


-
我国是一个储煤丰富的国家,以煤为主要原料经化学转化生产化学品对于我国减少石油的对外依赖度,维护能源和资源安全仍具有十分重要意义。现代煤气化技术在一定程度上改善了传统煤化工的能耗高、效率低、污染严重的现状。但煤气化过程中大量产生的煤气化细渣一直难以高效利用[1-2]。据统计,年生产气化渣超过3300万吨[3]。大量的气化残渣堆积,不但占用有限的土地资源,而且还会破坏当地的生态环境,危害人体健康。气化细渣的高效资源化利用已经成为我国现代煤化工可持续发展急需解决的重大问题[4]。
气化细渣中残碳的利用是其资源化利用的关键。建材化利用适应性好、消纳能力强,一直是煤矸石、粉煤灰等煤基固废资源化利用的重要利用途径。但气化细渣残碳含量高(15% — 50%),反应活性低,很难通过配料掺烧等常规办法进行充分利用,严重限制了其在建材化领域的大规模应用。北京低碳清洁能源研究所的赵永彬等研究了气化残渣的矿物组成和基本性质[5]。清华大学的王金福等开发了基于快床-湍床组合式循环流化床反应器技术,通过精密控温燃烧,将气化灰渣深度氧化脱碳,得到无碳灰分[6]。中国矿业大学的刘坤基等人探索了利用气化细渣中的残余碳制备高性能石墨材料的技术可行性[7]。如何提高气化细渣中残碳的反应活性,特别是中温段(500—650 ℃)的燃烧性能[8],是拓宽其在建材领域的资源化利用的关键。
本文采用机械活化法,为气化渣固废资源化综合利用提供一条新的途径。通过球磨机械力产生冲击、剪切、压缩、摩擦等作用,使气化细渣中的残碳晶格局部破坏,粒度变小、比表面积增大[9-10],产生大量微观缺陷,增加其表面反应位点,使其内能增大,反应活性增强[11-13],使其600 ℃的燃烧反应活化能从63.2 kJ·mol−1降低到28.4 kJ·mol−1,大大提高其在中温段的燃烧反应活性和能源转化效率。在球磨活化的基础上,不添加其它化学试剂,将其制备成环保轻质高强陶粒(堆积密度0.867 g·cm3,筒压强度7.67 MPa)。本文详细考察了机械活化对气化细渣中的残碳反应活性的影响,提出了气化细渣制备高强度轻质陶粒的制备条件,研究了气化细渣建材化利用过程中残碳燃烧反应动力学,相关研究可以为我国气化细渣的资源化利用提供一定的基础数据和技术支持。
-
本研究实验所用气化细渣由宁夏某能源集团提供,为水煤浆气化工艺过程中由煤气化气夹带离开煤气化炉,在气体净化过程中分离排出的残渣。气化细渣样品按照《工业固体废物采样制样技术规范》(HJ/T—20)采集,依据《固体废物腐蚀性测定-玻璃电极法》(GB/T15555.12)测定其pH值为5.87。气化细渣样品经105 oC下干燥24 h至恒重,确定其含水率为72%。再经600 ℃灼烧3h后冷却至室温,确定其热灼减率为22%。XRF检测分析得到的气化细渣的主要成份如表1所示。干燥后的气化细渣完全消解,采用ICP-MS和测汞仪检测得到的主要重金属元素如表2所示。
-
机械活化设备采用QM-3SP4行星式球磨机。将干燥后的60 g气化细渣置于不锈钢球磨罐中(Φ10 mm不锈钢磨球20个),球磨转速400 r·min−1,活化时间分别设定1、2、4、8 h。
碳的反应活化能采用动态 TGA分析[14]和Coats-Redfern模型测定,不同化学反应模型的微分和积分表达式[15]。根据气化细渣残碳的燃烧是一个气固缩核反应类型,碳的燃烧过程符合一级动力学反应方程,即利用一级化学反应公式对相关的1/T和G(x)积分函数进行回归即可得到反应活动能E。
-
以机械活化后的气化细渣为原料,加适量水制备陶粒生坯,然后将陶粒生坯置于干燥箱干燥12 h。烘干后的陶粒生坯置于马弗炉中,选择不同的烧胀程序,进行高温烧胀,最后冷却制得轻质高强环保陶粒。陶粒产品的表观密度及筒压强度根据《轻集料及其实验方法》(GB/T 17431—2010)进行测定。
-
气化渣的物相分析采用X射线衍射仪(Philips PW 1700),成分测定利用 X射线荧光光谱仪(Schimadu XRF-1800X),微观形貌观察采用发射扫描电子显微镜(Hitachi S-3000N),粒度测试采用激光粒度分析仪(Malvem Mastersizer 2000),热分析采用DTA-TG(Netzsch STA-409PC)。
-
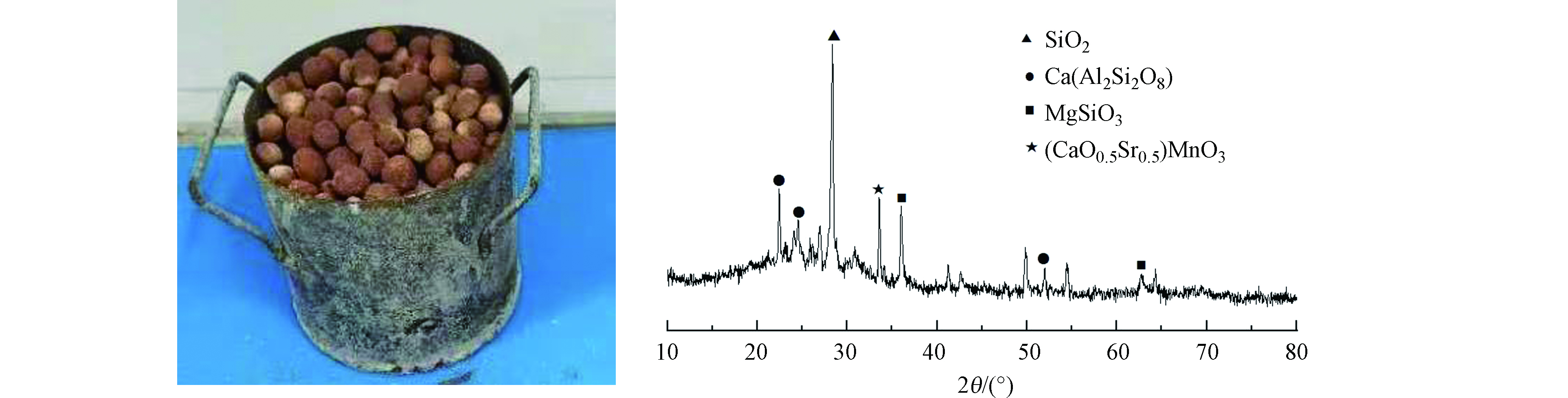
煤气化细渣样品的基本成分(表1)和物相分析图1(a)表明渣中的SiO2、Al2O3、Fe2O3和CaO占到气化细渣组分的60%以上,而且渣中重金属Cd、Cr、Pb、Zn、Cu和Hg的含量均比较低(表2)。
图1(a)显示细渣样品X射线衍射峰都有明显的包状起伏,表明样品由大量的非晶态的SiO2和少量的结晶硅铝酸盐矿物组成。图2(a)显示渣的粒径主要在55—290 μm之间。根据《固体废物-浸出毒性浸出方法(HJ557-2010)》进行浸出实验,发现渣中主要重金属浸出浓度均在0.05 μg·mL−1以下,表明该渣可作为潜在建材原料进行资源化利用。
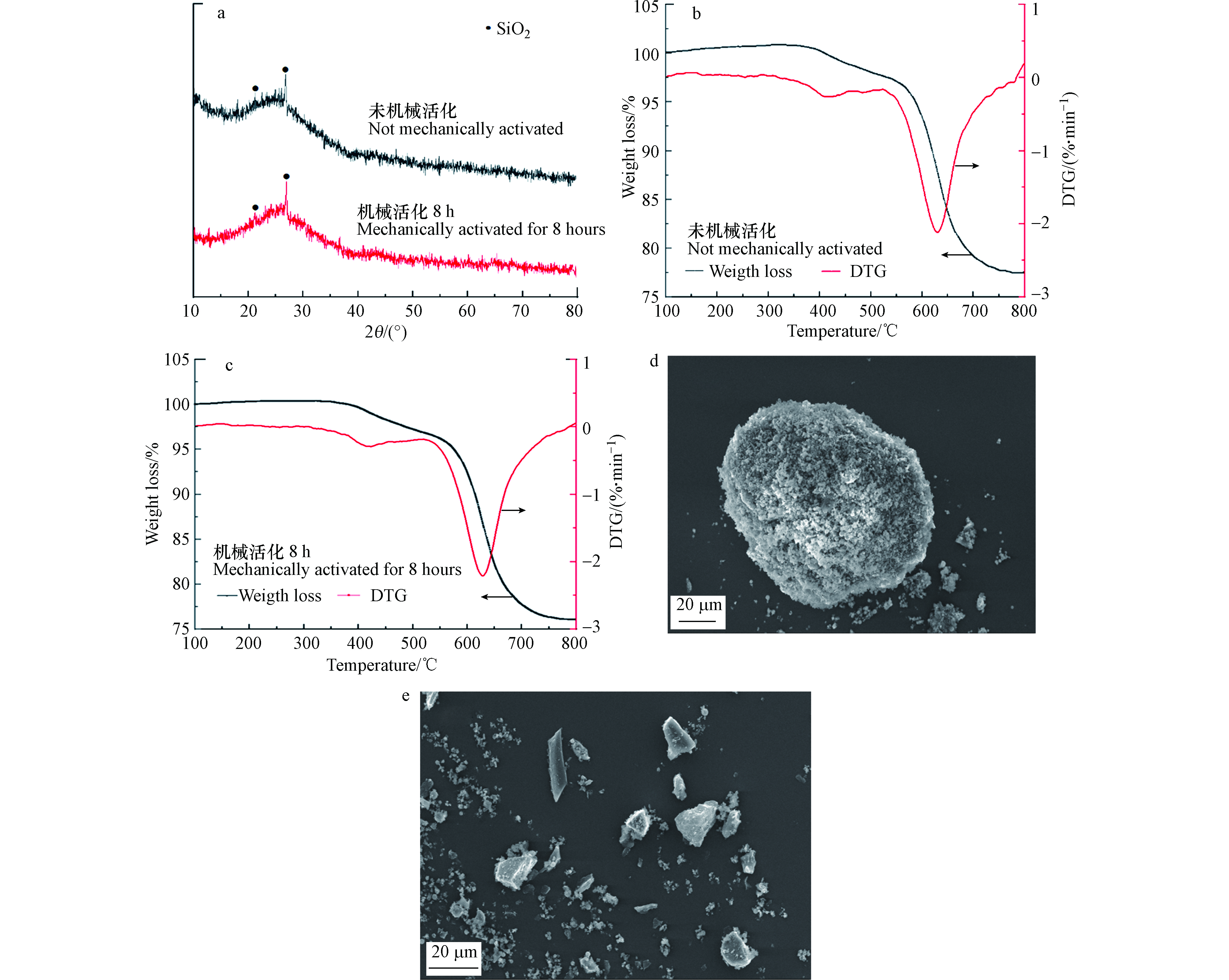
图1(d)细渣SEM形貌分析表明,细渣中存在较大形态的球形颗粒物。这主要是由于气化过程中的高温环境使得煤中的矿物质熔融,由于表面发生收缩呈现球状[3],其结构致密,表面附着有大量的小颗粒物质多为不规则多孔颗粒状态,主要是气化过程中煤发生膨胀和破碎形成的炭颗粒。图1(b、c)热重曲线表明该渣未机械活化由室温到800 ℃的减重约为22.4%,机械活化由室温到800 ℃的减重约为23.4%。样品的失重呈单峰出现,随着温度的升高,样品质量主要在以下三个方面发生改变:样品中的自由水与结合水挥发、残碳的分解以及碳酸盐的分解。
细渣的烧失量绝大部分来源于残碳,所以气化残渣的烧失量高低直接反映残碳含量的高低[5]。一般来说,制备建材过程中,希望残碳能够在450—650 ℃中温段充分燃烧,避免建材产品中含有残余碳而影响其性能。机械活化前细渣样品中的大量残碳被包裹在非晶态SiO2结构中,中温段反应性较差,不能完全燃烧,是制约气化细渣资源化利用的重要原因[16]。
-
图2(a)气化细渣的粒径分布表明,随着机械活化时间的增长,渣的粒径明显降低,比表面积显著增大。机械活化8 h以后,渣的粒径由55—290 μm降到2—30 μm,处理后平均粒径D10粒径、D50粒径、D90粒径均小于未处理样品。机械活化后,比表面积由614 m²·kg−1增大到1540 m²·kg−1。图1(a)表明机械活化后,细渣的物相组成未发生明显变化。
图1(e)细渣的粒径分析和微观形貌表明,机械球磨过程中气化渣受极大的冲击力发生破碎、断裂,从而使得颗粒、残碳和矿物颗粒由原来的块状和团状转化为由新产生的细小颗粒团聚成的松散颗粒,颗粒表面变得尖锐和晶粒不规则[18],残碳具有更多的活性位点、更大的比表面积、更无序的碳晶体结构,有利于增强固体颗粒的反应性[1,2,17-20]。处理后样品的热重分析也表明,细渣中残碳在中温段的燃烧反应速率增加约14%,800 ℃后样品中的不再存在未反应碳。机械活化可以破坏细渣中残碳颗粒的包裹结构,残碳大部分裸露出来,气固相反应阻力减小,燃烧反应速率增加。
气化细渣残碳的燃烧是一个气固缩核反应,由外向内逐步推进的过程,原始颗粒形成一种气体迁移相而分解,O2通过气相边界层到达气化渣表面,这是一个不可逆反应,主要发生气相边界层外扩散的反应和界面反应。活化能E是描述反应物的分子由初始稳定状态变为活化分子所需要吸收的能量,是用来表征反应进行的难易程度的反应动力学指标。
在不影响结果准确性的前提下,为简化分析,设碳的燃烧过程符合一级动力学反应方程,则有:
根据Arrhenius阿伦尼乌斯公式:
当加热速率
β=dTdt(∘C⋅min−1) ,(1)、(2)可改写为:对(3)式积分:
对上式取自然对数,
式中,A是指前因子(s−1),R是气体常数,T是开尔文温度(K),α是分解反应转化率,k是速率常数,n是反应级数,β是升温速率。
当n=1时,对于一级化学反应方程,
对一般反应温度区及大部分活化能,
2RTE 远小于1,因此忽略2RTE ,则上式化简为:以
1T和ln[−ln(1−α)T2] 分别作为X轴和Y轴作图,得到残碳的动力学反应模拟曲线,如图2(b-e)所示。拟合后可以得出机械活化1、2、4、8 h后的残碳反应活化能,如图2(f)所示。通过对比发现,活化能分布可以反映DTG曲线失重趋势,机械活化以后,气化细渣中残碳在500—650 ℃的燃烧反应活化能均明显降低,这是因为随着球磨时间的增长,煤样粒径变小,比表面积变大,受热时挥发分的析出速率越快,试样表面可燃物质的聚集程度也越大,同时暴露的煤分子活性物质就越多,更容易与氧气接触发生反应,进而产生了更多的热量,加速了反应的进行,同时相同条件下达到过热的温度也越低,经过球磨破碎的气化细渣活化能总是低于未经过任何处理的。在500—650 ℃期间,不同球磨状态下的活化能不是一直减少的状态,而是呈现了先增加再减少的趋势,由图2(f)可知,同一粒径粒子,随着温度的升高,活化能呈现先升高后降低的趋势,存在一个临界温度,临界温度阶段的活化能最大。低于临界温度时,反应消耗的是煤颗粒中存在的不稳定的活性官能团,没有破坏煤的结构,主要受水分蒸发吸热的影响。因此,低于临界温度时,活化能随温度的升高而升高。高于临界温度时,反应物中的大分子结构会加速断裂,使活性官能团增加,且水分已经完全蒸发,反应难度反而减小,故活化能随温度的升高而降低[21-22]。此外,也可以从产物的角度来理解。煤燃烧的产物有气相,如CO、CO2等,也有固相,如中间络合物。由现代反应速率理论可知,过渡态活化络合物具有比反应物分子和产物分子更高的势能,反应必须克服这一反应势垒,故可以认为反应的难易程度与活化络合物的含量密切相关。张玉龙等和Veselovski等都认为随着反应温度的升高,固相中间络合物的含量先增加后减少。活化能随着络合物含量的变化而变化,因此也呈现先升高后降低的趋势[23-24]。文献中报道煤的活化能范围27.2—76.2 kJ·mol−1[25],但有些活化能高于理论值,这主要与渣中灰分含量有关。灰分在燃烧过程中会形成包裹在未燃尽碳外面的灰壳,增加扩散阻力和传热阻力,从而导致活化能升高[26]。机械活化8 h后,其650 ℃燃烧反应活化能由63.2 kJ·mol−1降到28.4 kJ·mol−1,降低了55%,实验值处在理论值较低处,说明球磨活化可有效促进气化残碳的燃烧,使残碳更加容易分解。球磨处理气化渣能够明显降低反应活化能,降低反应的阻力,增大整体燃烧性能,增大残碳的反应性和细渣的可利用性。 -
陶粒主要以硅铝基成分为主要原料烧制而成,其中主要成分为SiO2和Al2O3。SiO2的作用是形成玻璃质使得陶粒光滑且增大强度,Al2O3的作用是形成莫来石,进而增大强度[27-31]。气化细渣主要由SiO2、Al2O3、Fe2O3和CaO组成,根据陶粒烧胀化学成分的Riley三角形范围,SiO2在40%—70%之间,Al2O3 在12%—26%之间,Fe2O3+MgO+CaO在8%—24%之间,是烧制膨胀陶粒的适宜原料化学成分。该渣成分处于Riley三角形内,不需要添加其它化学试剂,是制备高强度轻质陶粒的良好原料。
提高气孔产生率是制备高强度轻质陶粒的关键。一般来说,500—650 ℃中温段是形成气孔的关键阶段。为提高气孔率,降低陶粒的密度,在烧制陶粒过程中添加适宜发泡剂非常重要,如王川采用稻壳灰作为发泡剂,谢武明等[32]采用煤粉作为发泡剂,解小娟选择铵盐类作为发泡剂,在陶粒烧制过程中,随着温度的升高,生料球内部会产生熔融液相,从而形成致密和高强的结构。从图3(b)烧成陶粒的XRD分析图可以看出,烧制过程中会产生大量的Ca(Al2Si2O3)低熔点硅铝化合物,有利于降低液相生成温度。内部碳的快速反应和产气保证了烧成样品的气孔率和低密度。细渣中的大量的SiO2会增加熔体粘度,熔融相冷却会形成晶体和玻璃相,有效防止气孔的融合长大。
另外,烧成温度、保温平台和升温速率都是陶粒烧制的重要工艺参数。烧制温度过高,会导致烧结体收缩致密化,膨胀系数增加,进而密度降低。烧成温度小于950 ℃时,陶粒表面颜色较浅,熔融相较少,强度不足。当烧成温度达到1050 ℃时,陶粒表面由浅棕色转变为褐色且熔融物较多,陶粒内部产生较大孔隙,孔连通程度提高,产品开裂。而且,烧成陶粒会粘在坩埚壁上,难以取出。烧制温度在1000℃时,得到的陶粒强度高且无粘结。升温速度也会明显影响陶粒的外观形貌和强度。速率小于7 ℃·min−1时无发泡效果,速率过快大于11 ℃·min−1时会导致体积急剧膨胀,孔隙结构弱化,陶粒破坏。当采用10 ℃·min−1升温速度时,烧制的陶粒有光滑的釉质,并且陶粒表面没有粘连图3(a)。保温平台是保证气孔率的重要温度节点,通过机械活化后,保温平台设定在550—600 ℃容易形成均匀的气孔。保温时间5 min时,发泡效果不佳,体积膨胀较小,保温30 min时,烧制过度,胚体产生一定程度变形,选取保温时间10 min体积膨胀最佳。采用烧成温度1000 ℃,保温平台600 ℃下保温10 min和升温速率10 ℃·min−1,烧得陶粒的堆积密度0.867 g·cm−3,筒压强度7.67 MPa,达到国家高强度轻质陶粒产品标准的要求。
采用机械活化处理气化细渣,不仅可以提高其反应,直接烧制高强度轻质陶粒,而且残碳的燃烧可以为制备过程提供足够的热量,不需要外加燃料。以1050 ℃工艺为例,每吨陶粒烧制所需热量约为2.5×106 kJ,而煤气化细渣中22%的残碳充分燃烧可以提供约7.5×106 kJ的热量,完全满足烧制过程的热能需求。
-
现代煤化工行业的发展与我国能源安全密切相关,煤化工技术的发展对于保障能源和化工原料合理的自给率具有不可替代的作用,当前煤化工产业面临着经济与环保的双重压力,现对气化渣资源化利用技术应用前景分析如下:
(1)机械活化可以显著降低煤气化细渣中残渣的燃烧反应活化能。活化后气化细渣机械能总是小于未处理气化细渣,机械活化8 h后,残渣650 ℃时的燃烧反应活性明显增大,反应活化能由63.2 kJ·mol−1减小28.4 kJ·mol−1;
(2)机械活化后的煤气化细渣,粒径由55—290 μm降到2—30 μm,处理后平均粒径D10粒径、D50粒径、D90粒径均小于未处理样品,比表面积由614 m²·kg−1增大到1540 m²·kg−1;
(3)气化细渣用于烧制陶粒,不添加其他化学试剂,1000 ℃烧制高强度轻质陶粒,其堆积密度为0.867 g·cm−3,筒压强度达到7.67 MPa;
(4)资源化利用可以规模化消纳利用气化细渣,有效的减少环境污染,实现低成本增产增收,在气化细渣高附加值资源化利用中有较好的发展前景。
(5)机械活化后煤气化细渣中的残碳燃烧可以为陶粒烧制提供足够的热量,不需要外加燃料,完全满足烧制过程的热能需求。
机械活化煤气化细渣资源化利用及残碳燃烧反应动力学探究
Mechanical activation and combustion kinetics of residual carbon in coal-gasification fine slag
-
摘要: 随着我国煤气化技术的快速发展,气化渣的产生和排放量逐年增加。煤气化细渣的大量堆存,给当地的环境保护造成了巨大的压力,已经成为限制煤化工产业基地可持续发展急需解决的重大问题。本文针对气化细渣中残碳反应活性低和转化难度大的问题,研究了机械活化法对气化渣残碳反应动力学的影响。采用Coats-Redfern法分析了机械活化对残碳反应活化能的作用规律。结果表明,机械活化8 h,残碳燃烧反应活性显著提高,650 ℃下的活化能由63.2 kJ·mol−1降到28.4 kJ·mol−1。不添加其他化学试剂,1000 ℃烧制高强度轻质陶粒,其堆积密度为0.867 g·cm−3,筒压强度为7.67 MPa。相关研究可以为我国气化细渣的资源化利用提供一定的数据和技术基础。Abstract: With the rapid development of coal gasification technology in China, a large amount of coal-gasification fine slag (CGFS) generated and discharged recently. The CGFS has increased pressure on the local environmental protection and limited the sustainable development of coal chemical industry bases, which is just becoming a problem that needs to be solved urgently. To improve the reaction activity of residual carbon in CGFS and increase its value in resource recycling, this paper studied the influence of mechanical activation on the combustion kinetics of residual carbon. The Coats-Redfern method was used to analyze the effect of mechanical activation on the activation energy of residual carbon combustion reaction. The results showed that the combustion reaction activity of residual carbon in CGFS significantly increases after 8 hours of mechanical activation, and the activation energy decrease from 63.2 to 28.4 kJ·mol−1 at 650 ℃. Without other chemical reagents, high-strength lightweight ceramsite was prepared at 1000 ℃, with a bulk density of 0.867 g·cm−3 and a cylinder compressive strength of 7.67 MPa. This paper can provide valuable data and a technical route for the utilization of CGFS in China.
-
挥发性有机物(VOCs)是导致城市雾霾与光化学污染等大气复合污染的重要前体物,对人类健康和生态环境产生重大影响,已经引起了政府和公众的广泛关注[1-3]。因此,打好蓝天保卫战,VOCs治理是关键。在众多VOCs末端控制技术中,催化氧化技术因其具有高效性和彻底性等优势引起了学界的极大关注[4-5]。催化氧化技术的核心问题是开发出高效稳定且具有低温活性的新型催化剂[4-6]。一般而言,能够催化降解VOCs的催化剂有贵金属和过渡金属2大类,其中,贵金属催化剂具有催化活性高、稳定性差等特点,此外,贵金属催化剂因其价格昂贵和易中毒失活使其在工业应用中受到了极大的限制[5]。因此,研究开发低温高效的过渡金属氧化物催化剂成为了目前研究的热点[4-5, 7]。
近年来,Mn-Ce复合氧化物催化剂因其具有良好的催化氧化性能受到了研究人员的极大关注[7-11]。Mn-Ce复合氧化物一方面具备CeO2优异的储氧/释氧能力[12-13],另一方面MnOx具有环保、廉价易得且存在多种价态等优点[14-17]。然而传统制备工艺合成的Mn-Ce催化剂存在颗粒易团聚和形貌不可控等问题,这一定程度上限制了该类催化剂的改进和应用。本研究通过简单的水热合成法制备了一系列Mn-Ce复合氧化物催化剂,有望解决上述提及的问题。
目前研究结果表明,纳米材料在催化过程中具有形貌效应[15-17],将其应用于催化氧化VOCs方面也取得了大量的研究成果[6-7, 12]。纳米材料的催化性能与其形貌特性密切相关,LIAO等[7]通过水热法制备的具有纳米棒形貌的Mn-Ce复合氧化物催化剂在反应温度225 ℃下即可实现甲苯的完全降解。YU等[11]采用溶胶凝胶法制备了一系列MnOx/TiO2和MnOx-CeO2/TiO2催化剂,考察了Ce添加量对催化降解性能的影响,结果表明MnOx-CeO2/TiO2(Ce/Ti=0.05)催化剂活性最高(T90=180 ℃),高度分散的无定型Mn及催化剂表面存在大量活性氧物种是其具有优异低温催化氧化甲苯性能的关键。郑宽等[18]通过共沉淀法制备不同Mn/Ce比的复合氧化物,发现对Mn-Ce复合氧化物催化剂的甲苯催化降解性能高于单一MnOx和CeO2。大多数报道的催化剂均未通过对催化剂形貌进行控制从而调控催化剂的反应活性[5, 18]。因此,本研究以表面为纳米针的氧化锰微球为基础,通过在水热前驱液中加入不同含量的Ce,原位合成出具有一定形貌的Mn-Ce复合氧化物,通过SEM、XRD、H2-TPR、O2-TPD、BET及拉曼光谱等手段对所合成的复合材料进行表征,并深入研究分析催化剂催化氧化甲苯的构效关系。
1. 实验材料和方法
1.1 催化剂的制备
将2.700 g MnSO4·H2O和13.185 g (NH4)2S2O8溶解于60 mL去离子水中,待完全溶解后,定容至80 mL,转移到100 mL的聚四氟乙烯的反应釜中,80 ℃反应4 h后,自然冷却至室温,将所得沉淀物用去离子水洗涤至中性,最后在80 ℃条件下干燥后保存备用,所制备的样品记为MnO2。
其他过程与氧化锰微球的制备过程相同,不同之处是,在加入MnSO4·H2O和(NH4)2S2O8的过程中同时加入不同含量的Ce(SO4)2·4H2O(Ce的摩尔分数为5%、10%、20%和40%)。所制备样品分别记为Mn0.95Ce0.05Ox、Mn0.90Ce0.10Ox、Mn0.80Ce0.20Ox和Mn0.60Ce0.40Ox。
1.2 催化剂活性评价
催化剂的甲苯催化氧化性能测定在连续流动的固定床反应器中进行[15-16]。催化反应器为自制的内径为8 mm 的U型石英管,固体催化剂床层由0.13 g(40~60目)催化剂和0.52 g的石英砂组成。采用质量流量器控制气体流量,以甲苯为目标反应物,浓度为550 ppm,空气总流量为200 mL·min−1,重时空速(GHSV)约为92 300 mL·(g·h)−1。由气质联用仪(7890B-GC,5977B-MSD,Agilent Technologies)对甲苯降解前后的尾气进行在线检测。其中甲苯利用MS(分析柱为TG-BOND Q:30 mm×0.32 mm×20 μm)进行检测。配备的FID与镍转化炉连接,用于CO和CO2的检测,其分析柱为5A分子筛(2 m×2 mm)、Porapak Q(1.83 m×1.2 mm)和Porapak Q(0.91 m×2 mm)。
1.3 材料表征
SEM结果采用S-3400N电子显微镜(日立,日本)对催化材料的形貌进行观察。
XRD采用D8 ADVANCE X射线衍射仪(Bruker,德国)进行测定(Cu Kα射线,扫描角度为5°~90°)。
N2吸附-脱附采用日本BEL公司的BELSORP-miniⅡanalyzer全自动比表面积及微孔孔隙分析仪测定催化剂的比表面积和孔结构参数。测试时取110 mg样品,在120 ℃下脱气4 h,脱气完毕后,以N2为吸附质,于−196 ℃下进行测定。采用BET方法计算样品的比表面积[15-16],而计算样品的孔容和孔径采用BJH法。
H2-TPR在FINESORB-3010(浙江泛泰仪器有限公司)全自动物理化学分析仪上进行。将100 mg催化剂置于U型管中,以高纯Ar为载气(30 mL·min−1),10 ℃·min−1升至120 ℃,维持30 min。然后降到50 ℃,将气路切换为10% H2/Ar,等基线稳定后,从50 ℃程序升温至600 ℃,升温的速率为10 ℃·min−1。在升温过程中,利用TCD检测器对耗氢量[15-16]进行检测。O2-TPD的测定也是在FINESORB-3010全自动物理化学分析仪上进行的。将100 mg催化剂置于U型管中,以高纯Ar (30 mL·min−1)为载气,10 ℃·min−1的升温速率升至120 ℃,加热30 min。降温至50 ℃,将气路切换为10% O2/Ar(30 mL·min−1),吸附60 min。然后再把气路切换为高纯Ar,基线稳定后,以10 ℃·min−1程序升温至700 ℃。升温过程中氧的脱附量利用TCD检测器[15-16]进行检测。
拉曼光谱分析(Raman)在法国HJY公司LabRAM Aramis拉曼光谱仪上进行,主要技术参数如下:532 nm激光波长,扫描范围为100~1 000 cm−1,样品每次扫描采集时间为60 s。
2. 实验结果与讨论
2.1 Mn-Ce复合氧化物的可控制备
1)SEM分析。通过水热反应制备了一系列不同锰铈比例的氧化物催化剂,SEM结果如图1所示。以MnSO4·H2O为前驱体、(NH4)2S2O8为氧化剂条件下合成的MnO2呈现出表面为纳米针的海胆状微球结构,其直径为4.0~5.0 μm。而在海胆状MnO2微球制备的基础上,掺入一定比例的铈制备Mn-Ce复合氧化物,其SEM结果如图1(b)~图1(e)所示。当Ce掺杂量为5%时,形成的复合材料的形貌仍为海胆状微球结构,其微球颗粒粒径较纯的MnO2有所增加(7.0~8.5 μm)。但随着Ce掺杂量的增加,产物微球表面的针状结构逐渐消失,变为表面光滑的微球,其颗粒粒径呈现下降趋势,在10%时,微球直径为4.0~5.0 μm,且在微球的周围出现了较多无固定形态的物质(图1(c))。当含量增加到20%时,粒径进一步降低,约为3.59 μm(图1(d))。在含量为40%时,微球的粒径进一步降低至2.2 μm左右(图1(e))。但随着Ce含量的增加,微球的周围也生成了越来越多的表面碎片。同时,在水热反应过程中,通过对生成物产率的观察,发现随着Ce含量的增加,生成产物的量逐步减少,当不添加Mn时,利用Ce(SO4)2·4H2O与(NH4)2S2O8进行水热反应,在80 ℃反应4 h后,只有很少量淡黄色的物质生成,推测为CeO2,但产量极低,所以该方法不适合进行CeO2纳米材料的合成。
 图 1 不同Mn-Ce复合氧化物微球的SEM图Figure 1. SEM images of different Mn-Ce composite oxide microspheres
图 1 不同Mn-Ce复合氧化物微球的SEM图Figure 1. SEM images of different Mn-Ce composite oxide microspheres2)XRD分析。不同Mn-Ce复合氧化物的XRD谱图如图2所示。MnO2微球催化剂XRD谱图中在2θ=37.0°、42.0°,56.7°处出现的特征衍射峰归属于γ-MnO2(PDF#14-0644)[19]。但在所合成的含Ce微球催化剂XRD谱图中除37.0°处的峰没有变化外,其他对应的γ-MnO2特征峰均存在消失的现象。且没有观察到明显的CeO2的峰,这是因为在Ce的含量很低时(小于50%),在复合氧化物中仍以MnOx为主,因而无法探测到CeO2的衍射峰,这与之前研究报道的MnOx-CeO2催化剂所得出的结论[7]相似。
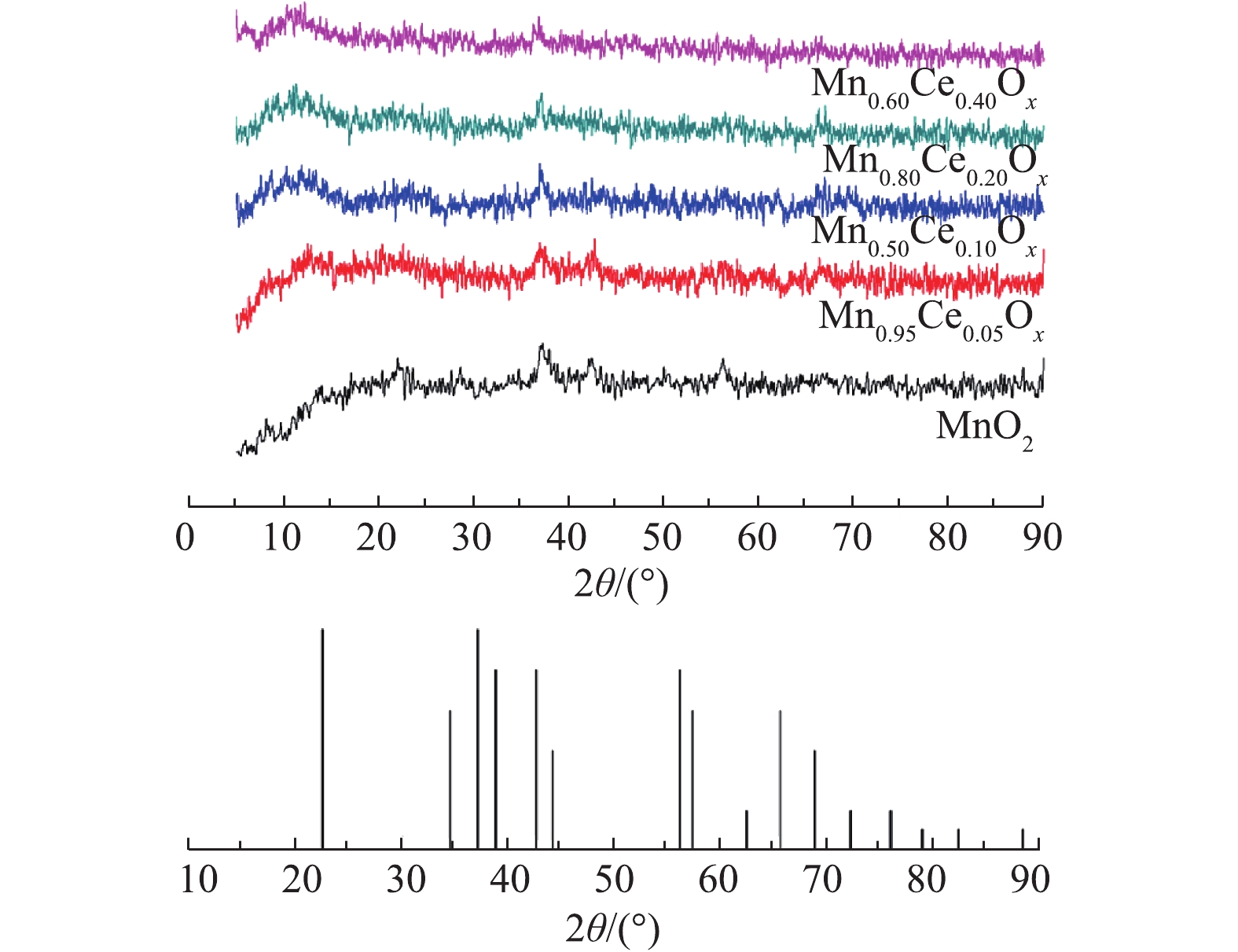 图 2 不同Mn-Ce复合氧化物微球的XRD谱图Figure 2. X-ray diffraction patterns of different Mn-Ce composite oxide microspheres
图 2 不同Mn-Ce复合氧化物微球的XRD谱图Figure 2. X-ray diffraction patterns of different Mn-Ce composite oxide microspheres结合SEM结果可知,在加入Ce以后,在微球的周围出现了大量的颗粒物,说明Ce的加入会对催化剂的形貌产生较大的影响,但从XRD的结果可看出,这些小的颗粒物可能是一些无定型的Mn-Ce复合氧化物。随着Ce含量的增加,2θ=42.0°、56.7°处的衍射峰的强度降低,说明MnO2的结晶度随着Ce含量的增加而下降。
2.2 Mn-Ce复合氧化物催化剂催化氧化甲苯性能
不同Mn-Ce比例的复合氧化物催化降解甲苯的性能如图3所示。由图3(a)可知,所制备的纯MnO2微球对甲苯的降解率随着反应温度上升呈现先降低后升高的趋势。在150 ℃降解率达到76.4%时,但当温度升高至175 ℃时却降低为65.3%,随后随着温度的进一步升高,降解率又开始增加,在200 ℃时,达到了89.8%,在225 ℃达到完全转化。这可能是由于MnO2催化剂在低温区间对甲苯主要为吸附作用,当吸附未达到饱和时,表现为甲苯的出口浓度在下降,因而甲苯的转化率偏高;当这个吸附作用达到饱和时,甲苯的出口浓度达到最低,随着反应温度的升高,吸附在催化剂表面的反应物逐渐脱附,甲苯的出口浓度增大,其降解率下降;当达到一定温度时,催化剂对甲苯的催化反应作用占主导地位,这种作用反应迅速,对甲苯的降解程度大,表现为甲苯的出口浓度迅速下降,因而甲苯的转化率急剧升高[7, 20]。LIAO等[7]和廖银念等[20]的研究也出现了类似的实验现象。
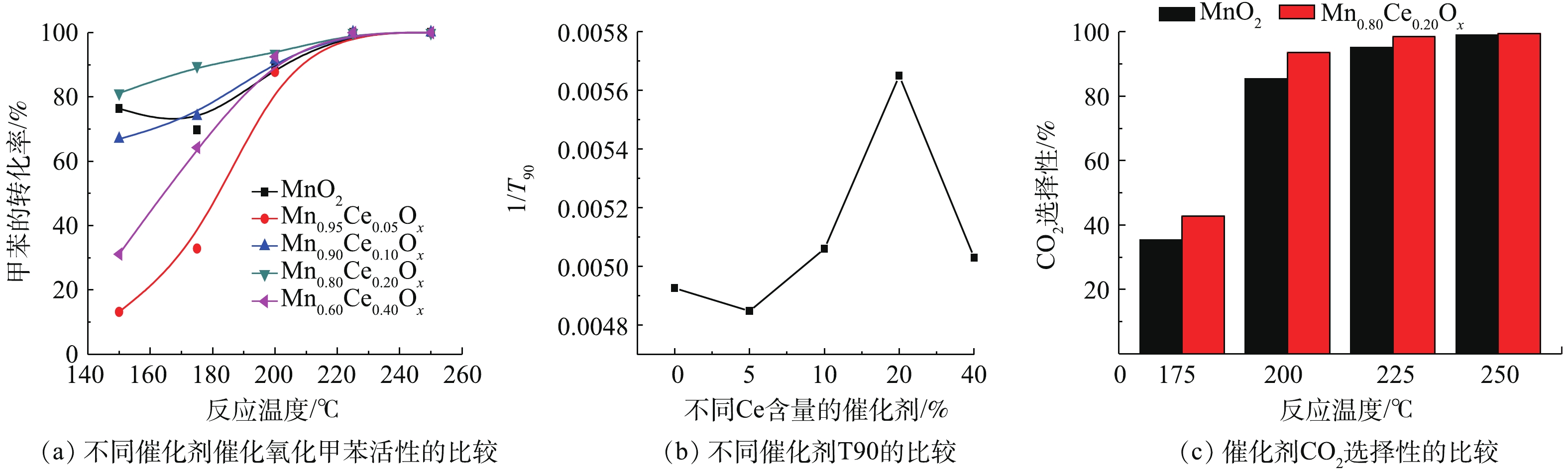 图 3 不同Mn-Ce复合氧化物微球对甲苯催化降解性能Figure 3. Catalytic performance of the different Mn-Ce composite oxide microspheres catalysts on toluene oxidation
图 3 不同Mn-Ce复合氧化物微球对甲苯催化降解性能Figure 3. Catalytic performance of the different Mn-Ce composite oxide microspheres catalysts on toluene oxidation当加入不同含量的Ce后,所形成的Mn-Ce复合氧化物对甲苯的降解率都随着温度的升高而升高,达到完全转化时的温度均为225 ℃,但含Ce的催化剂在催化降解甲苯时未出现甲苯脱附现象。在较低的反应温度区域(<200 ℃),不同的Ce含量的催化剂对甲苯的降解性能具有明显差异,随催化剂中Ce含量的增加呈现先降低后升高再降低的趋势,活性顺序为Mn0.80Ce0.20Ox>Mn0.90Ce0.10Ox>Mn0.60Ce0.40Ox>Mn0.95Ce0.05Ox。大多文献报道用T90(甲苯转化90%所需的温度)来表示催化剂的活性[15-16, 20]。T90越低,表明催化剂的活性越高。为了更直观反映不同Mn-Ce比例的复合氧化物和催化活性的关系,本研究采用插值法估算了各催化剂降解甲苯转化率达90%时所需温度(T90),利用1/T90来表示催化剂的活性,结果如图3(b)所示。结果发现Mn0.80Ce0.20Ox催化剂T90值最低,表明该催化剂具有最佳的低温催化氧化甲苯活性。此外,纯MnO2和Mn0.80Ce0.20Ox催化剂在催化氧化甲苯过程中对CO2的选择性存在一定的差异(图3(c)),Mn0.80Ce0.20Ox催化剂的CO2选择性略优于纯MnO2催化剂,且在反应温度200 ℃后,CO2生成率高于93.5%,由此说明,Mn0.80Ce0.20Ox催化剂实现完全甲苯催化氧化时,甲苯几乎都转化为CO2。
2.3 催化剂的表征
不同Mn-Ce复合氧化物催化剂的比表面积、孔容、孔径的结果见表1。由表1可知,单一MnO2和Mn-Ce复合氧化催化剂的比表面积随着Ce的含量的增加出现先减少后增加最后再减少的趋势。其中比表面积最大和最小的分别为Mn0.90Ce0.10Ox(162.29 m2·g−1)和Mn0.60Ce0.40Ox(68.07 m2·g−1)。催化剂的孔容最大和最小的分别为Mn0.95Ce0.05Ox(0.33 cm3·g−1)和Mn0.80Ce0.20Ox(0.15 cm3·g−1);而催化剂的最大和最小的平均孔径分别为Mn0.60Ce0.40Ox(17.57 nm)和Mn0.80Ce0.20Ox(5.48 nm)。由于甲苯催化氧化性能最优的为Mn0.80Ce0.20Ox催化剂,在所有合成的催化剂中,其具有最小的孔容和平均孔径,且具有适中的比表面积。这说明比表面积等物理性质并不是催化剂在低温催化氧化甲苯反应中起主导作用的影响因素。此外,由图3可知,Mn0.80Ce0.20Ox催化剂的低温催化氧化活性略优于MnO2催化剂,但具有少量Ce的Mn0.90Ce0.10Ox催化剂的活性却明显低于比表面积接近的MnO2催化剂,这可能是由于比表面积等物理性质并不是催化剂在低温催化氧化甲苯反应中起主导作用的因素,催化剂的低温催化氧化甲苯关键因素是催化剂的氧化还原性能和反应过程中的活性氧物种[15]。
表 1 不同Mn-Ce复合氧化物微球催化剂的比表面积、孔容、孔径Table 1. BET surface areas, pore volumes, and pore diameters of different Mn-Ce composite oxide microspheres catalysts催化剂 比表面积/(m2·g−1) 孔容/(cm3·g−1) 平均孔径/nm MnO2 77.97 0.29 14.91 Mn0.95Ce0.05Ox 74.62 0.33 8.19 Mn0.90Ce0.10Ox 162.29 0.26 6.48 Mn0.80Ce0.20Ox 109.54 0.15 5.48 Mn0.60Ce0.40Ox 68.07 0.30 17.57 | Show Table DownLoad:
CSV
DownLoad:
CSV
H2-TPR可以反映催化剂的氧化还原性能,其中氧物种伴随变价金属氧化还原循环与催化氧化反应密切相关[9, 15, 20]。不同比例的Mn-Ce复合氧化物催化剂的H2-TPR结果如图4所示。由此可以看出,纯的MnO2在246.4 ℃和290.4 ℃出现了2个还原峰,分别归属为MnO2→Mn2O3及Mn2O3→Mn3O4的还原,大约在400 ℃的还原峰归属为Mn3O4到MnO的还原峰[21-22]。据报道[7, 18],CeO2向Ce2O3的还原出现在380~450 ℃,而高于650 ℃才可能出现体相氧化铈的还原。深入分析Mn-Ce氧化物催化剂的TPR谱图可知,由于催化剂中Ce含量少,没有出现新的还原峰,说明在催化剂中Ce的还原峰面积比较小,极有可能被Mn2O3→Mn3O4的还原峰所掩盖,但随着Ce含量的增加,MnOx的还原温度向低温偏移。一般而言,催化剂还原峰中心所对应的温度越低,说明催化剂的氧化性越强[23-25],因此,催化剂的氧化性能依次为Mn0.90Ce0.10Ox>Mn0.80Ce0.20Ox>Mn0.95Ce0.05Ox>MnO2>Mn0.60Ce0.40Ox。结合各催化剂催化氧化甲苯活性的结果,可以看出,催化剂的氧化还原性能与其在低温区间催化氧化甲苯的性能存在一定的相关性,也间接说明催化氧化还原能力是影响催化剂催化氧化甲苯性能的重要指标。
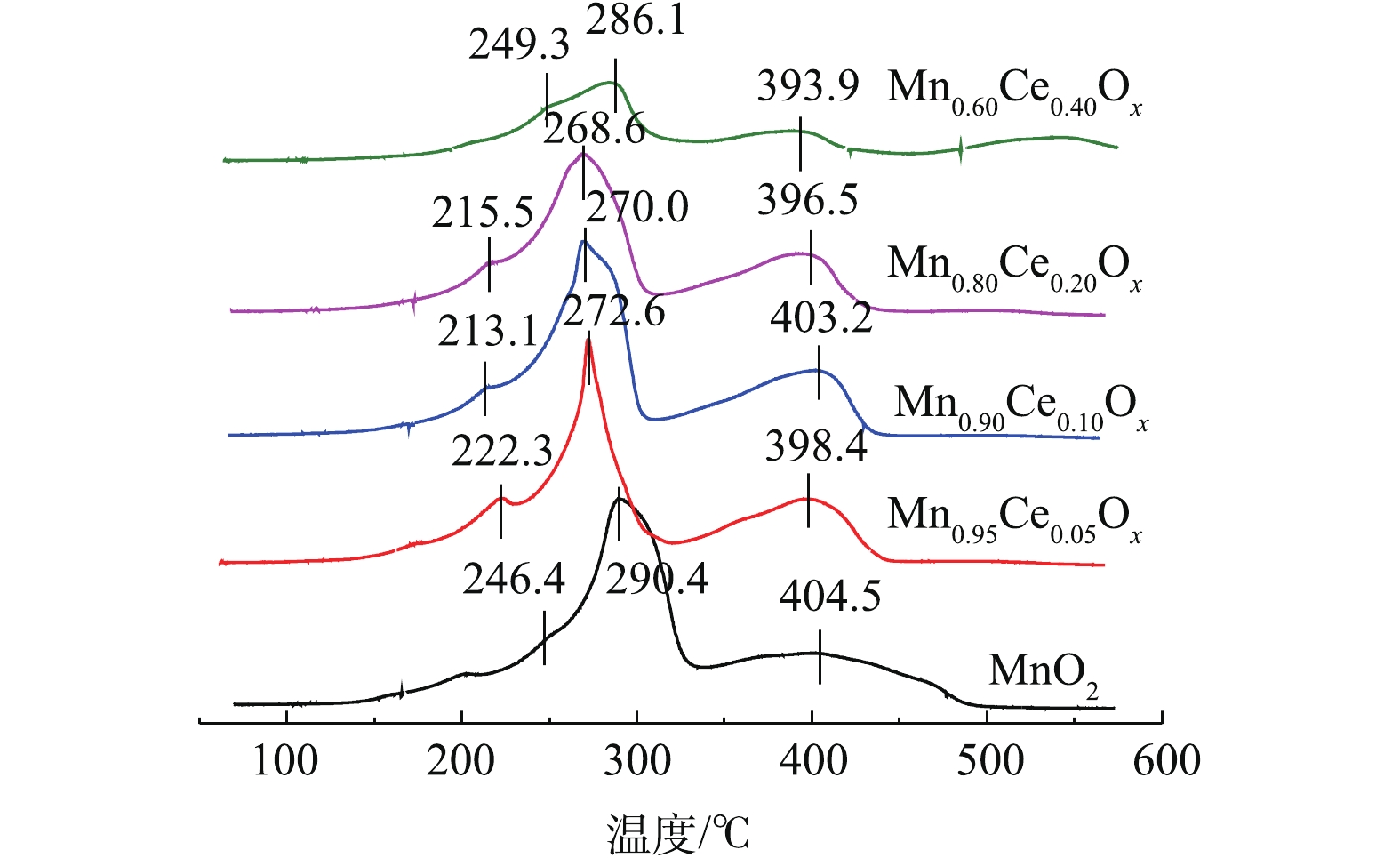 图 4 不同Mn-Ce复合氧化物微球催化剂的H2-TPR谱图Figure 4. H2-TPR profiles of different Mn-Ce composite oxide microspheres catalysts
图 4 不同Mn-Ce复合氧化物微球催化剂的H2-TPR谱图Figure 4. H2-TPR profiles of different Mn-Ce composite oxide microspheres catalystsO2-TPD 可对催化剂表面的氧空位以及物种存在状况[6, 7, 15, 26]进行测定。不同Mn-Ce复合氧化物的O2-TPD的结果如图5所示。其中在80 ℃、300~400 ℃和大于500 ℃出现的氧脱附峰可分别归属于弱的分子吸附氧(
O−2 )、化学吸附氧(O−)和晶格氧O2−的脱附峰[6, 7, 15]。如图5所示,不同比例的Mn-Ce复合氧化物催化剂在300~400 ℃的氧脱附峰峰面积大小依次为Mn0.80Ce0.20Ox>>Mn0.90Ce0.10Ox>Mn0.60Ce0.40Ox>Mn0.95Ce0.05Ox。这与催化氧化甲苯的活性评价结果一致,表明化学吸附氧的含量是低温催化氧化甲苯性能的重要因子。此外,在含Ce微球催化剂的低温催化氧化甲苯的活性存在差异的原因主要包括:1)催化剂还原峰中心所对应的温度越低,说明催化剂的氧化性越强,因此,催化剂的氧化性能依次为Mn0.90Ce0.10Ox>Mn0.80Ce0.20Ox>Mn0.95Ce0.05Ox>MnO2>Mn0.60Ce0.40Ox。结合活性评价的结果,可以看出,催化剂的氧化还原性能与其在低温区间催化氧化甲苯的性能存在一定的相符性;2)催化剂的氧化还原性能接近(Mn0.90Ce0.10Ox和Mn0.80Ce0.20Ox),而低温区间催化氧化甲苯的性能存在差异,这是由于不同比例的Mn-Ce复合氧化物催化剂在300~400 ℃的氧脱附峰峰面积大小依次为:Mn0.80Ce0.20Ox>>Mn0.90Ce0.10Ox>Mn0.60Ce0.40Ox>Mn0.95Ce0.05Ox。这与催化氧化甲苯的活性评价结果一致,表明化学吸附氧的含量是低温催化氧化甲苯性能的重要因子,这些化学吸附氧物种对于完成催化氧化循环起着关键作用。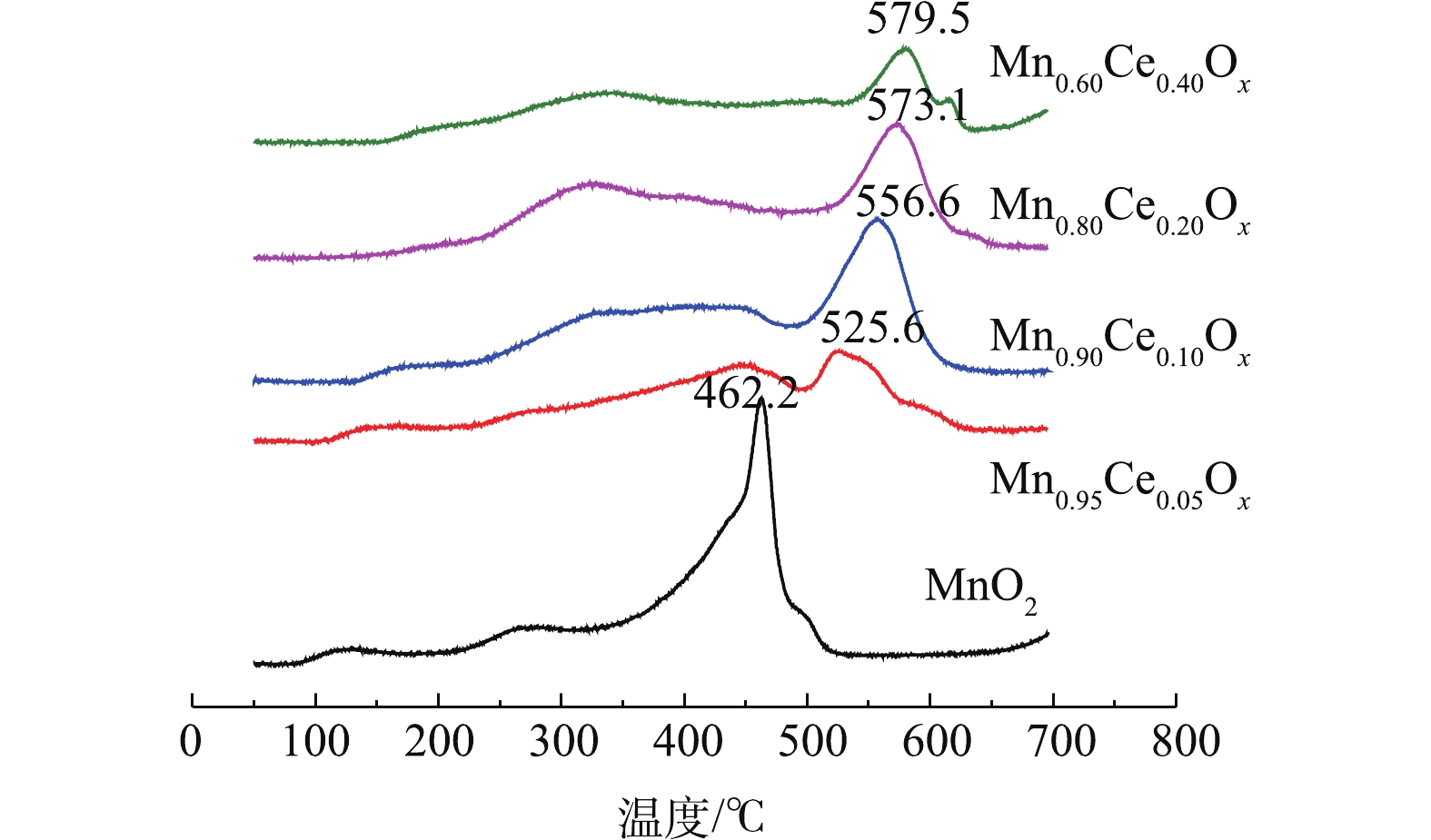 图 5 不同Mn-Ce复合氧化物微球催化剂的O2-TPD谱图Figure 5. O2-TPD profiles of different Mn-Ce composite oxide microspheres catalysts
图 5 不同Mn-Ce复合氧化物微球催化剂的O2-TPD谱图Figure 5. O2-TPD profiles of different Mn-Ce composite oxide microspheres catalysts图6是不同Mn-Ce复合氧化物微球催化剂的拉曼光谱图。其中纯MnO2和Mn-CeOx催化剂中都出现了500~700 cm−1的特征峰,该峰被认为是MnO2中[MnO6]八面体的伸缩振动,且纯MnO2所对应的特征峰正好与γ-MnO2的特征峰完全吻合[27],这一表征结果也更好地验证了前面的XRD结论。值得注意的是,与纯MnO2所对应的特征峰相比,Mn0.80Ce0.20Ox催化剂所对应的2个特征峰(509 cm−1和563 cm−1)及Mn0.60Ce0.40Ox催化剂所对应的1个特征峰(554 cm−1)都明显向低波数偏移,这表明Mn0.60Ce0.40Ox和Mn0.80Ce0.20Ox催化剂中都可能存在Mn-Ce固溶体,这与文献报道的结论[28]相符。而具有较低Ce含量的Mn0.90Ce0.10Ox和Mn0.95Ce0.05Ox所对应的特征峰并没有出现明显偏移现象,这说明当Ce掺杂量较低时,Mn-Ce催化剂中未形成固溶体。正由于Mn0.80Ce0.20Ox催化剂中形成了固溶体,使催化剂具有更优异的储氧能力,这与O2-TPD分析得出的该催化剂具有最高含量的化学吸附氧的结论相符。因此,Mn-Ce催化剂中存在固溶体也是其具有优异催化氧化甲苯性能的关键原因之一。值得一提的是,Mn0.60Ce0.40Ox催化剂中虽然也存在固溶体,但Mn0.80Ce0.20Ox催化剂具有比Mn0.60Ce0.40Ox催化剂更强的氧化还原性能和更高的化学吸附氧的含量,故Mn0.80Ce0.20Ox催化剂呈现更优异的低温催化氧化甲苯性能。
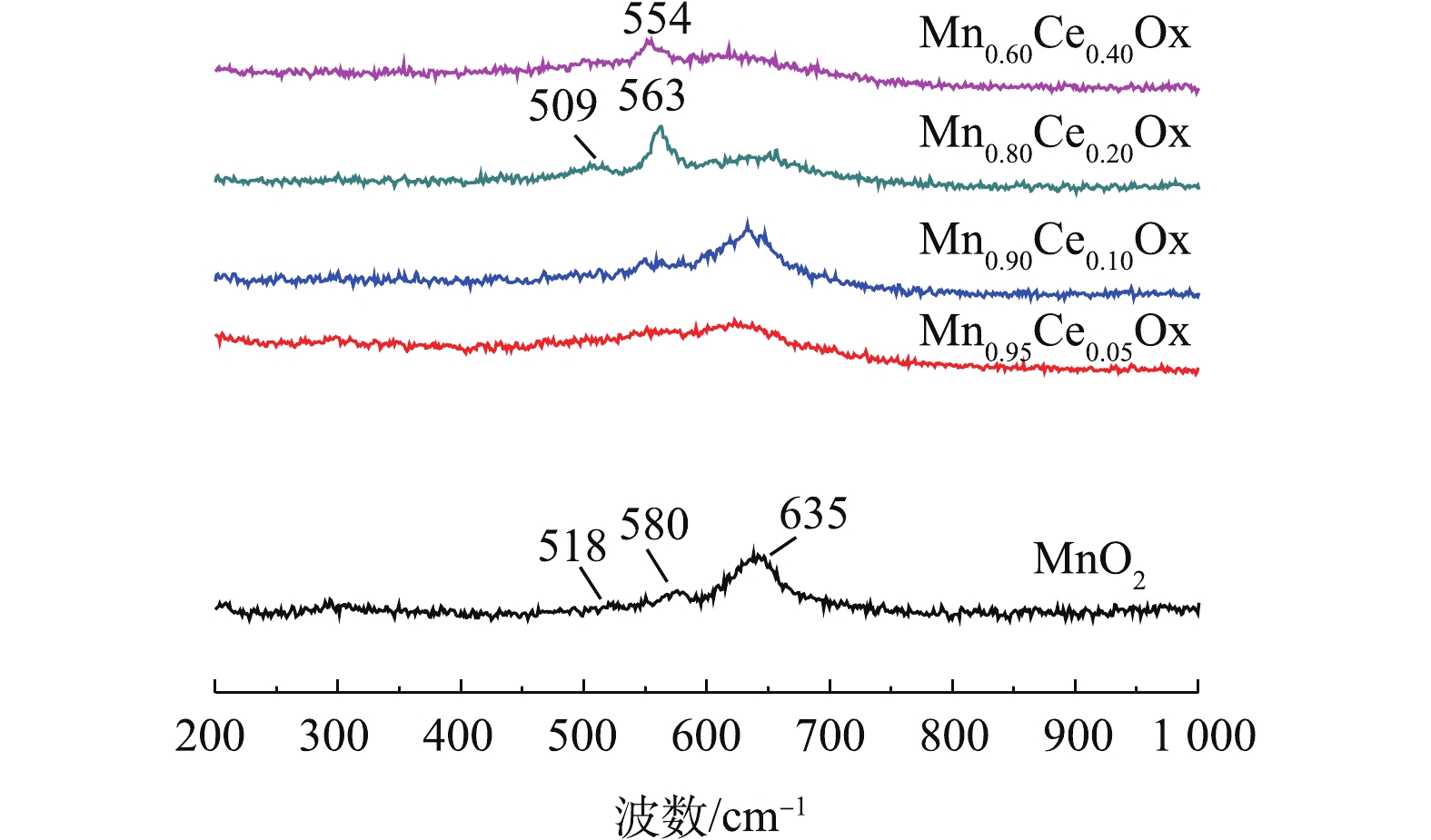 图 6 不同Mn-Ce复合氧化物微球催化剂的拉曼光谱图Figure 6. Raman spectra of different Mn-Ce composite oxide microspheres catalysts
图 6 不同Mn-Ce复合氧化物微球催化剂的拉曼光谱图Figure 6. Raman spectra of different Mn-Ce composite oxide microspheres catalysts3. 结论
1)采用简单的水热法成功合成出了Mn-Ce氧化物微球,且铈摩尔含量占5%、10%和20%的产物结晶都比较完整,而铈摩尔含量为20%时,其形貌为较光滑的微球,结晶较完整,颗粒尺寸较小。MnO2微球催化剂中存在γ-MnO2的特征峰,而随着Ce的加入,催化剂的结晶度有所下降。
2)Mn0.80Ce0.20Ox催化剂呈现最佳的催化氧化甲苯性能,较强的氧化还原性能和较高的化学吸附氧的含量是其具有优异的低温催化氧化甲苯性能的重要原因。
3)Mn0.80Ce0.20Ox催化剂中存在Mn-Ce固溶体,这也是其具有最佳催化氧化甲苯性能的关键原因之一。
-
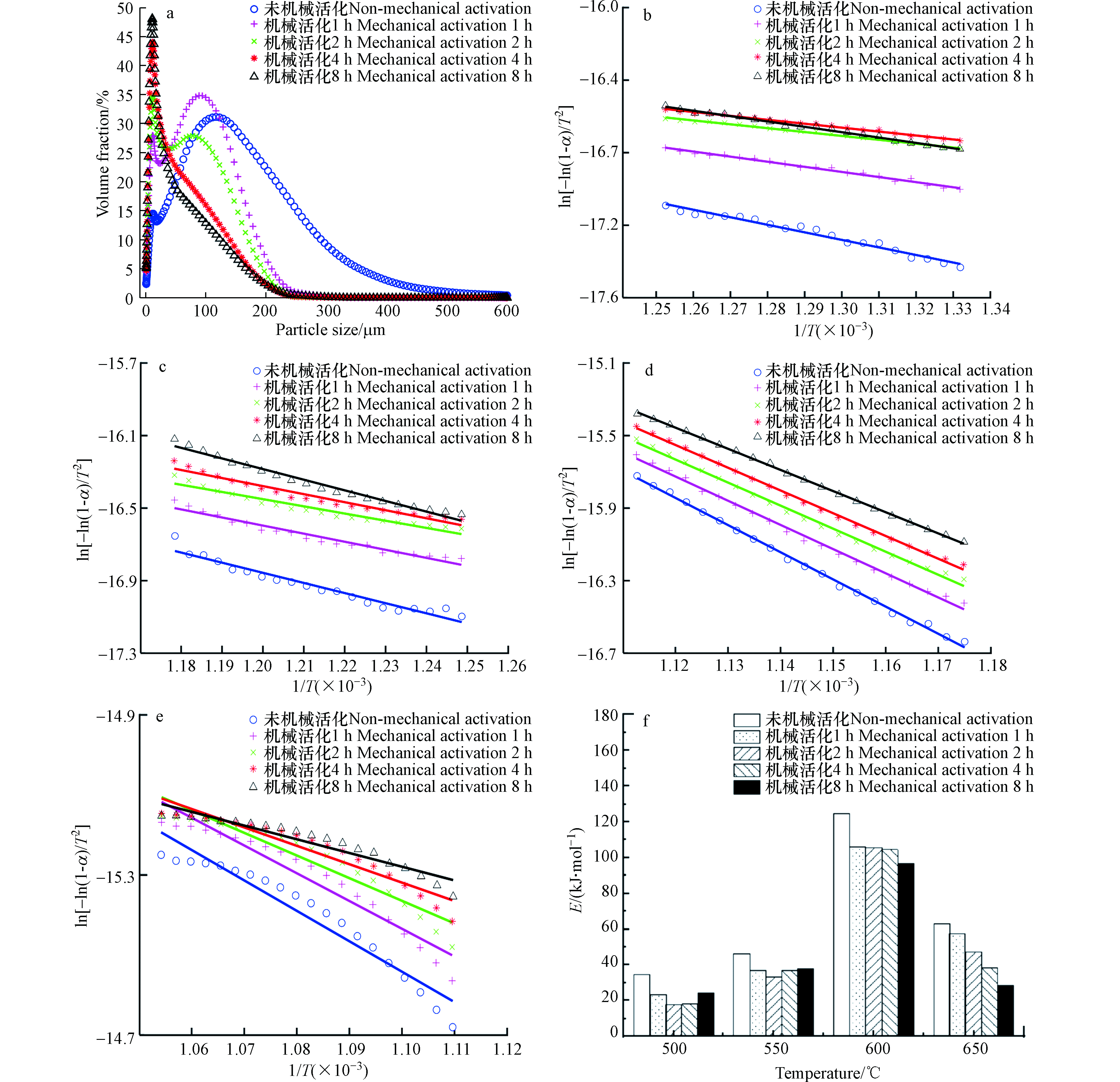
图 2 机械活化对气化细渣粒度及反应动力学的影响
Figure 2. Effect of mechanical activation on particle size and reaction kinetics of gasification fine slag
表 1 细渣中主要元素及组成成分
Table 1. The main elements and components of fine slag
组成成分 Composition SiO2 Al2O3 Fe2O3 CaO K2O MgO Na2O TiO2 SO3 SrO 其它 质量分数/% Quality score 51.88 15.93 10.34 10.14 2.94 2.79 1.76 1.26 1.12 0.48 1.36
下载: 导出CSV
表 2 细渣中部分重金属元素含量
Table 2. Content of main heavy metal elements of fine slag
元素 Element Cd Cr Pb Zn Cu Hg 含量/(mg·kg−1) Content <0.05 47.88 69.47 64 29 0.004
下载: 导出CSV
-
[1] WU S, HUANG S, JI L, et al. Structure characteristics and gasification activity of residual carbon from entrained-flow coal gasification slag [J]. Fuel, 2014, 122: 67-75. doi: 10.1016/j.fuel.2014.01.011 [2] WU S, HUANG S, WU Y, et al. Characteristics and catalytic actions of inorganic constituents from entrained-flow coal gasification slag [J]. Journal of the Energy Institute, 2015, 88(1): 93-103. doi: 10.1016/j.joei.2014.04.001 [3] 曲江山, 张建波, 孙志刚, 等. 煤气化渣综合利用研究进展 [J]. 洁净煤技术, 2020, 26(1): 184-193. QU J S, ZHANG J B, SUN Z G, et al. Research progress on comprehensive utilization of coal gasification slag [J]. Clean Coal Technology, 2020, 26(1): 184-193(in Chinese).
[4] STEFFAN J J, BREVIK E C, BURGESS L C, et al. The effect of soil on human health: an overview [J]. European Journal of Soil Science, 2018, 69(1): 159-171. doi: 10.1111/ejss.12451 [5] 赵永彬, 吴辉, 蔡晓亮, 等. 煤气化残渣的基本特性研究 [J]. 洁净煤技术, 2015, 21(3): 110-113. ZHAO Y B, WU H, CAI X L, et al. Basic characteristics of coal gasification residue [J]. Clean Coal Technology, 2015, 21(3): 110-113(in Chinese).
[6] 王金福, 唐强, 郑妍妍, 等. 一种煤气化灰渣氧化脱碳的组合循环流化床反应器: CN105396518 A[P].[2016-03-16]. WANG J F, TANG Q, ZHENG Y Y, et al. Combined circulating fluidized bed reactor for oxidative decarburization of coal gasification ash: CN105396518 A[P]. [2016-03-16] (in Chinese).
[7] 刘坤基. 气化细渣中残碳催化石墨化研究[D]. 北京: 中国矿业大学, 2019. LIU K J. Study on catalytic graphitization of residual carbon in gasification slag[D]. Beijing: China University of Mining and Technology, 2019 (in Chinese).
[8] ZHAO S W, YAO L Y, HE H B, et al. Preparation and environmental toxicity of non-sintered ceramsite using coal gasifi cation coarse slag [J]. Archives of Environmental Protection, 2019, 45(2): 84-90. [9] 陈志良, 陆胜勇, 毛琼晶, 等. 水平式球磨机用于POPs机械化学处置的能量传递 [J]. 环境化学, 2016, 35(10): 2134-2145. doi: 10.7524/j.issn.0254-6108.2016.10.2016031603 CHEN Z L, LU S Y, MAO Q J, et al. Energy transfer in mechanochemical treatment of POPs in a horizontal ball mill [J]. Environmental Chemistry, 2016, 35(10): 2134-2145(in Chinese). doi: 10.7524/j.issn.0254-6108.2016.10.2016031603
[10] BASTURKCU H, ACARKAN N, GOCK E. The role of mechanical activation on atmospheric leaching of a lateritic nickel ore [J]. International Journal of Mineral Processing, 2017, 163: 1-8. doi: 10.1016/j.minpro.2017.04.001 [11] 朱红青, 沈静, 张亚光. 升温速率和氧浓度对煤表观活化能的影响 [J]. 煤炭科学技术, 2015, 43(11): 49-53,106. ZHU H Q, SHEN J, ZHANG Y G. Temperature rising rates and oxygen concentrations influerced to apparent activation energy of coal [J]. Coal Science and Technology, 2015, 43(11): 49-53,106(in Chinese).
[12] GAO J C, CHANG M R, SHEN J. Comparison of bituminous coal apparent activation energy in different heating rates and oxygen concentrations based on thermo gravimetric analysis [J]. Journal of Thermal Analysis and Calorimetry, 2017(130): 1181-1189. [13] YU L Y, LI P S. Thermogravimetric analysis of coal and sludge co-combustion with microwave radiation dehydration [J]. Energy Institute, 2014,87(87): 220-226. [14] 徐东耀, 肖佩林, 庄亚辉, 等. 型煤燃烧特性初探 [J]. 环境化学, 1998, 17(6): 537-541. XU D Y, XIAO P L, ZHUANG Y H, et al. Preliminary study on combustion characteristics of briquette [J]. Environmental Chemistry, 1998, 17(6): 537-541(in Chinese).
[15] YOUSAF B, LIU G, ABBAS Q, et al. Systematic investigation on combustion characteristics and emission-reduction mechanism of potentially toxic elements in biomass and biocharcoal combustion systems [J]. Applied Energy, 2017, 208: 142-157. doi: 10.1016/j.apenergy.2017.10.059 [16] 吴思萍, 赵凯, 董永胜, 等. 气化细渣浮选脱碳研究进展 [J]. 华电技术, 2020, 42(7): 81-86. doi: 10.3969/j.issn.1674-1951.2020.07.011 WU S P, ZHAO K, DONG Y S, et al. Research progress on flotation decarburization of gasified fine slag [J]. Huadian Technology, 2020, 42(7): 81-86(in Chinese). doi: 10.3969/j.issn.1674-1951.2020.07.011
[17] TANG A, SU L, LI C, et al. Effect of mechanical activation on acid leaching of kaolin residue [J]. Applied Clay Science, 2010, 48(3): 296-299. doi: 10.1016/j.clay.2010.01.019 [18] LI X H, ZHANG Y J, PAN L P, et al. Effect of mechanical activation on dissolution kinetics of neutral leach residue of zinc calcine in sulphuric acid [J]. Transactions of Nonferrous Metals Society of China, 2013, 23(5): 1512-1519. doi: 10.1016/S1003-6326(13)62624-2 [19] LIAO Z, HUANG Z, HU H, et al. Microscopic structure and properties changes of cassava stillage residue pretreated by mechanical activation [J]. Bioresour Technology, 2011, 102(17): 7953-7958. doi: 10.1016/j.biortech.2011.05.067 [20] ZHOU X, WANG R, LI C, et al. Effect of high energy ball milling on the microstructure and properties of ultrafine gradient cemented carbides [J]. International Journal of Applied Ceramic Technology, 2020, 17(5): 2298-2306. doi: 10.1111/ijac.13551 [21] 邓军, 张宇轩, 赵婧昱, 等. 基于程序升温的不同粒径煤氧化活化能试验研究 [J]. 煤炭科学技术, 2019, 47(1): 214-219. DENG J, ZHANG Y X, ZHAO J Y, et al. Experiment study on oxidation and activated energy of different partical sizecoal based on programmed temperature rising [J]. Coal Science and Technology, 2019, 47(1): 214-219(in Chinese).
[22] WANG H, DLUGOGORSKI B Z, KENNEDY E M. Theoretical analysis of reaction regimes in low-temperature oxidation of coal [J]. Fuel, 1999, 78(9): 1073-1081. doi: 10.1016/S0016-2361(99)00016-2 [23] VESELOVSKI V S, TERPOGOSOVA E A. Dependence of oxidation of mineral fuels on temperature [J]. Otdl Tech Nauk, 1953, 4: 905-909. [24] 张玉龙. 基于宏观表现与微观特性的煤低温氧化机理及其应用研究[D]. 太原: 太原理工大学, 2014. ZHANG Y L. Study on low-temperature oxidation mechanism of coal based on macroscopic behavior and microscopic characteristics and its application[D]. Taiyuan: Taiyuan University of technology, 2014 (in Chinese).
[25] KOK M V. Simultaneous thermogravimetry–calorimetry study on the combustion of coal samples: Effect of heating rate [J]. Energy Conversion and Management, 2012, 53(1): 40-44. doi: 10.1016/j.enconman.2011.08.005 [26] MENG F, YU J, TAHMASEBI A, et al. Pyrolysis and combustion behavior of coal gangue in O2/CO2 and O2/N2 mixtures using thermogravimetric analysis and a drop tube furnace [J]. Energy & Fuels, 2013, 27(6): 2923-2932. [27] LI T, SUN T, LI D. Preparation, sintering behavior, and expansion performance of ceramsite filter media from dewatered sewage sludge, coal fly ash, and river sediment [J]. Journal of Material Cycles and Waste Management, 2016, 20(1): 71-79. [28] QIN J, CUI C, CUI X, et al. Preparation and characterization of ceramsite from lime mud and coal fly ash [J]. Construction and Building Materials, 2015, 95: 10-17. doi: 10.1016/j.conbuildmat.2015.07.106 [29] CHEN Y, SHI J, RONG H, et al. Adsorption mechanism of lead ions on porous ceramsite prepared by co-combustion ash of sewage sludge and biomass [J]. Science of the Total Environment, 2020, 702: 135017. doi: 10.1016/j.scitotenv.2019.135017 [30] XU G R, ZOU J L, LI G B. Stabilization of heavy metals in sludge ceramsite [J]. Water Research, 2010, 44(9): 2930-2938. doi: 10.1016/j.watres.2010.02.014 [31] CHENG F, WEN R, HUANG Z, et al. Preparation and analysis of lightweight wall material with expanded graphite (EG)/paraffin composites for solar energy storage [J]. Applied Thermal Engineering, 2017, 120: 107-114. doi: 10.1016/j.applthermaleng.2017.03.129 [32] 谢武明, 张文治, 周峰平, 等. 煤粉发泡剂对赤泥陶粒烧胀特性的影响 [J]. 环境工程学报, 2017, 11(12): 6458-6464. doi: 10.12030/j.cjee.201701130 XIE W M, ZHANG W Z, ZHOU F P, et al. Effect of pulverized coal on sintering expansion behavior of red mud ceramsite [J]. Chinese Journal of Environmental Engineering, 2017, 11(12): 6458-6464(in Chinese). doi: 10.12030/j.cjee.201701130
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 5370
- HTML全文浏览数: 5370
- PDF下载数: 85
- 施引文献: 0




