


-
沸石是一种水合铝硅酸盐的包合物,晶格内部孔穴比表面积大,晶格内部水分子与金属离子同矿物骨架的联结较松弛,使得骨架孔道内阳离子出入较为自由,形成较高的离子交换吸附容量,故对氨氮吸附能力较强,常用于富氨氮(NH4+)废水的快速处理[1]。然而,吸附过程因其存在饱和吸附容量,在实际应用中使用寿命较短,故吸附材料的广泛应用常受再生方法便利性及材料替换经济性的制约。传统的沸石再生方法常利用高盐或酸碱溶液[2-4]对吸附位点上的NH4+进行离子交换而实现吸附能力再生,但此类方法的效率有限,大量的药剂使用也使得再生成本较高,在我国已确立“双碳目标”的背景下,沸石的再生仍需寻找一种高效低耗的技术手段。有研究表明,影响沸石对氨氮吸附平衡过程的主要因素包括沸石粒径、沸石投加比、溶液pH、初始氨氮浓度、反应时间、反应温度等[5-6]。从实操性的角度考虑,粒径和沸石投加比受沸石选型和吸附能力的制约很难受人为调控[7-8];调节溶液pH会显著增加药剂消耗及再生成本并产生附加废弃物[9];初始氨氮浓度和反应时间受处理化合物自身特性及化学性质制约而不宜调控[10]。而反应温度调控属于可控性较高、副产物少且环境友好性较佳的再生手段,但能效的保障是主要难点,结合当下“双碳目标”的要求,值得进一步探索与优化[11-12]。
在传统的温度调控方式中,蒸汽、加热夹套或浸没式换热管等间接接触传热是大规模工程中较常用的手段,使用较为成熟,但间接传热常会发生2种以上相界面的传热过程,相间传热损失与环境热量散失较难忽略,热效率与热选择性不高[13]。传统的直接加热方法一般通过电阻加热元件加热接触介质达到升温目的,但电阻丝构型较固定,满足局部微环境需求的形态可调节性较差,在污水环境中电路接口密封性要求较高,故使用过程中也较难抑制加热元件在环境介质中的热损失,限制了吸附剂的再生效率及系统经济性[14]。较传统加热方法而言,微波具有直接快速加热内部物料的优点,微波能通过分子振动和摩擦转化为热量,能效高,但微波对场域内材料无差别作用,沸石再生时微波辐射区域内的水体不可避免被加热,能量损耗较大,可操作性大幅降低[15-16]。而光热技术利用光热转换材料可将光能转换为热能实现可控区域的加热,具有低功耗、光学简单等特点,但目前该技术尚处研发阶段,且光热转换材料制备价格昂贵,在环境领域的应用较为受限[17]。因此,通过温度调控沸石再生的方法仍需进一步优化,以寻求技术效率、经济与可操作性的平衡点。
磁热效应可以通过导体在交变磁场下产生涡旋电流而实现焦耳热快速加热的效果,是一种磁能高效实现热量转化的加热方式[18]。磁热效应加热较电阻加热有更强的构型灵活性和微型化潜力,较微波加热有更多的材料选择性,较光热技术有更佳的经济性。本研究拟将沸石与微型化磁热导体耦合成圈层结构,实现结构化的沸石载体局部磁致自辅热,通过对交变磁场的调控实现结构化沸石载体局部温度的精确调控,并辅助传质较好但导热系数较低的保温层材料设计,有效减少局部控温过程中的热量损失,提高沸石载体在自辅热系统中的热利用率、再生效率及经济性,实现低耗、高效、可控的沸石载体再生过程,从而可促进环境材料绿色循环利用。
本研究通过筛选优化的磁热导体与磁场条件,考察了结构化沸石载体的局部自辅热特性及水中保温特性,并在不同初始氨氮浓度条件下研究了磁致自辅热沸石在局部加热下的吸附及解吸动力学特征,探明了磁致自辅热效应对沸石再生过程的影响机制,以期为应用过程中实现低耗高效解决沸石再生难题提供参考。
-
实验选用商品级人造沸石,粒径为1.6~2.5 mm,沸石经去离子水洗涤后放入干燥箱中105 ℃烘干备用;磁热升温材料选用表面经防水处理的磁热导体,直径分别为2、4、6、7 mm,长度分别为4、6、8、10 cm;局部自辅热保温材料选用聚氨酯。
-
1)磁热导体的筛选及载体升温特性测试。在恒温(T=25℃)环境中,将磁热导体放置于不同磁场强度中,每隔5 min记录其在交变磁场中的温度直至温度达到平衡,其发热功率密度根据式(1)进行计算[19]。
式中:k为发热功率密度,表示单位体积发热功率,kW·m−3;c为磁热导体比热,J·(kg·℃)−1;m为磁热导体质量,kg;
ΔT 为平衡时温度变化,°C;t为达到平衡所需时间,s;Vc为磁热导体体积,m3;P为磁热导体发热功率,kW。称取人造沸石35 g,将其与筛选出的磁热导体及聚氨酯组装成如图1(a)所示载体形式。载体为圈层结构设计,最外层为传质性较好传热系数较低的聚氨酯保温层,中间层为用作氨氮吸附剂的人造沸石,最内层为磁热导体。导体在交变磁场中引起局部加热,通过热传递将温度传递至吸附剂层,导致吸附剂层升温,而外层含有隔热聚氨酯材料,因此,热量被保存在中间层,而外部溶液始终保持温度恒定状态,吸附剂在升温作用下进行氨氮吸附,达到局部加热的目的。将载体放置于装有1 L水的容器中,外设循环装置保持恒温状态(T=25 ℃),实验装置如图1(b),每隔10 min记录其在磁场中的升温情况至温度达到平衡。
2)结构化载体材料吸附特征。载体固定在盛有1 L初始质量浓度为300、400、500、600和700 mg·L−1(以N计)NH4Cl标准溶液的反应容器中,分别放置于交变磁场与恒温环境,连续搅拌(150 r·min−1)12 h,每1 h取1次样,采用纳氏试剂分光光度法测定氨氮质量浓度,实验重复3次。氨氮吸附量计算方法如式(2)所示[20]。
式中:qe为吸附平衡时的氨氮吸附量,mg·g−1;C0为溶液中氨氮初始质量浓度,mg·L−1;Ce为平衡时溶液中氨氮质量浓度,mg·L−1;Vl为反应溶液体积,L;m为吸附剂质量,g。
3)等温吸附实验分析。分别采用Langmuir[21](式(3))和Freundlich[22](式(4))方程对式(2)所得结果进行拟合。
式中:qmax为最大吸附能力,mg·g−1;KL为Langmuir常数;KF为Freundlich常数;
1n 是与吸附强度有关的参数,吸附强度随吸附材料的非均质性而变化。4)吸附动力学实验。载体固定在盛有1 L初始质量浓度为600 mg·L−1(以N计)NH4Cl标准溶液的反应容器中,分别放置于磁场与恒温环境,连续搅拌(150 r·min−1),吸附12 h,实验平行重复3次。分别采用准一级动力学方程(式(5))[23]、准二级动力学方程(式(6))[24]、Weber-Morris模型(式(7))[25-26]进行吸附动力学方程拟合。
式中:qt为t时刻单位吸附剂的吸附量,mg·g−1;K1为准一级吸附速率常数,min−1;K2为准二级吸附速率常数,g·(mg·min)−1;Ki为粒子吸附内扩散常数,mg·(min0.5·g)−1;C为常数。
5)载体解吸再生实验。将在不同质量浓度NH4Cl标准溶液中吸附饱和的载体放置于1 L纯水中,分别放置于交变磁场与恒温环境,连续搅拌(150 r·min−1),解吸12 h,每1 h取1次样,采用纳氏试剂分光光度法测定氨氮质量浓度,实验平行重复3次。氨氮解吸量根据式(8)进行计算[27]。
式中:qeʹ为氨氮解吸量,mg·g−1;C0ʹ为溶液中氨氮初始质量浓度,mg·L−1;Ceʹ为平衡时溶液中氨氮质量浓度,mg·L−1;m为吸附剂质量,g。
6)解吸动力学实验。将在600 mg·L−1(以N计)NH4Cl标准溶液中吸附饱和的载体放置于1 L去离子水中,分别放置于交变磁场与恒温环境,连续搅拌(150 r·min−1),解吸12 h,每1 h取1次样,采用纳氏试剂分光光度法测定氨氮浓度,实验平行重复3次。解吸动力学采用准一级(式(9))[23]、准二级(式(10))[24]动力学方程及Weber-Morris模型(式(11))[25-26]进行拟合。
式中:qtʹ为t时刻单位吸附剂的解吸量,mg·g−1;K1ʹ为准一级解吸速率常数,min−1;K2ʹ为准二级解吸速率常数,g·(mg·min)−1;Kiʹ为粒子解吸内扩散常数,mg·(min0.5·g)−1;Cʹ为常数。
-
不同规格磁热导体在不同交变磁场中发热功率密度情况如图2所示。X、Y、Z轴分别代表导体长度、直径及交变磁场强度,球体体积大小代表导体在磁场中发热功率密度数值。由图2可见,发热功率密度随着磁热导体长度增长而增加,10 cm导体发热功率密度最大值可达52.3 kW·m−3,较长度为8 cm的导体高8.9 kW·m−3,提升了20.5%。这表明导体长度是发热功率密度的关键影响要素之一。这是由于发热功率与加热导体的长度成正相关,相同条件,长度越长,涡流产热越大,发热功率越高[28]。同时,通过X-Y面数据可以发现,交变磁场强度亦与发热功率密度呈显著正相关性。这是由于涡流产热量与正弦交变磁场场强的平方成正比[29]。由图2可见,导体直径对发热功率密度未产生显著影响。这是由于发热功率密度计算中体积因素已被折算,因此,导体直径为4 mm时已能达到相同长度导体的最优发热功率密度。综上所述,在55 Gs条件下,直径4 mm、长度10 cm的磁热导体可达最大发热功率密度为52.3 kW·m−3。
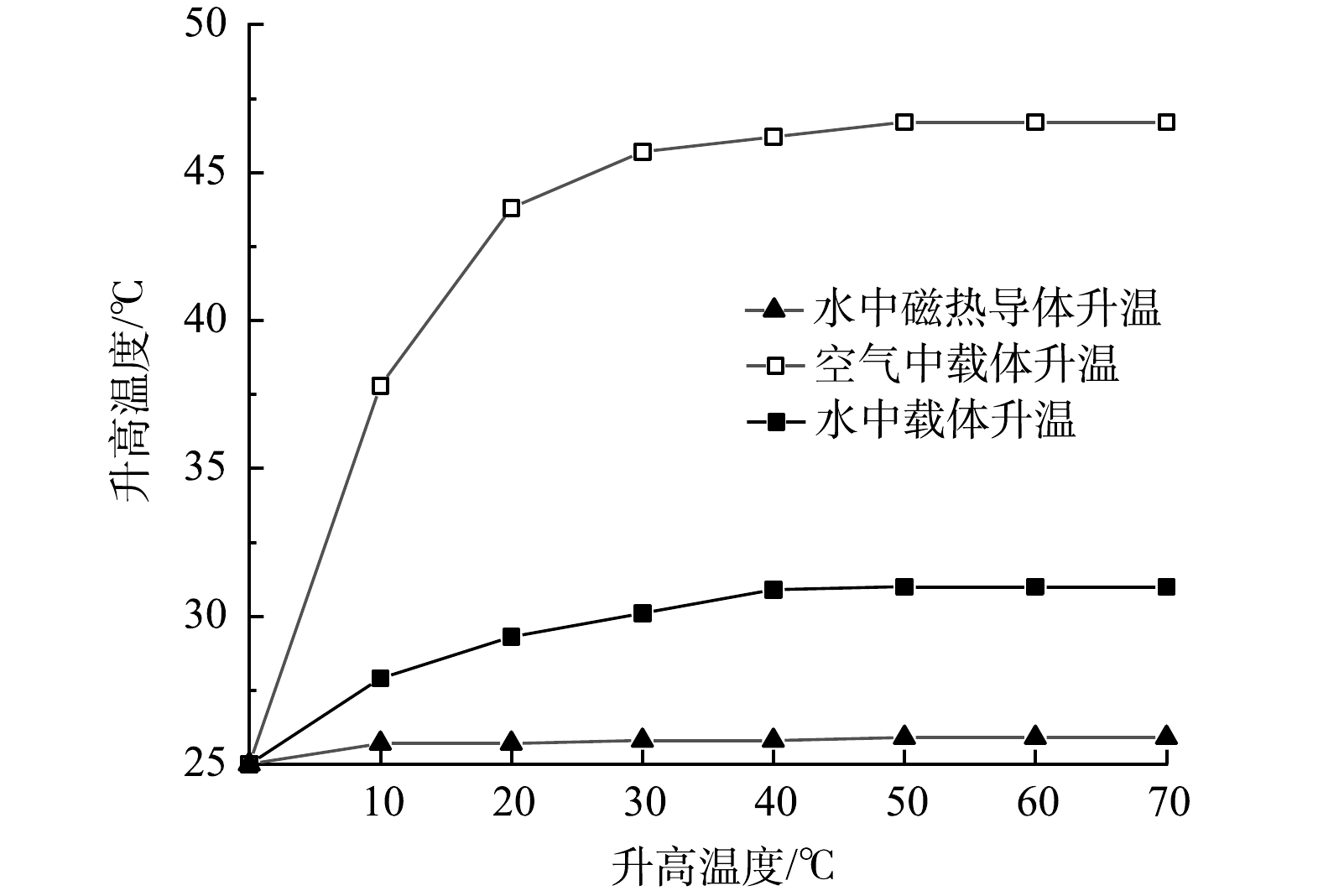
为识别结构化载体实现可保温的局部自辅热的可行性,载体磁热导体在空气与水中的升温情况与裸露磁热导体在水中的升温情况进行了对比。由图3可见,裸露磁热导体在交变磁场中产生的热量被周围水体迅速吸收,温度升高仅有0.9°C。此结果突破了以升温法为调控手段的主要技术难点。当磁热导体在结构化载体中时,磁热效应的保温效果得以实现,空气中载体磁热导体的最高升温可达46.7°C,水中载体磁热导体的升温幅度有所降低,但最高升温也可达31°C,结构化载体在水中的保温效果得以证实。水中的升温效率可在后续实验中进一步优化。
-
恒温及局部自辅热条件下结构化载体对不同氨氮初始质量浓度的吸附性能如图4所示。可见,随着氨氮初始质量浓度升高,恒温及局部自辅热的载体吸附量整体呈上升趋势,12 h恒温及局部自辅热载体对氨氮的吸附量分别可达12.18 mg·g−1和9.25 mg·g−1,且后者始终比前者的吸附量低。有研究表明,在溶液温度升高情况下,NH4+吸热运动速率加快,克服沸石表面阻力的能力随之增加,促使沸石表面吸附的氨氮向沸石内部迁移,导致沸石吸附量增大[30]。而磁致自辅热局部升温过程中,溶液温度并未升高,吸附量低于恒温状态。这可能是由于载体吸附过程中热传递方向发生改变,由吸热转换为放热,致使NH4+离子脱附到水溶液中[31],从而导致吸附量降低,说明载体局部自辅热作用对氨氮的吸附存在削弱作用。
-
为了更好地研究局部自辅热过程对载体吸附特性的影响,利用Langmuir及Freundlich模型对载体氨氮吸附量进行拟合,2种模型的相对参数分别由拟合方程的斜率和截距计算得到,拟合结果如图5所示,相应参数如表1所示。
Langmuir模型用于描述均匀的单分子层吸附,吸附剂的每个吸附位点均有相同的吸附能力且各位点之间相互不受影响,吸附类型为化学吸附,KL值反映吸附质吸附氨氮的能级,代表吸附质与氨氮之间的结合能,KL值越大,结合越稳定[32]。Freundlich模型则表示非均匀的多分子层吸附,代表物理吸附过程,KF表示吸附质吸附氨氮能力的大小,KF越大,吸附能力越强[32];
1n 的大小代表着吸附反应的发生的难易程度,1n 小于0.5说明吸附较容易进行,其值越大吸附越困难[33]。2种情况中Freundlich方程的R2均优于Langmuir方程,说明Freundlich模型拟合效果好,多分子层的物理吸附过程占主导。Freundlich模型中对比恒温中载体吸附,KF值较小且局部自辅热过程的1n >1,说明载体吸附能力较弱,物理吸附过程存在阻碍。这是由于磁场局部自辅热使载体内部温度升高,放热吸附的吸附量随温度的升高而降低,此外吸附强度KF较小,也证明其物理吸附能力受到影响。 -
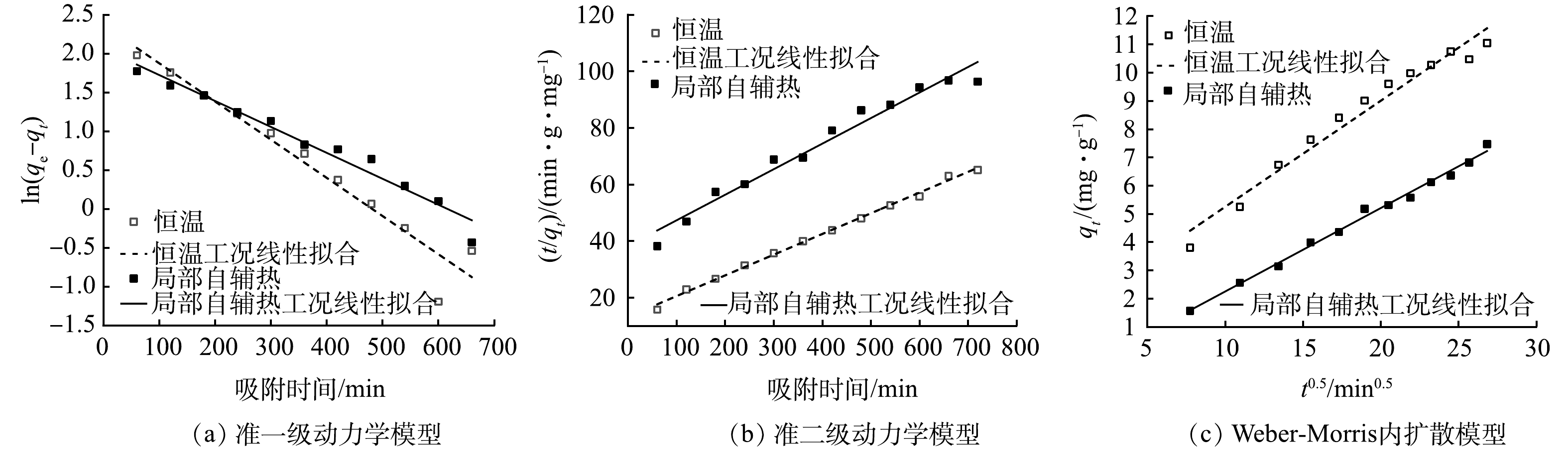
为更好了解在局部自辅热时载体对氨氮的吸附动态,采用准一级及准二级吸附动力学模型对吸附数据进行拟合分析。动力学模型拟合结果如图6所示,相应的参数如表2所示。
准一级动力学模型指吸附速率受扩散作用控制,适用于描述只存在物理吸附的吸附和释放行为[34]。准二级动力学模型指吸附速率受化学吸附控制,吸附过程中吸附质与吸附剂之间存在电子共用及电子转移作用[35]。由表2可见,恒温下载体的准二级模型R2=0.996 1高于准一级模型(R2=0.941 7)。这说明准二级方程对实验数据的拟合较好。添加磁场对载体进行局部升温后,准二级模型R2=0.964 7高于准一级模型(R2=0.963 3)。说明准二级模型能更好地描述局部自辅热过程中载体的吸附过程,吸附过程中存在着电子共用或电子转移的情况。
由于准一级及准二级动力学模型不能描述扩散机制,因此,采用Weber-Morris模型进行拟合。Weber-Morris模型用于表述吸附质的扩散类型,其定义粒子内扩散是速率限制的渐进吸附步骤,拟合直线如果经过原点,说明吸附过程主要受粒子内扩散控制[36],若不经过原点则说明存在液膜扩散并伴随着颗粒内扩散,截距的值与液膜扩散强度呈正相关[37]。如图6(c)和表2所示,恒温工况下内扩散模型的R2大于0.95,说明拟合程度较好。而局部自辅热工况下内扩散模型的R2大于0.99,说明拟合程度更好,粒子内扩散显著。但其拟合直线未经过原点(C≠0),证明扩散机制较为复杂,说明氨氮在孔隙中的扩散并不是控制吸附过程的唯一决定因素,实际吸附过程主要受粒子的内扩散和液膜扩散共同控制[38]且液膜扩散更为显著。
-
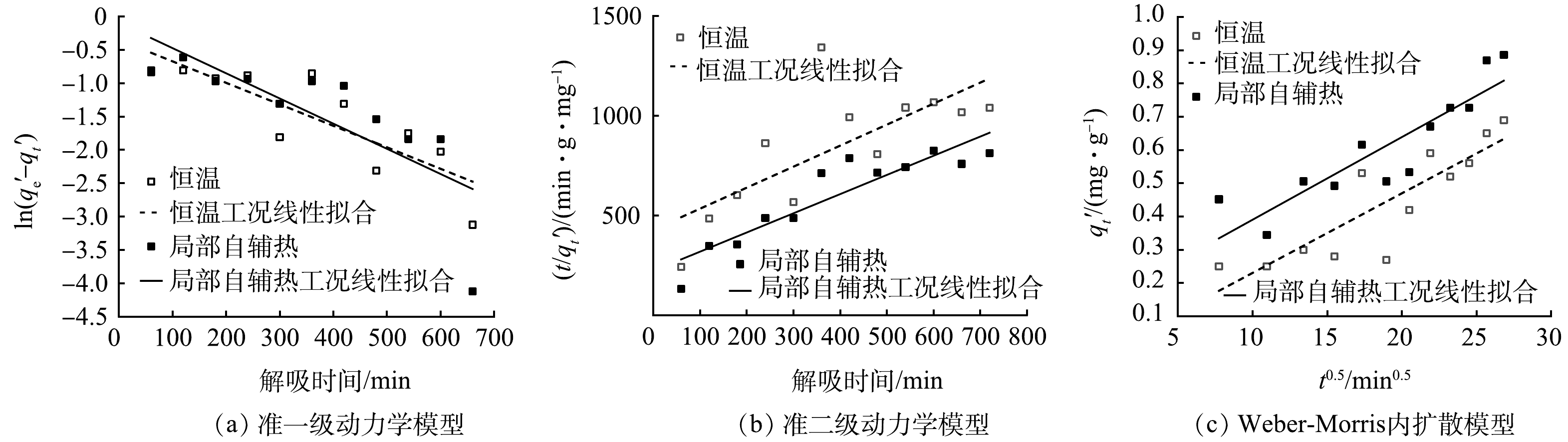
载体解吸过程中吸附量随时间的变化情况如图7所示。可见,解吸量随时间延长而波动。说明解吸过程是与吸附相结合的一个动态过程。同时,局部自辅热载体的解吸量始终比恒温下的解吸量高,12 h时的解吸量分别为0.89 mg·g−1和0.69 mg·g−1。由此证明载体的局部自辅热有利于加速沸石内部NH4+离子脱附到水溶液中,对沸石吸附解吸氨氮起到有效调控作用。
-
解吸动力学拟合情况及相关参数如图8及表3所示。准一级方程对恒温中载体实验数据拟合程度较好(R2=0.670 5);而局部自辅热工况下载体解吸动力学行为则与准二级模型(R2=0.819 5)更吻合,说明氨氮解吸过程主要受化学作用控制[27]。与吸附过程的Weber-Morris模型拟合结果相比,R2均小于0.95,说明2种工况解吸过程拟合程度较差。此外,局部自辅热中的线性拟合偏移原点较远,说明粒子的外部扩散在解吸过程中起主要作用。
-
1)规格4 mm×10 cm的磁热导体由于其合适的高度及直径发热功率密度较大,可达52.3 kW·m−3,以其做发热内核的结构化载体在聚氨酯包裹下放入水中能够起到良好的升温及保温效果,最大升温可达31°C。
2)局部自辅热情况下的载体由于放热作用,致使NH4+离子脱附到溶液中,吸附量相对减少,其吸附与Freundlich模型拟合较好,KF值较小且局部自辅热过程的
1n >1,说明载体吸附能力较弱,物理吸附过程存在阻碍。准二级模型能更好的诠释局部自辅热过程中载体的吸附行为,实际吸附过程主要受粒子的内扩散和液膜扩散共同控制。3)局部自辅热载体解吸过程符合准二级动力学方程,说明氨氮解吸过程主要受化学作用控制,载体的局部自辅热有利于NH4+离子脱附到溶液中。粒子的外部扩散在解吸过程中起重要作用,说明局部自辅热可以起到调控氨氮吸附的作用。
磁致自辅热沸石控温载体构建及其氨氮吸附再生调控机制
Preparation of a magnetically-induced self-heating zeolite carrier and the corresponding regulation mechanism on ammonia adsorption and regeneration
-
摘要: 为解决常规沸石再生过程药耗较高的问题,满足当下双碳目标的要求,构建了结构化磁致自辅热沸石控温载体,利用磁热效应实现沸石氨氮原位吸附过程调控,避免再生过程中的药剂消耗。在优化筛选磁场条件与发热内核构型基础上,应用Langmuir、Freundlich模型、动力学模型及粒子内扩散模型考察了局部自辅热对沸石氨氮原位吸附及解吸过程的影响。结果表明,局部自辅热使沸石氨氮吸附行为更满足Freundlich模型,以物理吸附为主,动力学过程为准二级动力学模型,以粒子内扩散和液膜扩散为主。同时,局部自辅热使沸石氨氮12 h解吸率提升约29 %,动力学拟合更满足准二级动力学模型,为化学解吸过程,扩散方式以以粒子外部扩散为主。最终证实,磁致自辅热可以有效调控沸石氨氮吸附解吸过程,可为沸石可控再生提供一种低耗高效途径。Abstract: In order to resolve the problems of high reagent-consuming in conventional regeneration of zeolite and meet the current global trends of emission peak and carbon neutrality, a layered-structure magnetically-induced self-heating zeolite carrier was built to regulate the in-situ ammonia adsorption/desorption process and avoid the extra reagents consumption for material regeneration. Langmuir model, Freundlich model, chemical kinetics, and intraparticle diffusion model were used to study the effect of local self-heating on the mechanism of in-situ ammonia adsorption/desorption process on zeolite. The result showed that local self-heating led to a better fit of ammonia adsorption behavior on the self-heating zeolite carrier by Freundlich model than others, which was dominated by physical adsorption process. The adsorption kinetics could be well fitted by pseudo-second-order dynamic model which is dominated by intraparticle and liquid-film diffusion. Meanwhile, the desorption capacity of the self-heating zeolite carrier increased by 29% within 12 h compared to the common zeolite carrier, the former could be better fitted by pseudo-second-order dynamic model, and the chemical desorption process was promoted, which implied that an elevated desorption process was controlled by external particle diffusion. In general, the self-heating zeolite carrier could effectively regulate in-situ ammonia adsorption/desorption process, provide a controllable regeneration approach with low-carbon and cost-effectiveness for zeolite.
-
短程硝化技术较现行污水处理厂普遍应用的传统全程硝化技术具有显著的经济优势,对于活性污泥工艺处理普遍面临的低碳氮比(C/N)污水具有可持续发展意义[1-2]。短程硝化即通过氨氧化细菌(AOB)的作用将氨氮氧化为亚硝酸盐;由亚硝酸盐氧化细菌(NOB)将亚硝酸盐进一步氧化为硝酸盐,通过以上2步过程完成全程硝化[2]。有研究表明,通过调节运行参数可以实现亚硝酸盐积累,如采用较低的溶解氧(DO)、高温、调节pH从而实现较高的游离氨(FA)或游离亚硝酸(FNA)浓度等。李培根等[3]采用较低的DO约0.3 mg·L−1,在pH为8.0的条件下,采用序批式反应器(SBR)成功实现亚硝酸盐积累率约80%。ZHENG等[4]将DO、温度和pH分别控制在0.4~0.6 mg·L−1、35 ℃和8~8.2,在无纺布生物转盘反应器内实现同步短程硝化、厌氧氨氧化和反硝化过程。SAUDER等[5]的研究表明,在21~33 ℃时,温度升高可提高亚硝酸盐积累的程度。此外,DURAN等[6]采用完全混合反应器(CSTR)进行了短程硝化,在pH为7.1~8.5进行调节,控制FA低于10 mg·L−1、FNA高于0.2 mg·L−1。然而实际中的低温地区或时节的大型污水处理厂若长期通过加热设备实现温度控制、或通过投加碱以提高pH所需费用较高,控制DO实现亚硝酸盐积累具有实际可行性。虽然已证实采用低DO可以实现亚硝酸盐积累,但是氨氮氧化速率随着DO降低也会随之下降[7-9]。
活性污泥生物污水处理工艺中功能菌的比例直接影响生物降解速率[7, 10]。著名的生物添加强化技术(BABE)正是基于此原理,通过在污水处理工艺的侧流反应器培养硝化细菌,将侧流中富有硝化细菌的污泥回流于主流工艺,从而强化主流工艺的硝化能力[11-12]。此外,已有研究表明载体挂膜、细胞固定化等方式也可以实现增加反应器内的目标功能菌。ANTILEO等[9]采用内有纺织材料载体的生物转盘反应器,将DO控制1.0 mg·L−1以下,通过pH控制以及在氨氮氧化完成时停止曝气的运行策略,实现AOB的优势增长,长期实验结果表明亚硝酸盐积累率稳定维持在84%~88%。徐浩等[13]的研究结果表明,添加立体弹性填料的序批式生物膜反应器(SBBR)利于AOB菌种的富集,亚硝酸盐积累率达到90%以上。DANIEL等[14]证实了固定化载体利于AOB菌体附着其上,促进亚硝酸盐积累的形成。因此,为加速在低DO条件下实现亚硝酸盐积累同时实现较高的氨氮氧化速率,向主流工艺中投加AOB以实现其相对于NOB的菌群优势,从而强化氨氮氧化至亚硝酸盐的能力,具有重要的现实意义。
在水污染控制领域,生物反应器通常采用连续流或间歇运行方式。已有研究采用此2种运行方式在30 ℃左右的温度条件下,实现了AOB的富集培养,历时一个月至数月[15-16]。本研究试图在常温(20 ℃)条件下,采用底物流加-间歇运行方式进行AOB富集培养,探讨了pH、FA、FNA、DO等因素对其的影响,并对富集过程中AOB进行了定性和定量分析,旨在为常温快速富集高纯度的AOB提供指导。
1. 材料与方法
1.1 实验装置和原水
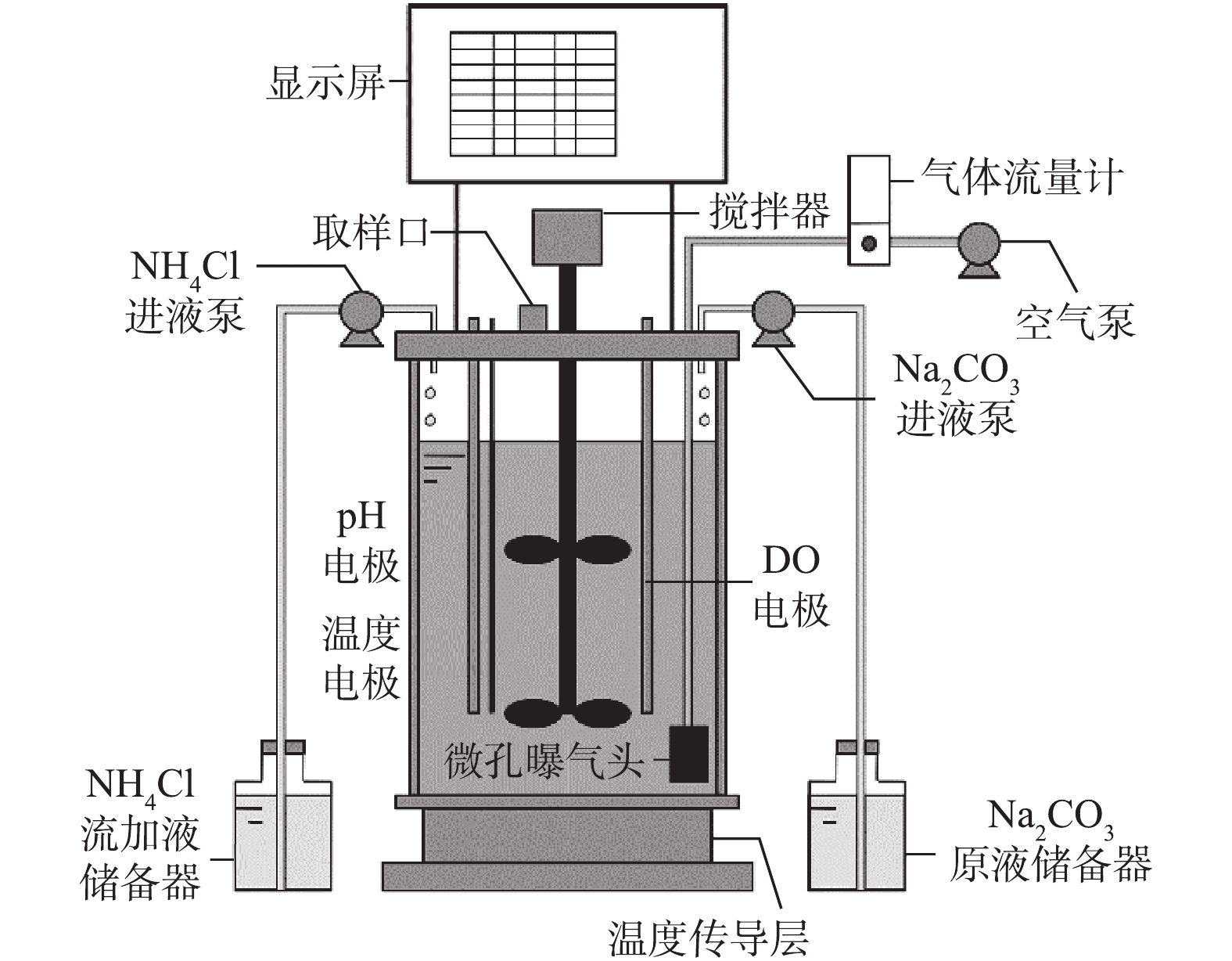
采用全自动发酵罐(BLBIO-5GJ,上海百仑生物科技有限公司,中国),为SBR反应器,有效容积为4 L,如图1所示。DO、pH和温度电极实时监测反应液的DO浓度,pH和温度;以50 r·min−1搅拌,使反应液与活性污泥完全混合;温度调节装置自动调节至预期温度(20±2) ℃;空气通过微孔曝气头均匀曝气,气体流量计控制曝气量;采用反应器自带的蠕动泵用于流加氨氧化反应底物
NH+4 (NH4Cl),流量为21 mL·h−1;另一蠕动泵根据预期pH自动补充碱溶液(Na2CO3 200 g·L−1);取样口用于加培养液、虹吸排水和取样。接种污泥取自北京高碑店污水处理厂A2/O工艺回流污泥。原水采用人工配水,由高浓度基础培养液和自来水配制成预期浓度,基础培养液具体组成成分为KH2PO4 7.02 g·L−1、MgSO4·7H2O 2.00 g·L−1、CaCl2 1.20 g·L−1、FeSO4·7H2O 2.00 g·L−1,微量元素(1 L)含ZnSO4·7H2O 0.50 g·L−1、MnCl2·4H2O 0.50 g·L−1、CoCl2·6H2O 0.40 g·L−1、CuSO4·5H2O 0.40 g·L−1、NiCl2·6H2O 0.20 g·L−1。
1.2 实验方法
采用底物流加-间歇运行方式[17-18],每个周期具体为四个阶段:加培养液、反应、污泥沉淀和排水。培养时间为18 d,其中第1~7天每天1周期(22 h),第8天开始每天2周期(8 h和12 h),共运行18天、29周期。具体步骤为:将自来水与计算好体积的基础培养液加入反应器,使得反应液初始浓度为80 mg·L−1
NH+4 -N (视情况添加),20 mg·L−1PO3−4 -P,25 mg·L−1 MgSO4·7H2O,15 mg·L−1 CaCl2,25 mg·L−1 FeSO4·7H2O 和1 mL·L−1微量元素;打开搅拌器、温度控制器、pH调节器、曝气装置、底物流加和补碱泵开始反应,预先配制底物和碱溶液于原液储备器中,根据前一周期的底物反应速率适当调整本周期的底物流加速率;反应结束,停止搅拌、温度控制、pH调节、曝气、底物流加和补碱装置;虹吸排水以确保下一周期培养液添加前剩余液体体积小于4 L,并视亚硝酸盐积累程度进行“污泥清洗”。采用此运行方式需在反应开始和结束时进行取样。通过调节反应液的pH、控制底物流加速率和实际反应速率的关系以实现体系内预期氨氮和亚硝酸盐浓度范围及预期的FA和FNA浓度,考察了pH、FA、FNA对AOB富集的影响。并结合污泥浓度以及采用不同DO浓度考察生物量及DO对反应速率的影响。
1.3 测定方法
进行化学分析前,样品首先采用中速定性滤纸进行过滤。
NH+4 -N浓度采用分光光度法测定,NO−2 -N和NO−3 -N浓度采用离子色谱法测定,污泥浓度(MLSS)、挥发性悬浮固体浓度(MLVSS)等按照国家环保总局规定的标准方法测定[19]。1)游离氨(FA)和游离亚硝酸(FNA)浓度根据公式(1)和(2)进行计算[20]。
CFA=1714×CNH+410pHe6344/(273+T)+10pH (1) CFNA=4614×CNO−2e−2300/(273+T)10pH (2) 式中:CFA为游离氨浓度,mg·L−1;CFNA为游离亚硝酸浓度,mg·L−1;
CNH+4 为氨氮浓度,mg·L−1;CNO−2 为亚硝酸盐浓度,mg·L−1;T为温度,℃。2)底物流加-间歇运行方式的每个周期氨氮氧化速率和底物原液储备器中底物质量分别采用式(3)和(4)进行计算[17-18, 21]。
vNH+4=CT,NH+4+vA,NH+4t−CR,NH+4t (3) 式中:
vNH+4 为每个周期氨氮氧化速率,mg·(L·h)−1;CT,NH+4 为每个周期初始氨氮浓度,mg·L−1;vA,NH+4 为每个周期氨氮流加速率至反应液,mg·(L·h)−1;CR,NH+4 为每个周期反应结束氨氮浓度,mg·L−1;t为每个周期氨氮流加时间也即反应时间,h。m=vA,NH+4tSV100 (4) 式中:m为每个周期所用NH4Cl的总质量,g;V为反应液体积,L;S为计算基准值,g:本研究取0.382,即0.382 g NH4Cl为1 L反应液提供氨氮浓度为100 mg·L−1。
3)每个周期亚硝酸盐积累率根据每周期反应开始和结束时亚硝酸盐和硝酸盐浓度生成量进行计算[16],按公式(5)进行计算。
η=CR,NO−2−CT,NO−2(CR,NO−2−CT,NO−2)+(CR,NO−3−CT,NO−3)×100% (5) 式中:η为每个周期亚硝酸盐积累率,%;
CT,NO−2 为每个周期初始亚硝酸盐浓度,mg·L−1;CR,NO−2 为每个周期结束亚硝酸盐浓度,mg·L−1;CT,NO−3 为每个周期初始硝酸盐浓度,mg·L−1;CR,NO−3 为每个周期结束硝酸盐浓度,mg·L−1。1.4 扫描电镜
采用扫描电镜观察AOB富集培养前后的污泥中细菌的形态。制样步骤如下:在4 ℃下使用戊二醇浸泡样品2 h以上,以固定细胞;吸出戊二醇固定液,加入去离子水浸泡样品数分钟;加入去离子水再浸泡2次;再分别加入50%、70%、85%、95%和100%乙醇浸泡。之后将样品置于纸包中进行临界点干燥,完成后取出样品并粘至金属台上,喷金处理。最后,在扫描电镜(SU8010,日立公司,日本)下进行观察。
1.5 高通量测序
取污泥样品进行DNA提取、PCR扩增(V3~V4)、琼脂糖凝胶电泳、DNA纯化,采用Illumina Miseq03测序平台进行16S rRNA高通量测序(生工生物工程股份有限公司,上海)。对结果进行过滤处理,得到优化序列。在97%相似度水平下进行操作分类单元(OTU)聚类分析,研究结果用于描述不同微生物群落在污泥中的丰度,明确了主要物种的比例。相应序列的原始数据存储于NCBI数据库中,https://www.ncbi.nlm.nih.gov/sra/PRJNA563303,编号:SRR10064655,SRR10064654,SRR10064653和SRR10064652(BioProject ID: PRJNA563303;BioSample编号:SAMN12675027, SAMN12675028, SAMN12675029, SAMN12675030)。
1.6 荧光定量PCR
对污泥样品采用荧光定量PCR法检测amoA基因的绝对含量(生工生物工程股份有限公司,上海)。引物序列为amoA-1F:GGGGTTTCTACTGGTGGT和amoA-2R:CCCCTCKGSAAAGCCTTCTTC。PCR反应体系:模板DNA 0.5 μL;引物 F (10 μmol·L−1 0.5 μL;引物 R (10 μmol·L−1) 0.5 μL;dNTP (10 mmol·L−1) 0.5 μL;Taq Buffer (10×) 2.5 μL;MgCl2 (25 mmol·L−1) 2 μL;Taq 酶 (5 U·μL−1) 0.2 μL;H2O 18.3 μL。反应条件:95 ℃预变性3 min;94 ℃变性30 s;57 ℃退火30 s;72 ℃延伸30 s;72 ℃修复延伸8 min;35个循环。PCR产物回收后进行克隆测序和定量质粒信息,之后DNA样品稀释50倍上机进行荧光定量PCR检测,得到平均拷贝数。
2. 结果与讨论
2.1 调节FA和FNA浓度抑制NOB
根据式(1)和(2),在一定温度下,反应体系的pH、
NH+4 -N和NO−2 -N浓度影响FA和FNA浓度。先前研究证实控制FA和FNA浓度是实现NOB抑制的有效方式,AOB对所能承受的FA和FNA的抑制浓度均高于NOB。先前有研究[22-23]表明,4~6 mg·L−1 FA可有效抑制NOB的生长,16 mg·L−1 FA未对AOB产生明显的抑制。对于FNA的抑制作用,有研究[22, 24]表明,其对NOB和AOB的抑制浓度分别为0.02 mg·L−1和0.10 mg·L−1,且达到0.40 mg·L−1时,对AOB的抑制作用严重。此外,AOB和NOB各有其适宜生长的pH范围,有研究[25-26]表明,适宜AOB和NOB生长的pH分别为7.0~8.5和6.0~7.5。本研究通过调节反应液的pH、控制底物流加速率和实际反应速率的关系以实现体系内预期
NH+4 -N和NO−2 -N浓度范围和预期的FA和FNA浓度,考察了pH、FA、FNA变化对AOB富集的影响。AOB富集过程中每周期开始和结束时NH+4 -N,NO−2 -N和NO−3 -N的浓度变化如图2(a)所示。根据式(1)和(2),温度以20 ℃计,结合反应体系内的pH波动范围(设定为±0.1),计算得出每周期开始和结束时体系内的FA和FNA浓度,结果如图2(b)和图2(c)所示。结果表明,在整个培养过程中,各周期反应体系内每一时刻的FA浓度基本保持在3~8 mg·L−1;因随着氨氮氧化能力的增强,每周期结束时体系内积累的NO−2 -N浓度逐渐升高,因此,体系内FNA浓度逐渐升高,各周期FNA的浓度基本保持在0.15 mg·L−1以下。符合先前已得到的对NOB产生抑制的浓度,且低于对AOB活性产生抑制的浓度。此外,通过在适宜AOB生长的pH范围内调节反应体系的pH,且随着AOB富集的进行,在保证FA和FNA浓度未对AOB活性产生抑制的前提下,灵活调节pH使其逐渐降低至近7.5,更接近于实际污水的pH。然而,降低pH会导致FNA升高,因此,可以通过缩短每周期的反应时间以减少周期反应结束时亚硝酸盐积累量,从而防止FNA对AOB活性产生抑制。 图 2 FA和FNA浓度调节及低DO环境Figure 2. Regulation on FA and FNA concentrations at low DO conditions
图 2 FA和FNA浓度调节及低DO环境Figure 2. Regulation on FA and FNA concentrations at low DO conditions2.2 AOB富集过程中的比氨氮氧化速率
根据图2(a)各周期初始
NH+4 -N、NO−2 -N和NO−3 -N浓度,反应结束NH+4 -N、NO−2 -N和NO−3 -N浓度,采用式(3)和式(5)计算各周期的氨氮氧化速率和亚硝酸盐积累率,考察AOB富集培养效果,结果如图3所示。约从第9天(第10周期)开始,AOB生长进入对数生长期,氨氮氧化速率由约15 mg·(L·h)−1大幅升高,在第15天(第22周期)达到最高,约为180 mg·(L·h)−1。与此同时,亚硝酸盐积累率逐渐升高至90%左右。在第1、10、20、29周期,氨氮氧化速率分别达到12.35、21.39、160.89、148.40 mg·(L·h)−1;污泥的MLSS分别为(4 414±92)、(5 441±211)、(6 008±162)、(4 583±51) mg·L−1;MLVSS分别为(2 776±48)、(2 836±174)、(2 812±48)、(2 725±79) mg·L−1。结合MLVSS计算比氨氮氧化速率,得出比氨氮氧化速率分别为4.45、7.54、57.22、54.46 mg·(g·h)−1。这表明活性污泥的生物量对AOB富集具有一定影响,保证较高的生物量对其快速富集具有积极意义。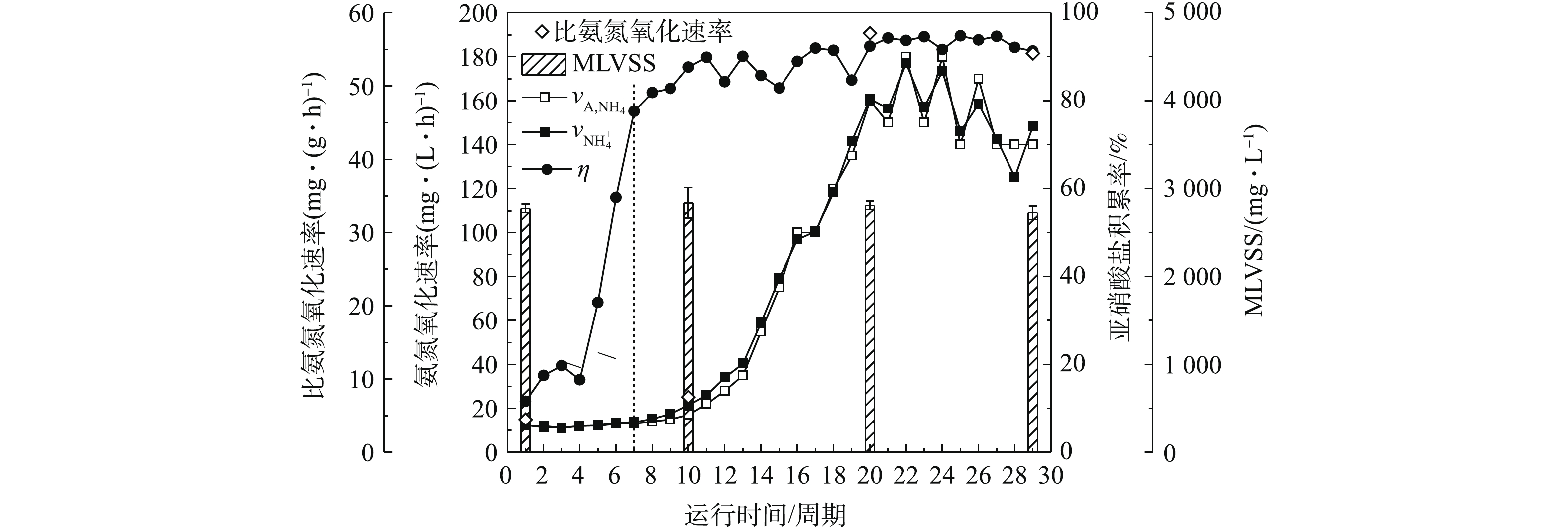 图 3 AOB富集培养期间比氨氮氧化速率和亚硝酸盐积累率Figure 3. The specific ammonia oxidation rate and nitrite accumulation ratio during the AOB enrichment period
图 3 AOB富集培养期间比氨氮氧化速率和亚硝酸盐积累率Figure 3. The specific ammonia oxidation rate and nitrite accumulation ratio during the AOB enrichment period2.3 DO浓度对氨氮氧化速率的影响
研究证实采用低DO可以实现亚硝酸盐积累,但是氨氮氧化速率随着DO降低也会随之下降[7-9]。由图2(a)可知,因室内曝气机所能提供的DO有限(若需提高DO,可采用室外大型曝气机[18]),在培养后期,体系内的DO已达到了极限(波动范围为±2%),仅为0.2~0.4 mg·L−1,但仍可以实现较高的氨氮氧化速率。
在AOB富集过程中,本研究采用间歇运行方式对DO的影响进行分析,结果如图4所示。由图4可知,DO浓度由0.8 mg·L−1左右降低至0.2 mg·L−1左右,对于相同的初始氨氮浓度,在低DO的条件下,氨氮氧化完全的历时较长,因此,氨氮氧化速率的确有所降低。然而在极低的DO条件下,因AOB的大量富集,仍可以实现高氨氮氧化速率。ANTILEO等[9]的实验结果表明,当DO浓度分别为1.0、0.8和0.6 mg·L−1时,氨氮氧化速率呈降低的趋势,分别为0.10、0.04、0.03 kg (m3·d)−1。WANG等[27]的研究结果同样表明,在DO分别为0.5 mg·L−1和1.5 mg·L−1,最大氨氮氧化速率增长约为50%。
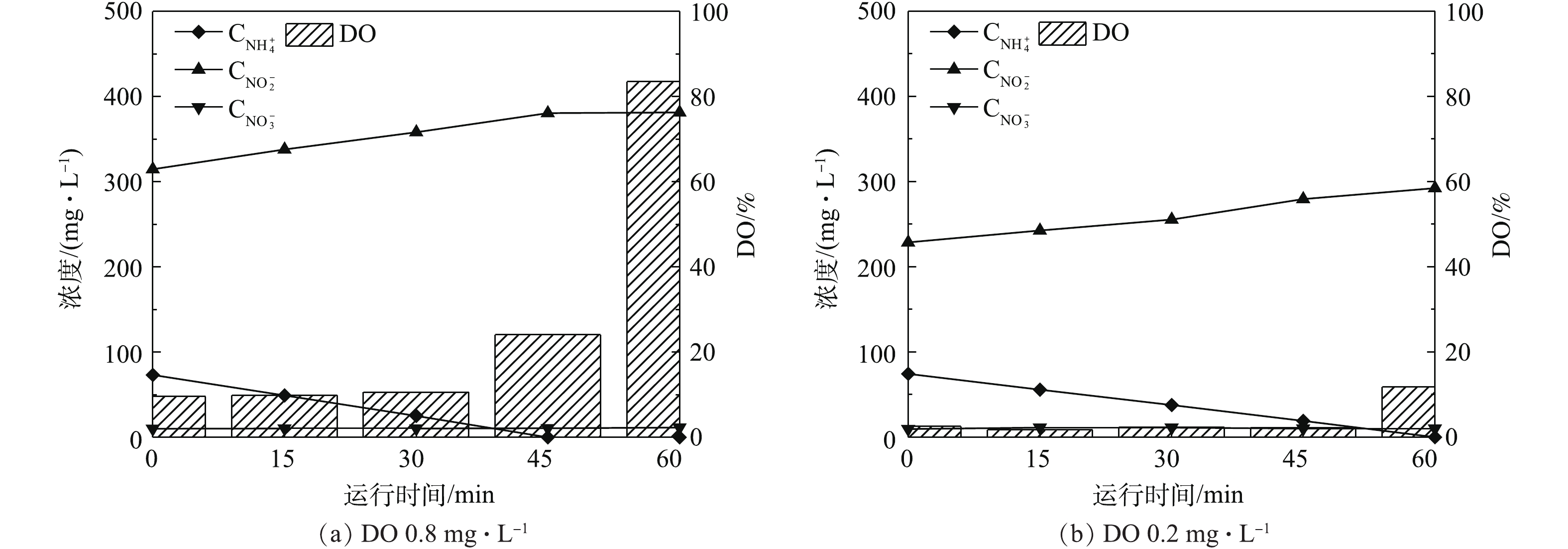 图 4 DO浓度对氨氮氧化速率的影响Figure 4. Effects of DO concentrations on the ammonia oxidation rate
图 4 DO浓度对氨氮氧化速率的影响Figure 4. Effects of DO concentrations on the ammonia oxidation rate2.4 定性分析微生物的种群分布
《伯杰氏细菌鉴定手册》中将AOB分为5个属,分别为亚硝化单胞菌属(Nitrosomonas)、亚硝化球菌属(Nitrosococcus)、亚硝化螺菌属(Nitrosospira)、亚硝化弧菌属(Nitrosovibrio)和亚硝化叶菌属(Nitrosolobus);将NOB分为4个属,分别为硝化杆菌属(Nitrobacter)、硝化球菌属(Nitrococcus)、硝化刺菌属(Nitrospina)和硝化螺菌属(Nitrospira)。有研究[28-29]表明,在污水处理厂经常出现的AOB主要是Nitrosomonas和Nitrosospira;经常出现的NOB主要是Nitrobacter和Nitrospira[30]。图5为属水平高通量测序结果,yrd1_1、yrd1_2、yrd1_3和yrd1_4分别为接种、富集培养第9、15和18天的污泥样品。接种污泥中微生物多样性较高,因AOB大量增殖,故随着AOB逐渐富集,其他细菌被淘洗出反应器,污泥中微生物多样性则随之降低。接种污泥Nitrosomonas的比例仅为0.23%,随着AOB富集的进行,在第9、15和18天,其比例逐渐上升至5.9%、51.61%和54.18%。从第9天至第15天增幅较大,结合图3的结果,原因为此期间AOB生长进入对数期。与Nitrosomonas属AOB相比,Nitrospira属的NOB一直处于非常低的水平,最高值仅为1.7%,最终比例仅为0.12%,并未完全淘洗出反应器。上述结果表明AOB在富集培养期间大量生长,与反应器接近180 mg·(L·h)−1的高氨氮氧化速率相符。此外,有研究[31]已证实采用含有有机物的实际污水也可以成功培养Nitrosomonas属的AOB,因此,有机物的存在不是AOB富集的限制因素。
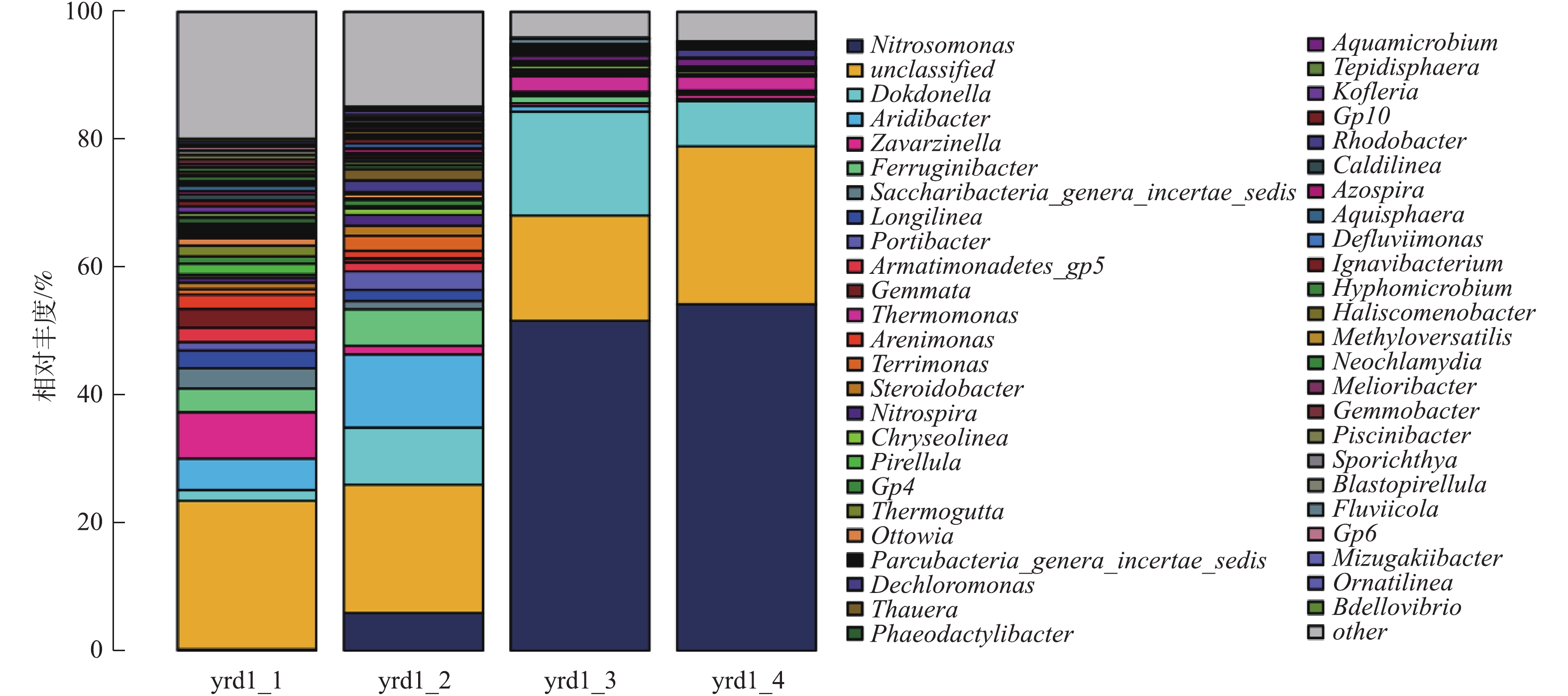 图 5 属水平高通量测序结果Figure 5. Distributions of bacterial community in the cultivated sludge and seed sludge at the genus level
图 5 属水平高通量测序结果Figure 5. Distributions of bacterial community in the cultivated sludge and seed sludge at the genus level此外,AOB形态多样,包括球状、杆状、螺旋状等,不同属的形态有所差异,已证实Nitrosomonas属的AOB为杆状或椭球状,Nitrosospira属的AOB为螺旋状[32]。本研究通过扫描电镜观察污泥样品中细菌的形态结构,如图6所示,结果显示富集培养后存在大量具有椭球状特征的细菌。
 图 6 扫描电镜微生物形态分析Figure 6. Microbial morphology analysis by scanning electron microscopy
图 6 扫描电镜微生物形态分析Figure 6. Microbial morphology analysis by scanning electron microscopy2.5 定量分析AOB富集效果
采用荧光定量PCR技术对AOB富集各阶段的污泥样品进行检测分析,以通过数量变化考察AOB的生长状况,结果如表1所示。amoA基因是AOB的特征序列,随着AOB富集的进行,可以看出活性污泥中amoA基因平均拷贝数显著增加。由于荧光定量PCR检测上机时DNA稀释50倍,且DNA提取时采用200 mg污泥样品,按照60 μL进行洗脱得到DNA溶液。因此,换算得出amoA基因平均拷贝数在接种、富集培养第9、15和18天分别为2.67×105、4.24×108、9.67×109、7.30×109 拷贝数·g−1。
表 1 amoA基因平均拷贝数Table 1. Average copy number of gene amoA for AOB样品 平均Ct值 平均拷贝数/(拷贝数·g−1) 接种污泥 32.238 64 17.799 9 第9天污泥 21.602 48 28 254.01 第15天污泥 17.185 25 644 733.5 第18天污泥 17.511 69 486 618 | Show Table DownLoad:
CSV
DownLoad:
CSV
结合比氨氮氧化速率(图3)和AOB群落占活性污泥中的比例(图5)的结果,由比氨氮氧化速率、AOB在活性污泥中的比例和功能基因数量等几个方面均可证实,AOB群落得到了高度增殖,逐渐成为活性污泥中的优势菌属。
3. 结论
1)在适宜AOB生长的pH范围内,调节反应体系内的pH、结合底物流加速率和实际反应速率关系的联合控制,实现了整个反应过程中预期的FA和FNA水平,以抑制NOB生长,而未对AOB活性产生抑制。在确保实现合理FA和FNA浓度范围的前提下,逐渐降低体系内的pH,至接近实际污水的pH范围,使得富集得到的AOB更利于实际应用。
2)低DO导致氨氮氧化速率降低,体系内高纯度的AOB可以确保在极低DO条件下,氨氮氧化速率维持在较高水平。
3)通过比氨氮氧化速率指标、高通量测序定性分析活性污泥中的菌群结构以及荧光定量PCR检测功能基因amoA平均拷贝数,综合证实AOB在富集期间得到了高度增殖。
-
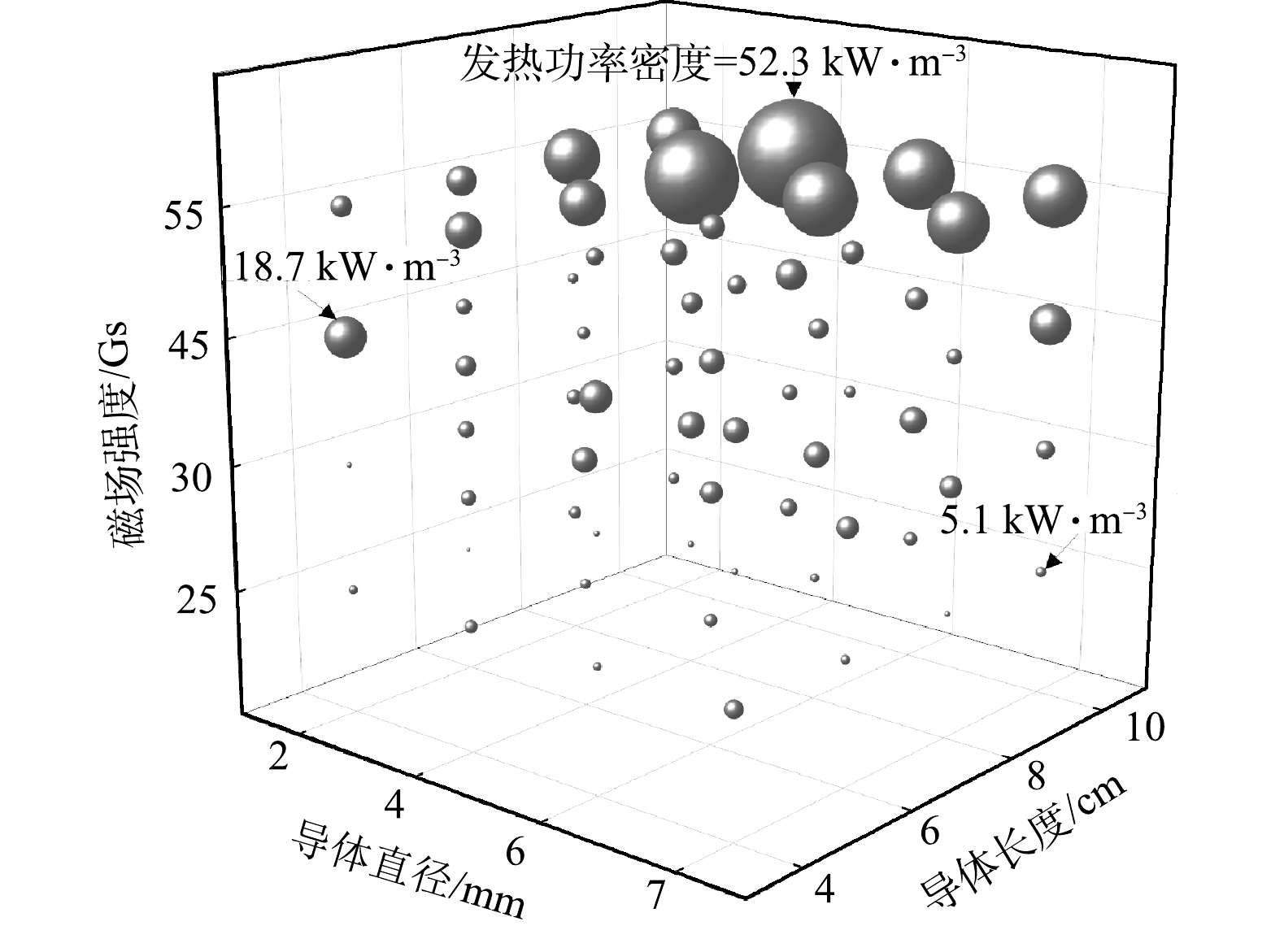
图 2 不同规格磁热导体在磁场中发热功率密度情况
Figure 2. Temperature rise of metal conductors in magnetic fields
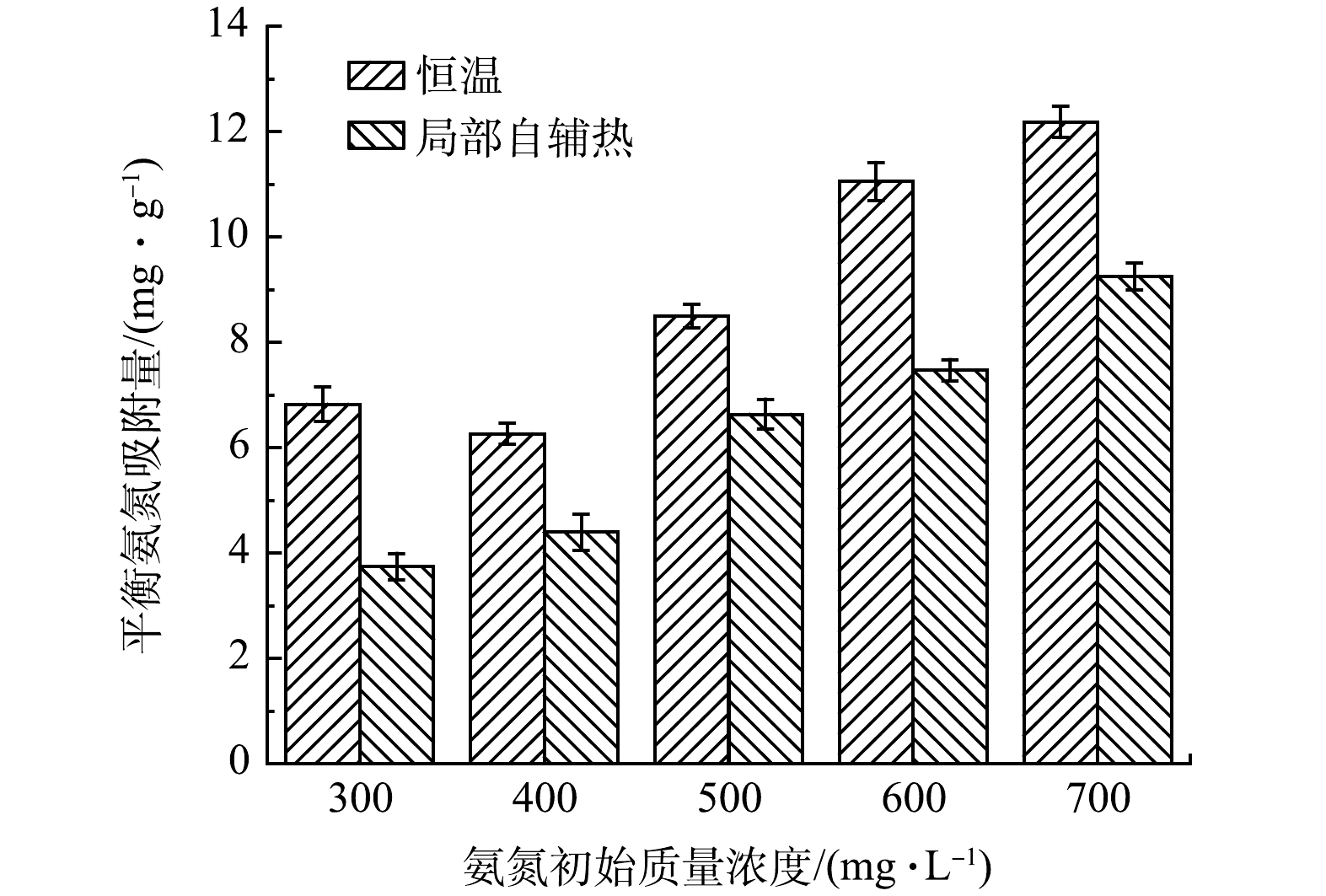
图 4 不同氨氮初始质量浓度下吸附量对比
Figure 4. Comparison of adsorption capacity at different initial concentration of ammonia nitrogen
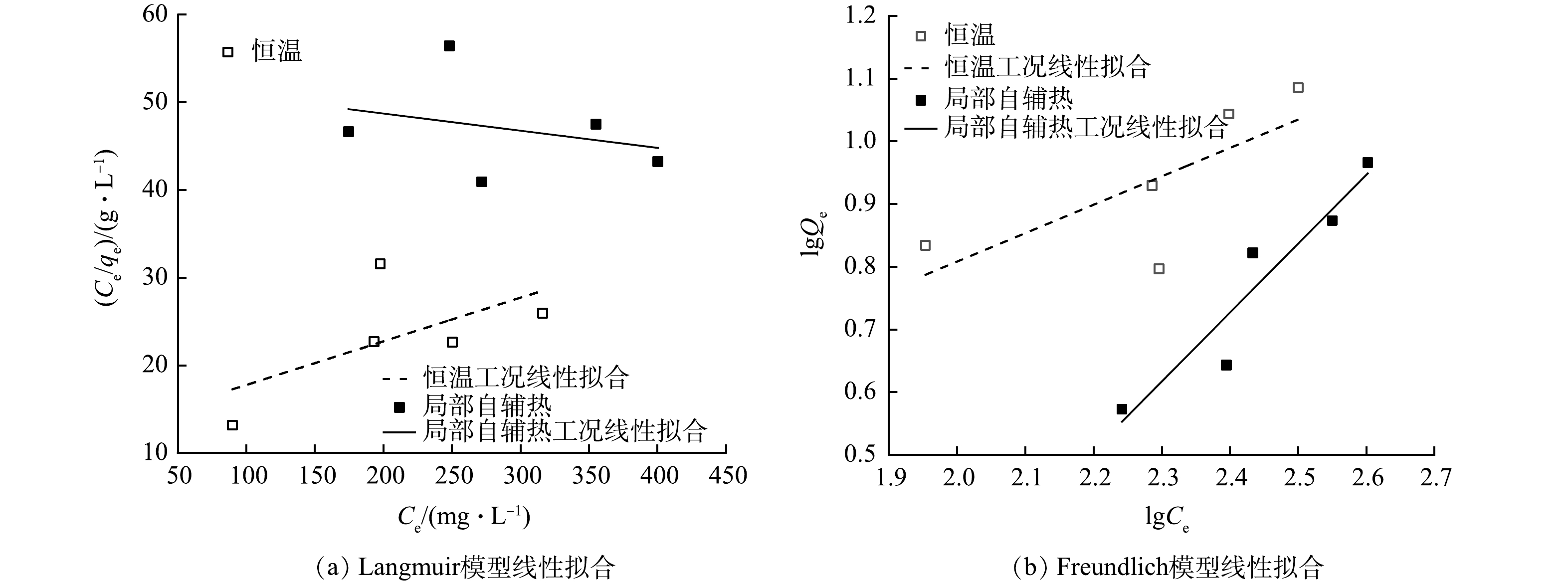
图 5 恒温与局部自辅热条件下氨氮等温吸附拟合曲线
Figure 5. Fitting curves of adsorption isotherm of NH3-N by two scenarios
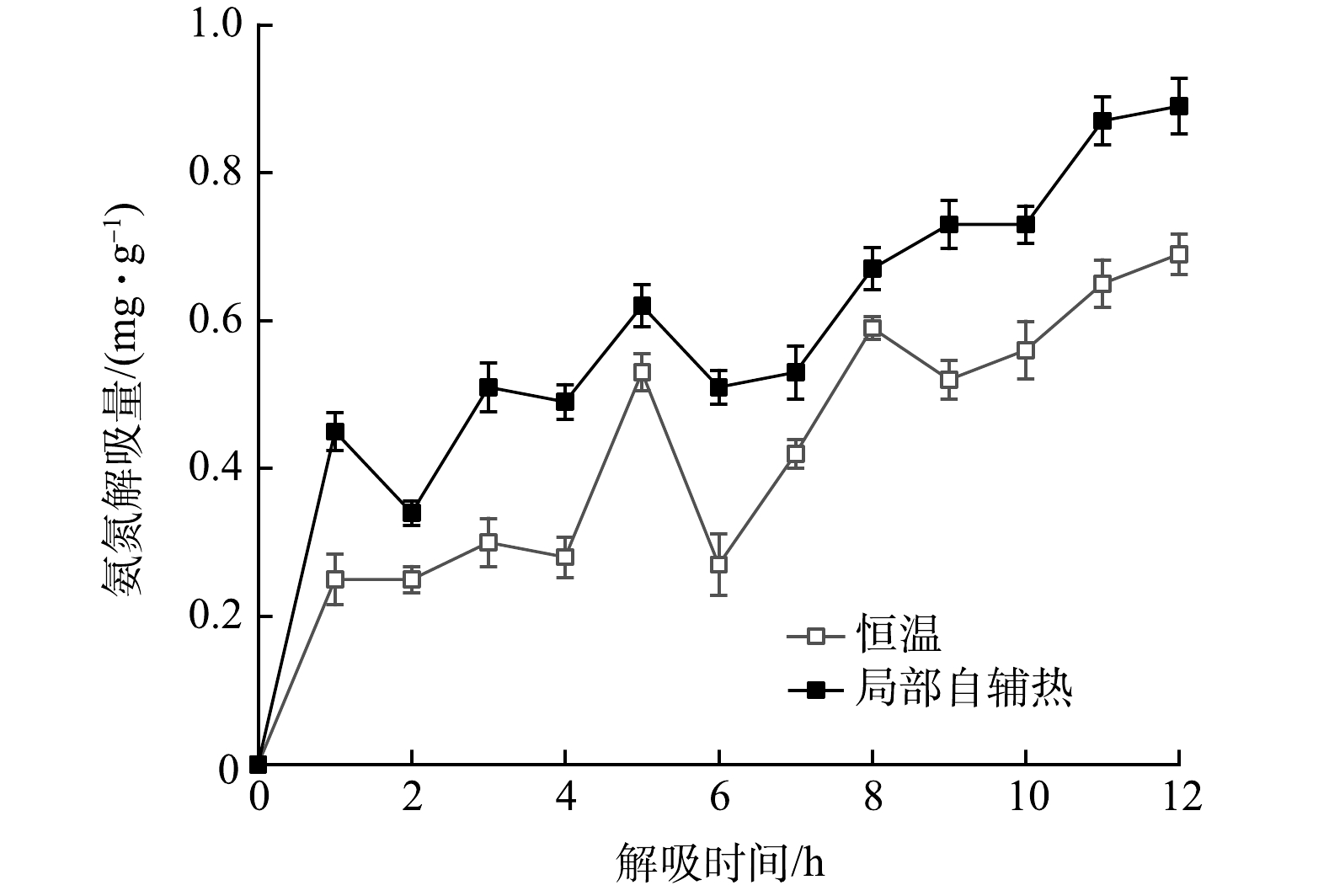
图 7 不同工况下氨氮解吸量随时间变化情况
Figure 7. Change of ammonia nitrogen desorption capacity with time under two scenarios
表 1 恒温与局部自辅热条件下吸附过程Langmuir及Freundlich模型相关参数
Table 1. Langmuir and Freundlich model parameters of adsorption process
类型 Langmuir参数 Freundlich参数 qmax/(mg·g−1) KL R2 KF 1/n R2 恒温 20.10 0.003 9 0.384 2 0.906 7 0.453 0.544 0 局部自辅热 — — 0.087 0 0.148 1 1.099 0.902 4
下载: 导出CSV
表 2 吸附动力学模型相关参数
Table 2. Parameters of adsorption kinetics model
类型 准一级动力学模型 准二级动力学模型 Weber-Morris模型 K1 qe/(mg·g−1) R2 K2 qe/(mg·g−1) R2 Ki R2 恒温 0.004 92 10.690 9 0.941 7 0.000 40 13.676 1 0.996 1 0.375 56 0.964 2 局部自辅热 0.003 34 7.848 0.963 3 0.000 21 11.080 3 0.964 7 0.295 44 0.992 3
下载: 导出CSV
表 3 解吸动力学模型相关参数
Table 3. Parameters of desorption kinetics model
类型 准一级解吸动力学模型 准二级解吸动力学模型 Weber-Morris模型 K1ʹ qeʹ/(mg·g−1) R2 K2ʹ qeʹ/(mg·g−1) R2 Kiʹ R2 恒温 0.003 22 0.704 5 0.670 5 0.002 59 0.949 3 0.495 2 0.023 94 0.732 9 局部自辅热 0.003 78 0.907 7 0.551 7 0.004 16 1.039 7 0.8195 0.024 85 0.777 9
下载: 导出CSV
-
[1] WIJESINGHE D T N, DASSANAYAKE K B, SCALES P, et al. Removal of excess nutrients by Australian zeolite during anaerobic digestion of swine manure[J]. Journal of Environmental Science and Health Part a-Toxic/Hazardous Substances & Environmental Engineering, 2018, 53(4): 362-372. [2] HUANG H, YANG L, XUE Q, et al. Removal of ammonium from swine wastewater by zeolite combined with chlorination for regeneration[J]. Journal of Environmental Management, 2015, 160: 333-341. [3] DU Q, LIU S, CAO Z, et al. Ammonia removal from aqueous solution using natural Chinese clinoptilolite[J]. Separation and Purification Technology, 2005, 44(3): 229-234. doi: 10.1016/j.seppur.2004.04.011 [4] JORGENSEN S E, LIBOR O, LEA GRABER K, et al. Ammonia removal by use of clinoptilolite[J]. Water Research, 1976, 10(3): 213-224. doi: 10.1016/0043-1354(76)90130-5 [5] HUANG H, XIAO X, YAN B, et al. Ammonium removal from aqueous solutions by using natural Chinese (Chende) zeolite as adsorbent[J]. Journal of Hazardous Materials, 2010, 175(1): 247-252. [6] CHEN Z, ZHENG X, CHEN Y, et al. Nitrite accumulation stability evaluation for low-strength ammonium wastewater by adsorption and biological desorption of zeolite under different operational temperature[J]. Science of the Total Environment, 2020: 704. [7] KARADAG D, KOC Y, TURAN M, et al. Removal of ammonium ion from aqueous solution using natural Turkish clinoptilolite[J]. Journal of Hazardous Materials, 2006, 136(3): 604-609. doi: 10.1016/j.jhazmat.2005.12.042 [8] HUANG H, XIAO X, YAN B, et al. Ammonium removal from aqueous solutions by using natural Chinese (Chende) zeolite as adsorbent[J]. Journal of Hazardous Materials, 2010, 175(1/2/3): 247-252. [9] SALTALI K, SAN A, AYDIN M. Removal of ammonium ion from aqueous solution by natural Turkish (Yildizeli) zeolite for environmental quality[J]. Journal of Hazardous Materials, 2007, 141(1): 258-263. doi: 10.1016/j.jhazmat.2006.06.124 [10] 崔天顺, 周文剑, 李云亭. 天然沸石处理氨氮废水的吸附性能及再生研究[J]. 能源与环境, 2011(1): 2. doi: 10.3969/j.issn.1672-9064.2011.01.001 [11] 张帅, 叶芳芳, 李长刚, 等. 饱和吸附氨氮沸石的化学再生方法研究[J]. 工业水处理, 2019, 39(8): 73-76. doi: 10.11894/iwt.2018-0815 [12] Experimental studies on 3A and 4A zeolite molecular sieves regeneration in TSA process: Aliphatic alcohols dewatering–water desorption[J]. Chemical Engineering Journal, 2015, 259: 232-242. [13] ZADRAZIL A, STEPANEK F. Remote control of desorption by radiofrequency heating: Single pellet experiments[J]. Chemical Engineering Science, 2013, 101: 382-389. doi: 10.1016/j.ces.2013.06.012 [14] ELLISON C, HOFFMAN J, SHEKHAWAT D. Comparison of microwave and conventional heating for CO2 desorption from zeolite 13X[J]. International Journal of Greenhouse Gas Control, 2021, 107: 103311. doi: 10.1016/j.ijggc.2021.103311 [15] SHEN H, FAN D, HUANG L, et al. Effects of microwaves on molecular arrangements in potato starch[J]. RSC Advances, 2017, 7(24): 14348-14353. doi: 10.1039/C6RA28048J [16] KUBOTA M, HANADA T, YABE S, et al. Water desorption behavior of desiccant rotor under microwave irradiation[J]. Applied Thermal Engineering, 2011, 31(8/9): 1482-1486. [17] LIN L, HILL E H, PENG X, et al. Optothermal manipulations of colloidal particles and living cells[J]. Accounts of Chemical Research, 2018, 51(6): 1465-1474. doi: 10.1021/acs.accounts.8b00102 [18] GREFF J, BABADAGLI T. Use of nano-metal particles as catalyst under electromagnetic heating for in-situ heavy oil recovery[J]. Journal of Petroleum Science and Engineering, 2013, 112: 258-265. doi: 10.1016/j.petrol.2013.11.012 [19] HAN M, WEE D. On the volume power density of radial thermoelectric generators[J]. International Journal of Energy Research, 2020, 44: 6049-6057. doi: 10.1002/er.5317 [20] 连神海, 张树楠, 刘锋, 等. 不同生物炭对磷的吸附特征及其影响因素[J/OL]. 环境科学: 1-10 2-06-17]. DOI:10.13227/j.hjkx.202109156.
[21] LANGMUIR I. The adsorption of gases on plane surfaces of glass, mica and platinum[J]. Journal of the American Chemical Society, 1917: 40. [22] MARAÑÓN E, ULMANU M, FERNÁNDEZ Y, et al. Removal of ammonium from aqueous solutions with volcanic tuff[J]. Journal of Hazardous Materials, 2006, 137(3): 1402-1409. doi: 10.1016/j.jhazmat.2006.03.069 [23] SUN N, SHI W, MA L, et al. Investigations on the mechanism, kinetics and isotherms of ammonium and humic acid co-adsorption at low temperature by 4A-molecular sieves modified from attapulgite[J]. RSC Advances, 2017, 7(28): 17095-17106. doi: 10.1039/C7RA00268H [24] KAVITHA D. Equilibrium, kinetics, thermodynamics and desorption studies of pentachlorophenol onto agricultural waste activated carbon[J]. Materials Today:Proceedings, 2020, 33: 4746-4750. doi: 10.1016/j.matpr.2020.08.357 [25] WEBER W, MORRIS J. Kinetics of adsorption on carbon from solution[J]. Journal of the Sanitary Engineering Division, 1963, 1: 1-2. [26] KUMAR P S, RAMALINGAM S, KIRUPHA S D, et al. Adsorption behavior of nickel(II) onto cashew nut shell: Equilibrium, thermodynamics, kinetics, mechanism and process design[J]. Chemical Engineering Journal, 2011, 167(1): 122-131. doi: 10.1016/j.cej.2010.12.010 [27] 谢妤, 宋卫军. 稻壳基A型沸石的表征及其对NH3-N的吸/脱附动力学研究[J]. 环境工程, 2017, 35(7): 43-48. [28] TARAN N, IONEL D, RALLABANDI V, et al. An overview of methods and a new 3D FEA and analytical hybrid technique for calculating AC winding losses in PM machines[J]. IEEE Transactions on Industry Applications, 2021, 57(1): 352-362. doi: 10.1109/TIA.2020.3034558 [29] 汤平华, 漆亚梅, 黄国辉, 等. 定子无铁心飞轮电机绕组涡流损耗分析[J]. 电工技术学报, 2010, 25(3): 27-32. doi: 10.19595/j.cnki.1000-6753.tces.2010.03.005 [30] 姜霞, 周小宁, 丁明玉, 等. 天然沸石及改性沸石去除低浓度氨氮的研究[J]. 环境科学研究, 2008(5): 37-42. doi: 10.13198/j.res.2008.05.39.jiangx.008 [31] LIU H, PENG S, SHU L, et al. Effect of Fe3O4 addition on removal of ammonium by zeolite NaA[J]. Journal of Colloid and Interface Science, 2013, 390(1): 204-210. doi: 10.1016/j.jcis.2012.09.010 [32] 赵桂瑜. 人工湿地除磷基质筛选及其吸附机理研究[D]. 上海: 同济大学, 2007. [33] 饶力. 天然沸石对氨氮废水吸附特性及再生工艺的研究[D]. 广州: 华南理工大学, 2016. [34] 黎铮. 紊动条件下泥沙纵向分布及氨氮吸附释放特性研究[D]. 重庆: 重庆交通大学, 2020. [35] 程宇. 混凝-吸附工艺对养猪废水处理效果的研究[D]. 武汉: 华中农业大学, 2017. [36] 杜兆林, 陈洪安, 秦莉, 等. 纤维素基吸附材料除污工艺及吸附模型研究进展[J]. 农业资源与环境学报, 2020, 37(1): 6-16. doi: 10.13254/j.jare.2018.0366 [37] LIU X, TU Y, LIU S, et al. Adsorption of ammonia nitrogen and phenol onto the lignite surface: An experimental and molecular dynamics simulation study[J]. Journal of Hazardous Materials, 2021, 416: 125966. doi: 10.1016/j.jhazmat.2021.125966 [38] AYDIN TEMEL F, KULEYIN A. Ammonium removal from landfill leachate using natural zeolite: Kinetic, equilibrium, and thermodynamic studies[J]. Desalination and Water Treatment, 2016, 57(50): 23873-23892. doi: 10.1080/19443994.2015.1136964 -

 点击查看大图
点击查看大图

计量
- 文章访问数: 3646
- HTML全文浏览数: 3646
- PDF下载数: 69
- 施引文献: 0






