


 下载:
下载:
-
氨氮是引起水体富营养化的主要原因之一。氨氮浓度过高会引起水生生物中毒死亡,形成黑臭水体;同时,氨氮自然氧化过程的中间产物可能会引起人体组织发生癌变[1-2]。利用电化学氧化法可以有效去除氨氮,故该方法在垃圾渗滤液[3]、养殖废水[4]、制革废水[5]等废水处理领域具有巨大的应用潜力。氨氮的电化学氧化一般分为2种路径:直接氧化与间接氧化。当溶液中有大量氯离子存在时,氨氮主要通过间接氧化去除,即氯离子先被氧化生成Cl2、ClO−等强氧化性产物,与氨氮结合生成氯胺,再进一步被氧化去除[6]。因此,电极析氯性能的优劣可直接影响到氨氮的电化学氧化效果。此外,初始氯离子浓度、初始氨氮浓度以及电流密度等因素均会对氨氮的电化学氧化产生不同程度的影响[7, 8]。
电极结垢是电化学水处理中的常见问题[9-12]。当采用电化学技术处理高硬度氨氮废水时,废水中的Ca2+、Mg2+等硬度离子会在阴极富集,并与水中的HCO3−和阴极析氢反应产生的OH−结合生成难溶性的CaCO3与Mg(OH)2垢层并覆盖在电极表面,阻碍电化学反应。WANG等[13]在研究燃煤电厂高盐氨氮废水时发现,电极表面的清洁度越高,氨氮去除效果越好。因此,若要保持较高的氨氮去除效率,需频繁对电极进行除垢处理。倒极脱垢是目前工业中常用的一种除垢方法。与机械刮擦脱垢、空气冲刷脱垢相比,倒极脱垢操作简单、脱垢效果佳,不需要另外增设阴极刮擦板,降低了设备的安装难度[14]。
形状稳定性电极(dimensionally stable anodes, DSA)是以钛为基体,表面涂覆金属氧化物制得的电催化电极[15]。工业上常用的DSA电极主要是钌系电极与铱系电极。钌系电极析氯活性高,耐腐蚀性强,成本相对较低,但在高电流密度下寿命短暂[16]。而与之相比,铱电极寿命可提高20倍[17],因此,在许多领域应用广泛[18-21]。然而,这类传统DSA电极通常只用作阳极,且工作时极性不发生变化,若经历倒极操作,活性涂层会脱落[22-23],电极迅速失效。因此,寻求一种在倒极工况下保持性能稳定的电极至关重要。为解决这一问题,在前期研究中,尝试在RuO2-IrO2体系中添加RhOx组分,制得了Ti/RuO2-IrO2-RhOx电极以用于对苯酚等有毒污染物的降解。前期研究中发现:与Ti/RuO2-IrO2相比,Ti/RuO2-IrO2-RhOx表面更加光滑致密,裂纹和孔隙较少,晶粒高度分散,涂层结构得到显著改善;Rh3+/Rhn+电对的氧化还原反应可逆,RhOx与传统的RuO2、IrO2可以产生协同效应,从而提高电极的倒极稳定性和氧化能力[24]。但前期研究并未涉及Ti/RuO2-IrO2-RhOx电极用于氨氮的电化学氧化。
为了解决上述问题,研制了一种倒极长寿命的Ti/RuO2-IrO2-RhOx新型电极用于处理氨氮废水。首先考察了其倒极稳定性、析氯电化学活性以及对氨氮的去除效果,并与传统的Ti/RuO2-TiO2、Ti/IrO2-Ta2O5等电极进行了对比;在此基础上,进一步将所研制的Ti/RuO2-IrO2-RhOx电极用于电解氨氮废水,考察了氨氮浓度、电流密度以及氯离子浓度等因素对氨氮电化学氧化效果的影响。
-
氯铱酸(H2IrCl6·xH2O,35% Ir),氯化钽(TaCl5,99.99%),氯化钌(RuCl3,45%~55% Ru),四氯化钛(TiCl4,AR),三水合氯化铑(RhCl3·3H2O),盐酸(HCl,AR),异丙醇(C3H8O,AR),氯化钠(NaCl,AR),碘化钾(KI,AR),淀粉指示剂(0.25%淀粉),冰乙酸(CH3COOH,AR),五水合硫代硫酸钠(Na2S2O3·5H2O,AR),氢氧化钠(NaOH,AR),碘化汞(Ⅱ)(HgI2,AR),酒石酸钾钠(NaKC4H4O6,AR),硫酸铵((NH4)2SO4,AR),氯化钙(CaCl2,AR),六水合氯化镁(MgCl2·6H2O,AR)。以上所有药品未经再次提纯,直接购买使用,实验用水为超纯水(电导率≤0.06 μS·cm−1)。
-
分别取用一定量的RuCl3、TiCl4、H2IrCl6·xH2O、TaCl5、RhCl3·3H2O溶于浓盐酸与异丙醇(体积比1∶1)的混合溶液,配制成0.25 mol·L−1的RuCl3、TiCl4、TaCl5、RhCl3·3H2O、H2IrCl6·xH2O溶液。将这些溶液分别按照RuCl3∶TiCl4=1∶1、H2IrCl6·xH2O∶TaCl5=1∶1、RuCl3∶H2IrCl6·xH2O∶RhCl3·3H2O=2∶1∶2的摩尔比混合,配制成不同组分的前驱液。以上前驱液均现用现配。
采用2 cm×3 cm的钛网为基底,制备电极前,钛网基底经喷砂、除油、煮沸的浓盐酸刻蚀2 min、自来水洗、去离子水超声洗10 min之后烘干称量,存放在无水乙醇中备用。采用热分解法制备电极。先于室温下将即时配制的前驱液均匀涂覆于钛网上,再用烘箱在80 ℃下烘干5 min,然后转移至450 ℃马弗炉中热解5 min,出炉冷却至室温,重复涂覆-烘干-热解-冷却操作13~18次,直至涂层覆载量达18 g·m−2,最后一次热解时间延长至1 h。
-
采用倒极加速寿命实验评估电极稳定性[25]。采用直流电源(MS303DS,东莞市迈豪电子科技有限公司)供电,用PLC控制器 (YBD-12,优博达电子)控制倒极频率。加速寿命实验条件:电解液为20 g·L−1 Na2SO4溶液,pH为7,温度为25 ℃,电流密度为2 000 A·m−2,倒极频率为20 min·次−1,倒极持续时长为20min·次−1。以电解槽电压上升5 V时作为评价电极失活的判据,评价加速寿命实验条件下的电极寿命。采用场发射电镜(FE-SEM)观察涂层形貌结构;采用X射线衍射仪(XRD)分析涂层晶体结构。
-
在经典三电极体系测试装置中,采用稳压/恒流电化学分析仪(CHI604E,上海辰华)测试循环伏安曲线(CV),工作电极为自制电极,对电极为铂电极,参比电极为饱和甘汞电极。分别在25 ℃,0.5 mol·L−1 NaCl溶液与10 mmol·L−1 [Fe(CN)6]3-/4-+0.2 mol·L−1 K2SO4溶液中得到阳极的循环伏安曲线。扫描电压分别为0.2~1.6 V、−0.2~0.8 V,扫描速率为100 mV·s−1。
-
通过电解NaCl溶液,在密闭电解槽中测定有效氯浓度,计算电流效率。实验在1 L恒温(25 ℃)密闭电解槽内进行,阴阳极均采用自制电极,NaCl质量浓度为15 g·L−1,电流密度为1 000 A·m−2。每隔30 min取样5 mL,采用碘量法测定有效氯浓度[26]。
有效氯浓度根据式(1)进行计算,电流效率根据式(2)进行计算。
式中:C为有效氯质量浓度,g·L−1;N为Na2S2O3溶液浓度,mol·L−1;V为滴定消耗的Na2S2O3溶液体积,mL。η为电流效率,%;V0为电解液总体积,L;I为反应电流,A;t为反应时间,h;1.323为每安培小时电量有效氯的理论生成量,g·(A·h)−1。
-
电解氨氮废水实验于200 mL的密闭恒温(25 ℃)电解槽内进行,阴阳极均采用自制电极。采用NaCl、(NH4)2SO4、去离子水配制电解液模拟氨氮废水,根据实验需要依次调节溶液初始氨氮浓度、电流密度、初始氯离子浓度等,每隔30 min取样250 μL,采用纳氏试剂分光光度法测定剩余氨氮浓度。采用CaCl2、MgCl2·6H2O按照摩尔比1∶1配制硬水,硬度以CaCO3质量(mg·L−1)计。
-
为便于比较,对本团队新研制的Ti/RuO2-IrO2-RhOx电极与传统的Ti/RuO2-TiO2、Ti/IrO2-Ta2O5电极进行了倒极加速寿命实验,结果如图1所示。Ti/RuO2-TiO2与Ti/IrO2-Ta2O5的电压变化情况相类似,短时间内电压迅速升高,电极随即失效,倒极加速寿命分别为30 h与60 h;而Ti/RuO2-IrO2-RhOx的电极电压上升是一个持续而缓慢的过程,1 000 h内电压总增幅仅2.5 V,加速寿命可达到1 400 h甚至更高,远远超过其他2种电极。ROGINSKAYA等[27]曾报道,RhOx在酸性溶液中比RuO2和IrO2稳定得多。HRUSSANOVA等[28]在研究RhOx电极在酸性溶液中的析氧电化学行为时也指出,在最初去除松散颗粒后,析氧不能降解RhOx电极。因此,惰性元素Rh组分的添加提高了电极的稳定性,使其适用于频繁颠倒阴阳极性的操作条件。
-
1)表面形貌。Ti/RuO2-IrO2-RhOx电极涂层在开展寿命实验前后的FE-SEM扫描结果见图2。由图2(a)与图2(b)可见,在开展寿命实验前新制备的Ti/RuO2-IrO2-RhOx电极涂层表面整体均匀致密但凹凸不平,伴随有簇状晶粒析出和少量裂纹。致密的表面结构使得溶液中的氧化活性粒子不易渗入基底,提高了电极稳定性;析出的簇状晶粒可以有效增大电极的比表面积,增加活性位点个数,更有利于电化学反应;裂纹的出现则是钛基体与涂层之间的热膨胀系数差异所致[29]。由图2(c)和图2(d)可见,Ti/RuO2-IrO2-RhOx电极失效后,电极表面凹凸不平,基底裸露,伴有大量气泡式细小孔隙。结合前期研究结果可推断,电极失效可能是由气体冲刷、涂层溶解、活性成分丧失所致[23-24]。
2) XRD分析。Ti/RuO2-IrO2-RhOx电极的XRD图谱以及主要氧化物的标准峰见图3。在27.8°、34.8°、38.1°、40.0°、52.9°、53.8°、57.5°、58.9°、65.1°、66.6°、69.0°、70.4°、73.9°、76.0°、77.2°等处检测到15个明显的衍射峰,与Ru、Ir、Rh等氧化物的标准峰进行比对,扣除1组基底钛标准峰,其他峰位置与RuO2、IrO2、RhO2的峰位置基本对应,但又有一定程度的偏移。这可能是因为Ti/RuO2-IrO2-RhOx涂层各组分充分混合,形成了均一的固溶体[30]。理论上,离子之间能否形成稳定固溶体取决于不同元素之间的离子半径差异。根据Hume-Rothery法则,当粒子之间的粒径差小于14%~15%时,有可能形成稳定的固溶体[31]。Ir4+、Ru4+、Rh4+和Rh3+的离子半径分别为0.062 5、0.062、0.06、0.066 5 nm,相互间的最大差值为10.8%,小于14%。因此,Ti/RuO2-IrO2-RhOx涂层元素很可能以固溶体形式存在。此外,Rh2O3的个别峰虽然能够被观测到,但并不能完全对应,因此,Rh2O3是否存在有待于深入探究。金华长等[17]在其研究中也得出了相似的结果。
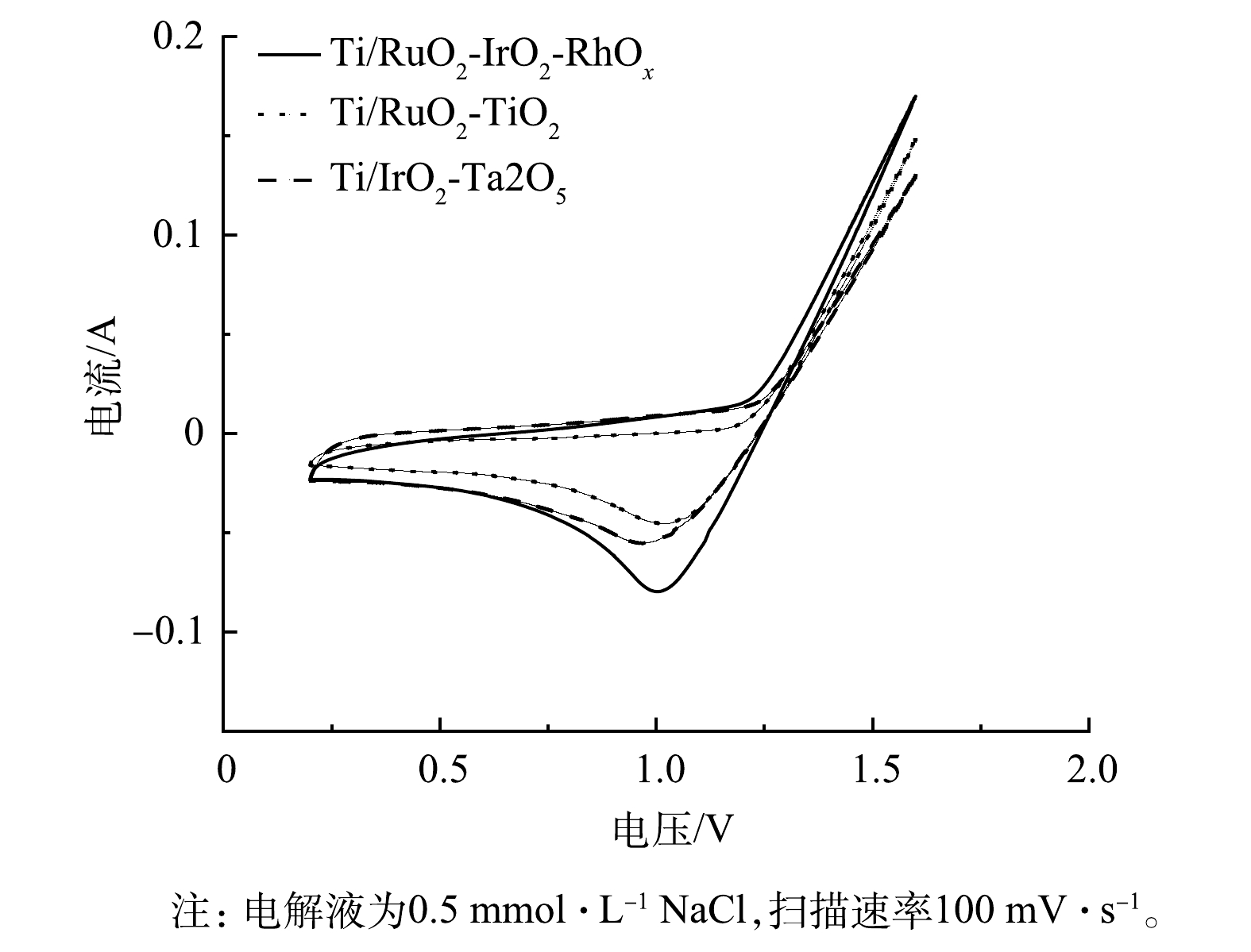
3)电化学性能。Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电极在0.5 mol·L -1 NaCl溶液中的CV测试结果如图4所示。3种电极均呈现出典型的伏安特性曲线。正向扫描时,曲线分别在1.19、1.24、1.21 V处由于发生析氯反应而出现转折,反应式为:2Cl− - 2e− = Cl2。在相同电位下,Ti/RuO2-IrO2-RhOx的电流明显大于Ti/RuO2-TiO2、Ti/IrO2-Ta2O5。这表明:与其他2种电极相比,Ti/RuO2-IrO2-RhOx具有最大的电容。
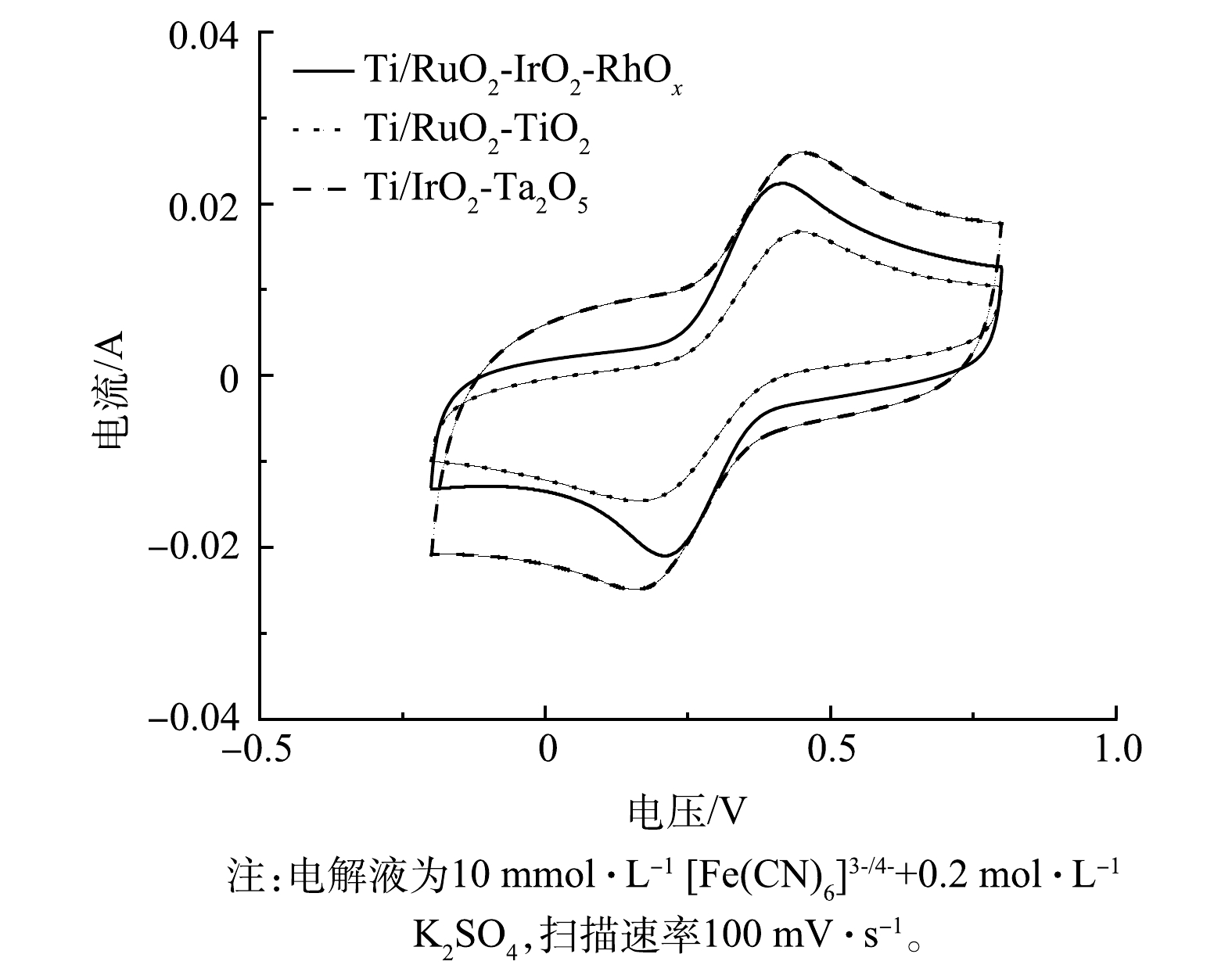
图5反映了Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电极在10 mmol·L−1[Fe(CN)6]3-/4-+0.2 mol·L−1 K2SO4溶液中的CV扫描结果。电极在[Fe(CN)6]3-/[Fe(CN)6]4-氧化还原电对中的电化学行为与涂层表面电子传递速率有关[32]。3种电极的CV曲线均出现1组氧化峰与还原峰。其中,Ti/RuO2-IrO2-RhOx电极氧化峰与还原峰对应的电流值大小基本相同,说明Ti/RuO2-IrO2-RhOx电极对于[Fe(CN)6]3-/[Fe(CN)6]4-电子对的氧化还原反应高度可逆。此外,Ti/RuO2-IrO2-RhOx两峰之间的距离最小,说明该电极拥有最快的电子转移速率。这说明:在电化学反应中,Ti/RuO2-IrO2-RhOx电极可能拥有最高的电流效率和电化学反应活性。
-
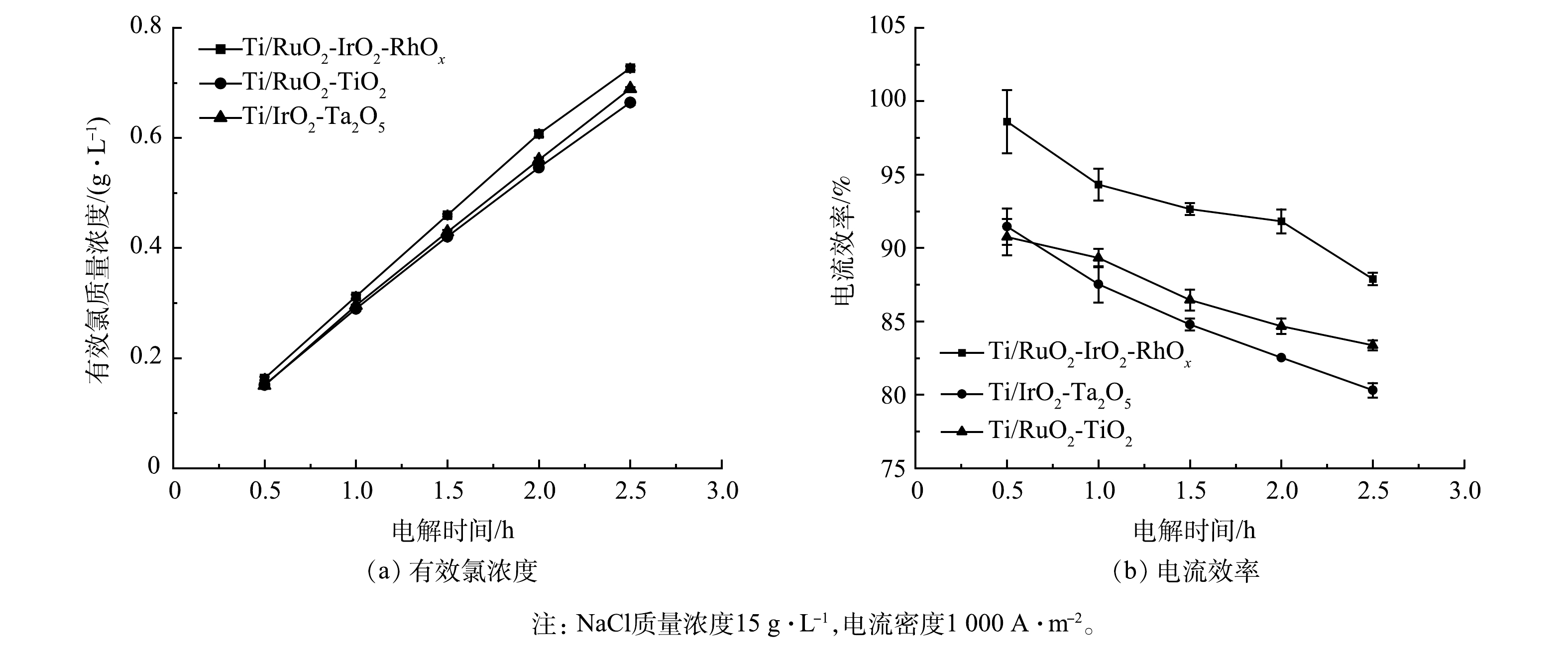
Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电极电解NaCl溶液时,有效氯浓度和电流效率随时间的变化情况见图6。如图6(a)所示,反应2.5 h后,Ti/RuO2-IrO2-RhOx电解产生0.73 g有效氯,高于Ti/RuO2-TiO2和Ti/IrO2-Ta2O5的0.69 g和0.66 g。如图6(b)所示,Ti/RuO2-IrO2-RhOx与其他2种电极相比,始终保持最高的电流效率。3种电极电解产生的有效氯浓度均随反应时间的延长而不断升高,但体系电流效率逐渐下降。这与体系中反应物氯离子浓度的下降及反应产物次氯酸根等的增加有关[33-34]。Ti/RuO2-IrO2-RhOx在单位时间内生成的有效氯浓度最高,这是因为Ti/RuO2-IrO2-RhOx电子传输速率快,电流效率高,可以有效催化氯离子氧化。这与2.2中电极的电化学性能测试所得结论一致。
-
1)不同电极去除氨氮效果对比。图7展示了采用Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电极电解氨氮时,氨氮浓度随时间变化情况。显而易见,3种电极都可用于电解去除氨氮,但去除效率不同。Ti/RuO2-TiO2和Ti/IrO2-Ta2O5电解2.5 h后,氨氮由最初的400 mg·L−1(以N计)分别降至146.4 mg·L−1和188.6 mg·L−1,去除率分别为63.4%和53.0%;电解4.5 h后,氨氮分别降至0.5 mg·L−1和28.6 mg·L−1,去除率分别为99.9%和92.9%。相比之下,Ti/RuO2-IrO2-RhOx电解2.5 h后,氨氮由原先的400 mg·L−1降至130 mg·L−1,去除率为67.6%;电解4.5 h后,氨氮降为0,去除率达100%。由此可见,Ti/RuO2-IrO2-RhOx具有更高的氨氮去除效率,这主要归因于该电极具有更好的析氯性能。
2)氨氮浓度的影响。不同初始氨氮浓度条件下Ti/RuO2-IrO2-RhOx电极电解氨氮时的实验结果如图8所示。为模拟实际常见氨氮废水,如皮革厂综合废水、氮肥厂氨氮出水及垃圾渗滤液等[35-37],将氨氮初始质量浓度设置为200~500 mg·L−1。当初始氨氮质量浓度为500 mg·L−1时,电解2.5 h后氨氮剩余质量浓度为234.8 mg·L−1,降解率为53.0%;当初始氨氮质量浓度为200 mg·L−1时,2 h内氨氮可全部降解,这说明Ti/RuO2-IrO2-RhOx对于氨氮的电化学氧化性能较佳,在短时间内能实现低浓度氨氮的彻底去除。电解5 h后,所有浓度的氨氮基本被完全电解,说明该电极对于高浓度氨氮废水亦有良好的处理效果。除此之外,氨氮浓度与反应时间呈现出线性关系,氨氮氧化速率随初始氨氮浓度的增加而下降,但幅度不大,斜率仅为0.015。这说明采用Ti/RuO2-IrO2-RhOx电极电解氨氮时,氨氮的氧化速率与其初始浓度关系不大,大致呈现为表观零级反应。这与文献[38-39]的研究结果一致。
3)电流密度的影响。图9展示了采用Ti/RuO2-IrO2-RhOx电极电解氨氮时电流密度对氨氮浓度的影响情况。当电流密度为250 A·m−2时,氨氮降解速率较慢,2.5 h后降解率仅为18.5%;当电流密度升高至1 500 A·m−2时,2.5 h后氨氮降解率可达76.0%。可见,增加电流密度有利于氨氮的降解,当废水电导率较高时,为了加快电极降解氨氮的速率,宜适当加大电流密度。
4)初始氯离子浓度的影响。图10显示了Ti/RuO2-IrO2-RhOx电极电解氨氮时初始氯离子浓度对氨氮浓度的影响。初始氯离子浓度对于氨氮的降解途径有很大影响。当氯离子浓度为0时,电解过程中,氨氮浓度变化不大,反应速率低,3 h后降解率仅为7.1%。此时,氨氮的氧化路径分为以下2种[40-41]:一方面,氨氮吸附于Ti/RuO2-IrO2-RhOx表面,与电极发生直接的电子传递被氧化去除;另一方面,在较高电势下,体系产生少量强氧化性的·OH与氨氮发生反应使之降解。加入氯离子后,反应速率迅速增大,且随着氯离子浓度增加,氨氮浓度下降速率明显加快。初始氯离子质量浓度为8 000 mg·L−1时,2.5 h后氨氮的降解率达74.2%,3.5 h后降解率高达98.3%,此时间接氧化反应占主导地位。这是因为氯离子在氨氮的电化学氧化反应中扮演了2个重要角色:一是作为电解质增加溶液的导电性,降低能耗;二是氯离子参与电化学反应,失去电子生成高价态的ClO−、Cl2等,发生以下反应:2NH4+ + 3ClO− → N2 + 3H2O + 2H+ + 3Cl−[42],进一步氧化氨氮。氯离子的添加有利于氨氮的电化学氧化过程,在一定范围内,氯离子浓度越高,氨氮氧化速率越快,但此时氨氮去除速率增幅有所递减。KIM等[43]发现,当氯离子质量浓度超过10 g·L−1后,只有部分氯离子与其他离子在电极上竞争吸附。过量的氯离子会产生一定程度的空间位阻效应和析氯效应,影响传质和反应。
5)硬度与倒极操作的影响。为模拟倒极操作和实际废水中的离子对反应体系的影响,向体系中添加Ca2+、Mg2+,分别在倒极与不倒极操作条件下,采用Ti/RuO2-IrO2-RhOx电极进行电解实验,氨氮浓度和电压的变化如图11(a)和图11(b)所示。在不倒极操作条件下,电解2.5 h后,硬水体系中氨氮去除率为60.1%,软水体系的氨氮去除率为59.3%,倒极操作下的氨氮去除率为66.4%,5 h内氨氮均完全反应。值得注意的是,在硬水体系中,倒极操作下反应5 h,电压由6.73 V上升至7.48 V,上升了11.1%;非倒极操作下反应5 h,电压由6.74 V上升至8.17 V,上升了21.2%。这说明倒极操作可以及时脱除电极表面垢层,维持系统电压稳定,降低反应能耗。因此,Ti/RuO2-IrO2-RhOx电极在处理高硬度氨氮废水领域具有广阔的应用前景。
-
1) Ti/RuO2-IrO2-RhOx新型电极表面均匀致密但凹凸不平,有簇状晶粒析出,有助于增大电极活性面积,有利于电极反应,体相元素高度固溶,结构致密。
2)与常用Ti/RuO2-TiO2、Ti/IrO2-Ta2O5电极相比,Ti/RuO2-IrO2-RhOx新型电极拥有最长的倒极加速寿命,适用于频繁倒极的实际工况,在20 g·L−1 Na2SO4、2 000 A·m−2电流密度,20 min·次−1倒极频率,20 min·次−1倒极持续时长下,加速寿命可达到1 400 h甚至更高,分别是Ti/RuO2-TiO2和Ti/IrO2-Ta2O5的47倍和23倍。
3)与Ti/RuO2-TiO2、Ti/IrO2-Ta2O5相比,Ti/RuO2-IrO2-RhOx电子传输速率高,拥有较好的析氯效果与最强的氨氮氧化能力,适用于电解法处理氨氮废水,且Ti/RuO2-IrO2-RhOx在频繁倒极操作下电解高硬度氨氮废水时,可保持较高的氨氮去除效率,同时维持电压稳定,降低反应能耗,在处理高硬度氨氮废水领域具有广阔的应用前景。
4)初始氨氮浓度、电流密度及初始氯离子浓度均可影响Ti/RuO2-IrO2-RhOx电极电化学氧化氨氮的效率。当体系中不存在其他杂质离子时,初始氨氮浓度基本不影响氨氮氧化速率,大致体现为表观零级反应;在一定范围内,氨氮氧化速率与电流密度正相关;初始氯离子浓度直接影响到氨氮电化学氧化途径,当存在氯离子时,氨氮主要通过间接氧化去除,适量的氯离子有助于氨氮的电化学氧化。
倒极长寿命的Ti/RuO2-IrO2-RhOx电极对氨氮废水的处理效能
Treatment performance of Ti/RuO2-IrO2-RhOx electrode with a long polarity inverted lifetime on ammonia nitrogen wastewater
-
摘要: 在电化学处理高硬度氨氮废水过程中,Ca2+、Mg2+等会在电场作用下富集并沉积在电极表面,引起电极结垢,最终影响氨氮去除效率。倒极除垢操作简单、脱垢效果佳,但会折损电极寿命。为了解决这一问题,研制了一种可在倒极操作下保持长寿命的Ti/RuO2-IrO2-RhOx新型电极,以用于处理上述高硬度氨氮废水。结果表明,在倒极操作下,Ti/RuO2-IrO2-RhOx电极加速寿命高达1 400 h,分别是Ti/RuO2-TiO2与Ti/IrO2-Ta2O5电极的47倍与23倍。物理化学表征结果表明,RuO2-IrO2-RhOx涂层为致密均匀固溶体,表面有簇状晶粒析出。将研制的Ti/RuO2-IrO2-RhOx新型电极用于电解氨氮废水,发现其在析氯与氨氮去除方面均优于Ti/RuO2-TiO2和Ti/IrO2-Ta2O5。此外,氨氮浓度、电流密度以及初始氯离子浓度对氨氮去除效率均有明显影响。以上研究结果可为Ti/RuO2-IrO2-RhOx新型电极在氨氮废水的处理中的应用提供参考。Abstract: In the process of electrochemical treating ammonia-nitrogen wastewater with high hardness, Ca2+ and Mg2+ can be enriched and deposited on the electrode surface under the action of electric field, which will result in electrode scaling, and ultimately affect the removal efficiency of ammonia-nitrogen. The polarity-reversal descaling is simple and effective, but it can shorten the electrode lifetime. In order to solve this problem, a novel Ti/RuO2-IrO2-RhOx electrode with a long lifetime under polarity reversal operation was prepared for above wastewater treatment. The experimental results show that, under the polarity reversal operation, the accelerated lifetime of Ti/RuO2-IrO2-RhOx could reach 1 400 h, which was 47 times that of Ti/RuO2-TiO2 electrode and 23 times that of Ti/IrO2-Ta2O5 electrode, respectively. Physiochemical characterization shows that Ti/RuO2-IrO2-RhOx was a type of uniform and compact solid solution with cluster grains precipitated on its surface. The prepared Ti/RuO2-IrO2-RhOx electrode was used to electrolyze ammonia wastewater. It was found that this novel electrode was better than the conventional Ti/RuO2-TiO2 and Ti/IrO2-Ta2O5 electrodes in chlorine evolution and ammonia nitrogen removal. In addition, it was demonstrated that the ammonia nitrogen concentration, current density and initial chloride ion concentration could influence the ammonia nitrogen removal remarkably. The above research results can provide a reference for the application of Ti/RuO2-IrO2-RhOx electrode in the treatment of ammonia-nitrogen wastewater.
-
氨氮是引起水体富营养化的主要原因之一。氨氮浓度过高会引起水生生物中毒死亡,形成黑臭水体;同时,氨氮自然氧化过程的中间产物可能会引起人体组织发生癌变[1-2]。利用电化学氧化法可以有效去除氨氮,故该方法在垃圾渗滤液[3]、养殖废水[4]、制革废水[5]等废水处理领域具有巨大的应用潜力。氨氮的电化学氧化一般分为2种路径:直接氧化与间接氧化。当溶液中有大量氯离子存在时,氨氮主要通过间接氧化去除,即氯离子先被氧化生成Cl2、ClO−等强氧化性产物,与氨氮结合生成氯胺,再进一步被氧化去除[6]。因此,电极析氯性能的优劣可直接影响到氨氮的电化学氧化效果。此外,初始氯离子浓度、初始氨氮浓度以及电流密度等因素均会对氨氮的电化学氧化产生不同程度的影响[7, 8]。
电极结垢是电化学水处理中的常见问题[9-12]。当采用电化学技术处理高硬度氨氮废水时,废水中的Ca2+、Mg2+等硬度离子会在阴极富集,并与水中的HCO3−和阴极析氢反应产生的OH−结合生成难溶性的CaCO3与Mg(OH)2垢层并覆盖在电极表面,阻碍电化学反应。WANG等[13]在研究燃煤电厂高盐氨氮废水时发现,电极表面的清洁度越高,氨氮去除效果越好。因此,若要保持较高的氨氮去除效率,需频繁对电极进行除垢处理。倒极脱垢是目前工业中常用的一种除垢方法。与机械刮擦脱垢、空气冲刷脱垢相比,倒极脱垢操作简单、脱垢效果佳,不需要另外增设阴极刮擦板,降低了设备的安装难度[14]。
形状稳定性电极(dimensionally stable anodes, DSA)是以钛为基体,表面涂覆金属氧化物制得的电催化电极[15]。工业上常用的DSA电极主要是钌系电极与铱系电极。钌系电极析氯活性高,耐腐蚀性强,成本相对较低,但在高电流密度下寿命短暂[16]。而与之相比,铱电极寿命可提高20倍[17],因此,在许多领域应用广泛[18-21]。然而,这类传统DSA电极通常只用作阳极,且工作时极性不发生变化,若经历倒极操作,活性涂层会脱落[22-23],电极迅速失效。因此,寻求一种在倒极工况下保持性能稳定的电极至关重要。为解决这一问题,在前期研究中,尝试在RuO2-IrO2体系中添加RhOx组分,制得了Ti/RuO2-IrO2-RhOx电极以用于对苯酚等有毒污染物的降解。前期研究中发现:与Ti/RuO2-IrO2相比,Ti/RuO2-IrO2-RhOx表面更加光滑致密,裂纹和孔隙较少,晶粒高度分散,涂层结构得到显著改善;Rh3+/Rhn+电对的氧化还原反应可逆,RhOx与传统的RuO2、IrO2可以产生协同效应,从而提高电极的倒极稳定性和氧化能力[24]。但前期研究并未涉及Ti/RuO2-IrO2-RhOx电极用于氨氮的电化学氧化。
为了解决上述问题,研制了一种倒极长寿命的Ti/RuO2-IrO2-RhOx新型电极用于处理氨氮废水。首先考察了其倒极稳定性、析氯电化学活性以及对氨氮的去除效果,并与传统的Ti/RuO2-TiO2、Ti/IrO2-Ta2O5等电极进行了对比;在此基础上,进一步将所研制的Ti/RuO2-IrO2-RhOx电极用于电解氨氮废水,考察了氨氮浓度、电流密度以及氯离子浓度等因素对氨氮电化学氧化效果的影响。
1. 材料与方法
1.1 实验原料
氯铱酸(H2IrCl6·xH2O,35% Ir),氯化钽(TaCl5,99.99%),氯化钌(RuCl3,45%~55% Ru),四氯化钛(TiCl4,AR),三水合氯化铑(RhCl3·3H2O),盐酸(HCl,AR),异丙醇(C3H8O,AR),氯化钠(NaCl,AR),碘化钾(KI,AR),淀粉指示剂(0.25%淀粉),冰乙酸(CH3COOH,AR),五水合硫代硫酸钠(Na2S2O3·5H2O,AR),氢氧化钠(NaOH,AR),碘化汞(Ⅱ)(HgI2,AR),酒石酸钾钠(NaKC4H4O6,AR),硫酸铵((NH4)2SO4,AR),氯化钙(CaCl2,AR),六水合氯化镁(MgCl2·6H2O,AR)。以上所有药品未经再次提纯,直接购买使用,实验用水为超纯水(电导率≤0.06 μS·cm−1)。
1.2 电极制备
分别取用一定量的RuCl3、TiCl4、H2IrCl6·xH2O、TaCl5、RhCl3·3H2O溶于浓盐酸与异丙醇(体积比1∶1)的混合溶液,配制成0.25 mol·L−1的RuCl3、TiCl4、TaCl5、RhCl3·3H2O、H2IrCl6·xH2O溶液。将这些溶液分别按照RuCl3∶TiCl4=1∶1、H2IrCl6·xH2O∶TaCl5=1∶1、RuCl3∶H2IrCl6·xH2O∶RhCl3·3H2O=2∶1∶2的摩尔比混合,配制成不同组分的前驱液。以上前驱液均现用现配。
采用2 cm×3 cm的钛网为基底,制备电极前,钛网基底经喷砂、除油、煮沸的浓盐酸刻蚀2 min、自来水洗、去离子水超声洗10 min之后烘干称量,存放在无水乙醇中备用。采用热分解法制备电极。先于室温下将即时配制的前驱液均匀涂覆于钛网上,再用烘箱在80 ℃下烘干5 min,然后转移至450 ℃马弗炉中热解5 min,出炉冷却至室温,重复涂覆-烘干-热解-冷却操作13~18次,直至涂层覆载量达18 g·m−2,最后一次热解时间延长至1 h。
1.3 倒极加速寿命评价及物理化学表征
采用倒极加速寿命实验评估电极稳定性[25]。采用直流电源(MS303DS,东莞市迈豪电子科技有限公司)供电,用PLC控制器 (YBD-12,优博达电子)控制倒极频率。加速寿命实验条件:电解液为20 g·L−1 Na2SO4溶液,pH为7,温度为25 ℃,电流密度为2 000 A·m−2,倒极频率为20 min·次−1,倒极持续时长为20min·次−1。以电解槽电压上升5 V时作为评价电极失活的判据,评价加速寿命实验条件下的电极寿命。采用场发射电镜(FE-SEM)观察涂层形貌结构;采用X射线衍射仪(XRD)分析涂层晶体结构。
1.4 循环伏安测试
在经典三电极体系测试装置中,采用稳压/恒流电化学分析仪(CHI604E,上海辰华)测试循环伏安曲线(CV),工作电极为自制电极,对电极为铂电极,参比电极为饱和甘汞电极。分别在25 ℃,0.5 mol·L−1 NaCl溶液与10 mmol·L−1 [Fe(CN)6]3-/4-+0.2 mol·L−1 K2SO4溶液中得到阳极的循环伏安曲线。扫描电压分别为0.2~1.6 V、−0.2~0.8 V,扫描速率为100 mV·s−1。
1.5 析氯性能测试
通过电解NaCl溶液,在密闭电解槽中测定有效氯浓度,计算电流效率。实验在1 L恒温(25 ℃)密闭电解槽内进行,阴阳极均采用自制电极,NaCl质量浓度为15 g·L−1,电流密度为1 000 A·m−2。每隔30 min取样5 mL,采用碘量法测定有效氯浓度[26]。
有效氯浓度根据式(1)进行计算,电流效率根据式(2)进行计算。
C=35.45/5NV (1) η=CV0/1.323It×100% (2) 式中:C为有效氯质量浓度,g·L−1;N为Na2S2O3溶液浓度,mol·L−1;V为滴定消耗的Na2S2O3溶液体积,mL。η为电流效率,%;V0为电解液总体积,L;I为反应电流,A;t为反应时间,h;1.323为每安培小时电量有效氯的理论生成量,g·(A·h)−1。
1.6 电解模拟氨氮废水
电解氨氮废水实验于200 mL的密闭恒温(25 ℃)电解槽内进行,阴阳极均采用自制电极。采用NaCl、(NH4)2SO4、去离子水配制电解液模拟氨氮废水,根据实验需要依次调节溶液初始氨氮浓度、电流密度、初始氯离子浓度等,每隔30 min取样250 μL,采用纳氏试剂分光光度法测定剩余氨氮浓度。采用CaCl2、MgCl2·6H2O按照摩尔比1∶1配制硬水,硬度以CaCO3质量(mg·L−1)计。
2. 结果与讨论
2.1 倒极寿命分析
为便于比较,对本团队新研制的Ti/RuO2-IrO2-RhOx电极与传统的Ti/RuO2-TiO2、Ti/IrO2-Ta2O5电极进行了倒极加速寿命实验,结果如图1所示。Ti/RuO2-TiO2与Ti/IrO2-Ta2O5的电压变化情况相类似,短时间内电压迅速升高,电极随即失效,倒极加速寿命分别为30 h与60 h;而Ti/RuO2-IrO2-RhOx的电极电压上升是一个持续而缓慢的过程,1 000 h内电压总增幅仅2.5 V,加速寿命可达到1 400 h甚至更高,远远超过其他2种电极。ROGINSKAYA等[27]曾报道,RhOx在酸性溶液中比RuO2和IrO2稳定得多。HRUSSANOVA等[28]在研究RhOx电极在酸性溶液中的析氧电化学行为时也指出,在最初去除松散颗粒后,析氧不能降解RhOx电极。因此,惰性元素Rh组分的添加提高了电极的稳定性,使其适用于频繁颠倒阴阳极性的操作条件。
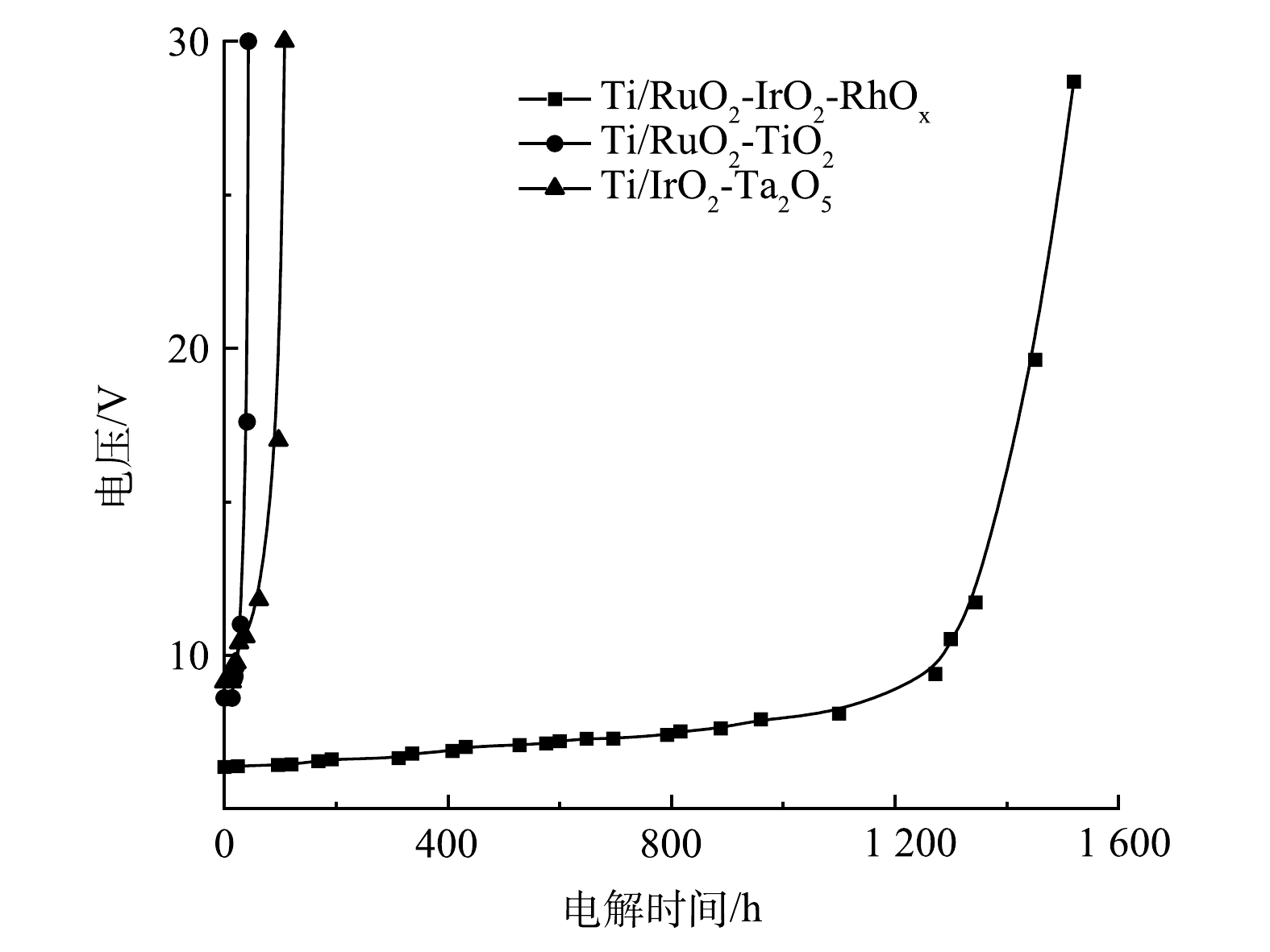 图 1 Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电压变化Figure 1. Voltage variation of Ti/RuO2-TiO2、Ti/IrO2-Ta2O5 and Ti/RuO2-IrO2-RhOx
图 1 Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电压变化Figure 1. Voltage variation of Ti/RuO2-TiO2、Ti/IrO2-Ta2O5 and Ti/RuO2-IrO2-RhOx2.2 物理化学表征与电化学性能分析
1)表面形貌。Ti/RuO2-IrO2-RhOx电极涂层在开展寿命实验前后的FE-SEM扫描结果见图2。由图2(a)与图2(b)可见,在开展寿命实验前新制备的Ti/RuO2-IrO2-RhOx电极涂层表面整体均匀致密但凹凸不平,伴随有簇状晶粒析出和少量裂纹。致密的表面结构使得溶液中的氧化活性粒子不易渗入基底,提高了电极稳定性;析出的簇状晶粒可以有效增大电极的比表面积,增加活性位点个数,更有利于电化学反应;裂纹的出现则是钛基体与涂层之间的热膨胀系数差异所致[29]。由图2(c)和图2(d)可见,Ti/RuO2-IrO2-RhOx电极失效后,电极表面凹凸不平,基底裸露,伴有大量气泡式细小孔隙。结合前期研究结果可推断,电极失效可能是由气体冲刷、涂层溶解、活性成分丧失所致[23-24]。
 图 2 Ti/RuO2-IrO2-RhOx的 FE-SEM图像Figure 2. FE-SEM images of Ti/RuO2-IrO2-RhOx electrode
图 2 Ti/RuO2-IrO2-RhOx的 FE-SEM图像Figure 2. FE-SEM images of Ti/RuO2-IrO2-RhOx electrode2) XRD分析。Ti/RuO2-IrO2-RhOx电极的XRD图谱以及主要氧化物的标准峰见图3。在27.8°、34.8°、38.1°、40.0°、52.9°、53.8°、57.5°、58.9°、65.1°、66.6°、69.0°、70.4°、73.9°、76.0°、77.2°等处检测到15个明显的衍射峰,与Ru、Ir、Rh等氧化物的标准峰进行比对,扣除1组基底钛标准峰,其他峰位置与RuO2、IrO2、RhO2的峰位置基本对应,但又有一定程度的偏移。这可能是因为Ti/RuO2-IrO2-RhOx涂层各组分充分混合,形成了均一的固溶体[30]。理论上,离子之间能否形成稳定固溶体取决于不同元素之间的离子半径差异。根据Hume-Rothery法则,当粒子之间的粒径差小于14%~15%时,有可能形成稳定的固溶体[31]。Ir4+、Ru4+、Rh4+和Rh3+的离子半径分别为0.062 5、0.062、0.06、0.066 5 nm,相互间的最大差值为10.8%,小于14%。因此,Ti/RuO2-IrO2-RhOx涂层元素很可能以固溶体形式存在。此外,Rh2O3的个别峰虽然能够被观测到,但并不能完全对应,因此,Rh2O3是否存在有待于深入探究。金华长等[17]在其研究中也得出了相似的结果。
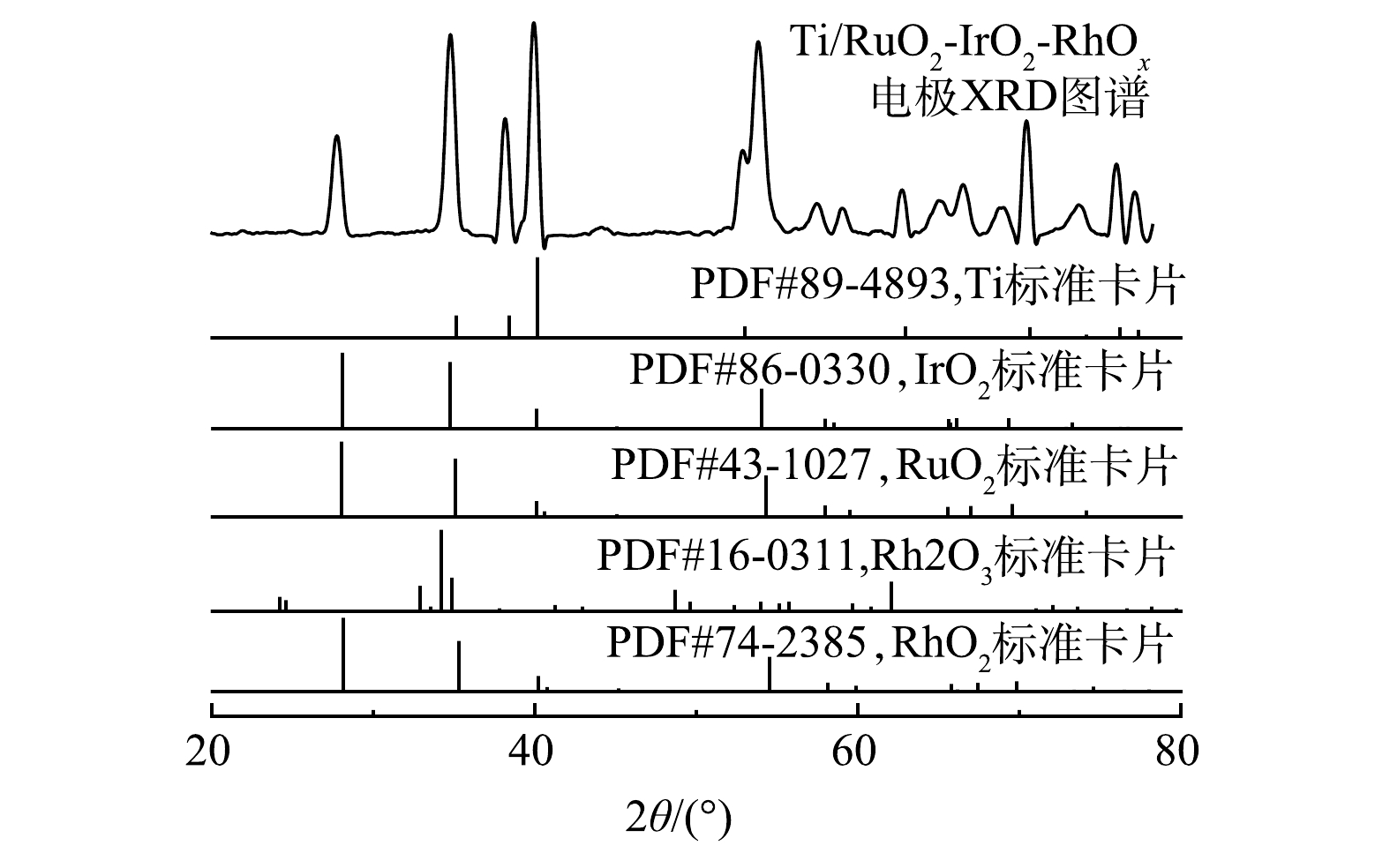 图 3 Ti/RuO2-IrO2-RhOx 的XRD图谱及主要氧化物标准衍射峰Figure 3. XRD partterns of Ti/RuO2-IrO2-RhOx and the diffraction peaks of the standard cards of key oxides
图 3 Ti/RuO2-IrO2-RhOx 的XRD图谱及主要氧化物标准衍射峰Figure 3. XRD partterns of Ti/RuO2-IrO2-RhOx and the diffraction peaks of the standard cards of key oxides3)电化学性能。Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电极在0.5 mol·L -1 NaCl溶液中的CV测试结果如图4所示。3种电极均呈现出典型的伏安特性曲线。正向扫描时,曲线分别在1.19、1.24、1.21 V处由于发生析氯反应而出现转折,反应式为:2Cl− - 2e− = Cl2。在相同电位下,Ti/RuO2-IrO2-RhOx的电流明显大于Ti/RuO2-TiO2、Ti/IrO2-Ta2O5。这表明:与其他2种电极相比,Ti/RuO2-IrO2-RhOx具有最大的电容。
图5反映了Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电极在10 mmol·L−1[Fe(CN)6]3-/4-+0.2 mol·L−1 K2SO4溶液中的CV扫描结果。电极在[Fe(CN)6]3-/[Fe(CN)6]4-氧化还原电对中的电化学行为与涂层表面电子传递速率有关[32]。3种电极的CV曲线均出现1组氧化峰与还原峰。其中,Ti/RuO2-IrO2-RhOx电极氧化峰与还原峰对应的电流值大小基本相同,说明Ti/RuO2-IrO2-RhOx电极对于[Fe(CN)6]3-/[Fe(CN)6]4-电子对的氧化还原反应高度可逆。此外,Ti/RuO2-IrO2-RhOx两峰之间的距离最小,说明该电极拥有最快的电子转移速率。这说明:在电化学反应中,Ti/RuO2-IrO2-RhOx电极可能拥有最高的电流效率和电化学反应活性。
2.3 析氯性能
Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电极电解NaCl溶液时,有效氯浓度和电流效率随时间的变化情况见图6。如图6(a)所示,反应2.5 h后,Ti/RuO2-IrO2-RhOx电解产生0.73 g有效氯,高于Ti/RuO2-TiO2和Ti/IrO2-Ta2O5的0.69 g和0.66 g。如图6(b)所示,Ti/RuO2-IrO2-RhOx与其他2种电极相比,始终保持最高的电流效率。3种电极电解产生的有效氯浓度均随反应时间的延长而不断升高,但体系电流效率逐渐下降。这与体系中反应物氯离子浓度的下降及反应产物次氯酸根等的增加有关[33-34]。Ti/RuO2-IrO2-RhOx在单位时间内生成的有效氯浓度最高,这是因为Ti/RuO2-IrO2-RhOx电子传输速率快,电流效率高,可以有效催化氯离子氧化。这与2.2中电极的电化学性能测试所得结论一致。
2.4 氨氮去除效果
1)不同电极去除氨氮效果对比。图7展示了采用Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电极电解氨氮时,氨氮浓度随时间变化情况。显而易见,3种电极都可用于电解去除氨氮,但去除效率不同。Ti/RuO2-TiO2和Ti/IrO2-Ta2O5电解2.5 h后,氨氮由最初的400 mg·L−1(以N计)分别降至146.4 mg·L−1和188.6 mg·L−1,去除率分别为63.4%和53.0%;电解4.5 h后,氨氮分别降至0.5 mg·L−1和28.6 mg·L−1,去除率分别为99.9%和92.9%。相比之下,Ti/RuO2-IrO2-RhOx电解2.5 h后,氨氮由原先的400 mg·L−1降至130 mg·L−1,去除率为67.6%;电解4.5 h后,氨氮降为0,去除率达100%。由此可见,Ti/RuO2-IrO2-RhOx具有更高的氨氮去除效率,这主要归因于该电极具有更好的析氯性能。
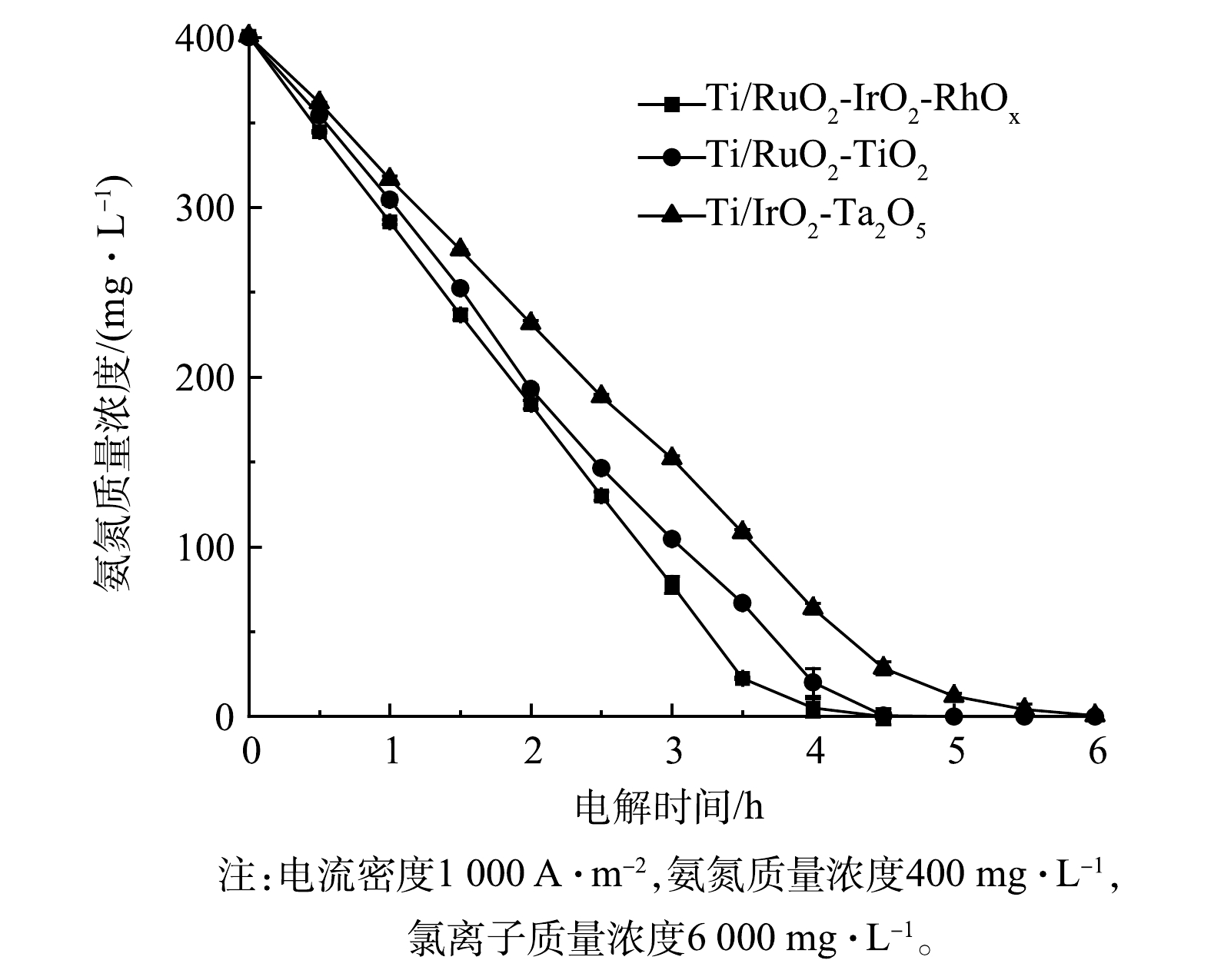 图 7 不同电极对氨氮降解的影响Figure 7. Effect of different electrodes on ammonia nitrogen degradation
图 7 不同电极对氨氮降解的影响Figure 7. Effect of different electrodes on ammonia nitrogen degradation2)氨氮浓度的影响。不同初始氨氮浓度条件下Ti/RuO2-IrO2-RhOx电极电解氨氮时的实验结果如图8所示。为模拟实际常见氨氮废水,如皮革厂综合废水、氮肥厂氨氮出水及垃圾渗滤液等[35-37],将氨氮初始质量浓度设置为200~500 mg·L−1。当初始氨氮质量浓度为500 mg·L−1时,电解2.5 h后氨氮剩余质量浓度为234.8 mg·L−1,降解率为53.0%;当初始氨氮质量浓度为200 mg·L−1时,2 h内氨氮可全部降解,这说明Ti/RuO2-IrO2-RhOx对于氨氮的电化学氧化性能较佳,在短时间内能实现低浓度氨氮的彻底去除。电解5 h后,所有浓度的氨氮基本被完全电解,说明该电极对于高浓度氨氮废水亦有良好的处理效果。除此之外,氨氮浓度与反应时间呈现出线性关系,氨氮氧化速率随初始氨氮浓度的增加而下降,但幅度不大,斜率仅为0.015。这说明采用Ti/RuO2-IrO2-RhOx电极电解氨氮时,氨氮的氧化速率与其初始浓度关系不大,大致呈现为表观零级反应。这与文献[38-39]的研究结果一致。
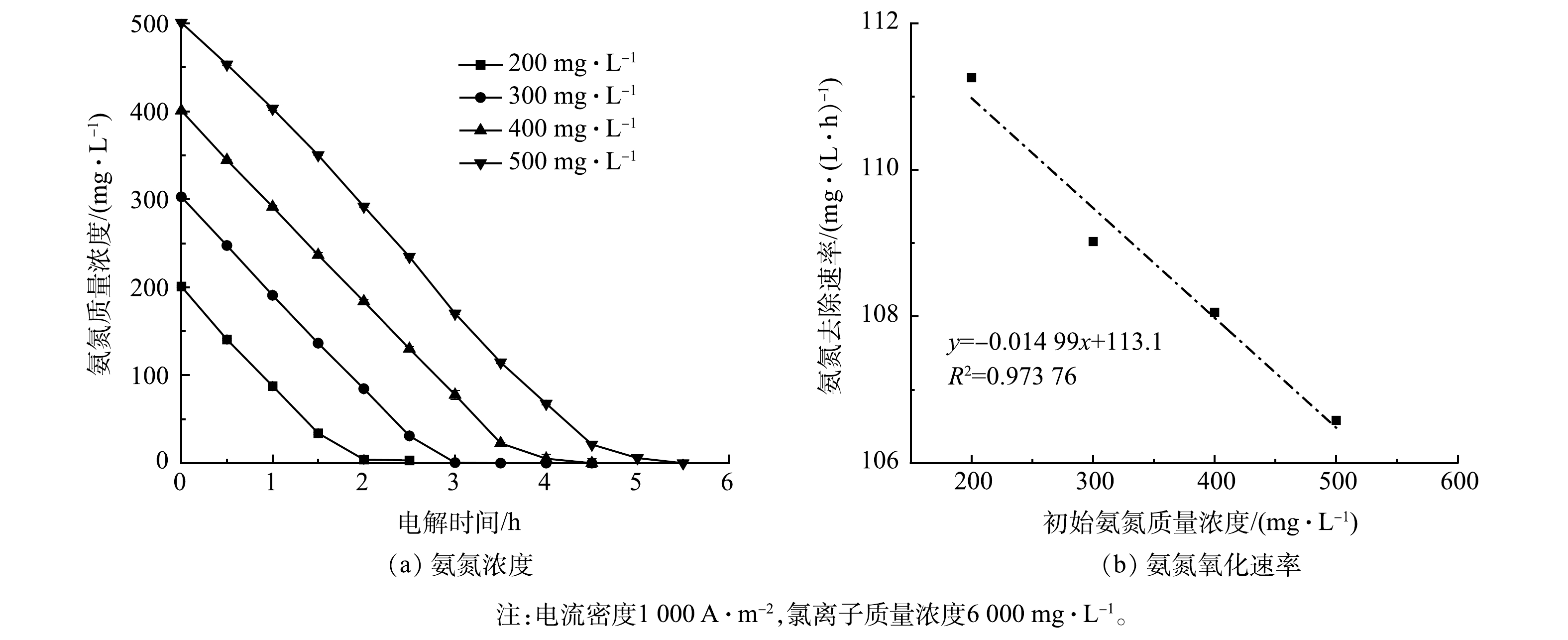 图 8 不同初始氨氮浓度对氨氮降解的影响Figure 8. Effects of different initial ammonia nitrogen concentrations on ammonia nitrogen degradation
图 8 不同初始氨氮浓度对氨氮降解的影响Figure 8. Effects of different initial ammonia nitrogen concentrations on ammonia nitrogen degradation3)电流密度的影响。图9展示了采用Ti/RuO2-IrO2-RhOx电极电解氨氮时电流密度对氨氮浓度的影响情况。当电流密度为250 A·m−2时,氨氮降解速率较慢,2.5 h后降解率仅为18.5%;当电流密度升高至1 500 A·m−2时,2.5 h后氨氮降解率可达76.0%。可见,增加电流密度有利于氨氮的降解,当废水电导率较高时,为了加快电极降解氨氮的速率,宜适当加大电流密度。
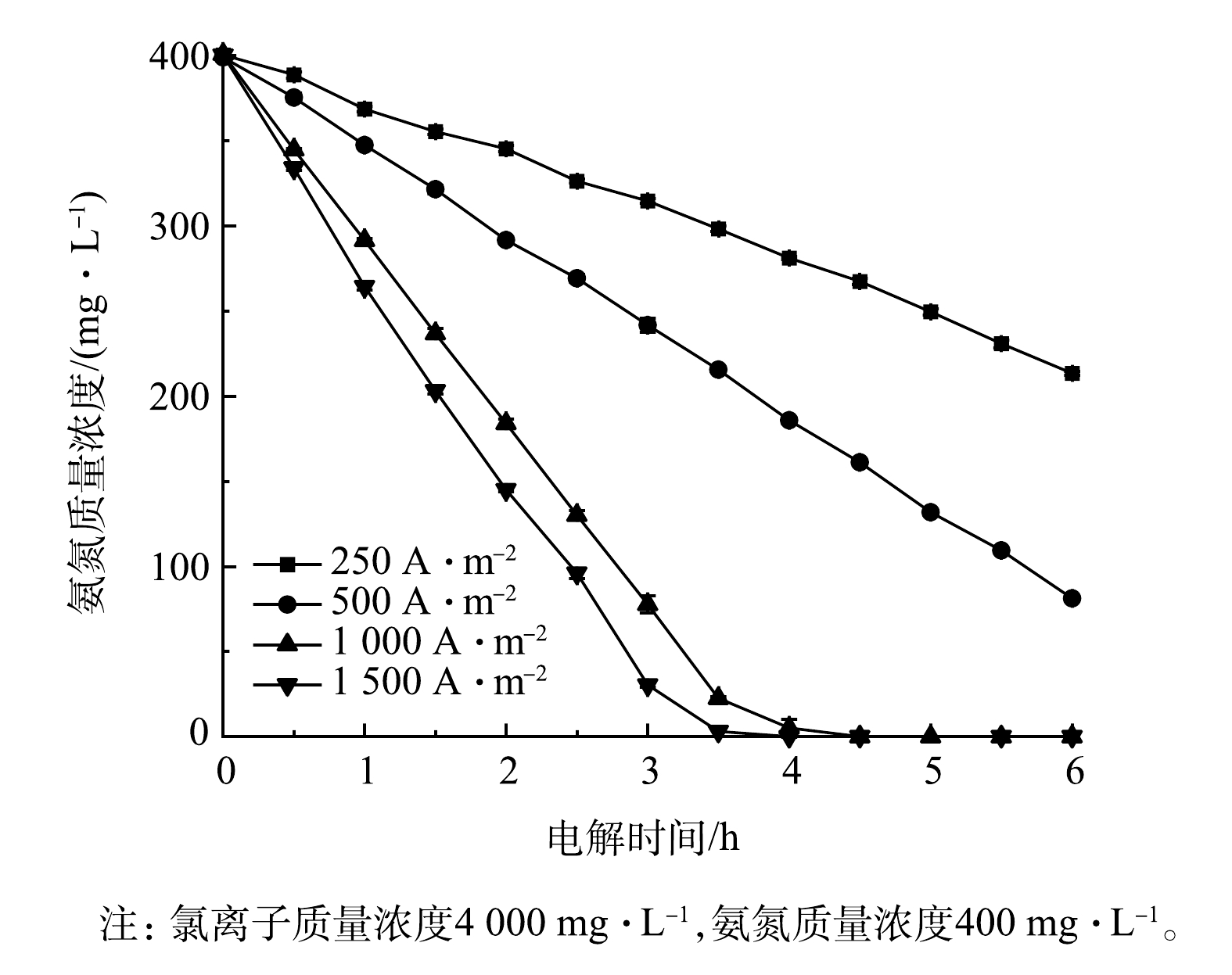 图 9 不同电流密度对氨氮降解的影响Figure 9. Effect of different current density on ammonia nitrogen degradation
图 9 不同电流密度对氨氮降解的影响Figure 9. Effect of different current density on ammonia nitrogen degradation4)初始氯离子浓度的影响。图10显示了Ti/RuO2-IrO2-RhOx电极电解氨氮时初始氯离子浓度对氨氮浓度的影响。初始氯离子浓度对于氨氮的降解途径有很大影响。当氯离子浓度为0时,电解过程中,氨氮浓度变化不大,反应速率低,3 h后降解率仅为7.1%。此时,氨氮的氧化路径分为以下2种[40-41]:一方面,氨氮吸附于Ti/RuO2-IrO2-RhOx表面,与电极发生直接的电子传递被氧化去除;另一方面,在较高电势下,体系产生少量强氧化性的·OH与氨氮发生反应使之降解。加入氯离子后,反应速率迅速增大,且随着氯离子浓度增加,氨氮浓度下降速率明显加快。初始氯离子质量浓度为8 000 mg·L−1时,2.5 h后氨氮的降解率达74.2%,3.5 h后降解率高达98.3%,此时间接氧化反应占主导地位。这是因为氯离子在氨氮的电化学氧化反应中扮演了2个重要角色:一是作为电解质增加溶液的导电性,降低能耗;二是氯离子参与电化学反应,失去电子生成高价态的ClO−、Cl2等,发生以下反应:2NH4+ + 3ClO− → N2 + 3H2O + 2H+ + 3Cl−[42],进一步氧化氨氮。氯离子的添加有利于氨氮的电化学氧化过程,在一定范围内,氯离子浓度越高,氨氮氧化速率越快,但此时氨氮去除速率增幅有所递减。KIM等[43]发现,当氯离子质量浓度超过10 g·L−1后,只有部分氯离子与其他离子在电极上竞争吸附。过量的氯离子会产生一定程度的空间位阻效应和析氯效应,影响传质和反应。
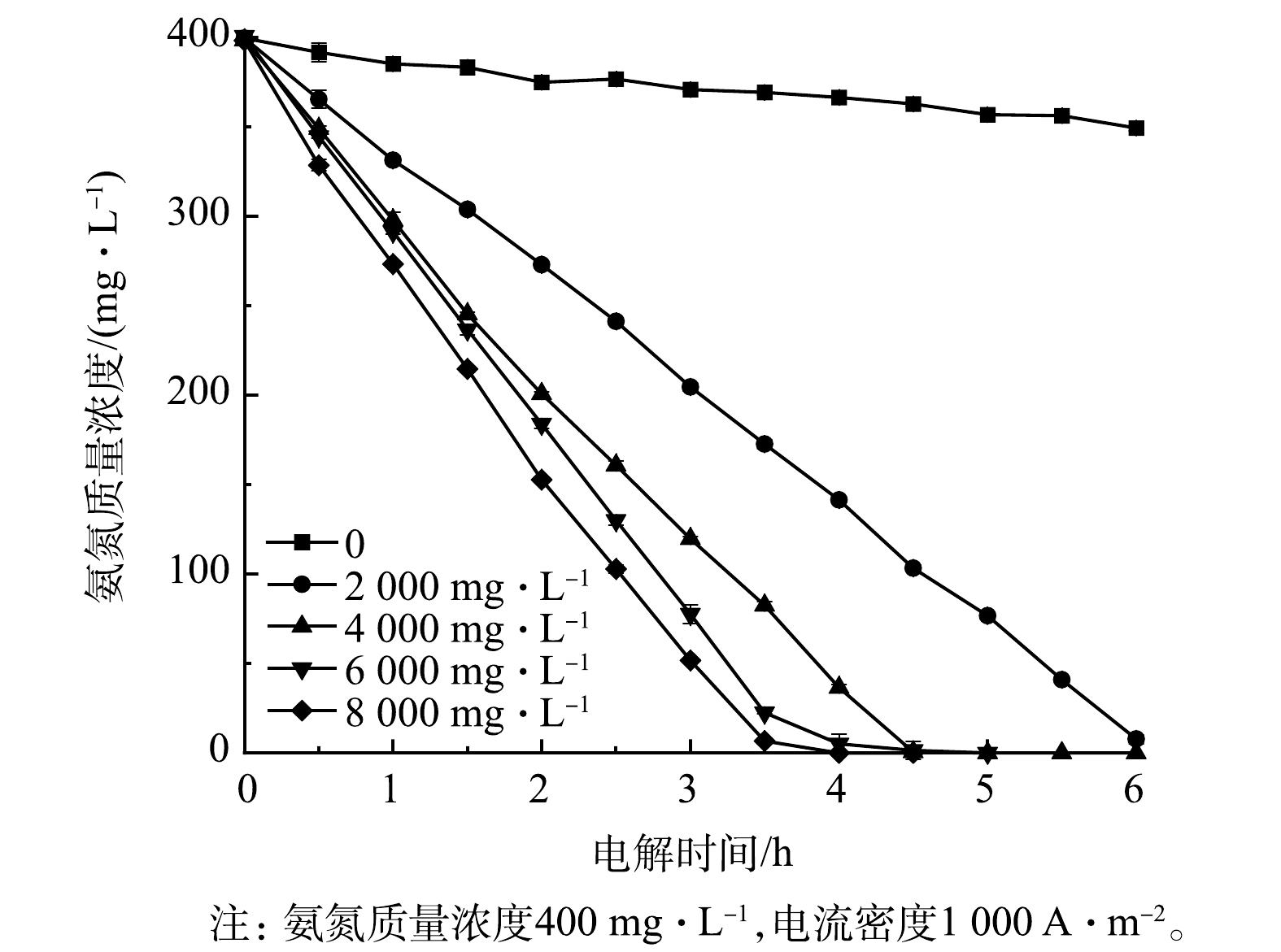 图 10 不同氯离子浓度对氨氮降解的影响Figure 10. Effect of different chloride ion concentration on ammonia nitrogen degradation
图 10 不同氯离子浓度对氨氮降解的影响Figure 10. Effect of different chloride ion concentration on ammonia nitrogen degradation5)硬度与倒极操作的影响。为模拟倒极操作和实际废水中的离子对反应体系的影响,向体系中添加Ca2+、Mg2+,分别在倒极与不倒极操作条件下,采用Ti/RuO2-IrO2-RhOx电极进行电解实验,氨氮浓度和电压的变化如图11(a)和图11(b)所示。在不倒极操作条件下,电解2.5 h后,硬水体系中氨氮去除率为60.1%,软水体系的氨氮去除率为59.3%,倒极操作下的氨氮去除率为66.4%,5 h内氨氮均完全反应。值得注意的是,在硬水体系中,倒极操作下反应5 h,电压由6.73 V上升至7.48 V,上升了11.1%;非倒极操作下反应5 h,电压由6.74 V上升至8.17 V,上升了21.2%。这说明倒极操作可以及时脱除电极表面垢层,维持系统电压稳定,降低反应能耗。因此,Ti/RuO2-IrO2-RhOx电极在处理高硬度氨氮废水领域具有广阔的应用前景。
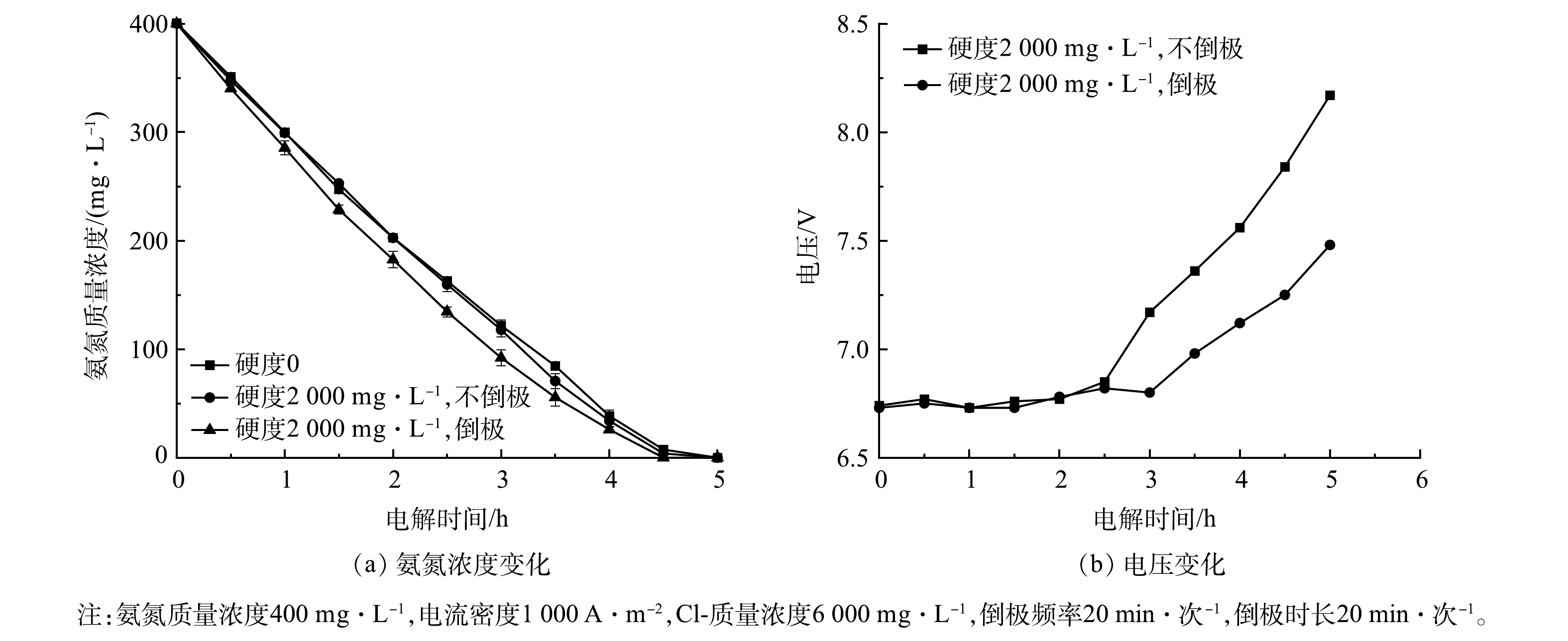 图 11 硬度与倒极操作对氨氮降解的影响Figure 11. Effects of hardness and polarity reversal operation on ammonia nitrogen degradation
图 11 硬度与倒极操作对氨氮降解的影响Figure 11. Effects of hardness and polarity reversal operation on ammonia nitrogen degradation3. 结论
1) Ti/RuO2-IrO2-RhOx新型电极表面均匀致密但凹凸不平,有簇状晶粒析出,有助于增大电极活性面积,有利于电极反应,体相元素高度固溶,结构致密。
2)与常用Ti/RuO2-TiO2、Ti/IrO2-Ta2O5电极相比,Ti/RuO2-IrO2-RhOx新型电极拥有最长的倒极加速寿命,适用于频繁倒极的实际工况,在20 g·L−1 Na2SO4、2 000 A·m−2电流密度,20 min·次−1倒极频率,20 min·次−1倒极持续时长下,加速寿命可达到1 400 h甚至更高,分别是Ti/RuO2-TiO2和Ti/IrO2-Ta2O5的47倍和23倍。
3)与Ti/RuO2-TiO2、Ti/IrO2-Ta2O5相比,Ti/RuO2-IrO2-RhOx电子传输速率高,拥有较好的析氯效果与最强的氨氮氧化能力,适用于电解法处理氨氮废水,且Ti/RuO2-IrO2-RhOx在频繁倒极操作下电解高硬度氨氮废水时,可保持较高的氨氮去除效率,同时维持电压稳定,降低反应能耗,在处理高硬度氨氮废水领域具有广阔的应用前景。
4)初始氨氮浓度、电流密度及初始氯离子浓度均可影响Ti/RuO2-IrO2-RhOx电极电化学氧化氨氮的效率。当体系中不存在其他杂质离子时,初始氨氮浓度基本不影响氨氮氧化速率,大致体现为表观零级反应;在一定范围内,氨氮氧化速率与电流密度正相关;初始氯离子浓度直接影响到氨氮电化学氧化途径,当存在氯离子时,氨氮主要通过间接氧化去除,适量的氯离子有助于氨氮的电化学氧化。
-
图 1 Ti/RuO2-TiO2、Ti/IrO2-Ta2O5与Ti/RuO2-IrO2-RhOx电压变化
Figure 1. Voltage variation of Ti/RuO2-TiO2、Ti/IrO2-Ta2O5 and Ti/RuO2-IrO2-RhOx
图 2 Ti/RuO2-IrO2-RhOx的 FE-SEM图像
Figure 2. FE-SEM images of Ti/RuO2-IrO2-RhOx electrode
图 3 Ti/RuO2-IrO2-RhOx 的XRD图谱及主要氧化物标准衍射峰
Figure 3. XRD partterns of Ti/RuO2-IrO2-RhOx and the diffraction peaks of the standard cards of key oxides
图 7 不同电极对氨氮降解的影响
Figure 7. Effect of different electrodes on ammonia nitrogen degradation
图 8 不同初始氨氮浓度对氨氮降解的影响
Figure 8. Effects of different initial ammonia nitrogen concentrations on ammonia nitrogen degradation
图 9 不同电流密度对氨氮降解的影响
Figure 9. Effect of different current density on ammonia nitrogen degradation
图 10 不同氯离子浓度对氨氮降解的影响
Figure 10. Effect of different chloride ion concentration on ammonia nitrogen degradation
-
[1] 刘香丽. 氨氮污染对生活用水的危害与对策[J]. 农家参谋, 2017(21): 194. [2] 路庆鹏, 金雪霞. 养殖池塘水体中氨氮的来源、危害及控制[J]. 科学养鱼, 2019(6): 87. doi: 10.3969/j.issn.1004-843X.2019.06.050 [3] RENOU S, GIVAUDAN J G, POULAIN S, et al. Landfill leachate treatment: Review and opportunity[J]. Journal of Hazardous Materials, 2008, 150(3): 468-493. doi: 10.1016/j.jhazmat.2007.09.077 [4] 欧阳超, 尚晓, 王欣泽, 等. 电化学氧化法去除养猪废水中氨氮的研究[J]. 水处理技术, 2010, 36(6): 111-115. [5] 刘志明, 王磊刚, 程天行, 等. 电催化氧化去除制革废水生物处理出水中氨氮的研究[J]. 工业用水与废水, 2013, 44(5): 18-20. doi: 10.3969/j.issn.1009-2455.2013.05.005 [6] DING J, ZHAO Q, JIANG J, et al. Electrochemical disinfection and removal of ammonia nitrogen for the reclamation of wastewater treatment plant effluent[J]. Environmental Science and Pollution Research, 2017, 24(6): 5152-5158. doi: 10.1007/s11356-016-6618-0 [7] 袁芳, 范洪波, 代晋国. 电化学氧化法处理高浓度氨氮废水的实验研究[J]. 东莞理工学院学报, 2011, 18(5): 99-102. doi: 10.3969/j.issn.1009-0312.2011.05.021 [8] JIANG Z, YANG G, LIU M. Electrochemical oxidation treatment of ammonia nitrogen wastewater from Mn3O4 production process[J]. Industrial Water Treatment, 2020, 40(3): 23-26. [9] HANSSEN B L, SIRAJ S, WONG D. Recent strategies to minimise fouling in electrochemical detection systems[J]. Reviews in Analytical Chemistry, 2016, 35(1): 1-28. doi: 10.1515/revac-2015-0008 [10] YANG X, KIRSCH J, FERGUS J, et al. Modeling analysis of electrode fouling during electrolysis of phenolic compounds[J]. Electrochimica Acta, 2013, 94: 259-268. doi: 10.1016/j.electacta.2013.01.019 [11] MOSSAD M, ZOU L. Study of fouling and scaling in capacitive deionisation by using dissolved organic and inorganic salts[J]. Journal of Hazardous Materials, 2013, 244: 387-393. [12] GUO J, CUI P, GUAN S, et al. Study on scaling mechanism of piezoelectric balance electrode of HVDC converter valve[J]. Surface Technology, 2019, 48(10): 285-291. [13] WANG L, QIAN X, SUN W, et al. Research on high salt ammonia nitrogen wastewater treatment from coal-fired power plant by electrochemical oxidation[J]. Technology of Water Treatment, 2020, 46(12): 94-99. [14] 於洋. 用于水软化的高性能电沉积反应器研究[D]. 杭州: 浙江大学, 2020. [15] SANCHEZ C D, CHIH-PIN H, JING-HUA T, et al. The recovery of sulfuric acid from spent piranha solution over a dimensionally stable anode (DSA) Ti-RuO2 electrode[J]. Journal of Hazardous Materials, 2021: 406. [16] 高显泽. RuO2基阳极涂层纳米结构的构建及其电催化析氧性能研究[D]. 哈尔滨: 哈尔滨工程大学, 2020. [17] JIN H, ZHANG Y, ZHANG X, et al. High-performance Ti/IrO2-RhOx-Ta2O5 electrodes for polarity reversal applications[J]. Industrial & Engineering Chemistry Research, 2021, 60(11): 4310-4320. [18] WANG H, WANG J, BO G, et al. Degradation of pollutants in polluted river water using Ti/IrO2-Ta2O5 coating electrode and evaluation of electrode characteristics[J]. Journal of Cleaner Production, 2020: 273. [19] MYBURGH D P, AZIZ M, ROMAN F, et al. Removal of COD from industrial biodiesel wastewater using an integrated process: electrochemical-oxidation with IrO2-Ta2O5/Ti anodes and chitosan powder as an adsorbent[J]. Environmental Processes:An International Journal, 2019, 6(4): 819-840. doi: 10.1007/s40710-019-00401-x [20] BABAEI T, ZAREI M, HOSSEINI M G, et al. Electrochemical advanced oxidation process of phenazopyridine drug waste using different Ti-based IrO2-Ta2O5 anodes[J]. Journal of the Taiwan Institute of Chemical Engineers, 2020, 117: 103-111. doi: 10.1016/j.jtice.2020.12.004 [21] FURUSHO Y, AMANO F. Effect of adding polyethylene glycol to the precursor solution of amorphous IrO2-Ta2O5 electrocatalysts for oxygen evolution reaction[J]. Electrochemistry, 2021, 89(3): 234-238. doi: 10.5796/electrochemistry.21-00001 [22] ELNATHAN F. The effect of current reversal on coated titanium electrodes[J]. Dissertations & Theses - Gradworks, 2012. [23] 王均涛, 韩严, 许立坤, 等. Ru-Ir-Ti氧化物阳极正反电流电解失效机理研究[J]. 电化学, 2005(4): 407-411. doi: 10.3969/j.issn.1006-3471.2005.04.010 [24] YU Y, JIN H, LI Q, et al. Pseudocapacitive Ti/RuO2-IrO2-RhOx electrodes with high bipolar stability for phenol degradation[J]. Separation and Purification Technology, 2021, 263(118395). [25] CHEN X M, CHEN G H, YUE P L. Stable Ti/IrOx-Sb2O5-SnO2 anode for O2 evolution with low Ir content[J]. Journal of Physical Chemistry B, 2001, 105(20): 4623-4628. doi: 10.1021/jp010038d [26] 中华人民共和国国家质量监督检验检疫总局, 中国国家标准化管理委员会. 工业循环冷却水中余氯的测定: GB/T 14424-2008[S]. 天津: 天津化工研究设计院, 2008. [27] ROGINSKAYA Y E, MOROZOVA O V, KAPLAN G I, et al. Thermally prepared Ti/RhOx electrodes: I structural, electronic and surface properties[J]. Electrochimica Acta, 1993, 38(16): 2435-2441. doi: 10.1016/0013-4686(93)85113-D [28] HRUSSANOVA A, GUERRINI E, TRASATTI S. Thermally prepared Ti/RhOx electrodes IV: O2 evolution in acid solution[J]. Journal of Electroanalytical Chemistry, 2004, 564: 151-157. doi: 10.1016/j.jelechem.2003.10.032 [29] ALIBEK R, ATAPOUR M, AGHAJANI A, et al. Microstructure and electrochemical properties of IrO2+RhOx+ZrO2 coated titanium anodes[J]. Electrochimica Acta, 2020, 329: 135158. doi: 10.1016/j.electacta.2019.135158 [30] 金华长. 用于循环冷却水软化的离子膜电沉积技术研究[D]. 杭州: 浙江大学, 2021. [31] MIZUTANI U, SATO H, MASSALSKI T B. The original concepts of the Hume-Rothery rule extended to alloys and compounds whose bonding is metallic, ionic, or covalent, or a changing mixture of these[J]. Progress in Materials Science, 2021, 120: 100719. doi: 10.1016/j.pmatsci.2020.100719 [32] JIN H, YU Y, CHEN X. Membrane-based electrochemical precipitation for water softening[J]. Journal of Membrane Science, 2020, 597(117639). [33] 郭静如, 张雪娇, 廖帅, 等. Ti/RuO2-IrO2-SnO2-Sb2O5阳极在农村饮用水消毒中的应用[J]. 电化学, 2021, 27(5): 549-557. [34] 张胜健, 辛永磊, 许立坤, 等. NaCl浓度对金属氧化物阳极电化学性能与失效影响研究[J]. 热加工工艺, 2015, 44(6): 60-63. [35] 姜维, 王小佳, 贾仁勇, 等. 吹脱法预处理皮革废水的实验研究[J]. 环境工程学报, 2010, 4(4): 789-794. [36] 梁蓓. 煤制氮肥厂废水氨氮预处理及回收研究[D]. 西安: 西安建筑科技大学, 2015. [37] 刘占孟, 徐礼春, 崔立杰, 等. 老龄垃圾渗滤液处理技术研究进展[J]. 工业水处理, 2017, 37(6): 19-24. doi: 10.11894/1005-829x.2017.37(6).019 [38] 王家宏, 秦静静, 蒋伟群, 等. 电化学氧化法处理低浓度氨氮废水的研究[J]. 陕西科技大学学报(自然科学版), 2016, 34(2): 12-15. [39] 李璇. 循环电解槽电化学氧化法处理氨氮废水的实验研究[D]. 长沙: 湖南大学, 2013. [40] 林学聪. 氨氮在无氯离子体系中的电化学反应特性及机理研究[J]. 广州: 暨南大学, 2019. [41] MU T, GUO X, SUN C. Removal of ammonia nitrogen in wastewater by three dimensional electrode method[J]. Journal of Northwest A & F University. Natural Science Edition, 2016, 44(11): 104-110. [42] 陈金銮, 施汉昌, 徐丽丽. pH值对氨氮电化学氧化产物与氧化途径的影响[J]. 环境科学, 2008, 29(8): 2277-2281. doi: 10.3321/j.issn:0250-3301.2008.08.032 [43] KIM K, KIM Y, KIM I, et al. Electrochemical conversion characteristics of ammonia to nitrogen[J]. Water Research, 2006, 40(7): 1431-1441. doi: 10.1016/j.watres.2006.01.042 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4950
- HTML全文浏览数: 4950
- PDF下载数: 118
- 施引文献: 0
