology, 2020, 54(1): 464-475. [32] HUANG Y, JIANG Q J, YU X B, et al. A combined radical and non-radical oxidation processes for efficient degradation of Acid Orange 7 in the homogeneous Cu(II)/PMS system: Important role of chloride [J]. Environmental Science and Pollution Research International, 2021, 28(37): 51251-51264. doi: 10.1007/s11356-021-14262-1 [33] LIU Y Q, HE X X, FU Y S, et al. Degradation kinetics and mechanism of oxytetracycline by hydroxyl radical-based advanced oxidation processes [J]. Chemical Engineering Journal, 2016, 284: 1317-1327. doi: 10.1016/j.cej.2015.09.034 [34] WANG S X, WANG H B, LIU Y Q, et al. Effective degradation of sulfamethoxazole with Fe2+-zeolite/peracetic acid [J]. Separation and Purification Technology, 2020, 233: 115973. doi: 10.1016/j.seppur.2019.115973 [35] WANG Z R, FU Y S, PENG Y L, et al. HCO3-/CO32- enhanced degradation of diclofenac by Cu(Ⅱ)-activated peracetic acid: Efficiency and mechanism [J]. Separation and Purification Technology, 2021, 277: 119434. doi: 10.1016/j.seppur.2021.119434 [36] DENG J W, WANG H B, FU Y S, et al. Phosphate-induced activation of peracetic acid for diclofenac degradation: Kinetics, influence factors and mechanism [J]. Chemosphere, 2022, 287: 132396. doi: 10.1016/j.chemosphere.2021.132396 期刊类型引用(4)
1. 汤崇锹,金丽丽,黄辉,任洪强. 基于过氧乙酸的高级氧化技术去除水中新污染物:现状与挑战. 环境化学. 2025(02): 395-404 .  本站查看
本站查看
2. 许冬梅,陈思颖,董冬吟,杨仕毅,陈铃心,林晓峰,崔航宇,邹景. 基于二价钴催化氧化脱色酸性橙7的分光光度法快速测定水中过氧乙酸浓度. 环境化学. 2024(09): 3165-3173 . 本站查看
3. 王辉锋,郭艳菲,郭壮,黄意涵,徐东耀,魏健. 活化过氧乙酸技术降解环境有机污染物的研究进展. 环境工程技术学报. 2023(06): 2154-2164 . 百度学术
4. 王华哲,乔成桓,战树岩,吴耀华,贾文瑞,梁永祺,郭婉茜. 过氧乙酸活化功能材料开发与水环境应用. 环境化学. 2023(11): 3613-3627 . 本站查看
其他类型引用(2)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 3807
- HTML全文浏览数: 3807
- PDF下载数: 107
- 施引文献: 6
