


-
寻求和利用可再生绿色清洁能源替代化石燃料,是解决能源与环境危机的重要途径。微生物燃料电池(microbial fuel cell, MFC)作为一种集污水处理与生物产电于一体的新型技术,以产电细菌为主体,可将化学能转化为电能,同时去除水体中的污染物[1-2]。电极材料是影响MFC性能的关键因素之一,也是MFC产电微生物的附着载体和生长场所[3]。因此,找到一种可供微生物大量附着和生长的载体,同时具有良好导电性能的材料至关重要。
MFC电极多采用碳质材料,拥有良好的生物相容性、导电性和化学稳定性[4]。碳质材料一般包括石墨烯、碳毡、碳布、生物炭等。其中石墨烯电极机械强度较好,但其材料表面相对光滑,不利于微生物附着,因而导致胞外电子传递效率低[5-6];碳毡电极柔韧性良好,但其在MFC运行时,由于材质较厚,生物膜会妨碍底物由外向内的扩散,影响对污染物的降解效率;碳布电极表面粗糙但机械强度较差,不适于投入大规模的实际工程应用中[7]。相比于传统电极材料,生物炭材料具有来源广泛、成本低廉、电化学性能较好、比表面积高和孔隙结构多等优点。2018年CHEN等[8]大麻槿秸秆通过简单的碳化处理制成MFC阳极,其电流密度达到了32.5 A·m−2,是对照组石墨棒电极的3倍,由此可见生物炭作为MFC电极材料是具有一定优势的。
据2020年中国统计年鉴统计,我国核桃栽培面积为5.54×1010 m²,约1.3×109株[9]。每年有大量的废弃核桃壳产生,如何有效处理这些固体废物,实现减量化和资源化是环境领域的研究热点。采用高温裂解法制备生物炭,再通过化学活化,可使其表面结构相对于碳基材料的平面结构更为粗糙,更有效的提升活性表面积[10-13]。常见的生物炭化学活化剂包括ZnCl2、HPO4、KOH等,其中ZnCl2活化制备的活性炭具有产率高、过渡孔发达、价廉易得等优点[14],JIANG等通过ZnCl2活化甘蔗渣发现,锌离子浓度越高,比表面积越大[15]。
目前,以改性核桃壳作为电极材料的研究鲜有报道。因此,本研究主要以改性核桃壳作为生物炭基电极材料,通过不同温度的碳化、不同浓度的ZnCl2活化、不同比例的材料复合制成微生物燃料电池电极,通过表征分析,考察不同制备方法制备出的材料的性能差异,分析其在MFC中产电性能的差异,以及最佳条件MFC去除污染物的能力,为微生物燃料电池的发展方向提供参考。
-
将市场上购买的核桃取果皮后粉碎,过40目分子筛后,置于石英舟中,再将其放入管式炉(OTF-1200 X),真空400 ℃炭化90 min后,得到黑色产物。称取一定量的黑色产物与氯化锌固体按质量比分别为5:1、5:3、5:5,置于烧杯中加去离子水刚好完全淹没,搅拌后,再将其置于105 ℃烘箱中烘干24 h。将烘干好的黑色产物放于管式炉中央,分别在400、600、800 ℃温度条件下真空煅烧2 h,反应结束后在真空保护下冷却至室温。煅烧好的样品先用10% HCl溶液洗涤,然后用去离子水洗涤,直至中性,最后将其置于105 ℃烘箱中烘干24 h,得到核桃壳碳化产物。
制备好的生物炭样品与聚苯胺和热熔胶按5:1:4和5:1:5质量比进行混合,然后将混合材料置于刚玉舟模具中压实,再放入200 ℃管式炉进行真空热熔,热熔30 min后,自然冷却至室温取出。制备后的电极材料样品尺寸为2 cm×3 cm×0.5(±0.05) cm。
-
实验采用扫描电镜(捷克TESCAN MIRA LMS),通过磨成粉末过0.4 mm筛网制样,对生物炭基电极材料表面形貌进行表征;采用拉曼光谱(激光器波长532 nm,扫描范围50~4 000 cm−1)分析电极材料的石墨化程度;采用孔隙及比表面积分析仪(康塔4000 e,脱气温度120 ℃)分析电极材料的比表面积、孔体积和孔径;采用电化学工作站(CHI 660 e),通过LSV、EIS测试,分析电极材料的导电性能的差异;采用HACH高量程(20~1 500 mg·L−1)消解法测定COD;采用国标纳氏试剂比色法测定氨氮;采用电流电压数据采集器(KEYSIGHT 34972A)测定MFC产电性能、采集电流电压及功率密度。
-
实验采用空气阴极单室MFC反应器,由有机玻璃制成,内径为10 cm,高度为14 cm,设置溢流堰用于出水,底部设置0.4 cm有机玻璃管用于进水。反应器阴阳极用尼龙螺栓固定,有效容积为377 mL。配制模拟废水(实验所用的去离子水为灭菌除氧后的水)用于MFC产电性能分析,每隔24 h进出水150 mL,其组成为1.356 g·L−1 C4H4Na2O4,0.15 g·L−1 (NH4)2SO4,0.253 5 g·L−1 KH2PO4,0.125 g·L−1 MgSO4·7H2O,0.125 g·L−1 NaCl,0.002 5 g·L−1 FeSO4·7H2O,0.002 g·L−1 MnSO4·H2O,1 mL·L−1 微量元素。其中微量元素包括1.5 g·L−1 FeC13·6H2O,0.02 g·L−1 CuC12·2H2O,0.18 g·L−1 KI,0.12 g·L−1 MnCl2·4H2O,0.01 g·L−1 ZnC12,0.06 g·L−1 Na2MoO4·2H2O,0.15 g·L−1 CoC12·6H2O,0.15 g·L−1 H3BO3,0.06 g·L−1 Na2MoO4·2H2O。
-
实验所用接种微生物为本研究组前期实验筛选出的异养硝化-好氧反硝化菌[16]。通过扩大培养,鉴定其异养硝化与好氧反硝化性能后接种至MFC反应器。具体过程如下:将菌种接种至LB液体培养基,在(30±2) ℃培养箱中培养24 h后,取菌种培养液与去离子水以1:9比例混合,用10 mL离心管离心去除上清液后加适量水摇匀,倒入好氧反硝化培养基,恒温振荡培养(160 min−1,30 ℃),每12 h测1次硝氮浓度和OD600;再取培养液置于异养硝化培养基,厌氧箱中恒温培养(30 ℃),每12 h测1次氨氮浓度和OD600。实验所用LB液体培养基含有3.0 g·L−1 牛肉膏、5.0 g·L−1 NaCl、10.0 g·L−1 蛋白胨(pH=7.0);好氧反硝化培养基(100 mL)含有1.356 g·L−1 C4H4 Na2O4、0.064 1 g·L−1 KNO3、0.253 5 g·L−1 KH2PO4、0.125 g·L−1 MgSO4·7H2O、0.125 g·L−1 NaCl、0.002 5 g·L−1 FeSO4·7H2O、0.002 g·L−1 MnSO4·H2O;异养硝化培养基(100 mL)含有1.356 g·L−1 C4H4Na2O4、0.253 5 g·L−1 KH2PO4、0.125 g·L−1 MgSO4·7H2O、0.125 g·L−1 NaCl、0.002 5 g·L−1 FeSO4·7H2O、0.002 g·L−1 MnSO4·H2O、0.15 g·L−1 (NH4)2SO4。
-
将目标菌株菌悬液接种至MFC反应器中,接种比例10%,上下电极间距4 cm。MFC置于恒温气候箱中,温度和湿度分别控制30 ℃、50%。进水pH为7.0±0.1,连接1 kΩ的外电阻,每5 min对MFC输出电流电压进行实时监控。在MFC电压达到稳定输出时,测量分析极化曲线和功率密度曲线(文中功率密度和电流密度以反应器有效容积为参比)。具体方法为:依次将外电阻由2 000 Ω调到100 Ω,每30 s记录1次MFC外阻的电流电压值,其中每更换一次电阻需等待3 min让电压值稳定。
-
为阐明ZnCl2用量对生物炭材料孔隙结构的影响,实验采用生物炭/氯化锌质量比分别为5:1、5:3、5:5的电极材料,在600 ℃煅烧后,通过比表面积和孔径分布来评估核桃壳生物炭的比表面积及相应孔径分布。由图1可知,ZnCl2活化后的生物炭孔结构的孔径主要集中在3.5 nm附近,在相对压力为0.1~1.0内出现较显著的滞后环,按照国际纯化学和应用化学联合会的定义,核桃壳生物炭是典型的Ⅰ型和Ⅱ型特性[17],说明核桃壳生物炭的结构属于尺寸较小的介孔结构。由表1可知,随着ZnCl2质量比的不断增加,改性核桃壳生物炭的比表面积由590 m2·g−1增加到883 m2·g−1,孔容由0.009 cm3·g−1逐渐增加到0.017 cm3·g−1。这说明随着ZnCl2用量的增加,活化后的核桃壳生物炭的比表面积也越大,可为微生物的生长提供更多的场所[18],做成MFC电极后其微生物负载量可得到提升,从而促进MFC的产电。
-
为阐明热处理温度对生物炭材料分子结构的影响规律,控制生物炭/氯化锌质量比为5:5,分别在400、600、800 ℃热处理条件下,对制备的生物炭进行拉曼光谱分析,实验结果如图2所示。由图2可见,核桃壳生物炭在1 316 cm−1和1 586 cm−1处有2个显著的拉曼峰,分别为炭材料的特征D峰和G峰[19-20]。其中D峰主要是芳香环之间的C—C结构,为环数大于6环的芳香环结构,是由炭材料缺陷引起;G峰与炭材料的C=C键Sp2杂化有关。D峰与G峰的强度比(ID/IG)可以在一定程度反映材料的缺陷程度,ID/IG值越高,代表材料的无序率越高;ID/IG值越低,说明材料的石墨化程度越高,导电性能越好[21]。根据拉曼光谱图Gauss拟合曲线方法可以得到ID/IG。由图2可以看出,随着热解温度的增加,ID/IG比值变小。这说明材料的石墨化程度增加,导电性能越好,制作的MFC电极性能越好。
-
聚苯胺与热熔胶比例也是考察MFC电极制作过程的因素之一。图3为在真空煅烧温度为600 ℃、生物炭与氯化锌活化质量比为5:3的条件下,生物炭/聚苯胺/热熔胶比例分别为5:1:4和5:1:5所制成的MFC复合电极的扫描电镜图。
由于当生物炭/聚苯胺/热熔胶的比例为5:1:1和5:1:2时,经过煅烧的生物炭电极几乎不成型,依旧保持粉末状态;当生物炭/聚苯胺/热熔胶添加比例为5:1:3时,经过煅烧的生物炭电极机械强度低,易碎,因此这些复合比例均不能到达作为MFC电极材料的要求,故实验选用的生物炭/聚苯胺/热熔胶比例为5:1:4和5:1:5。由图3可以看出,随着热熔胶中聚乙烯粉末和导电态聚苯胺的加入,其对生物炭的表面起到了修饰作用,但未对生物炭的多孔结构产生明显的影响。
-
图4反映了电解池三电极体系中生物炭/聚苯胺/热熔胶复合电极的电化学性能。以1 cm2的铂片作为辅助电极,以Ag/AgCl作为参比电极,将制备得到BPP 5:1:4和BPP 5:1:5的复合电极分别连接在电极夹上作为工作电极。所有的电化学性能测试均是在1 mmol·L−1 铁氰化钾混合溶液(0.1 mol·L−1 KCl)中完成。
如图4(a)所示,BPP 5:1:4材料在-0.8~0.8 V内的电流为0.305~-0.879 9 mA,且CV曲线有2对微弱的氧化还原峰;而BPP 5:1:5材料的电流为0.154~-0.546 mA,CV曲线没有氧化还原峰。材料氧化还原峰越多越明显,材料的电子传递能力越好[22],因此,BPP 5:1:4材料比BPP 5:1:5材料具有更良好的电子传递能力,更易促进氧化还原反应。
交流阻抗(EIS)曲线如图4(b)所示,其中,正弦信号频率为0.01~105 Hz,交流振幅为0.006。MFC中EIS的表征大多用于分析欧姆内阻和扩散内阻,由于低频区对扩散内阻的表征存在较大偏差,所以在数据拟合过程中未将低频区部分纳入拟合范围[23]。本次拟合使用软件Zview2,等效电路模型中RΩ为欧姆内阻,Rct为电荷转移内阻,电荷转移内阻与一个双电层电容并联,但因弥散效应的存在,该电容偏离理想双电层电容器,因而在本次拟合电路中使用常相位角原件代替传统双电层电容器。根据拟合结果,BPP 5: 1: 4欧姆内阻为37.17 Ω,电荷转移内阻为1 854 Ω,BPP 5:1:5欧姆内阻为46.54 Ω,电荷转移内阻为343 Ω。总的来看,Rct均大于RΩ,说明MFC系统内组主要受Rct控制。就Rct而言,BPP 5:1:4小于BPP 5:1:5,Rct主要反映电活性微生物与电极之间电子传递过程的内阻[24],Rct越小,其电子传递速率越快,因此,BPP 5:1:4生物电化学活性优于BPP 5:1:5。
-
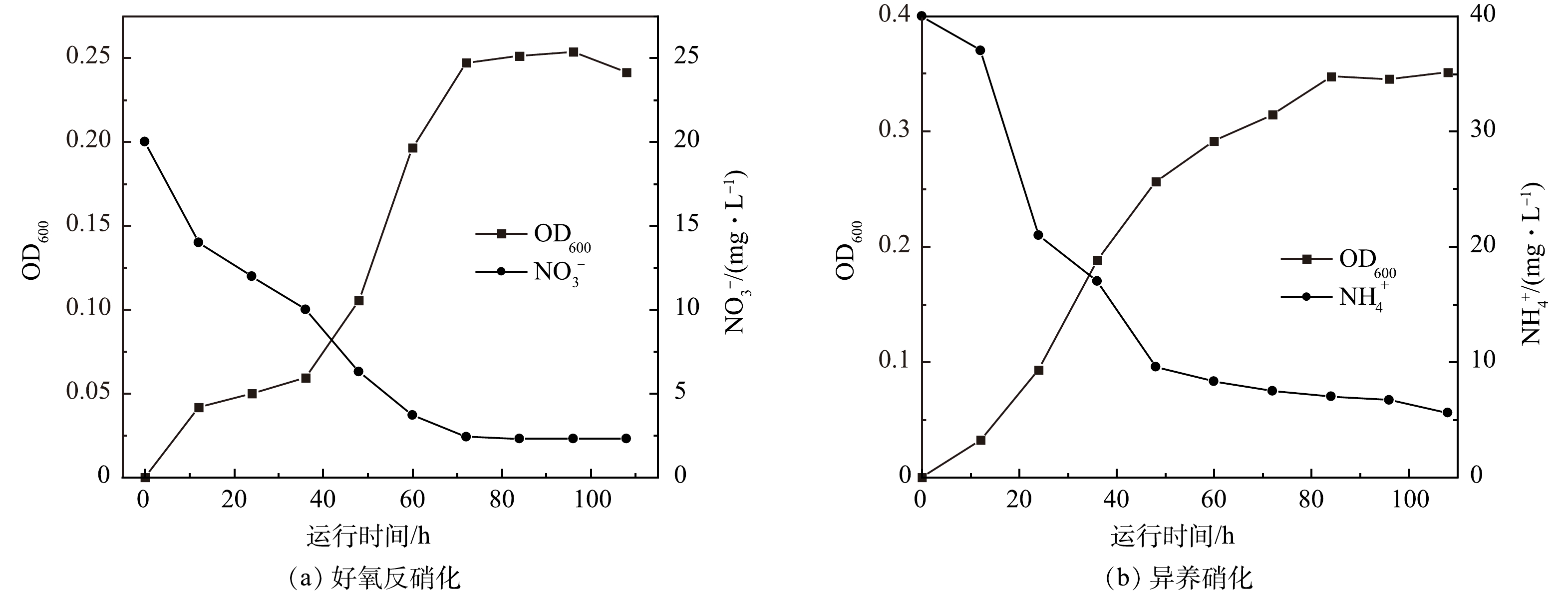
本研究组前期筛选出的异养硝化-好氧反硝化菌[16],通过扩大培养后,接种到异养硝化与好氧反硝化培养基中。由图5可见,在好氧反硝化、异养硝化培养基中菌株不断进行自我繁殖,并消耗培养基中的硝酸根与氨氮。这说明实验接种的微生物具有好氧反硝化与异养硝化能力,后续实验将采用此细菌做为MFC的产电菌。
-
实验构建单室MFC反应器,将脱氮菌株接种至MFC反应器中,连接不同条件下制备的改性核桃壳基生物炭电极材料,每5 min对MFC输出电流电压进行实时监控,其产电性能、功率密度与极化曲线如图6所示。
在真空煅烧温度为600 ℃,BPP为5:1:4的条件下,考察了不同浓度氯化锌对产电性能的影响,结果如图6(a)和图6(b)所示。当生物炭/氯化锌质量比为5:1时,MFC电极最大输出电压为0.103 V,随外电阻由大到小变化,反应器极化曲线电压由133 mV降至16 mV,最大体积功率为26 mW·m−3,电流密度为259 mW·m−3;当生物炭/氯化锌质量比为5:3时,最大输出电压为0.137 V,随外电阻由大到小变化,反应器极化曲线电压由171 mV降至28 mV,最大体积功率为51 mW·m−3,电流密度为406 mA·m−3;当生物炭/氯化锌质量比为5:5时,MFC电极最大输出电压为0.148 V,随外电阻由大到小变化,反应器极化曲线电压由181 mV降低至31 mV,最大功率密度为61 mW·m−3,电流密度为438 mA·m−3。根据极化曲线斜率可以得出MFC电极电阻,斜率越小MFC的内阻越大[25]。随着ZnCl2质量比的增加,MFC的产电能力增加,内阻逐渐减小,电极材料的产电性能越好。当生物炭/氯化锌质量比从5:3提升到5:5,MFC的产电性能提升不大,可能原因是ZnCl2对生物炭的造孔能力几乎达到饱和[26]。
在BPP为5:1:4,生物炭/氯化锌比为5:3条件下,考察了煅烧温度对电极性能的影响,结果如图7(a)和图7(b)所示。在400 ℃煅烧条件下,MFC电极最大输出电压为0.096 V,随外电阻由大到小变化,反应器极化曲线电压由119 mV降低至17 mV,最大功率密度为22 mW·m−3,电流密度为238 mA·m−3;在600 ℃煅烧条件下,最大输出电压为0.137 V,随外电阻由大到小变化,反应器极化曲线电压由171 mV降低至28 mV,最大体积功率为51 mW·m−3,电流密度为406 mA·m−3;在800 ℃煅烧条件下,MFC电极最大输出电压为0.143 V,随外电阻由大到小变化,反应器极化曲线电压由181 mV降低至29 mV,最大功率密度约为57 mW·m−3,此时的电流密度为436 mA·m−3。随着煅烧温度的增加,MFC的产电能力增加,内阻逐渐减小,电极材料的产电性能越好。结合图2可知,这是由于材料石墨化程度的增加,导致MFC的内阻减小。由图7可见,在600 ℃和800 ℃条件下,制备的电极性能相差不大。由此可见,当煅烧温度达到一定程度,MFC的产电性能提升不大,可能的原因是温度的增加破坏了部分生物炭的微孔和大孔,虽然生物炭石墨化程度增加,但是微生物的负载量减少[27]。
在生物炭/氯化锌比为5:3,真空煅烧温度为600 ℃条件下,考察了不同材料复合情况对电极产电性能的影响,结果如图8(a)和图8(f)所示。当生物炭/聚苯胺/热熔胶复合比例为5:1:4时,最大输出电压为0.137 V,随外电阻由大到小变化,反应器极化曲线电压由171 mV降至28 mV,最大体积功率为51 mW·m−3,电流密度为406 mA·m−3;当生物炭/聚苯胺/热熔胶复合比例为5:1:5时,最大输出电压为0.077 V,随外电阻由大到小变化,反应器极化曲线电压由100 mV降至13mV,最大体积功率为16 mW·m−3,电流密度为176 mA·m−3。由此可见,生物炭的含量对复合材料有显著影响。结合图3可知,在保证材料成型的前提下,生物炭含量越高,MFC的内阻越小,材料的产电性能越好[28]。
-
根据以上结果,确认电极制备的最佳条件为BPP 5:1:4、煅烧温度600 ℃、生物炭/氯化锌比5:3时,即节约了生产成本,又达到最大产电量的90%,为减少对环境的污染,选用此种方法制备的电极材料用来探讨改性核桃壳生物炭电极材料用于MFC反应器降解污染物的长期效果,结果如图9所示。可以看出,随着时间的推移,MFC中出水COD由685 mg·L−1降至100 mg·L−1。第1天时,出水COD大幅下降,这说明微生物在反应初期消耗废水中大量有机物用于增殖。随着微生物增殖所需能量减少,有机物需求也逐渐减少,最终COD稳定去除率为85%。出水氨氮质量浓度由38 mg·L−1降低至4.5 mg·L−1,在第4天达到稳定,改性核桃壳生物炭MFC对氨氮的去除率最终达到88%。水体中的硝氮在7 d内先上升后下降,可以看出硝化菌株产生的硝态氮会被好氧反硝化菌株利用,MFC具有较好的脱硝态氮能力。
-
1) MFC电极的最佳制备条件:活化时生物炭/氯化锌质量比5:3,真空煅烧温度为600 ℃,生物炭/聚苯胺/热熔胶复合电极比例为5:1:4,应用于MFC中最大的体积功率密度可达51 mW·m−3,对模拟废水中COD和氨氮的去除率分别为85%和88%。
2)相较于传统生物炭电极在水体中易碎,通过复合聚苯胺与热熔胶来制作生物炭电极可以在模拟废水中稳定运行。
3)用核桃壳生物炭通过简单的过程制备的MFC电极,为成本低廉,绿色清洁,操作简单的MFC发展方向提供了新的选择。
核桃壳生物炭电极在微生物燃料电池中的产电性能及其对污染物的去除性能
Electricity production and pollutant removal performance of walnut shell biochar electrode in microbial fuel cell
-
摘要: 微生物燃料电池(MFC)的电极材料是决定MFC性能的关键。本研究利用核桃壳生物炭制成MFC电极材料,对核桃壳生物炭基电极的制备条件、MFC的产电性能进行了探讨,利用比表面积分析、扫描电镜、拉曼光谱及电极电化学等方法对生物炭电极进行表征。结果表明: 最佳电极制备条件为活化时生物炭:氯化锌质量比5:3,真空煅烧温度600 ℃,生物炭:聚苯胺:热熔胶质量比5:1:4,在进水COD平均值为685 mg·L−1、氨氮平均值为38 mg·L−1、外电阻为1 000 Ω条件下,MFC的稳定输出电压为0.136 V,最大功率密度达到51 mW·m−3,内阻为762 Ω,运行7 d后,COD和氨氮的去除率分别可达到85%和88%,以上研究结果为制备有前景的MFC的电极材料提供了参考。Abstract: The electrode material is the key to determine the performance of microbial fuel cell (MFC). In this study, walnut shell biochar was used to prepare the MFC electrode. Both the preparation conditions of walnut shell biochar based electrode and MFC electricity production performance were discussed. The biochar electrode was characterized by specific surface area analysis scanning electron microscopy, Raman spectroscopy and electrochemistry. The experimental results showed that the optimum fabrication conditions were as follows: the mass ratio of biochar to zinc chloride was 5:3, the vacuum calcination temperature was 600 ℃, the mass ratio of biochar, polyaniline and hot melt glue was 5:1:4. At the average influent COD concentration of 685 mg·L−1, the average ammonia nitrogen concentration of 38 mg·L−1 and the external resistance of 1000 Ω, the stable output voltage of MFC was 0.136 V, the maximum power density reached 51 mW·m−3 and the internal resistance was 762 Ω. After 7 days of operation, the removal rates of COD and ammonia nitrogen could reach 85% and 88%, respectively. These results provide a reference for the preparation of the promising MFC electrode materials.
-
Key words:
- microbial fuel cell /
- walnut shell electrode /
- electricity generation /
- electrode /
- activization
-
铁电絮凝(Fe-EC)是在电场作用下,使金属铁阳极溶解产生铁离子,并在水中发生一系列物理化学反应,从而使水得到净化的技术。目前,铁电絮凝已被用于生活污水、工业废水以及饮用水等的净化和再生,对水中悬浮物、部分溶解性有机物以及重金属离子有较好的去除效果[1]。但铁电絮凝中污染物去除过程复杂,净化机理尚不明晰。以有机废水为例,铁电絮凝技术的主要作用被认为是吸附、沉淀和气浮作用[2]。除此之外,在铁电絮凝过程中,部分有机污染物可被氧化降解,其作用机制被认为有2种:其一,在含氯电解质中,Cl−在铁阳极表面氧化,形成活性氯(Cl2,HClO/ClO−),导致有机污染物氧化[3];其二,有机污染物在阳极表面直接失电子氧化[4-5]。但这2种氧化机制都是在电荷效率降低时阳极表面发生的副反应导致的。本课题组前期研究发现,铁电絮凝中存在Fe(Ⅱ)活化氧气产生羟基自由基而导致的有机污染物氧化机制[6],但这一过程的氧化效果较弱。
在工业生产中,许多有机配体(如草酸,柠檬酸和EDTA)被广泛用作金属螯合剂、食品添加剂等,最后这些物质通过各种途径进入污水处理厂或自然水体[7]。在天然水体及大气液相中,溶解态铁多以络合配体的形式存在,是水体净化中重要的氧化剂和还原剂[8]。近几年,铁-有机络合物在水处理中的应用也越来越多,Fe(Ⅱ)-有机络合物(如草酸盐,柠檬酸盐和丙酮酸盐)可以在有氧条件下还原氧气来产生活性氧物质(ROS),如·OH、H2O2和
O−2 ,且产生的ROS对多种有机物(如阿特拉津、4-氯苯酚和普萘洛尔)的氧化作用已经得到了证实[9]。YI等[10]也通过电子顺磁共振实验证明,EDTA有助于Fe(Ⅲ)/H2O2体系中产生·OH。因此,在使用铁电絮凝处理含有有机配体的有机污染废水或自然水体时,有机配体的存在可能会促进体系中·OH的产生,从而影响目标污染物的降解或转化。苯胺是化工生产中的原料和中间产物,在污废水中广泛存在。本研究采用苯胺作为目标污染物,在有机配体存在的铁电絮凝体系中证明这一作用机制的存在,继而以苯甲酸与·OH反应,生成对羟基苯甲酸为体系中·OH的定量手段[11],研究了有机配体对体系产·OH效率的影响以及不同有机配体含量时铁电絮凝体系中目标污染物的降解规律,研究结果为人们进一步了解铁电絮凝中污染物去除机制提供参考。1. 实验材料及方法
1.1 实验试剂
苯胺(C6H7N)、碳酸氢钠(NaHCO3)、甲酸(CH2O2)、四水合酒石酸钾钠(C4H12KNaO10)、丙酸(CH3CH2COOH)、二水合柠檬酸钠(Na3C6H5O7·2H2O)、乙二胺四乙酸二钠盐(C10H14N2Na2O8·2H2O)、苯甲酸钠(C7H5NaO2)、5,5-二甲基-1-吡咯啉-N-氧化物DMPO(C6H11NO)、三氟乙酸(CF3COOH)、乙醇等药品均为分析纯;甲醇(CH3OH)为色谱纯;铁电絮凝实验中电极材质为纯铁片(含铁量大于99.5%),尺寸为1.5 cm×8 cm;实验用水均为电阻率为18.25 MΩ·cm−1的超纯水。
1.2 实验仪器
高效液相色谱仪(浙江福立,FL2200),配套C18色谱柱(岛津,4.6 mm×250 mm,5 μm)。电子自旋共振谱仪(日本JEOL,JES-FA200),pH计(奥力龙,MODEL818),超纯水机(圣德利,SDLA-B-0501-P),电子天平(梅特勒-托利多,AL204),直流电源(台湾固纬,GPS-2303C),电流表(FLUKE,F15B+)等。
1.3 实验方法
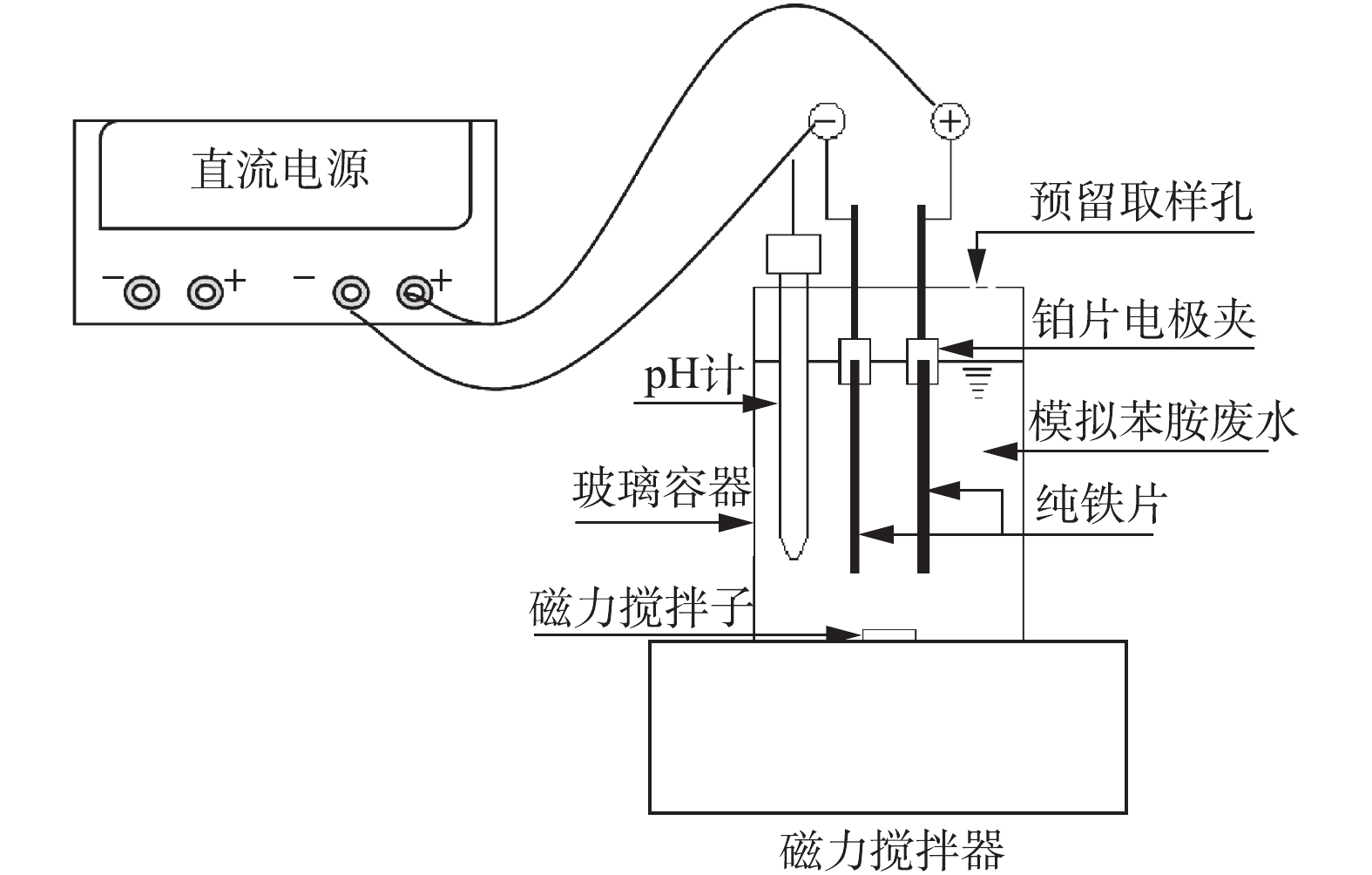
实验装置为600 mL圆柱形玻璃反应器,装有500 mL电解液,内置2片纯铁电极(1.5 cm×8 cm),电极间距为2 cm,实验装置如图1所示。由于碳酸氢根在地下水和地表水体中的广泛存在,本实验选择10 mmol·L−1 NaHCO3溶液为电解液,并在开始电解前搅拌30 min,使溶解氧饱和。实验初始电解液pH为8.5,4 h电解结束时上升至9.0。电解过程中采用磁力搅拌器使溶液保持完全混合,反应器开口与大气连通。采用稳压直流电源供电,并设置为恒电流模式,电流采用电流表进行监测,调节工作电流以达到所需的电荷投加速率。 根据法拉第电解定律计算相关指标,计算方法如式(1)所示。
D=Q(1ZF)=IV(1ZF) (1) 式中:D是电解过程中铁投加速率,mol·(L·min)−1;Q是电荷投加率,C·(L·min)−1;Z是所涉及的电子数;F是法拉第常数,取值96 485 C·mol−1;V为电解液体积,L;I为电流强度,A。电解过程中实验装置用锡纸包裹以避光,实验分别在0、30、60、120、180、240 min用注射器吸取一定量水样,并用0.22 μm尼龙滤膜过滤,用于溶液中苯胺或对羟基苯甲酸浓度分析。所有实验至少重复2次。
在苯胺初始浓度为1 mg·L−1、电解质采用10 mmol·L−1 NaHCO3溶液、初始pH为8.5、电流强度为30 mA(即电荷投加速率为3.6 C·(L·min)−1)、电解时间为240 min时,分别投加以下有机配体:10 mg·L−1的甲酸、丙酸、柠檬酸和酒石酸钾钠;30 mmol·L−1草酸钠;0.2 mmol·L−1 EDTA。在此条件下,研究各种有机配体对Fe-EC降解苯胺的影响。
在铁电絮凝实验中,电解质为10 mL的10 mmol·L−1 NaHCO3,加入20 mmol·L−1 DMPO和0.05 mmol·L−1 EDTA,电解反应5 min后取样并过滤,加入DMPO捕获羟基自由基,进行ESR测试,开展DMPO捕获羟基自由基实验。
为了测量铁电絮凝中羟基自由基产率,本研究选择10 mmol·L−1苯甲酸钠作为羟基自由基的捕获剂,通过检测氧化产物对羟基苯甲酸的浓度而换算成羟基自由基产量(C·OH=Cp-HBA×5.87)[11]。电解质采用5 mmol·L−1 NaHCO3溶液,初始pH为8.5,电流强度为10 mA(即电荷投加速率为1.2 C·(L·min)−1)。在此条件下,研究草酸钠、EDTA的浓度对铁电絮凝中产·OH的作用。
采用10 mmol·L−1苯甲酸钠为·OH捕获剂,电解质采用5 mmol·L−1 NaHCO3溶液,初始pH为8.5,分别在反应开始时向溶液内投加30 mmol·L−1草酸钠和0.2 mmol·L−1 EDTA。以此为基础,研究不同电荷投加速率对铁电絮凝中产·OH的作用。
苯胺初始浓度为5 mg·L−1,电解质采用5 mmol·L−1 NaHCO3溶液,初始pH为8.5,电流强度为20 mA(即电荷投加速率为2.4 C·(L·min)−1)。在此条件下,研究EDTA浓度对铁电絮凝中苯胺降解的影响。
1.4 分析方法
苯胺和对羟基苯甲酸浓度均采用高效液相色谱法(HPLC)测定;羟基自由基采用电子自旋共振谱仪(ESR)测定。
2. 结果与讨论
2.1 不同有机配体对电絮凝降解苯胺的影响
自然界中溶解态铁多以络合配体的形式存在,为研究有机配体在铁电絮凝中对有机污染降解的影响,实验中选取了几种水体中常见的有机酸,观察其在铁电絮凝中对有机污染物降解情况。 图2反映了溶液中存在不同有机配体时苯胺在铁电絮凝体系中的降解情况。在铁电絮凝中分别加入甲酸、丙酸、柠檬酸、酒石酸钾钠时,苯胺的残留率分别为74.9%、72.8%、70.3%、72.9%;在铁电絮凝中分别加入草酸钠和EDTA时,苯胺的残留率为17.5%和20.9%;不含有机配体时的对照实验中苯胺的残留率为61.4%。因此,甲酸、丙酸、柠檬酸和酒石酸钾钠在铁电絮凝体系中可使苯胺的降解率降低,而草酸钠和EDTA却对苯胺的去除有明显的促进作用。原因可能为:一方面,甲酸、丙酸、柠檬酸和酒石酸与Fe(Ⅱ)络合之后还原电位较高[12],没有明显提高其还原氧气的能力;另一方面,甲酸、丙酸、柠檬酸和酒石酸也能够与活性氧物质反应,进而影响苯胺的去除。
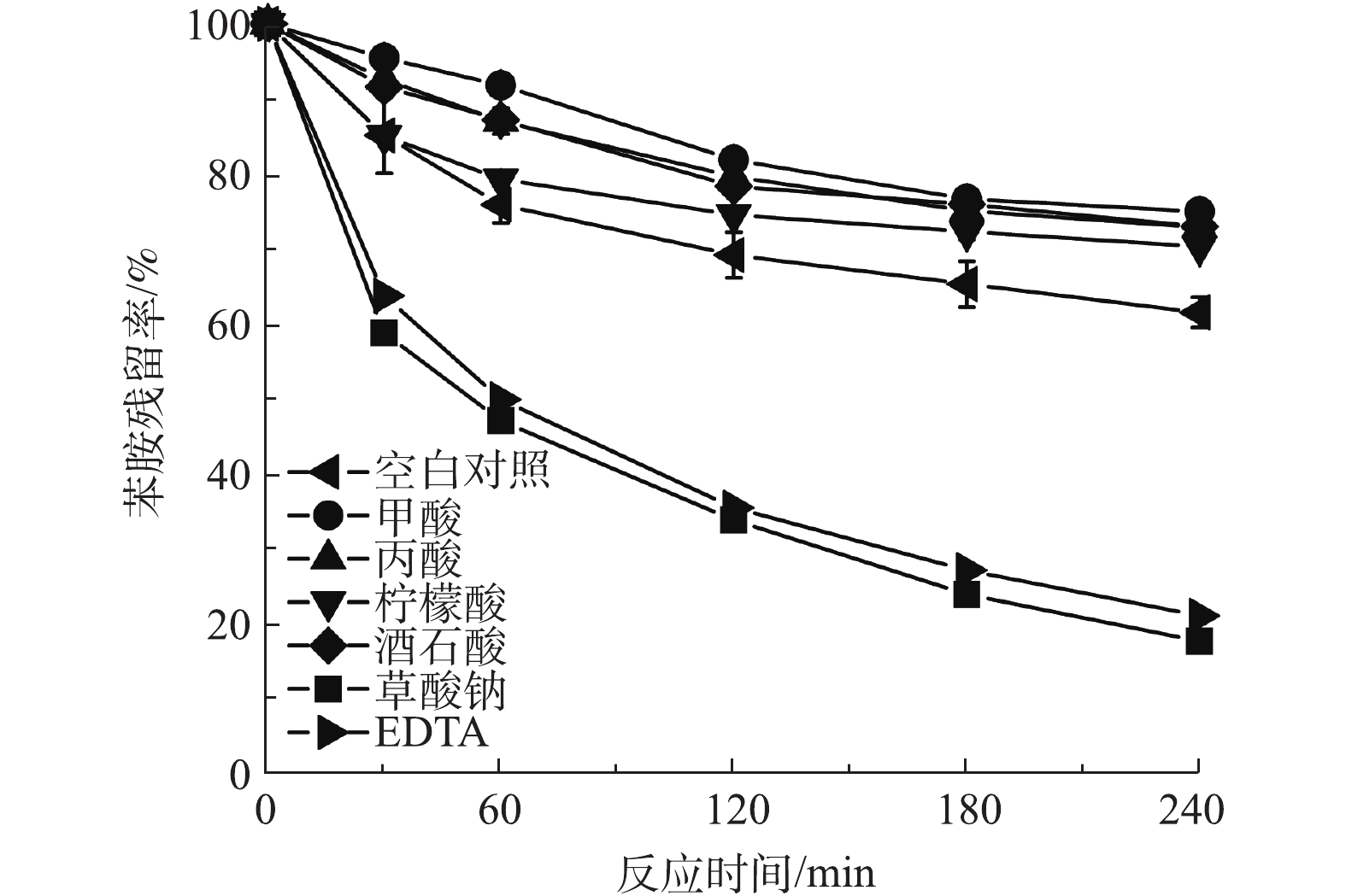 图 2 铁电絮凝中不同有机配体对苯胺降解的影响Figure 2. Effect of different organic ligands on the degradation of aniline in Fe-EC
图 2 铁电絮凝中不同有机配体对苯胺降解的影响Figure 2. Effect of different organic ligands on the degradation of aniline in Fe-ECCHEN等[13]发现,Fe(Ⅱ)-草酸络合物在氧气参与下可以发生一系列氧化还原反应生成·OH,这一过程和铁电絮凝生成·OH的机理类似。NORADOVN等[14]发现,Fe(Ⅱ)-EDTA同样能够与O2反应,生成·OH并导致有机物的氧化降解。可能发生的反应如式(1)~式(4)所示(L表示草酸和EDTA配体)。
Fe(Ⅱ)-L+O2→Fe(Ⅲ)-L+⋅O−2 (1) Fe(Ⅱ)-L+⋅O−2+2H+→Fe(Ⅲ)-L+H2O2 (2) Fe(Ⅱ)-L+H2O2→Fe(Ⅲ)-L+OH−+⋅OH (3) 有机物+⋅OH→氧化产物 (4) 因此,在草酸钠和EDTA体系中,能够生成还原电位较低的Fe(Ⅱ)-C2O4和Fe(Ⅱ)-EDTA络合物[12],极大地促进了活性氧物质的生成,虽然草酸钠和EDTA也会消耗一部分产生的活性氧物质,但总体效果表现为促进苯胺的氧化降解。
2.2 羟基自由基介导的氧化降解机制
在上述实验中,草酸和EDTA能够促进苯胺的降解,主要由于生成的活性氧物质所致。为进一步确认活性氧物质的主导作用,探究铁电絮凝体系中存在草酸钠和EDTA时苯胺的氧化降解机制,本研究做了乙醇淬灭实验和ESR测试。如图3(a)所示,在铁电絮凝体系中投加30 mmol·L−1草酸钠和0.2 mmol·L−1 EDTA时,反应240 min后,苯胺的残留率分别为17.5%和20.9%。乙醇与·OH的反应速率常数为1.9×109 L·(mol·s)−1,高浓度乙醇在铁电絮凝体系中可作为·OH的淬灭剂[15]。在上述实验体系中加入100 mmol·L−1乙醇后,苯胺的降解被极大地抑制,240 min时,苯胺的残留率为83.1%和93.6%。因此,淬灭实验结果证明了体系中导致苯胺氧化降解且起主导作用的活性中间氧化物种可能为·OH。
 图 3 乙醇对铁电絮凝中苯胺降解的影响和ESR波谱图Figure 3. Effect of ethanol on aniline degradation in Fe-EC and ESR spectra
图 3 乙醇对铁电絮凝中苯胺降解的影响和ESR波谱图Figure 3. Effect of ethanol on aniline degradation in Fe-EC and ESR spectra为进一步验证这一猜测,本研究采用DMPO作为·OH捕获剂,对含有EDTA的铁电絮凝体系进行了自由基检测(图3(b))。当只有DMPO存在时,只观察到较低的DMPO杂质波。当电絮凝反应体系中加入0.05 mmol·L−1 EDTA后,观察到相对较强的ESR信号,且这一信号与Fenton反应体系中检测到的信号峰相符。作为中间产物的·OH存在时间极短,浓度过低,很难被直接检测到,但它很容易与反磁性硝酮自旋捕获剂DMPO反应,形成稳定自旋加合物,因此,可以根据ESR光谱的磁参数进行识别[16]。波谱图包含4重峰的ESR信号,其具有1∶2∶2∶1的峰高模式和各向同性的超精细耦合常数,这是DMPO-OH加合物的特征。在图3(b)中,可观察到明显的DMPO-OH特征峰,从而证明体系中存在·OH。
2.3 草酸存在时铁电絮凝体系中·OH的产生机制
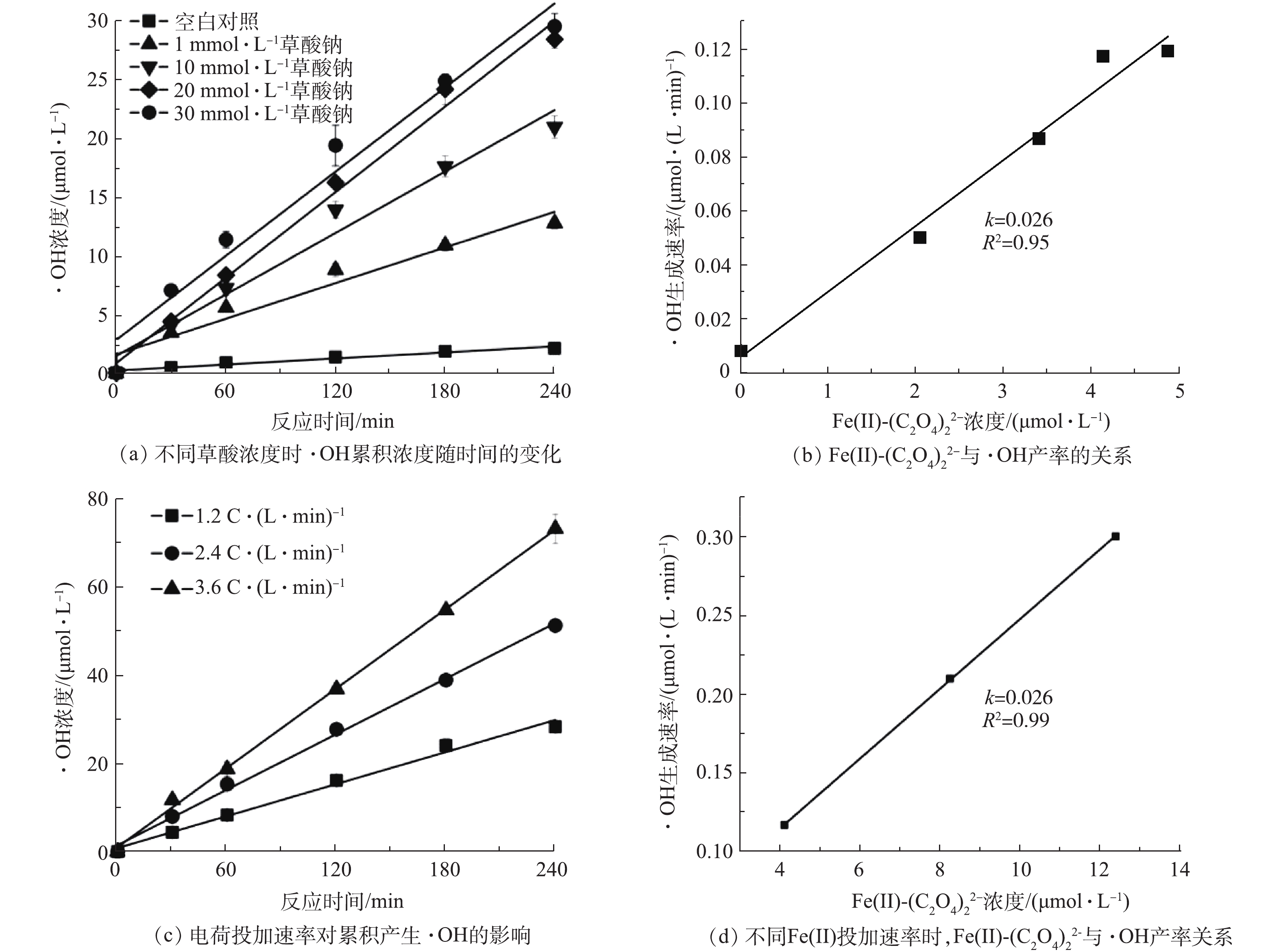
为了进一步探究有机配体存在时铁电絮凝体系中·OH的产生规律,本研究采用苯甲酸与·OH反应生成对羟基苯甲酸,作为·OH的定量分析手段[11],考察了不同条件下的·OH生成速率(见图4)。如图4(a)所示,铁电絮凝体系中·OH累积浓度随电解时间而增加,且符合拟零级动力学过程。当不存在有机配体时,铁电絮凝体系中的产生速率为0.008 6 μmol·min−1。当铁电絮凝中草酸浓度从10 mmol·L−1增加到50 mmol·L−1时,·OH产生速率从0.05 μmol·min−1增加至0.12 μmol·min−1。
根据法拉第电解定律[17],铁电絮凝中电荷投加速率为1.2 C·(L·min)−1时,铁阳极电解产生Fe(Ⅱ)的速率为6.22 μmol·(L·min)−1,反应240 min,产生的累积Fe(Ⅱ)浓度为1.5 mmol·L−1,按照Fe(Ⅱ)与草酸络合比1∶2计,10 mmol·L−1草酸是过量的。因此,当提高体系中草酸浓度时,Fe(Ⅱ)-草酸络合物的总量不变。但由图4(a)中的结果可知,体系中·OH产生速率随草酸浓度的增加而增加,可能是Fe(Ⅱ)-草酸络合物的形态发生变化所导致。为验证这一猜测,本研究采用Visual Minteq V3.1对反应体系中可能存在的Fe(Ⅱ)-草酸络合形态进行了计算,在反应体系中,Fe(Ⅱ)-草酸络合物的主要存在形式为Fe(Ⅱ)-(C2O4)0和Fe(Ⅱ)-
(C2O4)2−2 。随着草酸浓度的升高,Fe(Ⅱ)-草酸络合物的总含量几乎没有发生变化,而Fe(Ⅱ)-(C2O4)0含量降低,Fe(Ⅱ)-(C2O4)2−2 含量升高(表1)。利用计算得到的Fe(Ⅱ)-(C2O4)2−2 产生速率与实验结果中·OH的产生速率进行作图,二者呈现良好的线性关系(图4(b)),其比值为0.026。这一结果说明,体系中对·OH的产生起主要作用的是Fe(Ⅱ)-(C2O4)2−2 ,与已有研究结果[8, 12]一致,因为与Fe(Ⅱ)-(C2O4)0 相比,Fe(Ⅱ)-(C2O4)2−2 的还原电位更低。表 1 Visual MINTEQ 3.1模拟在5 mmol·L−1 NaHCO3中不同草酸浓度下Fe(Ⅱ)-C2O4络合物含量Table 1. Calculated concentrations of aqueous Fe(Ⅱ)-C2O4 species in 5 mmol·L−1 of NaHCO3 using Visual MINTEQ 3.1草酸/(mmol·L− 1) Fe(Ⅱ)-  /(μmol·L− 1)
/(μmol·L− 1)Fe(Ⅱ)-(C2O4)0/(μmol·L− 1) 总Fe(Ⅱ)-C2O4/(μmol·L− 1) 10 2.04 2.45 4.49 20 3.39 2.02 5.42 30 4.13 1.63 5.75 40 4.86 1.14 6.00 | Show Table DownLoad:
CSV
DownLoad:
CSV
为了进一步验证Fe(Ⅱ)-
(C2O4)2−2 产生速率与体系中·OH产生速率之间的关系,本研究在草酸钠浓度为30 mmol·L−1条件下,对电荷投加速率与·OH生成的关系进行了研究。如图4(c)所示,当电荷投加速率分别为1.2、2.4、3.6 C·(L·min)−1时,体系中·OH 浓度的增长都符合拟零级动力学过程,这是因为体系中投加的捕获剂苯甲酸钠对·OH而言是过量的。通过Visual Minteq V3.1计算得到的体系中Fe(Ⅱ)-(C2O4)2−2 产生速率与·OH产生速率的关系如图4(d)所示,二者同样符合线性关系,且比值为0.026,与图4(b)中的比值一致。需要指出的是,即使当电荷投加速率增加到3.6 C·(L·min)−1时,10 mmol·L−1草酸相对于电解产生的Fe(Ⅱ)依然是过量的。因此,可以认为在草酸过量且溶液pH不变时,铁电絮凝体系中·OH的生成只取决于体系中Fe(Ⅱ)-
(C2O4)2−2 含量,二者存在一个固定的比值,约为0.026,即每氧化1 mol Fe(Ⅱ)-(C2O4)2−2 所产生的·OH量为26 mmol。有研究[18]显示,在中性条件下,对于还原性矿物质,氧化1 mol Fe(Ⅱ)时,·OH的产量为20 mmol[19],对于1 mol FeS为7.420 mmol。因此,Fe(Ⅱ)-(C2O4)2−2 相对于前2个体系中有明显的提升。2.4 EDTA对铁电絮凝体系产·OH的作用
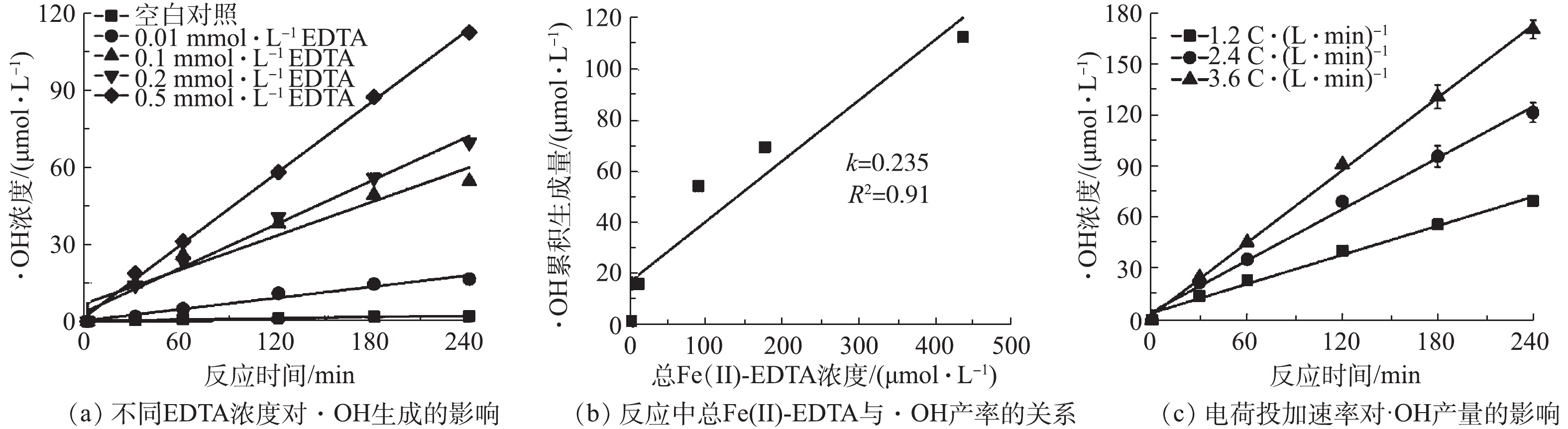
图5反映了EDTA对·OH产量的影响。图5(a)反映了不同EDTA浓度条件下铁电絮凝体系中·OH的生成量,其生成过程符合拟零级动力学过程。当电荷投加速率为1.2 C·(L·min)−1时,EDTA从0.01 mmol·L−1增加到0.5 mmol·L−1,反应进行240 min后,铁电絮凝反应中·OH的生成速率从0.07 μmol·(L·min)−1增加到0.47 μmol·(L·min)−1,相比不加配体时·OH生成速率有明显的提升。当电荷投加速率为1.2 C·(L·min)−1时,铁阳极电解产生Fe(Ⅱ)的速率为6.22 μmol·(L·min)−1,按EDTA最大浓度0.5 mmol·L−1且EDTA与Fe的络合比为1∶1计(图5(b)),80 min内,EDTA被完全络合,故与草酸钠体系不同,EDTA相对于Fe有所不足。为了解EDTA存在时铁电絮凝中主要起作用的铁络合物,本研究对反应体系中可能存在的铁络合形态进行了计算。由Visual Minteq V3.1拟合出EDTA在铁电絮凝中的络合形态,主要有Fe(Ⅱ)-EDTA2−、Fe(Ⅱ)-OHEDTA3−和Fe(Ⅱ)-HEDTA−,其中Fe(Ⅱ)-EDTA2−占87%左右(表2)。同时,在铁电絮凝反应中电荷投加速率为1.2 C·(L·min)−1时,单位时间形成的Fe(Ⅱ)-EDTA2−络合物含量极少(约为5.40 μmol·(L·min)−1),且EDTA浓度升高,其含量无明显变化。由表2可知,随着EDTA浓度升高,Fe(Ⅱ)-EDTA2−含量明显升高,Fe(Ⅱ)-OHEDTA3−含量缓慢变化,反应中起主要影响的物质是Fe(Ⅱ)-EDTA2−。这说明单位时间内反应中的EDTA浓度较低,难以定量Fe(Ⅱ)-EDTA2−对·OH影响,所以在研究EDTA体系时,以反应中累积生成的Fe(Ⅱ)-EDTA2−总量来衡量·OH生成速率。由图5(b)可知,1 mol Fe(Ⅱ)-EDTA2− 反应产生的·OH为235 mmol,这一产率为草酸体系的9倍。其主要原因是:Fe(Ⅱ)-EDTA还原电位(E0= 0.131 V)低于Fe(Ⅱ)-草酸络合物(E0=0.256 V)[12]。还原电位越低,还原能力越强,Fe(Ⅱ)-EDTA更容易与氧气发生系列反应生成·OH。
表 2 Visual MINTEQ V3.1模拟在5 mmol·L−1 NaHCO3中不同EDTA浓度下Fe(Ⅱ)-EDTA络合物含量Table 2. Calculated concentration of aqueous Fe(Ⅱ)-EDTA species in 5 mmol·L−1 of NaHCO3 using Visual MINTEQ 3.1EDTA/(mmol·L−1) Fe(Ⅱ)-EDTA2−/(μmol·L−1) Fe(Ⅱ)-HEDTA−/(μmol·L−1) Fe(Ⅱ)-OHEDTA3−/(μmol·L−1) 0.01 8.85 2.58×10−5 1.15 0.1 88.43 2.57×10−4 11.57 0.2 176.82 5.14×10−4 23.18 0.5 441.69 1.28×10−3 58.26 | Show TableDownLoad:
CSV
由图5(c)可知,·OH的产生速率随电荷投加速率的增加而增加,但由Fe(Ⅱ)与EDTA络合比1∶1可知,当电荷投加速率为3.6 C·(L·min)−1时,0.2 mmol·L−1 EDTA在 90 min左右能够被络合完全,因此,当EDTA量固定时,反应过程中产生的Fe(Ⅱ)-EDTA总量是不变的。而·OH的产生速率随电荷投加速率增加的原因可能是:一方面,电荷投加速率增加时Fe(Ⅱ)产生速率增加,降低了溶液的氧化还原电位[20];另一方面,电荷投加速率增加导致电压升高,促进Fe(Ⅲ)-EDTA在阴极表面还原为Fe(Ⅱ)-EDTA。
上述结果说明在铁电絮凝体系中EDTA浓度和Fe(Ⅱ)投加速率的增加均有利于·OH的生成,且·OH与形成的总Fe(Ⅱ)-EDTA含量呈正相关。
2.5 铁电絮凝中EDTA浓度对苯胺降解的影响
由于·OH与形成的Fe(Ⅱ)-EDTA含量呈正相关,电荷投加速率一定时,为探究不同有机配体浓度在铁电絮凝降解有机污染物时的影响,本实验采用苯胺作为目标污染物,以EDTA作有机配体进行了研究。图6(a)反映了在铁电絮凝过程中加入不同浓度的EDTA后苯胺的降解情况,苯胺的残留率分别为41%、20%、28%、42%;不加EDTA作为对照时,苯胺残留率为71%。随EDTA浓度的增加,铁电絮凝体系对苯胺的降解效果呈现先升高后降低的趋势。由图6(b)可知,当EDTA从0增加到0.5 mmol·L−1时,苯胺的氧化反应速率常数由0.002 8 min−1增加至0.010 7 min−1,而后降低至0.005 9 min−1;当EDTA为0.05 mmol·L−1时,苯胺的氧化反应速率常数达到最大值。苯胺、EDTA与·OH的反应速率常数分别为1.4×1010 L·(mol·s)−1[21]和5×108 L·(mol·s)−1[22]。因此,在二者含量相近时,反应中产生的Fe(Ⅱ)-EDTA和Fe(Ⅲ)-EDTA均能够与苯胺竞争·OH。EDTA与苯胺对·OH的竞争效率[23]可根据式(2)进行计算。
F=CEDTAKEDTA(CEDTAKEDTA+CANKAN) (2) 式中:F为竞争效率;CEDTA为EDTA浓度,mmol·L−1;KEDTA为EDTA与·OH反应速率常数,L·(mol·s)−1;CAN为苯胺浓度,mmol·L−1;KAN为苯胺与·OH反应速率常数,L·(mol·s)−1。当苯胺浓度为5 mg·L−1(即0.055 mmol·L−1),EDTA浓度为0.05 mmol·L−1时,其竞争效率为3.1%,即EDTA的竞争作用可忽略。但是当EDTA含量升高至0.5 mmol·L−1时,竞争效率达31%,影响显著,这一预测与以上实验结果(图6(a))相符。
为进一步证明EDTA对苯胺的竞争作用,本研究对体系中Fe(Ⅲ)-EDTA含量进行了检测,其结果如图6(c)所示。当Fe(Ⅲ)与EDTA按1∶1络合时,Fe(Ⅲ)-EDTA的最大含量为0.05 mmol·L−1,由于电荷投加速率为2.4 C·(L·min)−1时Fe的投加速率为12.44 μmol·(L·min)−1,因此,30 min内,EDTA会被络合完全,Fe(Ⅲ)-EDTA含量达到最大值。其后,Fe(Ⅲ)-EDTA含量随时间延长逐渐降低,验证了Fe(Ⅲ)-EDTA在铁电絮凝过程中被氧化,即Fe(Ⅲ)-EDTA与苯胺竞争·OH。
因此,虽然EDTA含量的升高促进了铁电絮凝中·OH的产生,但由于其对·OH的竞争作用,使得EDTA含量超过0.05 mmol·L−1时苯胺的降解效果减弱。
3. 结论
1)在铁电絮凝体系中甲酸、丙酸、柠檬酸和酒石酸钾钠会抑制苯胺的降解,而EDTA和草酸则能促进苯胺的降解。
2) EDTA存在时促进了铁电絮凝体系中·OH的产生,且·OH是导致苯胺降解的一种主要活性氧化物种。
3)铁电絮凝-草酸体系中产生的Fe(Ⅱ)-
(C2O4)2−2 是促进·OH产生的主要Fe(Ⅱ)物种,且1 mol Fe(Ⅱ)-(C2O4)2−2 反应可以产生26 mmol ·OH;而铁电絮凝-EDTA体系中具有活性的主要Fe(Ⅱ)物种为Fe(Ⅱ)-EDTA2−,1 mol Fe(Ⅱ)-EDTA2−反应可以产生235 mmol ·OH。4) EDTA对铁电絮凝体系中苯胺降解既有促进又有竞争作用,在其浓度为0.05 mmol·L−1时苯胺降解效率最佳。
-
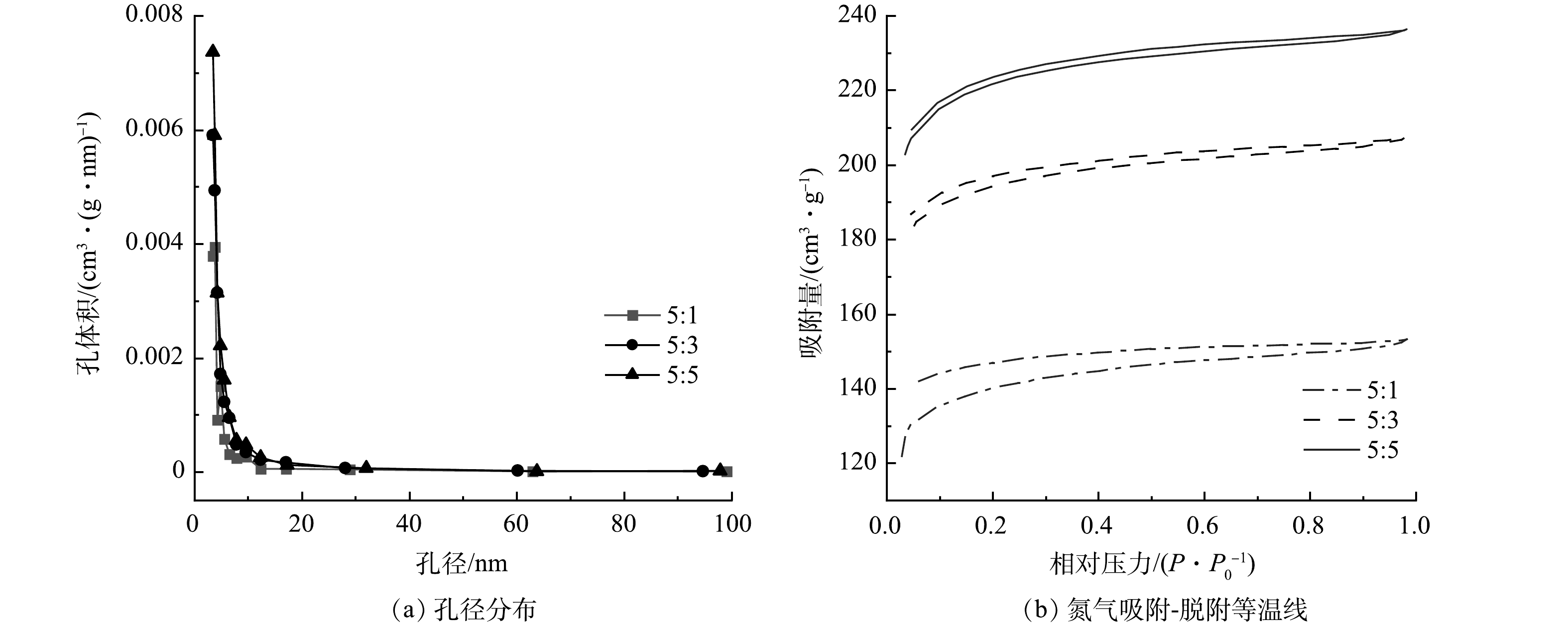
图 1 不同生物炭/氯化锌质量比条件下电极材料BET分析图
Figure 1. BET analysis chart of electrode materials at different biochar/zinc chloride mass ratios
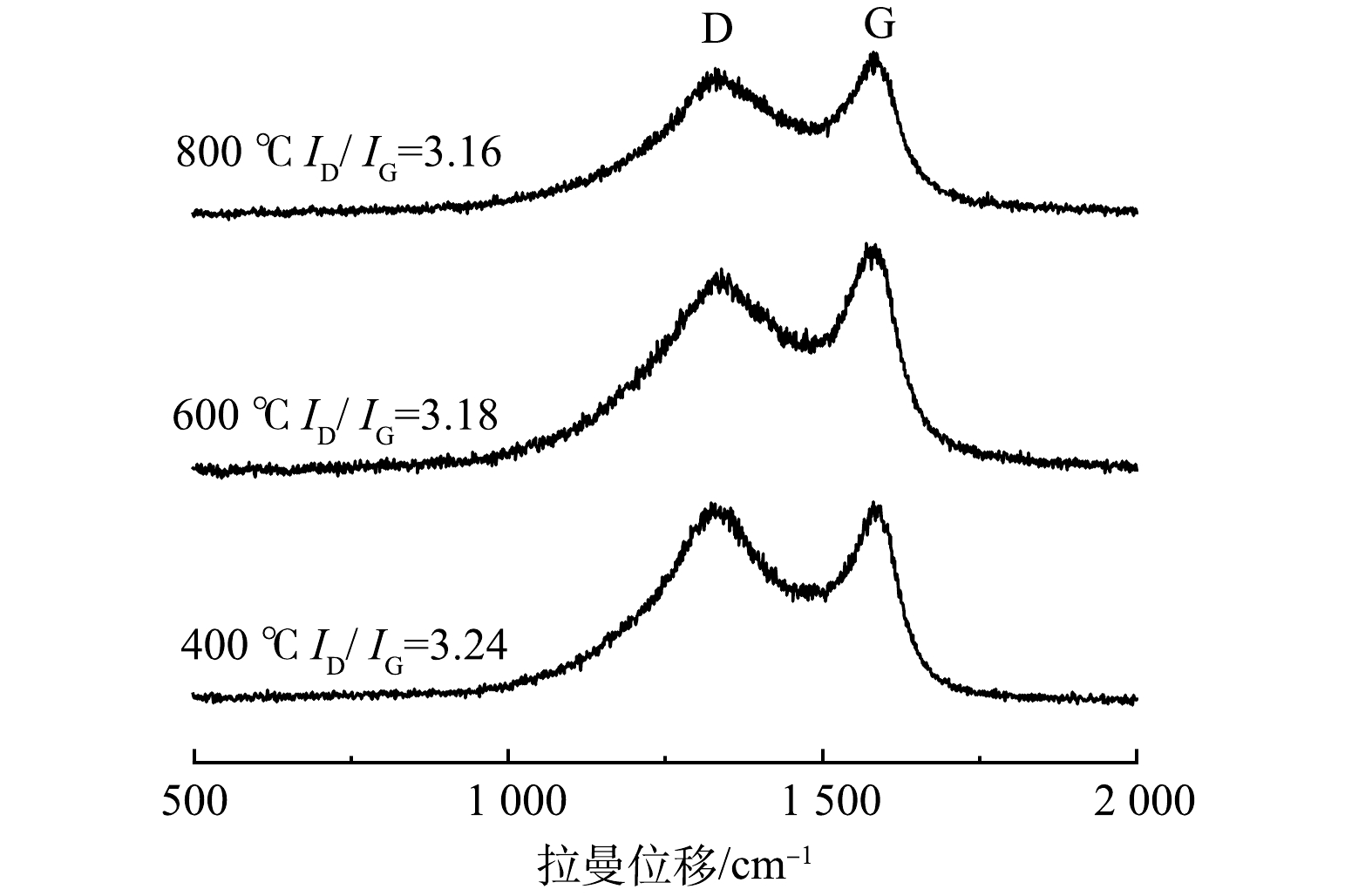
图 2 不同煅烧温度条件下改性核桃壳生物炭拉曼光谱分析
Figure 2. Raman spectra analysis of modified walnut shell biochar at different calcination temperatures

图 3 不同生物炭/聚苯胺/热熔胶复合比例条件下扫描电镜分析
Figure 3. SEM analysis at different composite ratios of biochar/polyaniline/hot melt adhesive
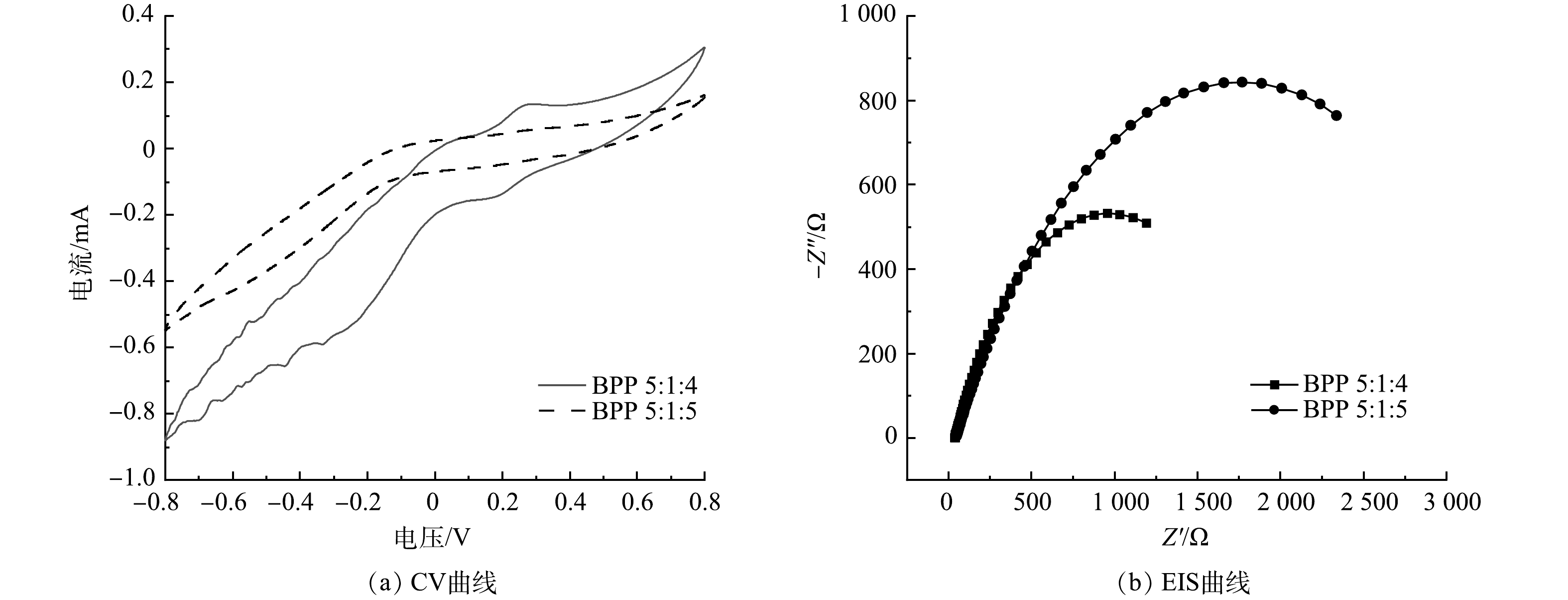
图 4 不同生物炭/聚苯胺/热熔胶复合比例电极电化学分析
Figure 4. Electrochemical analysis of electrodes with different composite ratios of biochar/polyaniline/hot melt adhesive
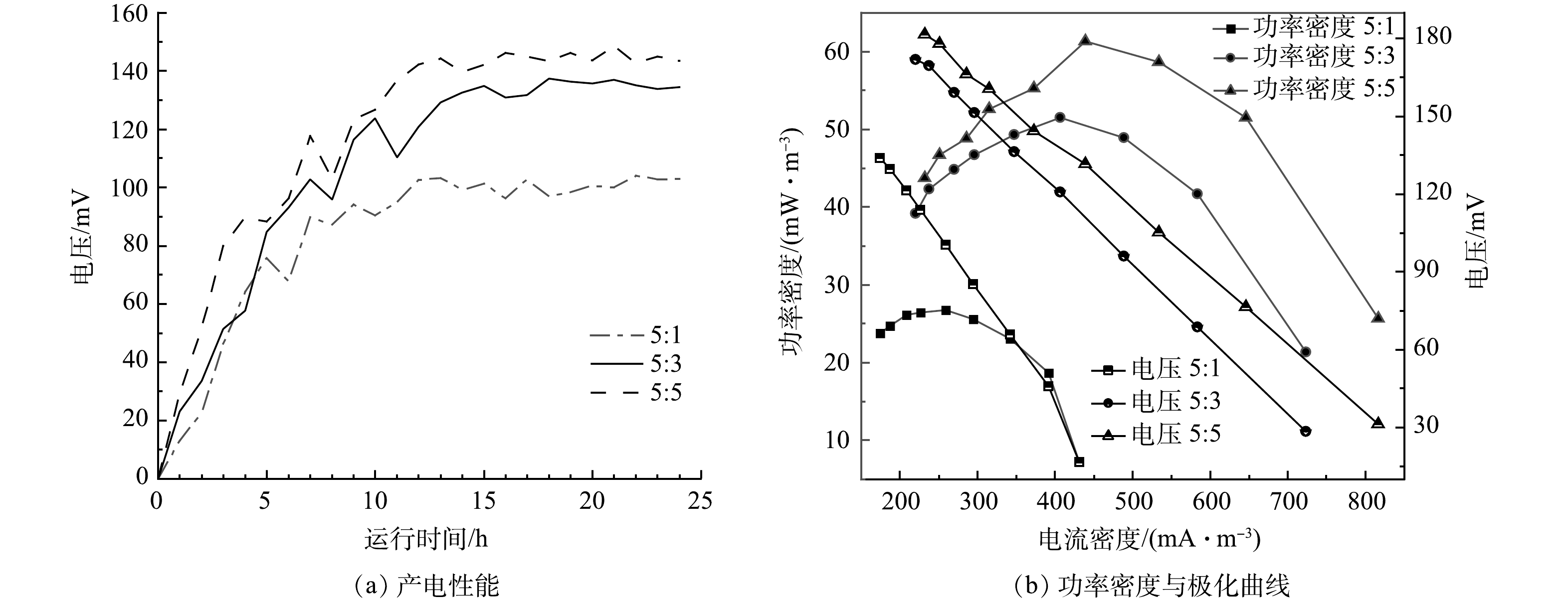
图 6 不同生物炭/氯化锌质量比条件下制备的电极材料
Figure 6. Electrode materials prepared at different biochar/zinc chloride mass ratios

图 7 不同煅烧温度条件下制备的电极材料
Figure 7. Electrode materials prepared at different calcination temperatures
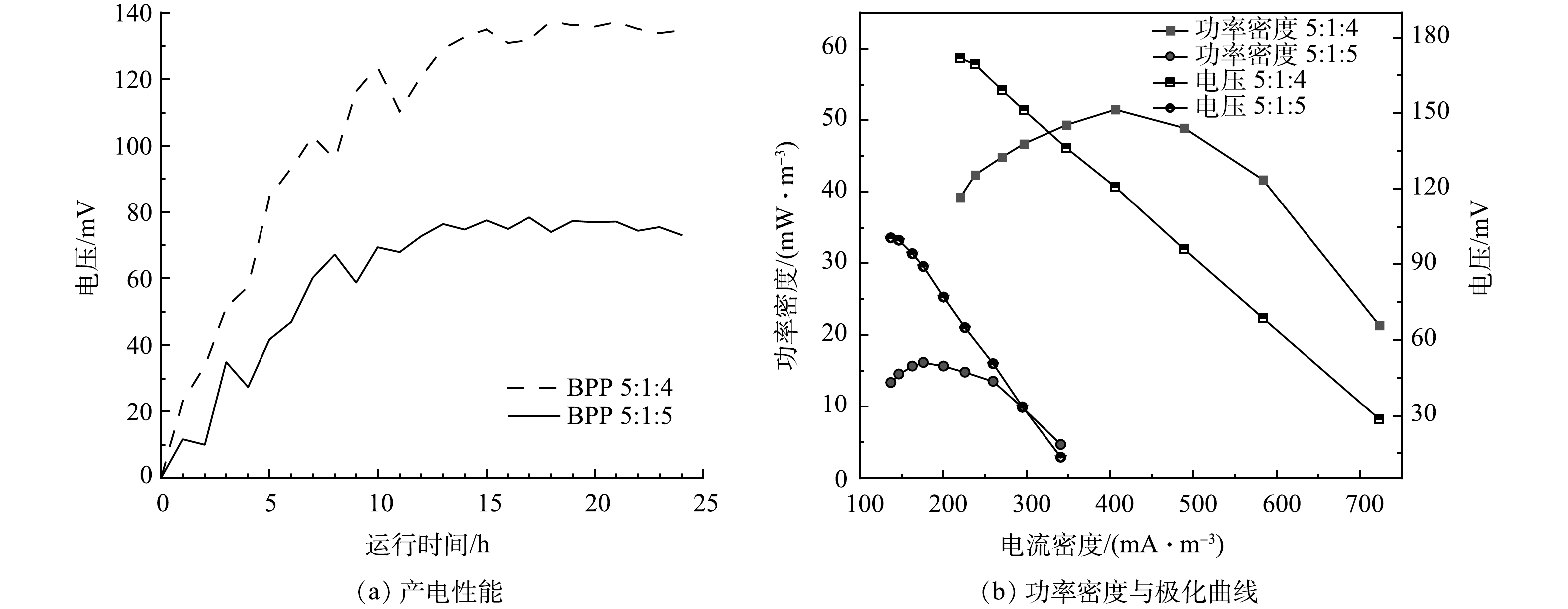
图 8 不同BPP条件下制备的电极材料
Figure 8. Electrode materials prepared at different BPP conditions
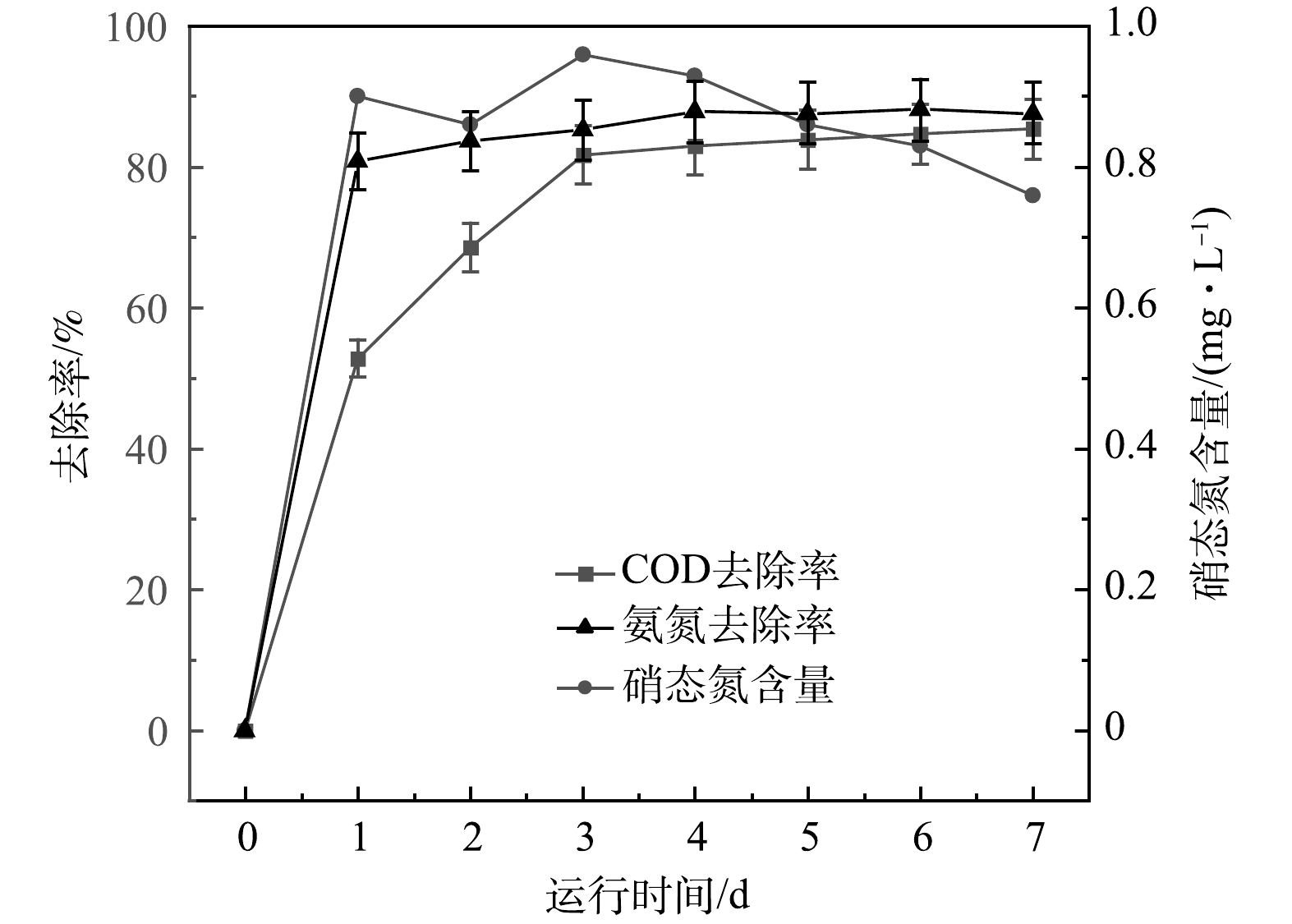
图 9 改性核桃壳生物炭MFC处理模拟废水中污染物的去除效果
Figure 9. Removal effect of pollutants in simulated wastewater treated by modified walnut shell biochar MFC
表 1 BET测量时获得的比表面积、孔径和孔容
Table 1. Specific surface area, pore size and pore volume determined by BET measurement
生物炭/氯化锌质量比 比表面积/(m2·g−1) 孔径/nm 孔容/(cm3·g−1) 5:1 590 3.818 0.009 5:3 657 3.424 0.015 5:5 883 3.421 0.017
下载: 导出CSV
-
[1] GOWTHAMI P, JUNG H Y, SADHASIVAM T, et al. A comprehensive review on microbial fuel cell technologies: Processes, utilization, and advanced developments in electrodes and membranes[J]. Journal of Cleaner Production, 2019, 221: 598-621. doi: 10.1016/j.jclepro.2019.02.172 [2] LI W Y, LIU Y X, REN R P, et al. Research progress on removal of nitrogen in water and wastewater by microbial fuel cell[J]. Chemical Industry and Engineering Progress, 2019, 38(2): 1097-1106. [3] ZHANG K X, MA Z K, SONG H H, et al. Macroporous carbon foam with high conductivity as an efficient anode for microbial fuel cells[J]. International Journal of Hydrogen Energy, 2020, 45(21): 12121-12129. doi: 10.1016/j.ijhydene.2020.02.123 [4] CHEN J F, HU Y Y, ZHANG L H, et al. Bacterial community shift and improved performance induced by in situ preparing dual graphene modified bioelectrode in microbial fuel cell[J]. Bioresource Technology, 2017, 238: 273-280. doi: 10.1016/j.biortech.2017.04.044 [5] LV W, LING Z J, DENG Y Q, et al. Graphene-based materials for electrochemical energy storage devices: Opportunities and challenges[J]. Energy Storage Materials, 2016, 2: 107-138. doi: 10.1016/j.ensm.2015.10.002 [6] CAI T, MENG L J, CHEN G, et al. Application of advanced anodes in microbial fuel cells for power generation: A review[J]. Chemosphere, 2020, 248(c): 125985. [7] LOGAN B, CHENG S, WATSON V, et al. Graphite fiber brush anodes for increased power production in air-cathode microbial fuel cells[J]. Environmental Science & Technology, 2007, 41(9): 3341-3346. [8] CHEN S L, HE G H, HU X W, et al. A three-dimensionally ordered macroporous carbon derived from a natural resource as anode for microbial bioelectrochemical systems[J]. Chemsuschem, 2012, 5(6): 1059-1063. doi: 10.1002/cssc.201100783 [9] 邓金龙. 我国核桃生产现状及发展策略[J]. 林产工业, 2016, 43(10): 56-58. doi: 10.3969/j.issn.1001-5299.2016.10.014 [10] JUAN J L, GUZMAN, MERYEM O, et al. Performance of electro-spun carbon nanofiber electrodes with conductive poly(3, 4-ethylenedioxythiophene) coatings in bioelectrochemical systems[J]. Journal of Power Sources, 2017, 356: 331-337. doi: 10.1016/j.jpowsour.2017.03.133 [11] LI Y Y, ZHU L H, SHEN F, et al. Highly conductive microfiber of graphene oxide templated carbonization of nanofibrillated cellulose[J]. Advanced Functional Materials, 2014, 24(46): 7366-7372. doi: 10.1002/adfm.201402129 [12] KARTHIKEYAN R, WANG B, XUAN J, et al. Interfacial electron transfer and bioelectrocatalysis of carbonized plant material as effective anode of microbial fuel cell[J]. Electrochimica Acta, 2015, 157: 314-323. doi: 10.1016/j.electacta.2015.01.029 [13] RAJAPAKSHA A U, GAO B, ZHANG M, et al. Engineered/designer biochar for contaminant removal/immobilization from soil and water: Potential and implication of biochar modification[J]. Chemosphere, 2016, 148: 276-291. doi: 10.1016/j.chemosphere.2016.01.043 [14] LI D, WANG Y, YANG J, et al. Activation characteristics comparison of activated carbons prepared from biomass and lignite[J]. Journal of Chemical Industry, 2013, 64(9): 3338-3347. [15] SI W J, WU X J, XING W, et al. Bagasse-based nanoporous carbon for supercapacitor application[J]. Journal of Inorganic Materials, 2010, 26(1): 107-112. [16] SONG S R, ZHANG M C, XU X Y, et al. Isolation of a heterotrophic nitrification-aerobic denitrification strain and identification of its potential electricity generation ability in microbial fuel cells.[J]. Environmental Technology, 2021, 1: 21-25. [17] SING K S W, EVERETT D H, HAUL R A W, et al. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (recommendations 1984)[J]. Pure and Applied Chemistry, 1985, 57(4): 603-619. doi: 10.1351/pac198557040603 [18] 王乐, 周娴娴, 陈良, 等. 核桃壳衍生活性炭孔结构的调控及其对锂硫电池性能的影响[J]. 太原理工大学学报, 2021, 52(6): 863-872. doi: 10.16355/j.cnki.issn1007-9432tyut.2021.06.003 [19] SONG H H, LI H, WANG H, et al. Chicken bone-derived N-doped porous carbon materials as an oxygen reduction electrocatalyst[J]. Electrochimica Acta, 2014, 147: 520-526. doi: 10.1016/j.electacta.2014.09.146 [20] YANG G H, LI X, WANG Y Y, et al. Three-dimensional interconnected network few-layered MoS2/N, S co-doped graphene as anodes for enhanced reversible lithium and sodium storage[J]. Electrochimica Acta, 2019, 293: 47-59. doi: 10.1016/j.electacta.2018.10.026 [21] 缺少内容 [22] LU M, QIAN Y J, YANG C C, et al. Nitrogen-enriched pseudographitic anode derived from silk cocoon with tunable flexibility for microbial fuel cells[J]. Nano Energy, 2017, 32: 382-388. [22]CHENG Z, DENG Y D, HU W B, et al. A review of electrolyte materials and compositions for electrochemical supercapacitors[J]. Chemical Society Reviews, 2015, 44(21): 7484-539. [23] HUTCHINSON A J, TOKASH J C, LOGAN B E. Analysis of carbon fiber brush loading in anodes on startup and performance of microbial fuel cells[J]. Journal of Power Sources, 2011, 196(22): 9213-9219. doi: 10.1016/j.jpowsour.2011.07.040 [24] ZHOU G, ZHOU Y, SHI H. Assessment of a novel overflow-type electrochemical membrane bioreactor (EMBR) for wastewater treatment, energy recovery and membrane fouling mitigation[J]. Bioresource Technology, 2015, 196: 648-655. doi: 10.1016/j.biortech.2015.08.032 [25] LIANG P, FANMZ, CAO X X, et al. Composition and measurement of the apparent internal resistance in microbial fuel cell[J]. Environmental Science, 2007, 28(8): 1894-1898. [26] 廖绍华, 杨晓梅, 黄毕生, 等. 氯化锌活化柚子皮制备生物炭及其对亚甲基蓝的吸附[J]. 大理大学学报, 2020, 5(12): 15-20. doi: 10.3969/j.issn.2096-2266.2020.12.003 [27] YE X F, ZHOU H J, YU X N, et al. Physiochemical properties and yields of corn-stalk-biochar under different pyrolyzed temperatures[J]. Journal of Plant Nutrients and Fertilizer, 2017, 23(5): 1268-1275. [28] WANG R F, WANG H, ZHOU T B, et al. The enhanced electrocatalytic activity of okara-derived N-doped mesoporous carbon for oxygen reduction reaction[J]. Journal of Power Sources, 2015, 274: 741-747. doi: 10.1016/j.jpowsour.2014.10.049 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5451
- HTML全文浏览数: 5451
- PDF下载数: 85
- 施引文献: 0
