


-
随着化石燃料中CO2、CH4、N2O等温室气体排放量逐年增加[1],温室效应带来的影响也日益严重,极端天气和自然灾害的出现也越来越频繁[2]。其中,CO2在大气中质量分数的增加对于温室效应的影响最大,造成了2/3的温室效应[3],CO2减排势在必行。CO2减排主要包括,改善能源效率,开发可再生能源,以及CO2碳捕获利用与封存技术 (carbon dioxide capture utilization and storage, CCUS) [4]。政府间气候变化专门委员会(IPCC)的一项评估指出,为了在2050年前将全球平均气温上升控制在2 ℃以内CO2排放量最低应减少50%[5]。国际能源署研究发现,为了实现这一目标,到2050年,CO2碳捕获利用与封存 (CCUS) 至少贡献需要达到全球减碳量的1/6[6]。
目前规模化CCUS技术主要是矿地质封存、海洋封存、矿化封存等。其中,地质封存可能改变原始地貌并且存在CO2泄漏的风险,海洋封存可能会破坏海洋环境[7],而利用工业固体废物进行矿化封存具有生成物稳定[8]、安全性高[9-10]、原料成本低和距离排放源近等优势[11],是一种有发展潜力的规模化CO2捕集封存利用技术。武鸽等[12]开展了典型工业固废矿化封存CO2的基础研究,揭示了电石渣和钢渣矿化封存CO2能力强的机理。张亚朋等[13]进一步开展了CO2矿化封存工艺 (包括干法和湿法) 路线实验研究,对比结果表明,电石渣和钢渣湿法矿化固碳性能一般优于干法,具有较强的应用潜力。然而,湿法矿化仍然存在能耗和成本较高等问题[14];与此同时,石化和煤化工生产排放大量污水及高盐水需要处理[15]。
多项研究使用化工含盐污水协同飞灰、钢渣、水泥窑粉尘等工业固体废物进行矿化反应[16]。LI等[17]用高质量浓度盐水与碱性炉渣进行矿化实验,研究溶液pH、Ca2+质量浓度和液固比等因素对矿化效率的影响,并且发现溶液中高质量浓度的Ca2+会阻碍炉渣中Ca2+的浸出。BANG等[18]用海水淡化厂中的浓缩海水作为反应介质替代去离子水矿化CO2,发现更高的pH有利于矿化反应的进行,Ca与Mg共存时不利于MgCO3的析出。MIGNARDI等[19]研究发现CO2在富Mg溶液中停留时间更长。由此可见,海水、化工盐水等同碱性灰渣协同矿化固碳具有可行性,需要进一步研究不同固废、高浓盐水同CO2的最佳工艺条件。本研究用实际含盐污水与电石渣进行矿化实验,探究温度、压力、液固比对矿化反应中CO2封存率的影响,寻找电石渣矿化反应的最优工艺条件,以期为研发含盐污水与电石渣协同处理的二次资源循环利用技术提供参考。
-
本研究所用电石渣采样于宁夏某工业园区。实验前将样品在研钵中研磨至均匀粉末状,随后在105 ℃下干燥至恒重状态,分别过120目和200目筛得到粒径为75~120 μm的样品干燥密封保存。实验所用CO2为99.9%高纯气体。化工含盐污水取自山西某化工园区。
-
反应装置主要由高压釜 (KTFD06-20型,烟台科立化工设备有限公司,可用体积为0.6 L,最高温度300 ℃,最高压力20 MPa) 、高纯CO2 钢瓶和在线记录控制箱3部分组成。
-
将溶液和处理后电石渣加入高压釜中,关闭出口阀门,在密闭条件下升温至设定温度。随后将高纯度CO2 气体从钢瓶注入反应器中,使高压釜内的压力稳定在设定压力。开启机械搅拌装置,转速200 r·min−1,同时开始计时。反应2 h后停止加热,打开出气阀门释放压力,等高压釜内温度降低到室温。然后对反应釜内的悬浮液用0.7 μm 滤纸进行过滤,将过滤后的反应物放入烘箱,在105 ℃下烘干12 h,并研磨均匀,后对反应产物进行分析表征。
-
矿化反应受温度、压力、液固比的影响较大,分别考察反应温度 (25、45、65、85 ℃) 、压力 (0.5、1.0、1.5、2.0 、2.5 MPa) 、液固比 (1、5、10、15 mL·g−1) 对电石渣协同含盐污水湿法矿化封存CO2能力的影响。基于单因素的实验结果设计实验,采用响应面曲线法 (Response surface methodology, RSM) 进行工艺条件优化[20-21],研究温度、压力、液固比之间的交互作用,确定矿化反应最佳工艺条件[22]。
-
CO2封存率计算:取适量样品,在N2流量为30 mL·min−1的条件下进行热重分析,温度为50~1 050 ℃,升温速率为50 ℃·min−1,通过测定样品在550~950 ℃的失重,计算样品中的CO2质量分数w (CO2) 和CO2封存量K,如式 (1)~式 (3) 所示。
式中:Δm550~950 ℃表示封存的CO2质量,g;w (CO2) 为矿化后电石渣中的CO2质量分数;K为CO2的封存率;QCO2为单位质量新鲜电石渣的CO2封存量,g·kg−1。
-
采用X 射线荧光光谱(X-ray fluorescence, XRF)分析电石渣化学组成;采用热重分析仪分析电石渣CO2封存量;采用Mettler Toledo-FE28 pH测试计测定实验水样pH;采用Dionex-320型离子色谱仪检测反应前后溶液中主要阴离子;采用电感耦合等离子体光谱仪 (ICP-AES) 测定阳离子。
-
分别使用实际化工含盐污水和去离子水作为反应介质进行矿化反应,图1是湿法矿化反应前后电石渣热重 (TG) 曲线。从图1中可以看出,反应前电石渣在100~540 ℃有明显质量损失,该质量损失对应Ca(OH)2分解。这表明,电石渣原样中含有Ca(OH)2,而矿化反应后的电石渣仅在550~950 ℃之间出现明显质量损失,100~540 ℃之间的失重消失说明矿化反应后电石渣中的Ca(OH)2转化为了其他物质。550~950 ℃的质量损失对应CaCO3的分解,表明矿化反应后电石渣中的Ca(OH)2转化成CaCO3。根据计算结果可知,使用化工含盐污水和去离子水作为反应介质的电石渣CO2封存率分别为59.59%和59.89%,封存率基本持平。这说明,化工含盐污水对电石渣湿法矿化封存CO2能力影响不大,可以使用化工含盐污水代替去离子水。
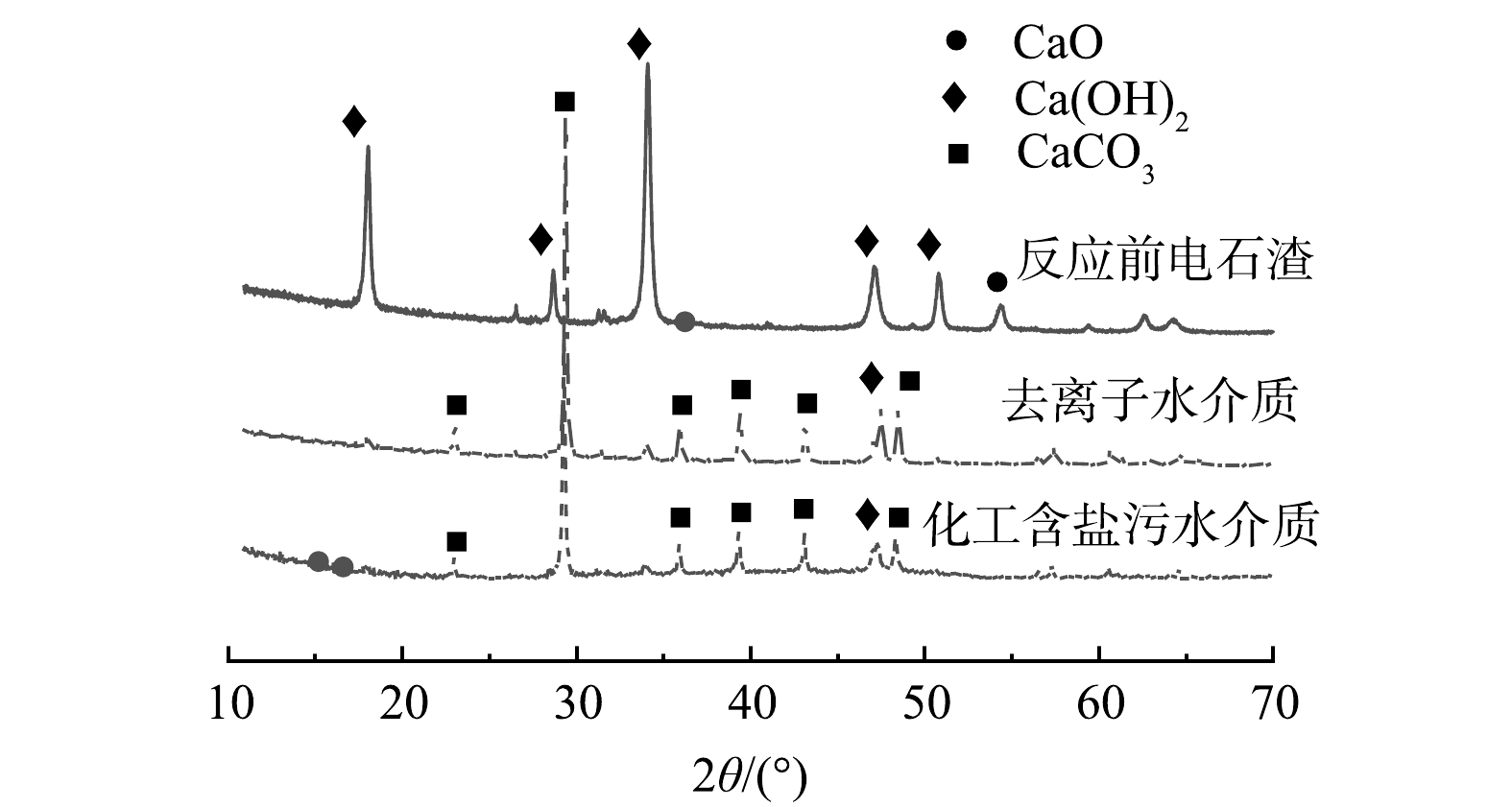
反应前后电石渣的XRD图谱如图2所示,反应前电石渣中主要矿物相为Ca(OH)2,此外还有少量的CaO。反应后电石渣样品的XRD图谱中还存在少量Ca(OH)2的衍射峰,使用化工含盐污水和去离子水作为反应介质的电石渣的矿物组成基本一致,这也与热重结果相符。随着反应的进行,生成的CaCO3颗粒聚集、附着或覆盖在原样的表面,使内部的活性钙未能完全发生反应,因此反应后样品中存在Ca(OH)2。经过矿化反应后的电石渣碱性降低,减少了环境风险,可以用作水泥、砂浆、混凝土或沥青的骨料[23-24],也可以通过过滤分离等手段获得高纯碳酸钙[25-26]。
对湿法矿化反应前后的化工含盐污水进行离子分析,如表1所示。从表中可以看出,反应后阳离子中Ca2+、Mg2+和Na+质量浓度出现了较明显的下降,分别从897、525、1 927 mg·L−1降至556、221、617 mg·L−1。阴离子中Cl−和SO42−质量浓度也出现了较明显的下降,分别从 4 484、3 565 mg·L−1降至4 087、617 mg·L−1。湿法反应前后电石渣的XRF结果如表2所示,反应后电石渣中Na、Cl、S元素含量增加。Ca2+和Mg2+质量浓度的降低可能的原因是反应过程中溶液中Ca2+和Mg2+与溶液中的CO32−发生了反应,消耗了Ca2+和Mg2+,导致其质量浓度下降。Na+、Cl−和SO42−不能与CO32−反应生成沉淀,其质量浓度下降一方面可能是电石渣的加入使溶液pH增加 (如图3所示) ,Ca2+与SO42−生成CaSO4沉淀;另一方面,这些离子可能吸附在电石渣表面,造成溶液中离子质量浓度的下降[27-28]。
对矿化过程中不同阶段的2种水样进行pH测试,其pH变化如图3所示。可以看出,2种水样pH不同,但是在反应过程中的pH变化趋势基本一致,反应前化工含盐污水pH为5.18,去离子水pH为7.03;加入电石渣、加热至实验温度并搅拌后,化工含盐污水滤液pH升至12.02,去离子水滤液pH为12.31,pH的升高源于电石渣中碱性物质如Ca(OH)2发生电离,产生大量OH−和Ca2+;通入CO2后,由于CO2与溶液中OH−和Ca2+ (或Mg2+) 发生矿化反应,OH−的消耗使溶液碱性降低,化工含盐污水滤液和去离子水滤液pH值分别下降至7.40和7.15。
-
1) 温度对电石渣CO2封存率的影响。CO2封存率随温度变化关系如图4 (a) 所示。由图可见,随着温度的升高,CO2封存率先增大后减小,在65 ℃达到最大封存率47.56%。随着温度升高,分子运动会随之变得剧烈,电石渣中钙更容易浸出形成Ca2+溶解于液相中,但是温度的升高会降低CO2气体在溶液中的溶解度。在65 ℃之前,Ca2+的浸出是反应控制步骤,温度升高,Ca2+的浸出量增大,CO2封存率升高。在温度高于65 ℃后,CO2的溶解是反应控制步骤,随着温度升高,CO2气体在溶液中的溶解度下降[29],导致封存率下降。矿化反应为放热反应,温度升高,反应平衡向左移动,也会导致封存率下降。
2) 压力对电石渣CO2封存率的影响。CO2封存率随初始压力变化关系如图4 (b) 所示。由图可见,CO2封存率随初始压力的变化较小,随着初始压力的升高,CO2封存率先增大后减小,在压力为1.0 MPa时K达到最大封存率52.79%。压力主要对CO2在溶液中的溶解度产生影响。在一定的温度下,溶液中CO2的溶解量与压力成正比,压力差越大,推动力越大,CO2在溶液中的溶解量越大[30]。随着压力从0.5 MPa增大到1.0 MPa,CO2在溶液中的溶解量增大,产生的CO32+离子增加,CO2封存率增大。当压力从1.0 MPa增大到2.5 MPa时,溶液中的CO2质量浓度继续增加,与化工含盐污水中的Ca2+快速反应,生成的CaCO3颗粒附着于电石渣表面,阻碍了电石渣中Ca的浸出[31],同时干扰CO2气体在电石渣中的扩散,限制反应的进行[32] 。
3) 液固比对电石渣CO2封存率的影响。CO2封存率液固比变化关系如图4 (c) 所示。由图可见,随着液固比的升高,CO2封存率先增大后减小,在液固比为5 mL·g−1时K达到最大63.47%。溶液中的Ca2+、CO32−质量浓度对矿化反应过程有很大的影响,而液固比会影响电石渣中碱性物质和CO2在水中的溶解程度,进而影响溶液中Ca2+、CO32−质量浓度及矿化封存CO2性能[33]。在液固比小于5 mL·g−1时,虽然提供了电石渣中碱性物质与气体发生反应的液体介质,但是液固比太小,不足使电石渣中钙充分浸出,CO2在水中的溶解量也较少。当液固比达到一定值时,电石渣中碱性物质溶解基本完全,继续增大液固比,碱性物质的溶解量不再增加,相反,液体的增加会使溶液中Ca2+质量浓度下降,进而矿化反应平衡向左移动,封存量降低。
-
1) 模型建立与检验。根据单因素影响的实验结果设计响应面曲线工业条件优化实验,实验结果如表3所示。
根据实验结果拟合模型,由Design-Expert软件拟合出经验模型如式 (4) 所示。模型的P值为0.008 722<0.05,意味着预测模型是显著的。
2) 因素交互作用。采用响应面曲线法研究3个变量之间的交互作用,图5为CO2封存率的响应面图和等高线图。图5 (a) 、图5 (b) 表示,温度和压力之间的交互作用对CO2封存率的影响,在液固比不变的情况下,温度与压力之间的交互作用较小,压力的改变对CO2封存率的影响很微弱,这也与单因素试验的结果吻合。图5 (c) 、图5 (d) 为压力一定时温度与液固比的交互作用对CO2封存率的影响,由于温度和液固比的影响,CO2封存率增加,等高线的形状为椭圆形,说明温度和液固比的交互作用显著,是影响封存率最显著的参数之一。图5 (e) 、图5 (f) 为温度一定时压力与液固比的交互作用CO2封存率的影响,三维图基本呈拱形,说明液固比对于反应的影响更加明显,而2者的交互作用对于CO2的封存来说并不显著。
3) 最佳工艺条件。基于响应面曲线拟合结果,确定CO2封存率最大的反应条件为:85 ℃、0.5 MPa、液固比7.5 mL·g−1,在此条件下,模型预测值为66.9%。同时,在上述条件下进行含盐污水协同电石渣湿法矿化封存CO2实验,3次平行实验所得CO2封存率的平均值为66.1%,电石渣实际固碳量为661 g·kg−1,实验值与预测值的误差小于1.5%。因此,通过上述方法可高效准确获得矿化封存CO2实验的最佳工艺条件。
-
1) 化工含盐污水和去离子水为介质的电石渣矿化封存CO2封存率分别为59.59%和59.89%,基本持平,可以使用化工含盐污水代替去离子水作为反应介质。
2) 温度和液固比对CO2封存率的影响较大,压力影响较小。单因素优化实验条件下,在温度65 ℃、压力1.0 MPa和液固比5 mL·g−1时CO2封存率分别达到47.56%、52.79%和63.47%。
3) 温度和液固比之间的交互作用最为显著,温度和压力、液固比和压力的交互作用不显著。同时确定在反应温度85 ℃、初始反应压力0.5 MPa、液固比7.5 mL·g−1的最优工艺条件下,含盐污水协同电石渣矿化的CO2封存率为66.1%。
响应面曲线法优化含盐污水协同电石渣矿化封存CO2
Optimization of carbon dioxide sequestration by carbide slag mineralization with chemical salty wastewater by response surface methodology
-
摘要: 针对电石渣湿法矿化消耗水资源较多的问题,采用实际化工含盐污水作为反应介质,开展电石渣湿法矿化封存CO2实验。探究温度、压力、液固比等单因素对电石渣矿化固碳封存率的影响,并采取Box-Behnken响应面曲线法对工艺条件进行优化。结果表明,污水和去离子水为介质的电石渣矿化封存CO2封存率分别为59.59%和59.89%。温度和液固比对CO2封存率的影响较大,压力影响较小,且在温度65 ℃、压力1.0 MPa和液固比5 mL·g−1的优化实验条件下的CO2封存率分别达到47.56%、52.79%和63.47%。响应面曲线法实验结果表明,温度和液固比之间的交互作用最为显著,温度和压力、液固比和压力的交互作用不显著。同时确定,在反应温度85 ℃、初始反应压力0.5 MPa、液固比7.5 mL·g−1的最优工艺条件下,含盐污水协同电石渣矿化的CO2封存率为66.1%。本研究结果可为研发“气 (CO2) - 液 (含盐污水) - 固 (电石渣) ”协同的二次资源循环利用技术提供参考。Abstract: In view of the problem that the wet mineralization of carbide slag consumes a lot of water resource, an experimental study on carbon dioxide sequestration by carbide slag wet mineralization was carried out using actual chemical salty wastewater as the reaction medium. The effect of temperature, pressure, liquid-solid ratio and other single factors on carbon sequestration rate of calcium carbide slag was studied, and then Box-Behnken response surface curve method was used to optimize the process conditions. The results showed that the CO2 sequestration rates of calcium carbide slag mineralization with wastewater and deionized water as media were 59.59% and 59.89%, respectively, basically unchanged. The experiment revealed that temperature and liquid-solid ratio had a great influence on CO2 sequestration rates, while pressure had a small influence. Under the optimized experimental conditions of temperature 65 ℃, pressure 1.0 MPa and liquid-solid ratio 5 mL·g−1, the CO2 sequestration rate reached 47.56%, 52.79% and 63.47%, respectively. The results of response surface curve method showed that the interaction between temperature and liquid-solid was the most significant, while temperature and pressure, liquid-solid and pressure were not significantly interacted. At the same time, under the optimal conditions of reaction temperature 85 ℃, initial reaction pressure 0.5 MPa and liquid-solid ratio 7.464 mL·g−1, the CO2 sequestration rate of salt wastewater coordinated calcium carbide slag mineralization reached 66.1%. This study can provide a reference for the further development of synergy technology of the secondary resource cycle of the industrial enterprise “three pollutants emissions”, for example CO2, salty wastewater and calcium carbide slag.
-
随着化石燃料中CO2、CH4、N2O等温室气体排放量逐年增加[1],温室效应带来的影响也日益严重,极端天气和自然灾害的出现也越来越频繁[2]。其中,CO2在大气中质量分数的增加对于温室效应的影响最大,造成了2/3的温室效应[3],CO2减排势在必行。CO2减排主要包括,改善能源效率,开发可再生能源,以及CO2碳捕获利用与封存技术 (carbon dioxide capture utilization and storage, CCUS) [4]。政府间气候变化专门委员会(IPCC)的一项评估指出,为了在2050年前将全球平均气温上升控制在2 ℃以内CO2排放量最低应减少50%[5]。国际能源署研究发现,为了实现这一目标,到2050年,CO2碳捕获利用与封存 (CCUS) 至少贡献需要达到全球减碳量的1/6[6]。
目前规模化CCUS技术主要是矿地质封存、海洋封存、矿化封存等。其中,地质封存可能改变原始地貌并且存在CO2泄漏的风险,海洋封存可能会破坏海洋环境[7],而利用工业固体废物进行矿化封存具有生成物稳定[8]、安全性高[9-10]、原料成本低和距离排放源近等优势[11],是一种有发展潜力的规模化CO2捕集封存利用技术。武鸽等[12]开展了典型工业固废矿化封存CO2的基础研究,揭示了电石渣和钢渣矿化封存CO2能力强的机理。张亚朋等[13]进一步开展了CO2矿化封存工艺 (包括干法和湿法) 路线实验研究,对比结果表明,电石渣和钢渣湿法矿化固碳性能一般优于干法,具有较强的应用潜力。然而,湿法矿化仍然存在能耗和成本较高等问题[14];与此同时,石化和煤化工生产排放大量污水及高盐水需要处理[15]。
多项研究使用化工含盐污水协同飞灰、钢渣、水泥窑粉尘等工业固体废物进行矿化反应[16]。LI等[17]用高质量浓度盐水与碱性炉渣进行矿化实验,研究溶液pH、Ca2+质量浓度和液固比等因素对矿化效率的影响,并且发现溶液中高质量浓度的Ca2+会阻碍炉渣中Ca2+的浸出。BANG等[18]用海水淡化厂中的浓缩海水作为反应介质替代去离子水矿化CO2,发现更高的pH有利于矿化反应的进行,Ca与Mg共存时不利于MgCO3的析出。MIGNARDI等[19]研究发现CO2在富Mg溶液中停留时间更长。由此可见,海水、化工盐水等同碱性灰渣协同矿化固碳具有可行性,需要进一步研究不同固废、高浓盐水同CO2的最佳工艺条件。本研究用实际含盐污水与电石渣进行矿化实验,探究温度、压力、液固比对矿化反应中CO2封存率的影响,寻找电石渣矿化反应的最优工艺条件,以期为研发含盐污水与电石渣协同处理的二次资源循环利用技术提供参考。
1. 材料与方法
1.1 实验原料
本研究所用电石渣采样于宁夏某工业园区。实验前将样品在研钵中研磨至均匀粉末状,随后在105 ℃下干燥至恒重状态,分别过120目和200目筛得到粒径为75~120 μm的样品干燥密封保存。实验所用CO2为99.9%高纯气体。化工含盐污水取自山西某化工园区。
1.2 实验装置
反应装置主要由高压釜 (KTFD06-20型,烟台科立化工设备有限公司,可用体积为0.6 L,最高温度300 ℃,最高压力20 MPa) 、高纯CO2 钢瓶和在线记录控制箱3部分组成。
1.3 实验方法
将溶液和处理后电石渣加入高压釜中,关闭出口阀门,在密闭条件下升温至设定温度。随后将高纯度CO2 气体从钢瓶注入反应器中,使高压釜内的压力稳定在设定压力。开启机械搅拌装置,转速200 r·min−1,同时开始计时。反应2 h后停止加热,打开出气阀门释放压力,等高压釜内温度降低到室温。然后对反应釜内的悬浮液用0.7 μm 滤纸进行过滤,将过滤后的反应物放入烘箱,在105 ℃下烘干12 h,并研磨均匀,后对反应产物进行分析表征。
1.4 实验设计
矿化反应受温度、压力、液固比的影响较大,分别考察反应温度 (25、45、65、85 ℃) 、压力 (0.5、1.0、1.5、2.0 、2.5 MPa) 、液固比 (1、5、10、15 mL·g−1) 对电石渣协同含盐污水湿法矿化封存CO2能力的影响。基于单因素的实验结果设计实验,采用响应面曲线法 (Response surface methodology, RSM) 进行工艺条件优化[20-21],研究温度、压力、液固比之间的交互作用,确定矿化反应最佳工艺条件[22]。
1.5 数据处理
CO2封存率计算:取适量样品,在N2流量为30 mL·min−1的条件下进行热重分析,温度为50~1 050 ℃,升温速率为50 ℃·min−1,通过测定样品在550~950 ℃的失重,计算样品中的CO2质量分数w (CO2) 和CO2封存量K,如式 (1)~式 (3) 所示。
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) stringUtils.convertMath(!{formula.content}) (3) 式中:Δm550~950 ℃表示封存的CO2质量,g;w (CO2) 为矿化后电石渣中的CO2质量分数;K为CO2的封存率;QCO2为单位质量新鲜电石渣的CO2封存量,g·kg−1。
1.6 分析与表征
采用X 射线荧光光谱(X-ray fluorescence, XRF)分析电石渣化学组成;采用热重分析仪分析电石渣CO2封存量;采用Mettler Toledo-FE28 pH测试计测定实验水样pH;采用Dionex-320型离子色谱仪检测反应前后溶液中主要阴离子;采用电感耦合等离子体光谱仪 (ICP-AES) 测定阳离子。
2. 结果与讨论
2.1 化工含盐污水协同电石渣矿化封存CO2 性能评价
分别使用实际化工含盐污水和去离子水作为反应介质进行矿化反应,图1是湿法矿化反应前后电石渣热重 (TG) 曲线。从图1中可以看出,反应前电石渣在100~540 ℃有明显质量损失,该质量损失对应Ca(OH)2分解。这表明,电石渣原样中含有Ca(OH)2,而矿化反应后的电石渣仅在550~950 ℃之间出现明显质量损失,100~540 ℃之间的失重消失说明矿化反应后电石渣中的Ca(OH)2转化为了其他物质。550~950 ℃的质量损失对应CaCO3的分解,表明矿化反应后电石渣中的Ca(OH)2转化成CaCO3。根据计算结果可知,使用化工含盐污水和去离子水作为反应介质的电石渣CO2封存率分别为59.59%和59.89%,封存率基本持平。这说明,化工含盐污水对电石渣湿法矿化封存CO2能力影响不大,可以使用化工含盐污水代替去离子水。
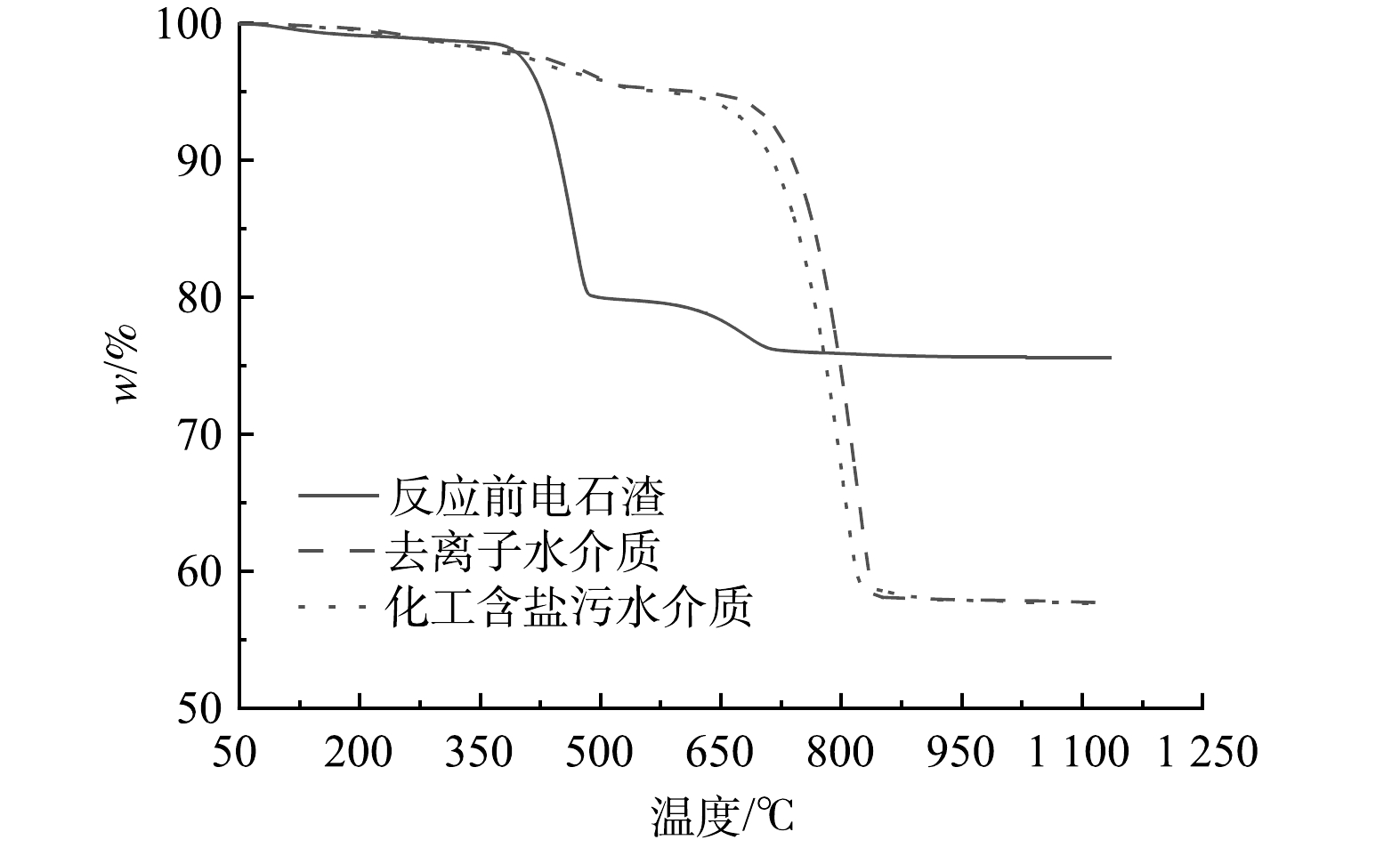 图 1 湿法矿化反应前后热重 (TG) 曲线Figure 1. Thermogravimetric (TG) curves before and after wet mineralization
图 1 湿法矿化反应前后热重 (TG) 曲线Figure 1. Thermogravimetric (TG) curves before and after wet mineralization反应前后电石渣的XRD图谱如图2所示,反应前电石渣中主要矿物相为Ca(OH)2,此外还有少量的CaO。反应后电石渣样品的XRD图谱中还存在少量Ca(OH)2的衍射峰,使用化工含盐污水和去离子水作为反应介质的电石渣的矿物组成基本一致,这也与热重结果相符。随着反应的进行,生成的CaCO3颗粒聚集、附着或覆盖在原样的表面,使内部的活性钙未能完全发生反应,因此反应后样品中存在Ca(OH)2。经过矿化反应后的电石渣碱性降低,减少了环境风险,可以用作水泥、砂浆、混凝土或沥青的骨料[23-24],也可以通过过滤分离等手段获得高纯碳酸钙[25-26]。
对湿法矿化反应前后的化工含盐污水进行离子分析,如表1所示。从表中可以看出,反应后阳离子中Ca2+、Mg2+和Na+质量浓度出现了较明显的下降,分别从897、525、1 927 mg·L−1降至556、221、617 mg·L−1。阴离子中Cl−和SO42−质量浓度也出现了较明显的下降,分别从 4 484、3 565 mg·L−1降至4 087、617 mg·L−1。湿法反应前后电石渣的XRF结果如表2所示,反应后电石渣中Na、Cl、S元素含量增加。Ca2+和Mg2+质量浓度的降低可能的原因是反应过程中溶液中Ca2+和Mg2+与溶液中的CO32−发生了反应,消耗了Ca2+和Mg2+,导致其质量浓度下降。Na+、Cl−和SO42−不能与CO32−反应生成沉淀,其质量浓度下降一方面可能是电石渣的加入使溶液pH增加 (如图3所示) ,Ca2+与SO42−生成CaSO4沉淀;另一方面,这些离子可能吸附在电石渣表面,造成溶液中离子质量浓度的下降[27-28]。
表 1 反应前后溶液组分变化Table 1. Change of solution composition before and after reactionmg·L−1 样品 Cl− NO3− SO42 Ca2+ K+ Mg2+ Na+ 化工含盐污水 4 484 102 3 565 897 231 525 1 927 反应后滤液 4 087 93 617 556 171 221 617 | Show Table DownLoad:
CSV
表 2 反应前后电石渣化学组成Table 2. Chemical composition of calcium carbide slag before and after reaction
DownLoad:
CSV
表 2 反应前后电石渣化学组成Table 2. Chemical composition of calcium carbide slag before and after reaction% 样品 Na2O MgO Al2O3 SiO2 SO3 Cl CaO TiO2 Fe2O3 SrO K2O NiO 反应前电石渣 0.020 0.170 0.950 2.700 0.470 — 90.900 — 0.170 — — — 反应后电石渣 0.549 0.523 0.658 1.900 1.990 0.615 93.100 0.072 0.192 0.079 0.102 0.177 | Show TableDownLoad:
CSV
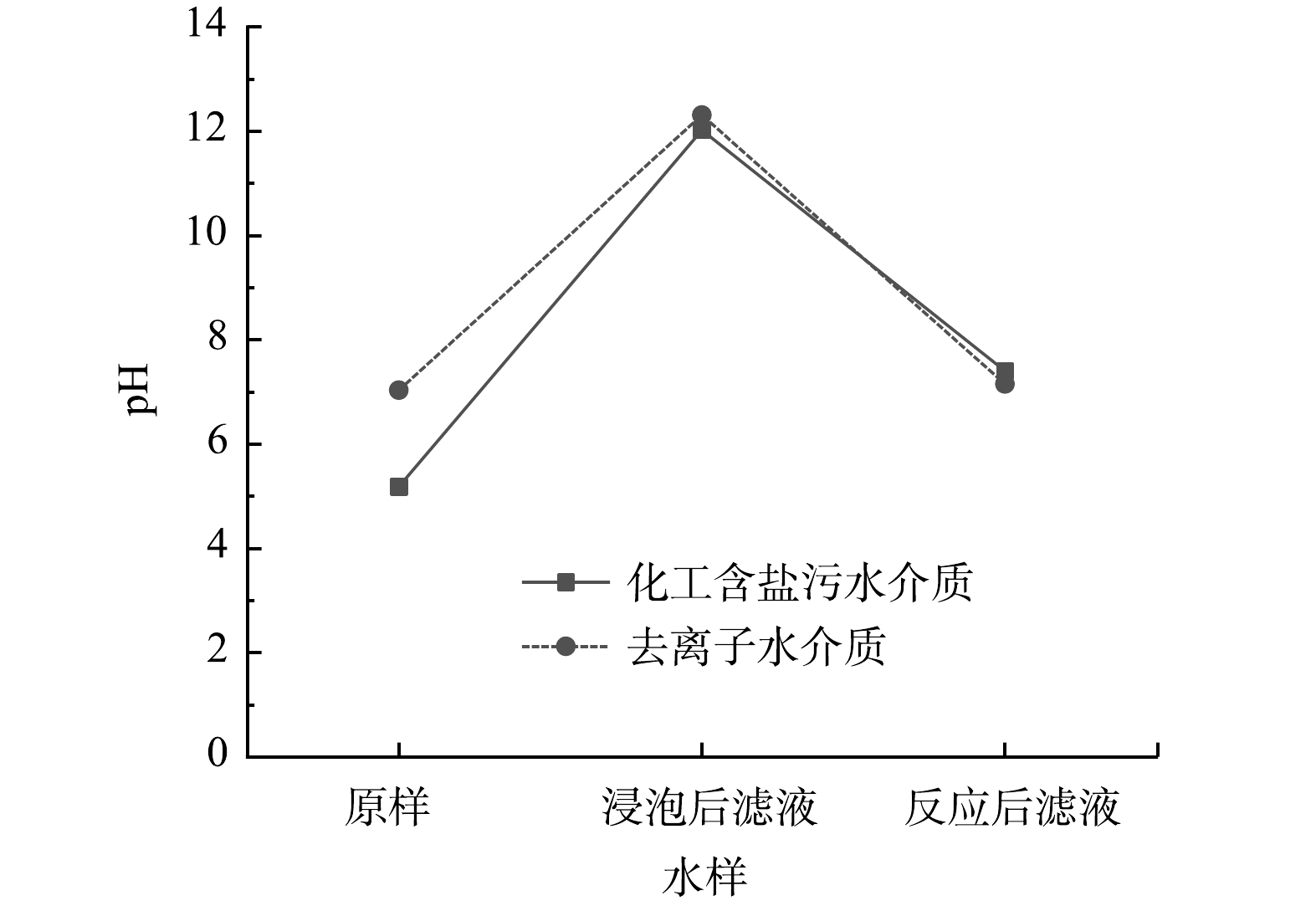 图 3 湿法矿化过程水样pH变化Figure 3. Change of pH value of water samples during wet mineralization
图 3 湿法矿化过程水样pH变化Figure 3. Change of pH value of water samples during wet mineralization对矿化过程中不同阶段的2种水样进行pH测试,其pH变化如图3所示。可以看出,2种水样pH不同,但是在反应过程中的pH变化趋势基本一致,反应前化工含盐污水pH为5.18,去离子水pH为7.03;加入电石渣、加热至实验温度并搅拌后,化工含盐污水滤液pH升至12.02,去离子水滤液pH为12.31,pH的升高源于电石渣中碱性物质如Ca(OH)2发生电离,产生大量OH−和Ca2+;通入CO2后,由于CO2与溶液中OH−和Ca2+ (或Mg2+) 发生矿化反应,OH−的消耗使溶液碱性降低,化工含盐污水滤液和去离子水滤液pH值分别下降至7.40和7.15。
2.2 化工含盐污水协同电石渣矿化封存CO2反应特性
1) 温度对电石渣CO2封存率的影响。CO2封存率随温度变化关系如图4 (a) 所示。由图可见,随着温度的升高,CO2封存率先增大后减小,在65 ℃达到最大封存率47.56%。随着温度升高,分子运动会随之变得剧烈,电石渣中钙更容易浸出形成Ca2+溶解于液相中,但是温度的升高会降低CO2气体在溶液中的溶解度。在65 ℃之前,Ca2+的浸出是反应控制步骤,温度升高,Ca2+的浸出量增大,CO2封存率升高。在温度高于65 ℃后,CO2的溶解是反应控制步骤,随着温度升高,CO2气体在溶液中的溶解度下降[29],导致封存率下降。矿化反应为放热反应,温度升高,反应平衡向左移动,也会导致封存率下降。
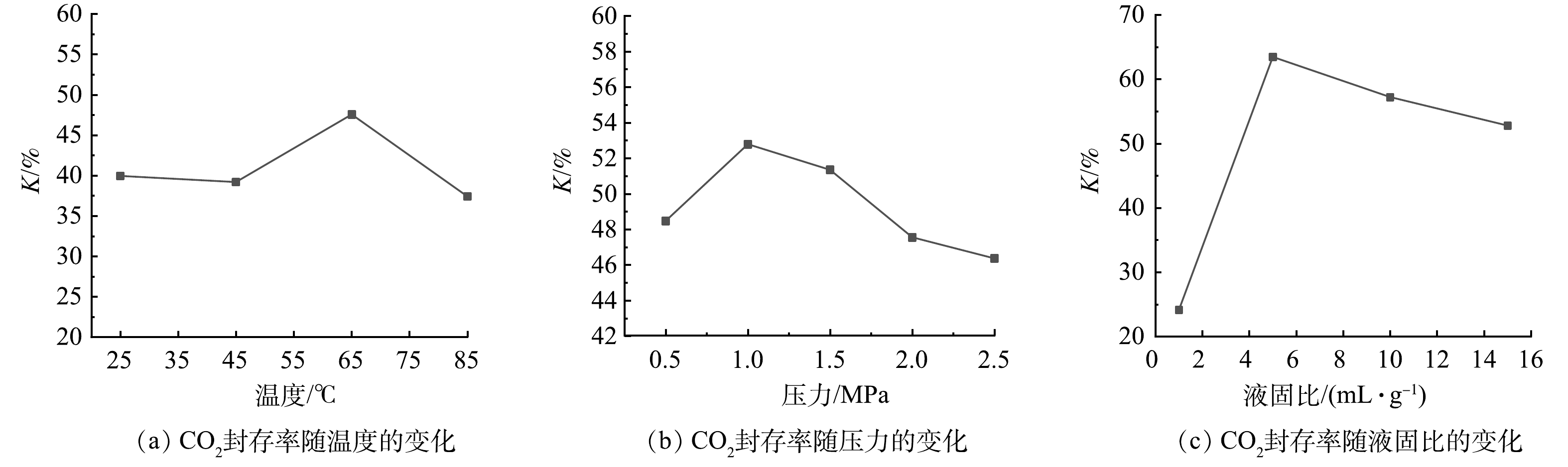 图 4 CO2封存率随不同因素的变化关系Figure 4. Variation of CO2 sequestration rate K with different factors
图 4 CO2封存率随不同因素的变化关系Figure 4. Variation of CO2 sequestration rate K with different factors2) 压力对电石渣CO2封存率的影响。CO2封存率随初始压力变化关系如图4 (b) 所示。由图可见,CO2封存率随初始压力的变化较小,随着初始压力的升高,CO2封存率先增大后减小,在压力为1.0 MPa时K达到最大封存率52.79%。压力主要对CO2在溶液中的溶解度产生影响。在一定的温度下,溶液中CO2的溶解量与压力成正比,压力差越大,推动力越大,CO2在溶液中的溶解量越大[30]。随着压力从0.5 MPa增大到1.0 MPa,CO2在溶液中的溶解量增大,产生的CO32+离子增加,CO2封存率增大。当压力从1.0 MPa增大到2.5 MPa时,溶液中的CO2质量浓度继续增加,与化工含盐污水中的Ca2+快速反应,生成的CaCO3颗粒附着于电石渣表面,阻碍了电石渣中Ca的浸出[31],同时干扰CO2气体在电石渣中的扩散,限制反应的进行[32] 。
3) 液固比对电石渣CO2封存率的影响。CO2封存率液固比变化关系如图4 (c) 所示。由图可见,随着液固比的升高,CO2封存率先增大后减小,在液固比为5 mL·g−1时K达到最大63.47%。溶液中的Ca2+、CO32−质量浓度对矿化反应过程有很大的影响,而液固比会影响电石渣中碱性物质和CO2在水中的溶解程度,进而影响溶液中Ca2+、CO32−质量浓度及矿化封存CO2性能[33]。在液固比小于5 mL·g−1时,虽然提供了电石渣中碱性物质与气体发生反应的液体介质,但是液固比太小,不足使电石渣中钙充分浸出,CO2在水中的溶解量也较少。当液固比达到一定值时,电石渣中碱性物质溶解基本完全,继续增大液固比,碱性物质的溶解量不再增加,相反,液体的增加会使溶液中Ca2+质量浓度下降,进而矿化反应平衡向左移动,封存量降低。
2.3 化工含盐污水协同电石渣湿法矿化封存CO2工艺条件优化
1) 模型建立与检验。根据单因素影响的实验结果设计响应面曲线工业条件优化实验,实验结果如表3所示。
表 3 响应面实验结果Table 3. Experimental results of response surface methodology实验编号 A/ ℃ B /MPa C/(mL·g−1) K/% 1 45 0.5 5 62.6 2 85 0.5 5 63.1 3 45 1.5 5 63.9 4 85 1.5 5 56.0 5 45 1.0 2 59.8 6 85 1.0 2 36.0 7 45 1.0 8 51.9 8 85 1.0 8 56.4 9 65 0.5 2 47.5 10 65 1.5 2 51.1 11 65 0.5 8 64.9 12 65 1.5 8 59.2 13 65 1.0 5 62.4 14 65 1.0 5 63.6 15 65 1.0 5 61.7 注:A—温度;B—初始压力;C—液固比。 | Show TableDownLoad:
CSV
根据实验结果拟合模型,由Design-Expert软件拟合出经验模型如式 (4) 所示。模型的P值为0.008 722<0.05,意味着预测模型是显著的。
stringUtils.convertMath(!{formula.content}) (4) 2) 因素交互作用。采用响应面曲线法研究3个变量之间的交互作用,图5为CO2封存率的响应面图和等高线图。图5 (a) 、图5 (b) 表示,温度和压力之间的交互作用对CO2封存率的影响,在液固比不变的情况下,温度与压力之间的交互作用较小,压力的改变对CO2封存率的影响很微弱,这也与单因素试验的结果吻合。图5 (c) 、图5 (d) 为压力一定时温度与液固比的交互作用对CO2封存率的影响,由于温度和液固比的影响,CO2封存率增加,等高线的形状为椭圆形,说明温度和液固比的交互作用显著,是影响封存率最显著的参数之一。图5 (e) 、图5 (f) 为温度一定时压力与液固比的交互作用CO2封存率的影响,三维图基本呈拱形,说明液固比对于反应的影响更加明显,而2者的交互作用对于CO2的封存来说并不显著。
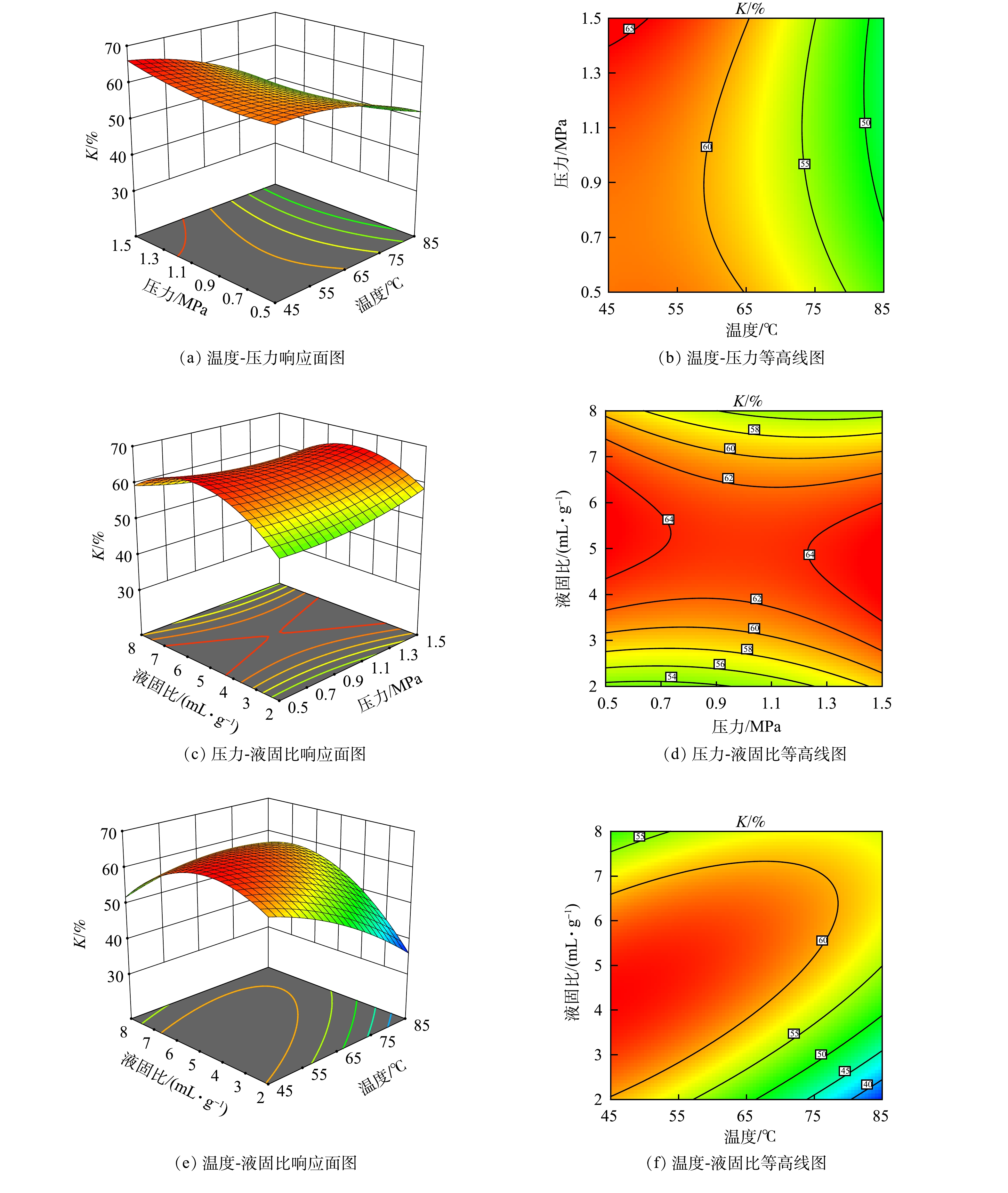 图 5 CO2封存率响应面曲线和等高线图Figure 5. Response surface plot and contour plot of CO2 sequestration rate
图 5 CO2封存率响应面曲线和等高线图Figure 5. Response surface plot and contour plot of CO2 sequestration rate3) 最佳工艺条件。基于响应面曲线拟合结果,确定CO2封存率最大的反应条件为:85 ℃、0.5 MPa、液固比7.5 mL·g−1,在此条件下,模型预测值为66.9%。同时,在上述条件下进行含盐污水协同电石渣湿法矿化封存CO2实验,3次平行实验所得CO2封存率的平均值为66.1%,电石渣实际固碳量为661 g·kg−1,实验值与预测值的误差小于1.5%。因此,通过上述方法可高效准确获得矿化封存CO2实验的最佳工艺条件。
3. 结论
1) 化工含盐污水和去离子水为介质的电石渣矿化封存CO2封存率分别为59.59%和59.89%,基本持平,可以使用化工含盐污水代替去离子水作为反应介质。
2) 温度和液固比对CO2封存率的影响较大,压力影响较小。单因素优化实验条件下,在温度65 ℃、压力1.0 MPa和液固比5 mL·g−1时CO2封存率分别达到47.56%、52.79%和63.47%。
3) 温度和液固比之间的交互作用最为显著,温度和压力、液固比和压力的交互作用不显著。同时确定在反应温度85 ℃、初始反应压力0.5 MPa、液固比7.5 mL·g−1的最优工艺条件下,含盐污水协同电石渣矿化的CO2封存率为66.1%。
-
图 1 湿法矿化反应前后热重 (TG) 曲线
Figure 1. Thermogravimetric (TG) curves before and after wet mineralization
图 3 湿法矿化过程水样pH变化
Figure 3. Change of pH value of water samples during wet mineralization
图 4 CO2封存率随不同因素的变化关系
Figure 4. Variation of CO2 sequestration rate K with different factors
图 5 CO2封存率响应面曲线和等高线图
Figure 5. Response surface plot and contour plot of CO2 sequestration rate
表 1 反应前后溶液组分变化
Table 1. Change of solution composition before and after reaction
mg·L−1 样品 Cl− NO3− SO42 Ca2+ K+ Mg2+ Na+ 化工含盐污水 4 484 102 3 565 897 231 525 1 927 反应后滤液 4 087 93 617 556 171 221 617
下载: 导出CSV
表 2 反应前后电石渣化学组成
Table 2. Chemical composition of calcium carbide slag before and after reaction
% 样品 Na2O MgO Al2O3 SiO2 SO3 Cl CaO TiO2 Fe2O3 SrO K2O NiO 反应前电石渣 0.020 0.170 0.950 2.700 0.470 — 90.900 — 0.170 — — — 反应后电石渣 0.549 0.523 0.658 1.900 1.990 0.615 93.100 0.072 0.192 0.079 0.102 0.177
下载: 导出CSV
表 3 响应面实验结果
Table 3. Experimental results of response surface methodology
实验编号 A/ ℃ B /MPa C/(mL·g−1) K/% 1 45 0.5 5 62.6 2 85 0.5 5 63.1 3 45 1.5 5 63.9 4 85 1.5 5 56.0 5 45 1.0 2 59.8 6 85 1.0 2 36.0 7 45 1.0 8 51.9 8 85 1.0 8 56.4 9 65 0.5 2 47.5 10 65 1.5 2 51.1 11 65 0.5 8 64.9 12 65 1.5 8 59.2 13 65 1.0 5 62.4 14 65 1.0 5 63.6 15 65 1.0 5 61.7 注:A—温度;B—初始压力;C—液固比。
下载: 导出CSV
-
[1] KAPILA S, ONI A, GEMECHU E, et al. Development of net energy ratios and life cycle greenhouse gas emissions of large-scale mechanical energy storage systems[J]. Energy, 2019, 170: 592-603. doi: 10.1016/j.energy.2018.12.183 [2] MYSIAK J, SURMINSKI S, THIEKEN A, et al. Brief communication: Sendai framework for disaster risk reduction – success or warning sign for Paris?[J]. Natural Hazardsand Earth SystemSciences, 2016, 16(10): 2189-2193. doi: 10.5194/nhess-16-2189-2016 [3] NYAMBURA M G, MUGERA G W, FELICIA P L, et al. Carbonation of brine impacted fractionated coal fly ash: implications for CO2 sequestration[J]. Journal of environmental management, 2011, 92(3): 655-664. doi: 10.1016/j.jenvman.2010.10.008 [4] 王丹. 二氧化碳捕集、利用与封存技术全链分析与集成优化研究[D]. 北京: 中国科学院大学(中国科学院工程热物理研究所), 2020. [5] NORHASYIMA R S, MAHLIA T. Advances in CO2 utilization technology: A patent landscape review[J]. Journal of CO2 Utilization, 2018, 26: 323-335. doi: 10.1016/j.jcou.2018.05.022 [6] CUéLLAR-FRANCA R, AZAPAGIC A. Carbon capture, storage and utilisation technologies: A critical analysis and comparison of their life cycle environmental impacts[J]. Journal of CO2 Utilization, 2015, 9(1): 82-102. [7] 王建行, 赵颖颖, 李佳慧, 等. 二氧化碳的捕集, 固定与利用的研究进展[J]. 无机盐工业, 2020, 52(4): 6. [8] PANDEY S, SRIVASTAVA V C, KUMAR V. Comparative thermodynamic analysis of CO2 based dimethyl carbonate synthesis routes[J]. The Canadian Journal of Chemical Engineering, 2021, 99(2): 467-478. doi: 10.1002/cjce.23893 [9] WANG F, DREISINGER D, JARVIS M, et al. Quantifying kinetics of mineralization of carbon dioxide by olivine under moderate conditions[J]. Chemical Engineering Journal, 2019, 360: 452-463. doi: 10.1016/j.cej.2018.11.200 [10] BOBICKI E R, LIU Q, XU Z, et al. Carbon capture and storage using alkaline industrial wastes[J]. Progress in Energy and Combustion Science, 2012, 38(2): 302-320. doi: 10.1016/j.pecs.2011.11.002 [11] ZHANG Z, PAN S Y, LI H, et al. Recent advances in carbon dioxide utilization[J]. Renewable and Sustainable Energy Reviews, 2020, 125: 109799. doi: 10.1016/j.rser.2020.109799 [12] 武鸽, 刘艳芳, 崔龙鹏, 等. 典型工业固体废物碳酸化反应性能的比较[J]. 石油学报(石油加工), 2020, 36(1): 169-178. [13] 张亚朋, 崔龙鹏, 刘艳芳, 等. 3种典型工业固废的CO2矿化封存性能[J]. 环境工程学报, 2021, 15(7): 2344-2355. doi: 10.12030/j.cjee.202101003 [14] ROMANOV V, SOONG Y, CARNEY C, et al. Mineralization of Carbon Dioxide: A Literature Review[J]. ChemBioEng Reviews, 2015, 2(4): 231-256. doi: 10.1002/cben.201500002 [15] 刘展, 郭瑞亚, 李娜, 等. 高含盐废水资源化利用技术的研究进展[J]. 应用化工, 2020, 49(10): 2657-2661. doi: 10.3969/j.issn.1671-3206.2020.10.055 [16] MUSTAFA J, MOURAD A A H I, AL-MARZOUQI A H, et al. Simultaneous treatment of reject brine and capture of carbon dioxide: A comprehensive review[J]. Desalination, 2020, 483: 114386. doi: 10.1016/j.desal.2020.114386 [17] LI Y, PEI S, PAN S Y, et al. Carbonation and utilization of basic oxygen furnace slag coupled with concentrated water from electrodeionization[J]. Journal of CO2 Utilization, 2018, 25: 46-55. doi: 10.1016/j.jcou.2018.03.003 [18] BANG J H, YOO Y, LEE S W, et al. CO2 mineralization using brine discharged from a seawater desalination plant[J]. Minerals, 2017, 7(11): 207. doi: 10.3390/min7110207 [19] MIGNARDI S, DE V C, FERRINI V, et al. The efficiency of CO2 sequestration via carbonate mineralization with simulated wastewaters of high salinity[J]. Journal of Hazardous Materials, 2011, 191(1): 49-55. [20] BEHBAHANI M, MOGHADDAM M R A, ARAMI M. Techno-economical evaluation of fluoride removal by electrocoagulation process: Optimization through response surface methodology[J]. Desalination, 2011, 271(1-3): 209-218. doi: 10.1016/j.desal.2010.12.033 [21] KHEDMATI M, KHODAII A, HAGHSHENAS H F. A study on moisture susceptibility of stone matrix warm mix asphalt[J]. Construction and Building Materials, 2017, 144: 42-49. doi: 10.1016/j.conbuildmat.2017.03.121 [22] ZHANG L, ZENG Y, CHENG Z. Removal of heavy metal ions using chitosan and modified chitosan: A review[J]. Journal of Molecular Liquids, 2016, 214: 175-191. doi: 10.1016/j.molliq.2015.12.013 [23] WANG B, PAN Z, CHENG H, et al. A review of carbon dioxide sequestration by mineral carbonation of industrial byproduct gypsum[J]. Journal of Cleaner Production, 2021, 302: 126930. doi: 10.1016/j.jclepro.2021.126930 [24] 任国宏, 廖洪强, 吴海滨, 等. 粉煤灰、电石渣及其配合物碳酸化特性[J]. 环境工程学报, 2018, 12(08): 2295-2300. doi: 10.12030/j.cjee.201803119 [25] 颜鑫, 魏义兰. 含钙镁废渣综合利用的现状及展望[J]. 无机盐工业, 2022, 54(1): 7-11. doi: 10.19964/j.issn.1006-4990.2021-0227 [26] 赵立文, 朱干宇, 李少鹏, 等. 电石渣特性及综合利用研究进展[J]. 洁净煤技术, 2021, 27(3): 13-26. doi: 10.13226/j.issn.1006-6772.21010601 [27] PAN S, CHANG E, CHIANG P C. CO2 capture by accelerated carbonation of alkaline wastes: a review on its principles and applications[J]. Aerosol Air Quality Research, 2012, 12(5): 770-791. doi: 10.4209/aaqr.2012.06.0149 [28] SOONG Y, FAUTH D L, HOWARD B H, et al. CO2 sequestration with brine solution and fly ashes[J]. Energy conversion and Management, 2006, 47(13-14): 1676-1685. doi: 10.1016/j.enconman.2005.10.021 [29] NI P, XIONG Z, TIAN C, et al. Influence of carbonation under oxy-fuel combustion flue gas on the leachability of heavy metals in MSWI fly ash[J]. Waste Management, 2017, 67: 171-180. doi: 10.1016/j.wasman.2017.05.023 [30] 倪鹏. 垃圾焚烧飞灰矿化解毒一体化的研究[D]. 武汉: 华中科技大学, 2018. [31] BACIOCCHI R, COSTA G, DI BARTOLOMEO E, et al. The effects of accelerated carbonation on CO2 uptake and metal release from incineration APC residues[J]. Waste Management, 2009, 29(12): 2994-3003. doi: 10.1016/j.wasman.2009.07.012 [32] UKWATTAGE N L, RANJITH P, YELLISHETTY M, et al. A laboratory-scale study of the aqueous mineral carbonation of coal fly ash for CO2 sequestration[J]. Journal of Cleaner Production, 2015, 103: 665-674. doi: 10.1016/j.jclepro.2014.03.005 [33] HO H J, IIZUKA A, SHIBATA E. Utilization of low-calcium fly ash via direct aqueous carbonation with a low-energy input: Determination of carbonation reaction and evaluation of the potential for CO2 sequestration and utilization[J]. Journal of Environmental Management, 2021, 288: 112411. doi: 10.1016/j.jenvman.2021.112411 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4789
- HTML全文浏览数: 4789
- PDF下载数: 47
- 施引文献: 0
