







































-
溴酸盐(
BrO−3 )是含溴离子(Br−)水体在臭氧或氯气消毒过程中生成的一种消毒副产物 [1]。在经臭氧消毒的饮用水中检测到BrO3−的质量浓度可达到120 μg·L−1[2]。BrO−3 具有较强的DNA损伤能力,可引起肾肿瘤和腹膜间皮瘤等疾病[3]。因此,世界卫生组织(WHO)规定饮用水中BrO−3 质量浓度应低于10 μg·L−1。目前,很多技术被应用于水体中BrO−3 的去除,包括生物修复[4-5]、化学/催化还原[6-7]、光催化还原[8]、物理分离[9]等。然而这些技术存在二次污染、运行和维护成本高以及操作复杂等缺点。近年来,电化学还原技术因其反应速度快、无污染、操作便捷等优点,在控制水体中BrO3−污染方面已有较多应用[10]。在电化学还原
BrO−3 体系中,阴极材料处于“心脏”地位,是实现高效率和低能耗去除BrO3−的关键。常用的电极材料有贵金属基(Pd、Ir、Pt等)[11-13]和非贵金属基(Fe、Cu等)[6,14]两大类。MAO等采用氧化还原石墨烯修饰碳纸负载钯纳米颗粒阴极(Pd-rGO/C),并耦合活性炭负载钯(Pd/GAC)颗粒电极,在恒定电流条件下可将BrO−3 还原成为Br−离子[12]。然而贵金属价格昂贵,目前对其研究仅仅停留在实验室阶段。因此,开发具有高活性且廉价的非贵金属基电极材料已受到更广泛的关注。BrO−3 的电化学还原包括2种作用,即电子的直接还原和活性氢(H*)介导的间接还原[13]。泡沫钴(cobalt foam, CF)由于成本低廉、比表面积大、稳定性高等优点,被认为是优良的电极材料[15]。但CF电子传递速率差和催化产生H*的活性较低,导致其电化学还原BrO−3 的性能较差。因此,能否在保留CF自身优点的基础上提升其催化产生H*的性能和电子传递速率是一个值得研究的课题。磷化钴(CoP)具有独特的电子结构,不仅可以作为电子媒介促进电子传递,还能作为催化剂电催化产生H*[16-18]。LIU等在以CoP作为阴极开展氟苯尼考电化学脱卤的研究中发现CoP确实能促进电子传递和H*的形成[16]。受上述结果启发,将CoP与CF结合有望得到一种活性高和稳定性佳的复合电极。通常,研究者会利用聚偏氟乙二烯、聚四氟乙烯等高分子化合物粘结剂将催化剂负载于电极表面,但粘结剂的使用会严重影响活性物质的催化性能和电极的稳定性[19-20]。为此,无粘结剂的催化剂和电极耦合技术是制备复合电极的热门方法[21-23]。本研究以CF作为基底,通过原位生长制备CoP与CF耦合自支撑电极(CoP/CF),该电极不使用粘结剂,增加了电子传递速率和电催化产H*性能,具备较好的电还原
BrO−3 性能。本研究通过优化电极制备条件,调控其还原BrO−3 性能,并探讨其机制,以期为电催化还原BrO−3 提供参考。 -
溴酸钠(NaBrO3)、硫酸钠(Na2SO4)、丙酮(CH3COCH3)、无水乙醇、碳酸钠(Na2CO3)、碳酸氢钠(NaHCO3)、磷酸二氢钠(NaH2PO4)等药剂均从国药集团化学试剂有限公司购买,药品均为分析纯且在使用前未经任何前处理。泡沫钴(CF)从北京鹏达科技有限公司采购,其面密度为600 g·m2,孔径为450 μm,厚度约0.5 mm。
-
CoP/CF自支撑电极以CF作为钴源和载体,以NaH2PO4作为磷源,通过高温原位一步磷化法制备。具体方法:取一小片有效面积为5 cm2 (5 cm ×1 cm)的CF,用乙醇、盐酸(1 mol·L−1)、丙酮以及超纯水依次清洗数次去除其表面的杂质,在鼓风干燥箱中60 ℃烘干12 h。将 0.5 g NaH2PO4、CF、0.5 g NaH2PO4依次按照5 cm的间距排列后,N2氛围保护下在管式炉(OTF-1200X, HF-Kejing, China)中进行高温磷化(300~450 ℃),并将在300、350、400和450 ℃下制备的复合电极分别记作CoP/CF-300、CoP/CF-350、CoP/CF-400和CoP/CF-450。
-
电化学还原BrO3−实验均在直流电源(ATTEN, PPS3005T-3S, China)提供的恒定电流下开展(电流密度2.5~10 mA·cm−2),槽压为~18.0 V,50 mM Na2SO4为电解质,
BrO−3 为目标污染物,反应温度为室温(20±5) ℃。电化学反应器是采用质子交换膜(Nafion 117, Dupont, USA)隔开的双室电解池,阴极和阳极室的有效容积均为100 mL。其中,阴极采用CoP/CF自支撑电极,阳极为同等面积的Ir-Ru/Ti电极。电化学反应过程中使用磁力搅拌器将溶液混匀(600 r·min−1),每间隔12 min从阴极室取样2 mL用于检测溶液中BrO−3 和Br−的浓度。 -
溶液中
BrO−3 和Br−采用离子色谱仪(ICS-900, Dionex, USA)进行检测。检测使用的分析柱为IonPac® AS 23 4 mm×250 mm,检测器为电导型检测器(ASRS 300 4 mm),保护柱为IonPac® AG 23 4 mm×50 mm、抑制器型号DZS1106397,进样体积为500 μL。淋洗液为碳酸盐混合溶液(9.4 mmol·L−1 Na2CO3和1.8 mmol·L−1 NaHCO3),流动速度为1 mL·min−1,抑制器施加的电流为40 mA,BrO−3 的最低检测限为1 μg·L−1。电极中的Co和P的价态采用X-射线光电子能谱仪(XPS, Escalab 250Xi, Thermol scientific, USA)对Co 2p和P 2p进行分析;电极的表面形貌和形态利用扫描电子显微镜(SEM, FEI QuANTA 200 CZ, Netherlands)进行观察;晶体结构采用X射线衍射(XRD, Ultima IV, Rigaku, Japan)进行分析,以JCPDS (Joint Committee on Powder diffraction Standards) 数据库为参考,采用MDI (MDI) Jade 5.0对衍射峰和晶相进行分析。
电化学阻抗谱(EIS)奈奎斯特图中弧半径大小可以表示电解过程中电极表面电子传输速率的快慢[24]。本研究采用特定大小的CoP/CF(1 cm2)为工作电极在电化学工作站(辰华CHI7760E,中国上海)上利用三电极体系进行EIS图谱和循环伏安曲线(CV)分析,其中EIS的扫描频率为105~10−2 Hz,对电极为Pt片,参比电极是Ag/AgCl。CV的扫描范围为−1.2~0 V(以Ag/AgCl为参比电极)。
BrO−3 的去除率根据式(1)进行计算;电化学还原BrO−3 的过程利用准一级动力学模型进行分析,动力学方程如式(2)所示;去除每毫克BrO−3 需要消耗的电能根据式(3)进行计算。式中:R为
BrO−3 的去除率,%;Ct和C0是指不同反应时间下电解液中BrO−3 质量浓度,μg·L−1;k为电化学去除BrO−3 的速率常数,min−1;U为系统运行的平均电压,V;I为电流,mA;m为BrO−3 的去除量,μg;t为运行时间,min。 -
本研究以NaH2PO4为磷源,在高温下将CF直接磷化合成CoP/CF自支撑电极。在此过程中CF不仅作为CoP的Co源,还作为CoP的载体或支撑体。显然,这种原位生长方式在不使用粘合剂的条件下可实现CoP和CF的紧密接触,提高了电化学氧化还原反应中的电子转移速率,从而改善了电极的电化学性能[15]。如图1(a)和图1(b)所示,原始CF具有多孔骨架结构且表面光滑。高温磷化后的CF保持了其原有的宏观结构,但在其表面生长出了均匀的纳米颗粒(图1(c)和图1(d))。该变化可能是由于高温下CF表面发生结构塌陷和再固结形成CoP所致[25]。
为了证明CoP成功地原位生长在CF表面,利用X射线衍射(XRD)对CoP/CF自支撑电极表面的晶相和主要物质成分进行分析(图2(a))。在CoP/CF的XRD图谱中,在衍射角为44.51°和47.37°处有2个明显的衍射峰,这2个峰与金属钴的标准特征峰的位置一致,分别对应钴的(002)和(101)晶面(JCPDS 89-7373)。此外,在衍射角为40.81°、50.27°和51.4°处观察到3个峰,分别对应CoP的(112)、(301)和(020)晶面(JCPDS 65-2380)。上述结果证明,经过高温磷化后CoP纳米颗粒成功地原位生长在CF表面,CoP/CF自支撑电极制备成功。
为了进一步分析电极的表面元素组成及相应的价态,采用X射线光电子能谱(XPS)对CoP/CF进行了表征,结果如图2(b)和图2(c)所示。Co2p图谱中结合能为793.0 eV和778.1 eV处对应金属钴的特征峰。此外,结合能为781.2 eV和797.5 eV以及位于786.1 eV和803.3 eV的2个肩峰分别对应钴的氧化物[15]。从P2p的图谱中可以看出,结合能在129.5 eV和130.3 eV处的峰对应磷化物[26-27]。而位于134.1 eV的峰对应的是磷的氧化物。钴和磷的氧化物的出现主要是材料在制备过程中被空气氧化所致。上述结果进一步证明:高温磷化的方法能够直接将CoP原位生长在CF的表面,从而获得CoP/CF自支撑电极。虽然在电极表面不可避免的存在少量磷和钴的氧化物,但不影响自支撑电极的结构和组成。
-
在本研究中,CoP/CF自支撑电极的理化和电化学特性主要受制备的磷化温度所影响。为优化制备条件,本研究使用在不同温度下(300~450 ℃)得到的CoP/CF自支撑电极作为阴极开展电化学还原
BrO−3 的实验,结果如图3所示。不同磷化温度下制备的自支撑电极均表现出较好的电化学还原BrO−3 的活性。其中CoP/CF-350电催化性能最佳,BrO−3 去除率(R)和降解动力学常数(k)分别达到97.6%和0.059 min−1;相比之下,CF电极电化学还原BrO−3 的性能最差,相应的R和k分别仅有32.9%和0.006 min−1。有研究[16]表明,CoP在电化学反应过程中不仅可以作为电子传递媒介或桥梁促进电子传递,还能作为催化剂电催化还原H+产生H*。H*作为一种强还原物质可以将
BrO−3 通过加氢还原为Br−[12]。电化学阻抗谱(EIS)奈奎斯特图中弧半径大小可以表示电解过程中电极表面电子传输速率的快慢[24]。由图4(a)可以看出,CF磷化之后具有了较快的电子传递速率和还原BrO−3 的活性。因此,在CF表面原位生长CoP有利于BrO−3 的电化学还原。此外,当温度为300 ℃时,CF表面只有小部分的钴被磷化成CoP,BrO−3 去除率和k分别只有63.7%和0.017 min−1;当磷化温度达到450 ℃时,尽管CF电极表面CoP含量有所增加,但其电子传递速率却相对于CoP/CF-300、CoP/CF-350和CoP/CF-400降低,BrO−3 去除率和k分别仅为78.0%和0.024 min−1。因此,在适中的磷化温度(350 ℃)下得到的CoP/CF-350电极具有较高电化学还原活性。 -
本研究中,电化学还原
BrO−3 是在恒定电流下开展的,故电流密度对于电化学系统的影响至关重要。不同电流密度(2.5、5.0、7.5和10.0 mA·cm−2)下CoP/CF-350电化学还原BrO−3 的效果如图5(a)和图5(b)所示。当CoP/CF-350电极表面的电流密度由2.5 mA·cm−2增加到10.0 mA·cm−2时,系统中BrO−3 去除率由95.2%增加到100%,相应的降解动力学常数k也由0.052 min−1增加到0.081 min−1。电流密度越高,产生的电子越多,进而导致更高的去除效率和更快的降解速率。然而,相应的去除BrO−3 的所需电能却从0.007 kWh·mg−1上升到0.036 kWh·mg−1。这是由于在高的电流密度下水被电解,从而浪费的电能也相应地增加。值得注意的是,虽然在电流密度为2.5 mA·cm−2和5.0 mA·cm−2时,系统对于BrO−3 去除率差别不大,但当电流密度为2.5 mA·cm−2时,系统出水中BrO3−质量浓度约为12 μg·L−1,该值大于WHO规定的10 μg·L−1限值。因此,综合考虑系统对BrO−3 的去除率和能耗,5.0 mA·cm−2是CoP/CF-350电极电化学还原BrO−3 的最佳值。 -
在电化学反应过程中,溶液中溶解氧(DO)会与
BrO−3 竞争电子,从而影响BrO−3 的去除效率[10]。不同初始DO浓度对CoP/CF-350电极电化学还原BrO−3 的影响如图5(c)和图5(d)所示。当初始DO质量浓度由1.5 mg·L−1增加到8.0 mg·L−1时,BrO−3 的去除率由100%略微下降到97.6%。此外,去除BrO−3 需要的电能没有明显变化(0.013~0.014 kWh·mg−1)。值得注意的是,BrO−3 的去除速率由0.086 min−1降至0.059 min−1,下降了31.3%。这是由于DO在反应过程中与BrO−3 争夺电子,从而导致BrO−3 的去除速率下降。综上所述,CoP/CF自支撑电极可以在较广的DO范围内开展电催化还原BrO−3 ,但较低的DO更有利于BrO−3 的去除。 -
在实际水体中,
BrO−3 质量浓度存在较大差异。本研究在120~1 000 μg·L−1内开展了BrO−3 初始质量浓度对CoP/CF-350电化学还原BrO−3 的影响实验,结果如图6(a)和图6(b)所示。BrO−3 初始质量浓度对CoP/CF-350电化学去除BrO−3 系统的影响较明显。低质量浓度(120 μg·L−1)条件下,系统的反应速率常数(k)为0.068 min−1;然而在高质量浓度1 000 μg·L−1下k仅仅只有0.021 min−1。但是,值得注意的是,在高质量浓度下BrO−3 的绝对去除量较高。当BrO−3 初始质量浓度为120 μg·L−1时,CoP/CF-350电化学反应60 min后去除的BrO−3 总量为11.69 μg。然而,当BrO3−初始质量浓度提高到1 000 μg·L−1时,去除的BrO−3 总量高达74.12 μg,相应的能耗分别为0.032 kWh·mg−1和0.004 kWh·mg−1。以上结果表明:在高浓度下在体系中更多的BrO−3 被去除,并且电能的利用效率更高。如图6(c)所示,
BrO−3 的还原峰电流随着BrO−3 浓度的升高而增加,并且峰电流与BrO−3 浓度呈线性关系(图6(d))。这表明电化学还原BrO−3 是一个由扩散控制的过程[28-29]。通常情况下,溶液中污染物浓度越高,扩散或吸附到电极表面的污染物也就越多[30]。因此,在高的BrO−3 初始质量浓度条件下更多的BrO−3 被吸附到CoP/CF表面,从而使得BrO−3 绝对去除量和电能利用效率增大。此外,虽然在BrO−3 初始质量浓度为1 000 μg·L−1时电化学反应60 min后其去除率只有70.6%,但当反应继续延长到180 min后,BrO−3 的去除率高达98%。上述结果证明,CoP/CF在较宽的BrO−3 浓度范围内均展现出较高的电化学活性以及处理含有不同浓度BrO−3 水体的能力。 -
电化学还原
BrO−3 是一个复杂的过程,其中溴的价态从+5价变为−1价[31]。如图3(b)所示,在不同的电极构建的电化学还原BrO−3 体系中Br−的生成率高达90%。这表明被去除的BrO−3 几乎全部转化成为Br−。此外,有研究[13]表明,BrO−3 不仅能够被电极表面的电子直接还原,还可以被电化学反应中产生的H*通过加氢还原转化成为Br−。在本研究中,由图6(c)可见,BrO−3 能够从CoP/CF自支撑电极表面直接获得电子被还原成为Br−。如图7(a)和图7(b)所示,BrO−3 的还原峰电流随着扫描速率的增加升高,并且其与扫描速率的0.5次方呈线性关系。这表明CoP/CF电化学还原BrO−3 的电子传递过程存在质子扩散控制的过程[28]。CoP在电化学反应过程中不仅能够促进电子传递,还可以电催化还原H+产生H*。因此,本研究以CoP/CF-350和CF电极分别作为阴极,在添加一定浓度叔丁醇(TBA)下开展电化学还原
BrO−3 的实验(淬灭实验)。TBA能够有效地淬灭电化学反应过程中产生的H*[32],并将其转化成为惰性的2-甲基-2-丙醇自由基[33]。如图7(c)所示,当溶液中的TBA浓度为20 mmol·L−1时,CF电极构建的电化学系统中BrO−3 的去除率并没有发生显著变化。这表明CF电化学还原BrO−3 主要是通过电子传递而不是H*介导的加氢还原实现。然而,CoP/CF电极对BrO−3 的去除率因为TBA的加入降低了32.9%。因此,可以推测,在CoP/CF构建的电化学系统中BrO−3 的去除是通过电子直接还原和H*介导的间接还原的协同作用。综上所述,CoP/CF自支撑电极电化学还原
BrO−3 的机理如图8所示。在CoP/CF电极为阴极构建的电化学系统中,BrO−3 首先通过浓差扩散吸附到电极表面(式(4),BrO−3ads ),BrO−3ads 通过电子传递直接被还原为Br−(式(5))。在此过程中CoP作为电子传递媒介或桥梁促进反应的进行。此外,CoP还可以电催化产生H*(式(6)~(8))。由于金属磷化物中有磷的存在会改变H*在电极表面的吸附能,使其不倾向于Heyrovsky或Tafel过程生成H2[34-35],而是作为强还原剂参与加氢还原反应。因此,BrO−3 可以通过加氢还原转化为Br−(式(9))。 -
电极的稳定性是电化学反应的一个重要指标,其不仅包括电极的性能稳定,也包括电极在反应过程是否会有溶出或渗出从而导致水体的二次污染。为了有效地评估电极的可持续利用性能,本研究在最优条件下(350 ℃磷化CoP/CF电极,电流密度5.0 mA·cm−2,溴酸盐初始质量浓度250 μg·L−1),在反应60 min内开展了CoP/CF-350电极电化学还原
BrO−3 的循环实验,结果如图7(d)所示。相对于第1次反应,第2次循环反应后BrO−3 去除率由97.6%降低到90.5%,但在后续的3次循环反应中BrO−3 去除率基本保持在90%左右。溶液中溶出的Co离子基本保持在0.1 mg·L−1左右。此外,5次循环后的SEM和XRD表征结果表明(图(9)),电极的表面结构和磷化钴的晶体结构并未发生明显的变化。这表明CoP/CF-350在多次循环后仍能保持较好的稳定性。与其他的电极比较(表1),CoP/CF-350在电极制备、稳定性和对溴酸盐去除等方面具有较大优势。 -
1)当磷化温度为350 ℃时,CoP/CF自支撑电极对
BrO−3 展现出最佳的还原性能,对初始质量浓度为250 μg·L−1的BrO−3 ,去除率最高可达97.6%。2) CoP/CF自支撑电极电化学还原
BrO−3 时,BrO−3 初始浓度越高,去除的绝对量越大,同时还原1 mgBrO−3 需要的电能越低;电流密度越大,BrO−3 去除率越高,但相应的能耗也越高;DO对CoP/CF自支撑电极影响不大。3) CoP/CF自支撑电极电化学还原
BrO−3 的产物为Br−,并没有其他副产物的积累;在以CoP/CF为阴极构建的电化学系统中,BrO−3 的去除包括直接电子还原和H*介导的间接还原;CV分析表明BrO−3 的还原是一个受扩散传质控制的过程;在还原过程中CoP不仅作为电子传递媒介或桥梁促进电子传递,还可以作为电催化剂催化产生H*并用于BrO−3 的加氢还原。4) CoP/CF自支撑电极在循环5次后仍然保持较高的电化学活性。
磷化钴-泡沫钴自支撑电极电化学还原溴酸盐
Efficient electrochemical bromate reduction using cobalt phosphide self-supported cobalt foam electrode
-
摘要: 开发活性高和价格低廉的电极材料是溴酸盐(
BrO−3 )电化学还原技术的关键。贵金属电极因其高活性受到广泛关注,但贵金属储量低且价格昂贵导致其推广应用受限。为此,本研究在不使用粘结剂的情况下,通过直接在高温条件下将磷化泡沫钴原位生长转化为磷化钴,从而制备出磷化钴-泡沫钴自支撑电极(CoP/CF),并将其用于电化学还原BrO−3 。结果表明:350 ℃下磷化制备的电极CoP/CF-350性能最优;当BrO−3 初始质量浓度为250 μg·L−1、电流密度为5.0 mA·cm−2时,该电极对BrO−3 去除率为97.6%,相应的能耗为0.014 kWh·mg−1。循环伏安测试(CV)和淬灭实验结果表明,BrO−3 的去除归因于直接电子传递和活性氢(H*)间接还原的协同作用。原位生长的CoP作为双功能催化剂,起到了电子传递媒介和桥梁的作用,强化了BrO−3 的还原。经过5次循环实验后,CoP/CF-350电极仍保持良好电催化活性,说明其具有良好的稳定性。Abstract: Exploring highly active and inexpensive electrode are the keys in electrochemical reduction of bromate (BrO−3 ). Although the noble metal-based electrode has attracted intensive attention for its high activity, low abundance and high price of noble metal seriously impede its practical application. In this study, a binder-free composite electrode (CoP/CF) was prepared by direct growth of cobalt phosphide(CoP) on cobalt foam(CF) and then was used to perform electrochemical reduction ofBrO−3 . The results showed that CoP/CF-350 electrode prepared at 350 ℃ had a better electrochemical performance on highBrO−3 reduction(97.6%) and lower energy consumption (0.014 kWh·mg−1) at the initialBrO−3 concentration of 250 μg·L−1 and current density of 5.0 mA·cm−2. The cyclic voltammetry(CV) and quenching experiments demonstrated thatBrO−3 removal should be ascribed to the synergistic effect of direct electron transfer and active hydrogen (H*) indirect reduction. The in-situ grown CoP served as the bifunctional catalyst and played the dual roles of electron mediator and bridge, further enhanced theBrO−3 reduction. Even after five cyclic experiments, the CoP/CF-350 electrode still remained a good electrocatalytic activity, this indicated that this electrode had a good stability.-
Key words:
- CoP /
- self-supported electrode /
- electrochemical reduction /
- BrO−3 /
- reaction mechanism
-
镉 (Cd) 是人体非必需的有剧毒的重金属元素[1-2],极易在小麦等农作物中富集,并通过食物链对人体健康产生威胁[3-5]。近年来河南[6]、甘肃[7]和陕西[8]等地均相继出现不同程度的小麦Cd含量超标现象。SONG等[9]指出小麦对我国北方人群Cd摄入量的贡献高达29.8%。土壤-小麦系统Cd污染风险识别与评估是小麦Cd污染防治策略制定的前提和重要参考。
近年来国内外研究人员对土壤-小麦污染风险进行了大量研究,多集中于应用不同评价指标对Cd污染土壤和小麦进行污染等级划分与定性评估[5,10-11]。其中,MULLER开发的地积累指数法被广泛用于土壤及河道沉积物Cd污染风险研究[12]。HAKANSON提出的潜在生态风险指数综合考虑了Cd毒性效应和相对贡献度等参数,在农田土壤Cd累积研究中应用较多[13]。ZHUANG等[14]应用地积累指数法对我国主要小麦产区土壤Cd生态风险进行了量化评估。李艳玲等[15]应用潜在生态指数对济源市平原区小麦田Cd污染特征进行了评估分析。美国环保署开发的健康风险评估模型建立了Cd暴露途径与人体Cd摄入量的定量关系,被广泛用于评估污染小麦摄入风险评估的研究中[8,16]。然而土壤Cd累积周期长,影响因素繁杂,不同区域农田土壤Cd空间异质性显著[1,11,17-18]。贵州土壤Cd背景值 (0.66 mg·kg−1) 约为内蒙古土壤Cd背景值 (0.05 mg·kg−1) 的13.2倍[17]。上述评价模型多使用固定评价因子和参考标准,难以对区域农田Cd污染风险进行量化分析[19-20]。同时,我国幅员辽阔,不同地区农田土壤理化性质、社会经济水平和人群生活习惯与国外研究差别较大,固定参数的直接应用使评价结果存在较大偏差[21-22],难以充分反映区域土壤-小麦系统Cd污染风险变化趋势。
基于以上问题,本研究以河南省某地为研究对象,基于部分区域大面积调查和不确定性理论,将Monte-Carlo随机模拟方法引入到区域农田Cd污染风险综合评估中,获取该地区土壤和小麦Cd污染风险概率分布情况,探讨有效可行的污染防控目标,以期为区域小麦Cd污染风险管理提供理论指导。
1. 材料与方法
1.1 研究区概况和样点采集
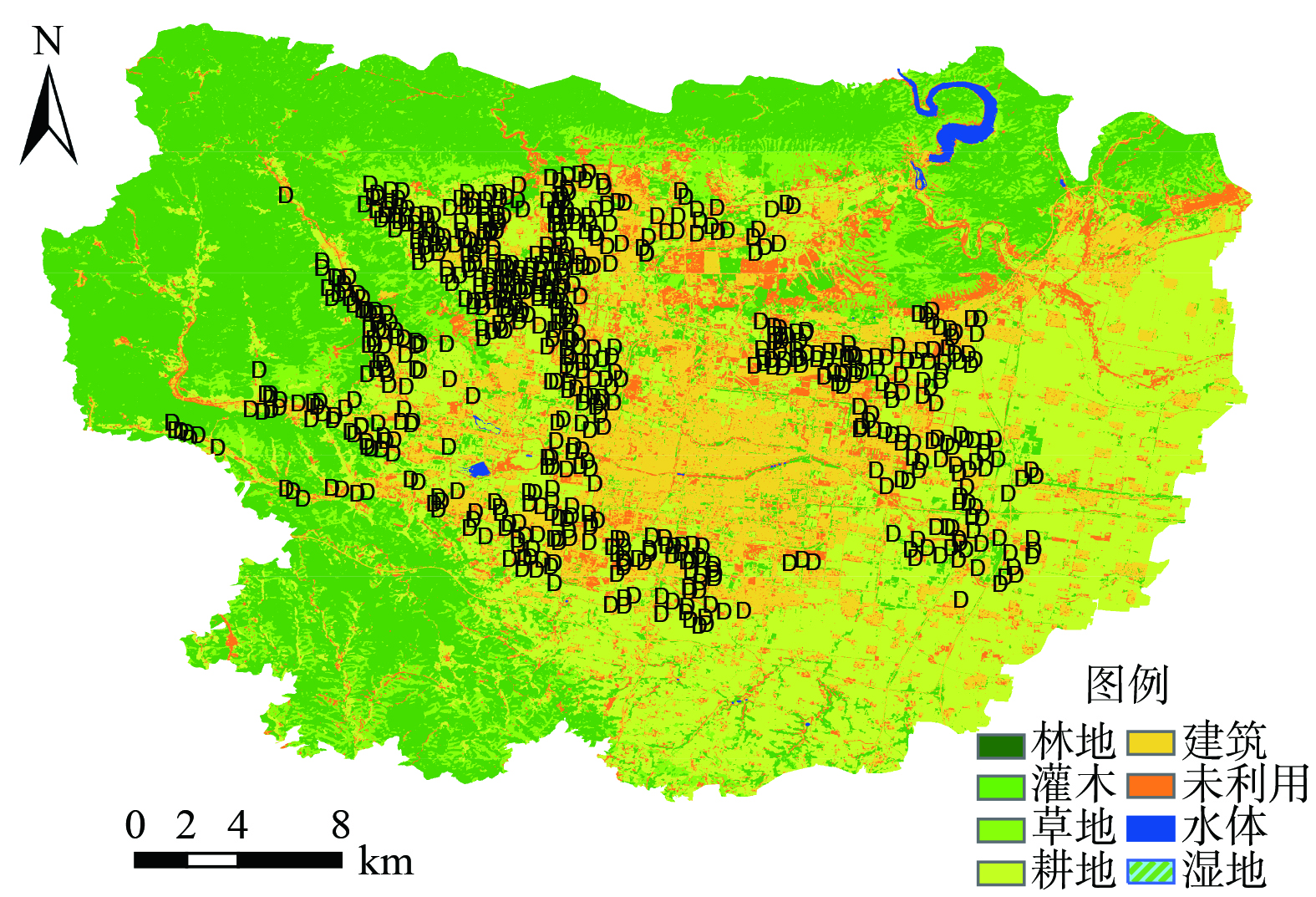
研究区位于河南省某地,年平均温度为15.5 ℃,年平均降水量为546.6~686.2 mm[2],研究区概况如图1所示。区域调查共布设635个采样点位,样点具体分布见图1。另外,10个土壤背景样品采集自研究区远离工矿企业和人类活动干扰的山区[6]。每个采样点应用五点混合采样法采集耕作层土壤 (0~20 cm) 样品1.0 kg左右,同时采集5~10株对应小麦样品,样品标记后带回实验室进行处理分析。
1.2 实验室分析
土壤样品经风干、研磨和过筛后置于自封袋密封备用。小麦样品经去泥、洗净分出籽粒后再用去离子水冲洗2~3遍,晾干后置于烘干箱中105 ℃杀青30 min,60 ℃烘干至恒重,脱壳后研磨粉碎,置于自封袋密封备用。土壤pH使用电极法测定,土水比为1∶2.5。土壤有机质 (SOM,乙二胺四乙酸—乙酸铵溶液浸提法) 和阳离子交换量 (CEC,盐酸浸提法) 具体测定方法方法参见文献[23]。应用四酸法HCl-HNO3-HF-HCIO4消解土壤样品,应用HNO3-HClO4消解小麦样品[6,24]。使用ICP-MS (7500A,Agilent,USA) 测定样品Cd含量,测定过程中应用国家标准物质 (GBW 07427华北平原土壤;GBW 10046河南小麦) 进行质量控制,回收率在91.8%~108.7%之间。
1.3 数据处理
小麦Cd富集系数 (BCF) 常被用来分析土壤-小麦系统Cd富集特征,并可在一定程度上消除区域环境因子影响差异[13,21]。其计算公式见式(1)。
BCF=WCd/SCd (1) 式中:BCF表示小麦籽粒Cd富集因子;WCd表示小麦籽粒Cd含量,mg·kg−1;SCd表示土壤Cd含量,mg·kg−1。
Moran’s I指数多用于空间自相关分析,可有效衡量同一区域内观测值间的依存效应[25],具体计算公式见式(2)~式(3)。
Moran’sI=∑ni∑nj≠iwij(xi−ˉx)(xj−ˉx)/∑ni∑nj≠iwij(xi−ˉx)(xj−ˉx)(S2×∑ni∑nj≠iwij)(S2×∑ni∑nj≠iwij) (2) S2=(1/n)×∑ni(xi−ˉx)2 (3) 式中:n表示空间对象数量;wij表示空间对象i和空间对象j的空间权重矩阵,如果i和j是相邻的,则权重为1,否则为0;xi和xj代表在空间对象i和空间对象j的属性值 (i,j=1,2,3……n) ;
ˉx 地积累指数 (Igeo) 考虑了自然地质条件下土壤Cd背景值的影响,是区分和快速判断人类活动影响程度的重要参数[12,14],具体计算公式见式(4)。
Igeo=log2[SCd/(1.5×Bn)] (4) 式中:SCd表示土壤Cd含量 (mg·kg−1) ,Bn表示土壤Cd背景值,本研究采用研究区土壤Cd背景值 ((0.324±0.105) mg·kg−1) [6]。地积累指数评价结果划分为7个等级,具体等级见表1。
表 1 土壤风险指数分级Table 1. Grade of of soil risk index分级 0 1 2 3 4 5 6 Igeo值 <0 0~1 1~2 2~3 3~4 4~5 >5 污染程度 无 轻度 轻~中 中度 中~重 重度 极严重 Er值 — <40 40~80 80~160 160~320 >320 — 生态风险 — 低 中等 中~高 高 极高 — | Show Table DownLoad:
CSV
DownLoad:
CSV
潜在生态风险指数 (Er) 考虑了土壤Cd毒理效应和环境生态效应对生态环境造成的影响[13,15],其具体计算公式见式(5)。
Er=Tr×SCd/Bn (5) 式中:Tr表示Cd生物毒性系数 (Tr-Cd=30) ,SCd表示土壤Cd含量 (mg·kg−1) ,Bn表示土壤Cd背景值,本研究采用研究区土壤Cd背景值 (0.324±0.105 mg·kg−1) [6]。潜在生态风险指数评价结果分为5个等级,具体分级情况见表1。
健康风险评估模型建立了暴露途径与人体健康风险的定量关系[6,16],计算公式见式(6)。
ADD−WCd=(WCd×IR×f)/BW (6) 式中:ADD-WCd为区域成人经摄食小麦摄入的Cd日均暴露剂量,mg·kg−1·d−1;WCd为小麦Cd含量,mg·kg−1;IR为研究区成人日均小麦食用量,g·d−1;f为河南省居民食用自产小麦的比例,%;BW为研究区成人平均体重,kg。
耦合公式(1)和(6),人体健康评估模型可进一步用来表征土壤-小麦系统Cd富集量与人体健康风险的量化关系,其计算公式见式(7)。
ADD−SCd=(SCd×BCF×IR×f)/BW (7) 暴露参数对健康风险评价的结果具有关键性影响[9,21]。本研究中,成人日均小麦食用量 (IR) 和平均体重 (BW) 来自于区域人群健康调查,分别设定为 (307.6±98.8) g·d−1的对数正态分布变量和 (58.6±5.6) kg的正态分布变量[6]。河南省居民食用自产小麦的比例 (f) 参考中国人群暴露参数手册河南部分设定为80.2%[26]。蒙特卡洛 (Monte-Carlo) 模拟是一种基于概率抽样的数理统计方法[19],通过对观测样本已知分布内的随机抽样,获取目标变量的概率分布[20,22]。本研究应用monte carlo模拟方法来降低土壤-小麦系统Cd污染综合风险评估结果的不确定性。
1.4 数据分析
应用SPSS 25.0进行农田Cd富集特征统计分析,应用ArcGIS 10.8和Origin 2021进行空间分析和制图,应用Matlab R2021a进行Monte-Carlo随机模拟,设定随机模拟的迭代次数为10000次。
2. 结果与讨论
2.1 土壤-小麦系统Cd富集特征分析
研究区土壤-小麦系统Cd富集特征统计结果如表2所示。区域土壤pH在5.44~8.60之间,平均值为8.05,其中94.7%土壤采样点pH均为碱性。耕作层土壤Cd平均含量为1.45 mg·kg−1,为农用地土壤风险筛选值 (0.6 mg·kg−1,《土壤环境质量农用地土壤污染风险管控标准 (试行) 》GB 15618-2018) [27]的2.42倍,约92.8%的土壤样点超过农用地土壤风险筛选值。土壤Cd变幅高达3.78 mg·kg−1,属强变异水平。以上结果说明区域土壤Cd污染形势严峻且受人为活动影响较为剧烈。
表 2 区域土壤基本性质及土壤-小麦Cd含量特征Table 2. Descriptive statistics of soil properties and Cd concentration in the soil-wheat system being investigated检测参数 最小值 最大值 平均值 中值 标准差 土壤pH 5.44 8.60 8.05 8.23 0.56 土壤有机质/(g·kg−1) 10.90 72.90 23.80 21.80 8.82 黏粒/% 16.50 47.30 28.50 28.00 5.54 阳离子交换量 10.60 27.50 18.20 17.8 3.35 土壤Cd/(mg·kg−1) 0.11 3.89 1.45 1.40 0.63 小麦Cd/(mg·kg−1) 0.04 0.51 0.18 0.17 0.09 小麦Cd-BCF 0.022 3.24 0.16 0.12 0.18 | Show TableDownLoad:
CSV
研究区小麦籽粒Cd平均含量为0.18 mg·kg−1,为《食品安全国家标准食品中污染物限量》GB 2762-2022 (0.1 mg·kg−1) 1.84倍[28],显著高于我国河北 (0.05 mg·kg−1) [29]和陕西 (0.02 mg·kg−1) 等[8]小麦产区的小麦籽粒Cd含量,略低于甘肃地区 (0.23 mg·kg−1) [7]小麦籽粒Cd含量。区域小麦籽粒样品超标率高达84.0%,可见区域小麦富Cd现象广泛存在且较为严重。小麦Cd富集系数 (BCF) 可以量化Cd在土壤-小麦的转移过程,表征小麦吸收积累Cd的能力[24,29]。研究区BCF范围为0.02~3.24,平均值为0.16。Gaussian分布方程[19]拟合结果显示区域小麦BCF服从对数正态分布 (图2) ,拟合结果显著 (p<0.001) 。进一步对中国和美国小麦Cd-BCF拟合发现研究区小麦Cd-BCF显著低于美国小麦Cd-BCF (美国小麦Cd-BCF平均值为3.98±5.29,中值为1.96,图2) [19]。另外,中国小麦Cd-BCF大田观测结果 (小麦Cd-BCF平均值为0.28±0.35,中值为0.19) [10,29]和温室观测结果 (小麦Cd-BCF平均值为0.43±0.22,中值为0.44) [30]与区域小麦Cd-BCF累积分布较为相似,可见研究区小麦Cd富集特征具备一定的典型性。
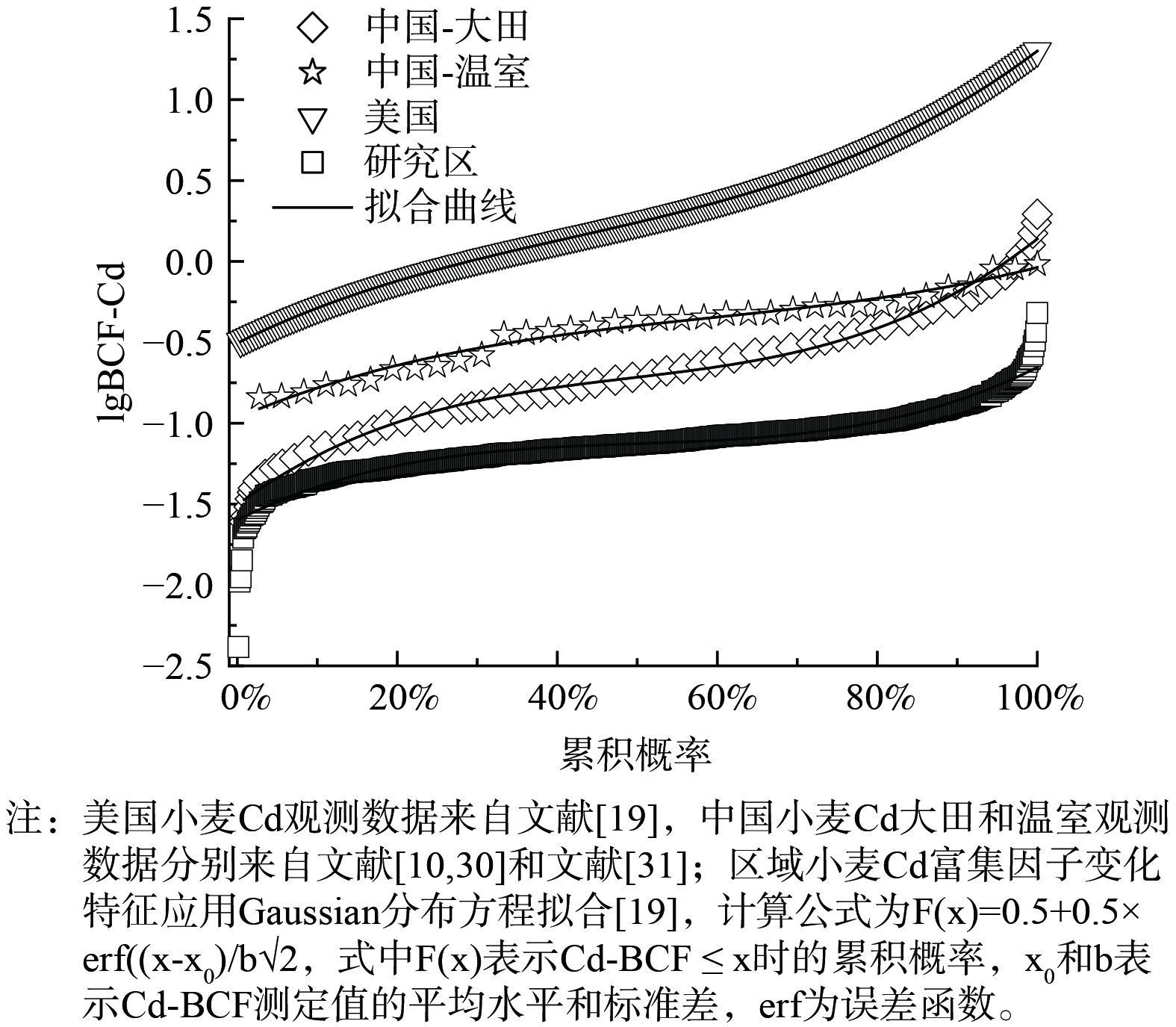 图 2 小麦Cd富集因子累积分布特征Figure 2. Cumulative probability distributions of the Cd BCF for wheat plants
图 2 小麦Cd富集因子累积分布特征Figure 2. Cumulative probability distributions of the Cd BCF for wheat plants2.2 空间异质特征分析
研究区土壤-小麦Cd含量空间自相关分析结果如图3所示。区域土壤-小麦Cd含量Moran’s I指数在0~20 km内均为正值,p值均<0.05,Z值均>2,超过20 km后p值>0.05,说明在该范围内 (0~20 km) 区域土壤-小麦Cd含量存在显著空间自相关关系[25]。土壤Cd含量Moran’s I指数在0~10 km范围内变化较大,整体上呈现先显著下降,再轻微上升,再持续下降的趋势。小麦Cd含量Moran's I指数在0~10 km内下降幅度较大,之后趋于平缓。由此可知区域土壤-小麦系统Cd分布特征存在较强的空间聚集性,这种现象可能是由于当地的工矿企业作业和交通运输活动的干扰所致[6]。整体来看,区域土壤-小麦系统Cd含量Moran’s I指数在0~20 km内均表现出随距离增加而逐渐降低的趋势,这表明可以进一步进行空间插值分析。
 图 3 研究区土壤-小麦 Cd含量 Moran's I指数Figure 3. Moran’s I coefficient of Cd concentration in soil-wheat system in the study area
图 3 研究区土壤-小麦 Cd含量 Moran's I指数Figure 3. Moran’s I coefficient of Cd concentration in soil-wheat system in the study area采用反距离加权法对研究区土壤-小麦Cd含量进行空间插值,空间分布如图4所示。由图4(a)可知,土壤Cd含量整体由西北向东南逐渐降低。土壤Cd高值区 (土壤Cd>1.8 mg·kg−1) 主要分布在研究区西北部和中东部区域。LI等[6]对研究区麦田土壤Cd污染进行溯源分析,指出区域西北部靠近铅锌冶炼厂区,中东部靠近人口密集区,频繁的人类活动容易造成土壤Cd累积。同时,区域土壤Cd含量高值区靠近当地主要河流上游,存在通过河流进行Cd迁移的风险。由图4(b)可知,研究区西北部和中部出现小麦Cd富集现象。空间尺度上,区域土壤和小麦Cd含量在研究区西北部表现出一定的相关性,均出现了较高水平的Cd富集现象。但研究区中东部区域存在一定空间差异,出现土壤Cd含量较高而小麦Cd含量较低现象。由以上结果可知区域工矿企业活动对于小麦Cd富集趋势影响显著,针对该地区的污染风险评估是确保研究区耕地安全利用的关键。
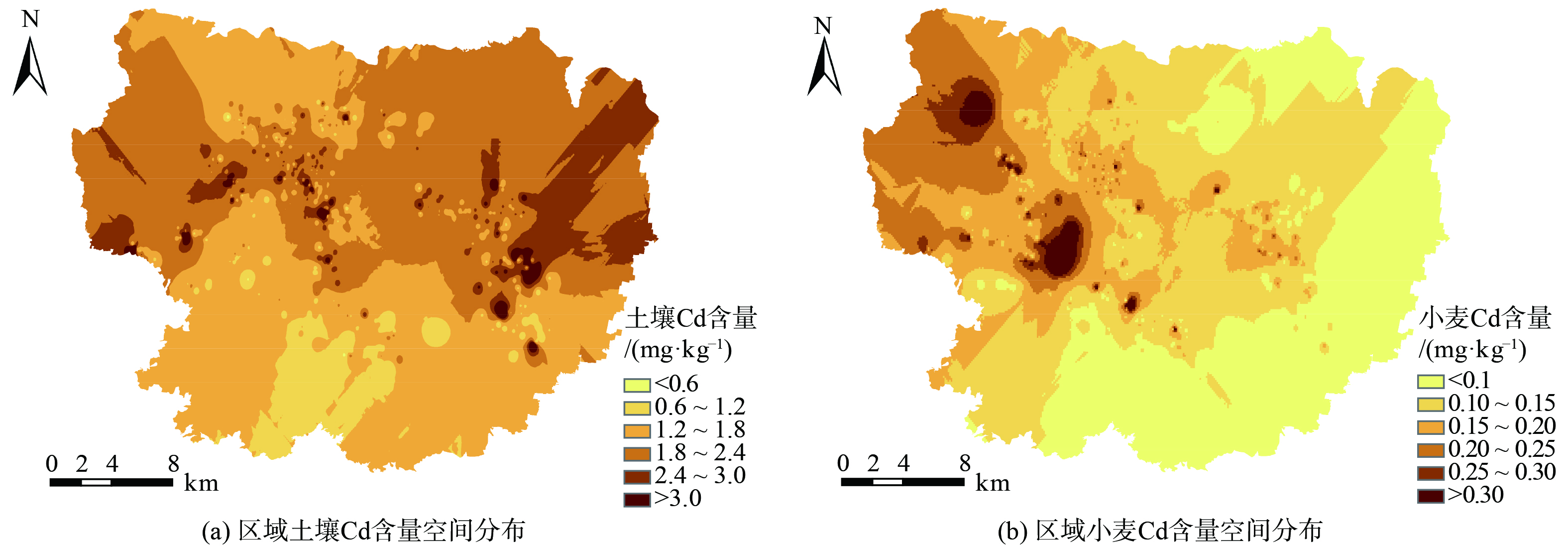 图 4 研究区土壤和小麦Cd含量空间分布特征Figure 4. Spatial distribution of Cd concentration in soil and wheat grain of the study area
图 4 研究区土壤和小麦Cd含量空间分布特征Figure 4. Spatial distribution of Cd concentration in soil and wheat grain of the study area2.3 综合风险评估
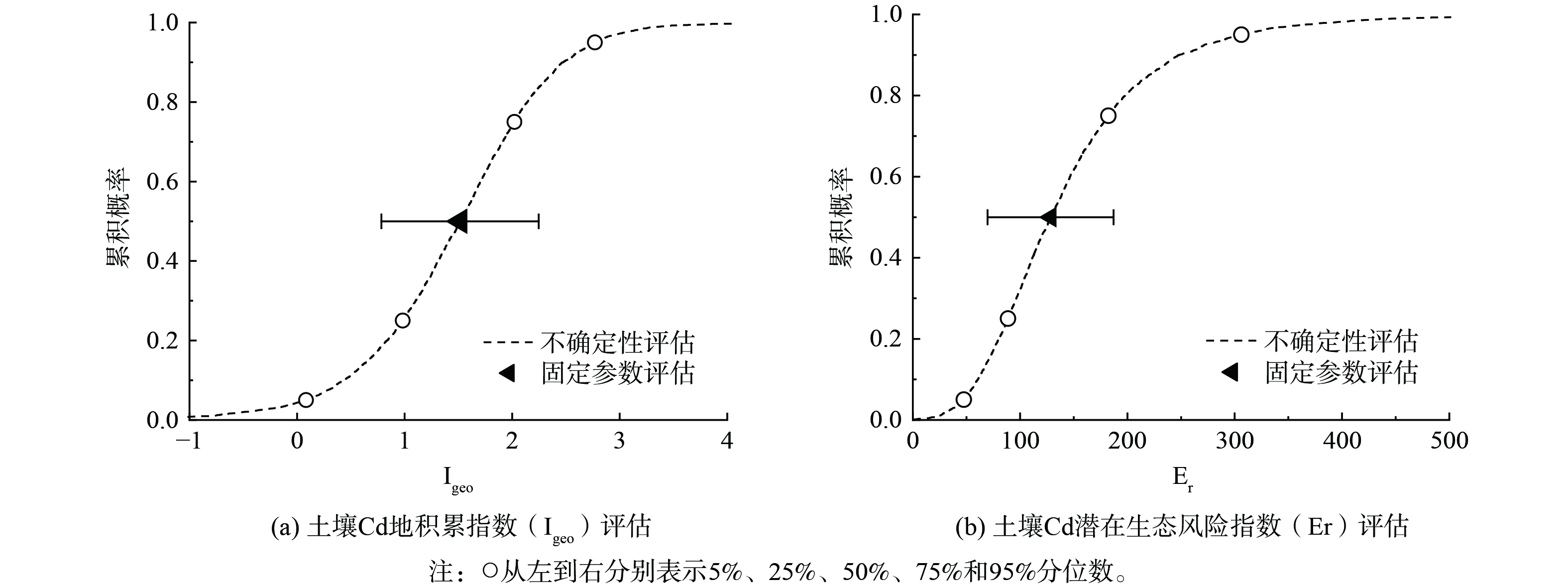
应用地积累指数 (Igeo) 和潜在生态风险指数 (Er) 开展基于固定参数的区域土壤Cd污染风险评估,评价结果见图5。由图5(a)可知,区域土壤Cd地积累指数平均值为1.42±0.73,隶属于轻中度污染等级 (表1) 。由图5(b)可知,区域土壤Cd潜在生态风险指数平均值为134±58.7,属中高等污染水平 (表1) 。考虑到区域土壤Cd含量的强空间异质性水平,进一步开展基于不确定性理论的区域土壤Cd污染风险评估 (图5) 。由图5(a)可知,区域土壤Cd地积累指数主要集中在1.01~1.89之间 (25%~75%分布) ,所调查样点隶属于无污染、轻度、轻中度、中度、中重度污染等级的比例分别为4.4%、21.5%、48.1%、23.2%和2.5%。由图5(b)可知,区域土壤Cd潜在生态风险指数主要集中在90.9~167之间 (25%~75%分布) ,所调查样点隶属于低污染、中等、中高等、高等和极高等污染等级的比例分别为3.2%、1.7%、4.7%、29.2%和4.3%。可见区域土壤Cd生态风险普遍处在高位,需对耕地上风向和河流上游的工矿企业排污进行治理 (图4) ,并避免出现新的污染源以加重区域土壤Cd累积趋势。
基于固定参数的小麦籽粒Cd人体健康风险评估结果如图6所示。结果显示,研究区成人Cd日均摄入量 (ADD) 为0.71±0.35μg BW·kg−1 d−1,略低于世界卫生组织 (WHO) 推荐的成人Cd摄入量安全值 (0.83 μg BW·kg−1·d−1) [31]。SONG等[9]对我国成人Cd摄入量的研究指出饮食结构的不同、小麦Cd含量差异等因素会促进健康风险评估结果的变异。简单的标准对比难以充分反映区域人群Cd摄入风险变化趋势。进一步开展基于不确定性理论的区域人群Cd摄入风险评估 (图6) 。由图6(a)可知区域人群经食用小麦Cd平均摄入量 (ADD) 主要集中在0.41~0.90 μg BW·kg−1·d−1之间 (25%~75%分布) ,近31.6%的区域人群Cd摄入量超过WHO推荐的安全值 (0.83 μg BW·kg−1·d−1) 。在小麦Cd含量较高的研究区西北部,该风险概率显著上升至79.3%,该区域ADD均值为1.12 μg BW·kg−1·d−1,为研究区人群经食用小麦摄入Cd量的1.58倍。考虑到研究区部分乡镇食用自产小麦比例接近100%,因此造成的危害也较大,在风险管理时应给予足够重视。
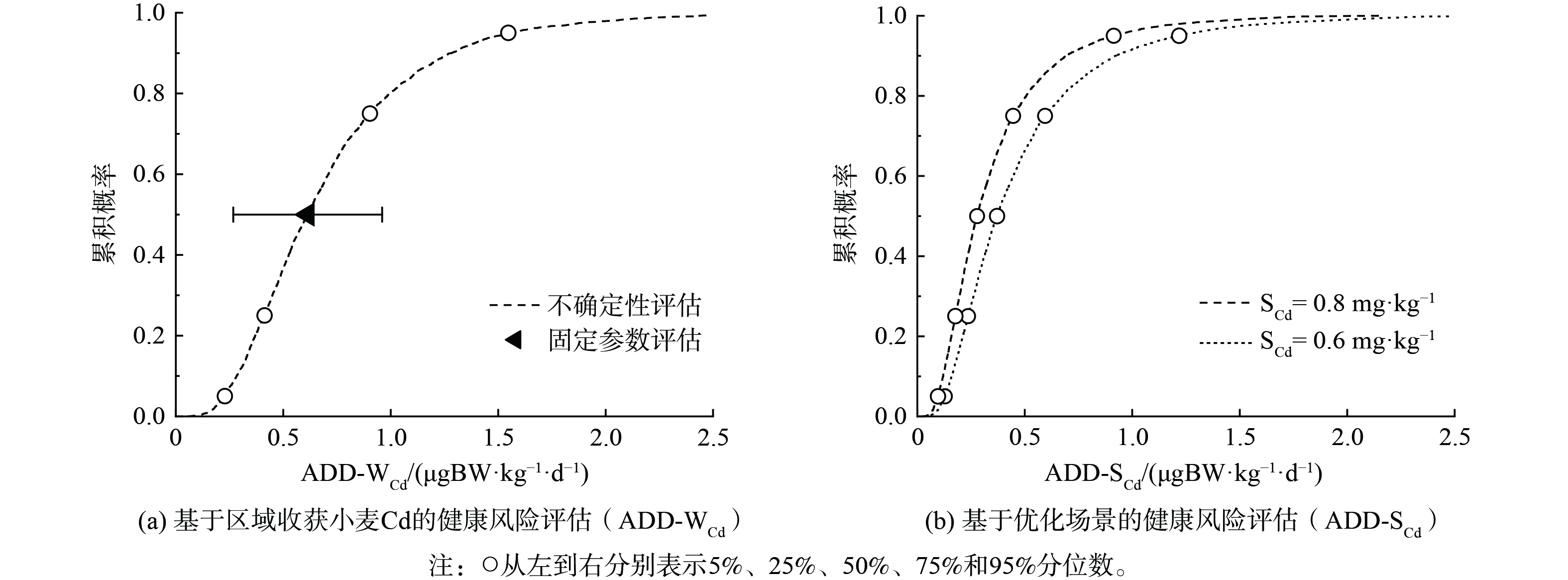 图 6 研究区成人Cd日均暴露风险评估Figure 6. Assessment of daily average exposure risk of adult Cd in the study area
图 6 研究区成人Cd日均暴露风险评估Figure 6. Assessment of daily average exposure risk of adult Cd in the study area2.4 不同场景评估
国务院于2016年印发了《土壤污染防治行动计划》 (简称“土十条”) ,提出我国受污染耕地安全利用率到2030年要达到95%左右[4]。近年来,当地政府积极落实“土十条”各项指标,采取了诸如关闭重污染企业、优化产业格局、减少秸秆还田等手段以降低区域土壤Cd累积趋势,但是不同措施应用下区域人群Cd摄入风险仍不明确。基于不确定性理论应用所构建的人体健康风险评估模型 (公式7) 开展多场景评估,评价结果如图6(b)所示。由图6(b)可知,当区域小麦土壤Cd含量下降到0.80 mg·kg−1 (区域土壤Cd含量25%分布) 时,区域人群Cd摄入量由0.71 μg BW·kg−1·d−1显著下降至0.37 μg BW·kg−1·d−1,超过WHO推荐安全值 (0.83 μg BW·kg−1·d−1) [31]的风险概率由31.6%显著下降至14.1%。ÅKESSON和CHANEY[3]指出人体经饮食摄入Cd含量造成的危害随着Zn摄入量的增加而减小。增加牛奶、鸡蛋等高Zn食物在区域人群日常饮食中的比例[3,9]可进一步降低区域人群Cd摄入风险。
土壤酸化是影响Cd在土壤-小麦系统中富集的主要因子[5-6]。在本研究中,有9.61%的土壤pH<7.5,最小值 (pH=5.44,表2) 低于酸化土壤阈值 (pH=5.5,GB 15618-2018) [27]。为保证区域95%农田的安全利用,研究区土壤Cd含量应控制在0.6 mg·kg−1以下 (《土壤环境质量农用地土壤污染风险管控标准 (试行) 》GB 15618-2018) [27]。该场景下区域人群Cd摄入量为0.28 μg BW·kg−1·d−1,超标风险概率可进一步下降至7.3%。在这些土壤酸化区域增施石灰、生物炭等土壤调理剂可有效控制小麦Cd富集[24,32-33],提升小麦质量水平并降低人群Cd摄入风险。众多研究指出土壤-小麦系统Cd累积量还受到距工业区、矿区和城镇区的距离,不同种类肥料的投入及气候条件等多种因素影响[5,34-35]。充分考虑这些因素的影响,基于不确定性分析原理评估区域Cd污染风险水平及相应概率,使评价过程更加稳健可靠,该方法与空间分析的联合使用更有助于风险决策的科学性和合理性。
3. 结论
将Monte-Carlo随机模拟方法引入到地积累指数、潜在风险因子指数和健康风险评价研究中,综合评估区域土壤-小麦系统Cd污染风险。案例分析说明研究区土壤-小麦系统Cd含量空间异质性强,污染风险较高,以区域西北部Cd富集现象最为显著。研究区成人通过食用当地小麦存在一定的健康风险,需引起当地政府足够重视,并根据不同地区概况采取适当防范措施以减少Cd污染危害,保障区域小麦的安全和高质生产。基于不确定性理论开展模拟分析,结果可同步展示不同场景下区域土壤Cd生态风险和小麦Cd健康风险的风险水平及其相应概率,为当地Cd污染防治提供决策支撑,对其他用地类型的重金属污染防治也有一定借鉴作用。
-
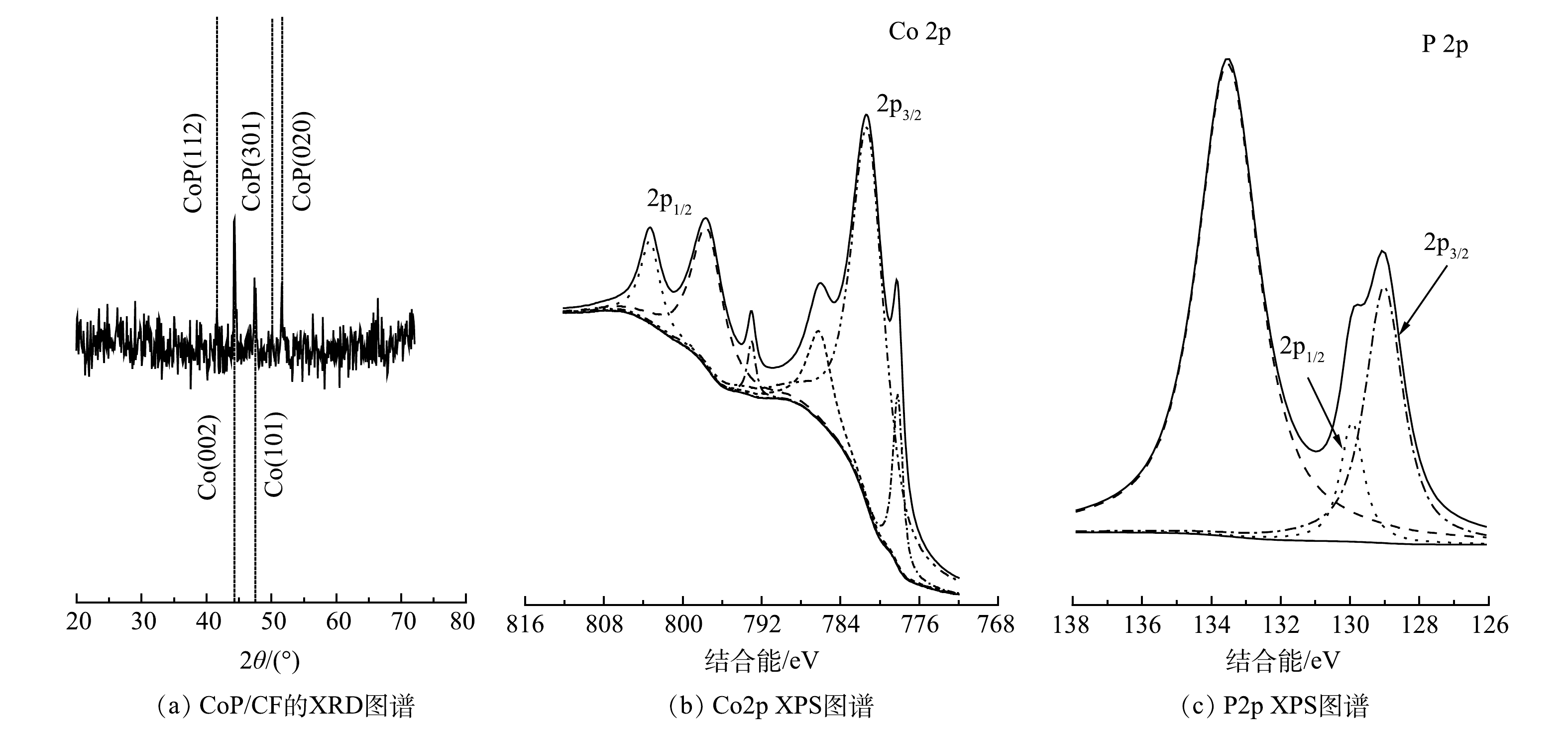
图 2 350 ℃磷化CoP/CF的XRD图谱,Co2p和P2p XPS图谱
Figure 2. XRD pattern of CoP/CF and XPS spectra of Co2p and P2p
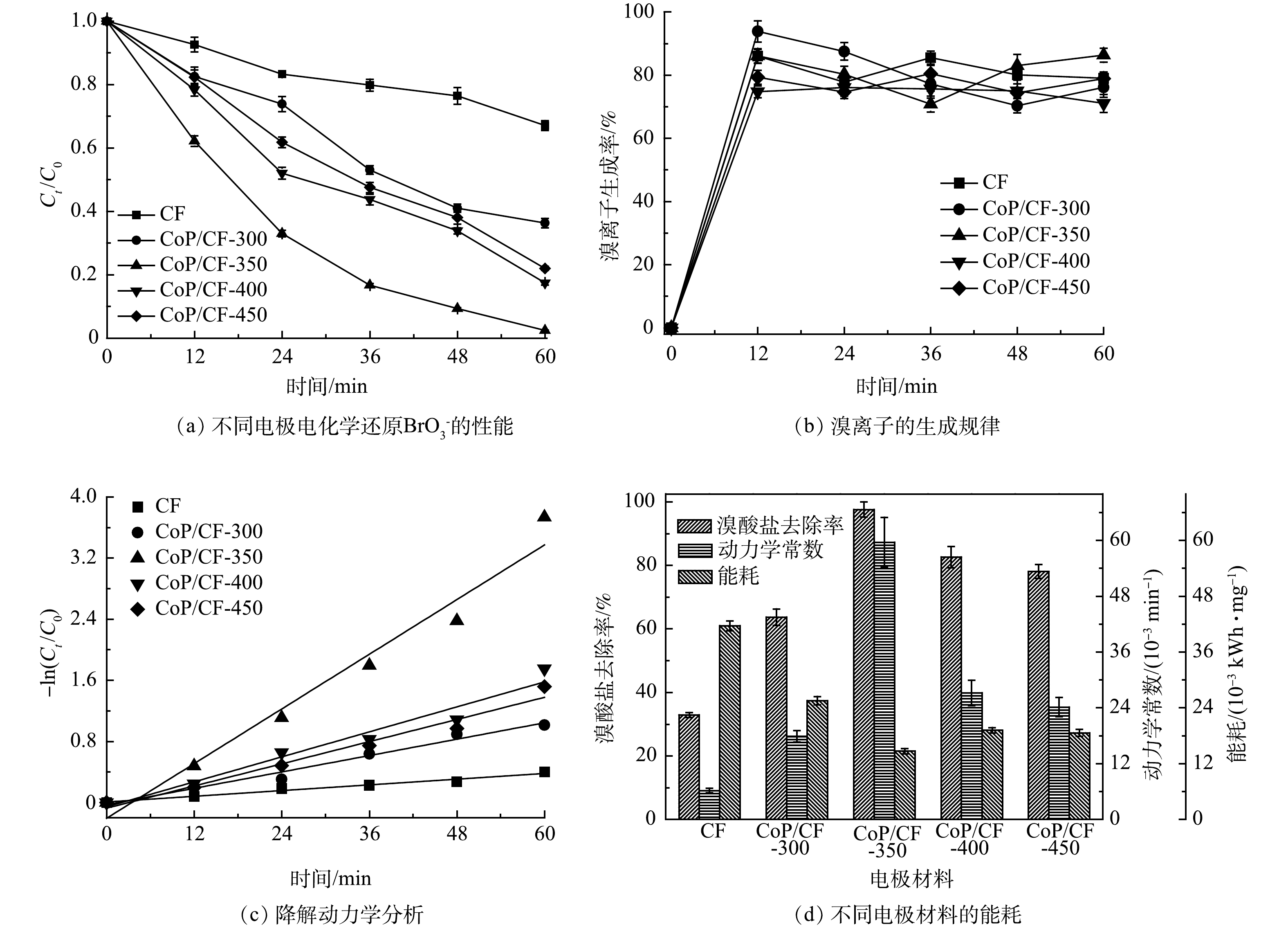
图 3 不同电极电化学还原
BrO−3 的性能Figure 3. Electrochemical
BrO−3 reduction performance of different electrodes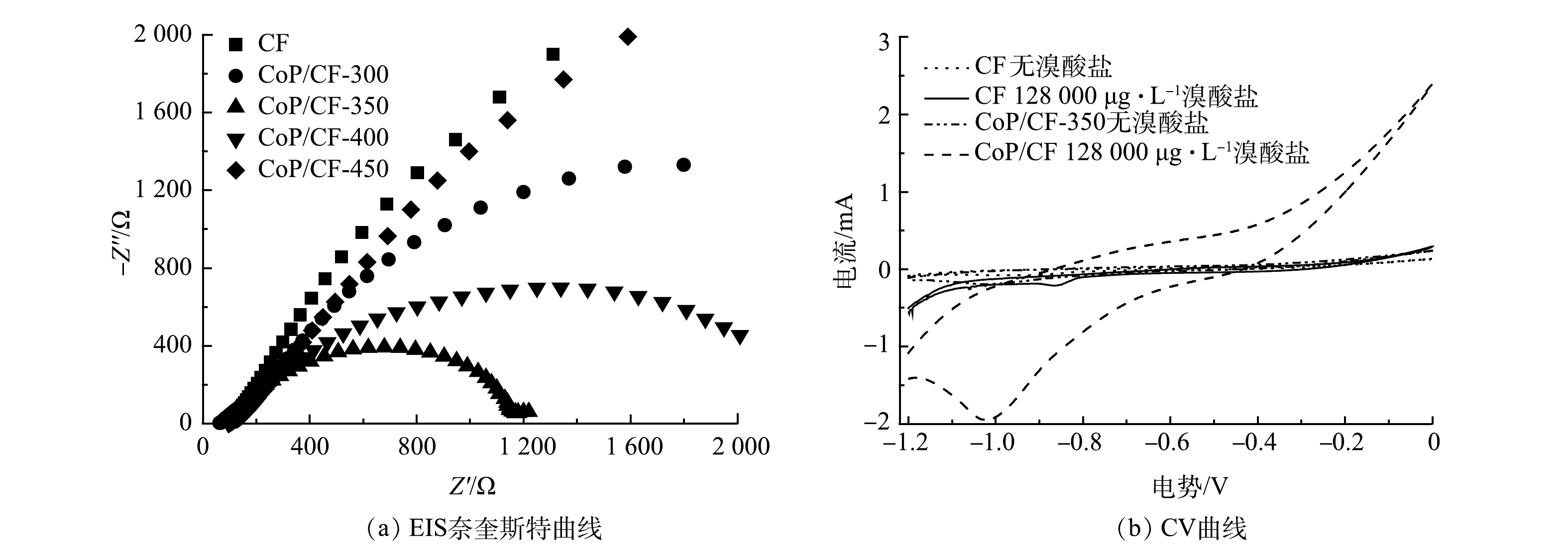
图 4 不同电极的EIS奈奎斯特曲线和CV曲线
Figure 4. The EIS Naquist curves and CV curves of different electrodes
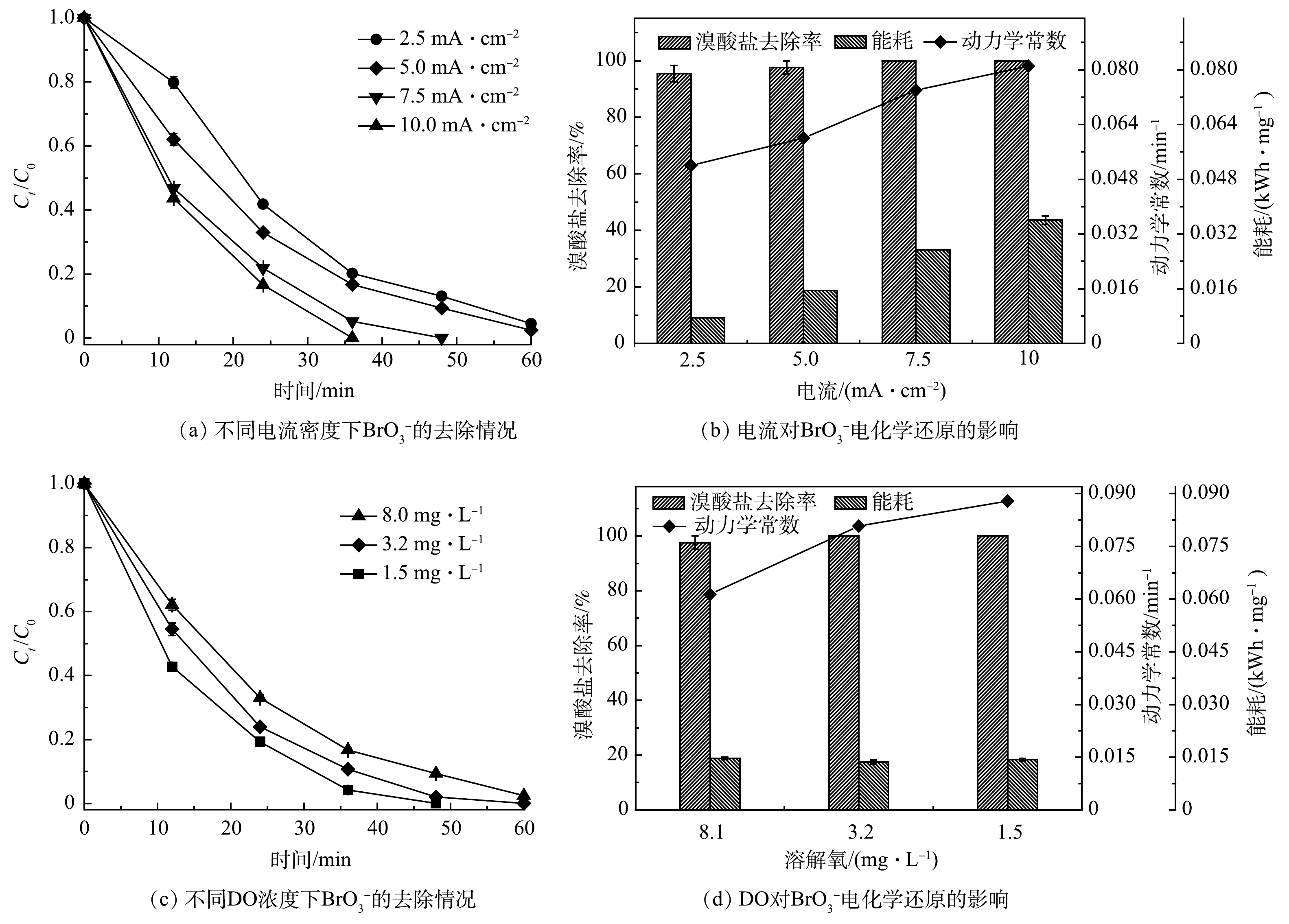
图 5 电流密度和DO对于电化学还原
BrO−3 的影响Figure 5. Effect of current density and DO on the electrochemical
BrO−3 reduction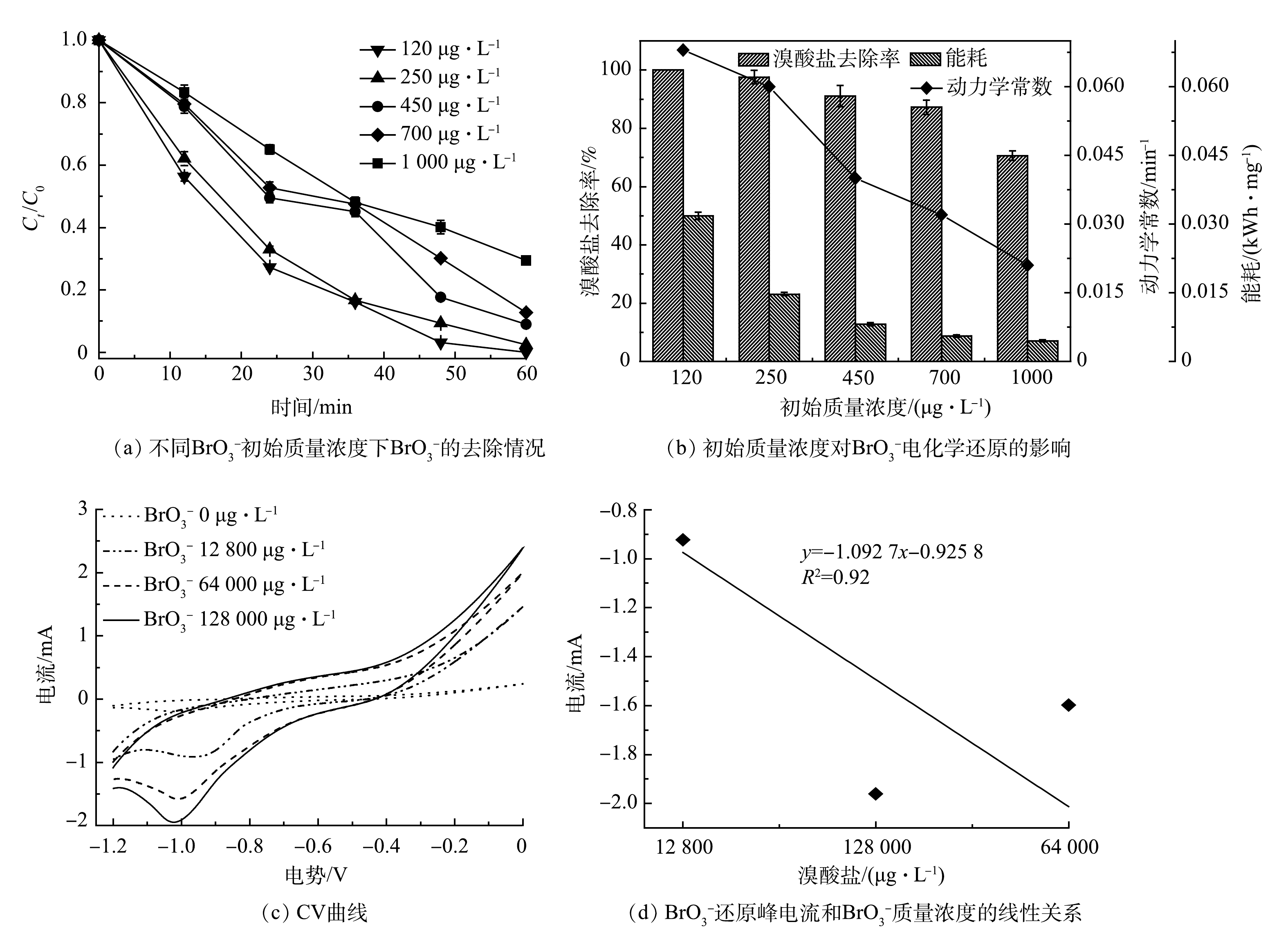
图 6 不同
BrO−3 初始质量浓度对CoP/CF-350电化学还原BrO−3 的影响Figure 6. Effect of initial
BrO−3 concentration on the electrochemicalBrO−3 reduction by CoP/CF-350 electrode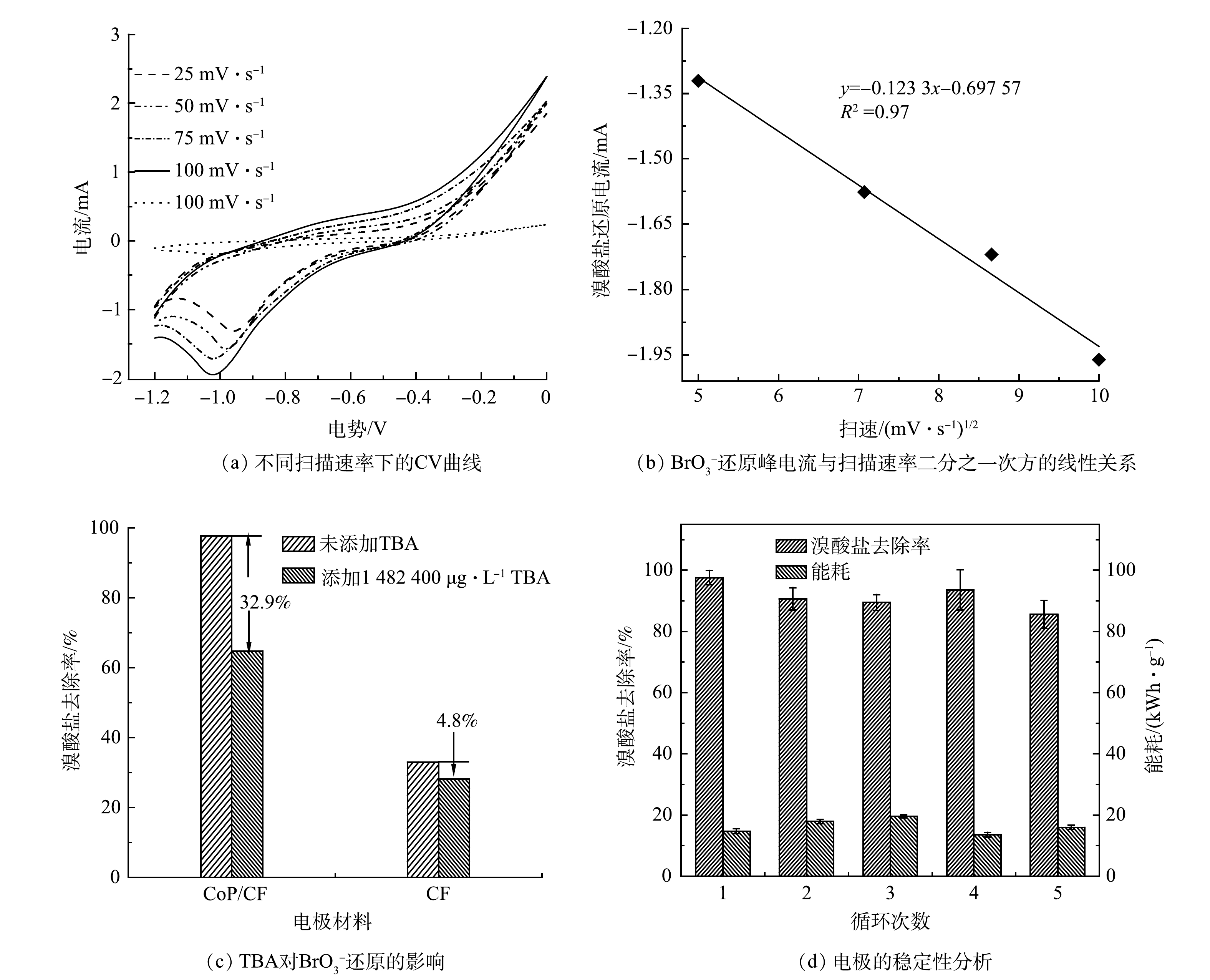
图 7
BrO−3 还原峰电流和电极的稳定性分析Figure 7. The reduction current peak of
BrO−3 and the stability of CoP/CF electrode
图 8 CoP/CF自支撑电极电化学还原
BrO−3 机理图Figure 8. Mechanism of electrochemical
BrO−3 reduction by CoP/CF self-supported electrode
图 9 5次循环后CoP/C-350的SEM图和XRD图
Figure 9. SEM image and XRD pattern of CoP/CF-350 after five cycles
-
[1] 武琳, 杨宏伟, 杨少霞, 等. 催化臭氧氧化过程中溴酸盐的生成机制研究[J]. 环境科学, 2011, 32(8): 2281-2283. [2] LI Y K, SHEN W H, FU S J, et al. Inhibition of bromate formation during drinking water treatment by adapting ozonation to electro- peroxone process[J]. Chemical Engineering Journal, 2017, 264: 322-328. [3] 王执伟, 刘冬梅, 张文娟, 等. 溴酸盐对水生生物的急性毒性效应[J]. 环境科学, 2016, 37(2): 756-764. [4] 钟宇, 杨麒, 李小明, 等. RBER生物降解溴酸盐及微生物群落结构分析[J]. 中国环境科学, 2017, 37(5): 1945-1953. doi: 10.3969/j.issn.1000-6923.2017.05.043 [5] ZHONG Y, LI X M, YANG Q, et al. Complete bromate and nitrate reduction using hydrogen as the sole electron donor in a rotating biofilm-electrode reactor[J]. Journal of Hazardous Materials, 2016, 307: 82-90. doi: 10.1016/j.jhazmat.2015.12.053 [6] WU X Q, YANG Q, XU D C, et al. Simultaneous adsorption/reduction of bromate by nanoscale zerovalent iron supported on modified activated carbon[J]. Industrial & Engineering Chemistry Research, 2013, 52: 12574-12581. [7] YURANOVA T, KIWI-MINSKER L, FRANCH C, et al. Nanostructured catalysts for the continuous reduction of nitrates and bromates in Water[J]. Industrial & Engineering Chemistry Research, 2013, 52: 13930-13937. [8] CHEN F, YANG Q, ZHONG Y, et al. Photo-reduction of bromate in drinking water by metallic Ag and reduced graphene oxide (RGO) jointly modified BiVO4 under visible light irradiation[J]. Water Research, 2016, 101: 555-563. doi: 10.1016/j.watres.2016.06.006 [9] 安东, 宋佳秀, 乐林生, 等. 溴离子和溴酸盐活性炭竞争吸附及溴酸盐生成影响[J]. 环境科学, 2008, 29(4): 948-953. doi: 10.3321/j.issn:0250-3301.2008.04.018 [10] 李昂臻, 冒冉, 赵旭. 泡沫铜电极电还原去除溴酸盐研究[J]. 中国科学, 2014, 44(10): 1675-1681. [11] MAO R, ZHAO X, QU J H, et al. Electrochemical reduction of bromate by a Pd modified carbon fiber electrode: Kinetics and mechanism[J]. Electrochemical Acta, 2014, 132: 151-157. doi: 10.1016/j.electacta.2014.03.170 [12] MAO R, ZHAO X, LAN H C, et al. Graphene-modified Pd/C cathode and Pd/GAC particles for enhanced electrocatalytic removal of bromate in a continuous three-dimensional electrochemical reactor[J]. Water Research, 2015, 77: 1-12. doi: 10.1016/j.watres.2015.03.002 [13] LAN H C, MAO R, TONG Y T, et al. Enhanced electroreductive removal of bromate by a supported Pd-In bimetallic catalyst: Kinetics and mechanism investigation[J]. Environmental Science & Technology, 2016, 50: 11872-11878. [14] SHEN W J, LIN F J, JIANG X, et al. Efficient removal of bromate with core-shell Fe@Fe2O3 nanowires[J]. Chemical Engineering Journal, 2017, 308: 880-888. doi: 10.1016/j.cej.2016.09.070 [15] YUAN C Z, ZHONG S L, JIANG Y F, et al. Direct growth of cobalt-rich cobalt phosphide catalysts on cobalt foil: an efficient and self-supported bifunctional electrode for overall water splitting in alkaline media[J]. Journal of Materials Chemistry A, 2017, 5: 10561-10566. doi: 10.1039/C7TA01776F [16] LIU H L, HAN J L, YUAN J L, et al. Deep dehalogenation of florfenicol using crystalline CoP nanosheet arrays on a Ti plate via direct cathodic reduction and atomic H[J]. Environmental Science & Technology, 2019, 53: 11932-11940. [17] MISUDOME T, SHENG M, NAKATA A, et al. A cobalt phosphide catalyst for the hydrogenation of nitriles[J]. Chemical Science, 2020, 11: 6682-6689. doi: 10.1039/D0SC00247J [18] LIU D N, CHEN T, ZHU W X, et al. Cobalt phosphide nanowires: An efficient electrocatalyst for enzymeless hydrogen peroxide detection[J]. Nanotechnology, 2016, 27: 33LT01. doi: 10.1088/0957-4484/27/33/33LT01 [19] LIU Z W, DONG S S, ZOU D, et al. Electrochemically mediated nitrate reduction on nanoconfined zerovalent iron: Properties and mechanism[J]. Water Research, 2020, 173: 115595. [20] SU L, HAN D D, ZHU G J, et al. Tailoring the assembly of iron nanoparticles in carbon microspheres toward high-performance electrocatalytic denitrification[J]. Nano Letters, 2019, 19: 5423-5430. doi: 10.1021/acs.nanolett.9b01925 [21] CHEN X T, ZHANG T, KAN M, et al. Binderless and oxygen vacancies rich FeNi/graphitized mesoporous carbon/Ni foam for electrocatalytic reduction of nitrate[J]. Environmental Science & Technology, 2020, 54: 13344-13353. [22] YAO Q F, ZHOU X F, XIAO S Z, et al. Amorphous nickel phosphide as a noble metal-free cathode for electrochemical dechlorination[J]. Water Research, 2019, 165: 114930. doi: 10.1016/j.watres.2019.114930 [23] YAO F B, YANG Q, YAN M, et al. Synergistic adsorption and electrocatalytic reduction of bromate by Pd/N-doped loofah sponge-derived biochar electrode[J]. Journal of Hazardous Materials, 2020, 386: 121651. doi: 10.1016/j.jhazmat.2019.121651 [24] ZHANG X, WANG Y T, LIU C B, et al. Recent advances in non-noble metal electrocatalysts for nitrate reduction[J]. Chemical Engineering Journal, 2021, 403: 126269. doi: 10.1016/j.cej.2020.126269 [25] PFEIFFER H, TANCRET F, BICHAT M P, et al. Air stable copper phosphide (Cu3P): A possible negative electrode material for lithium batteries[J]. Electrochemistry Communications, 2004, 6: 263-267. doi: 10.1016/j.elecom.2003.12.012 [26] ZHUANG M H, OU X W, DOU Y B, et al. Polymer-embedded fabrication of Co2P nanoparticles encapsulated in N, P-doped graphene for hydrogen generation[J]. Nano Letters, 2016, 16: 4691-4698. doi: 10.1021/acs.nanolett.6b02203 [27] JIANG N, YOU B, SHENG M L, et al. Electrodeposited cobalt-phosphorous-derived films as competent bifunctional catalysts for overall water splitting[J]. Angewandte Chemie-International Edition, 2015, 54: 6251-6254. doi: 10.1002/anie.201501616 [28] SHIH Y J, WU Z L, HUANG Y H, et al. Electrochemical nitrate reduction as affected by the crystal morphology and facet of copper nanoparticles supported on nickel foam electrodes (Cu/Ni)[J]. Chemical Engineering Journal, 2020, 383: 123157. doi: 10.1016/j.cej.2019.123157 [29] YAO F B, JIA M C, YANG Q, et al. Highly selective electrochemical nitrate reduction using copper phosphide self-supported copper foam electrode: Performance, mechanism, and application[J]. Water Research, 2021, 193: 116881. doi: 10.1016/j.watres.2021.116881 [30] KISHIMOTO N, MATSUDA N. Bromate ion removal by electrochemical reduction using an activated carbon felt electrode[J]. Environmental Science & Technology, 2009, 43: 2054-2059. [31] MAO R, ZHAO X, LAN H C, et al. Efficient electrochemical reduction of bromate by a Pd/rGO/CFP electrode with low applied potentials[J]. Applied Catalysis B:Environmental, 2014, 160: 179-187. [32] GAO J N, JIANG B, NI C C, et al. Enhanced reduction of nitrate by noble metal-free electrocatalysis on P doped three- dimensional Co3O4 cathode: Mechanism exploration from both experimental and DFT studies[J]. Chemical Engineering Journal, 2020, 382: 123034. doi: 10.1016/j.cej.2019.123034 [33] GAO J N, JIANG B, NI C C, et al. Co3O4-TiO2/Ti cathode based electrocatalytic nitrate reduction: Preparation, performance and mechanism[J]. Applied Catalysis B: Environmental, 2019, 254: 391-402. [34] LIU C, ZHANG A Y, PEI D N, et al. Efficient electrochemical reduction of nitrobenzene by defect engineered TiO2-x single crystals[J]. Environmental Science & Technology, 2016, 50: 5234-5242. [35] LI Y H, LIU P F, PAN L F, et al. Local atomic structure modulations activate metal oxide as electrocatalyst for hydrogen evolution in acidic water[J]. Nature Communications, 2015, 6: 8064 [36] YAO F B, YANG Q, JIAN S, et al. Electrochemical reduction of bromate using noble metal-free nanoscale zero-valent iron immobilized activated carbon fiber electrode[J]. Chemical Engineering Journal, 2020, 389: 123588 -

 点击查看大图
点击查看大图

计量
- 文章访问数: 4690
- HTML全文浏览数: 4690
- PDF下载数: 91
- 施引文献: 0










































































































