




-
NOx是有害的大气污染物之一,普遍存在于烟气中,其中汽车尾气和固定污染源为关键目标污染物,它对环境造成了酸雨、光化学烟雾、臭氧层空洞等诸多问题[1-3]. 目前,选择性催化还原技术(SCR)是最主要脱除NOx的方法,而SCR催化剂是该技术的核心要素[4-5]. 由于传统的V2O5-WO3/TiO2催化剂最佳脱硝温度窗口为中高温区间 (300—400 ℃),难以直接适用于众多非电行业的中低温(< 280℃). 此外,由于金属钒为危险废物,废弃后危害环境且不易处理,限制了其工业应用范围[6-7]. 因此,对于非钒系中低温SCR脱硝催化剂的研究与开发成为近十年来的热点.
近年来,负载型锰系催化剂由于在低温下具有较高的催化活性被广泛研究[8-9]. 有大量文献记载锰的氧化形态、制备方法等对锰系催化剂的催化性能影响显著[10-11]. Yu等[12]使用改进的分子设计分散法制备了MnOx/SAPO-34低温SCR催化剂,在160 ℃具有近90%的NOx转化率. 并且该催化剂表面MnOx在载体表面均匀分布,且具有较大的比表面积、Mn4+含量以及丰富的酸性位点. Zuo等[13]采用不同方法合成了几种负载于热改性白云石-坡缕石(MnOx/M-DPC)上的氧化锰催化剂,研究了制备方法对NH3在低温条件下选择性催化还原NOx的影响. 实验结果表明,沉淀法制备样品的催化活性比浸渍法和柠檬酸法更高,在120 ℃时NOx转化率可达83%.
累托石(REC)作为一种特殊的规则粘土矿物,具有二维片状结构,由二八面体云母层(不可膨胀) 和二八面体蒙脱石层(可膨胀)按1∶1的比例堆叠而成[14]. 云母层决定了该矿物的结构稳定性和热稳定性,而蒙脱石层则赋予了该矿物材料的结构可变性. 层间丰富的硅羟基使其可与绝大多数金属阳离子载体产生键合作用,并且具有很好的离子交换性能. 同时,这种天然硅酸盐矿物由于具有比表面积大,稳定性好,表面电位粗糙以及成型后抗压强度高等优势,可作为负载型金属氧化物催化剂的载体材料[15-16].
目前,在各类载体上负载MnOx制备低温SCR脱硝催化剂的方法主要有等体积浸渍法[17],共沉淀法[18],溶胶-凝胶法[19]等. 文献表明,制备方法对催化剂的脱硝性能有很大影响,并且不同类型的载体与催化剂制备方法间存在显著的匹配效应,这表明活性组分的负载与载体结构密切相关[20]. 而对于催化剂制备方法的研究目前大多数文献报道集中于传统的零维微尺度结构载体,如TiO2、Al2O3等[21-22]. 对于二维片层结构载体上制备方法对催化剂结构及性能的影响研究较少.
本文以累托石为载体,采用等体积浸渍法、共沉淀法和实验室前期研究中开发的原位生长法[23-24]制备锰基累托石NH3-SCR催化剂,考察不同负载方法对催化剂微观结构及其低温SCR脱硝性能的影响,探索不同制备方法下锰氧化物在载体表面的生成机理及活性组分与载体间协同效应的物理化学本质.
-
累托石(REC,湖北钟祥名流累托石开发有限公司),主要化学组成为(质量分数):42.02%. SiO2,28.95% Al2O3,4.11% CaO,2.32% Na2O,1.39% K2O;乙酸锰 Mn(CH3COO)2;高锰酸钾KMnO4;碳酸铵(NH4)2CO3;均由国药集团化学试剂有限公司提供,纯度为AR,模拟烟气由南京上元工业气体厂提供.
-
等体积浸渍法:称取20 g累托石粉末,加入溶解好的乙酸锰溶液(浓度为17.82—53.45 g·mL−1,固液比为1∶1),用玻璃棒均匀搅拌后于室温下浸渍24 h. 再将其置于干燥箱中60 ℃干燥6 h、110 ℃干燥12 h,最后将干燥后的样品于马弗炉中300 ℃煅烧3 h,所得催化剂记为Mnα/REC-IP(其中α表示Mn元素所占催化剂的质量分数(%),下同).
共沉淀法:将20 g累托石粉末加入完全溶解的乙酸锰溶液(浓度为17.82—53.45 g·mL−1,固液比为1∶1),用玻璃棒搅拌均匀后浸渍24 h. 再取一定量的碳酸铵完全溶解,与预浸渍好的累托石乙酸锰溶液充分混合,搅拌均匀. 室温下浸渍24 h后,用去离子水抽滤洗涤6—8次. 置于干燥箱中60 ℃干燥6 h、110℃干燥12 h,最后将干燥后的样品于马弗炉300 ℃中煅烧3 h后制得催化剂,记为Mnα/REC-CP.
原位生长法:取称量好的乙酸锰与高锰酸钾分别完全溶解 摩尔比为3∶2),再称取20 g累托石粉末加入溶解好的乙酸锰溶液中,固液比为1∶1,用玻璃棒搅拌均匀后浸渍24 h. 将浸渍好的累托石乙酸锰溶液与高锰酸钾溶液充分混合,搅拌均匀.室温下浸渍24 h后,用去离子水多次抽滤洗涤. 置于干燥箱中60 ℃干燥6 h、110 ℃干燥12 h后制得催化剂. 催化剂标记为Mnα/REC-SP.
-
催化剂的脱硝活性评价测试在高80 cm,内径15 mm的竖式石英玻璃常压固定床反应器中进行,活性评价装置由模拟烟气(1000 mg·L−1 NO、1000 mg·L−1 NH3、3% O2、N2为平衡气混合而成,总流量为350 mL·min−1),固定床反应器和分析检测仪器3大部分组成. 反应空速为105000 h−1,反应温度为100—300 ℃. 反应器进出口烟气组成采用德国MRU(名优)OPTIMA 7烟气分析仪进行在线测量. 催化剂的NH3-SCR脱硝性能评价装置如图1所示.
催化剂的活性计算NO转化率(XNO):
式中,[NO]in、[NO]out分别是NO进口、出口浓度.
-
采用D/max2500V(Rigaku,Japan)仪器测定催化剂的晶型结构,测试条件为:Cu Kα射线,管电流为100 mA,管电压为40 kV,扫描速率为4o∙min−1,测试范围为2θ=5o—90o. 采用新型的热场发射扫描电子显微镜(SU8020,Hitachi,Japan)观察催化剂的表面微观结构,电压范围为0. 1—30 kV. 采用日本电子制造JEM-2100F型场发射透射电子显微镜对催化剂的微观形貌进行表征. 采用管电压为1486.6 eV,ESCALAB250型号X射线光电子能谱仪进行测试,测试条件为Al Kα射线,1486.6 eV的管电压. 采用全自动Quantachrome Nove(2200e)比表面积测定仪来测定样品的比表面积、孔容和孔径. 采用质谱在线测试(Hiden QIC-200)催化剂对反应气体NH3的吸附活化能力. 具体步骤为:将0.5 g催化剂放入石英反应管中,首先在120 ℃恒温下通入N2吹扫,吹扫结束后,使反应体系降温至50 ℃并保持50 ℃恒温,通入1000 mg·L−1 NH3进行吸附(N2与NH3总流量为100 mL∙min−1),至基线平稳后关闭气体并将N2流量调至100 mL∙min−1吹扫除去未吸附气体. 最后,从50 ℃开始进行脱附,升温速率为5 ℃∙min−1,终止温度为900 ℃.
-
催化剂的活性组分负载量与其脱硝活性密切相关,负载量的过高与过低会导活性组分在催化剂表面的分布,进而影响其催化活性[25]. 图2(a)—(c)为锰负载量对Mnα/REC-IP, Mnα/REC-CP和Mnα/REC-SP催化剂脱硝活性的影响. 实验过程温度控制在100—300 ℃,每个温度点保温30—40 min,每25 ℃测3次NO出口浓度,分别在保温时间剩余16、8、2 min时进行测试,取3次测量结果的平均值作为每个温度点最终NO出口浓度,且测量误差均控制在1%左右,整个活性测试测试持续约8 h. 同时,每个样品的活性测试过程重复2—3次,取测量的平均值作为每个温度点的最终NO转化率,测量误差控制在2%左右. 从图2可以看出,REC在100—200 ℃温度区间内NO转化率几乎为0,使用不同制备方法引入Mn后,催化剂的NO转化率明显提升. 对比3种制备方法在Mn负载量为4%—12%的NO转化率,如图(a)—(c)所示. 其中在Mn负载量为10%时,Mn10/REC-CP和Mn10/REC-SP催化剂的NO转化率最高. 因此,在固定α=10的情况下对比3种催化剂的NO转化率(图2d),Mn10/REC-IP催化剂的NO转化率明显较低,在100—200 ℃温度范围内无催化活性,200—300 ℃温度范围内NO转化率逐渐提升. 相反地,Mn10/REC-CP和Mn10/REC-SP催化剂在150—250 ℃的温度范围内NO转化率可达98%以上,而Mn10/REC-SP催化剂的NO转化率略高于Mn10/REC-CP催化剂. 因此,为深入探究不同制备方法下Mn/REC催化剂NO转化率不同的原因,对REC及3种催化剂进行了如下表征.
-
图3为REC及3种不同制备方法下催化剂的XRD谱图. 其中,2θ=7.24°、19.94°、28.35°、36.81°、62.17°为REC的特征衍射峰(PDF#29-1495) . 2θ=7.24°的002晶面的衍射峰强度最强,这是具有高度有序和取向硅酸盐层结构的累托石所必需的[26]. 同时,从XRD谱图中可以看出,负载MnOx后3种催化剂均仍呈现出典型REC的晶型结构,这表明Mn的引入并未破坏REC的片层结构. 但3种催化剂样品与原始REC相比,REC整体特征衍射峰强度有所下降. 其中REC 002晶面的特征衍射峰下降的最为明显,这是由于MnOx生长在REC表面削弱了其原衍射峰强度. 因此,可初步说明活性组分MnOx成功负载在REC上[27].
-
为了直观地研究制备方法对催化剂表面负载MnOx形态的影响,对REC及催化剂进行SEM表征,如图4所示. 由图4(a)可以看出,REC表面较为光滑平整,由不同形状的单一片层堆叠而成. 采用不同制备方法引入Mn后REC整体片层状结构并未被破坏,而MnOx呈不同形态分散在REC表面.图4(b)中Mn10/REC-IP催化剂表面活性组分呈颗粒状分布在REC表面且表面依旧光滑平整. 相反地,Mn10/REC-CP和Mn10/REC-SP催化剂表面MnOx则呈团簇状分布在REC片层与片层间的空隙中,片层间的间隙被略微打开且表面较为粗糙. 可见,不同制备方法下REC与MnOx结合方式不同,并且MnOx呈不同形态与REC相结合.
-
为进一步观察不同制备方法下催化剂表面局部MnOx分布情况,对4个样品进行了TEM以及EDS表征,如图5所示. 从图5(a)和(e)可以清晰地看出REC是由片层堆积而成的层状结构,在采用不同制备方法引入Mn后,催化剂表面MnOx的分散度以及颗粒形态不同 (图b—d) . 其中,图(b)、(f)、(i)中Mn10/REC-IP催化剂上MnOx呈块状团聚分布于累托石片层上,并表现出小颗粒的团聚现象,这与SEM结果表现一致. 而在图(c)、(g)、(j)和图(d)、(h)、(k)中可以看到,Mn10/REC-CP和Mn10/REC-SP催化剂表面生长有海绵状的MnOx,且十分均匀地生长在REC局部表面. 同时,结合催化剂的脱硝性能图可以看出,活性组分MnOx在REC表面的分散程度对催化剂的SCR脱硝性能影响较大,MnOx在REC表面分散度越高越有利于催化活性的提升,而催化剂表面活性组分的团聚结块现象则会显著降低其催化活性.
-
为了进一步说明不同制备方法对催化剂脱硝性能的影响,对REC及3种催化剂的比表面积、孔体积和孔径进行分析,结果如表1所示. 可以看出,REC在采用不同制备方法负载活性组分后呈不同程度地增大或减小,REC及催化剂的比表面积大小排序为:Mn10/REC-SP(62 m2∙g−1)> Mn10/REC-CP (52 m2∙g−1)> REC(18 m2∙g−1)> Mn10/REC-IP(12 m2∙g−1). 其中,Mn10/REC-IP催化剂相对于原始累托石比表面积和孔体积均有明显降低. 结合TEM可知,这是由于Mn10/REC-IP催化剂表面活性组分分散不均匀且发生明显的团聚现象,使得表面孔道结构堵塞所致. 相反地,其他2种催化剂的比表面积和孔体积均有明显提升,这与其表面活性组分载体表面的分布及颗粒大小密切相关. 结合SEM和TEM可以看出,Mn10/REC-SP和Mn10/REC-CP 催化剂中REC与MnOx的结合使海绵状MnOx生长在片层间,使催化剂的比表面积和孔体积均明显增加. 同时,结合催化活性测试结果可证实,这种较大的比表面积和孔容,更有利于反应过程中的物理传质过程,从而直接促进了催化剂的SCR催化性能[28].
-
NH3-TPD验证了REC及3种催化剂的表面酸性强度,它是影响催化剂低温NH3-SCR催化活性的重要因素之一. REC和Mn/REC的NH3-TPD结果如图6所示. REC在400—800 ℃存在很强的解吸峰,这归属于强酸性位点上的配位NH3的解吸,热稳定性较强[29]. 但是,在采用不同制备方法负载活性组分MnOx后,3种催化剂样品在强酸性位点上对配位NH3解吸温度明显降低. 这是由于负载MnOx后与REC的相互作用改变了该峰的位置,使强酸性位点上吸附的配位NH3在较低的温度下被脱附,热稳定性降低. 同时,在100—350 ℃新增出不同面积大小的峰,该位置的峰主要归因于催化剂表面MnOx弱酸性位点Lewis酸位点对NH3的解吸,这些位点对催化剂的低温活性起着重要作用[30- 31],其大小顺序为:Mn10/REC-SP > Mn10/REC-CP > Mn10/REC-IP. 而其解吸峰面积与酸性位点数量成正比,峰面积越大,NH3吸附量越大,催化剂上的酸性位点越多,催化活性越好[32]. 可见Mn10/REC-SP催化剂在100—350℃区间内具有最多的酸性位点,NH3吸附量最大,催化剂的低温催化活性最高,Mn10/REC-CP次之,Mn10/REC-IP峰面积最小,酸性位点最少,因此催化活性明显最低. 这与催化剂的脱硝活性图结果表现一致,是其具有较高低温活性的重要因素之一.
-
为探究3种催化剂的表面Mn元素价态比例差异以及表面氧的吸附形态,对Mn 2p和O 1s谱图进行了分峰拟合面积计算,如图7和表2所示. 从图7(a)中3种催化剂在640—660 eV之间均观察到Mn 2p1/2和Mn 2p3/2的2个主峰. 表2列出了不同制备方法下Mn 2p3/2位于641.17—641.43 eV、642.36—642.55 eV、643.64—644.32 eV的峰, 分别归属于 Mn2+、Mn3+ 和 Mn4+[33]. 有研究表明,Mn4+所占比例越高越有益于MnOx更好地参与氧化还原反应的循环过程,Mn4+物种数量直接影响催化剂低温脱硝性能[34]. 因此,比较3种催化剂的Mn4+所占比例,其大小顺序为Mn10/REC-SP > Mn10/REC-CP > Mn10/REC-IP,Mn4+所占比例分别为31.49%,29.75%和25.62%,这是Mn10/REC-SP催化剂的催化活性高于其他2种催化剂的原因之一.
图7(b)为O 1s的分峰拟合结果,531.2—531.4 eV和532.1—532.3 eV处的峰分别归属于晶格氧Oβ(O2−)和化学吸附氧(
O2−2 或O−)[35]. 化学吸附氧被认为是最活跃的氧种类,在氧化还原反应中发挥重要作用,是影响催化剂催化活性的关键因素之一[36]. 由图(b)的计算结果可以得出,3种催化剂中Mn10/REC-SP的Oα/Oβ的比例最高为3.54,而Mn10/REC-CP和Mn10/REC-IP的比例分别为1.53和1.83. 这与3种催化剂的低温SCR催化性能结果表现一致. -
(1)以累托石为载体,采用等体积浸渍法、共沉淀法和原位生长法负载活性组分锰氧化物制备Mn/REC催化剂. 在空速为105000 h−1时,100—300 ℃温度范围内,Mn10/REC-IP催化剂的NO转化率很低. 而Mn10/REC-CP和Mn10/REC-SP催化剂在高空速下均能够保持80%以上的NO转化率. 其中Mn10/REC-SP催化剂的NO转化率最高,在150—250 ℃温度范围内的NO转化率可达98%以上.
(2)累托石与催化剂制备方法间存在显著的匹配效应,原位生长法和共沉淀法对累托石片层具有显著的柱撑效应使活性组分在层间生长,且具有较高分散性的团簇状MnOx使得催化剂的比表面积成倍增长.
(3)制备方法对催化剂活性组分形貌、分散性、锰氧化物价态分布、载体结构及比表面积等理化性质影响显著. 原位生长法得到的催化剂活性组分在累托石局部表面具有最好的分散性和最高的Mn4+比例,这是其具有良好的中低温SCR催化活性的主要原因.
锰基累托石低温NH3-SCR催化剂的制备方法
The preparation method of manganese-based rectorite low-temperature NH3-SCR catalyst
-
摘要: 本文以天然累托石 (REC) 为载体,采用等体积浸渍法、共沉淀法和原位生长法负载MnOx制备Mn/REC催化剂用于烟气的低温SCR脱硝. 采用XRD、SEM、TEM、BET、NH3-TPD和XPS等手段对催化剂的理化结构特性进行表征. 考察制备方法及主要制备参数对催化剂理化性能及脱硝活性的影响,探讨影响催化剂低温SCR脱硝活性的结构因素. 结果表明,在标准反应条件下(空速为105000 h−1、反应温度在150—250 ℃),原位生长法所制备的Mn10/REC-SP催化剂的NO转化率最高,可达近100%,共沉淀法制备的Mn10/REC-CP催化剂NO转化率次之,而等体积浸渍法制备的Mn10/REC-IP催化剂催化活性最低. 表征结果表明,制备方法对催化剂活性组分形貌、分散性、MnOx价态分布、酸性强度及比表面积等理化性质影响显著. 原位生长法得到的催化剂活性组分在催化剂局部表面具有最好的分散性和最高的Mn4+比例,这是其具有良好的中低温SCR催化活性的主要原因. 原位生长法和共沉淀法对REC具有显著的柱撑效应,使得活性组分在层间生长而具有更好的分散性,催化剂的比表面积成倍增长.Abstract: The Mn/REC catalysts were prepared by incipient-wetness impregnation, co-precipitation and in-situ growth method loading MnOx with natural rectorite (REC) as carrier for low-temperature NH3-SCR of NO from flue gas. The physi-chemical properties of catalysts were characterized by XRD, SEM, TEM, BET, NH3-TPD and XPS. Effects of preparation methods and parameters on catalyst’s properties and SCR activity were investigated with the attempt to explore the structure-activity relationship (SAP) of the Mn/REC catalyst. The experimental results showed that the NO conversion of Mn10/REC-SP catalyst maintain nearly 100% in a wide temperature window (150—250 ℃) under gas hourly space velocity (GHSV) of 105000 h−1. The NO conversion of the catalyst prepared by co-precipitation method is slightly lower, and the catalyst prepared by impregnation method showed the lowest catalytic activity. It was found that preparation method had a significant impact on the physi-chemical properties of the catalysts including active component morphology, dispersion, valence distribution of MnOx and even the support structure and specific surface area. The active component of catalyst obtained by in-situ growth method had the best surface dispersion and highest Mn4+ ratio which were suggested to be main reasons for its good low temperature SCR activity. Compared to incipient-wetness impregnation, in-situ growth and co-precipitation method may easily lead to the layer pillaring of REC support, enabling the better dispersion of avtive species between the layers of REC support and the result of drastic increase in specific surface area of the catalyst.
-
Key words:
- rectorite /
- MnOx /
- preparation method /
- NH3-SCR
-
地下水修复是污染场地修复的核心内容,然而我国场地修复行业长期存在“重土轻水”的问题。污染物在土壤中的赋存分布呈现高度的非均质性,而地下水是一个动态开放系统,故地下水监测更容易反映出地层的污染状况,对于挥发性有机物 (VOCs) 污染场地来说更为突出[1]。因此,笔者长期呼吁在污染地块管理领域对地下水环境质量的监管应提升到与土壤环境质量相同的地位。上述观点及其科学机制已在本系列专论1《我国挥发性有机污染地块调查评估中存在的问题及对策建议》和专论3《地下水监测在污染场地管理中的重要作用、存在问题与对策建议》中已经进行过详细的讨论[1-2]。在近期发表的专论4《我国挥发性有机污染场地修复中存在的问题及对策建议》中,笔者总结了我国VOCs污染场地修复管控及效果评估方面存在的5个问题[3]:1) 污染修复深度止步于土壤层,忽视了基岩层中的污染;2) 表层土采样可能会高估VOCs污染土壤的修复效果;3) 修复效果评估中地下水监测数据的指示作用未受到足够重视;4) 地下水修复目标值的制定流程不规范,部分项目存在修复目标值过于宽松的问题;5) 少数复杂高风险场地未能彻底修复即进行敏感用地类型开发。本文则基于问题4展开关于制定地下水修复目标值的讨论。
1. 地下水修复目标值设定方面存在的问题及解决问题总思路
污染场地修复效果评估是污染场地环境管理工作的关键环节,在管理链条中起最终把关作用。修复目标值是评判污染场地修复是否合格,以及修复后地块能否安全利用的唯一标准,因此制定合理的修复目标值对于污染场地修复治理和安全利用起着至关重要的作用。目标值设定过严会导致过度修复,而目标值设定过松则无法保障地块的风险可控和安全利用。
污染场地修复一般同时包括土壤和地下水的修复。2022年12月,生态环境部发布了《建设用地土壤污染修复目标值制定指南 (试行) 》,然而地下水修复目标值制定的指南标准长期缺位。目前,国内常以GB/T14848的III类或者IV类地下水标准作为修复目标,当难以达到IV类地下水标准时一般利用风险评估计算一套更加宽松的目标值。然而,少数项目基于风险评估计算出的地下水修复目标值过于宽松,无法保障地块未来的安全利用。特别是对于挥发性有机物 (volatile organic compounds,VOCs,如苯系物、氯代烃等) 污染场地,项目制定的VOCs的地下水修复目标值超过了这些化合物的有效溶解度,这意味着地层中存在该化合物的的纯相 (即非水相液体,nonaqueous phase liquids,NAPL) ,即修复后场地地层中仍存在NAPL相污染物亦会被认为修复达标(合格),且该地块达到了安全利用标准,这显然是极其不合理的。NAPL是污染场地最重要的污染源形态,如果污染源没有清除干净,则说明修复治理的效果不佳,应继续进行修复治理。关于地层中是否存在NAPL,典型错误是认为“高密度的土壤钻孔没有发现NAPL则说明地层中不存在NAPL”。然而,“Absence of evidence is not evidence of absence”,由于NAPL赋存分布的高度非均质性,土壤钻孔可能无法勘测到NAPL(特别是DNAPL)[2],但并不代表地层中没有NAPL。而且跟土壤监测相比,地下水监测更容易指示污染源的存在[1]。
解决上述问题的关键是借鉴土壤环境管理的思路,制定一个类似于《土壤环境质量 建设用地土壤污染风险管控标准 (试行) 》(GB36600-2018)中“土壤管制值”的“地下水修复目标上限值 (风险管制值) ”。在任何修复项目中,地下水修复目标值的设定不应超过这个上限值,以避免“一放就乱”。同时参考《建设用地土壤污染修复目标值制定指南 (试行) 》中的思路,允许地下水修复目标值根据项目的实际情况进行适度的调整,避免“一收就死”。随着科学研究的进展、修复技术的进步、经济的发展及社会文明程度的提升,修复目标上限值还应做到不断地调整优化,从而实现对于我国地下水环境质量的动态管理和不断提升。
2. 制定地下水修复目标上限值的思考和建议
目前,在实际项目中,常以GB/T14848的III类或者IV类地下水标准作为修复目标,当难以达到IV类地下水标准时则通过风险评估计算一套更加宽松的目标值。然而,少数项目基于风险评估计算得出的地下水修复目标值过于宽松,无法保障地块未来的安全利用。笔者认为地下水修复目标上限值 (或管制值) 的制定不能只基于风险评估计算的结果,而应充分考虑每种污染物的理化性质、迁移转化归趋机制、环境赋存特征、健康和环境风险、国外同类标准的取值、检测方法的准确度和成本、经济社会承受能力等因素,力争做到合规合法、科学严谨、综合平衡。以下将从风险评估方法的可靠性、有效溶解度的影响、不同污染物的迁移归趋暴露毒性的差异、现行地下水标准存在的问题、现行标准与国外同类标准的差距等方面展开讨论。
2.1 制定修复目标值时不应完全参照风险评估计算结果,因其具有较大的主观性和不确定性
我国已建立起基于风险的污染场地管理制度和技术体系,然而“基于风险”并不意味着“仅仅依赖风险评估计算”。因为风险评估计算存在较大的主观性和不确定性,其结果并不是唯一的、确定的、完全客观公正的。风险评估模型计算结果的误差有多个来源,包括概念模型误差、数学模型误差、输入参数误差等。《污染场地VOCs蒸气入侵风险评估与管控》第六章第五节对于挥发吸入室内空气 (蒸气入侵) 暴露途径的风险评估模型误差进行了详细介绍[4]。本文仅以输入参数误差为例对风险评估计算的不确定性进行介绍。风险评估计算需输入包括环境介质理化性质 (土、水、气) 、污染物理化性质、毒性参数、暴露参数等4大类共计几十种输入参数。任何一种输入参数的变化都会影响风险计算结果,部分关键参数的微小变化可能导致计算结果几倍甚至数量级的变化 (模型敏感性高) 。在实际场地中,很多参数都存在不同程度的时间和空间变异性,在同一场地部分参数的时间-空间波动可能高达几个数量级,然而在风险计算时 (如我国HJ 25.3方法或者美国ASTM方法) 每种输入参数只取一个数值,因此风险评估计算结果具有较高的不确定性 (模型不确定性高) 。另外,风险评估计算的输入参数是人为选择的,甚至部分输入参数选取不同的数值都能找到一定的合理性都能自圆其说,因此风险评估计算具有一定的主观性,很难做到完全精准和绝对客观公正。
2.2 有效溶解度决定了污染物在地下水中的浓度上限
水中溶解度是指每升水中所能溶解的溶质的总质量,单位为mg·L−1。有效溶解度是指混合物中特定组分在每升水中所能溶解的溶质的总质量。如苯在水中的溶解度是1 800 mg·L−1,而常规汽油中苯在水中的有效溶解度只有20~40 mg·L−1。这是由于苯仅是汽油上千种化学组分中的一种 (质量占比约为汽油的1%~2%),因此汽油中苯的有效溶解度远低于其纯物质的水中溶解度。有效溶解度构成了污染物地下水浓度的上限,如果地下水中检测到的有机物浓度超过其有效溶解度,则说明地层中存在有机物的纯相 (即NAPL相) 。
而实际上,由于地下水流动往往很缓慢,只有与NAPL源紧邻区域的地下水中污染物质量浓度才能达到或接近其有效溶解度。当污染物溶解进入地下水后,在对流、弥散、扩散等机制作用下其质量浓度会不断被稀释而降低,另外在地下水采样时污染物在监测井中可能被进一步稀释。因此,即使在地层中存在NAPL,采样监测得到的污染物质量浓度可能也远低于其有效溶解度。BRUCE等[5]认为测得的地下水中苯质量浓度超过其有效溶解度的20%即可认为地层中存在LNAPL。经验值显示,汽油污染地下水中的苯质量浓度超过5 mg·L−1,或者总石油烃浓度超过30 mg·L−1时,认为存在LNAPL。也有研究认为,油污染地下水中的苯质量浓度超过1 mg·L−1即可认为存在LNAPL[6]。综合以上研究成果和国外实际经验,笔者认为地下水修复目标值不应超过有效溶解度 (注意不是水中溶解度) 的20%。
2.3 制定修复目标值时需考虑不同类型污染物的迁移、归趋、暴露和毒性等方面的差异
不同类型的污染物 (如重金属、VOCs、SVOCs) 在迁移转化行为、环境归趋机制、暴露途径及毒性方面的差异巨大。易溶于水的化合物泄漏进入地层后往往迅速溶于地下水,因此会出现土壤中无检出而地下水中浓度较高的现象。对于这类物质,在制定地下水修复目标值时要特别关注。对于再开发场地,重金属和SVOCs都可相对容易地通过风险管控措施切断人体暴露途径,而VOCs由于其易挥发和易迁移的特点,很难通过风险管控完全阻断人体暴露。土壤或者地下水中的VOCs可通过挥发进入室内空气 (蒸气入侵) 和挥发进入室外空气产生人体暴露。国内已经有若干再开发场地的蒸气入侵实际案例[2, 7]。因此,对于VOCs的地下水污染及其修复目标值应给与额外的关注。
2.4 GB/T 14848中的与地下水环境管理需求不完全匹配,地下水修复目标值制定应聚焦环境管理需求
原国土资源部和水利部共同制定的《地下水质量标准》 (GB/T 14848) 是现阶段地下水环境管理工作的主要依据,而生态环境部尚未发布基于环境管理需求的地下水环境质量标准。这给我国地下水以及污染场地环境管理工作带来了一系列问题。目前,大部分场地调查项目通常选择GB/T 14848中的39项常规指标作为监测指标,然而这39项中的大部分指标并非污染因子。其中,与污染场地关系密切的仅包括4项VOCs (三氯甲烷、四氯化碳、苯、甲苯) 和6项金属 (汞、砷、硒、镉、六价铬、铅) ,而实际场地的污染物种类远多于这4+6种。另外,部分场地调查项目选择GB36600中的45项基本项目作为地下水监测指标,而GB 36600是土壤风险管控标准并未考虑污染物在土壤和地下水中赋存特征的差异。综上所述,笔者建议生态环境管理部门尽快制定满足环境管理需求的地下水监测指标及其标准值。
GB/T 14848用于污染场地和地下水环境管理时存在的其他问题还有:1) 部分高毒性和常用污染物的指标偏高,未必能有效管控其环境和健康风险,如氯乙烯、二氯甲烷;2) 个别指标不明确,如 1,2-二氯乙烯未规定顺式、反式或是总量;3) 一些污染场地中的常见指标缺乏,如C6-C9石油烃、C10-C40石油烃、甲基叔丁基醚等;4) 一些重点行业的典型特征污染物缺乏,如杂环芳烃、酚类等。
综上所述,笔者建议管理部门应尽快对地下水监测因子进行扩容,在扩容时应综合考虑国内外地下水标准、国内已有的监测方法标准、国内场地调查实践检出频率较高的特征污染物。本文提出了一些具体的补充建议:C6-C9石油烃、C10-C40石油烃、三甲基苯类、异丙苯、甲基叔丁基醚、四乙基铅、1,2-二溴乙烷、烷基萘类、丙酮、甲醛、乙醛、苯酚类、烷基酚类、氯酚类、苯胺类、甲苯胺类、氯苯胺类、硝基苯胺类、硝基苯类、硝基甲苯类、硝基氯苯类、二硫化碳、苊、菲、芘、芴、含氮杂环芳烃类、含硫杂环芳烃类、含氧杂环芳烃类、顺-1,2-二氯乙烯、反1,2-二氯乙烯、1,2,3-三氯丙烷、1,1-二氯乙烷、四氯乙烷类、六氯丁二烯、六氯乙烷。
2.5 地下水环境质量标准的制定应跟上国家整体的发展态势以及其他环保行业的发展步伐
环境质量标准的核心功能在于为环境质量状况提供比对依据,与援引环境质量标准的法律规定、行政规划等要求共同发挥设定目标、考核激励、督政问责的作用[8]。近十年来,我国大气污染防治取得了举世瞩目的成就,我国大气污染物排放量大幅下降,主要空气指标显著改善,并赢得了国际社会高度评价。2012年被认为是这一切变化的起点,当年原环境保护部修订了《环境空气质量标准 (GB3095-2012) ,从而开启了大气污染防治的“黄金十年”[9]。中国现行的环境空气质量标准及大气污染物排放标准中的部分指标已达到甚至比发达国家更加严格,正是严格的环境标准推动中国成为全球空气质量改善速度最快的国家,蓝天保卫战的成就极大地提升了全体国民的获得感和幸福感。在地下水环境标准的宽严程度方面,我国与发达国家的差距仍十分明显,环境监管的提升空间很大。第2个百年目标要求2049我国要建成富强、民主、文明、和谐、美丽的社会主义现代化强国。展望未来的26年,我国的地下水环境质量管理要跟上国家整体的发展态势及其他环保领域的发展步伐,以全面推进美丽中国建设。
-

图 2 锰负载量及制备方法对催化剂NO转化率的影响
Figure 2. The effect of manganese loading and preparation method on NO conversion of catalysts
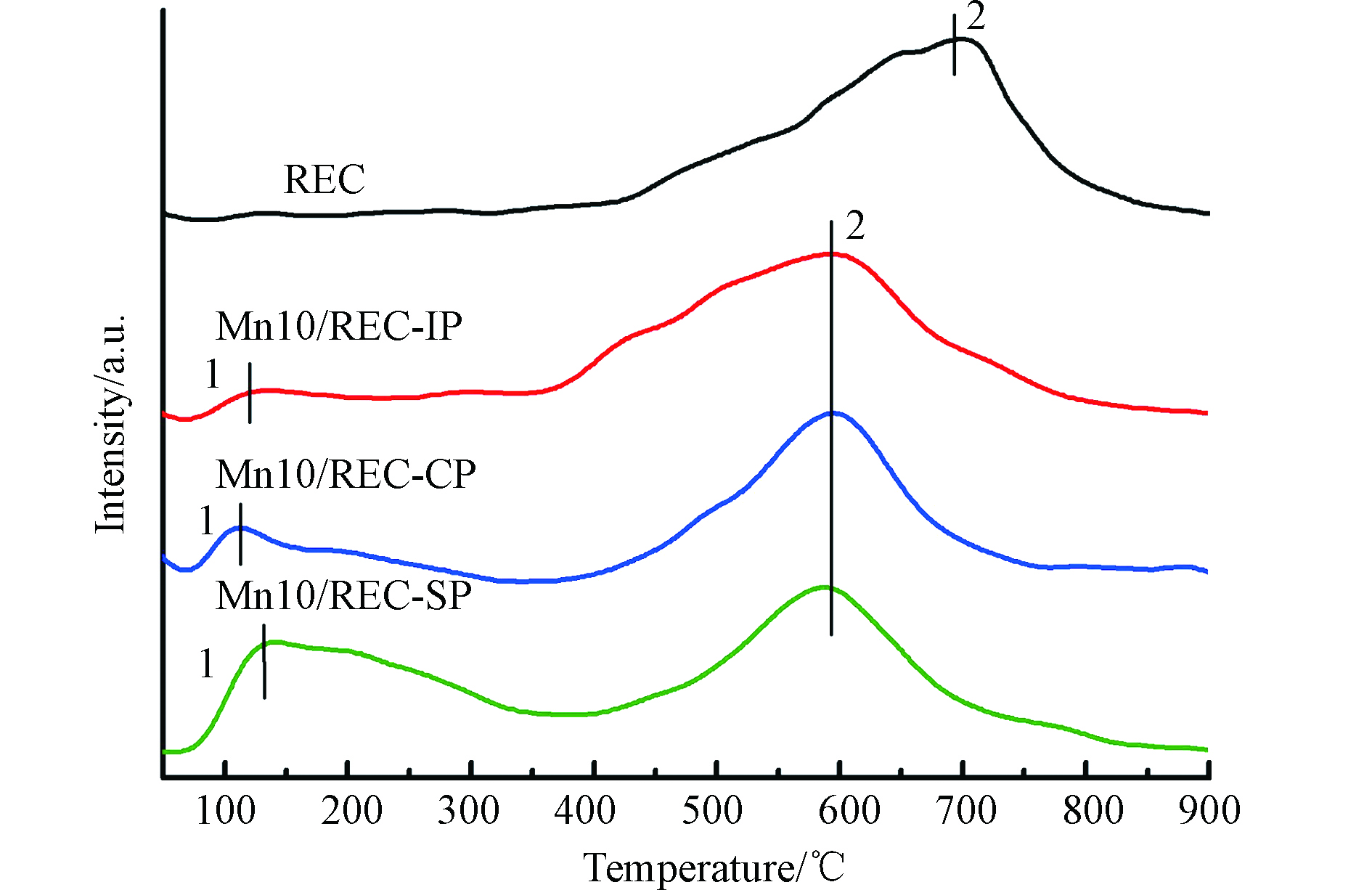
图 6 不同制备方法下催化剂的NH3-TPD图谱
Figure 6. NH3-TPD patterns of catalysts with different preparation methods
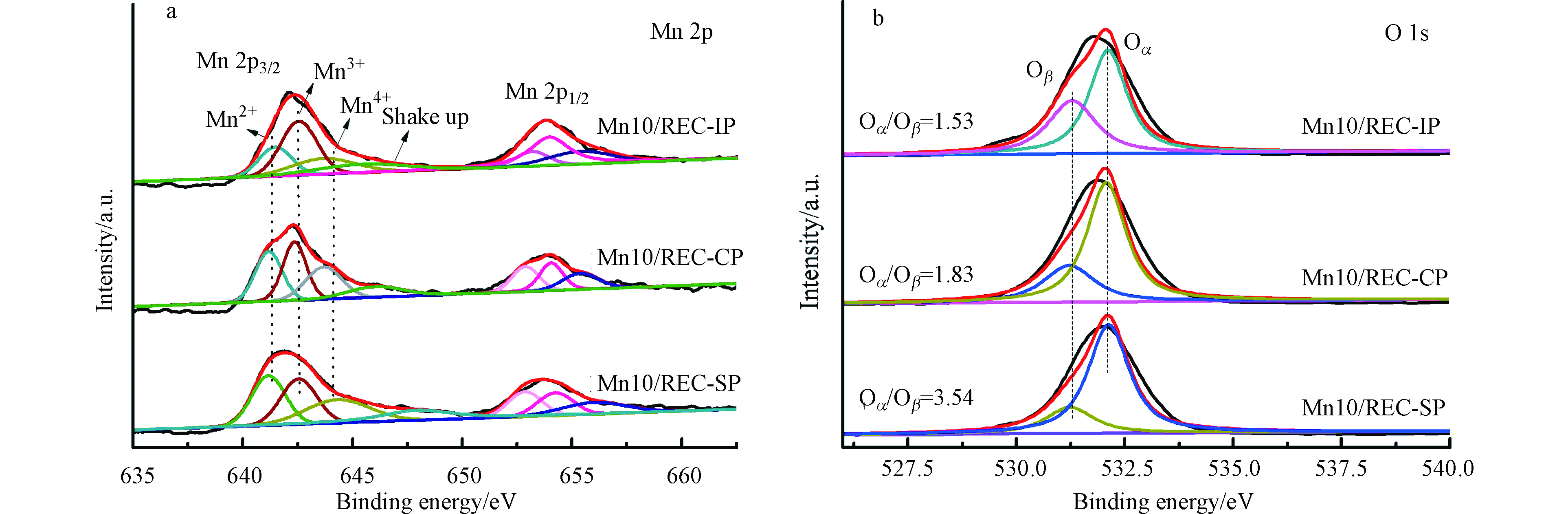
图 7 不同制备方法下催化剂XPS图谱
Figure 7. XPS patterns of catalysts with different preparation methods:(a) Mn2p XPS,(b) O1s XPS
表 1 REC及三种催化剂的比表面积及孔道结构
Table 1. Specific surface area and pore structure of REC and three catalysts
样品Samples 比表面积/(m 2 ∙g−1)Specific surface area 孔容 /(cm3 ∙g−1)Pore volume 孔径/nm Pore diameter REC 18 0.06 4.05 Mn10/REC-IP 12 0.03 4.05 Mn10/REC-CP 52 0.16 7.32 Mn10/REC-SP 62 0.18 4.04  下载: 导出CSV
下载: 导出CSV
表 2 不同制备方法下的Mn10/REC催化剂的Mn 2p XPS结合能
Table 2. Mn 2p XPS binding energy of Mn10 / REC catalyst under different preparation methods
样品Samples 结合能/eVBinding energy 表面浓度比值/%Surface concentration ratio Mn2+ Mn3+ Mn4+ Mn4+/(Mn4++Mn3++Mn2+) Mn10/REC-IP 641.43 642.50 643.64 25.62 Mn10/REC-CP 641.18 642.36 643.69 29.75 Mn10/REC-SP 641.17 642.55 644.32 31.49
下载: 导出CSV
-
[1] WEI L, CHENG Z, XIN L, et al. Ho-modified Mn-Ce/TiO2 for low-temperature SCR of NO with NH3: Evaluation and characterization [J]. Chinese Journal of Catalysis, 2018, 39(10): 1653-1663. doi: 10.1016/S1872-2067(18)63099-2 [2] SHAO J, LIN F, HUANG Y, et al. MnO2 fabrication with rational design of morphology for enhanced activity in NO oxidation and SO2 resistance[J]. Applied Surface Science, 2020, 503: 144064. [3] ZHANG H, ZHOU C, GALVEZ M E, et al. MnOx-CeO2 mixed oxides as the catalyst for NO-assisted soot oxidation: The key role of NO adsorption/desorption on catalytic activity [J]. Applied Surface Science, 2018, 462: 678-684. doi: 10.1016/j.apsusc.2018.08.186 [4] LIU J, WEI Y, LI P Z, et al. Experimental and theoretical investigation of mesoporous MnO2 nanosheets with oxygen vacancy for high-efficiency catalytic deNOx [J]. ACS Catalysis, 2018, 8(5): 3865-3874. doi: 10.1021/acscatal.8b00267 [5] KWON D W, NAM K B, HONG S C. The role of ceria on the activity and SO2 resistance of catalysts for the selective catalytic reduction of NOx by NH3 [J]. Applied Catalysis B Environmental, 2015, 166/167: 37-44. doi: 10.1016/j.apcatb.2014.11.004 [6] XU Y, WU X, LIN Q, et al. SO2 promoted V2O5-MoO3/TiO2 catalyst for NH3-SCR of NO, at low temperatures [J]. Applied Catalysis A:General, 2018, 570: 42-50. [7] ZHANG G D, HUANG X S, TANG Z C. Enhancing water resistance of a Mn-based catalyst for low temperature SCR reaction by modifying super hydrophobic layer [J]. Applied Materials & Interfaces, 2019, 11(40): 36598-36606. [8] YAO X, KONG T, YU S, et al. Influence of different supports on the physicochemical properties and denitration performance of the supported Mn-based catalysts for NH3-SCR at low temperature [J]. Applied Surface Science, 2017, 402: 208-217. doi: 10.1016/j.apsusc.2017.01.081 [9] HUANG C Y, GUO R T, PAN W G, et al. SCR of NOx by NH3 over MnFeOx@TiO2 catalyst with a core-shell structure: The improved K resistance [J]. Journal of the Energy Institute, 2018, 92(5): 1364-1378. [10] GONG P J, LIN J, FANG D, et al. Effects of surface physicochemical properties on NH3-SCR activity of MnO2 catalysts with different crystal structures [J]. Chinese Journal of Catalysis, 2017, 38(11): 1925-1934. doi: 10.1016/S1872-2067(17)62922-X [11] LIU Y, WEN J, ZANG P Y, et al. Nitric acid-treated birnessite-type MnO2: An efficient and hydrophobic material for humid ozone decomposition [J]. Applied Surface Science, 2018, 442: 640-649. doi: 10.1016/j.apsusc.2018.02.204 [12] YU C, DONG L, CHEN F, et al. Low-temperature SCR of NOx by NH3 over MnOx/SAPO-34 prepared by two different methods: A comparative study [J]. Environmental Technology, 2017, 38(5/8): 1030-1042. [13] ZUO H, XU D, LIU W, et al. Heat-treated dolomite-palygorskite clay supported MnOx catalysts prepared by various methods for low temperature selective catalytic reduction (SCR) with NH3 [J]. Applied Clay Science, 2018, 152(2): 276-283. [14] YANG H, WEI J, LIU W Q, et al. Synthesis of a carbon@Rectorite nanocomposite adsorbent by a hydrothermal carbonization process and their application in the removal of methylene blue and neutral red from aqueous solutions [J]. Desalination & Water Treatment, 2016, 57(29): 13574-13585. [15] WU D, ZHENG P, CHAN P R, et al. Preparation and characterization of magnetic rectorite/iron oxide nanocomposites and its application for the removal of the dyes [J]. Chemical Engineering Journal, 2011, 174(1): 489-494. doi: 10.1016/j.cej.2011.09.029 [16] EZZATAHMADI N, GODWIN A, MILLAR G, et al. Clay-supported nanoscale zero-valent iron composite materials for the remediation of contaminated aqueous solutions: A review [J]. Chemical Engineering Journal, 2017, 312: 336-350. doi: 10.1016/j.cej.2016.11.154 [17] XU W, ZHANG G, CHEN H, et al. Mn/beta and Mn/ZSM-5 for the low-temperature selective catalytic reduction of NO with ammonia: Effect of manganese precursors [J]. Chinese Journal of Catalysis, 2018, 39(1): 118-127. doi: 10.1016/S1872-2067(17)62983-8 [18] PUDUKUDY M, YAAKOB Z. Synthesis, characterization, and photocatalytic performance of mesoporous α-Mn2O3 microspheres prepared via a precipitation route [J]. Journal of Nanoparticles, 2016, 2016: 1-7. [19] HWANG S, JO S H, KIM J, et al. Catalytic activity of MnOx/TiO2 catalysts synthesized with different manganese precursors for the selective catalytic reduction of nitrogen oxides [J]. Reaction Kinetics, Mechanisms and Catalysis, 2016, 117(2): 583-591. doi: 10.1007/s11144-015-0948-7 [20] ZHANG Q, ZHANG Y, ZHANG T, et al. Influence of preparation methods on iron-tungsten composite catalyst for NH3-SCR of NO: The active sites and reaction mechanism [J]. Applied Surface Science, 2019, 503: 144190. [21] HUANG X, LI S N, QIU W G, et al. Effect of organic assistant on the performance of ceria-based catalysts for the selective catalytic reduction of NO with ammonia [J]. Catalysts, 2019, 9(4): 357-357. doi: 10.3390/catal9040357 [22] ILIOPOULOU E F, EVDOU A P, LEMONIDOU A A, et al. Ag/alumina catalysts for the selective catalytic reduction of NOx using various reductants [J]. Applied Catalysis A General, 2004, 274(1/2): 179-189. [23] 吴琼, 张先龙, 马康, 等. Mnx /SEP (海泡石) 催化剂低温NH3-SCR脱硝性能 [J]. 环境化学, 2018, 37(7): 1609-1618. doi: 10.7524/j.issn.0254-6108.2017092802 WU Q, ZHANG X L, MA K, et al. Mnx/SEP (sepiolite) catalysts for low temperature NH3-SCR catalytic reduction of NO [J]. Environmental Chemistry, 2018, 37(7): 1609-1618(in Chinese). doi: 10.7524/j.issn.0254-6108.2017092802
[24] ZHANG X L, WAN P M, WU X P, et al. Application of MnOx/HNTs catalysts in low-temperature NO reduction with NH3 [J]. Catalysis Communications, 2016, 83: 18-21. doi: 10.1016/j.catcom.2016.05.003 [25] 吴彦霞, 房晶瑞, 雷本喜, 等. 制备条件对Mn-Fe/TiO2催化剂低温脱硝性能的影响 [J]. 化工环保, 2014, 34(4): 380-384. doi: 10.3969/j.issn.1006-1878.2014.04.017 WU Y X, FANG J R, LEI B X, et al. Effect of preparation conditions on low-temperature denitrification performance of Mn-Fe/TiO2 catalyst [J]. Chemical Industry and Environmental Protection, 2014, 34(4): 380-384(in Chinese). doi: 10.3969/j.issn.1006-1878.2014.04.017
[26] NAING H H, WANG K, TUN P P, et al. Enhanced broad spectrum (vis-NIR) responsive photocatalytic performance of Ag2O/rectorite nanoarchitectures [J]. Applied Surface Science, 2019, 491: 216-224. doi: 10.1016/j.apsusc.2019.06.079 [27] ZANG S M, ZHANG G Z, QIU W G, et al. Resistance to SO2 poisoning of V2O5/TiO2-PILC catalyst for the selective catalytic reduction of NO by NH3 [J]. Chinese Journal of Catalysis, 2016, 37(6): 888-897. doi: 10.1016/S1872-2067(15)61083-X [28] WANG X, ZHOU J, JIANG C J, et al, Precursor and dispersion effects of active species on the activity of Mn-Ce-Ti catalysts for NO abatement[J]. Korean Journal of Chemical Engineering, 2019, 36(12): 1991-1999. [29] WANG L, LI W, SCHMIEG S J, et al. Role of Brønsted acidity in NH3 selective catalytic reduction reaction on Cu/SAPO-34 catalysts [J]. Journal of Catalysis, 2015, 324: 98-106. doi: 10.1016/j.jcat.2015.01.011 [30] ZUO J, CHEN Z, WANG F, et al. Low-temperature selective catalytic reduction of NOx with NH3 over novel Mn-Zr mixed oxide catalysts [J]. Industrial & Engineering Chemistry Research, 2014, 53(7): 2647-2655. [31] HU X N, HUANG L, ZHANG J P, et al. Facile and template-free fabrication of mesoporous 3D nanospheres-like MnxCo3-xO4 as highly effective catalysts for low temperature SCR of NOx with NH3 [J]. Journal of Materials Chemistry A, 2018, 6(7): 1925-2963. doi: 10.1039/c7ta08000j [32] JIANG Y, CHENG G, YANG R, et al. Influence of preparation temperature and acid treatment on the catalytic activity of MnO2 [J]. Journal of Solid State Chemistry, 2019, 272: 173-181. doi: 10.1016/j.jssc.2019.01.031 [33] YANG Q, LI W, CHEN Z H, et al. Ceria modified FeMnOx-enhanced performance and sulphur resistance for low-temperature SCR of NOx [J]. Applied Catalysis B:Environmental, 2017, 206: 203-215. doi: 10.1016/j.apcatb.2017.01.019 [34] GU S, GUI K, REN D, et al. The effects of manganese precursors on NO catalytic removal with MnOx/SiO2 catalyst at low temperature [J]. Reaction Kinetics, Mechanisms and Catalysis, 2020, 130(1): 195-215. doi: 10.1007/s11144-020-01772-1 [35] SUN P, HUANG S X, GUO R P, et al. The enhanced SCR performance and SO2 resistance of Mn/TiO2 catalyst by the modification with Nb: A mechanistic study [J]. Applied Surface Science, 2018, 447: 479-488. doi: 10.1016/j.apsusc.2018.03.245 [36] QI K, XIE J L, ZHANG Z, et al. Facile large-scale synthesis of Ce-Mn composites by redox-precipitation and its superior low-temperature performance for NO removal [J]. Powder Technology, 2018, 338: 774-782. doi: 10.1016/j.powtec.2018.07.073 -

 点击查看大图
点击查看大图

计量
- 文章访问数: 4123
- HTML全文浏览数: 4123
- PDF下载数: 94
- 施引文献: 0

