


-
近年来,工业用地土壤和地下水常受到持久性有机物的污染,已对人群健康及生态环境造成威胁[1-2]。其中,多环芳烃(Polycyclic aromatic hydrocarbons, PAHs)具有致癌变、致畸变及致突变的特点,成为一类备受关注的持久性有机污染物[3-4]。PAHs是含有2个以上苯环的碳氢化合物,属于疏水性有机污染物,在土壤环境中容易积累。有研究表明,环境中的PAHs可以通过土壤进入人体,危害人体健康[5]。目前,PAHs污染土壤是我国乃至全球亟待解决的环境问题之一。
国内外PAHs污染土壤修复技术主要包括热脱附技术、化学氧化修复技术、微生物修复技术和植物修复技术等[6-9]。近年来,化学氧化修复技术由于具有易操作、修复效果好和周期短的特点,已成为PAHs污染土壤修复领域的主流实用技术[2]。化学氧化修复技术是向污染土壤中添加氧化剂,利用氧化作用将土壤中有机污染物转化为毒性更小或无毒的物质。常用的氧化剂包括臭氧、双氧水、过硫酸钠和高锰酸钾[4, 10]。高锰酸钾由于具有氧化还原电位高、反应速率易控及反应过程易监测的特点而受到关注[11]。目前,国内外已有大量关于高锰酸钾氧化修复PAHs污染土壤的研究,诸多案例表明,高锰酸钾可有效去除土壤中PAHs[12-14]。LIAO等[13]研究并比较了不同化学氧化剂对煤气厂和焦化厂污染场地土壤中PAHs的去除效果,结果发现,高锰酸钾对土壤中PAHs的去除效率最高,达到90%以上。LEMAIRE等[14]的研究考察了不同土壤有机质类型、老化时间、PAHs有效性及碳酸盐质量分数等条件下,不同化学氧化剂对土壤中PAHs的去除效果,结果表明,在不同条件下高锰酸钾均是最有效的氧化剂。同时,高锰酸钾氧化修复技术运用过程中引发的环境问题也越来越受到国内外学者的关注[15-16]。例如,高锰酸钾氧化处理会降低土壤的渗透性,进而使土壤质量退化[16-17]。锰(Mn)虽然是一种土壤中常见的微量元素,但当质量分数过高时,也会对生态环境和人体健康造成危害。因此,针对高锰酸钾氧化修复技术应用过程中Mn元素迁移转化规律及其潜在的环境风险进行研究非常重要,但目前鲜见相关研究报道。
本研究通过室内模拟实验,采用某焦化厂PAHs污染土壤作为研究材料,系统研究高锰酸钾投加量对土壤Mn质量分数、赋存形态分布及有效态质量分数的影响,探讨高锰酸钾氧化修复PAHs污染土壤过程中Mn元素的迁移转化规律,旨在为高锰酸钾氧化修复技术应用工艺优化提供科学依据。
-
供试土样采自某焦化厂0~20 cm表层土壤,去除杂物后混合均匀,土壤样品自然风干过2 mm筛网后置于棕色玻璃瓶中备用。土壤各项基本理化性质为:pH为7.73、总有机碳质量分数为720.00 mg·kg−1、阳离子交换量为12.16 cmol·kg−1、总氮质量分数为30.00 mg·kg−1、有效磷质量分数为2.85 mg·kg−1、土壤中PAHs的质量分数为275.02 mg·kg−1、土壤中Mn的质量分数为418.97 mg·kg−1。
-
称取过2 mm筛的风干土壤样品 (30.000±0.001) g于150 mL三角瓶中,按照表1设计剂量添加高锰酸钾溶液和去离子水,液固比为2∶1,盖上封口膜,并用铝箔将三角瓶包裹避光,避免污染物光解和挥发。将三角瓶置于180 r·min−1、温度为25 ℃的恒温摇床中,分别在反应第1、3、5、7 d时取样,将样品转入100 mL离心管中,在4 000 r·min−1条件下离心10 min,取100 μL上清液测定高锰酸钾的浓度。土壤置于冷冻干燥机中,在−48 ℃下冷冻干燥48 h,取10.0 g土壤样品过0.015 mm筛,用于PAHs质量分数测定。分析氧化处理7 d土壤样品中Mn质量分数及其赋存形态。
-
1)高锰酸钾的测定。高锰酸钾浓度采用紫外分光光度法 (UV-5200,上海元析仪器有限公司) 测定[18]。高锰酸钾消耗量按式 (1) 计算。
式中:Q为高锰酸钾消耗量,mmol·g−1;C0为高锰酸钾初始浓度,mmol·g−1;Ct为实验结束后高锰酸钾浓度,mmol·g−1。
2) 土壤中PAHs的测定。准确称取过0.015 mm筛的土壤样品 (0.500±0.001) g,无水硫酸钠2.0 g,将土壤样品与无水硫酸钠置于100 mL烧杯中混和均匀,随后转移到快速溶剂萃取仪 (ASE350,美国赛默飞世尔科技有限公司) 的萃取池 (池底事先填充5.0 g无水硫酸钠) 中,加入25 μL 40 mg·L−1的5种氘代物混合液 (Naphthalene-d8、Acenaphthene-d10、Phenanthrene-d10、Chrysene-d12、Perylene-d12,百灵威科技有限公司) 作为回收率指示物,再向萃取池中加入2.0 g无水硫酸钠,然后盖紧萃取池盖子置于加速溶剂萃取仪中萃取,萃取溶剂采用二氯甲烷∶丙酮=1∶1的混合溶剂。萃取条件设置为:温度100 ℃、压力10 342 kPa、静态提取时间为5 min、循环次数为3次、溶剂淋洗体积分数为70%、氮气吹扫100 s。萃取液净化采用的方法和仪器分析条件见参考文献[19]。实验过程中,除Naphthalene-d8的回收率低于70%,其他4种回收率指示物回收率在72.21%~104.79%。
3) 土壤中Mn的测定。土壤中Mn的质量分数测定采用HClO4-HNO3消解法[20],土壤中Mn赋存形态分析采用Tessier五步连续提取法[21],土壤消解液及提取液中Mn的质量分数采用电感耦合等离子体质谱仪 (7500cx,美国安捷伦科技有限公司) 测定。实验过程中,Mn质量分数分析采用土壤标准物质 (GSS-1,购自中国地质科学院地球物理地球化学勘察研究所) 进行质量控制,Mn元素的回收率在88.83%~114.37%之间。土壤中有效态Mn的质量分数的测定方法参照文献[22]。
4) 数据统计与分析。采用SPSS18.0统计软件,对高锰酸钾消耗量及土壤中Mn质量分数数据的正态性进行检验,应用K-S检验判断数据是否符合正态分布,采用Pearson相关系数法对符合正态分布的数据进行相关性分析。采用one-way ANOVA进行方差分析。
-
各处理组土壤中PAHs质量分数的变化规律如图1所示。与对照组相比,0.05、0.10、0.20和0.40 mmol·g−1的高锰酸钾氧化处理7 d后,PAHs去除率分别达到32.30%、68.84%、87.01%和89.61%,这与赵丹等[10]的研究结果相似。高锰酸钾投加量在0.05~0.20 mmol·g−1时,PAHs的去除率随投加量增加而明显增高。当高锰酸钾投加量增加至0.40 mmol·g−1时,土壤中PAHs去除率无明显变化。以上实验结果表明,在一定的条件下,随高锰酸钾投加量的增加PAHs去除效率呈先升高后趋于平稳的趋势。郭涓等[23]的研究也发现了类似规律。高锰酸钾投加量增加不能持续提高土壤中PAHs去除率。这可能是因为,残留的PAHs存在于土壤颗粒内部,被土壤颗粒紧密吸附,导致其难以被高锰酸钾氧化降解[4, 24]。本实验体系确定的最佳高锰酸钾投加量为0.20 mmol·g−1。
-
不同处理下高锰酸钾消耗量的变化规律如图2所示。0.05、0.10、0.20和0.40 mmol·g−1的高锰酸钾氧化处理7 d后,高锰酸钾消耗量分别为0.05、0.10、0.19和0.27 mmol·g−1。相关性分析结果显示,高锰酸钾消耗量和投加量之间呈显著相关关系 (P<0.05) 。这表明,高锰酸钾消耗量与投加量密切相关,该结果与已有研究结果相似[25-26]。此外,当高锰酸钾投加量从0.20 mmol·g−1增加至0.40 mmol·g−1时,尽管土壤中PAHs质量分数基本保持不变,但高锰酸钾消耗量仍明显增加。高锰酸钾是一种非选择性氧化剂,除了土壤中目标污染物能与其反应外,土壤中还原性物质(如天然有机质、还原性金属 (铁、锰) 、硫等)也可同其发生反应,增加高锰酸钾的消耗量[26]。LIAO等[18]研究发现,土壤中PAHs对高锰酸钾消耗量的贡献是主要的;同时,土壤中其他还原性物质也会消耗高锰酸钾。
-
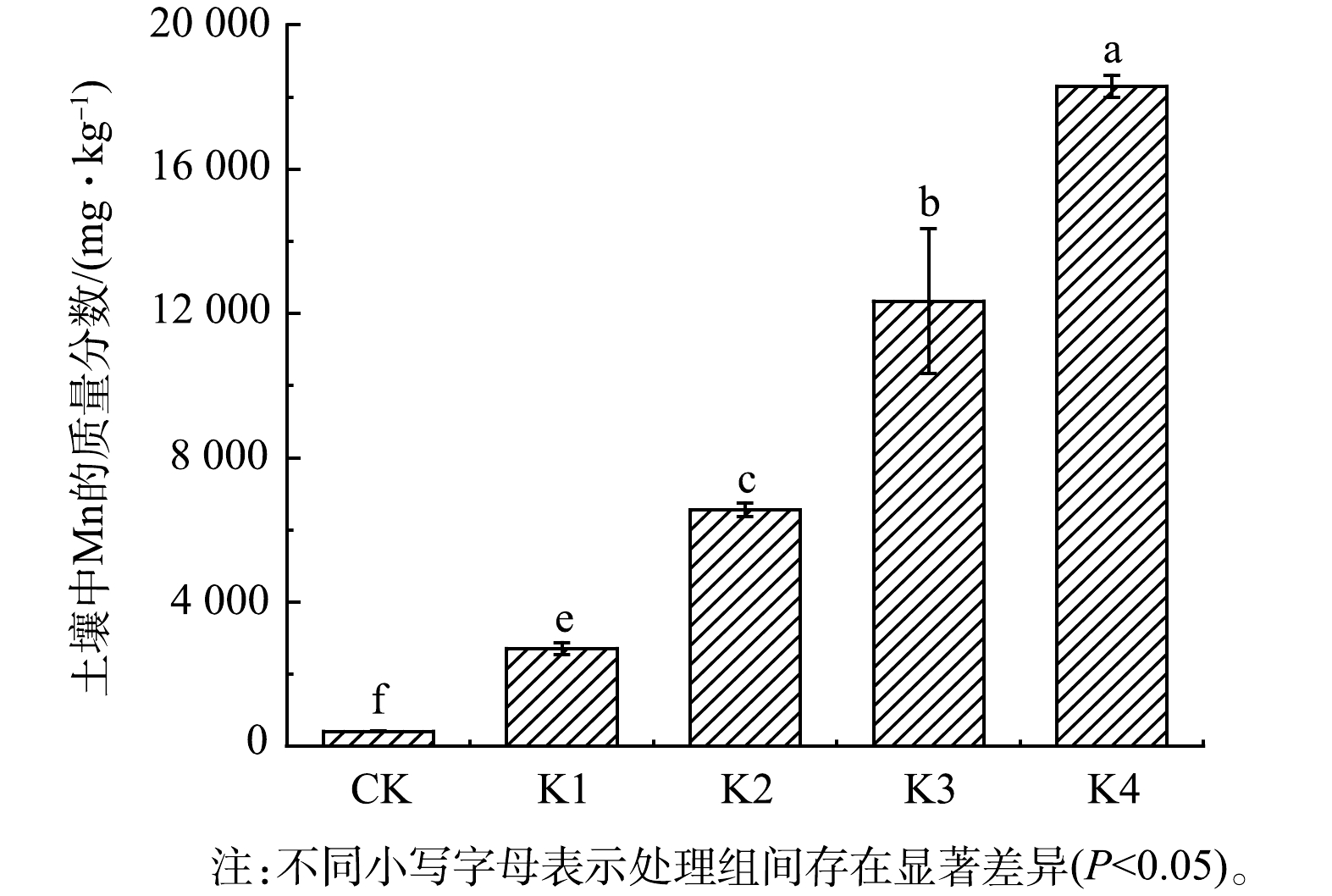
由图3可看出,与对照组相比,高锰酸钾在投加量为0.05、0.10、0.20、0.40 mmol·g−1的条件下氧化处理7 d后,土壤Mn的质量分数明显增加,分别升高了5.5、14.7、28.5、42.7倍。这表明,高锰酸钾氧化处理会使土壤中Mn质量分数升高。土壤中Mn的质量分数与高锰酸钾消耗量之间呈显著相关关系 (P<0.01) 。有研究表明,高锰酸钾氧化处理过程中,消耗的高锰酸钾会转化成MnO2并以固体颗粒物形式残留在土壤中,进而使土壤中Mn质量分数升高[17, 27]。
-
土壤中Mn赋存形态的分布如图4所示。对照组土壤中Mn的赋存形态主要以碳酸盐结合态、铁锰氧化物结合态和残渣态为主,分别占总量的26.62%、30.09%和32.69%。而在0.05~0.40 mmol·g−1高锰酸钾氧化处理后,土壤中Mn的赋存形态主要以铁锰氧化物结合态为主,所占比例为77.04%~92.17%。
由表2可看出,与对照组相比,高锰酸钾氧化处理土壤中可交换态Mn的质量分数均显著降低 (P<0.05) ;而土壤中铁锰氧化物结合态、有机结合态和残渣态Mn的质量分数均显著增加 (P<0.05) ,且随高锰酸钾投加量的增加而升高。研究表明,土壤pH是影响土壤可交换态Mn质量分数的重要因素,其随土壤pH上升而降低[28]。由图5可知,与对照组相比,高锰酸钾处理后土壤pH明显升高,这可能是土壤可交换态Mn质量分数降低的主要原因。在投加低剂量 (0.05和0.10 mmol·g−1) 的高锰酸钾氧化处理中,碳酸盐结合态Mn的质量分数增加可能是由于可交换态Mn转化而来。土壤pH的升高会使游离态的Mn2+沉淀,并与土壤中碳酸钙的表面结合形成碳酸盐结合态Mn[29]。但当高锰酸钾投加量增加至0.20 mmol·g−1以上时,碳酸盐结合态Mn的质量分数明显降低。这可能是因为,过量的高锰酸钾会使碳酸盐结合态Mn向难迁移态Mn转化 (铁锰氧化物结合态、有机结合态、残渣态) ,从而导致土壤中碳酸盐结合态Mn的质量分数下降[30]。总体而言,高锰酸钾氧化处理后土壤中Mn赋存形态的分布与高锰酸钾投加量密切相关。
-
土壤中有效态Mn的质量分数如图6所示。对照组土壤中有效态Mn的质量分数为26.24 mg·kg−1。在高锰酸钾投加量为0.05 mmol·g−1氧化处理下,土壤中有效态Mn的质量分数明显高于对照组,增加了0.94倍。这可能是因为,碳酸盐结合态Mn的质量分数升高导致 (如表2所示) ,因为Tessier法的碳酸盐结合态是土壤中有效态的主要组成部分[31]。但当高锰酸钾投加量为0.10~0.40 mmol·g−1时,土壤中有效态Mn的质量分数明显低于对照组,下降了77.65%~99.09%。上述结果表明,高锰酸钾氧化处理后土壤中有效态Mn的质量分数与高锰酸钾投加量密切相关,合适的高锰酸钾投加量可以降低处理后土壤中Mn的环境风险。
-
1) 高锰酸钾氧化技术可有效去除土壤中PAHs,当高锰酸钾投加量为0.20 mmol·g−1时,PAHs去除率最高。
2) 高锰酸钾氧化处理过程中,高锰酸钾的消耗量与高锰酸钾投加量有关,随投加量的增加而升高;土壤中Mn的质量分数与高锰酸钾的消耗量之间呈显著相关关系,随消耗量的升高而增加。
3) 高锰酸钾氧化处理后土壤中Mn主要以铁锰氧化物结合态存在;高锰酸钾投加量是影响土壤中Mn赋存形态分布的关键因素。
4) 高锰酸钾投加量会影响氧化处理后土壤中有效态Mn的质量分数,是高锰酸钾氧化修复技术应用的关键调控因子。
高锰酸钾修复PAHs污染土壤过程中Mn迁移转化规律
Migration and transformation of Mn during remediation process of soil contaminated with PAHs by potassium permanganate oxidation
-
摘要: 为探究高锰酸钾氧化修复技术应用过程中Mn元素迁移转化规律及其潜在的环境风险,通过室内模拟实验,采用某焦化厂PAHs污染土壤作为研究材料,探究了高锰酸钾修复技术中不同药剂投加量对PAHs去除、高锰酸钾消耗量、土壤中Mn质量分数、Mn赋存形态分布及有效态Mn质量分数等的影响。结果表明,高锰酸钾氧化处理可有效去除土壤中PAHs;当高锰酸钾投加量为0.20 mmol·g−1、反应时间为1 d时,PAHs去除率最高,可达89.61%。氧化处理过程中,高锰酸钾消耗量和土壤Mn质量分数均与高锰酸钾的投加量有关,随投加量增加而升高。其中,土壤Mn的质量分数与高锰酸钾消耗量呈显著正相关关系。高锰酸钾氧化处理后土壤中Mn主要以铁锰氧化物结合态存在,所占比例为77.04%~92.17%。土壤经0.05 mmol·g−1高锰酸钾氧化处理后,土壤有效态Mn的质量分数比对照组增加了0.94倍;而在高锰酸钾投加量为0.10~0.40 mmol·g−1的处理条件下,土壤有效态Mn的质量分数下降了77.65%~99.09%。药剂投加量是影响高锰酸钾氧化修复PAHs污染土壤过程中Mn环境行为的关键因子。本研究结果可为高锰酸钾氧化修复技术应用工艺优化提供参考。Abstract: Potassium permanganate oxidation is an effective soil remediation technology being used widely for removing PAHs from soil. In order to investigate the migration and transformation of Mn, and its potential environmental risk during remediation process of soil contaminated with PAHs by potassium permanganate oxidation, a simulation experiment was carried out with PAHs contaminated soil from a coking site. The effects of different doses of potassium permanganate on amount of consumed potassium permanganate, content of manganese (Mn), speciation of Mn and extractable Mn content in soil were investigated. Then, the migration and transformation of Mn during the remediation process of PAHs contaminated soil by potassium permanganate oxidation was explored. The results showed that potassium permanganate may effectively remove PAHs from the soil of the coking plant. When the dosage of potassium permanganate was 0.20 mmol·g−1 and the reaction time was 1 d, the removal rate of PAHs was the highest, reaching 89.61%. In the remediation process, potassium permanganate consumption and contents of Mn in soil increased with the dose level of potassium permanganate. The result of correlation analysis demonstrated that there is a significant positive correlation between the content of Mn in soil and the amount of consumed potassium permanganate. Moreover, Mn in soil existed mainly in Fe-Mn oxides bound fraction, and its proportions were 77.04%~92.17%. Compared to the treatment without potassium permanganate, the content of extractable Mn in soil increased by 0.94 times after treatment with potassium permanganate at 0.05 mmol·g−1. However, extractable Mn content in soil declined by 77.65% to 99.09% after treatment with potassium permanganate oxidation at 0.10~0.40 mmol·g−1. In conclusion, the dose of oxidant is a key factor for the environmental behaviors of Mn in soil during the remediation process of PAHs contaminated soil by potassium permanganate oxidation. The results of this study can provide a reference for the process optimization of application of potassium permanganate oxidation.
-
Key words:
- potassium permanganate oxidation /
- PAHs /
- remediation /
- manganese /
- migration /
- transformation
-
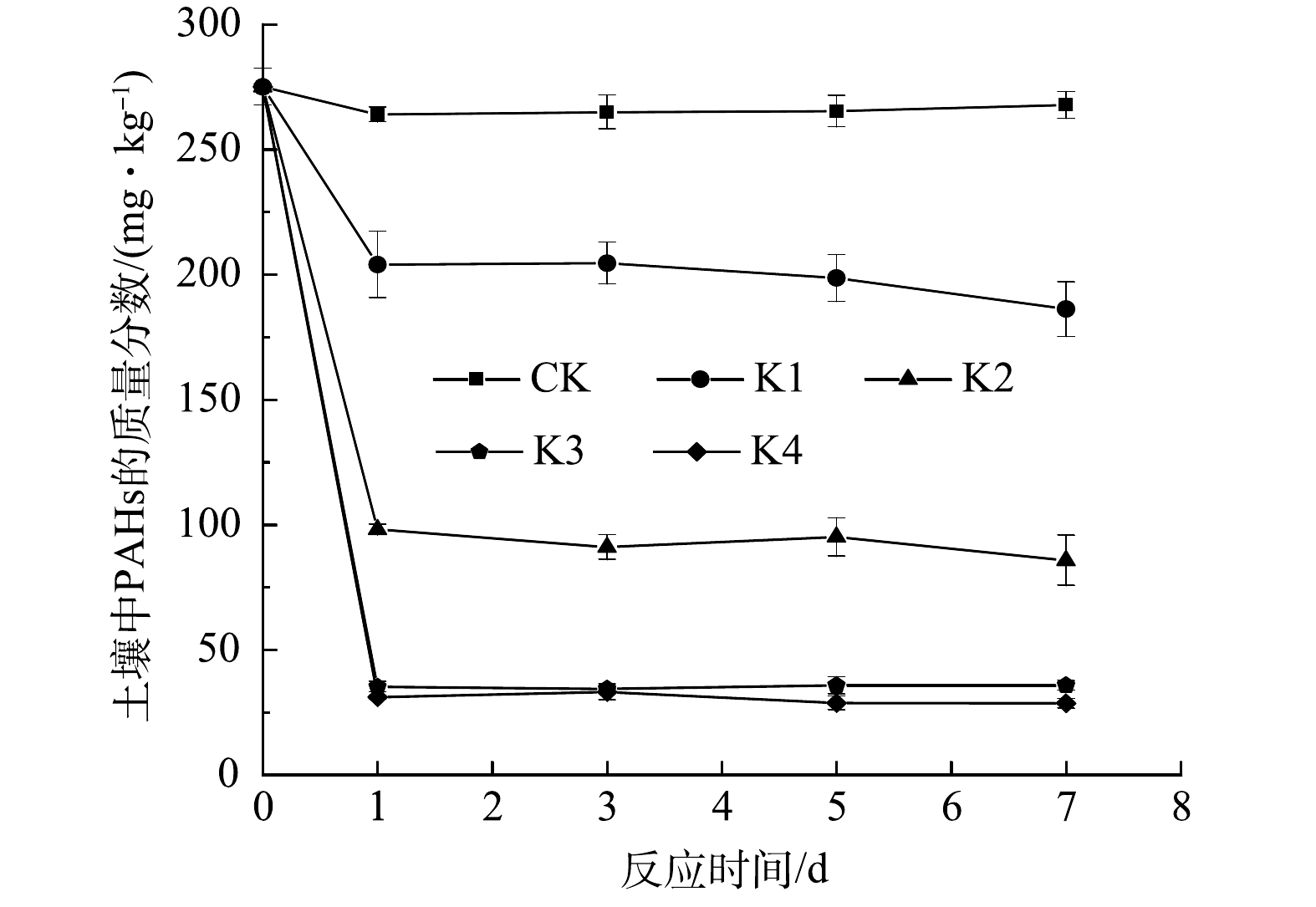
图 1 不同处理下土壤PAHs质量分数的变化
Figure 1. Change of residual PAHs concentration in soil under different treatments
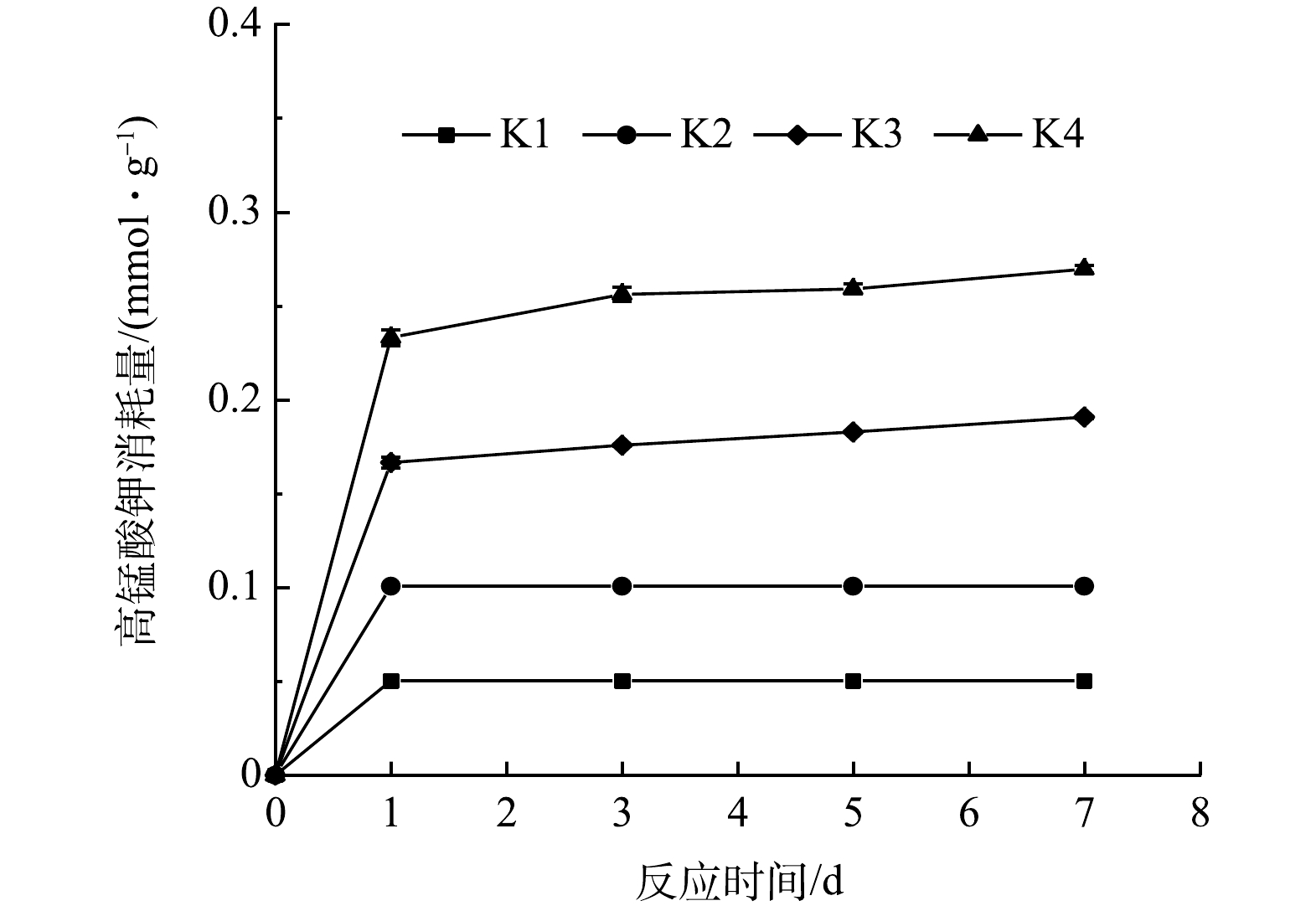
图 2 不同处理下高锰酸钾消耗量的变化
Figure 2. Changes of the consumption of potassium permanganate under different treatments
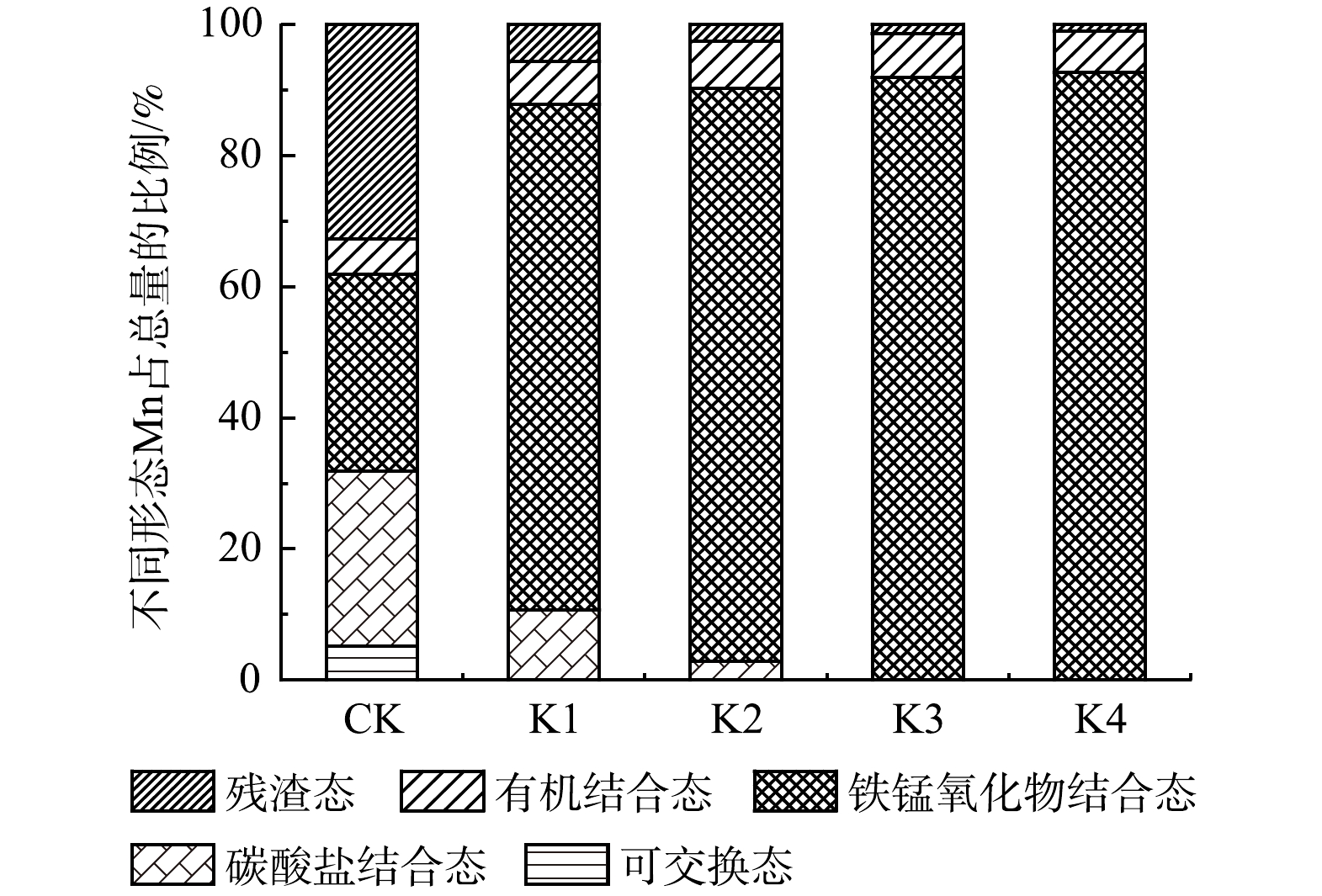
图 4 不同处理下土壤Mn的赋存形态分布
Figure 4. Distribution of Mn chemical forms in soil under different treatments
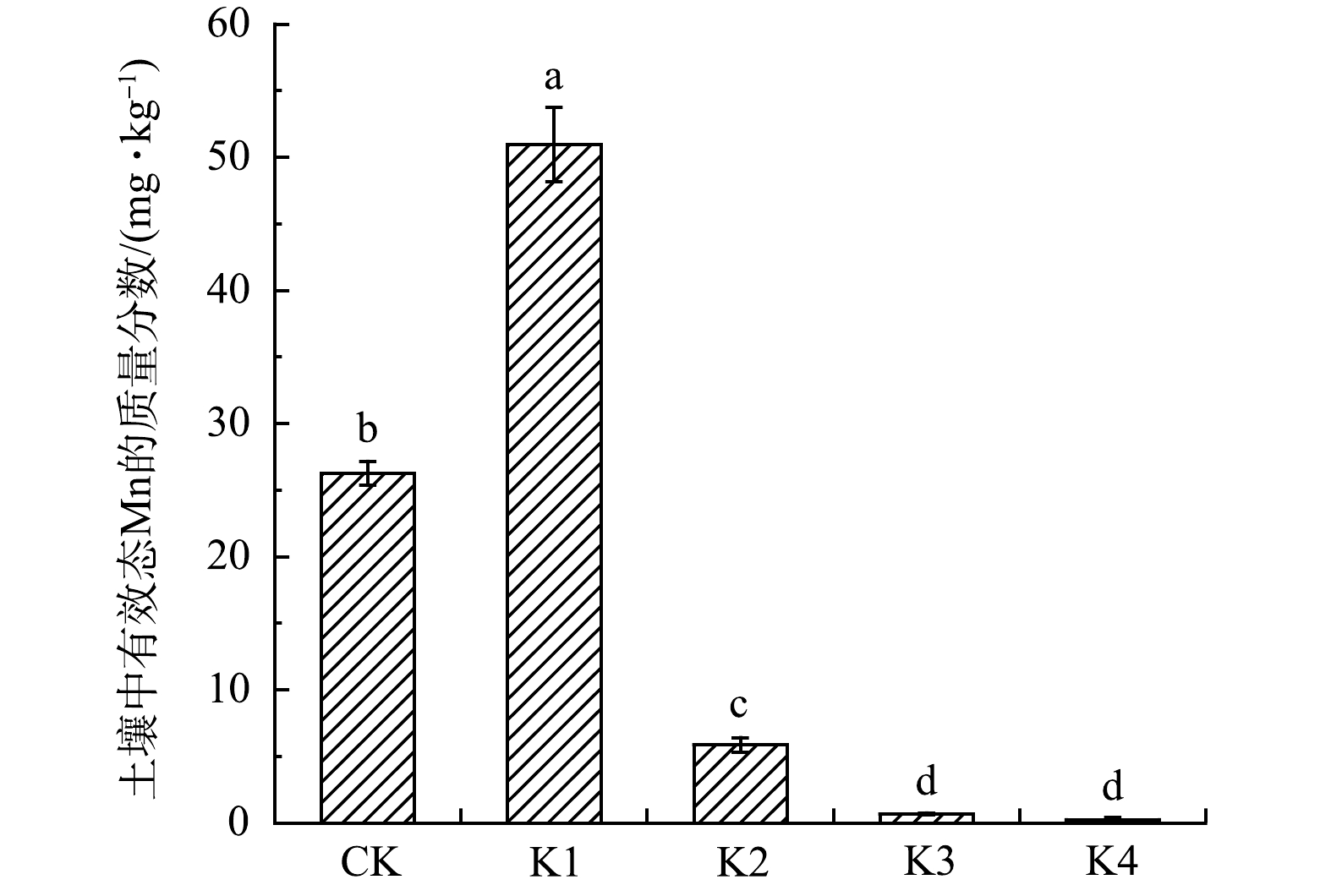
图 6 不同处理下土壤中有效态Mn的质量分数
Figure 6. Contents of available Mn in soil under different treatments
表 1 实验处理
Table 1. List of treatments
处理组 高锰酸钾投加量/(mmol·g−1) 氧化剂溶液浓度/(mol·L−1) 加入氧化剂溶液体积/mL 去离子水体积/mL CK 0 0 0 60.00 K1 0.05 0.20 7.50 52.50 K2 0.10 0.20 15.00 45.00 K3 0.20 0.20 30.00 30.00 K4 0.40 0.20 60.00 0  下载: 导出CSV
下载: 导出CSV
表 2 土壤中不同赋存形态Mn的质量分数
Table 2. Contents of Mn chemical forms in soil
mg·kg−1 处理组 可交换态 碳酸盐结合态 铁锰氧化物结合态 有机结合态 残渣态 CK 21.36±1.00 108.91±5.10 123.11±5.62 21.98±2.64 133.74±9.30 K1 2.00±0.24 295.60±18.15 2 138.21±246.13 181.84±38.68 157.86±5.34 K2 0.29±0.06 208.58±8.51 6 180.25±312.15 513.73±32.19 182.19±5.22 K3 ND 15.52±2.82 12 715.20±8.28 923.56±24.06 200.03±3.33 K4 ND 3.05±0.43 18 302.56±1208.37 1 238.72±99.39 205.65±9.91 注:ND表示未检出。
下载: 导出CSV
-
[1] SANDU C, POPESCU M, ROSALES E, et al. Electrokinetic-Fenton technology for the remediation of hydrocarbons historically polluted sites[J]. Chemosphere, 2016, 156: 347-56. doi: 10.1016/j.chemosphere.2016.04.133 [2] YUE S, GUO D F, CHANG Y Z, et al. Zero-valent iron activated persulfate remediation of polycyclic aromatic hydrocarbon-contaminated soils: An in situ pilot-scale study[J]. Chemical Engineering Journal, 2019, 355: 65-75. doi: 10.1016/j.cej.2018.08.126 [3] RONALD W P. Toxicity of polyaromatic hydrocarbons other than benzo (a) pyrene: a review[J]. Journal of Toxicology:Cutaneous and Ocular Toxicology, 2000, 19(1): 55-67. doi: 10.3109/15569520009051478 [4] RANC B, FAURE P, CROZE V, et al. Selection of oxidant doses for in situ chemical oxidation of soils contaminated by polycyclic aromatic hydrocarbons (PAHs): A review[J]. Journal of Hazardous Materials, 2016, 312: 280-297. doi: 10.1016/j.jhazmat.2016.03.068 [5] 程家丽, 黄启飞, 魏世强, 等. 我国环境介质中多环芳烃的分布及其生态风险[J]. 环境工程学报, 2007, 1(4): 138-144. doi: 10.3969/j.issn.1673-9108.2007.04.032 [6] ZHAO C, DONG Y, FENG Y, et al. Thermal desorption for remediation of contaminated soil: A review[J]. Chemosphere, 2019, 221: 841-855. doi: 10.1016/j.chemosphere.2019.01.079 [7] 杨勇, 张蒋维, 陈恺, 等. 化学氧化法治理焦化厂PAHs污染土壤[J]. 环境工程学报, 2016, 10(1): 427-431. doi: 10.12030/j.cjee.20160171 [8] 苏梦缘, 王红旗, 李艺, 等. 多环芳烃降解菌菌群构建及其适宜降解环境条件的确定[J]. 环境工程学报, 2017, 11(2): 1192-1198. doi: 10.12030/j.cjee.201509056 [9] FENG N X, YU J, ZHAO H M, et al. Efficient phytoremediation of organic contaminants in soils using plant-endophyte partnerships[J]. Science of the Total Environment, 2017, 583: 352-368. doi: 10.1016/j.scitotenv.2017.01.075 [10] 赵丹, 廖晓勇, 阎秀兰, 等. 不同化学氧化剂对焦化污染场地多环芳烃的修复效果[J]. 环境科学, 2011, 32(3): 857-863. doi: 10.13227/j.hjkx.2011.03.033 [11] WALDEMER R H, TRATNYEK P G. Kinetics of contaminant degradation by permanganate[J]. Environmental Science and Technology, 2006, 40(3): 1055-1061. doi: 10.1021/es051330s [12] BENDOUZ M, DIONNE J, TRAN L H, et al. Polycyclic aromatic hydrocarbon oxidation from concentrates issued from an attrition process of polluted soil using the Fenton reagent and permanganate[J]. Water, Air, & Soil Pollution, 2017, 228(3): 115. [13] LIAO X Y, WU Z Y, LI Y, et al. Effect of various chemical oxidation reagents on soil indigenous microbial diversity in remediation of soil contaminated by PAHs[J]. Chemosphere, 2019, 226: 483-491. doi: 10.1016/j.chemosphere.2019.03.126 [14] LEMAIRE J, MORA V, FAURE P, et al. Chemical oxidation efficiency for aged, PAH-contaminated sites: An investigation of limiting factors[J]. Journal of Environmental Chemical Engineering, 2019, 7(3): 103061. doi: 10.1016/j.jece.2019.103061 [15] SCHROTH M H, OOSTROM M, WIETSMA T W, et al. In-situ oxidation of trichloroethene by permanganate: effects on porous medium hydraulic properties[J]. Journal of Contaminant Hydrology, 2001, 50(1): 79-98. [16] CATHERINE S, PAULA T D S E S, CHRISTOPHE S, et al. Impact of chemical oxidation on soil quality[J]. Chemosphere, 2008, 72(2): 282-289. doi: 10.1016/j.chemosphere.2008.01.027 [17] SILVA P, SILVA V, NETO B D B, et al. Potassium permanganate oxidation of phenanthrene and pyrene in contaminated soils[J]. Journal of Hazardous Materials, 2010, 168(2-3): 1269-1273. [18] LIAO X Y, ZHAO D, YAN X L. Determination of potassium permanganate demand variation with depth for oxidation-remediation of soils from a PAHs-contaminated coking plant[J]. Journal of Hazardous Materials, 2011, 193: 164-170. doi: 10.1016/j.jhazmat.2011.07.045 [19] XIONG B J, ZHANG Y C, HOU Y W, et al. Enhanced biodegradation of PAHs in historically contaminated soil by M. gilvum inoculated biochar[J]. Chemosphere, 2017, 182: 316-324. doi: 10.1016/j.chemosphere.2017.05.020 [20] LEE C S, LI X D, SHI W Z, et al. Metal contamination in urban, suburban, and country park soils of Hong Kong: A study based on GIS and multivariate statistics[J]. Science of the Total Environment, 2006, 356(1/2/3): 45-61. [21] TESSIER A, CAMPBELL P G C, BISSON M. Sequential extraction procedure for the speciation of particulate trace-metals[J]. Analytical Chemistry, 1979, 51(7): 844-851. doi: 10.1021/ac50043a017 [22] GRYSCHKO R, KUHNLE R, TERYTZE K, et al. Soil extraction of readily soluble heavy metals and As with 1 M NH4NO3-solution-evaluation of DIN 19730 (6 pp)[J]. Journal of Soils and Sediments, 2005, 5(2): 101-106. doi: 10.1065/jss2004.10.119 [23] 郭涓, 杨玉盛, 杨红玉, 等. 高锰酸钾氧化修复污染土壤中菲和芘的研究[J]. 农业环境科学学报, 2010, 29(3): 471-475. [24] SUN H W, YAN Q S. Influence of pyrene combination state in soils on its treatment efficiency by Fenton oxidation[J]. Journal of Environmental Management, 2008, 88(3): 556-63. doi: 10.1016/j.jenvman.2007.03.031 [25] URYNOWICZ M A, BALU B, UDAYASANKAR U. Kinetics of natural oxidant demand by permanganate in aquifer solids[J]. Journal of contaminant hydrology, 2008, 96(1/2/3/4): 187-194. [26] DANGI M B, URYNOWICZ M A, UDAYASANKAR U. Assessment of the experimental conditions affecting natural oxidant demand of soil by permanganate[J]. Journal of Environmental Chemical Engineering, 2018, 6(4): 5160-5166. doi: 10.1016/j.jece.2018.07.048 [27] SIEGRIST R L, URYNOWICZ M A, CRIMI M L, et al. Genesis and effects of particles produced during in situ chemical oxidation using permanganate[J]. Journal of Environmental Engineering, 2002, 128(11): 1068-1079. doi: 10.1061/(ASCE)0733-9372(2002)128:11(1068) [28] 朱端卫, 万小琼, 耿明建, 等. 酸化及施碳酸钙对土壤各形态锰的影响[J]. 植物营养与肥料学报, 2001, 7(3): 325-330. doi: 10.3321/j.issn:1008-505X.2001.03.013 [29] 黄兴星, 朱先芳, 唐磊, 等. 密云水库上游某铁矿区土壤重金属含量及形态研究[J]. 中国环境科学, 2012, 32(9): 1632-1639. doi: 10.3969/j.issn.1000-6923.2012.09.014 [30] 唐铭, 丁亮, 颜亮, 等. 高锰酸钾法降低自来水锰含量的技术运用[J]. 给水排水, 2003, 29(2): 30-32. doi: 10.3969/j.issn.1002-8471.2003.02.010 [31] 岳聪, 汪群慧, 袁丽, 等. TCLP法评价铅锌尾矿库土壤重金属污染: 有效剂的选择及其与重金属形态的关系[J]. 北京大学学报(自然科学版), 2015, 51(1): 109-115. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5207
- HTML全文浏览数: 5207
- PDF下载数: 73
- 施引文献: 0
