









-
近年来,随着重污染工业企业的关闭和搬迁,产生了大量土壤和地下水严重污染的工业企业遗留场地,1,2-二氯乙烷(1,2-DCA)是化工污染场地典型污染物之一。1,2-DCA作为一种重要的化工原料,具有致癌、致畸、致突变“三致”效应[1-2],同时其具有易迁移和难降解的特点,会对生态环境产生危害。1,2-DCA污染地下水主要修复方法有异位修复、原位修复、监测自然衰减等。异位修复耗时长费用高,难以彻底降解1,2-DCA。原位修复包括热脱附[3]、蒸汽浸提[4]、原位化学氧化技术[5]。其中高级氧化技术能够在短时间内高效处理地下水中污染物,使其达到可接受的水平。
基于活化过硫酸盐(PS)的高级氧化技术被广泛应用于有机污染土壤和地下水治理中[6]。在碱、热、光、过渡金属等活化下,PS可分解产生氧化性更强的硫酸根自由基(
SO⋅−4 ,E0=2.5~3.1 eV)。SO⋅−4 通过吸氢、加成和电子转移与有机分子发生反应[7],从而可降解氯代烃、多环芳烃等多种有机污染物[8-11]。SO⋅−4 可由过硫酸盐阴离子(S2O2−8 )与过渡金属的反应生成(式(1)~式(2)),在碱性环境下,还会诱发链式反应产生HO·反应(式(3))。纳米零价铁(nZVI)具有粒径较小、比表面积较大、反应活性强等特点,既是良好的还原剂,又可作为PS活化剂,因此,被广泛应用于土壤和地下水修复工程[12-13]。nZVI活化PS具有较窄的pH适应范围,在碱性环境下活化效果不理想,在酸性环境下有较好的活化效果[14]。但在酸性环境下,nZVI活化PS反应快速,迅速产生大量的Fe2+,而过量的Fe2+易与自由基反应生成Fe3+,对自由基产生清除作用,不利于自由基的生成。有研究表明,螯合剂(2,2'-联吡啶、1,10-二氮菲、乙二胺四乙酸、柠檬酸等)可与Fe2+结合,降低溶液中游离Fe2+浓度,减少自由基的清除,进而提高PS对污染物氧化降解的效率[15-19]。李明等[20]发现,加入柠檬酸后,60 min内nZVI/PS体系对TCE降解率高达94.7%。ZHANG[21]的研究表明,EDTA的强化性能优于柠檬酸,这是因为EDTA与铁的螯合能力高于柠檬酸,Fe2+(EDTA)的高稳定性使得较少的
SO⋅−4 被清除。目前,nZVI/PS降解地下水中1,2-DCA尚存在诸多问题,1,2-DCA去除效果和药剂利用效率有待提高。因此,本研究拟以水中1,2-DCA为目标污染物,以nZVI为活化剂,EDTA为稳定剂,建立nZVI/PS/EDTA还原/氧化体系,以提高1,2-DCA降解效果和药剂有效利用率,考察了EDTA对nZVI/PS体系降解1,2-DCA强化效果、作用机制及影响因素,以期为1,2-DCA污染地下水化学修复提供参考。
-
实验试剂:1,2-二氯乙烷(上海凌峰化学试剂有限公司)、过硫酸钠(捷润化学)、纳米零价铁(上海麦克林生化科技有限公司,粒径50 nm,纯度>99.9%)、氢氧化钠和氯化钠(无锡市晶科化工有限公司)、氯化铵、氯化钾、乙二胺四乙酸(天津科密欧化学试剂有限公司)、碳酸氢钠(国药集团化学试剂有限公司),以上试剂均为分析纯。实验用水为超纯水,电导率为0.055 μs·cm−1。
实验仪器:气相色谱质谱联用仪(7890B GC system,5977A MSD)、离子色谱仪(Thermo-ICS-1100)、总有机碳分析仪(耶拿MultiN/C3100)、紫外可见分光光度计(岛津UV-2600)、电子分析天平(德国赛多利斯BSA124S)、水质多参数检测仪(梅特勒托利多Multiparameter)、超纯水机(Milli-Q)、数显水浴恒温振荡器(THZ-82A)、固相萃取仪(NAI-YXCQY-24A)。
-
1) EDTA强化nZVI/PS降解1,2-DCA实验。用超纯水配置初始浓度为1.8 mmol·L−1的1,2-DCA溶液至250 mL锥形瓶中,依次加入1.8 mmol nZVI、3.6 mmol PS和1.8 mmol EDTA,建立nZVI、nZVI/PS、nZVI/PS/EDTA 3种氧化/还原反应体系,分析不同反应体系下1,2-DCA的降解效果。将反应瓶放置在恒温振荡器上,转速设置为150 r·min−1,从0 min开始,分别于不同时间间隔取样,测定1,2-DCA、Fe2+、Fe3+、总有机碳(TOC)、氯离子(Cl−)等指标。每组实验取3个样品,结果数据取三者平均值。实验过程中设置只加1,2-DCA的空白实验,结果表明,实验过程中1,2-DCA损失率小于5%,1,2-DCA的挥发可以忽略不计。
为了阐明EDTA对nZVI/PS体系降解1,2-DCA的影响,定义了以nZVI计的比降解率,即单位质量nZVI降解的1,2-DCA的质量(式(4))。
式中:Sq为比降解率,mg·g−1;V为反应溶液体积,L;C0为溶液中1,2-DCA初始质量浓度,mg·L−1;Ct为t时溶液中1,2-DCA质量浓度,mg·L−1;m为nZVI的投加质量,g。
TOC是水中有机物所含碳的总量,能够直接反映1,2-DCA被彻底降解程度,矿化度根据式(5)进行计算。
式中:M为矿化度,%;T0和Tt分别为溶液中初始和t时刻的总有机碳质量浓度,mg·L−1。
利用阿伦尼乌斯公式不同温度下的表观速率常数计算1,2-DCA降解反应所需要的活化能(式(6))。
式中:k为温度T时的反应速度常数;Ea为表观活化能,J·mol−1;R为摩尔气体常数,8.314 J·mol−1·K−1;T为绝对温度,K;A0为吉布斯自由能因子。
2) EDTA强化降解1,2-DCA影响实验。向nZVI/PS体系中分别投加0.5、0.9、1.2、1.8、3.6、5.4 mmol EDTA,调节pH分别为3、7、11,考察不同pH下nZVI/PS体系和nZVI/PS/EDTA体系的1,2-DCA降解率。配置含有10 mmol阴阳离子的模拟地下水,考察不同离子对EDTA强化nZVI/PS降解1,2-DCA效果的影响。
-
溶液中1,2-DCA采用HJ 810-2016《水质 挥发性有机物的测定顶空/气相色谱-质谱法》进行检测。气相色谱条件:色谱柱型号:HP-5MS;进样口温度250 ℃;进样模式为分流进样(分流比20∶1);柱流量(恒流模式)为1.0 mL·min−1;升温程序为40 ℃保持2 min,以8 ℃·min−1的速率升至100 ℃,保持4 min,再以15 ℃·min−1的速率升至120 ℃,保持5 min。
溶液中TOC采用总有机碳分析仪(耶拿MultiN/C3100)进行测定。测定原理:NDIR,进样体积1 mL,进样次数3 次;溶液中铁离子浓度采用HJ/T 345-2007《水质 铁的测定 邻菲啰啉分光光度法》进行测定,测定波长为510 nm;溶液中Cl−浓度采用GB/T 39305-2020《再生水水质 氟、氯、亚硝酸根、硝酸根、硫酸根的测定 离子色谱法》进行检测。
-
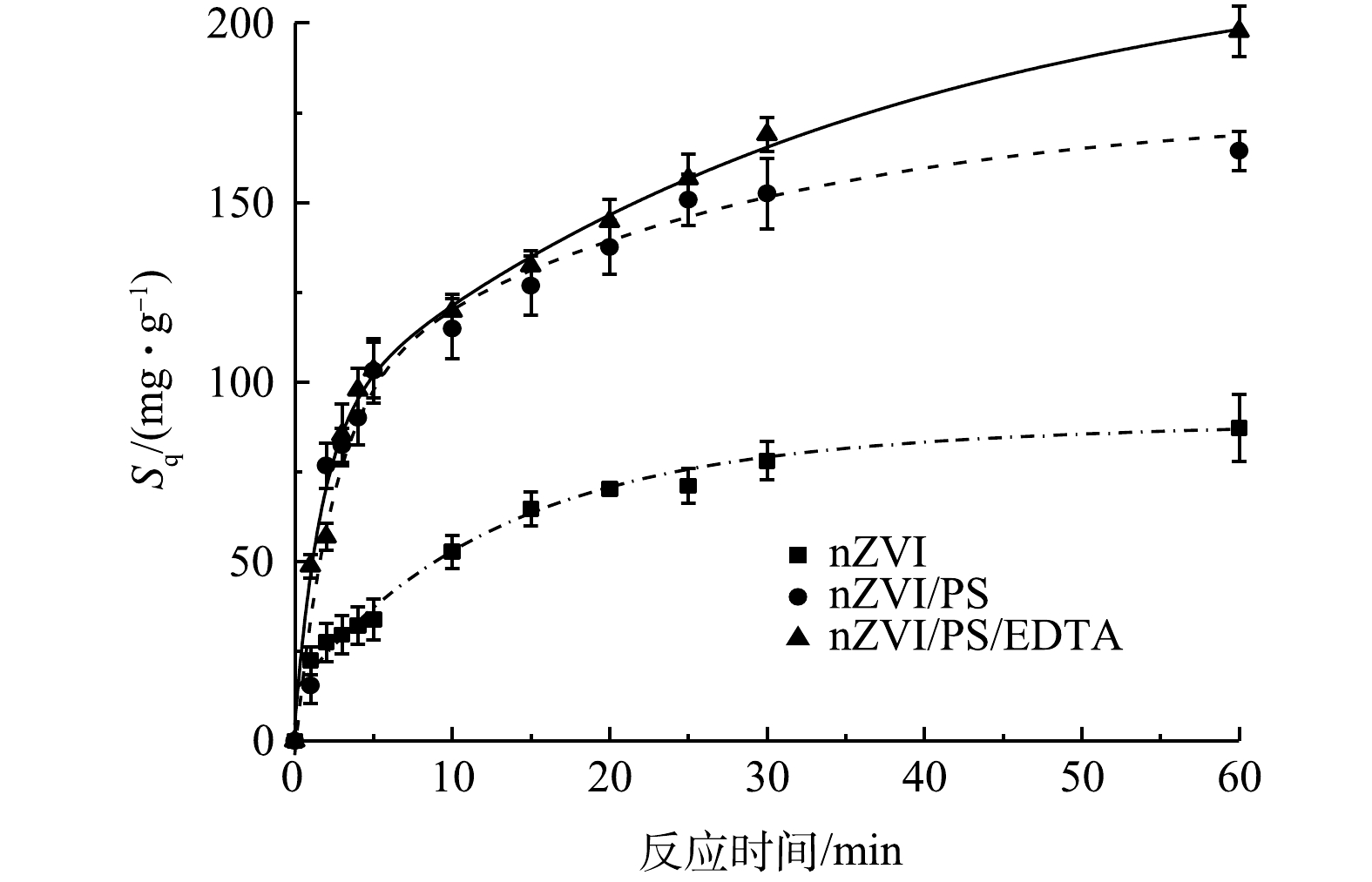
由图1可以看出,单独nZVI对1,2-DCA有一定的还原降解效果,60 min时,Sq约为90 mg·g−1;nZVI/PS体系对1,2-DCA降解效果较单独nZVI体系有显著提升,Sq约为165 mg·g−1,提高了83%;加入EDTA后,Sq进一步提升至约200 mg·g−1,较nZVI/PS体系提高了21%。在不同体系中,Sq均呈现先快速增长后缓慢增长的趋势,说明污染物降解速度逐渐变缓。这是因为随着反应进行,溶液中污染物浓度和化学药剂浓度均有所降低,导致化学反应速度逐渐变缓。
对比nZVI/PS体系和nZVI/PS/EDTA体系的Sq可知,反应初始阶段,2种体系中Sq增加速度相差不大。这说明2种体系中1,2-DCA降解速度基本相同。EDTA的加入并未在反应初始阶段快速提高1,2-DCA降解效率。这是因为:一方面,EDTA是一种弱酸,可促进nZVI转化为Fe2+,有利于活化PS产生自由基[22];另一方面,EDTA易与Fe2+发生螯合反应,可降低溶液中Fe2+质量浓度;2种作用相互拮抗,最终导致2种体系的初始反应速度基本相同。随着反应的进行,nZVI/PS/EDTA体系中1,2-DCA比降解率逐渐大于nZVI/PS体系,且Sq增长速度比nZVI/PS体系更快,EDTA对nZVI活化PS的强化效果逐渐显现。这是因为:一方面,EDTA可与Fe2+形成配合物,减小了Fe2+与自由基反应机率,降低了清除自由基的负面作用;另一方面,EDTA的弱酸性能够促进Fe2+产生,抑制氢氧化物沉淀产生[23]。EDTA的加入,提高了Fe2+在体系中的稳定性,同时提供了一种持续的弱酸环境,有利于反应过程中Fe2+的持续产生,从而提高了nZVI对PS活化的持续有效性。
-
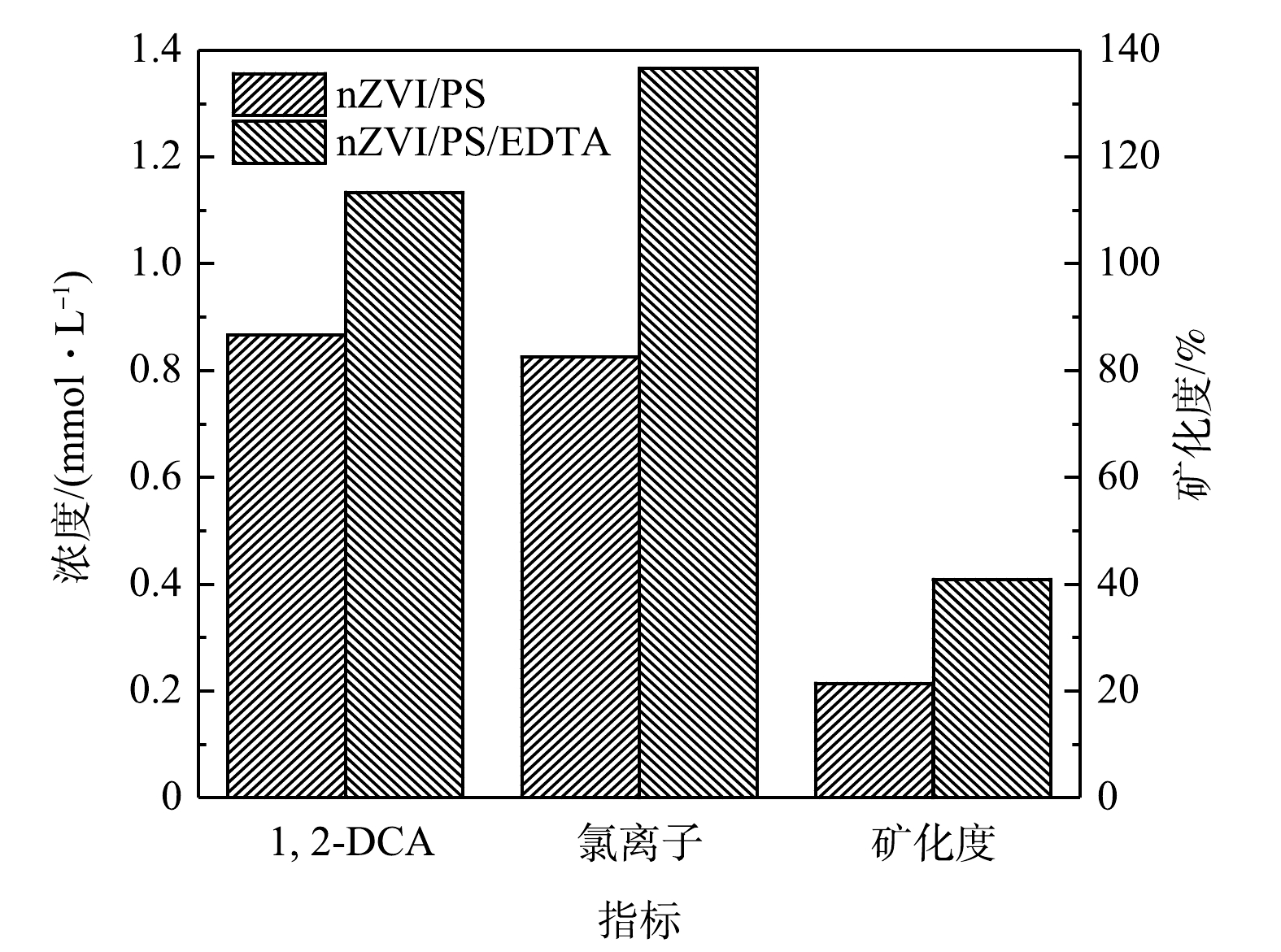
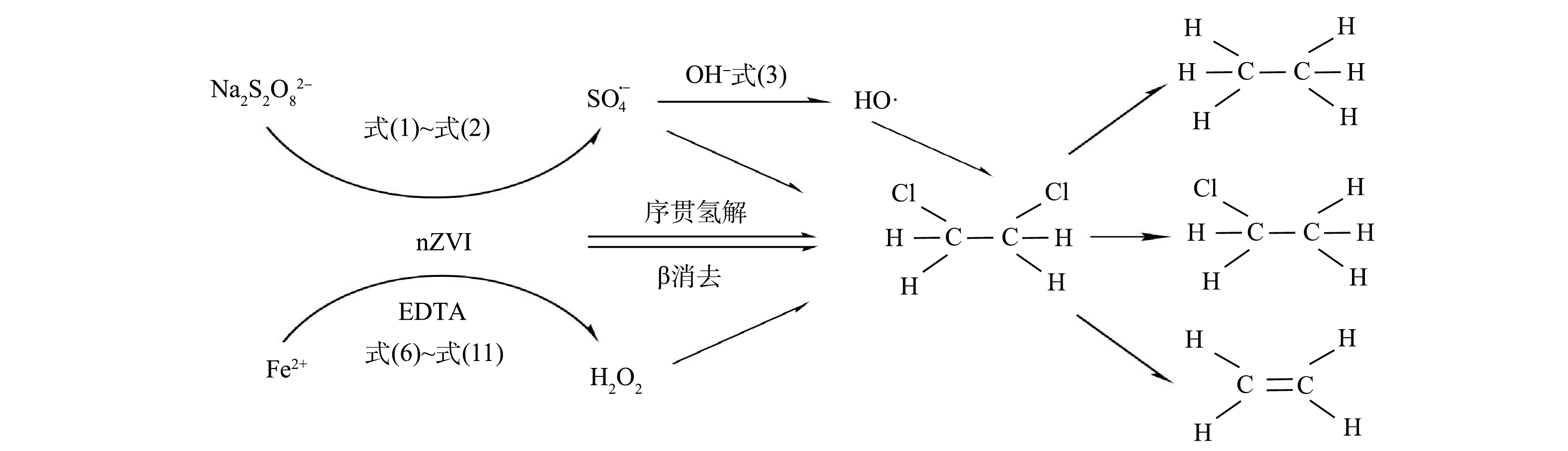
TOC是水中有机物所含碳的总量,能够直接反映1,2-DCA被彻底降解程度。另外,溶液中Cl−的浓度变化可以反映1,2-DCA的脱氯程度,也从侧面反映1,2-DCA降解的彻底程度。在1,2-DCA降解过程中,1,2-DCA去除量、Cl−脱出量和TOC的变化如图2所示。根据1,2-DCA分子组成,理论上每降解1 mmol 1,2-DCA应有2 mmol Cl−生成。由图2可以看出,在反应60 min后,nZVI/PS/EDTA体系中1,2-DCA去除量、Cl−脱出量以及1,2-DCA矿化度均较nZVI/PS体系有所升高。nZVI/PS体系中Cl−生成与1,2-DCA去除摩尔质量比约为1∶1。加入EDTA后,Cl−生成与1,2-DCA去除摩尔质量比约为1.2∶1,但仍未达到二者理论摩尔质量比(2∶1)。nZVI/PS/EDTA体系矿化度为40.84%,较nZVI/PS体系矿化度(21.43%)提高了约1.9倍。这说明1,2-DCA降解过程中存在含氯中间产物。降解途径如图3所示:首先,PS与nZVI发生如式(1)和式(2)的反应生成
SO⋅−4 ,在碱性环境下,还会诱发式(3)的反应生成HO·,进而促进1,2-DCA降解。还原脱氯过程中碳原子与氯原子断裂一般是由于序贯氢解或是β消去反应,序贯氢解是由于1个氢原子取代1个氯离子从而释放1个氯离子,β消去反应是指释放2个氯原子,由烷烃转化为烯烃的过程[24]。在这个过程中,序贯氢解占主要地位,因此,nZVI/PS和nZVI/PS/EDTA体系中乙烷均为主要的产物,并有少量的乙烯和氯乙烷产生。YPABC等[19]发现,Fe2+可以与EDTA发生如式(7)~式(11)的反应,最终生成H2O2,从而促进1,2-DCA的降解。加入EDTA后,提高了1,2-DCA的降解、还原脱氯和矿化度。主要原因如前所述,EDTA提高了nZVI活化PS的有效性和持续性,促进了1,2-DCA的持续降解,提高了其矿化程度。 -
在不同温度条件下1,2-DCA降解动力学曲线见图4,动力学参数见表1。由图4可以看出,nZVI/PS体系与nZVI/PS/EDTA体系中1,2-DCA降解动力学均符合准一级动力学模型。由于EDTA的加入,在不同温度条件下,反应速率常数均有提高。当温度为20、30、40 ℃时,nZVI/PS/EDTA体系的反应速率常数较nZVI/PS体系分别提高了约18.75%、22.95%和10.06%。这主要是因为,EDTA的加入有效减缓了Fe2+对自由基的清除,同时可为反应提供弱酸环境,因此,反应速率常数比不加EDTA时有所提高。随着温度的升高,温度逐渐成为影响反应的主导因素[25],nZVI/PS/EDTA体系反应速率常数较nZVI/PS体系的提高有所减弱。
活化能可用于区分速率限制步骤,其中表面控制反应具有较大的活化能(>29 kJ·mol−1);相反,溶液扩散控制反应具有相对较低的活化能(8~21 kJ·mol−1)[26]。由表1可看出,在相同温度下nZVI/PS/EDTA体系较nZVI/PS体系反应活化能更低,说明降解反应更容易进行。这是因为PS的水解作用会随着pH的降低和温度的升高而增强。EDTA呈弱酸性,可为反应提供H+,从而促进了PS的水解作用并降低了体系反应所需能量[27]。两者反应活化能均大于29 kJ·mol−1,说明nZVI/PS降解1,2-DCA反应为表面控制反应[28]。
-
由图5可以看出,在反应过程中,游离Fe2+和Fe3+质量浓度总体呈现先增加后稳定的趋势,nZVI/PS体系中游离Fe2+质量浓度小于nZVI/PS/EDTA体系,游离Fe3+质量浓度大于nZVI/PS/EDTA体系。游离铁离子质量浓度取决于其生成和消耗。加入EDTA后,使反应体系呈弱酸性,促进了Fe2+的产生;另一方面,EDTA>与部分Fe2+发生络合作用生成Fe2+(EDTA)络合物,降低了Fe2+与自由基反应机率,从而减少了Fe2+与自由基反应导致的无效消耗(式(12)),使反应体系保留较高质量浓度的游离Fe2+和较高的降解效率[29]。这与前述章节EDTA对1,2-DCA降解效果影响的结果一致。EDTA与Fe2+和Fe3+均可发生络合作用,nZVI/PS/EDTA体系中EDTA与Fe3+的络合化合物是导致游离Fe3+浓度低于nZVI/PS体系的主要原因[30]。其次,与nZVI/PS体系相比,nZVI/PS/EDTA体系中Fe2+与自由基反应减少,Fe3+的产生也相应减少,因此,总体浓度低于nZVI/PS体系。
-
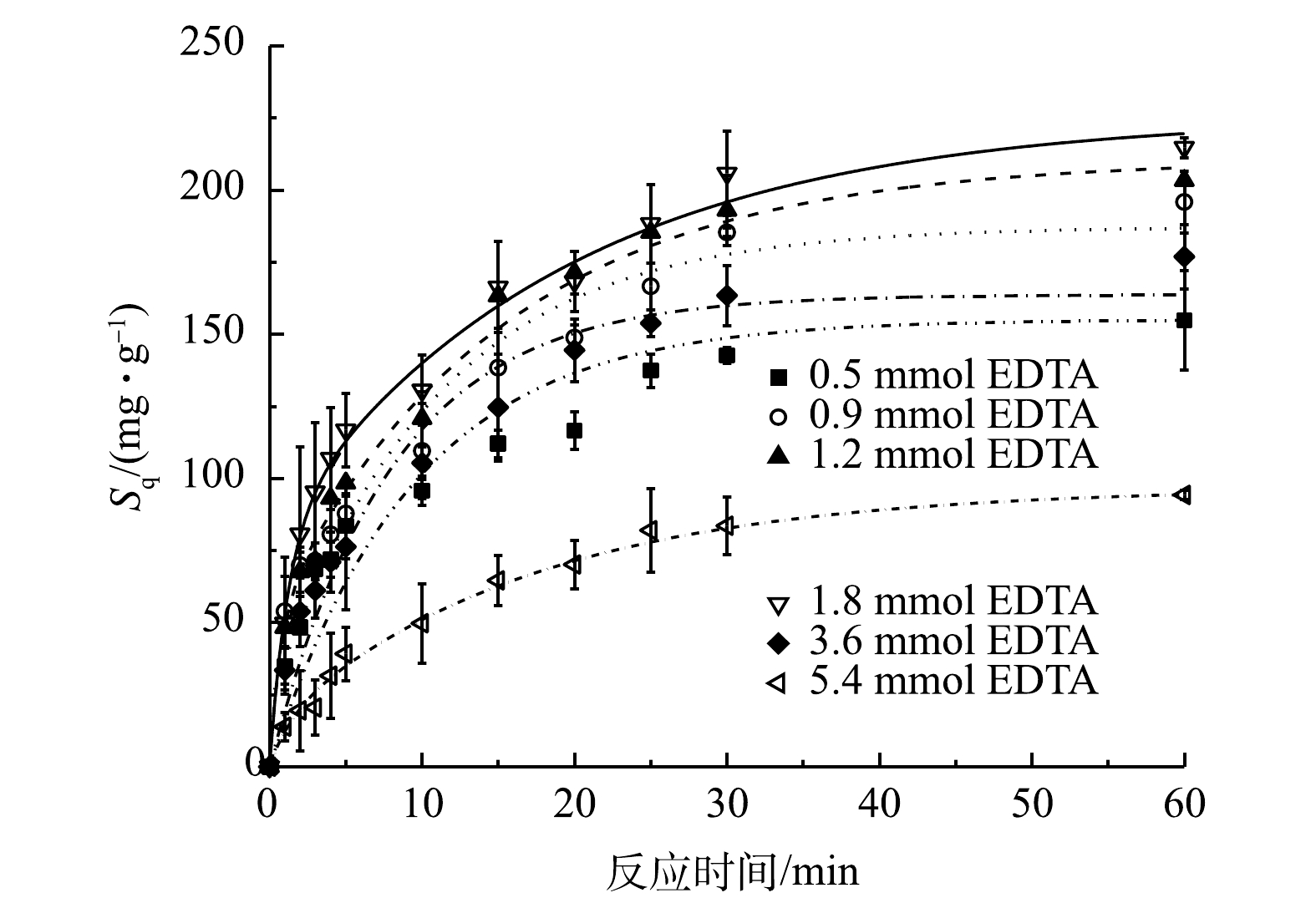
由图6可以看出,随着EDTA投加量的增加,Sq呈现先增加后减小的趋势,当EDTA投加量为1.8 mmol时达到最大值,约为215 mg·g−1。这表明适量的EDTA促进nZVI/PS降解1,2-DCA,过量的EDTA则会起到抑制作用。这是因为:当EDTA投加量较低时,EDTA与Fe2+产生少量络合,增加了体系中游离Fe2+质量浓度,有利于强化nZVI/PS体系对1,2-DCA降解。当EDTA过量时,大量的Fe2+与EDTA形成络合物,体系中游离Fe2+质量浓度降低,对PS的活化效果降低,不利于体系中自由基的产生。另一方面,过量的EDTA存在会与1,2-DCA竞争体系中的HO·,反应速率常数达到8.6×106 mol·(L·s)−1 ,消耗体系中的自由基,从而抑制了nZVI/PS对1,2-DCA降解[31]。
-
由图7可以看出,酸性环境更有利于nZVI/PS对1,2-DCA的降解。HUSSAIN等[32]提出,酸性环境比中性和碱性环境更有利于五氯苯甲醚的降解,当pH高于4时,Fe2+对
S2O2−8 活化效率将会降低,这与本实验研究结论一致。LIANG等[33]发现,pH影响SO⋅−4 和HO·的生成,在酸性环境下SO⋅−4 是主要自由基;而在强碱环境中,HO·成为主要自由基。SO⋅−4 较HO·具有更高的氧化还原电位(ESO4·-=2.5~3.1 V,EHO·=1.9~2.7 V)[34-35],因此,在酸性环境下1,2-DCA具有更高的降解效率。当pH为3、7、11时,反应60 min后,nZVI/PS/EDTA体系的Sq较nZVI/PS体系分别提高了约41.76、36.31和24.80 mg·g−1。这说明EDTA在酸性和碱性环境下均有一定的强化效果,但酸性环境更有利于EDTA强化nZVI活化PS降解1,2-DCA。这主要是因为:一方面,在碱性环境下,Fe2+容易与氢氧根形成氢氧化物;另一方面,氢氧根与EDTA产生中和作用,削弱了EDTA弱酸性对反应的积极作用。
-
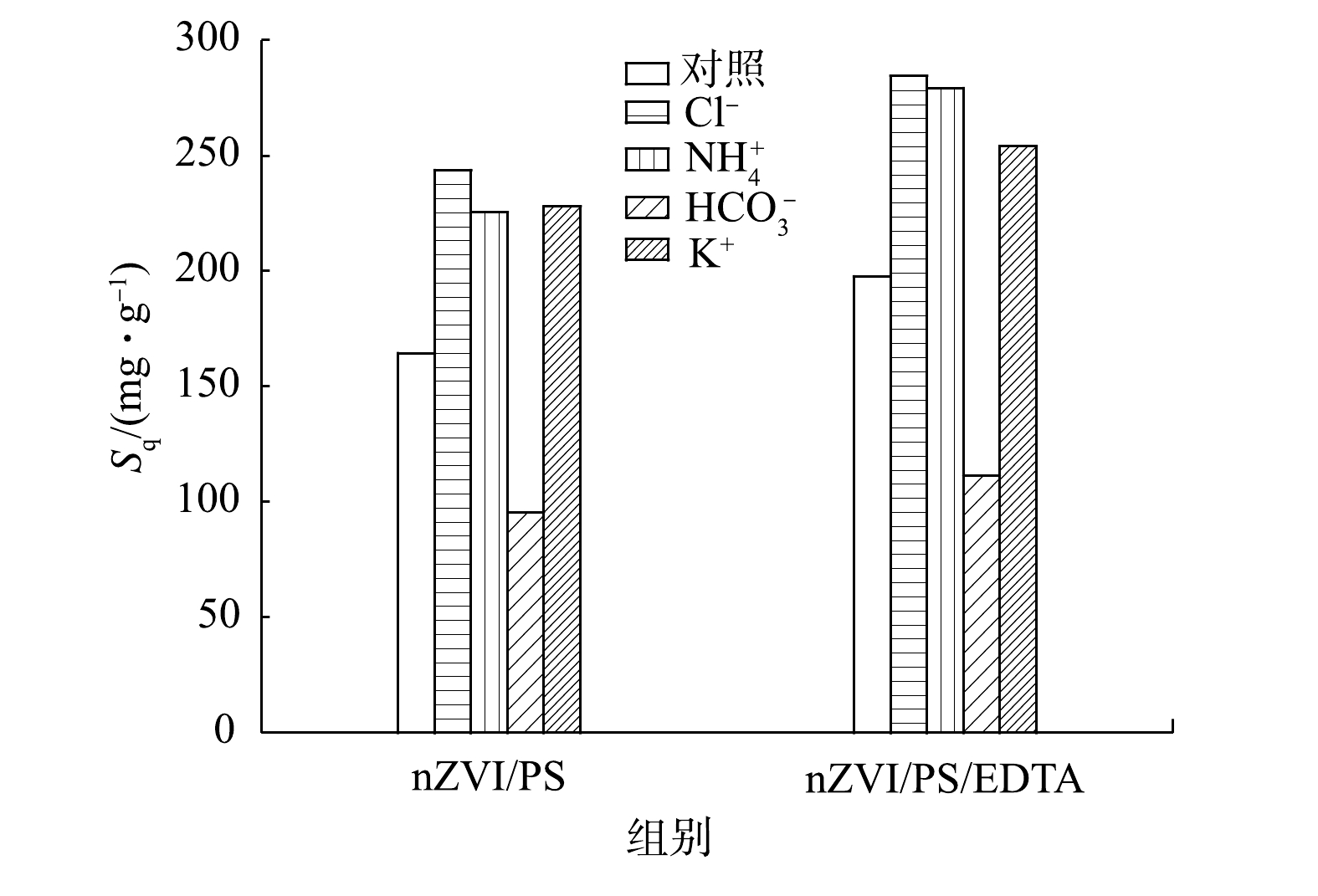
对照组为在反应体系中不含阴阳离子。由图8可以看出,Cl−、NH4+和K+使nZVI/PS体系中Sq分别提高了约79.31、60.94和63.54 mg·g−1,使nZVI/PS/EDTA体系中Sq分别提高了约86.75、81.54和56.43 mg·g−1。
HCO−3 分别使nZVI/PS体系和nZVI/PS/EDTA体系中Sq降低了68.99和86.28 mg·g−1。这说明离子对nZVI/PS体系和nZVI/PS/EDTA体系呈现相同作用规律,即Cl−、NH+4 和K+均提高了1,2-DCA去除效率。这是因为Cl−能够通过位点腐蚀的方式进入钝化层内部与Fe2+结合形成易溶于水的FeCl2,并且Cl−的加入能够增加反应体系的电导率,因而促进nZVI腐蚀加速进行;K+和NH+4 存在会在有限范围提高1,2-DCE的降解率。这可能是由于K+和NH+4 的存在提高了反应体系的电导率,加快了nZVI腐蚀[36-37]。而HCO−3 则会抑制1,2-DCA的去除。这是因为:HCO−3 的加入会显著提高体系pH,该体系在碱性环境下降解率会有所削弱;此外,HCO−3 会与铁离子或亚铁离子反应生成铁的碳酸盐沉淀及其络合物,导致nZVI钝化并阻隔活性位点[25]。另外,对照组为在nZVI/PS反应体系中加入EDTA,其对Sq的提升率为21%。与对照组相比,引入
NH+4 后Sq由225.3 mg·g−1增至279.2 mg·g−1,提升率为23.9%。除NH+4 外,引入Cl−、K+和HCO−3 对Sq的提升率均低于对照组。尤其当K+存在时,Sq由227.92 mg·g−1增至254.08 mg·g−1,提升率仅为11.5%,EDTA对nZVI/PS降解1,2-DCA的强化效果降低约50%。这说明EDTA在实际地下水修复中可能因复杂的水质环境而难以达到理想效果。 -
1) EDTA对nZVI/PS降解1,2-DCA具有良好的强化效果,Sq和矿化度分别提高了21%和190%,在30 ℃时反应速率常数提高了22.95%,反应活化能降低了8.91 kJ·mol−1。
2) EDTA强化nZVI/PS降解1,2-DCA作用机制主要有2点:一是为反应提供了弱酸环境,促进Fe2+生成;二是EDTA与Fe2+反应生成螯合物,降低了Fe2+与自由基反应机率,提高了nZVI活化PS的持续有效性。
3) EDTA投加量、pH和水中阴阳离子均影响nZVI/PS/EDTA对1,2-DCA的降解效果。Sq随EDTA投加量先增加后减小,EDTA最佳投加量为1.8 mmol,Sq达到最大值214.798 mg·g−1;酸性环境更有利于EDTA强化nZVI/PS降解1,2-DCA;Cl−、K+和
HCO−3 均抑制EDTA强化作用,EDTA在实际复杂的水质环境条件下可能难以达到理想效果。
EDTA强化nZVI/PS降解地下水中1,2-二氯乙烷的作用机制及影响因素
Mechanism and influencing factors of 1,2-dichloroethane degradation in groundwater by EDTA-enhanced nZVI/PS
-
摘要: 采用乙二胺四乙酸(EDTA)强化纳米零价铁(nZVI)活化过硫酸钠(PS)降解地下水中1,2-二氯乙烷(1,2-DCA)。通过分析1,2-DCA比降解率(Sq)、矿化度、1,2-DCA降解动力学及游离Fe2+和Fe3+质量浓度变化规律,阐明了EDTA强化效果及作用机制;考察了EDTA投加量、pH、阴阳离子对EDTA强化效果的影响。结果表明:经EDTA强化后,1,2-DCA的Sq和矿化度分别提高了21%和190%,反应速率常数提高了10.06%~22.95%,活化能降低了6.98~8.91 kJ·mol−1;EDTA为反应提供了弱酸性环境,促进Fe2+生成,EDTA与Fe2+生成螯合物,降低了Fe2+与自由基反应的机率,提高了nZVI/PS的持续有效性;Sq随EDTA投加量的加大先增加后减小,EDTA投加量为1.8 mmol时Sq达到最大值214.80 mg·g−1。酸性环境下EDTA的强化效果更好。除
NH+4 外,Cl−、K+和HCO−3 均抑制EDTA的强化作用,说明EDTA在实际地下水修复中可能因多种离子存在而难以达到理想的强化效果。以上研究结果可为EDTA强化nZVI/PS降解地下水中1,2-DCA提供参考。Abstract: Ethylenediaminetetraacetic acid (EDTA) was used to strengthen nano-zero-valent iron (nZVI) activated sodium persulfate (PS) for 1,2-dichloroethane (1,2-DCA) degradation in groundwater. Through analyzing the specific degradation rate (Sq), mineralization, degradation kinetics of 1,2-DCA and mass concentration change rules of Fe2+ and Fe3+, the enhancement effect of EDTA and its mechanism were clarified. The effects of EDTA dosage, pH, anion and cation on the strengthening effect of EDTA were investigated. The results showed that after EDTA strengthening, Sq and the degree of mineralization of 1,2-DCA increased by 21% and 190%, respectively. Reaction rate increased by about 10.06%~22.95% and activation energy decreased by 6.98~8.91 kJ·mol−1. EDTA provided a weak acid environment for the promotion of Fe2+ formation. Then EDTA formed a chelate with Fe2+ which reduced the probability of the reaction between Fe2+ and free radicals, and improved the sustained effectiveness of nZVI/PS. Sq first increased and then decreased with the increase of EDTA dosage. Sq reached the maximum value of 214.80 mg·g−1 at EDTA dosage of 1.8 mmol. The strengthening effect of EDTA was better in acidic environment. ExceptNH+4 had a promoting effect, Cl−, K+ andHCO−3 all inhibited the strengthening effect of EDTA, indicating that EDTA might be difficult to achieve an ideal strengthening effect in remediation of actual groundwater due to the existence of multiple ions. The research results provide basic parameters and technical supports for degradation of 1,2-DCA in groundwater by EDTA-enhanced nZVI/PS.-
Key words:
- groundwater /
- 1,2-dichloroethane /
- PS advanced oxidation /
- EDTA enhancement /
- specific degradation rate
-
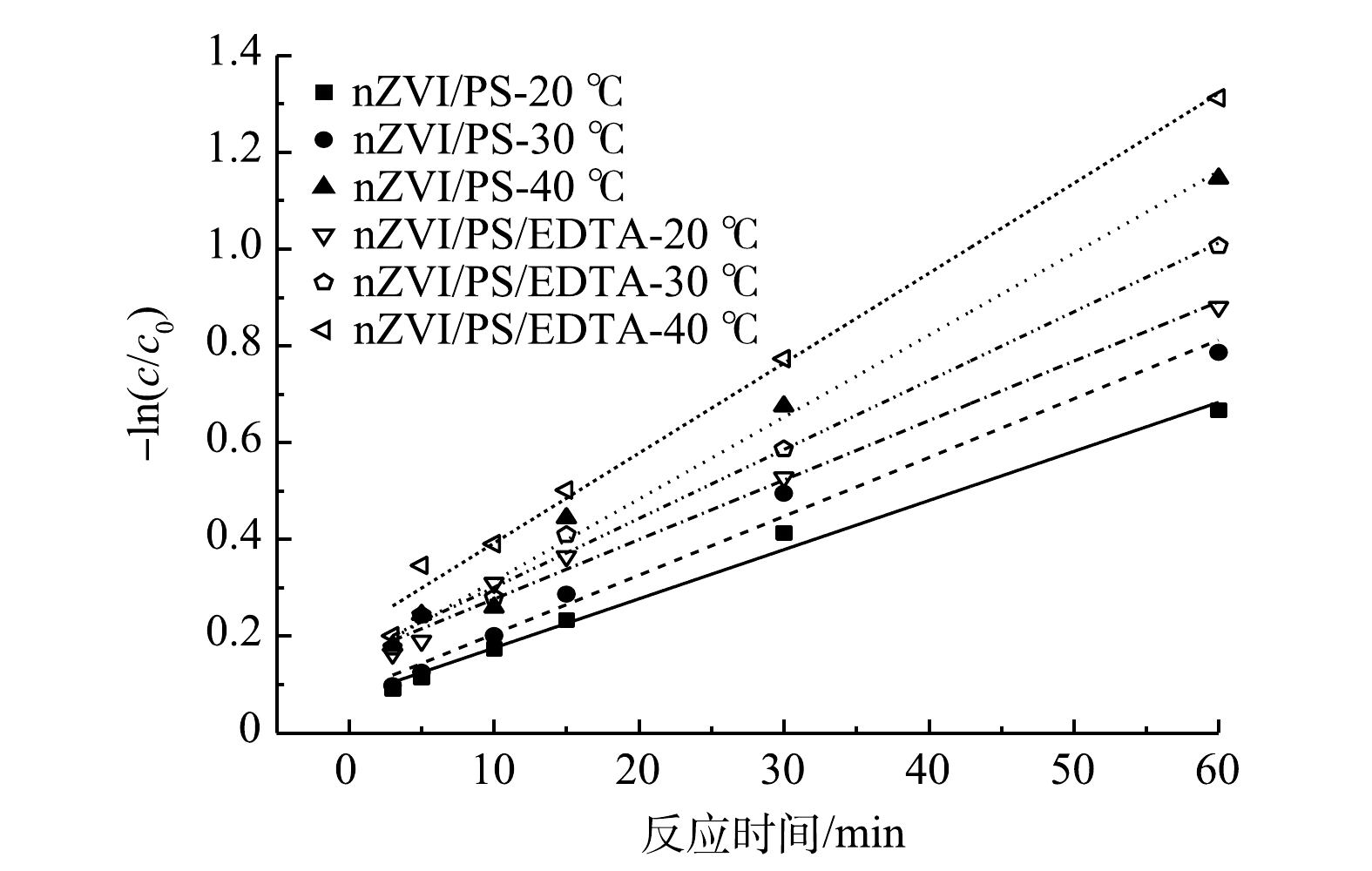
图 4 不同温度下1,2-DCA降解动力学曲线
Figure 4. Degradation kinetic curve of 1,2-DCA at different temperatures
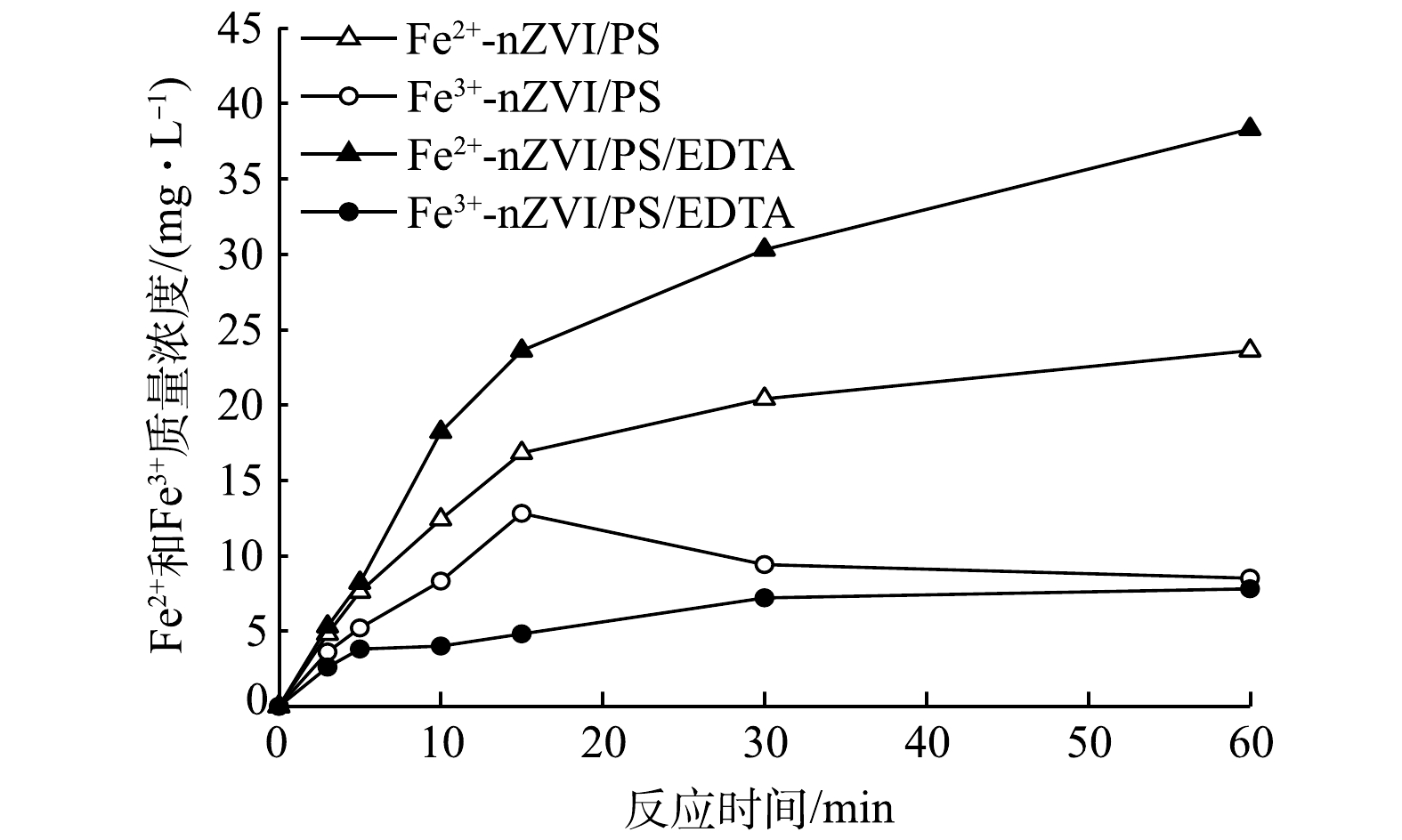
图 5 不同体系Fe2+、Fe3+的质量浓度变化
Figure 5. Changes in the mass concentration of Fe2+ and Fe3+ in different systems
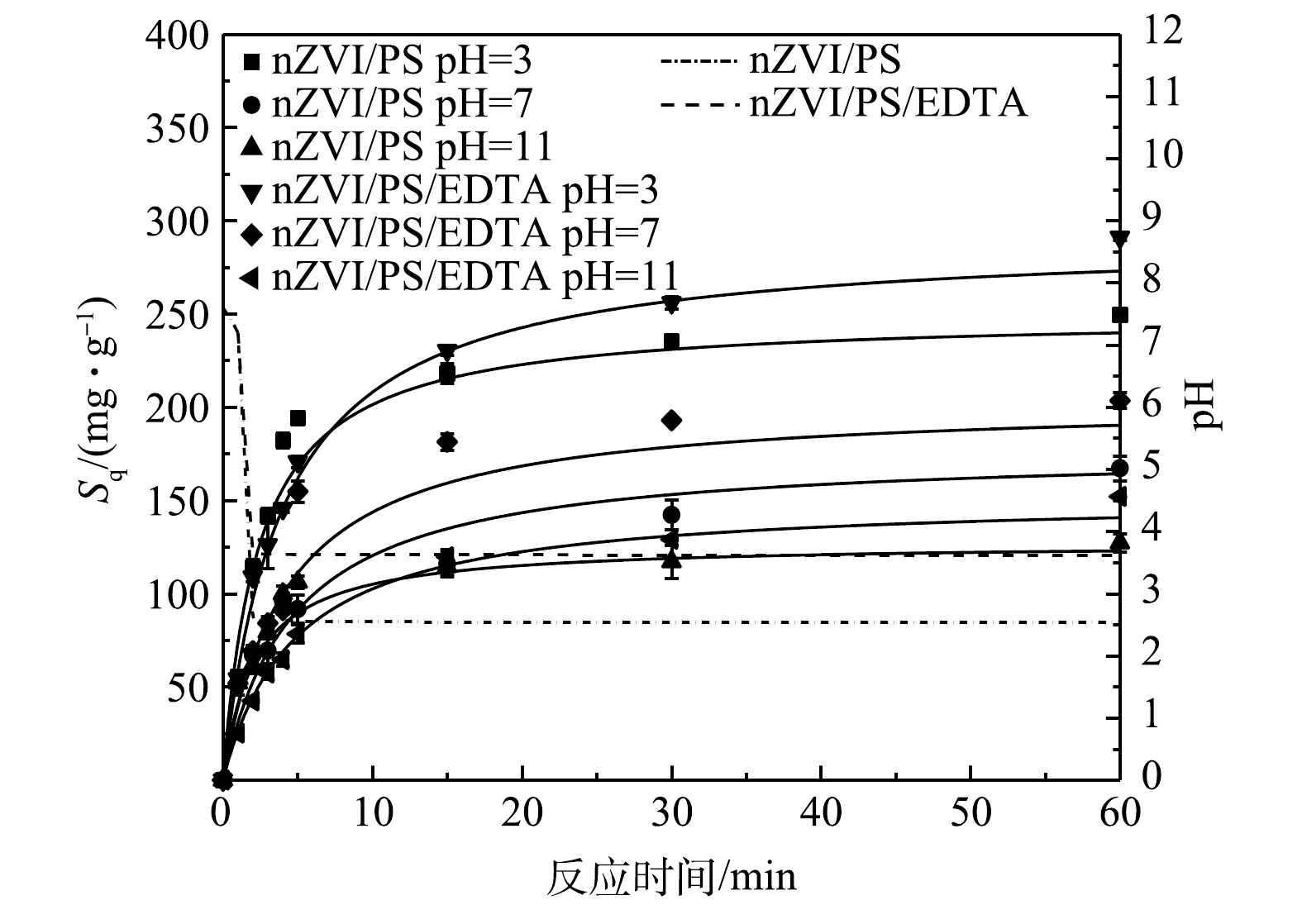
图 7 体系pH变化及pH对不同反应体系Sq的影响
Figure 7. pH change and the influence of pH on Sq of different reaction systems
表 1 动力学参数
Table 1. Kinetic parameters
体系 温度/℃ k/min−1 R2 Ea/(kJ·mol−1) nZVI/PS 20 0.003 2 0.992 8 64.22 30 0.012 2 0.988 0 63.04 40 0.016 9 0.990 8 62.27 nZVI/PS/EDTA 20 0.003 8 0.990 6 57.24 30 0.015 0 0.963 3 54.13 40 0.018 6 0.992 1 53.64  下载: 导出CSV
下载: 导出CSV
-
[1] DOHERTY R E. A history of the production and use of carbon tetrachloride, tetrachloroethylene, trichloroethylene and 1, 1, 1-trichloroethane in the United States: part 1: historical background; carbon tetrachloride and tetrachloroethylene[J]. Environmental Forensics, 2000, 1(2): 69-81. doi: 10.1006/enfo.2000.0010 [2] HUANG B, CHAO L, WEI C, et al. Chlorinated volatile organic compounds (Cl-VOCs) in environment-sources, potential human health impacts, and current remediation technologies[J]. Environment International, 2014, 71(10): 118-138. [3] 孟宪荣, 葛松, 许伟, 等. 原位电阻热脱附修复氯代烃污染土壤[J]. 环境工程学报, 2021, 15(2): 669-676. doi: 10.12030/j.cjee.202009077 [4] 张明, 赵怡阳, 徐辰. 气相抽提与氧化压裂技术结合在有机污染土壤修复工程中的应用[J]. 环境与发展, 2020, 32(6): 88-90. [5] UNGTAE K, PARKER, BORDEN. Stochastic cost-optimization and risk assessment of in situ chemical oxidation for dense non-aqueous phase liquid (DNAPL) source remediation[J]. Stochastic Environmental Research and Risk Assessment, 2019, 33(1): 73-89. doi: 10.1007/s00477-018-1633-y [6] 蒋梦迪, 季跃飞, 陆隽鹤. 碳酸盐活化过硫酸盐降解对乙酰氨基酚的研究//清华大学持久性有机污染物研究中心[J]. 持久性有机污染物论坛2017暨第十二届持久性有机污染物学术研讨会论文集. 武汉, 2017: 223-225. [7] NETA P, MADHAVAN V, ZEME H, et al. Rate constants and mechanism of reaction of SO4·- with aromatic compounds[J]. Chemischer Informationsdienst, 1977, 8(14): 163-164. [8] YANG X, DING X, ZHOU L, et al. Oxygen dependent oxidation of trimethoprim by sulfate radical: kinetic and mechanistic investigations[J]. Chinese Chemical Letters, 2021, 32(10): 3164-3168. doi: 10.1016/j.cclet.2021.02.062 [9] USHANI U, LU X, WANG J, et al. Sulfate radicals-based advanced oxidation technology in various environmental remediation: a state-of-the-art review[J]. Chemical Engineering Journal, 2020, 402: 126232. doi: 10.1016/j.cej.2020.126232 [10] WANG W, CHEN M, WANG D, et al. Different activation methods in sulfate radical-based oxidation for organic pollutants degradation: catalytic mechanism and toxicity assessment of degradation intermediates[J]. Science of the Total Environment, 2021, 772: 145522. doi: 10.1016/j.scitotenv.2021.145522 [11] 郝慧茹, 张倩, 李孟, 等. 改性生物炭负载纳米零价铁活化过硫酸盐降解活性蓝19的机理及老化研究[J]. 环境科学学报, 2021, 41(2): 477-485. [12] WANG R, LI C, LV N, et al. Deeper insights into effect of activated carbon and nano-zero-valent iron addition on acidogenesis and whole anaerobic digestion[J]. Bioresource Technology, 2021, 324: 124671. doi: 10.1016/j.biortech.2021.124671 [13] XU H, LIU Y, YANG B, et al. Inhibitory effect of released phosphate on the ability of nano zero-valent iron to boost anaerobic digestion of waste-activated sludge and the remediation method[J]. Chemical Engineering Journal, 2020, 405: 126506. [14] KIM C, AHN J Y, KIM T Y, et al. Activation of persulfate by nanosized zero-valent iron (nZVI): mechanisms and transformation products of nZVI[J]. Environmental Science & Technology, 2018, 52(6): 3625-3633. [15] LIANG C J, LIANG C P, CHEN C C. pH dependence of persulfate activation by EDTA/Fe(III) for degradation of trichloroethylene[J]. Journal of Contaminant Hydrology, 2009, 106(3/4): 173-182. [16] LIANG C J, BRUELL C J, MARLEY M C, et al. Persulfate oxidation for in situ remediatin of TCE. II activated by chelated ferrous ion[J]. Chemosphere, 2004, 55(9): 1225-1233. doi: 10.1016/j.chemosphere.2004.01.030 [17] WU X L, GU X G, LU S G, et al. Degradation of trichloroethylene in aqueous solution by persulfate activated with citric acid chelated ferrous ion[J]. Chemical Engineering Journal, 2014, 255: 585-592. doi: 10.1016/j.cej.2014.06.085 [18] YPABC D, MZBC D, QI W, et al. EDTA, oxalate, and phosphate ions enhanced reactive oxygen species generation and sulfamethazine removal by zero-valent iron-sciencedirect[J]. Journal of Hazardous Materials, 2020, 391: 12200. [19] DONG H, HE Q, ZENG G, et al. Degradation of trichloroethene by nanoscale zero-valent iron (nZVI) and nZVI activated persulfate in the absence and presence of EDTA[J]. Chemical Engineering Journal, 2017, 316(Complete): 410-418. [20] 李明, 孙勇, 顾梦斌, 等. 柠檬酸强化纳米零价铁活化过硫酸钠体系降解水溶液中的三氯乙烯[J]. 环境工程学报, 2020, 14(3): 569-578. doi: 10.12030/j.cjee.201905182 [21] ZHANG K J, ZHOU X Y, ZHANG T Q, et al. Degradation of the earthy and musty odorant 2, 4, 6-tricholoroanisole by persulfate activated with iron of different valences[J]. Environmental Science and Pollution Research, 2018, 25(4): 3435-3445. doi: 10.1007/s11356-017-0452-x [22] 余文杰. 亚铁离子活化过硫酸盐氧化降解普施安蓝MX-R的过程强化研究[D]. 重庆: 重庆大学, 2019. [23] 钟燕清, 张永清, 陈宪方, 等. 不同螯合剂对零价铁活化过硫酸盐降解对氯苯胺的影响[J]. 环境化学, 2015, 34(4): 685-691. doi: 10.7524/j.issn.0254-6108.2015.04.2014101302 [24] 王薇. 包覆型纳米铁的制备及用于地下水污染修复的实验研究[D]. 天津: 南开大学, 2008. [25] 王胜军. 零价金属强化氧化工艺去除水中邻苯二甲酸二丁酯的研究[D].哈尔滨: 哈尔滨工业大学, 2010. [26] 张良波, 许春红, 祝慧娜, 等. Fe2+/过硫酸钠体系降解盐酸四环素的研究[J]. 环境污染与防治, 2017, 39(7): 776-779. [27] 陈希. 基于过硫酸盐高级氧化法的精对苯二甲酸生产废水处理及机理研究[D]. 天津: 河北工业大学, 2019. [28] 金晓英, 李任超, 陈祖亮. 纳米零价铁活化过硫酸钠降解2, 4-二氯苯酚[J]. 环境化学, 2014, 33(5): 812-818. doi: 10.7524/j.issn.0254-6108.2014.05.007 [29] HE J, YANG X, MEN B, et al. EDTA enhanced heterogeneous Fenton oxidation of dimethyl phthalate catalyzed by Fe: kinetics and interface mechanism[J]. Journal of Molecular Catalysis A-Chemical, 2015, 408: 179-188. doi: 10.1016/j.molcata.2015.07.030 [30] MUSHTAQUE A, TEEL A L, OLHA S, et al. Oxidative and reductive pathways in iron-ethylenediaminetetraacetic acid-activated persulfate systems[J]. Journal of Environmental Engineering, 2012, 138(4): 411-418. doi: 10.1061/(ASCE)EE.1943-7870.0000496 [31] GDS A, EOM A LLB B, et al. Contrasting the performance of photo-fenton at neutral pH in the presence of different organic iron-complexes using hydrogen peroxide or persulfate as oxidants for naproxen degradation and removal of antimicrobial activity[J]. Process Safety and Environmental Protection, 2021, 147: 798-807. doi: 10.1016/j.psep.2021.01.005 [32] HUSSAIN I, ZHANG Y, HUANG S, et al. Degradation of p-chloroaniline by persulfate activated with zero-valent iron[J]. Chemical Engineering Journal, 2012, 203(5): 269-276. [33] LIANG C, SU H W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate[J]. Industrial & Engineering Chemistry Research, 2009, 48(11): 5558-5562. [34] BUXTON G V, GREENSTOCK C L, HELMAN W P, et al. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O) in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17(2): 513-886. doi: 10.1063/1.555805 [35] NETA P, HUIERE, ROSS A B. Rate constants for reactions of inorganic radicals in aqueous solution[J]. National Bureau of Stan, 1979, 17(3): 1027-1284. [36] 董书玉. 零价铁活化过硫酸盐降解水中萘普生的研究[D]. 吉林: 东北电力大学, 2020. [37] 张珍. 纳米级双金属体系对水中氯苯和多氯联苯的催化还原脱氯研究[D]. 杭州: 浙江大学, 2013. -

 点击查看大图
点击查看大图
图( 8) 表( 1)
计量
- 文章访问数: 4784
- HTML全文浏览数: 4784
- PDF下载数: 100
- 施引文献: 0





















