














-
20世纪90年代中期,研究人员[1]在地球的多种基质中均检测到了人类或兽用抗生素的残留,这些残留的抗生素会给海洋和陆地上的各种生物带来潜在的危害。因此,研究抗生素废水的处理技术对于控制环境中抗生素的来源具有重要意义。各种抗生素废水的处理技术已见报道,如臭氧净化技术[2]、Fenton氧化-活性炭吸附协同处理等高级氧化技术[3]、微藻生物处理技术等[4]。其中,高级氧化技术已在抗生素废水处理领域中显现出巨大的发展潜力[5],但是,高级氧化技术处理抗生素废水成本高,且在去除机理、动力学等方面的研究尚未成熟,以致不能实际应用于废水处理工程[6],因此,须亟待开发新的抗生素废水处理技术。
空化效应是在液体中产生的一种物理现象,是由于液体中的压力低于液体的饱和蒸汽压而导致的气泡的形成、发展和溃灭的过程[7-8]。空泡在溃灭的过程中会使局部压力和局部温度瞬时急剧上升,产生强烈的冲击波和高微射流[9-10],出现的这种高温、高压、高微射流等极端条件能在局部产生足以打断有机物分子链的能量,从而达到降解污水中有机物的效果。利用空化效应降解废(污)水中的有机物是近年来研究的热点[11-13]。水力空化技术主要包括涡流空化技术和射流空化技术。射流空化技术与涡流空化技术相比,由于流体在进入射流装置前必须获得很高的初速度,射流空化要求设备更为复杂,耗能也更多,而涡流能够在较低的进口压力的情况下形成空化,因此,涡流空化效应在有毒、难降解有机废水的处理方面显示出巨大的应用潜力。
国内外学者设计了不同类型的涡流空化器[14-16],但将其用于降解染料等有机物的效果不大理想[10, 17]。本课题组前期设计了一种由涡流腔与螺旋线流道相组合的新型旋流式涡流空化器[18],本研究在此基础上进行流场仿真分析,论证了涡流空化效应的产生过程,并探讨了不同条件下降解抗生素的性能,以期为抗生素废水的处理提供新的途径。
-
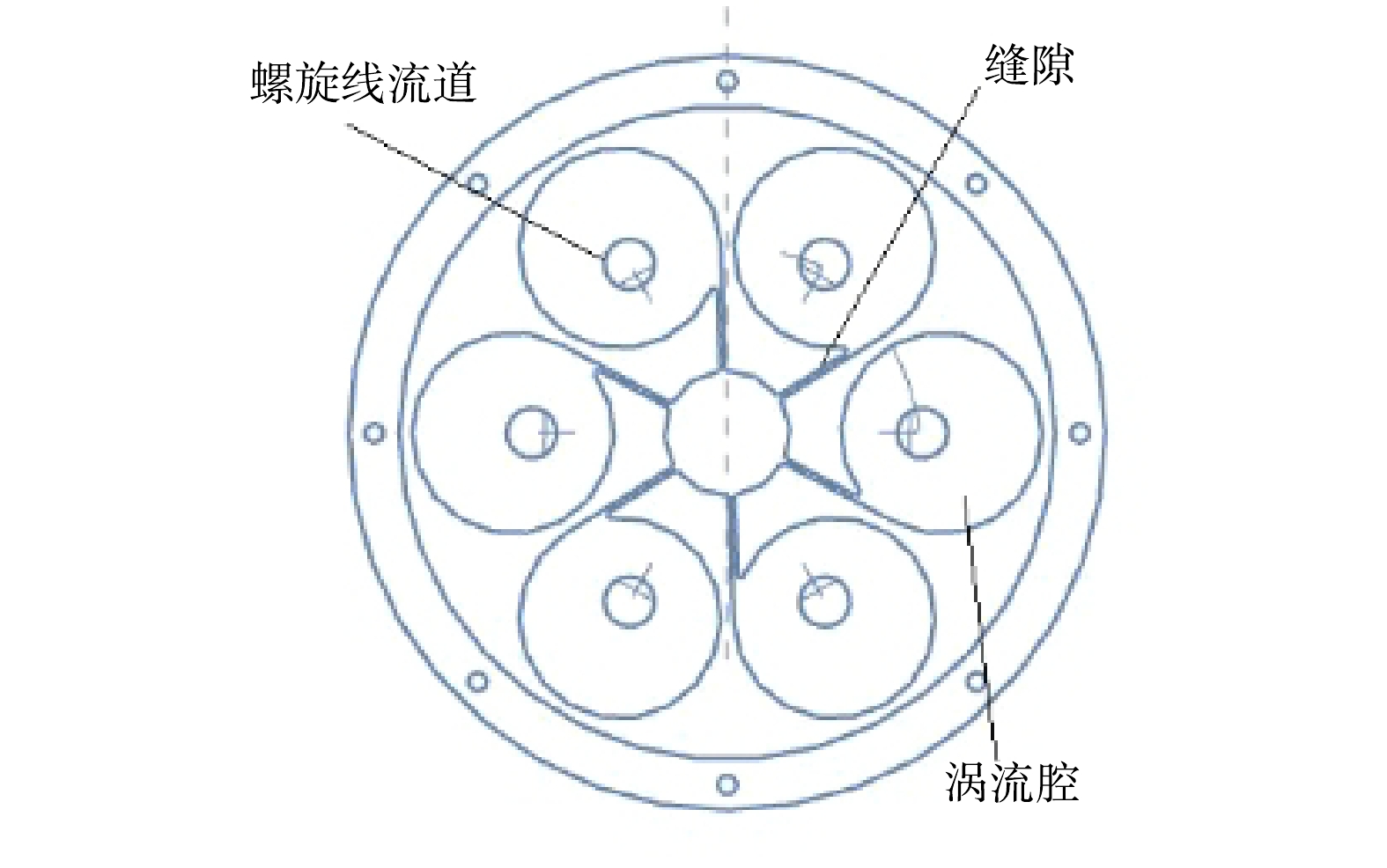
旋流式涡流空化器核心部件的立体剖视图和涡流空化部件的俯视图如图1和图2所示。该旋流式涡流空化器主要由接头、上端盖、涡流空化部件、射流腔,下挡板组成。该涡流空化部件的创新性结构为涡流腔与螺旋线流道的组合,设计有6个涡流腔,每个涡流腔连接1条带孔道的螺旋线流道,流道直径逐渐降低。该旋流式涡流空化器的基本结构尺寸为:涡流腔的直径为65 mm,涡流腔缝隙宽度为5 mm,流道入口直径为9 mm,流道出口直径为3 mm。每条螺旋线流道上孔道数为4,孔直径为3 mm。
-
1)软件:运用ANSYS仿真软件对涡流空化器进行仿真分析,运用Creo三维设计软件创建零件模型。
2)解决方案:求解时应用了多相流模型中的mixture模型,相应的控制方程为多相流形式下的NS守恒方程;湍流模型采用了realizeble k-ε双方程湍流模型。
3)边界条件:入口采用压力入口边界条件,给定入口总压为0.3 MPa,出口设置为压力出口边界条件,给定出口背压为0 Pa,操作压力设置为101 325 Pa。
-
1)土霉素溶液的制备。准确称取土霉素标准样品(C22H24N2O9,USP级,上海源叶生物科技有限公司)10 mg置于100 mL棕色容量瓶中,用0.1 mol·L−1的盐酸溶液定容、摇匀,作为标准贮备液。样品测定前,取2 mL的土霉素溶液,0.22 μm滤膜过滤到色谱瓶中,备用。
2)土霉素降解实验。配置pH 7.0、初始浓度为2 mg·L−1的土霉素溶液10 L于储水箱中,放置8个冰袋以防止运行时间过长导致溶液温度过高。启动水泵(设置水泵的入口压力值0.15 MPa),降解一定时间后取样,到达设定更换冰袋的时间,暂停水泵,更换冰袋,暂停10 min;继续降解一定时间,取样,测定降解后的土霉素浓度。倒出水箱中和水泵中的土霉素溶液,导入去离子水清洗水泵和水箱3次。所有实验均重复3次。
3)土霉素浓度的测定。土霉素浓度采用高效液相色谱法测定。色谱条件为:色谱柱为Extend-C18(150 mm×4.6 mm,3.5 μm),流动相为乙腈-水(体积比为80∶20),流速为1.0 mL·min−1,检测波长为353 nm,进样量为20 μL,柱温为室温。
4)羟自由基浓度的测定。羟基自由基浓度的测定采用亚甲基蓝法[19],计算方法见式(1)。
式中:C·OH为羟自由基摩尔浓度,μmol·L−1;ΔA为降解前后溶液中亚甲基蓝吸光度差值(测定波长为664 nm)。
5)降解率的计算。降解率的计算方法见式(2)。
式中:X为降解率;C前为降解前土霉素质量浓度, mg·L−1;C后为降解后土霉素质量浓度, mg·L−1。
-
1)旋流式涡流空化器空化效应的形成。由旋流式涡流空化器的结构图(图1和图2)可以看出,涡流空化器部件中设计了6个涡流发生的涡流腔,流体通过上端盖上方入口后,进入该涡流空化部件中心,随后进行发散运动。流体在涡流空化部件中心边缘缝隙的作用下,会产生较大的入口速度并进入各自的涡流腔。流体将会绕涡流腔体进行涡流运动,螺旋线流道的横截面大小随流体去向方向逐渐缩小。根据流速与通量面积的关系,流体在流道出口处会产生极大的流速,结合伯努利方程可知,流场在此处会形成局部低压,空泡随之形成。流体冲出流道后截面积骤升,压力恢复,空泡内外压力无法维持其形态而导致溃灭,空化效应发生。每个涡流腔都设计1个通向射流腔的螺旋线流道,并驱使多个流道里流体与位于涡流部件下方的挡板发生激烈冲撞,使小气泡溃灭更加彻底,从而提高空化效应。为了产生更多的空化次数,在每条螺旋线流道上均布有4个通向射流腔的孔道,蓄备更多可发生空化效应的条件。
2)旋流式涡流空化器的流场仿真。利用Creo三维设计软件创建旋流式涡流空化组件的三维模型(图3),并利用Gambit软件对涡流空化部件进行网格划分,划分好的流体区域的网格模型如图4所示。因仿真的流体区域结构比较复杂,拐角的位置较多,故采用四边形的网格划分策略。
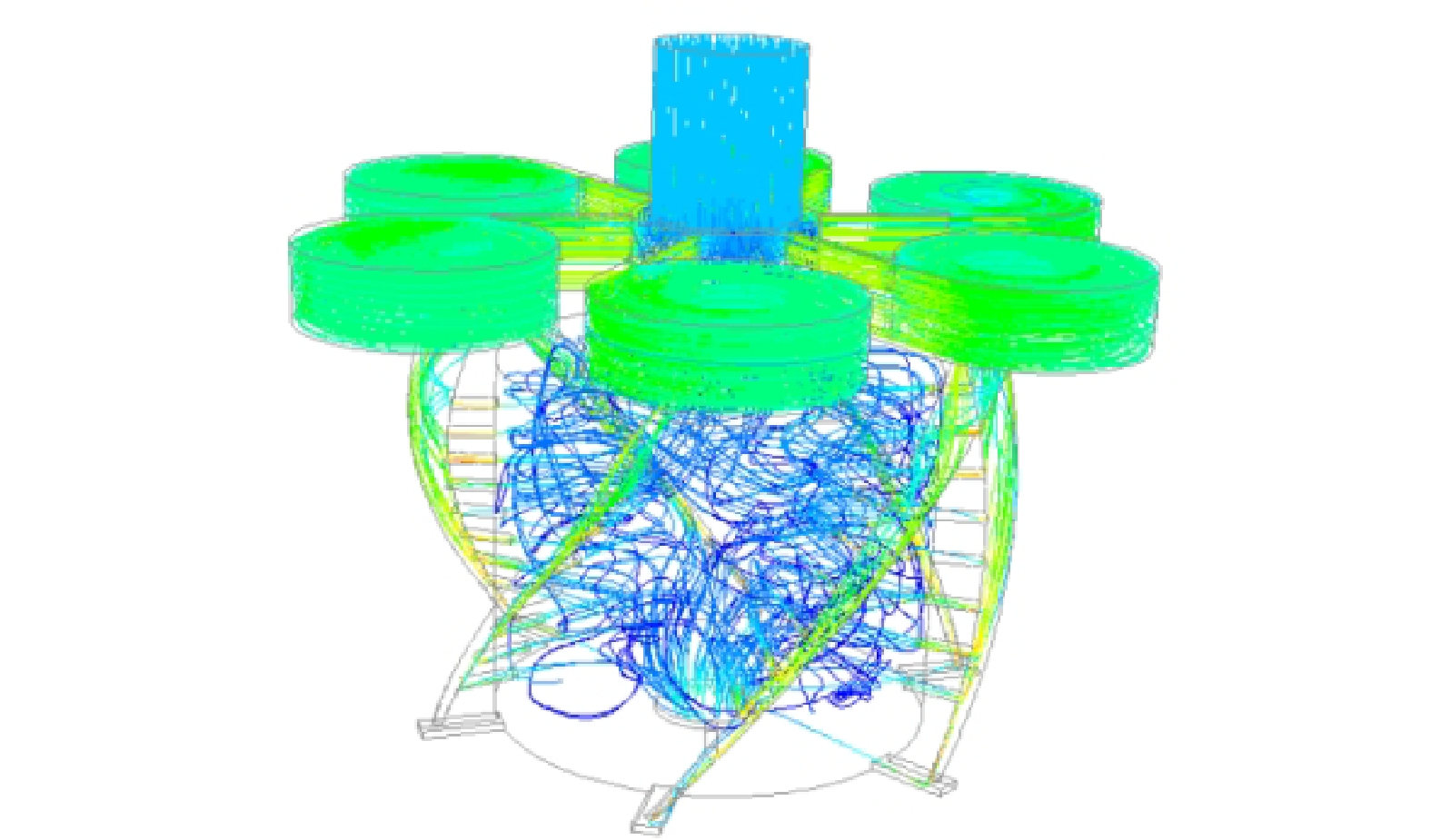
由图5可看出,流体在装置内的运动情况,流体从涡流空化部件上方的缝隙进入涡流腔中,在涡流腔内进行旋转运动,随后进入螺旋线流道。
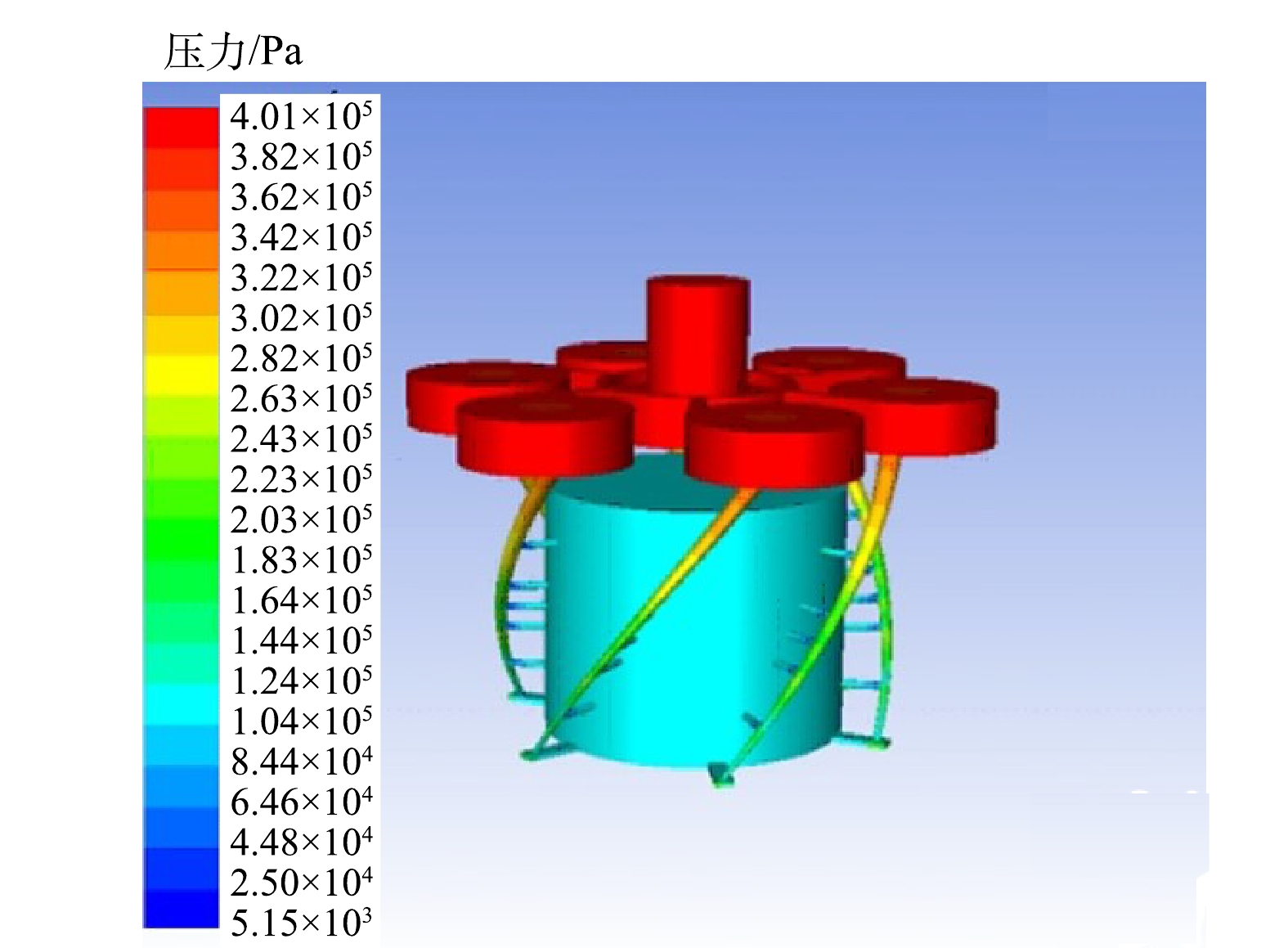
图6是仿真后的涡流空化器的流体压力分布图。可以看出,低压区主要分布在射流腔中,在低压区域会产生一定数量的空泡,根据压力云图数据得到该装置流体的最低压力约为5 465.737 Pa。实验温度控制在55 ℃以下,纯水在55 ℃时的饱和蒸汽压为15 737 Pa,该涡流空化器产生的最低压力值远低于该温度下的饱和蒸汽压,因此,实验条件下能产生明显涡流空化效应。
对比图6压力云图和图5的流体流线图可以发现,流体从流道出口冲出前会形成一个局部低压的区域,当流体经出口流出后截面积骤升,此时由于压力恢复,致空泡内外压力差增大,空泡无法维持其形态而导致溃灭,空化效应发生。
-
在土霉素溶液初始pH值为5~9、降解30 min时,土霉素的降解率如图7所示。由图7可知,随着反应初始pH的升高,土霉素的降解率呈下降趋势。当pH在5~7时,土霉素的降解率变化不大,降解率分别为70.47%、71.76%、68.84%;当pH在7~9时,土霉素的降解率下降较明显;pH为8和9时,土霉素的降解率为57.77%和57.96%。这与钱盛[20]利用钛基PbO2电催化降解土霉素废水得出的研究结果类似:在pH为4~6时,土霉素降解率未发生明显变化;在pH为6~10时,土霉素的降解率逐渐下降。据报道[21],酸性条件有利于促进涡流空化效应,产生强氧化性的羟基自由基,有利于提高涡流空化器中抗生素的降解效果;而碱性条件会抑制自由基的产生。因此,在酸性和中性条件下更有利于涡流空化器中土霉素的降解。本涡流空化器在碱性条件下对土霉素的降解率仍在55%以上,说明本涡流空化器在较宽的pH范围内降解效果均较佳。后续实验选择土霉素溶液初始pH值为7。
-
图8为土霉素初始质量浓度为2 ~10 mg·L−1条件下,涡流空化器对土霉素的降解率。由图8可以看出,随着土霉素初始质量浓度的增大,土霉素的降解率逐渐下降,当土霉素质量浓度为10 mg·L−1时,降解率为30.08%。因为在较低的初始质量浓度下,涡流空化产生的羟自由基与土霉素溶液充分反应,故降解率较大,但涡流空化产生的羟自由基量是恒定的,当土霉素的初始质量浓度继续增大时,则降解土霉素的量有限,土霉素的降解率随之降低,尽管如此,土霉素的绝对降解量因高浓度条件下自由基利用充分而比低浓度时要大。
-
由于本实验装置没有恒温装置,因此,要利用更换冰袋的间隔时间控制体系的温度范围。每10 min和30 min换1次冰袋,控制温度分别为25~50 ℃和25~60 ℃。在一定温度范围内,不同降解时间下涡流空化器中土霉素的降解率如图9所示。
由图9可以看出,25~50 ℃下的降解率比25~60 ℃下的降解率稍大。反应50 min后,土霉素的降解率分别为83.12%和76.45%,增加了6.67%。这说明温度升高对土霉素溶解的降解的影响具有双重的作用。一方面,随着温度的升高,液体的饱和蒸气压升高,流体的压力与饱和蒸气压的差距变大,更容易形成涡流空化发生所需的压力降;同时,饱和蒸汽压的提高还有利于气核的形成和长大,从而可提高涡流空化效应的强度,温度的升高还有利于促进土霉素自身的水解。另一方面,在自然状态下,流体中溶有一定数量的气核,在一定条件下,气核数与涡流空化强度成正比。随着温度的提高,气核数量相应减少,从而降低了涡流空化的强度[22]。因此,涡流空化效应的强度存在最佳温度条件。
此外,由图9可以看出,不同时间下土霉素降解规律为:随着反应时间的进行,土霉素溶液的降解率逐渐增大,在反应50 min后,土霉素溶液的降解率达到了最高的76.45%;在反应10 min内,土霉素的降解率达到了53.06%,降解速度较快;在反应10 ~50 min内,降解速度变慢。与反应10 min相比,50 min时的土霉素降解率仅增加了23.39%。产生这种现象的原因与体系温度和降解中间产物2大因素有关:第一,在反应初期,涡流空化产生大量的羟自由基与土霉素快速发生反应,但随着反应时间的增加,降解土霉素时会逐渐积累中间产物,中间产物会与土霉素母体竞争结合羟自由基,导致土霉素的降解速率变慢;第二,降解体系温度的增高和饱和溶解氧的增大更易产生涡流空化效应,但气核含量的降低反过来会削弱涡流空化效应,因而导致降解速率不会线性增长。
-
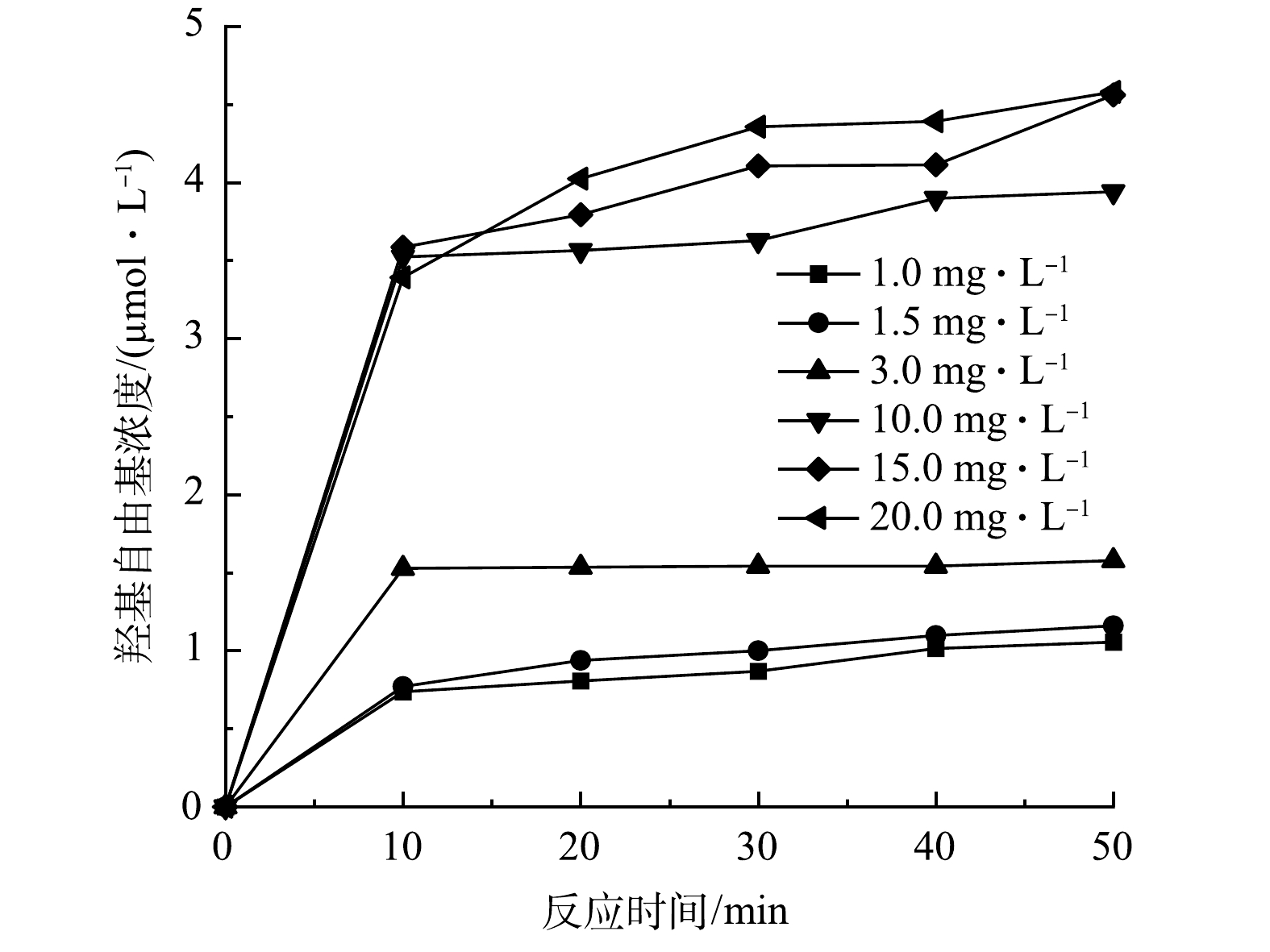
一般而言,涡流空化器中土霉素降解的机理是由于涡流空化效应产生的羟基自由基的强氧化作用。利用亚甲基蓝法间接测定了不同时间下的羟基自由基浓度,结果如图10所示。由图10可以看出,本研究设计的旋流式涡流器能够产生羟基自由基。当亚甲基蓝浓度较低时,对羟基自由基的捕捉率较低,随着亚甲基蓝溶液质量浓度的增加,对羟基自由基的捕捉率也随之增加;当亚甲基蓝溶液质量浓度达到20 mg·L−1时,则羟基自由基的捕捉率基本不变,说明此时涡流空化产生的羟基自由基已充分被亚甲基蓝捕捉,质量浓度为4.58 μmol·L−1。张晓冬等[19]利用亚甲基蓝测定水力空化产生的羟基自由基,反应70 min后,在最佳条件下产生的羟基自由基浓度不到0.3 μmol·L−1,远远小于本涡流空化装置产生的羟基自由基浓度。可见,本旋流式涡流空化器能够产生较高浓度的羟基自由基,而土霉素的降解可能主要在羟基自由基的氧化作用下完成。
-
1)本研究中的新型旋流式涡流空化器,可在流场局部区域形成远低于饱和蒸汽压的低压区域,具有较明显的涡流空化效应。
2)将溶液温度控制在25~60 ℃时,酸性和中性条件有利于涡流空化器中土霉素的降解,碱性条件下土霉素的降解率仍在55%以上;随土霉素质量浓度的增大,土霉素的降解率相应下降,在前10 min内降解速率较大,前50 min内对2 mg·L−1的土霉素溶液的降解率为76.45%。控制溶液温度在25~50 ℃时,对降解土霉素有利。
3)旋流式涡流空化器产生的羟基自由基浓度为4.58 μmol·L−1。
新型旋流式涡流空化器的流场仿真及该空化器对废水中土霉素的去除效果
Flow field simulation in a novel swirling vortex cavitator and its performance on oxytetracycline removal from wastewater
-
摘要: 为有效去除废水中的残留抗生素,设计了一种由涡流腔与螺旋线流道相组合的新型旋流式涡流空化器。运用ANSYS流场计算软件仿真该涡流空化器核心部件流场的绝对压力;研究了不同溶液初始pH值、土霉素初始质量浓度、降解时间及温度控制范围等条件下该旋流式涡流空化器对水中土霉素的去除效果;用亚甲基蓝法检测该旋流式涡流空化器产生的羟基自由基质量浓度,初步推断其降解土霉素的机理。结果表明:本研究中设计的新型旋流式涡流空化器在局部区域可形成远低于饱和蒸汽压的低压区域,具有较明显的涡流空化效应;酸性和中性条件有利于涡流空化器中土霉素的降解,但总体上,溶液初始pH值对降解率影响不大;随土霉素初始浓度的增大,土霉素的降解率相应下降;在最初10 min内土霉素的降解速率较快,随后变慢,至50 min时初始质量浓度2 mg·L−1的土霉素溶液的降解率为76.45%;控制溶液温度在25~50 ℃时,对土霉素的降解更有利;该旋流式涡流空化器产生的羟基自由基质量浓度为4.58 μmol·L−1,由此可推断,土霉素的降解可能主要在羟基自由基的强氧化作用下完成。涡流空化法降解抗生素技术可为废水中抗生素的去除提供一种新的途径。Abstract: To effectively remove antibiotics residues in wastewater, a novel swirling vortex cavitator composed of vortex cavity and spiral channel was designed. ANSYS software was used to simulate the absolute pressures of flow fields in the vortex cavitator core components. Under the different conditions including initial solution pH values, initial concentrations of oxytetracycline, degradation times and temperature control ranges, the removal efficiencies of oxytetracycline in wastewater were examined. The concentrations of hydroxyl radical were determined by methylene blue method, and the mechanism of oxytetracycline degradation was deduced. The results showed that the local area in the vortex cavitator can form a low-pressure area with far lower pressure than the saturated vapor pressure, and has an obvious vortex cavitation effect. The acidic and neutral pHs were conducive to oxytetracycline degradation in the vortex cavitator. However, the initial pH value of the solution had slight effect on the degradation efficiency. The degradation efficiencies decreased with the increase of oxytetracycline concentration. The degradation rate was fast within 10 minutes, and then slowed down. The degradation efficiency of 2 mg·L−1 oxytetracycline solution reached 76.45% at 50 minutes. 25~50 ℃ for the solution temperature was conducive to oxytetracycline degradation. The concentration of hydroxyl radical produced by the swirling vortex cavitation device was 4.58 μmol·L−1. It was concluded that the possible oxytetracycline degradation pathway was mainly the oxidation of hydroxyl radicals. This provides a new way for antibiotics treatment by the swirling vortex cavitation degradation technology.
-
Key words:
- swirling vortex cavitator /
- flow field simulation /
- oxytetracycline /
- degradation
-
氰化提金所带来的环境污染隐患是黄金冶炼行业面临的共性关键难题。据统计,我国每年产出的氰化尾渣数量约为2.45×107 t[1]。氰化尾渣中含有大量的氰化物和有价金属,如果仅仅进行堆存或填埋处理,不仅会污染环境,而且也浪费了资源。因此,氰化尾渣的无害化处理是黄金冶炼行业节能减排及可持续发展面临的关键问题。
氰化尾渣的综合利用主要包括预处理-二次提取金银,综合回收铜、铁、锌,以及无害化处理3大类。前2类侧重于有价资源的综合回收,无害化处理则侧重于氰化物的破坏及重金属离子的去除。综合回收一般流程长、工艺较为复杂,以浮选法为主;无害化处理工艺则比较简单,是解决氰化提金高污染问题最直接最有效的方法之一。目前,氰化尾渣的无害化处理主要包括化学氧化法、电解氧化法、微生物分解法及自然净化法。采用SO2-空气氧化法处理氰化尾渣,以铜为催化剂,SO2和空气的混合物在碱性条件下能将复合氰化物(CNWAD)氧化为氰酸盐(CNO−),同时沉淀除去金属和氰化铁[2-3]。MANORANJAN等[4]的研究结果表明,硫代硫酸盐和活性炭的混合物能够去除废水中的总氰化物和CNWAD。ADJEI等[5]发现,假单胞菌属、芽孢杆菌属等菌种都对氰化物有一定的降解能力。SAARELA等[2]发现,在电化学氧化过程中,氰化物和金属氰络合物首先在阳极被氧化成氰酸根离子,然后进一步分解成无毒的CO2和N2,释放出的金属阳离子在阴极处被还原成金属单质析出。一般情况下,氰化尾渣的无害化处理主要可分为矿浆直接氧化和矿渣洗涤后含氰废水氧化2种。前者氧化剂消耗量大,金属离子以沉淀形式进入渣中,氰化尾渣中的有价金属并没有得到有效的去除;而后者增加了洗涤、液固分离程序,处理过程比较繁杂,洗水量大,处理难度相对增加。因此,研究开发一种工艺简单、成本低、效果好的处理方法是黄金行业创新发展的迫切需要。
本研究中采用矿浆电解技术无害化处理氰化尾渣,将氰化尾渣的洗涤、氰化物的电解氧化、金属离子的电解沉积和矿物的氧化分解集成在同一反应器中进行,利用阳极反应生成的氯气/次氯酸根的强氧化性,破坏含氰离子,氧化包裹矿物,提高氰化尾渣中矿物的单体解离度,以期为后续有价金属的综合利用创造有利条件。
1. 材料与方法
1.1 实验原料
本研究中所用原料为陕西省太白金矿浮选-金精矿炭浆提金后的氰化尾渣,其主要组成为CNT 1 748.00 g·t−1、CN− 327.40 g·t−1、Cu 184.80 g·t−1、Fe 3 632.04 g·t−1、Zn 15.84 g·t−1,即该氰化尾渣中总氰与铁氰络合离子含量较高。
1.2 实验装置
矿浆电解无害化处理氰化尾渣实验装置如图1所示。阴、阳极板均为TC4钛合金板,规格为20 mm×15 mm×2 mm,采用一阴两阳三极板体系。电压由直流稳压电源控制,型号为LP2002D型稳压电源,搅拌装置采用78-1型磁力搅拌器。
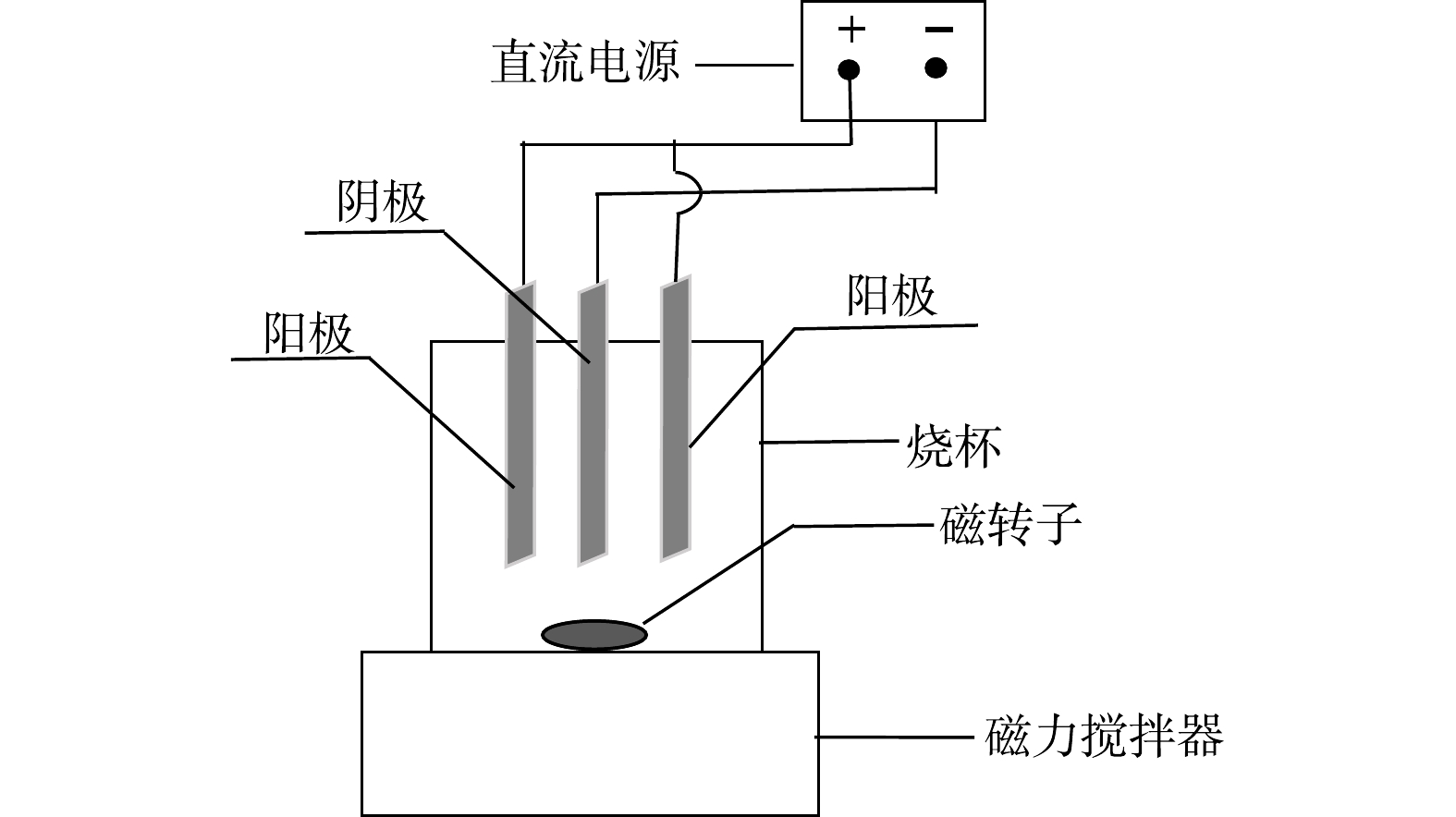 图 1 矿浆电解氰化尾渣实验装置Figure 1. Slurry electrolysis cyanide tailings experimental arrangement diagram
图 1 矿浆电解氰化尾渣实验装置Figure 1. Slurry electrolysis cyanide tailings experimental arrangement diagram1.3 实验方法
室温条件下(约25 ℃),取50 g氰化尾渣、一定量的NaCl以及200 mL的去离子水加入到500 mL烧杯中,用磁力搅拌器搅拌,同时控制极板间距。电解一定时间后固液分离,取滤液测定总氰、游离氰及各金属离子含量,并计算各离子去除率,尾渣用去离子水反复冲洗使pH稳定在7左右。
1.4 分析方法
氰化尾渣中CNT、CN−的含量按照GB 5085.3-2007[6]所示硝酸银滴定法进行测定,金属离子采用化学滴定法测定,离子去除率按式(1)计算。
φ=Ca−CbCa⋅0.5×10−350×10−6×100% (1) 式中:Ca为氰化尾渣中各离子的浓度,mg∙L−1;Cb为处理后渣洗液中各离子的浓度,mg∙L−1。
实验中采用FEI MLA 250型高精度工艺矿物学参数自动测试系统(MLA)测定金属矿石的解离程度和连生关系。
2. 结果与讨论
2.1 NaCl用量对各离子去除效果的影响
矿浆电解中,Cl−在阳极氧化生成的Cl2/ClO−是主要的氧化物,NaCl的添加量是影响氰化尾渣氧化效果的重要因素之一。在极板间距10 mm、电压8 V、电解时间4 h的条件下进行矿浆电解实验,结果如图2所示。随着NaCl添加量的增加,CNT、CN−及Cu、Zn离子的去除率逐渐增大,至NaCl添加量大于0.5 g后上述离子的去除率几乎不再增加,最大去除率分别为94.05%、98.25%、85.61%、73.92%。电解过程中,Cl−定向迁移至阳极表面,发生阳极氧化反应产生有效氯(ClO−和Cl2)(式(2)~式(4))。其中,有效氯将游离氰化物、铜氰及锌氰络合离子氧化为氰酸盐及金属离子(式5~式10),氰酸盐进一步被氧化为无毒的N2和碳酸盐。同时,金属离子定向迁移至阴极附近,在阴极板上发生金属离子的沉积反应(式(11)~式(14))。随着NaCl用量的增大,各离子的氧化去除效果不断增强的原因是矿浆中的Cl−含量增加,使得电极的电流效率和有效氯浓度增大[7],能够不断氧化去除氰离子和金属氰络离子。NaCl添加量大于0.5 g后,离子去除率几乎不再变化的原因是,NaCl添加量的增大造成溶液中离子浓度增加,离子间距减小,传质作用降低[8],导致有效氯的生成量不会有大幅度提高,离子去除率便不再增加。
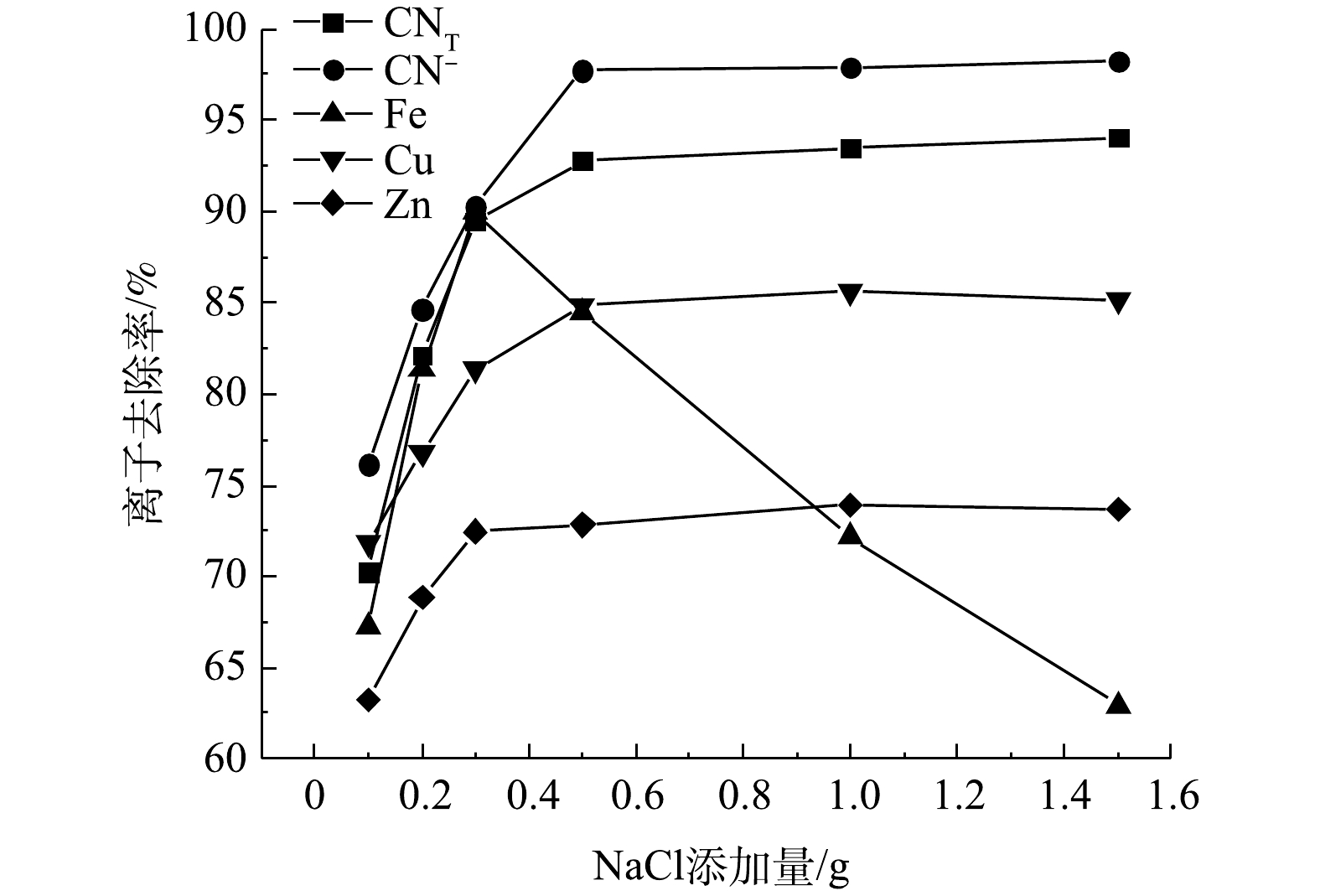 图 2 不同NaCl添加量下氰化尾渣中的氰化物及部分金属离子的去除率Figure 2. Removal rates of cyanide and some metal ions in cyanide tailings treated under different dosages of NaCl
图 2 不同NaCl添加量下氰化尾渣中的氰化物及部分金属离子的去除率Figure 2. Removal rates of cyanide and some metal ions in cyanide tailings treated under different dosages of NaClCl−−2e−=Cl2 (2) Cl−+2OH−−2e−=ClO−+H2O (3) Cl2+H2O=ClO−+Cl−+2H+ (4) CN−+ClO−=CNO−+Cl− (5) CN−+Cl2+2OH−=CNO−+2Cl−+H2O (6) 2CNO−+3ClO−+H2O=2HCO−3+N2+3Cl− (7) Zn(CN)2−4+4ClO−=Zn2++4CNO−+4Cl− (8) Cu(CN)−2+2ClO−=Cu2++2CNO−+2Cl− (9) Cu(CN)2−3+3ClO−=Cu2++3CNO−+3Cl− (10) Cu2++2e=Cu (11) Zn2++2e=Zn (12) Fe3++e=Fe2+ (13) Fe2++2e=Fe (14) Fe1−xS+(2−x/2)ClO−+2xH2O=(1−x)Fe2++SO2−4+4xH++(2−x/2)Cl− (15) Fe2+−e=Fe3+ (16) 值得注意的是,Fe的去除率在NaCl添加量大于0.3 g时,随着NaCl添加量的增大而大幅度降低,最大仅为84.46%。这是因为有效氯的含量随着Cl−的增加而增加,ClO−会氧化部分黄铁矿和磁黄铁矿(式(15)~式(16)),使得矿石中的铁以离子形态再次回到矿浆液中,导致Fe3+去除率降低。
2.2 外加电压对各离子去除效果的影响
电压的大小直接影响NaCl的电解反应速率以及矿浆中CN−的迁移过程,进而影响氰化物的去除效果。在极板间距10 mm、NaCl添加量0.5 g、电解时间4 h的条件下进行矿浆电解实验,结果如图3所示。随着电压的升高,CNT、CN−的去除率先增大,在8 V之后趋于平缓,去除率最大为95.25%、98.97%;Cu、Fe、Zn离子的去除率随电压增大而增大,在10 V时达到最大值85.86%、84.76%和73.99%,随后有所下降。根据Faraday第一定律,当电流通过电解质溶液时,在电极界面上发生化学反应的物质量与通过电极的电量呈正比,这说明电压是电化学反应过程的一种驱动力[9],随着电压的增大,电流密度逐渐增大,阳极析氯量增加,氧化反应加剧,各离子的去除率均不断上升。电压大于8 V以后,随着电压的增大,各离子去除几乎不再增加的原因:一方面,是由于电压过大会导致电极表面极化现象过大[10-11],造成电流密度减小,不利于氧化反应的发生;另一方面,是难降解的铁氰络离子会与其他金属离子结合转化成更加稳定的金属铁氰络合物沉淀(式(17)~式(18)),不易被氧化去除。此外,金属离子的去除率在大于10 V时呈现降低趋势,其原因可能与尾渣中矿物的氧化溶解(式(15)、式(19)、式(20))有关。
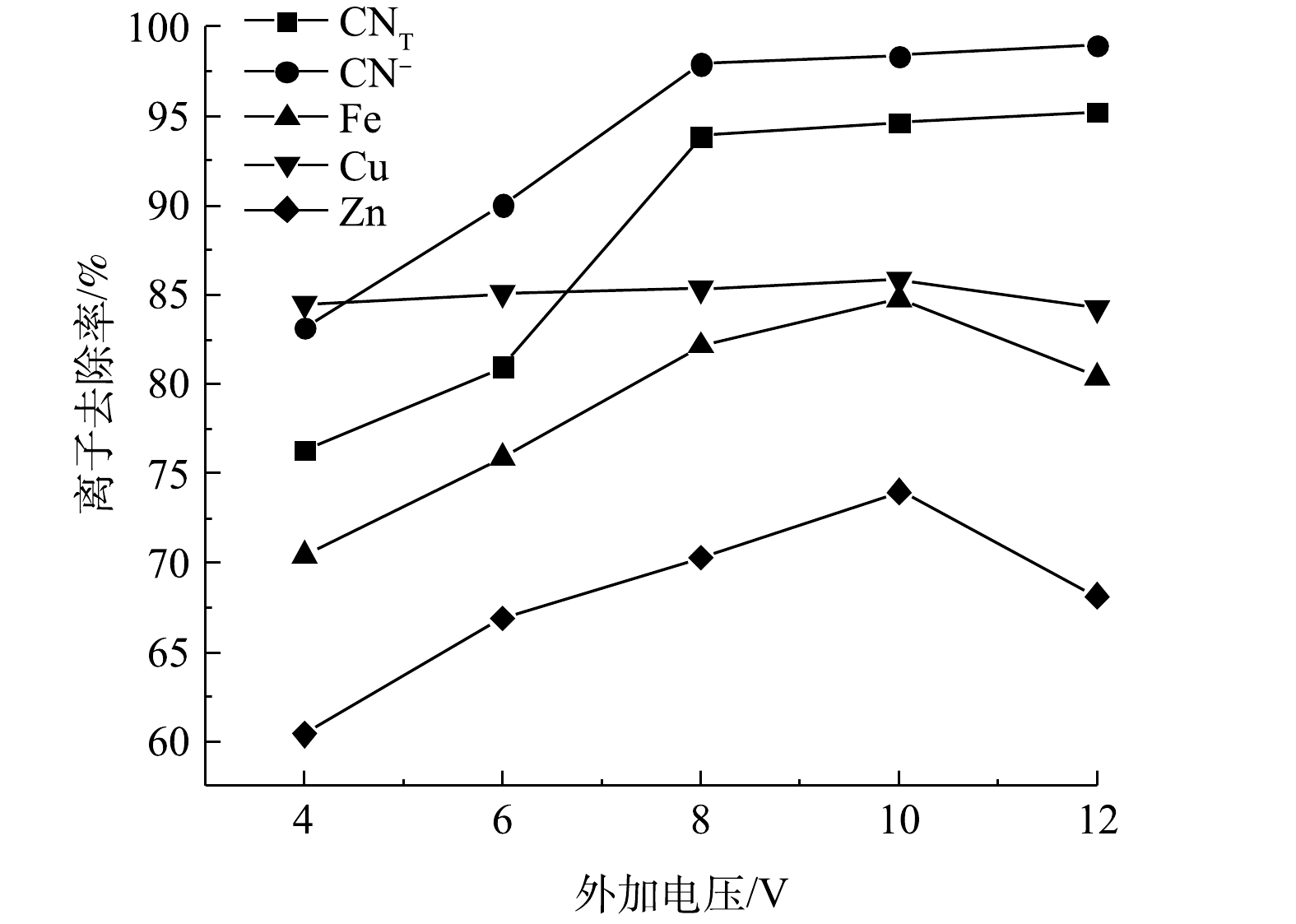 图 3 不同电压条件下氰化尾渣中的氰化物及部分金属离子的去除率Figure 3. Removal rates of cyanide and some metal ions in cyanide tailings treated under different tension
图 3 不同电压条件下氰化尾渣中的氰化物及部分金属离子的去除率Figure 3. Removal rates of cyanide and some metal ions in cyanide tailings treated under different tension2Zn2++Fe(CN)4−6=Zn2Fe(CN)6↓ (17) 3Cu2++2Fe(CN)3−6=Cu3[Fe(CN)6]2↓ (18) 2CuFeS2+17ClO−+2H+=2Cu2++2Fe3++4SO2−4+H2O+17Cl− (19) ZnS+4ClO−=Zn2++SO2−4+4Cl− (20) 2.3 电解时间对各离子去除效果的影响
电解时间是影响氰化尾渣中离子去除效果的一个重要因素。在极板间距10 mm、NaCl添加量0.5 g、电压8 V的条件下,改变电解时间进行矿浆电解实验,结果如图4所示。随着电解时间的增长,CNT、CN−及金属离子的去除率不断增大,CNT与CN−的去除率在4 h之后趋于平缓。随着电解时间的增长,溶液中不断生成有效氯用于氧化氰化物和金属氰化物;电解4 h时,CNT与CN−去除率已经达到93.48%、97.84%,而Cu、Zn、Fe离子去除率分别为83.74%、77.52%和90.21%;随后金属离子去除率的增幅开始变小。考虑到能耗的影响,选择4 h为最佳的电解时间。
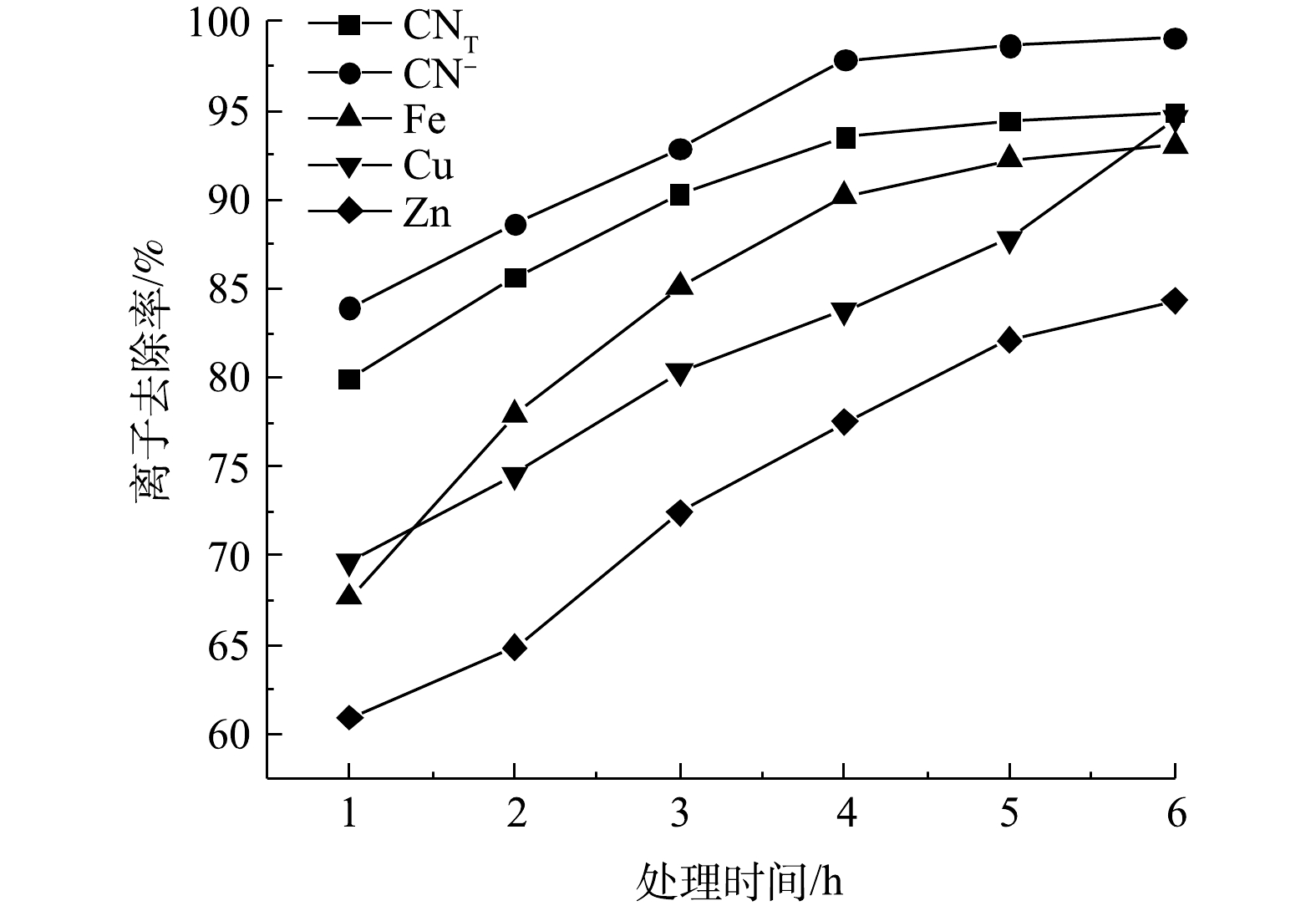 图 4 不同电解时间条件下的氰化尾渣中的氰化物及部分金属离子的去除率Figure 4. Removal rate of cyanide and some metal ions in cyanide tailings treated under different electrolysis time
图 4 不同电解时间条件下的氰化尾渣中的氰化物及部分金属离子的去除率Figure 4. Removal rate of cyanide and some metal ions in cyanide tailings treated under different electrolysis time2.4 极板间距对各离子去除效果的影响
极板间距会影响电路电流大小、电解反应速率和传质效率。在NaCl添加量0.5 g、电压8 V、电解时间4 h的条件下,改变极板间距进行矿浆电解实验,结果如图5所示。随着极板间距增大,CNT与CN−的去除率先增大后减小,在极板间距为10 mm时达到最大,分别为95.25%和98.95%;Cu、Zn、Fe的离子去除率随极板间距增大而减小。极板间距过小时,极板间的电压差增大,易形成瞬时强大的电流,从而造成短路现象。随着极板间距的逐渐增大,极板上的析气反应逐渐正常,电解液流速提升,传质作用不断增强,氧化效果和离子去除率随之增大。但有研究者报道,当极板间距过大时,会导致槽电压和电能消耗不断增加[12],电解反应速率降低;随着反应的进行,离子浓度下降,导致浓差极化作用增强,亦造成金属单质在阴极表面沉积厚度增加,使得阴极电位降低[13],H+在金属单质表面的析出电位增高,最终氧化还原效果降低,从而导致氰化物及金属离子的去除率不断降低。
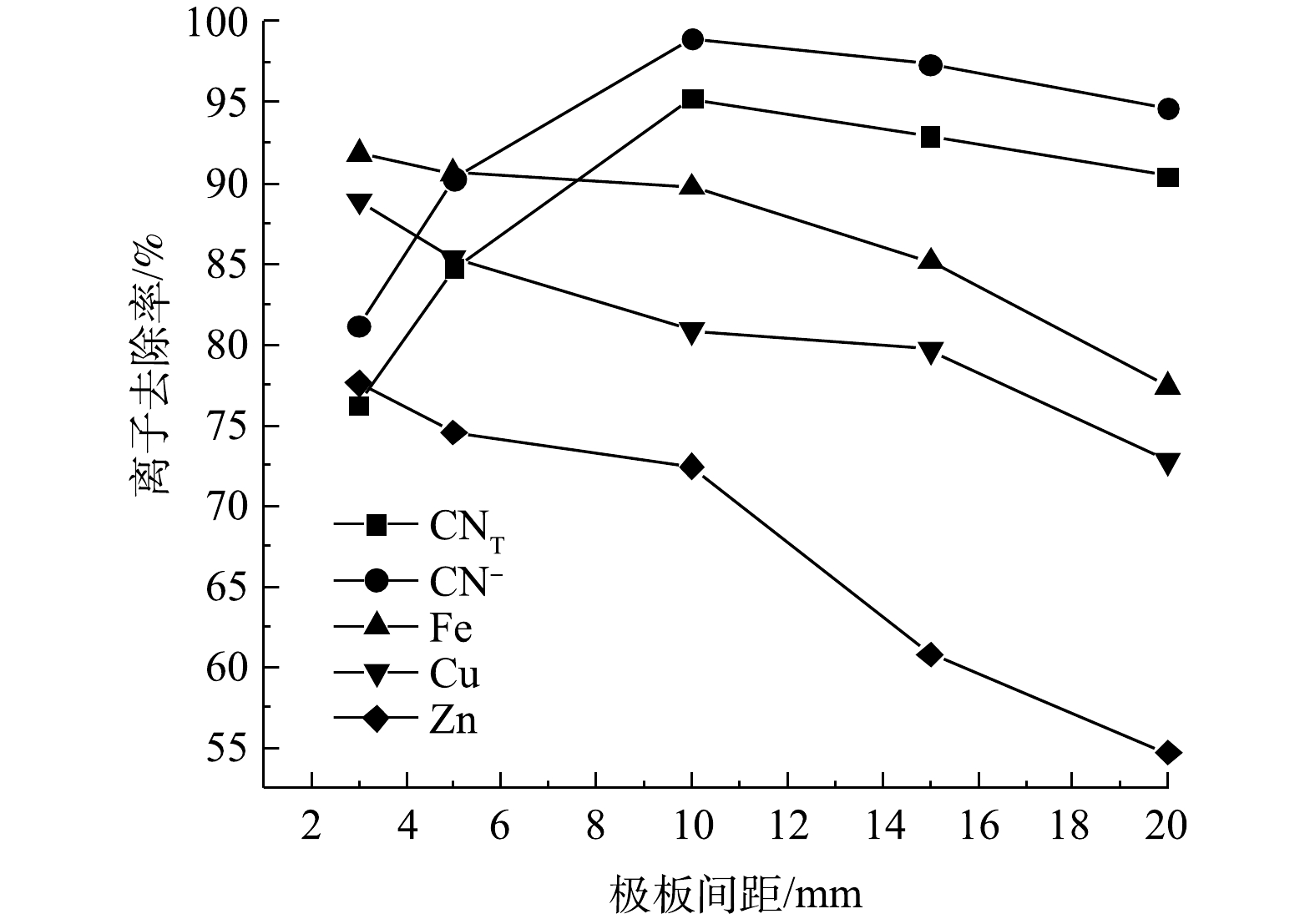 图 5 不同极板间距氰化尾渣中的氰化物及部分金属离子的去除率Figure 5. Removal rates of cyanide and some metal ions in cyanide tailings treated under different plate spacing
图 5 不同极板间距氰化尾渣中的氰化物及部分金属离子的去除率Figure 5. Removal rates of cyanide and some metal ions in cyanide tailings treated under different plate spacing2.5 氰化尾渣中矿物解离度分析
根据MLA分析结果统计所得的电解氯氧化处理前后氰化尾渣主要矿物的解离度,如图6所示。黄铁矿、磁黄铁矿与黄铜矿、闪锌矿的连生比例如表1所示。当NaCl添加量为0.1 g时,尾渣中与黄铜矿、闪锌矿连生的黄铁矿、磁黄铁矿的比例存在小幅度的下降,表明电解产生的有效氯能在一定程度上氧化矿石,添加量较少时氧化效果不太明显。随着NaCl添加量的增加,矿石的解离度不断增加,与黄铜矿、闪锌矿连生的黄铁矿、磁黄铁矿比例大幅下降,同时有部分矿物被氧化溶解,矿物自由面积百分比含量也存在一定程度的降低。NaCl添加量为1.0 g时,矿粒被氧化的程度继续加深,解离度进一步提高,但是,解离度过高使得更多的黄铁矿、磁黄铁矿、黄铜矿和闪锌矿暴露并被氧化,反而导致溶液中金属离子含量有所增加。
 图 6 氰化尾渣和不同NaCl添加量电解后尾渣解离情况Figure 6. Dissociation situation of cyanide tailings and tailings after electrolysis with different NaCl additions表 1 黄铁矿、磁黄铁矿与闪锌矿、黄铜矿之间的连生比例Table 1. Relationship between pyrite, pyrrhotite and sphalerite, chalcopyrite
图 6 氰化尾渣和不同NaCl添加量电解后尾渣解离情况Figure 6. Dissociation situation of cyanide tailings and tailings after electrolysis with different NaCl additions表 1 黄铁矿、磁黄铁矿与闪锌矿、黄铜矿之间的连生比例Table 1. Relationship between pyrite, pyrrhotite and sphalerite, chalcopyrite矿物 实验条件 连生比例/% 自由面积百分比/% 与闪锌矿 与黄铜矿 黄铁矿 原氰化尾渣 35.16 20.91 49.67 0.1 g NaCl 30.80 16.91 57.54 0.5 g NaCl 14.99 8.87 40.27 1.0 g NaCl 10.69 5.40 28.26 磁黄铁矿 原氰化尾渣 4.35 11.55 57.50 0.1 g NaCl 3.76 9.55 62.43 0.5 g NaCl 1.47 3.91 32.22 1.0 g NaCl 0.51 1.79 19.80 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.6 矿浆电解氰化尾渣平行验证实验
在极板间距10 mm、外加电压8 V、NaCl用量0.5 g、电解时间4 h的最佳工艺条件下进行了A、B、C 3组平行验证实验,结果如表2所示。由表2可以看出,最佳工艺条件下CNT、CN−以及Cu、Fe、Zn离子的去除率均值分别为94.83%、98.94%、85.65%、84.51%、73.85%,说明氰化尾渣中大部分氰化物及金属离子已被去除。氰化尾渣中游离氰含量达到了国家黄金行业氰渣污染控制技术规范(HJ 943-2018)的要求。
表 2 最佳条件下氰化尾渣中氰化物及部分金属离子的去除率Table 2. Removal rates of cyanide and some metal ions in cyanide tailings treated under best conditions离子种类 滤渣中平均离子含量/(g·t−1) 平均离子去除率/% CNT 90.31 94.83 CN- 3.34 98.94 Cu 26.52 85.65 Zn 949.78 73.85 Fe 2.45 84.51 | Show TableDownLoad:
CSV
2.7 矿浆电解氧化氰化尾渣过程分析
从热力学角度分析,氰化尾渣矿浆液中的还原性物质的氧化还原电极电位均小于氯的氧化还原电极电位,这些物质是可以被氯氧化的。各物质被氧化分解的顺序为:CN−>>
Zn(CN)2−4 Cu(CN)2−3 Fe(CN)4−6 Cu(CN)2−3 Fe(CN)4−6 Zn(CN)3−4 Cu(CN)2−3 Fe(CN)4−6 Fe(CN)4−6 Fe(CN)3−6 Cu(CN)2−3 Zn(CN)3−4 3. 结论
1)采用一步矿浆电解技术可有效处理氰化尾渣中的氰化物,当极板间距10 mm、外加电压8 V、NaCl添加量0.5 g、电解时间4 h时,CNT、CN−、Cu、Fe以及Zn离子的去除率分别可达到94.83%、98.94%、85.65%、84.51%和73.85%。
2)矿浆电解处理氰化尾渣过程中阳极附近主要发生的是氰化物的直接氧化与间接氧化,阴极板主要是Cu、Fe、Zn等重金属离子的电解沉积。随着电解电压、NaCl添加量以及电解时间的增大,金属矿物的解离度不断增加,矿物之间的连生比例不断降低,尾渣中黄铁矿、磁黄铁矿的大颗粒连生体被分解为小颗粒的单体态并不断被电解氧化溶解。
-
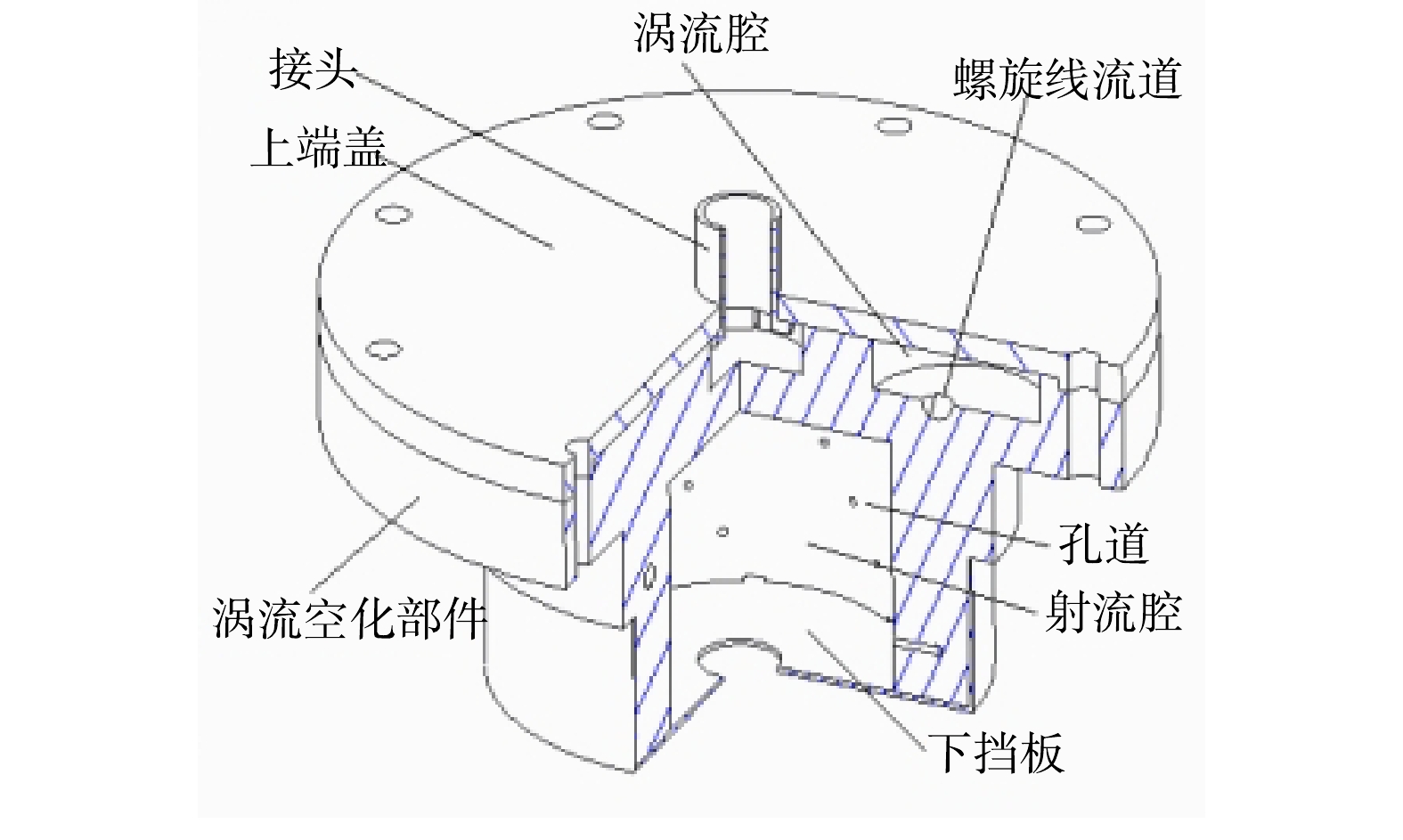
图 1 涡流空化核心部件立体剖视结构图
Figure 1. Three-dimensional sectional structure of vortex cavitation core components
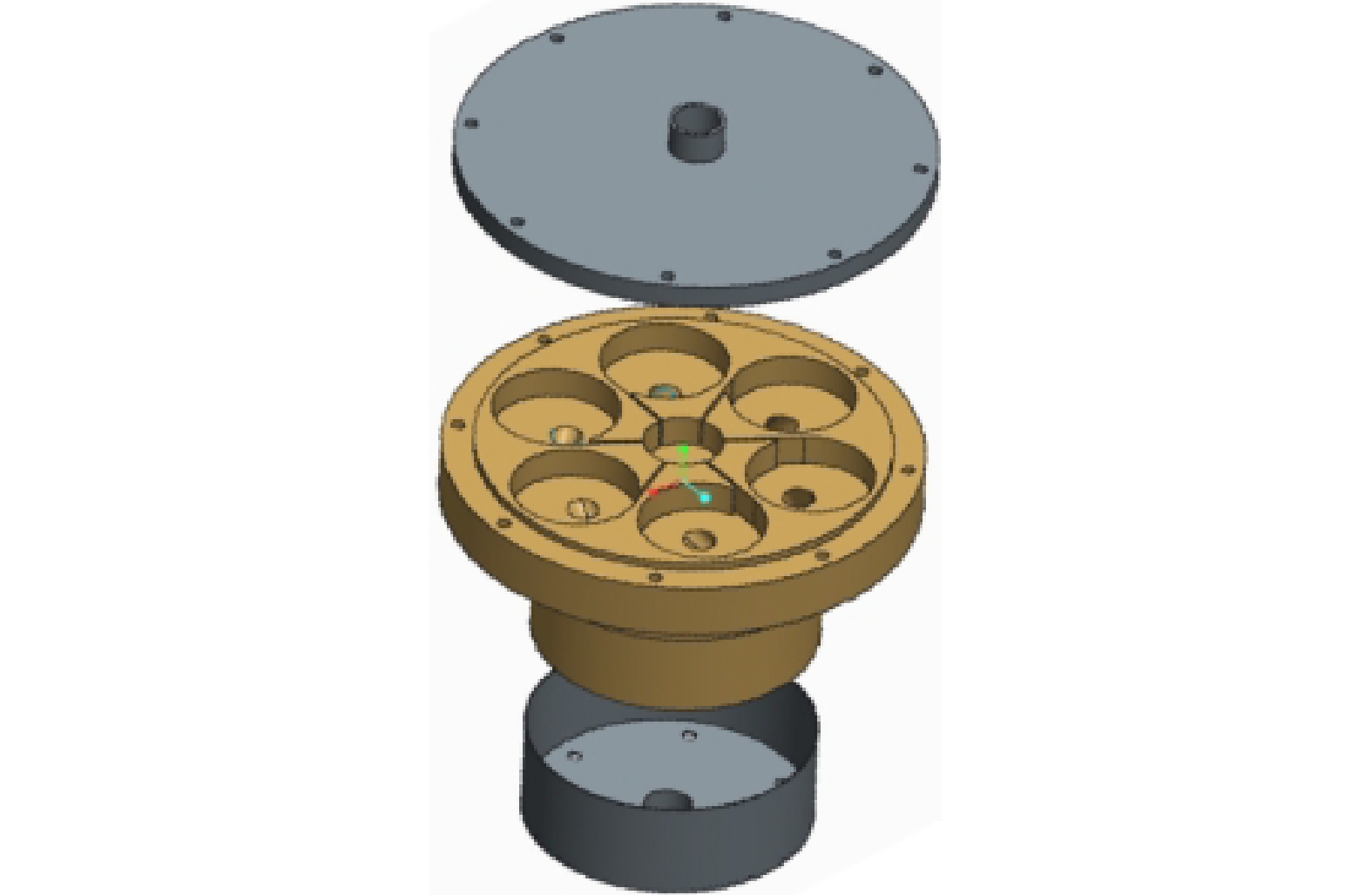
图 3 涡流空化装置零件三维结构
Figure 3. Three-dimensional structure of the swirling vortex cavitator
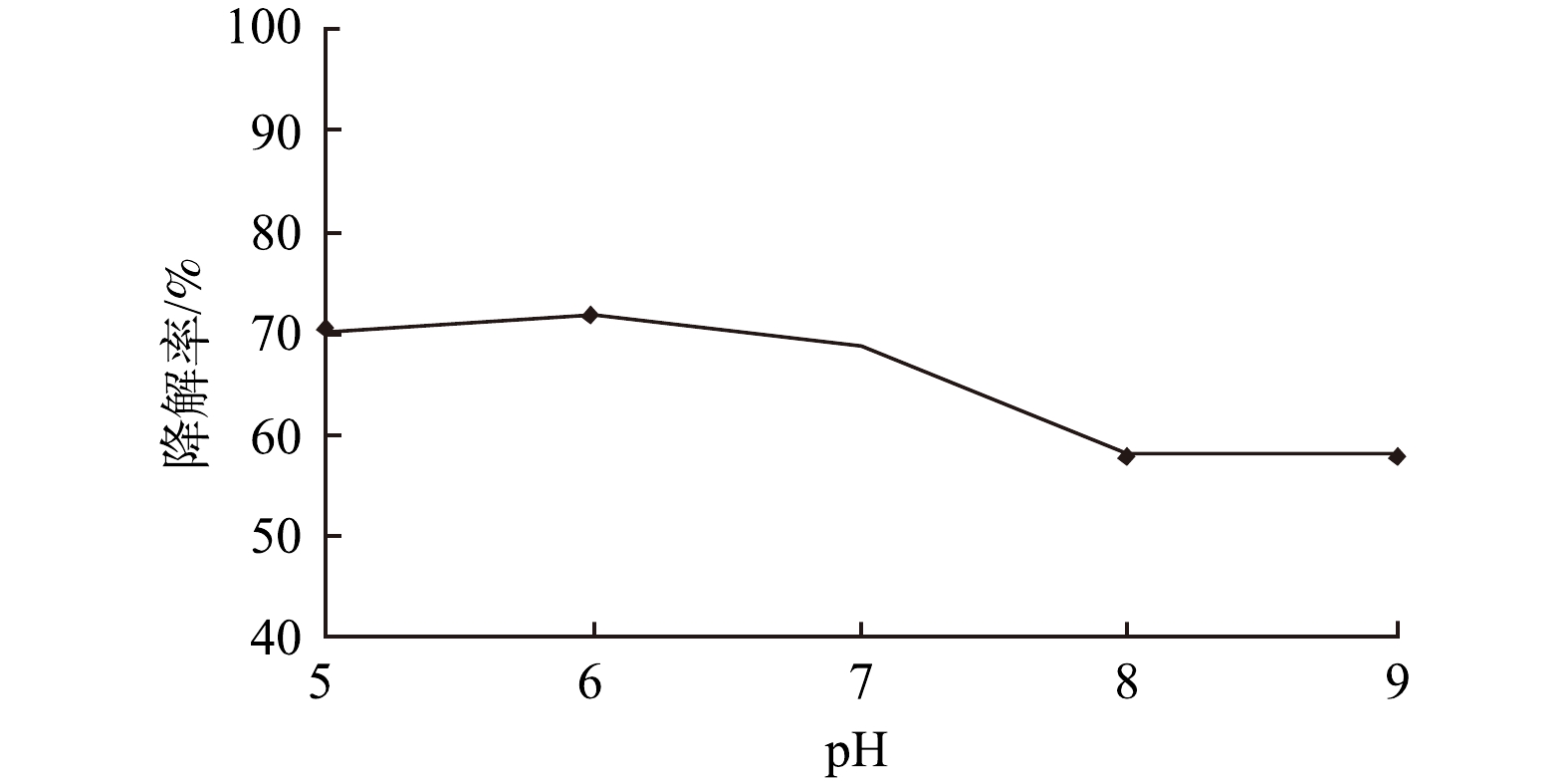
图 7 不同溶液初始pH值下的土霉素降解率
Figure 7. Degradation of oxytetracycline at different initial pH
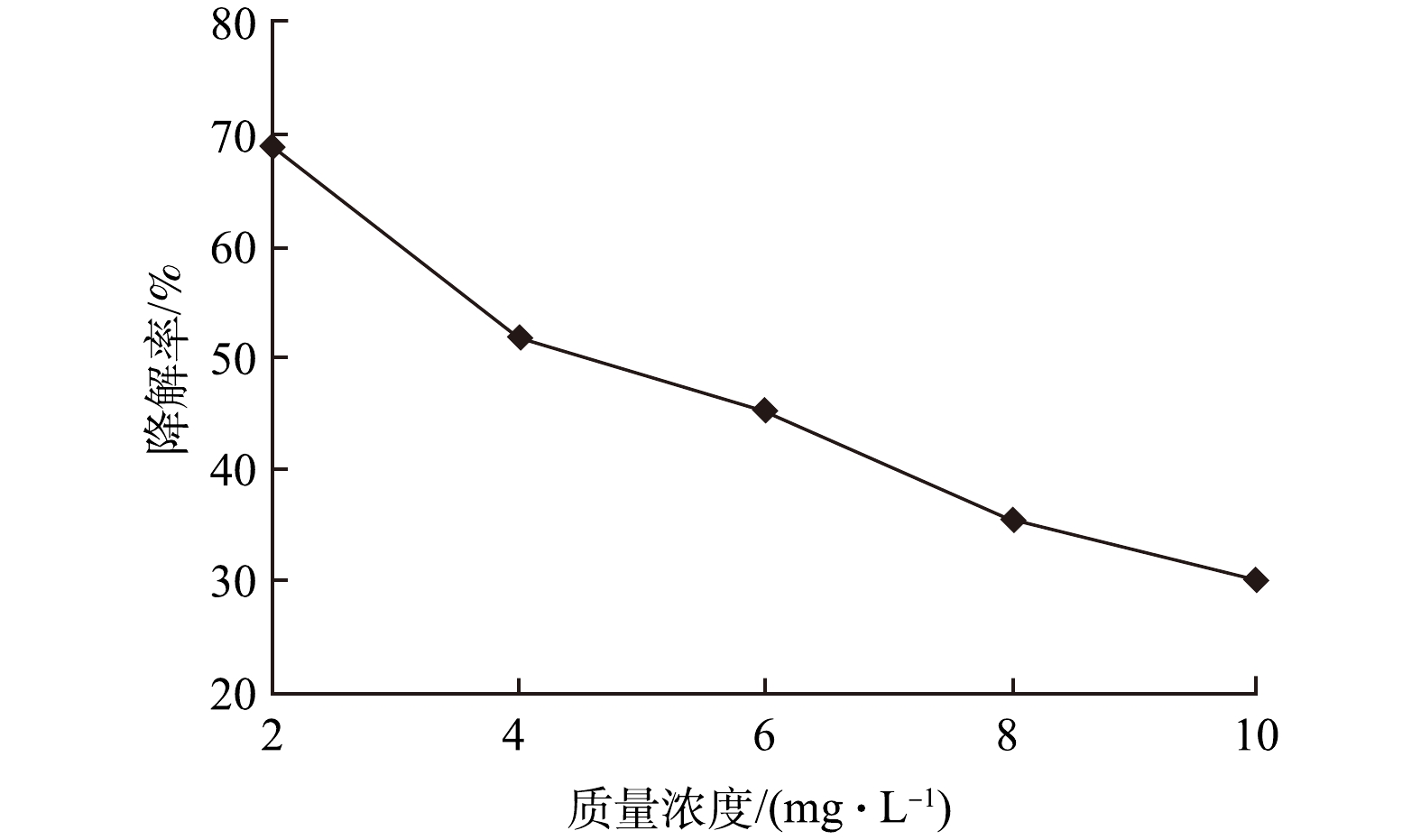
图 8 不同土霉素初始浓度下的降解率
Figure 8. Degradation of oxytetracycline at different initial oxytetracycline concentrations

图 9 不同温度条件下对土霉素的降解率
Figure 9. Degradation of oxytetracycline at different temperature ranges
-
[1] 张玮玮, 弓爱君, 邱丽娜, 等. 废水中抗生素降解和去除方法的研究进展[J]. 中国抗生素, 2013, 38(6): 401-410. [2] 李玉冰, 张凡建, 蔡泽川, 等. 臭氧净化技术治理猪场废水中兽用抗生素残留的研究[J]. 黑龙江畜牧兽医, 2017(4): 184-187. [3] 祁佩时, 王娜, 刘云芝, 等. Fenton氧化-活性炭吸附协同深度处理抗生素制药废水研究[J]. 净水技术, 2008, 27(6): 38-41. doi: 10.3969/j.issn.1009-0177.2008.06.010 [4] LENG L, WEI L, XIONG Q, et al. Use of microalgae based technology for the removal of antibiotics from wastewater: A review[J]. Chemosphere, 2020, 238: 1-14. [5] ANJALI R, SHANTHAKUMAR S. Insights on the current status of occurrence and removal of antibiotics in wastewater by advanced oxidation processes[J]. Journal of Environmental Management, 2019, 246: 51-62. [6] 黄昱, 李小明, 杨麒, 等. 高级氧化技术在抗生素废水处理中的应用[J]. 工业水处理, 2006, 26(8): 13-19. doi: 10.3969/j.issn.1005-829X.2006.08.004 [7] 武君, 张晓冬, 刘学武, 等. 水力空化及应用[J]. 化学工业与工程, 2003, 20(6): 387-391. doi: 10.3969/j.issn.1004-9533.2003.06.015 [8] 倪汉根. 气核空化空蚀[M]. 成都: 成都科技大学出版社, 1993. [9] 钱光毅. 基于空化原理的孔板污水处理设备性能模拟[J]. 四川建筑科学研究, 2010, 36(2): 262-265. doi: 10.3969/j.issn.1008-1933.2010.02.067 [10] 张婵, 郑爽英. 超声空化效应及其应用[J]. 水资源与水工程学报, 2009, 20(1): 136-138. [11] CHAHINE G L, KALUMUCK K M. Swirling fluid jet cavitation method and system for efficient decontamination of liquids: US6221260B1 [P]. 2001-01-04. [12] WU Z, ONDRUSCHKA B, BRAEUTIGAM P. Degradation of chlorocarbons driven by hydrodynamic cavitation[J]. Chemical Engineering & Technology, 2007, 30(5): 642-648. [13] WANG X, ZHANG Y. Degradation of alachlor in aqueous solution by using hydrodynamic cavitation[J]. Journal of Hazardous Materials, 2009, 161(1): 202-207. doi: 10.1016/j.jhazmat.2008.03.073 [14] 张斌, 陈银银, 李育敏, 等. 新型涡流空化器的实验研究[J]. 轻工机械, 2017, 35(2): 58-62. doi: 10.3969/j.issn.1005-2895.2017.02.012 [15] 李祥, 任旭东, 袁寿其, 等. 一种用于有机水污染降解系统中的涡流空化装置: CN106587256A [P]. 2017-04-26. [16] CURT H T, MORTEN O M. Vortex generator with vortex chamber: US. F15D1/00. 20120097280A1 [P]. 2012-04-26. [17] BRAEUTIGAM P, WU Z, STARK A, et al. Degradation of BTEX in aqueous solution by hydrodynamic cavitation[J]. Chemical Engineering & Technology, 2009, 32(5): 745-753. [18] 王宝娥, 张日红, 练晓明. 一种旋流式涡流空化器: ZL201821724174.2 [P]. 2019-09-27. [19] 张晓冬, 杨会中, 李志义. 水力空化强度与空化自由基产量的关系[J]. 化工学报, 2007, 58(1): 32-37. [20] 钱盛. 钛基PbO2电极电催化降解废水中的四环素类抗生素[D]. 重庆: 西南大学, 2018. [21] 陶跃群. 水力空化降解废水中有机污染物的理论与实验研究[D]. 北京: 中国科学院大学(中国科学院工程热物理研究所), 2018. [22] 杨会中. 水力空化强化效应实验研究[D]. 大连: 大连理工大学, 2006. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4410
- HTML全文浏览数: 4410
- PDF下载数: 79
- 施引文献: 0
