



-
城市生活垃圾是影响生态环境的严重污染源,其无害化处理已经成为环境治理的重要问题之一[1]。根据国家统计局发布的《2021中国统计年鉴》[2],我国2020年生活垃圾清运量达到963 460 t·d−1,其中焚烧日处理量已达567 804 t·d−1,占总垃圾无害化处理量的58.9%。生活垃圾进入焚烧炉前通常需要采用堆酵5~7 d的方式进行熟化,降低含水率,从而提高垃圾热值[3-4]。垃圾焚烧厂渗滤液是一种水质复杂且有毒有害的高浓度有机废水,具有一定的处理难度。如果收集和处理处置不当,将会对周边自然环境和人体健康造成严重影响。已有研究[5-6]表明,采用厌氧膜生物反应器 (anaerobic membrane bioreactor,AnMBR) 和负压原位碱度脱氨工艺处理垃圾焚烧厂渗滤液可分别实现COD和氨氮的高效脱除。然而出水水质依然无法满足相应的排放标准,因此还需要对脱氨出水进一步深度处理。当前采用的主流深度处理工艺为NF、RO等,存在运行能耗较大,膜污染严重以及残留大量难处理浓缩液等问题[7]。
与以压力作为驱动力的NF、RO等工艺相比,正渗透 (forward osmosis,FO) 工艺是一种以膜两侧的渗透压差作为驱动力的膜处理工艺,具有能耗低、膜污染小、出水水质好、浓缩液少等优势[8]。目前,针对正渗透深度处理垃圾焚烧厂渗滤液的研究较少,主要集中于垃圾填埋场渗滤液处理。AFTAB等[9]采用CTA膜组件的正渗透工艺直接对垃圾填埋场渗滤液原水进行处理,并研究生物炭 (BC) 和粉末活性炭 (PAC) 在线吸附减轻膜污染对正渗透性能的影响,结果表明,加入在线吸附后,过滤体积分数为57%以上,污染物截留率>80%。IBRAR等[10]研究了不同清洗方式对正渗透处理垃圾填埋场渗滤液生化出水膜性能的影响,结果表明物理清洗方法中35 ℃热水物理清洗和1.5 mol NaCl渗透反冲洗具有较好效果;化学清洗方法中,碱洗比酸洗更有效,可以达到100%通量回收。ISKANDER等[11]对正渗透回收垃圾填埋场渗滤液进行能耗分析,结果表明,污染物浓度升高,循环次数降低,汲取液浓度提高,可以使能耗从 (0.276±0.033) kWh·m−3下降到 (0.005±0.000) kWh·m−3。以上研究表明,正渗透作为垃圾焚烧厂渗滤液深度处理具有一定的研究价值和应用潜力。在应用FO处理垃圾渗滤液方面的报道主要集中于处理垃圾填埋场渗滤液,在处理垃圾焚烧渗滤液方面的研究却鲜有报道,在FO处理垃圾焚烧渗滤液的运行效能、工艺条件以及膜污染特性方面的研究较少,因此,采用FO工艺深度处理垃圾焚烧厂渗滤液负压原位碱度脱氨出水具有重要的理论和现实意义。
本研究采用FO工艺深度处理垃圾焚烧厂渗滤液负压原位碱度脱氨出水,考察FO在不同膜朝向、不同汲取液浓度和不同错流速率下的浓缩效果和污染物截留率,在满足相关排放标准的情况下确定最佳运行参数,进行连续实验并分析FO处理负压原位碱度脱氨出水的可行性,利用三维荧光 (EEM) 结合平行因子分析方法 (PARAFAC) 对膜污染成分和膜污染特征进行分析,旨在为后续FO处理垃圾焚烧渗滤液方面的研究与应用提供参考。
-
研究所用废水取自无锡某垃圾焚烧发电厂,经AnMBR和负压原位碱度脱氨工艺处理后,使用0.45 μm滤膜过滤并在4 ℃下保存,进水水质指标如下:COD为1 950~2 050 mg·L−1,氨氮为56.5~63.1 mg·L−1,总氮质量浓度为64.6~72.2 mg·L−1,总磷质量浓度为2.1~2.3 mg·L−1,电导率为19.2~19.8 mS·cm−1,Ca2+质量浓度为12.1~12.9 mg·L−1,Mg2+质量浓度为19.2~20.0 mg·L−1。在正渗透操作前,将原料液pH调节到8左右,汲取液为配置不同摩尔浓度的NaCl溶液。
-
本研究采用的实验室规模正渗透实验装置如图1所示。主要由汲取液罐、原料液罐、正渗透膜组件、蠕动泵、温度控件和电导率仪组成。汲取液罐、原料液罐容量均为1.5 L;通过温度监测装置在线监测汲取液和原料液温度变化;两侧液体分别从汲取液罐和原料液罐底部流出,在2台蠕动泵 (上海卡川尔流体有限公司,UIP WIFI-S183,中国) 的驱动下在膜两侧进行独立循环,2台蠕动泵转速相同,流量相同,形成稳定的错流循环;通过罐体表面的刻度实时读取两侧液面变化;通过电导率仪实时监测汲取液浓度变化,控制循环泵使浓盐水进入汲取液罐,保持电导率不变。
正渗透膜组件由2块有机玻璃材质的内凹膜室组成,膜室之间夹有一片正渗透膜,正渗透膜组件内置CTA膜 (HTI,美国) ,是由活性层和支撑层组成的双层结构。CTA膜将膜组件隔开为2个独立膜室。膜组件规格为:长20 cm,宽11 cm,厚5 cm,单个膜室体积为28.4 cm3。膜有效面积为94.5 cm2,2个膜室内部设有均匀布水板,以提高正渗透效率。实验前将CTA膜放入蒸馏水在4 ℃下保存。
-
实验分为4个部分。第1部分,研究膜朝向对正渗透浓缩性能和膜通量恢复情况的影响。实验时改变运行过程中CTA膜活性层的朝向,2个朝向分别为活性层朝向汲取液 (AL-DS) 模式和活性层朝向原料液 (AL-FS) 模式。保持温度不变,错流速率为8 cm·s−1。每15 min读取一次原料液体积变化并计算膜通量。当连续2次原料液体积变化值均小于5 mL时结束实验。第2部分,研究汲取液浓度对正渗透浓缩性能的影响。分别设置5个汲取液浓度水平,考察膜通量变化情况和通量恢复情况。第3部分,研究错流速率对正渗透运行过程中膜通量的影响。在选定最适汲取液浓度前提下,将错流速率分别设置为4 、8 、12 cm·s−1。第4部分,研究连续实验对正渗透效果影响。实验过程中保持汲取液电导率不变,每30 min记录一次原料液体积变化,每12 h为一次循环,每次循环期间需对原料液进行一次更换,并对膜进行在线物理清洗。清洗方式为将剩余原料液更换为去离子水,其他条件不改变,继续运行30 min。在第3个循环结束后,进行在线化学清洗。在线化学清洗方式步骤如下:将剩余原料液先后更换为0.01 mol·L−1 NaOH (清洗15 min) 和0.01 mol·L−1 HCl (清洗15 min) ,再更换为去离子水,运行5 min,清洗表面残留化学药剂。化学清洗后,再进行一次循环,观察膜通量恢复情况。
-
COD采用重铬酸钾法测定,用硫酸汞掩蔽Cl−;氨氮、总氮、总磷、电导率参照文献中的方法[12]进行测定:氨氮、总氮和总磷采用纳氏试剂光度法进行测定,电导率采用水质分析仪 (奥豪斯AB23EC) 进行测定;采用荧光分光光度仪 (F-7000) 对所有膜清洗水样进行EEM测定;膜污染分析采用MATLAB 2017b软件和DOM Flour工具箱对所有样品的EEM数据集进行PARAFAC平行因子分析 (parallel factor analysis,PARAFAC) 建模;纯水通量JW的测定可参考文献中的方法[9]。
-
1) 膜朝向对FO运行效能的影响。采用AL-FS和AL-DS 2种不同的膜朝向进行正渗透实验,通量变化曲线如图2所示。AL-DS模式下的初始通量为17.55 L·(m2·h)−1,而AL-FS模式下的初始通量仅为14 L·(m2·h)−1。此现象与VU等[13]的研究相似,主要原因是支撑层产生的内浓差极化类型不同:在AL-DS模式下,影响膜通量的主要因素是浓缩型内浓差极化,而AL-FS模式下主要为稀释型内浓差极化。二者相比,浓缩型浓差极化对水通量的影响较小[14],所以AL-DS模式下的初始通量大于AL-FS模式下的初始通量。ZHAO等[15]认为,AL-DS模式初始通量更好,这也可归因于膜表面与污染物的相互作用,由于CTA膜支撑层比活性层带有更多的负电荷,导致膜表面与垃圾渗滤液污染物之间的静电排斥更大。AL-FS模式在实验过程中的膜通量较为平稳,而AL-DS模式下的膜通量下降速度明显,在第6小时,下降到 14 L·(m2·h)−1以下。这归因于AL-DS模式下支撑层朝向原料液,粗糙度更高的支撑层表面使膜污染程度更高,从而导致通量的快速下降。
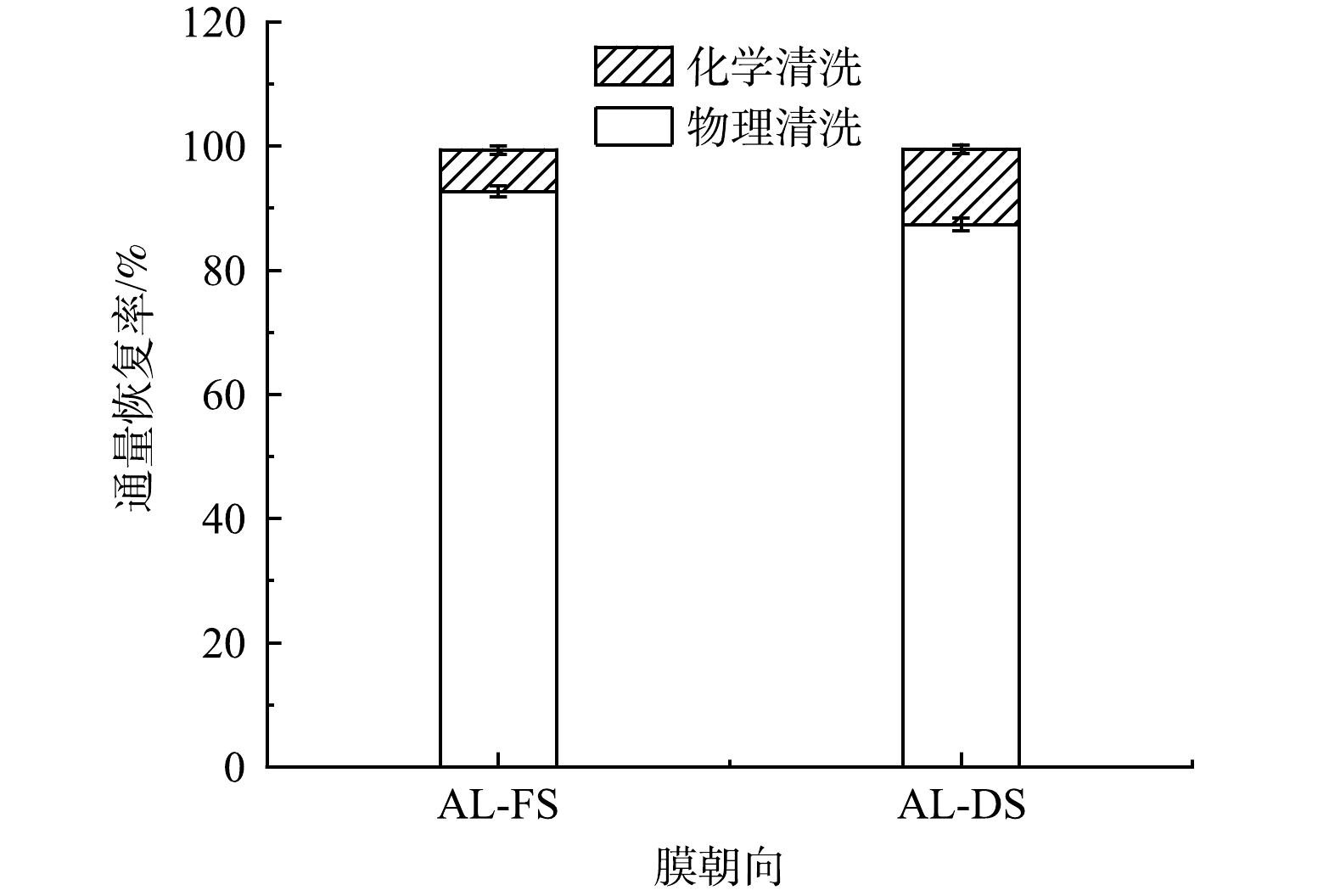
对不同膜朝向实验后的FO膜分别进行物理清洗和化学清洗,通量恢复情况如图3所示。AL-FS和AL-DS模式的水通量经过物理清洗后分别为初始纯水通量的86.35%和81.50%,经过化学清洗后水通量均恢复到98%以上。结果表明,AL-DS模式下不仅会加速膜污染,从而导致通量迅速下降,物理清洗效果也相对较差,膜污染更难去除。IBRAR等[10]对不同膜朝向运行的正渗透膜进行去离子水冲洗,发现AL-DS模式下的膜表面残留有更多的污染物,进一步证实了AL-DS模式下物理清洗效果相对较差,膜污染更难去除。因此,综合通量变化和清洗后通量恢复情况,确定后续实验以AL-FS模式运行。
2) 不同汲取液浓度对FO运行效能的影响。正渗透的动力来源为汲取液和原料液间产生的渗透压差,因此汲取液浓度对正渗透系统的运行效率和浓缩性能具有重大影响。在两侧水温为25 ℃、错流速率为8 cm·s−1的条件下,分别考察1.0、1.5、2.0、2.5、3.0 mol·L−1 5组不同NaCl汲取液浓度条件下的膜通量随时间的变化情况,结果如图4所示。由于初始原料液侧浓缩倍数较低,膜两侧的渗透压差变化不明显,且此时膜污染程度较轻,故膜通量下降不明显;而随着原料液不断被浓缩导致渗透压差减少和膜污染的加剧,膜通量急剧下降。因此,各组不同汲取液浓度条件下的膜通量均呈先缓慢下降后迅速下降的趋势。由各组比较分析结果可以看出,随着汲取液浓度的上升,初始通量也呈现逐渐增加的趋势。在汲取液为1.0 mol·L−1 NaCl的条件下,初始通量仅为7.41 L·(m2·h)−1;而当NaCl浓度提升到3.0 mol·L−1时,初始通量为18.62 L·(m2·h)−1。此外,汲取液浓度越高,膜通量的稳定运行时间越短,迅速下降的时间越快,在汲取液浓度为1.0 mol·L−1条件下,膜通量在13.5 h时出现明显下降;而当汲取液浓度提升到3.0 mol·L−1时,膜通量在3.5 h便出现迅速下降趋势。结果表明,较高的汲取液浓度使膜两侧渗透压差增大,驱动力增大,从而带来更高的初始通量,但由此导致渗透压差迅速降低,稳定运行时间缩短,迅速下降的拐点更早出现。同时,汲取液浓度提高也会导致膜污染的迅速累积。方舟[16]研究表明,高汲取液浓度会通过加快原料液中污染物在膜表面的累积速率从而加剧膜污染;与此同时,汲取液浓度的提高也会使内浓差极化现象更加严重,而膜表面污染层的积累又会进一步加深内浓差极化现象,导致更严重的膜污染。
原料液的浓缩倍数是表征正渗透浓缩性能的重要指标,可间接表征运行过程中的膜处理量,在工程应用中具有重要意义。若浓缩倍数过低,则说明浓缩不彻底,浓缩性能较差,在处理过程中将产生大量浓水,提高后续处理成本。不同汲取液浓度下原料液的浓缩倍数情况如图5所示。随着汲取液浓度从1.0 mol·L−1上升到2.0 mol·L−1,原料液的浓缩倍数从2.93倍迅速上升到10.00倍。由于垃圾焚烧厂渗滤液脱氨出水具有较高的电导率,随着浓缩倍数的提高,两侧的渗透压差会迅速降低,从而导致浓缩过程缺少推动力,膜通量迅速下降,在浓度为1.0~2.0 mol·L−1时,提高汲取液浓度可以明显提升正渗透浓缩性能。在汲取液浓度为2.0~3.0 mol·L−1时,浓缩倍数没有出现明显变化,说明此范围内汲取液浓度继续提升对于单次浓缩过程中的膜处理量没有进一步的促进作用。由图4可以看出,随着汲取液浓度从1.0 mol·L−1上升到3.0 mol·L−1,浓缩所耗时间分别为16、13、11、11 和9 h。这表明,汲取液浓度在2.0 mol·L−1之后,浓度进一步提高,对膜处理量没有促进作用,但可以在一定程度上减少浓缩时间,使正渗透过程更快完成。
通量恢复率计算方法见式 (1) 。
式中:η为通量恢复率;J为物理清洗和化学清洗后的通量;JW为清水通量。
如图6所示,随着汲取液浓度的上升,物理清洗后的通量恢复率η呈现缓慢下降趋势,由91.36%降至83.68%;化学清洗后各组的通量恢复率η均为96%~97%,各组之间的η数值变化不大。这说明正渗透具有较低的膜污染趋势,膜表面的污染层与膜表面的结合并不紧密,仅靠物理清洗就可以恢复绝大部分的膜通量。通过化学清洗,不可恢复通量仅为初始通量的3%~4%。这说明正渗透过程中产生的不可逆污染水平较低,绝大部分污染为可逆污染,与其他膜处理工艺中产生的膜污染相比,正渗透过程中产生的膜污染更易控制,具有更高的运行寿命。通量恢复情况分析不仅可反映正渗透处理垃圾焚烧厂渗滤液脱氨出水后的膜污染情况方面,也可表征正渗透工艺的运行寿命及其应用中的可行性。
3) 不同错流速率对FO运行效能的影响。错流速率是正渗透工艺运行的重要参数之一,低错流速率会导致外部浓差极化上升,影响膜通量,同时导致膜污染更容易形成。在汲取液浓度为2 mol·L−1、温度为25 ℃的条件下,控制不同错流速率 (4、8、12 cm·s−1) 进行单轮正渗透实验,分析膜通量变化情况,实验结果如图7所示。在错流速率为4 cm·s−1时,初始膜通量仅为12.69 L·(m2·h)−1,且随着系统运行膜通量逐步减少,导致单次浓缩时间延长,在第16 小时,膜通量为1.69 L·(m2·h)−1,浓缩仍未结束。错流速率为8 cm·s−1时,初始通量显著上升至14.81 L·(m2·h)−1,呈先稳定运行后迅速下降趋势。且单次浓缩在11 h完成。错流速率进一步提升至12 cm·s−1时,膜通量变化情况与8 cm·s−1条件下的膜通量变化没有明显区别。实验结果表明,错流速率为4 cm·s−1时,速率过低,不利于正渗透的运行。这可能是由于错流速率下降,水流速率变慢,导致对膜的冲刷作用减弱,使得膜污染更容易形成。当错流速率提升至8 cm·s−1后,较高的错流速率增强了膜面冲刷,减缓了膜污染,从而使得初始通量升高且单次浓缩时间缩短。当进一步提高错流速率至12 cm·s−1时,对膜通量并没有明显的促进作用,说明错流速率的提高对膜通量的提升是有限度的。故在后续实验中,将错流速率控制在8 cm·s−1条件下运行。
-
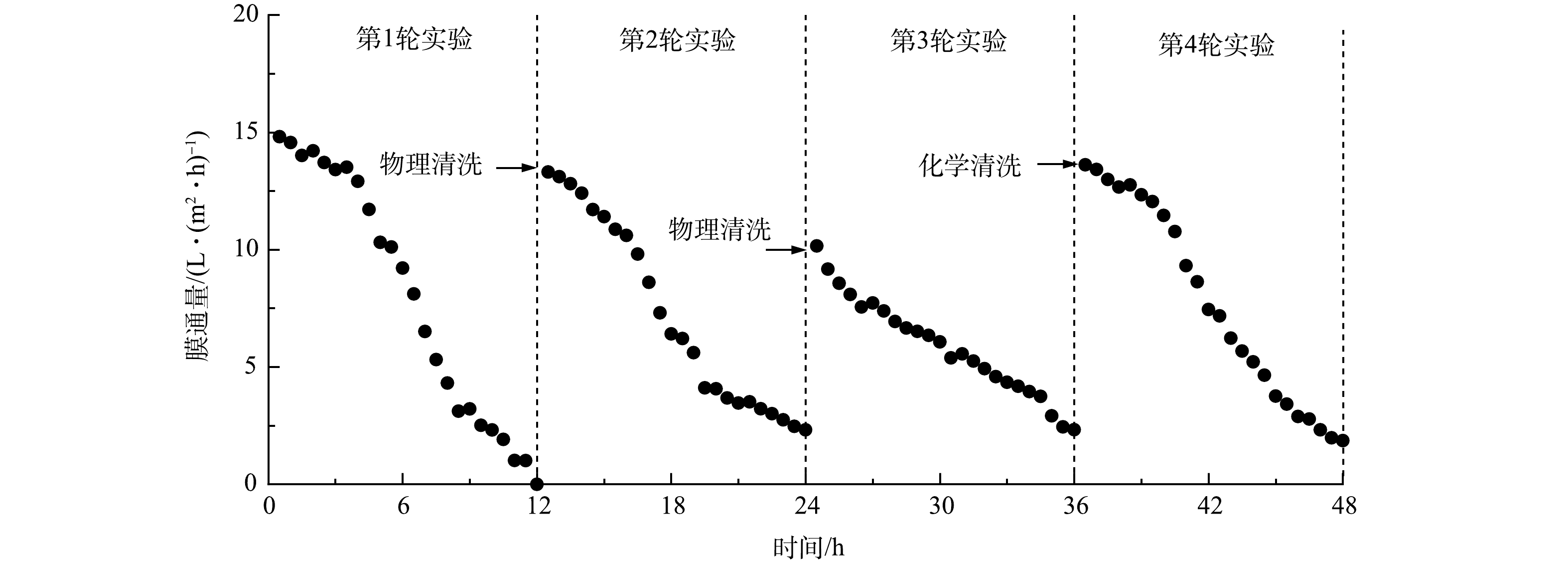
为进一步探究连续运行过程中的浓缩性能和出水水质变化,在汲取液浓度为2 mol·L−1、错流速率为8 cm·s−1的条件下,进行为期36 h的连续实验。实验共分为3轮,每轮实验结束后更换原料液,并对FO膜进行物理清洗。第3轮实验结束后,为探究化学清洗对膜通量的恢复情况,采用化学清洗并再进行一轮实验。连续运行的实验结果如图8所示。
在第1轮运行过程中,初始通量为14.81 L·(m2·h)−1,前4 h的膜通量变化较缓慢,仅下降至13.01 L·(m2·h)−1,随后由于浓缩倍数的提高,导致渗透压差减少和膜污染产生,膜通量迅速下降。通过物理清洗,第2轮的初始通量为13.31 L·(m2·h)−1,由于物理清洗不能有效去除膜上的有机污垢,导致初始通量随着运行周期的延长而下降。同时,第2轮的膜通量下降程度相比第1轮,迅速下降的节点更早,运行至第4 小时,膜通量已降至10.61 L·(m2·h)−1。这可能是由于膜上残留的无机和有机污垢提高了膜面粗糙度,导致运行过程中膜污染迅速加剧,从而影响浓缩性能[17]。运行至第3轮,初始通量降至10.16 L·(m2·h)−1,此阶段通量变化与前2阶段有明显区别,且膜通量呈逐步下降趋势,可能是经过前2轮浓缩实验后,膜污染累积导致通量的进一步下降。第3轮实验后,对膜表面采用化学清洗,第4轮的初始通量显著提升至13.62 L·(m2·h)−1,膜通量变化趋势与第1轮相似,说明化学清洗可以较好地去除膜表面的可逆污染。虽然化学清洗对膜性能的恢复效果更好,但其成本相对较高,且会产生化学废水。因此,将物理清洗与化学清洗方案相结合可实现高效去除FO表面的膜污染操作。这将最大限度地减少对频繁清洗膜的要求,减少化学清洗对膜完整性和膜寿命相关的损害,并降低与化学清洗相关的运营成本。
经过连续浓缩过程后,出水污染物浓度如表1所示。可以看出,经过正渗透浓缩后,出水中的主要污染物均达到了《生活垃圾渗滤液排放标准》 (GB 16889-2008) 提出的排放限制。正渗透膜对COD的截留率均达到95%以上,出水质量浓度为90 mg·L−1左右,对TP基本达到了完全截留。有研究[18]表明,较小晶体半径和较大水合半径的离子,表面的水合层强度更高,更容易被膜排斥。因此,在本研究中,Ca2+和Mg2+的截留率均达到90%以上,出水质量浓度分别为0.65 mg·L−1和1.64 mg·L−1。然而,FO膜对氨氮的截留率较低,仅有60%~65%,出水的氨氮质量浓度仅稍低于排放限制。胡涛战[19]采用厌氧正渗透膜生物反应器 (AnMF-OMBR) 处理模拟废水时也发现FO对氨氮的截留率较低,并将其归因于膜两侧发生了唐南平衡现象。已有研究[20]表明,正渗透膜对一价离子的截留率较低。当以NaCl作为汲取液溶质时,由于Na+扩散系数大于NH4+[21],导致Na+不断透过正渗透膜,从汲取液一侧进入原料液侧,发生溶质反渗。溶质反渗导致两侧电荷不平衡,原料液一侧的NH4+不断进入汲取液,从而导致截留率下降。
-
采集所有单次实验结束后的膜清洗水样,进行 EEM分析并整理数据,导入MATLAB 2017b,利用DOM Flour工具箱进行PARAFAC平行因子分析。通过PARAFAC对整个EEM进行建模,共解析出4种不同荧光成分的组合,可以描述获得的所有膜清洗水样样本。其组分及其峰强度如图9所示。组分1 (C1) 在250 nm的激发波长 (Ex) 和350 nm的发射波长 (Em) 范围内显示其主峰。组分2 (C2) 的主峰和次峰分别位于230 nm/400 nm和290 nm/400 nm的Ex/Em范围内。组分3 (C3) 呈现3个荧光峰,其中310 nm/480 nm和360 nm/480 nm的Ex/Em范围处的峰较强,265 nm/480 nm 的Ex/Em处的峰较弱。而组分4 (C4) 呈现2个荧光峰,分别为245 nm/445 nm的Ex/Em范围的主峰和345 nm/445 nm的Ex/Em范围的二级峰。
对通过PARAFAC分析出的4种组分进行成分分析,其表征的主要成分如表2所示。根据已有研究[22-24]中描述的峰值位置和分配,C1表现出类似于色氨酸类物质的荧光特性,C3更接近于长波类腐殖质的荧光特性,故C3主要为腐殖质类物质。C2和C4与短波类腐殖质荧光特性接近 (主要为富里酸) ,说明C2和C4的组分主要为富里酸类物质。本研究中富里酸类物质被分为C2和C4组分,这可能与垃圾焚烧厂渗滤液中存在较多富里酸类物质、其具体成分的疏水性不同有关,从而使其Em值有差异[25]。垃圾焚烧厂渗滤液的EEM也主要由这3类物质组成,说明这3种物质在正渗透过程中也会附着在膜表面,造成膜污染。
各组分的荧光强度可以表征其浓度变化。采用PARAFAC分别对物理清洗和化学清洗的单独组分荧光强度进行分析,结果如图10所示。化学清洗后的各组荧光组分峰强均高于物理清洗,说明化学清洗对膜污染缓解的贡献更高,膜表面清洗更彻底。此外,物理清洗和化学清洗后膜污染提取物中各组分荧光强度从大到小排序均为C1>C4>C2>C3,即以污染物浓度为基准得出的膜污染贡献从大到小排序为色氨酸类物质>富里酸类物质>腐殖酸类物质,色氨酸类物质是膜污染的主要成分,分别占物理和化学清洗的44.49%和45.93%。已有研究[9]表明,膜污染贡献大小与物质分子质量有关,各荧光组分的分子质量从小到大排序为C1<C2/C4<C3,且Em的高低与有机物疏水性强度密切相关[25]。色氨酸类物质分子质量较小且亲水性更强,更容易与膜表面接触并附着在膜表面结合,形成污染层,而相对分子质量高且疏水性较强的腐殖酸类物质膜污染潜力较低,与膜表面的结合较为松散,更容易在膜清洗过程中去除。
-
采用FO处理垃圾焚烧厂渗滤液负压原位碱度脱氨出水,FO出水中的COD<100 mg·L−1,NH4+-N<25 mg·L−1,TP基本为0,主要污染物均达到了《生活垃圾渗滤液排放标准》 (GB 16889-2008) 规定的排放限制。与常规反渗透膜 (RO) 深度处理垃圾渗滤液工艺相比,FO不仅具有较高的污染物截留率,同时也具有浓缩倍数高、浓缩液少、能耗较低、膜污染潜力低等优势。故FO在垃圾焚烧厂渗滤液深度处理领域具有一定的应用潜力。然而,目前相对于常规RO膜工艺,FO的实际应用仍面临一些关键瓶颈,主要包括正渗透膜成本较高、初期投资成本较高、汲取液溶质需进一步分离回收等。因此,在未来的研究中,开发低成本、高质量FO膜材料仍是FO膜进一步推广应用的关键。
-
1) AL-DS与AL-FS模式相比,可以取得更高的初始通量,但膜通量下降速率更快,且具有更强的膜污染,物理清洗恢复率更低。而AL-FS的膜通量更稳定,且具有较低的膜污染趋势。汲取液浓度为 2 mol·L−1时,初始通量为14.81 L·(m2·h)−1,运行10.5 h后对垃圾焚烧厂渗滤液脱氨出水的浓缩倍数为10倍,出水水质可以达到排放标准。且物理清洗后通量恢复率为90%,膜污染程度较轻。在错流速率较低的情况下,膜污染会更容易形成,还会影响膜性能。所以错流速率地提升有利于提高膜通量,进一步提高错流速率至12 cm·s−1时对膜通量并没有明显的促进作用。
2) 长期运行结果表明,将物理清洗与化学清洗方案相结合以实现高效的正渗透操作是可行的。
3) EEM-PARAFAC实验结果表明,膜污染主要成分为色氨酸类物质、腐殖质类物质和富里酸类物质,以污染物浓度为基准得出的膜污染贡献从大到小排序为色氨酸类物质>富里酸类物质>腐殖酸类物质。
FO深度处理垃圾焚烧厂渗滤液的运行效能及膜污染特性
Operational performance and membrane fouling characteristics of FO for deep treatment of the leachate from the waste incineration plant
-
摘要: 针对垃圾焚烧厂渗滤液负压原位碱度脱氨出水中COD和氨氮浓度无法满足现行排放标准的问题,采用正渗透 (FO) 对出水进行进一步处理;探究膜朝向、汲取液浓度和错流速率对正渗透运行性能的影响,确定最佳运行参数;通过FO连续运行实验,确定正渗透连续运行下的膜清洗方案;采用三维荧光 (EEM) 结合平行因子分析方法 (PARAFAC) 对膜污染物质和特性进行分析。结果表明:活性层朝向汲取液 (AL-DS) 与活性层朝向原料液 (AL-FS) 模式相比,可以取得更高的初始通量,但膜通量下降速率更快,且具有更强的膜污染,物理清洗恢复率更低;而AL-FS的膜通量更稳定,且具有较低的膜污染趋势;汲取液浓度为2 mol·L−1时,初始通量为14.81 L·(m2·h)−1,运行10.5 h后,脱氨出水的浓缩倍数为10倍,出水水质可以达到排放标准;物理清洗后,通量恢复率为90%,膜污染程度较轻;提高错流速率至8 cm·s−1可以减缓膜污染,而进一步提高错流速率对膜通量并没有明显的促进作用;连续运行实验结果证明将物理清洗与化学清洗相结合以实现高效的正渗透操作是可行的;膜污染主要成分为色氨酸类物质、腐殖质类物质和富里酸类物质,膜污染贡献大小为色氨酸类物质>富里酸类物质>腐殖质类物质。由此可知,采用FO深度处理垃圾焚烧厂渗滤液负压原位碱度脱氨出水,可以使出水达到排放标准,并且在FO运行过程中,通过选取合适的运行措施可以适当减缓膜污染,确保FO稳定运行。该研究结果可为FO处理垃圾焚烧厂渗滤液工业化规模提供理论基础和应用参考。Abstract: In view of the problem that the concentrations of COD and ammonia nitrogen in the effluent of ammonia removal from incineration leachate after in-situ alkalinity vacuum stripping process cannot meet the current discharge standards, forward osmosis (FO) was used to deeply treat the effluent in this study. The effects of membrane orientation, different draw solution concentrations and cross-flow rates on the performance of FO operation were investigated and the optimal operating parameters were determined. Then the membrane cleaning scheme was proposed under the continuous operation of FO. Moreover, the membrane fouling characteristic was analyzed by three-dimensional fluorescence (EEM) combined with parallel factor analysis (PARAFAC). The results showed that AL-DS (Active layer oriented towards draw solution) mode could achieve a higher initial flux compared with AL-FS (Active layer oriented towards feed solution) mode. However, a faster decrease rate of membrane flux and a more serious fouling with lower flux recovery rate after physical cleaning occurred for AL-DS mode. On the contrary, the AL-FS mode presented a more stable flux and lower membrane fouling tendency. When the concentration of the aspirate was 2 mol·L−1, the initial flux of the FO was 14.81 L·(m2·h)−1, the concentration multiple was up to 10 times and the effluent quality of ammonia removal could meet the discharge standards after 10.5 h running. Furthermore, the fouling was relatively light and the flux recovery rate was 90% after physical cleaning. Additionally, the increase of cross flow rate up to 8 cm·s−1 could slow down the membrane fouling, but the membrane flux had no significant improvement as the cross-flow rate further increased. Continuous operational tests showed that it was feasible to combine physical cleaning with chemical cleaning protocols for highly efficient FO operation. EEM-PARAFAC experimental results showed that the main components of membrane fouling were tryptophan-like substances, humic-like substances and fulvic-like substances. Their order of pollution degree was tryptophan-like substances > fulvic-like substances > humic-like substances. It can be seen that the effluent of ammonia removal by in-situ alkalinity vacuum stripping process can meet the discharge standard after FO deep treatment. And in the FO operation process, appropriate operation strategies could effectively alleviated membrane fouling and ensured the stable FO operation. The results of this study can provide a theoretical basis and application reference for the industrial scale of FO treating the incineration leachate.
-
Key words:
- incineration leachate /
- forward osmosis /
- concentrate /
- membrane fouling /
- parallel factor analysis
-
气溶胶是指固体或液体颗粒物均匀地分散在气体中形成的相对稳定的悬浮体系,其动力学直径在1 nm到100 μm之间。生物气溶胶是含有生物性粒子的气溶胶,包括细菌、病毒以及致敏花粉、霉菌孢子、蕨类孢子和寄生虫卵等,具有传染性和致敏性。当病人在进行呼吸活动的时候,会产生携带大量病原体的飞沫。飞沫在空气中悬浮的过程中会失去水分,剩下的蛋白质和病原体组成核,就形成了生物气溶胶。生物气溶胶可在空气中悬浮较长时间,造成远距离传播。人类大部分活动都是在室内进行的,由生物气溶胶引发的疾病越来越多。全球约20%的呼吸道感染疾病是由其引起的[1]。曾经肆虐全球的非典病毒(SARS病毒)就是一种通过气溶胶形式进行传播的典型传染性病毒[2]。此后,国内外相继出现了禽流感、甲型和乙型流感和手足口病等通过气溶胶进行传播的传染性疾病[3]。
目前,新型冠状病毒肺炎(COVID-19)疫情正在全球范围内大规模爆发。研究发现,导致这次疫情的新型冠状病毒与SARS-CoV具有相似性,并且该病毒比SARS更具传染性[4]。新型冠状病毒的主要传播途径包括呼吸道飞沫传播和接触传播。而气溶胶传播作为一种可能的传播途径也备受关注。国家卫生健康委员会在发布的《新型冠状病毒肺炎诊疗方案(试行第七版)》中提到:“在相当密闭的环境中长时间暴露于高浓度气溶胶情况下存在经气溶胶传播的可能。”因此,认识生物气溶胶来源、危害及其传播规律,有助于采取有效措施降低室内环境的传染以及交叉感染风险。本研究通过比较主要消杀技术的优势及局限性,总结SARS冠状病毒疫情期间有效消杀技术的经验,提出室内空气高效滤除与杀灭病原体措施的方案建议。
1. 室内空气中生物气溶胶的危害及常用的防控措施
1.1 生物气溶胶种类和来源
居室、医院、办公室、船舶、高铁和飞机等室内环境空气中主要有病毒、细菌和霉菌气溶胶。不同室内环境的空气中,生物气溶胶的种类、数量以及所占的比例均不相同。医院是生物气溶胶浓度最高的地方。室内生物气溶胶的来源分为室内和室外源,主要来自室内人和动物的活动、物体表面(马桶、地毯、吸尘器、水面墙壁等)、空调、室外植物花粉、废水和固废处理等[5]。
1.2 生物气溶胶的传播方式
当病人在进行呼吸活动的时候,会产生携带大量病原体的飞沫。随后飞沫会在空气中传播,即空气飞沫传播,是空气传播的一种方式。一般情况下,空气飞沫传播只有与传染源近距离接触才可能实现,而距离传染源1 m以外是相对安全的,距离2 m以上安全性更高。因为没有外部条件(如风力)的帮助,飞沫喷射到2 m以外的可能性非常小。然而,飞沫在空气传播过程中会雾化成生物气溶胶,该过程会受到各种环境因素的影响。随着时间的推移,生物气溶胶逐渐丧失活性。影响飞沫中病原体生存的因素包括飞沫中的介质、温度、湿度、氧敏感性以及紫外、臭氧或电磁辐射等[6]。相关学者也开展了飞沫颗粒大小与传播距离的关联性的研究。研究[7]表明,直径大的飞沫会很快落到地面上,中等大小的飞沫,尤其是直径小于5 μm的飞沫会扩散到空气中,以气溶胶的方式传播,传播路径见图1。如果病原体在传播过程中没有丧失活性且被易感染人员吸入一定剂量,那么就可能造成疾病感染。
 图 1 致病微生物的传播路径示意图Figure 1. Schematic diagram of transmission path of pathogenic microorganisms
图 1 致病微生物的传播路径示意图Figure 1. Schematic diagram of transmission path of pathogenic microorganisms1.3 室内空气中生物气溶胶的危害
室内生物气溶胶病原体对人体健康具有很大的危害,可引起人体自身免疫系统和超敏反应[8],重则导致死亡。近年来病毒性疾病暴发频繁,如冠状病毒导致SARS疫情的爆发;呼吸道合胞病毒造成的流感肆虐;源于啮齿类动物粪便的汉坦病毒引发的肺炎等[9],导致高的死亡率。细菌性气溶胶致人感染的风险也很高,人类即使暴露在较低浓度的军团菌病、化脓性链球菌结核病、炭疽热等细菌生物气溶胶也容易感染。芽枝霉菌、曲霉菌、青霉菌以及链格孢菌这4类真菌气溶胶大量存在于室内空气中,容易引起哮喘和鼻炎等疾病。
1.4 对生物气溶胶的防控措施
生物气溶胶的源头是飞沫。飞沫中病原体的存活受多种因素影响。直径小的飞沫容易以气溶胶的方式传播,并且积累到一定浓度容易造成人的感染。根据生物气溶胶的传播特征,生物气溶胶最常用的防控措施主要有如下3个方面:一是要正确佩戴口罩、护目镜、穿隔离服等,堵断生物气溶胶的飞沫传播;二是要通风,降低生物气溶胶的浓度。由于空气的稀释作用,生物气溶胶致病的几率大大降低;三是要定期消毒。室内需要定期用含氯类(如84消毒液、漂白粉)消毒剂进行喷洒消毒;用酒精类的如75%酒精擦拭常用物品表面进行消毒。然而,上述防控措施存在操作的局限性:长期佩戴口罩会非常不舒服,还可能导致面部皮肤损伤;有些办公、居家、公共交通工具等环境内不具备良好的通风和消毒条件等等;尤其在医院及公共场所,人员流动频率高,在无人情况下的消毒方法十分有局限性且没有持续性。因此,这就需要采取其他高效、安全、可行的消杀措施,实现在有人员流动的情况下对室内空气中病原体的消杀,确保人们的健康安全。
2. 室内生物气溶胶消杀的主要技术
空气中细菌和病毒的消杀技术,大体上可分为物理和化学方法两大类,包括热、过滤截留、紫外线、臭氧及催化技术等。
2.1 热消杀
通过加热的方法可以使微生物中的蛋白质和核酸物质变性,从而有效地杀灭各种病原体,如各类细菌、真菌和病毒。热消杀技术应用广泛,并且效果可靠。不同微生物对温度的耐受性存在差异,微生物繁殖的温度范围为−10~90 ℃。当加热温度达到56 ℃时,每15 min会有10 000个SARS冠状病毒被杀灭[10]。新型冠状病毒对温度也很敏感,56 ℃下加热30 min即可灭活[11]。但受物体的耐高温性和应用场所的限制,热消杀方式应用起来也存在诸多缺点。
2.2 过滤消毒
空气过滤消毒指用物理阻留的方法去除空气中的病原体,包括细菌与病毒。针对不同病原体的尺寸,通常用不同孔径滤材对生物气溶胶进行过滤,其过滤效率与滤材的孔径密切相关。通常孔径越小,过滤效率越高,但是引起的空气阻力也会变大。与细菌相比,病毒的直径要小很多。因此,为了高效过滤病毒,必须使用孔径小的高效滤材[12-13]。近年来,除了广泛使用的HEPA等过滤材料外,静电纺丝也成为能有效过滤消毒的新型材料,广泛应用于含有滤材的口罩、空气净化器和新风过滤系统等。该技术的优点是在室温下就可以去除空气中的病原体,操作简易;缺点是只能起到过滤的作用,并不能杀灭微生物,可能为微生物繁殖提供温床,造成二次污染。
2.3 紫外线消毒
紫外线消毒指利用波长220~300 nm的紫外线破坏微生物机体细胞中的DNA或RNA的分子结构[14],将各种细菌、病毒、寄生虫以及其他病原体直接杀死。这是最常用的密闭空间内生物气溶胶消杀技术。紫外线消毒速度快、效率高,并且几乎对所有的细菌、病毒都能有效灭活。紫外杀灭病原体的作用取决于照射剂量、空气的水分含量、空气运动模式以及密闭空间的大小。常规应用需要照射无死角并且达到照射剂量才能达到杀菌消毒的效果。据报道,新型冠状病毒等各种病毒也对紫外线敏感。紫外线由于其良好杀毒性能及功效,已经在多种场景下取得了广泛应用,例如在医院病房、手术室、无菌实验室等对环境要求较高的场所。紫外线消毒技术的应用已经相对纯熟,但是紫外线对人体有害,不能直接暴露应用在有人员活动的场所。
2.4 臭氧消毒
臭氧是一种强氧化剂,可直接氧化胞内有机物,遇到水蒸气可以产生强氧化性的羟基等自由基间接氧化病原体,从而达到杀菌消毒目的。在20 m3的气雾室内产生50 mg·m−3的臭氧可对人工污染的空气中的白色葡萄球菌杀灭99.96%以上;在60 m3的密闭房间产生10 mg·m−3的臭氧可对空气中的自然菌杀灭93%以上[15]。臭氧消毒为溶菌级方法,杀菌彻底,具有杀菌广谱、高效的特点,对所有细菌和病毒都有明显的杀灭效果。臭氧在应用于室内消毒杀菌取得很好效果的同时,残余的臭氧浓度过高,会对人体造成危害,因此需采取有效措施抑制臭氧释放与残留。臭氧消毒技术具有高效性、广谱性和便捷性的特点。
2.5 催化消杀技术
催化消杀技术是利用活性高、寿命长的催化剂进行杀菌消毒,包括光催化和非光催化杀菌技术。光催化杀菌技术主要是利用光催化剂在光的作用下产生活性氧物种(reactive oxygen species,ROS)和自由基氧化微生物达到杀灭效果。众多的研究表明,TiO2光催化杀灭微生物技术具有效果好、作用迅速等特点。但是由于TiO2带隙较宽,只能利用占太阳光不到5%的紫外光,同时活性受光照强度等条件影响。SAWAI等[16-17]连续多年对MgO和CaO等氧化物的常温杀菌消毒性能进行研究,取得了很多有价值的成果。他们否定了CaO与MgO的杀菌机理主要是利用其碱性的观点,证明这些氧化物的杀菌机理与活性氧的产生有关;并通过在CaO和MgO粉末胶体中加入超氧歧化酶或同时加入超氧歧化酶和过氧化氢酶等活性氧捕捉剂,采用荧光法检测到的活性氧信号的变化,充分证明了活性氧自由基·O2-和H2O2在杀菌过程中发挥的重要作用,为非光催化氧化杀菌机理提供了有力的支持[16-17]。
2.6 耦合消杀技术
通过耦合两种或两种以上技术可以提高室内空气中细菌和病毒的杀灭效率,减少二次污染等技术弊端。国内已有学者着手开展了复合技术用于室内空气病原体消杀的探索研究,发现在同样的条件下,臭氧耦合二氧化钛复合技术的消杀效率比单一技术效果好[18]。紫外线联合臭氧催化技术在生物气溶胶模拟现场以及储物室和卫生间等实际现场测试的结果表明,该耦合技术的室内杀菌消毒效果优异,具有很好应用前景[1]。
3. 非典疫情时期针对SARS冠状病毒消杀的非光催化技术研究案例及启示
2003年,为抗击“非典”疫情,控制SARS病毒的传播,中国科学院大连化学物理研究所刘中民等与大连医科大学病原学教研室合作成立了“抗病毒纳米催化材料研究”攻关小组。攻关组成员利用流感病毒等开展了“用于呼吸道病毒阻隔、吸附及灭活的纳米催化材料及相关作用机制研究”的工作,发现多种无机多孔固体催化材料对吸附在其表面的病毒具有明显的灭活作用,说明催化灭活微生物是普遍存在的现象[19]。这是国内外最早利用催化材料灭活SARS病毒的研究。
中国科学院生态环境研究中心贺泓院士课题组在非典期间开发的催化剂也被证实对SARS病毒具有高效杀灭效果[20],结果见表1。在空气中无需外加光电能源的人居条件下,Ag/Al2O3催化剂可在5 min内即可有效杀灭吸附于其表面的初始量为2×106 cfu.mL−1的多形德巴利酵母菌和大肠杆菌、初始量为105PFU (plaque forming unit)的杆状病毒和SARS冠状病毒。Cu/Al2O3催化剂在20 min内也可以有效杀灭大肠杆菌、杆状病毒和SARS冠状病毒。作为对照的Al2O3载体和滤纸则没有任何消杀效果。将该催化剂涂覆到空气净化器模块上,在30 m3的标准测试舱中测试后,发现其除菌率可以达到95%以上,极具推广应用潜力。在前期研究的基础上,课题组又开发了一系列不同晶型、不同形貌的载银催化剂,无需引入外加光源就呈现出优异的室温接触杀菌活性[21-28]。这一系列催化剂的杀菌机制为:通过活化分子氧产生具有高氧化性的ROS,同时结合溶出的银离子的毒性作用,导致细胞内活性氧的产生、细胞的破损乃至死亡。红外测量结果进一步显示,上述杀菌过程有CO2生成,证实了ROS参与的催化氧化杀菌及矿化过程(见图2),最终微生物脱离材料表面,进而重新露出具有杀菌消毒作用的催化材料表面,从而实现材料的重复使用。该催化材料在对空气杀菌和净化过程中不会对人体和生态环境构成任何伤害,具有广谱和高效等优点,非常适合室内的杀菌消毒。而且这种消杀方法的实施过程不会损耗光电能源,在室温条件即可实现,很具应用前景。
表 1 不同催化剂对不同微生物的灭活效果Table 1. Inactivation efficiencies of different materials to various microorganisms材料 SARS冠状病毒 杆状病毒 大肠杆菌/(cfu·mL−1) 多形德巴利酵母菌/(cfu·mL-1) Ag/Al2O3 未测到 未测到 未测到 未测到 Cu/Al2O3 未测到 未测到 未测到 102 Al2O3 不可计 不可计 不可计 105 滤纸 典型 典型 106 105 | Show Table DownLoad:
CSV
DownLoad:
CSV
 图 2 室温下载银催化剂高效的杀菌活性与机理Figure 2. Excellent sterilization ability and mechanism of silver-based catalysts
图 2 室温下载银催化剂高效的杀菌活性与机理Figure 2. Excellent sterilization ability and mechanism of silver-based catalysts日本“钻石公主”号邮轮上1位乘客确诊感染了新型冠状病毒,之后邮轮上所有乘客均在海上隔离了十几天。2020年3月5日的新闻证实,隔离后仍然有696人感染。在我国湖南省,也发生了公共交通工具内新型冠状病毒肺炎聚集性疫情事件[29]。这些事例表明新型冠状病毒在通风不畅的室内环境中进行了传播与交叉感染。相比于室外具备阳光照射等不利于细菌和病毒存活的因素,室内环境中生物气溶胶的浓度较高,室内空气中的细菌和病毒通常也会存活得更久。因此,在居室、医院、隔离场所、火车、飞机、轮船和办公室等室内环境,应在采取通风、防护和消毒等措施的同时,加强室内空气中生物气溶胶的消杀,阻断病原体感染风险,从而控制病原体以及疫情的传播,保障人们的身心健康。目前,主要采用消毒剂以及紫外线的方式来杀灭新型冠状病毒。然而,这些方法仅适合用于物体表面或者特殊场所的消毒,亟需开发高效复合技术,比如能实现病毒高效过滤和原位灭活的复合技术、防护产品与净化装置,形成室内空气病原体消杀的整体解决方案。
现有空气净化设备和空调粗效或中效过滤材料对直径较小的病毒滤除效果不高,建议采用高效过滤材料和紫外、臭氧、室温高效催化剂等相结合的复合技术,实现对病毒的高效滤除和原位杀灭(见图3)。普通空气净化设备一般采用H11~H12等级的HEPA滤网,其对直径为0.3 μm以上的微粒去除效率可达到98%以上,是去除细颗粒物污染物最为有效的过滤材料。然而,病毒直径更小,并且要求的滤除效率更高,所以需要H13~H14等级的HEAP滤网。通过对室内生物气溶胶消杀技术的优劣势分析发现,紫外、臭氧和高效催化剂具有优异的杀灭病源微生物的能力。耦合紫外、臭氧和高效催化剂技术的优势,实现在高风量条件下对滤除病毒的高效杀灭,具有很好的可行性。基于此原理,贺泓院士团队与合作企业对传统空气净化器的结构进行改造,采用H13等级HEPA滤网,微生物拦截率可达99.9%;再将紫外灯管或臭氧发生装置置于HEPA滤网迎风面实现对过滤下来的微生物高效杀灭;同时末端采用高效催化分解材料分解残留臭氧,避免二次污染,为病原体的消杀和疫情的防控起到了积极的作用。近期在30 m3的监测舱内的检测结果证实,基于过滤、紫外光和臭氧耦合技术改造的空气净化器对空气中噬菌体病毒单位小时去除率达到99.9%。若将此净化器应用于新冠病毒的消杀,还需要在国家生物安全四级实验室进行新冠病毒滤除和灭活效果的验证。为提高技术的实用性与时效性,建议将复合技术涉及的材料进行集成与模块化,组装到现有的空气净化器、新风系统和空调系统等设备中。另外,过滤和室温高效消杀催化材料也可以进行复合,并与无纺布等材料进行成型处理,应用到口罩等防护产品中。依托复合消杀技术研发的净化设备与防护产品需尽快投入使用,在助力打赢新冠病毒疫情防控狙击战的同时,也为今后室内空气病原体消杀设备的研发与推广应用提供技术支持。
 图 3 高效滤除与原位杀灭室内空气中病原体的技术方案Figure 3. Technical scheme for high-efficiency filtration and in situ inactivation of pathogen in indoor air
图 3 高效滤除与原位杀灭室内空气中病原体的技术方案Figure 3. Technical scheme for high-efficiency filtration and in situ inactivation of pathogen in indoor air4. 结语
针对家居、办公室、疫区医院或公共场所等室内环境的空气净化,开发高效细菌和病毒等病原体的过滤和原位灭活技术与装置,并应用到中央空调、新风系统、空气净化器、分体空调或特殊场所的排风系统,实现对病原体的高效过滤和杀灭,将为室内空气生物气溶胶消杀和疫情的防控提供技术支撑,对于疫情的防控具有重要意义。目前,测试结果表明,该方案与建议具有很好的效果,是行之有效的。同时,与空气净化器、新风系统、空调系统和口罩等集成的过滤、消杀复合技术与设备,也非常值得室内环境中推广应用。
-
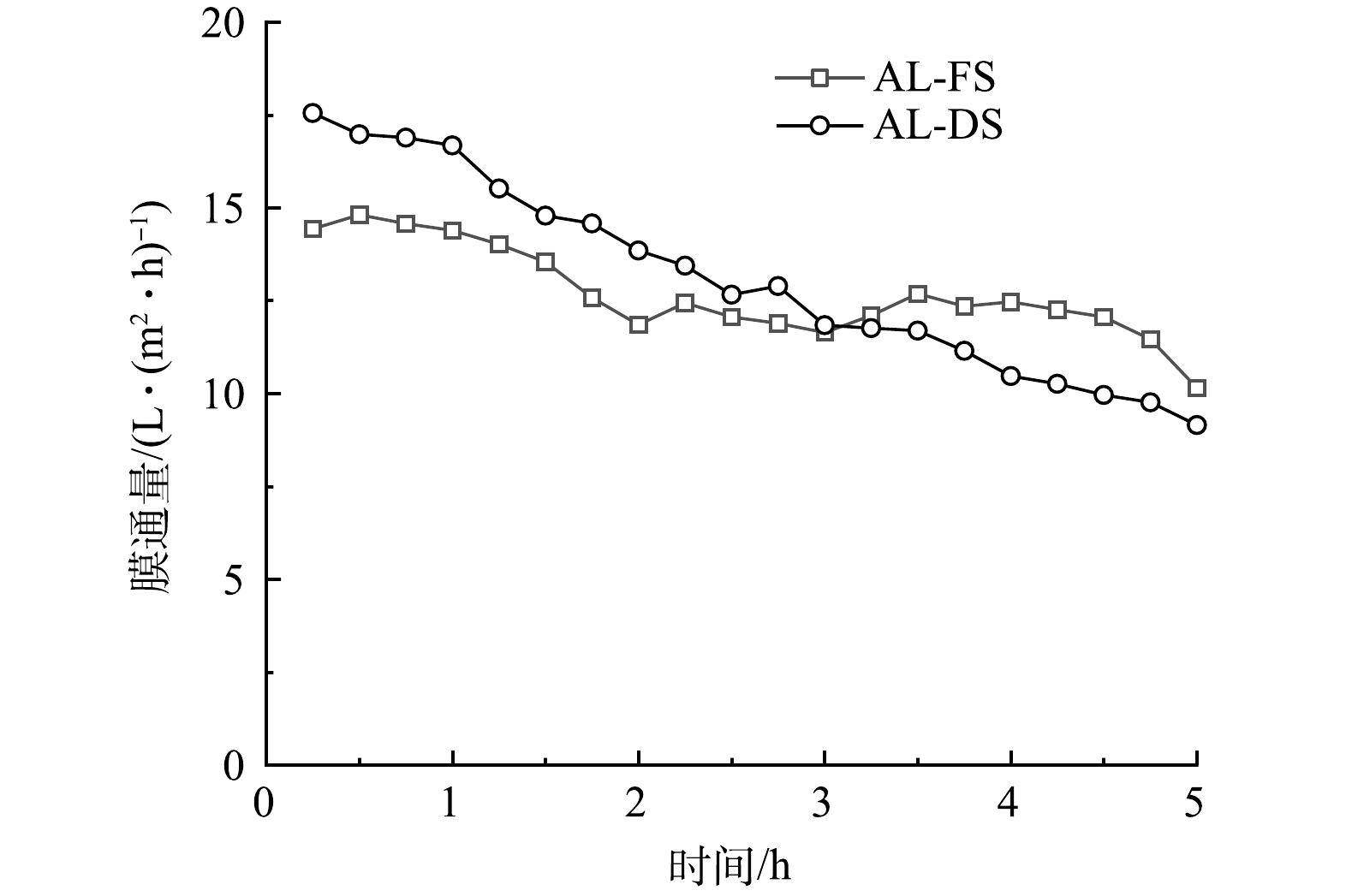
图 2 AL-DS模式和 AL-FS模式下的膜通量
Figure 2. Membrane flux for the AL-DS mode and the AL-FS mode

图 4 不同汲取液浓度对 FO 膜通量的影响
Figure 4. Influence of draw solution concentration on membrane flux of FO membrane
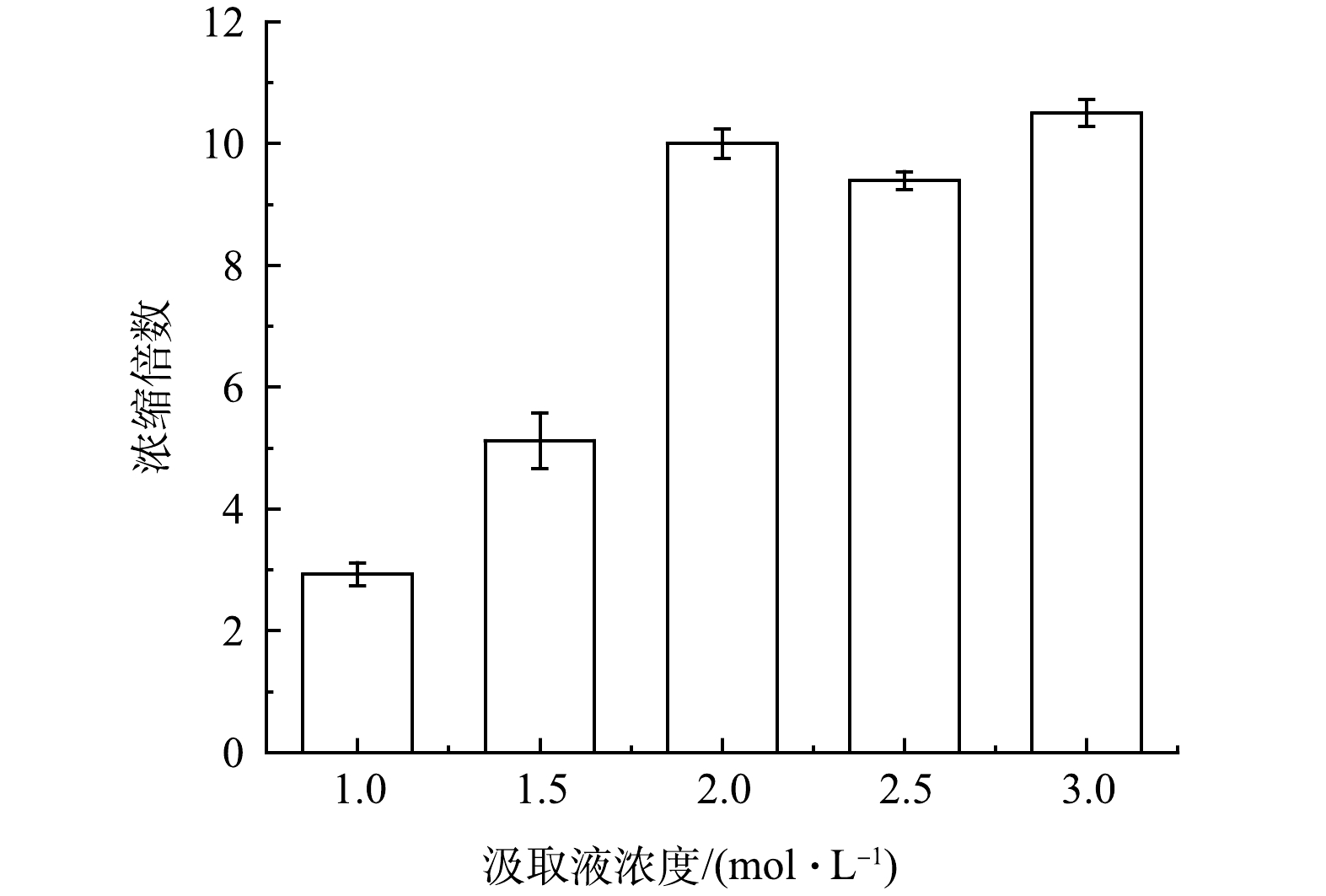
图 5 不同汲取液浓度的正渗透浓缩倍数
Figure 5. Forward osmosis concentration ratio at different draw solution concentrations
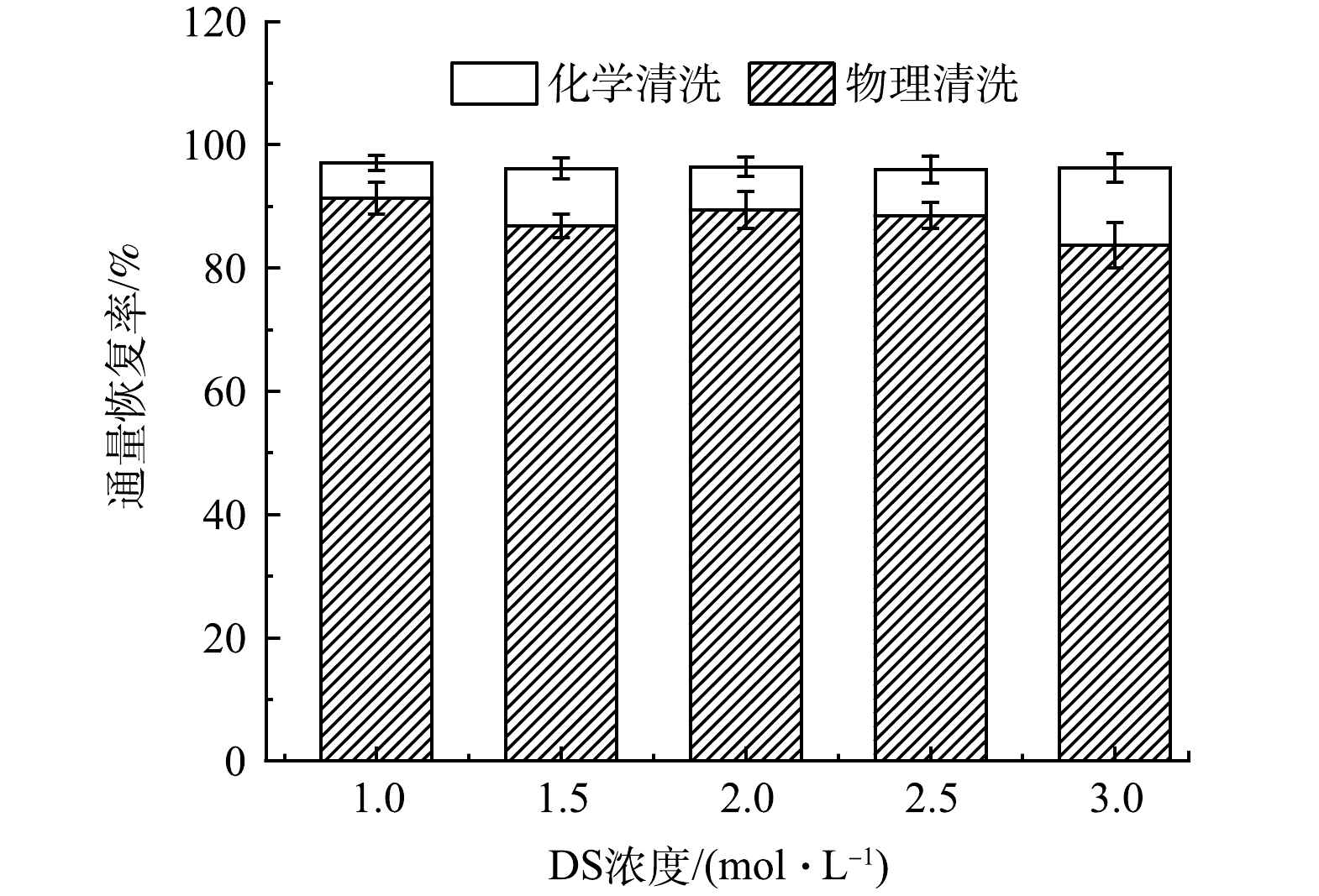
图 6 不同汲取液浓度下通量恢复情况
Figure 6. Flux recovery at different concentrations of draw solution
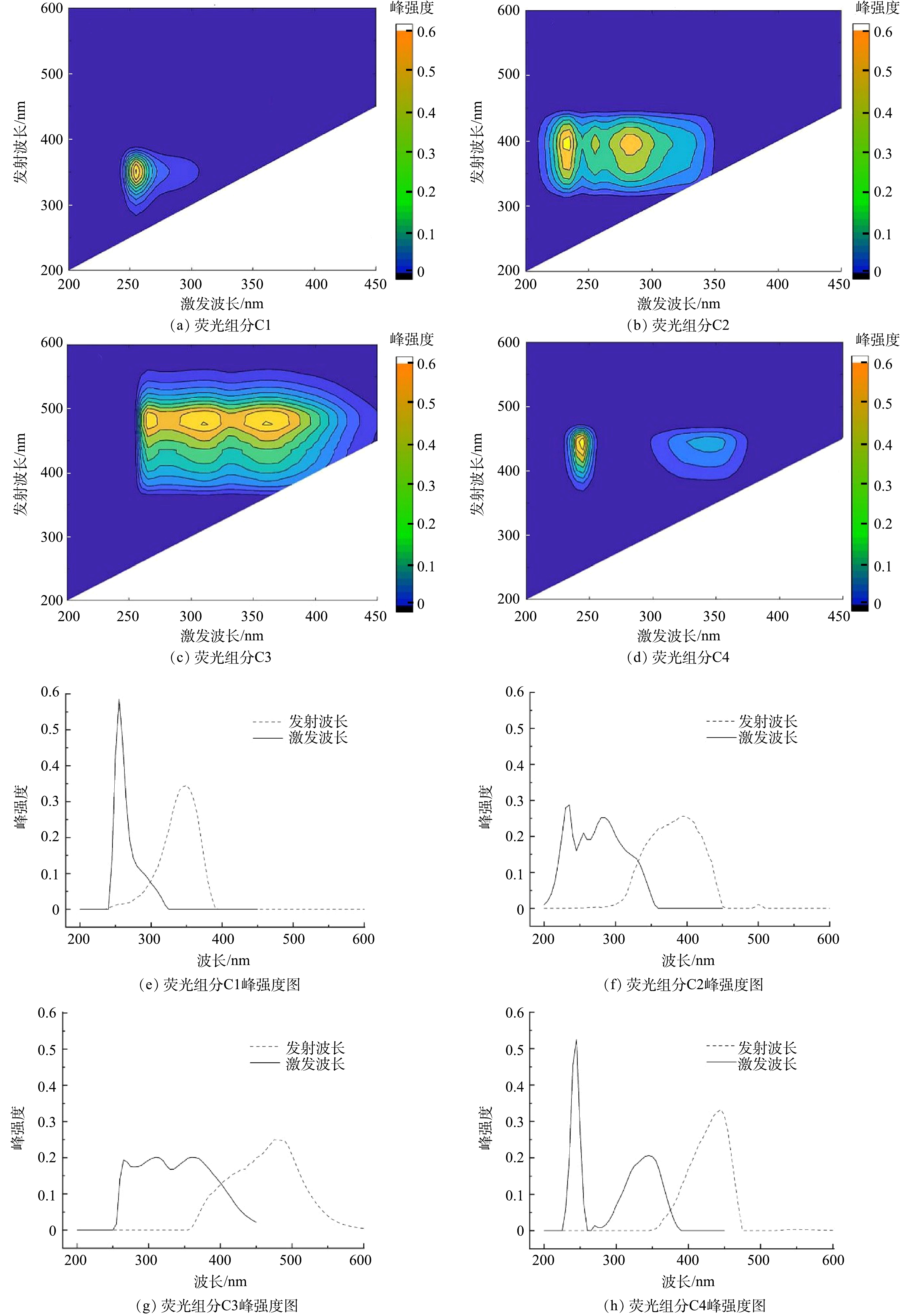
图 9 PARAFAC模型识别出的4个不同的荧光组分及其对应峰强度图
Figure 9. Four fluorescence components and the corresponding peak intensity plots identified by the PARAFAC
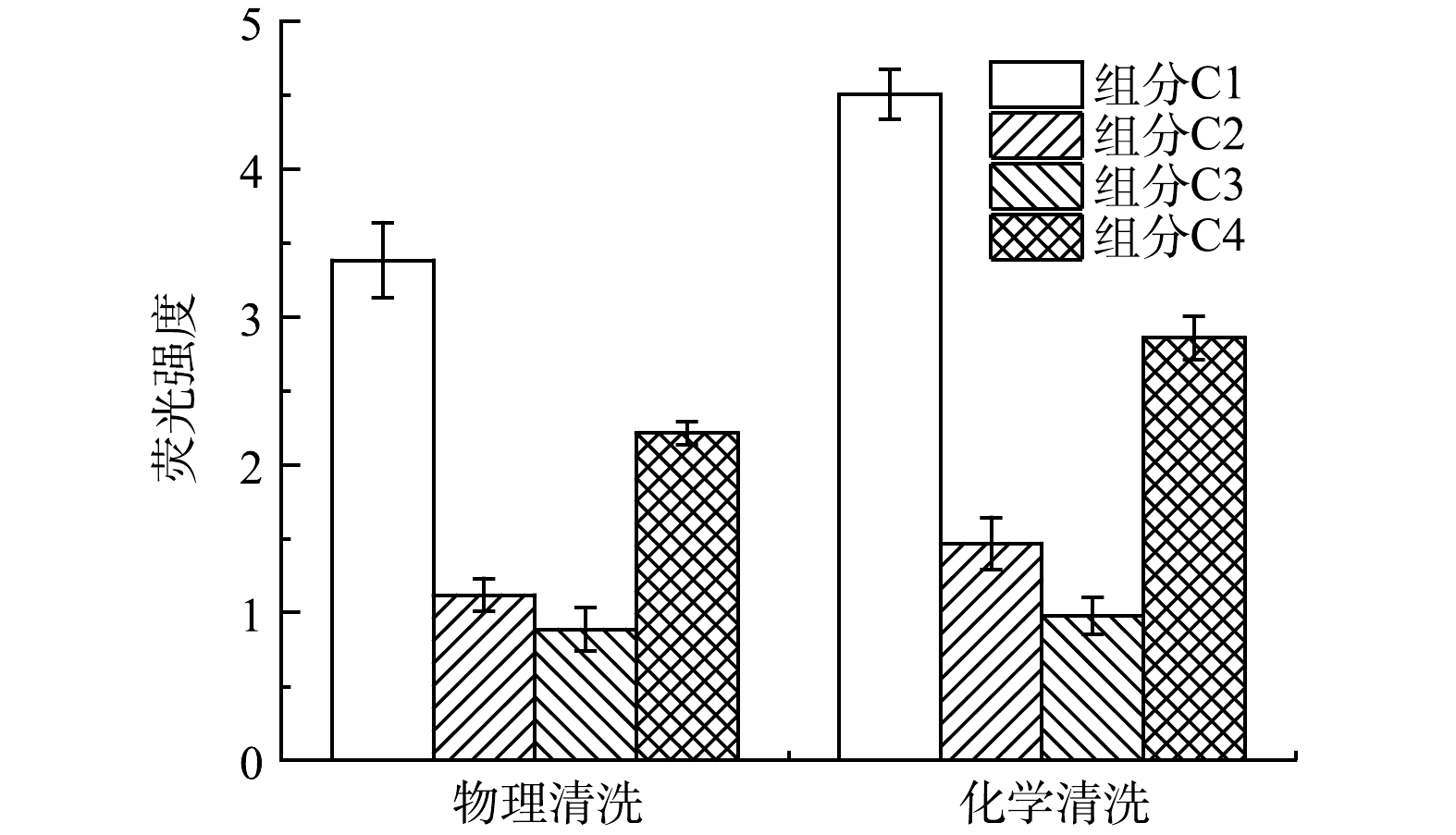
图 10 4个不同的荧光组分在膜污染中的强度
Figure 10. Intensities of four fluorescent components in the extracted membrane foulants
表 1 出水污染物指标
Table 1. Index of pollutants in effluent
项目 COD/(mg·L−1) 氨氮/(mg·L−1) TP/(mg·L−1) Ca2+/(mg·L−1) Mg2+/(mg·L−1) 实测值 89.43±2.65 22.7±0.65 0 0.65±0.12 1.64±0.32 排放标准(GB 16889-2008) 100 25 3 — —
下载: 导出CSV
-
[1] 张亚楠, 沈海滨. 论城市生活垃圾现状、管理及对策[J]. 世界环境, 2014(2): 28-29. [2] 中华人民共和国国家统计局. 中国统计年鉴2021[M]. 北京: 中国统计出版社, 2021. [3] CHEN Y M, XIAO Y T, WANG G P, et al. A pilot-scale test on the treatment of biological pretreated leachate by the synergy of ozonation-biological treatment-catalytic ozonation[J]. Environmental Engineering Research, 2021, 26(4): 200349. [4] SHI J Y, DANG Y, QU D, et al. Effective treatment of reverse osmosis concentrate from incineration leachate using direct contact membrane distillation coupled with a NaOH/PAM pre-treatment process[J]. Chemosphere, 2019, 220: 195-203. doi: 10.1016/j.chemosphere.2018.12.110 [5] BURMAN I, SINHA A. Anaerobic hybrid membrane bioreactor for treatment of synthetic leachate: Impact of organic loading rate and sludge fractions on membrane fouling[J]. Waste Management, 2020, 108: 41-50. doi: 10.1016/j.wasman.2020.04.031 [6] 赵贤广, 杨世慧, 邱明建, 等. 负压蒸发-吹脱组合新技术处理垃圾沥滤液高浓度氨氮实验研究[J]. 现代化工, 2020, 40(2): 187-190. [7] 孙燕. 正渗透处理城市污水的膜污染特性及控制研究[D]. 哈尔滨: 哈尔滨工业大学, 2019. [8] 祁伟健, 张胜寒, 王若彤, 等. 正渗透膜研究进展及其在电厂水处理中的应用[J]. 现代化工, 2022, 42(1): 85-89. [9] AFTAB B, OK Y S, CHO J, et al. Targeted removal of organic foulants in landfill leachate in forward osmosis system integrated with biochar/activated carbon treatment[J]. Water Research, 2019, 160: 217-227. doi: 10.1016/j.watres.2019.05.076 [10] IBRAR I, YADAV S, ALTAEE A, et al. Treatment of biologically treated landfill leachate with forward osmosis: Investigating membrane performance and cleaning protocols[J]. Science of the Total Environment, 2020, 744: 140901. doi: 10.1016/j.scitotenv.2020.140901 [11] ISKANDER S M, ZOU S Q, TING T, et al. Energy consumption by forward osmosis treatment of landfill leachate for water recovery[J]. Waste Management, 2017, 63: 284-291. doi: 10.1016/j.wasman.2017.03.026 [12] 国家环境保护总局. 水和废水监测分析方法[M]. 4版. 北京: 中国环境科学出版社, 2002. [13] VU M T, ANSARI A J, HAI F I, et al. Performance of a seawater-driven forward osmosis process for pre-concentrating digested sludge centrate: Organic enrichment and membrane fouling[J]. Environmental Science Water Research & Technology, 2018, 4(7): 1047-1056. [14] HONDA R, RUKAPAN W, KOMURA H, et al. Effects of membrane orientation on fouling characteristics of forward osmosis membrane in concentration of microalgae culture[J]. Bioresource Technology, 2015, 197: 429-433. doi: 10.1016/j.biortech.2015.08.096 [15] ZHAO B, GAO B Y, YUE Q Y, et al. The performance of forward osmosis in treating high-salinity wastewater containing heavy metal Ni2+[J]. Chemical Engineering Journal, 2016, 288: 569-576. doi: 10.1016/j.cej.2015.12.038 [16] 方舟. 厌氧正渗透膜生物反应器污水资源化及膜运行特性研究[D]. 北京: 清华大学, 2015. [17] BOWEN W R, DONEVA T A. Atomi force microscopy studies of membranes: Effect of surface roughness on double-layer interactions and particle adhesion[J]. Journal of Colloid & Interface Science, 2000, 229(2): 544-549. [18] TANSEL B, SAGER J, RECTOR T, et al. Significance of hydrated radius and hydration shells on ionic permeability during nanofiltration in dead end and cross flow modes[J]. Separation & Purification Technology, 2006, 51(1): 40-47. [19] 胡涛战. 厌氧正渗透膜生物反应器的膜污染机理及其控制措施的研究[D]. 无锡: 江南大学, 2017. [20] HE X, SAITO T, HICKNER M A. Zeta potential of ion-conductive membranes by streaming current measurements.[J]. Langmuir:The ACS Journal of Surfaces and Colloids, 2011, 27(8): 4721-4727. doi: 10.1021/la105120f [21] BIAN L X, FANG Y Y, WANG X L. Experimental investigation into the transmembrane electrical potential of the forward osmosis membrane process in electrolyte solutions[J]. Membranes, 2014, 4(2): 275-286. doi: 10.3390/membranes4020275 [22] LIU R X, LEAD J R, ZHANG H. Combining cross flow ultrafiltration and diffusion gradients in thin-films approaches to determine trace metal speciation in freshwaters[J]. Geochimica et Cosmochimica Acta, 2013, 109(1): 14-26. [23] OLOIBIRI V, CONINCK S D, CHYS M, et al. Characterisation of landfill leachate by EEM-PARAFAC-SOM during physical-chemical treatment by coagulation-flocculation, activated carbon adsorption and ion exchange[J]. Chemosphere, 2017, 186: 873-883. doi: 10.1016/j.chemosphere.2017.08.035 [24] BALLESTEROS S G, COSTANTE M, VICENTE R, et al. Humic-like substances from urban waste as auxiliaries for photo-Fenton treatment: a fluorescence EEM-PARAFAC study[J]. Photochemical and Photobiological Sciences, 2017, 16(1): 38-45. doi: 10.1039/C6PP00236F [25] LEE B M, SEO Y S, HUR J. Investigation of adsorptive fractionation of humic acid on graphene oxide using fluorescence EEM-PARAFAC[J]. Water Research, 2015, 73: 242-251. doi: 10.1016/j.watres.2015.01.020 -

 点击查看大图
点击查看大图

计量
- 文章访问数: 4179
- HTML全文浏览数: 4179
- PDF下载数: 60
- 施引文献: 0
