

</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M12">$ {K}_{\mathrm{L}} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" colspan="3" align="center" valign="middle">Langmuir模型</td><td rowspan="2" class="table_top_border" style="width:0.5%" align="center" valign="middle"></td><td class="table_top_border" colspan="3" align="center" valign="middle">Freundlich模型</td></tr><tr><td class="table_top_border2" align="center" valign="middle"><i>q</i><sub><i>m</i></sub>/(mg·g<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M12">$ {K}_{\mathrm{L}} $</tex-math><alternatives><img class="graphic" src="202111108-liujie_M12.jpg"><img class="graphic" src="202111108-liujie_M12.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td> <td class="table_top_border2" align="center" valign="middle"><i>n</i></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M13">$ {K}_{\mathrm{F}} $</tex-math><alternatives><img class="graphic" src="202111108-liujie_M13.jpg"><img class="graphic" src="202111108-liujie_M13.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td></tr></thead>
<tbody><tr><td class="table_top_border2 table_bottom_border" align="center" valign="middle">17.14</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.372</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.895</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle"></td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">2.729</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">17.760</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.904</td></tr> </tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td> <td class="table_top_border2" align="center" valign="middle"><i>n</i></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M13">$ {K}_{\mathrm{F}} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" colspan="3" align="center" valign="middle">Langmuir模型</td><td rowspan="2" class="table_top_border" style="width:0.5%" align="center" valign="middle"></td><td class="table_top_border" colspan="3" align="center" valign="middle">Freundlich模型</td></tr><tr><td class="table_top_border2" align="center" valign="middle"><i>q</i><sub><i>m</i></sub>/(mg·g<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M12">$ {K}_{\mathrm{L}} $</tex-math><alternatives><img class="graphic" src="202111108-liujie_M12.jpg"><img class="graphic" src="202111108-liujie_M12.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td> <td class="table_top_border2" align="center" valign="middle"><i>n</i></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M13">$ {K}_{\mathrm{F}} $</tex-math><alternatives><img class="graphic" src="202111108-liujie_M13.jpg"><img class="graphic" src="202111108-liujie_M13.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td></tr></thead>
<tbody><tr><td class="table_top_border2 table_bottom_border" align="center" valign="middle">17.14</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.372</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.895</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle"></td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">2.729</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">17.760</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.904</td></tr> </tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_top_border2" align="center" valign="middle"><i>R</i><sup>2</sup></td></tr></thead>
<tbody><tr><td class="table_top_border2 table_bottom_border" align="center" valign="middle">17.14</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.372</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.895</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle"></td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">2.729</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">17.760</td><td class="table_top_border2 table_bottom_border" align="center" valign="middle">0.904</td></tr> </tbody>
</table></div></foreignObject></svg>)
-
淡水危机一直是人类生存面临的主要挑战之一[1-2],而脱盐是解决淡水危机的重要途径之一。目前被广泛应用的脱盐技术包括反渗透[3]、蒸馏[4]、电渗析[5]等,但这些技术仍存在着二次污染、能耗高、成本高等弊端。基于双电层理论的电容去离子技术(capacitive deionization, CDI)是一种操作简单、能耗低(其运行电压一般在1.2 V以下)、绿色环保的新型电化学脱盐技术,已经被越来越多的研究者所关注。
但CDI在运行过程中,由于同离子效应的存在,系统的电荷效率很低(60%左右)。为了避免同离子效应,提高电荷利用率,在CDI装置中正/负电极表面分别增加阴/阳离子交换膜,利用阴阳离子交换膜的选择透过性来提升脱盐系统电荷效率,形成了膜电容去离子技术(membrane capacitive deionization, MCDI)[6]。有研究[7]表明,MCDI脱盐性能除了与运行参数有关外,电极材料也是影响MCDI脱盐性能的关键因素。目前,应用较多的电极活性吸附材料有活性炭[8]、炭气凝胶[9]和石墨烯[10]等。其中,活性炭因廉价易得,具有丰富的孔径和良好的稳定性而被研究者们重点关注。但因其导电性差,在应用过程中通常需要添加导电剂炭黑制备混合电极,并利用粘结剂来增强混合电极的机械性能,以提升MCDI装置的脱盐能力。而混合电极中各组分(活性炭、炭黑及粘结剂)的质量比对其脱盐性能具有重要的影响,但此方面的研究尚鲜见报道。
本研究考察了MCDI混合电极中材料质量比对脱盐性能的影响,并探究了加电电压、进水流速以及进水盐浓度等运行参数对MCDI系统脱盐性能的影响; 进一步利用循环伏安法和电化学阻抗谱测试对不同质量比的电极进行了电化学表征;最后利用热力学和动力学方程对不同组分电极的吸附过程进行了模拟,以期得到此混合电极的最佳运行条件及动力学热力学参数,为后期工程化应用提供参考。
-
电容性活性炭(activated carbon, AC)购自福州益环碳素有限公司,炭黑(carbon black, CB)购自阿法埃莎(中国)化学有限公司,聚四氟乙烯(polytetrafluroethylene, PTFE)购自东莞市展阳高分子材料有限公司,用于制作电极片的粘结剂。无水乙醇和氯化钠购自天津市永大化学试剂有限公司,所用试剂均为分析纯,且未经二次处理。
-
AC∶CB∶PTFE按照质量比为8∶1∶1的比例,分别称取0.288 g AC、0.036 g CB和0.036 g PTFE置于烧杯中。加入适量无水乙醇,并搅拌使其混匀成橡皮泥状后均匀涂覆在4.5 cm×5 cm的钛网上。施加0.1 MPa压力将材料与钛网压紧,压制后的电极片静置几分钟,待表面乙醇挥发后,置于50 ℃恒温干燥箱中干燥5~10 min,即得MCDI电极,此电极命名为8-1-1。按照同样的方法制备AC∶CB∶PTFE质量比分别为9∶0∶1、6∶3∶1、0∶9∶1的电极片,分别命名为9-0-1、6-3-1、0-9-1。
-
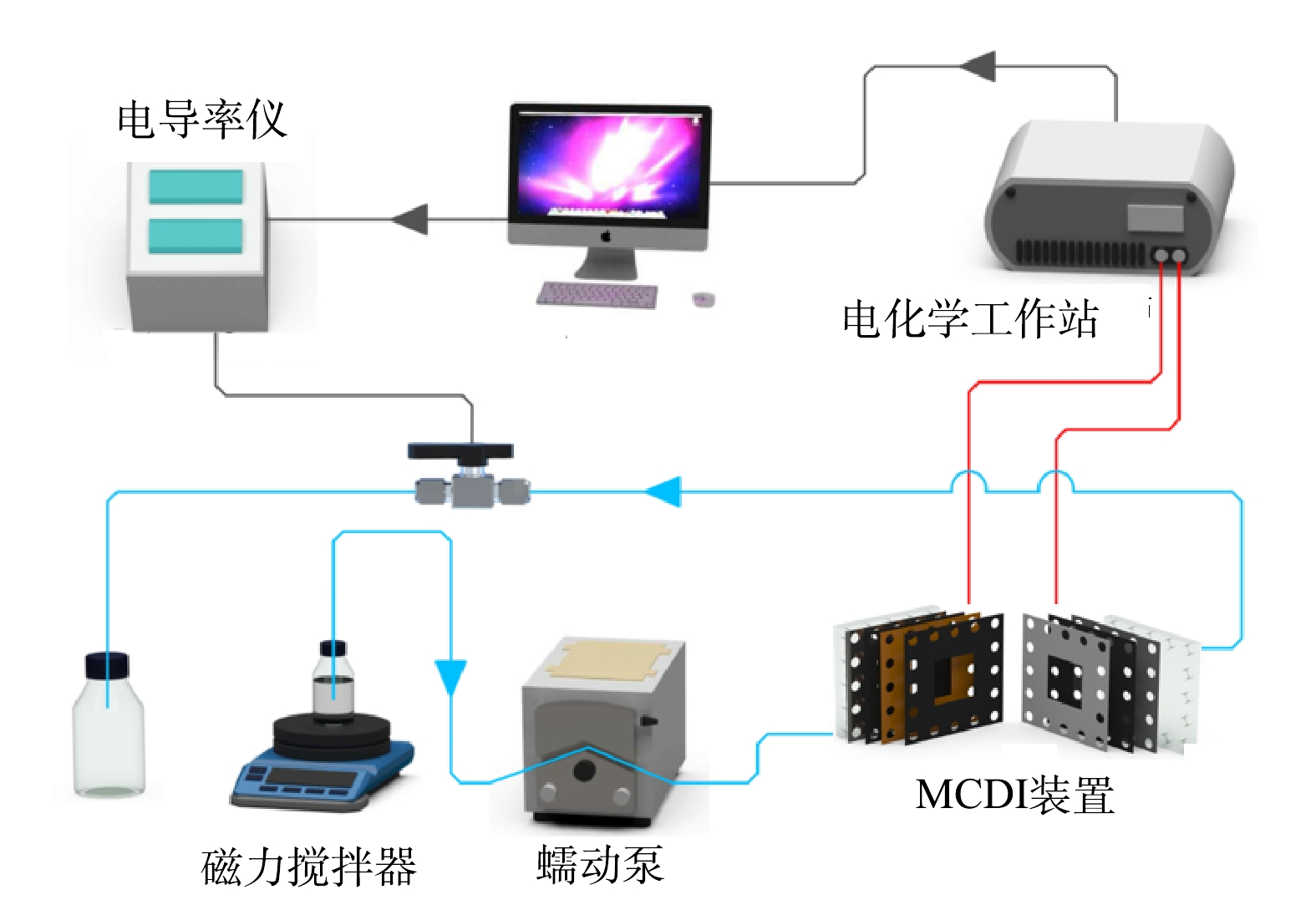
MCDI脱盐系统如图1所示。该系统由蠕动泵(LongerPump BT100-1L)、电导率仪(Mettler Toledo SG3)、MCDI脱盐装置和电化学工作站(瑞士万通MAC90589)组成。MCDI装置由2个电极、1对阴阳离子交换膜和塑料格网组成,每2层之间用硅胶垫圈密封,两端用亚克力塑料板进行加固。MCDI脱盐系统的有效容积为5 cm×4.5 cm×0.5 mm。
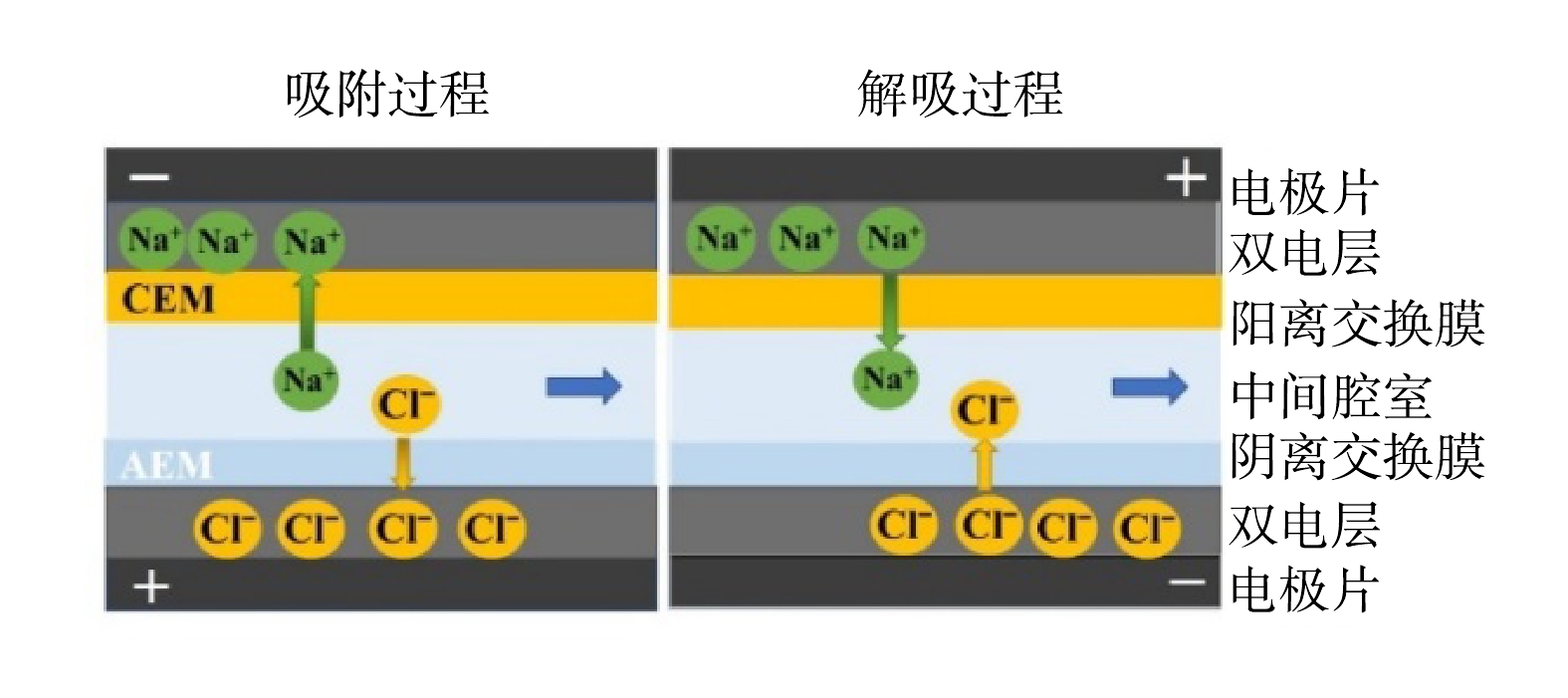
进水为人工配置的NaCl溶液,进水模式为连续流(single pass)模式,出水电导率由电导率仪实时监测记录。采用单一变量法,探究电极材料的质量比,不同运行电压(0.6、1.2、1.8 V),不同进水流速(1、2、4、6 mL·min−1)和不同进水盐质量浓度(0.2、0.4、0.6 g·L−1)对MCDI装置脱盐性能的影响。脱盐基本原理如图2所示。施加正向电压600 s后,将电压反向,电极解吸600 s,即完成1个循环。所有实验均至少重复2次以上。
系统的脱盐性能通过脱盐率、电荷效率、吸附容量、平均脱盐速度和单位能量脱盐量进行评估[11-14]。脱盐率根据式(1)进行计算,电荷效率根据式(2)进行计算,吸附容量根据式(3)进行计算,平均脱盐速度根据式(4)进行计算,单位能量脱盐量根据式(5)进行计算。
式中:
η为脱盐率 ,%;△c为进水与时间t时出水盐浓度的差值,mol·L−1;c0 为初始盐浓度,mol·L−1;t为吸附时间,s。式中:
Λ 为电荷效率,%;v 为进水流速,L·s−1;F 为法拉第常数(F=96485 C·mol−1);∫Idt 为整个吸附阶段消耗的电荷量。式中:
qe 为吸附容量,mg·g−1;MNaCl 为NaCl的摩尔质量,g·mol−1;m为电极片的质量,g。式中:α为平均脱盐速度,μmol·(cm2·min)−1;s为电极片的面积,cm2;t为电吸附的总时间,s。
式中:β为单位能量脱盐量,μmol·J−1;V为MCDI装置的施加电压,V。
-
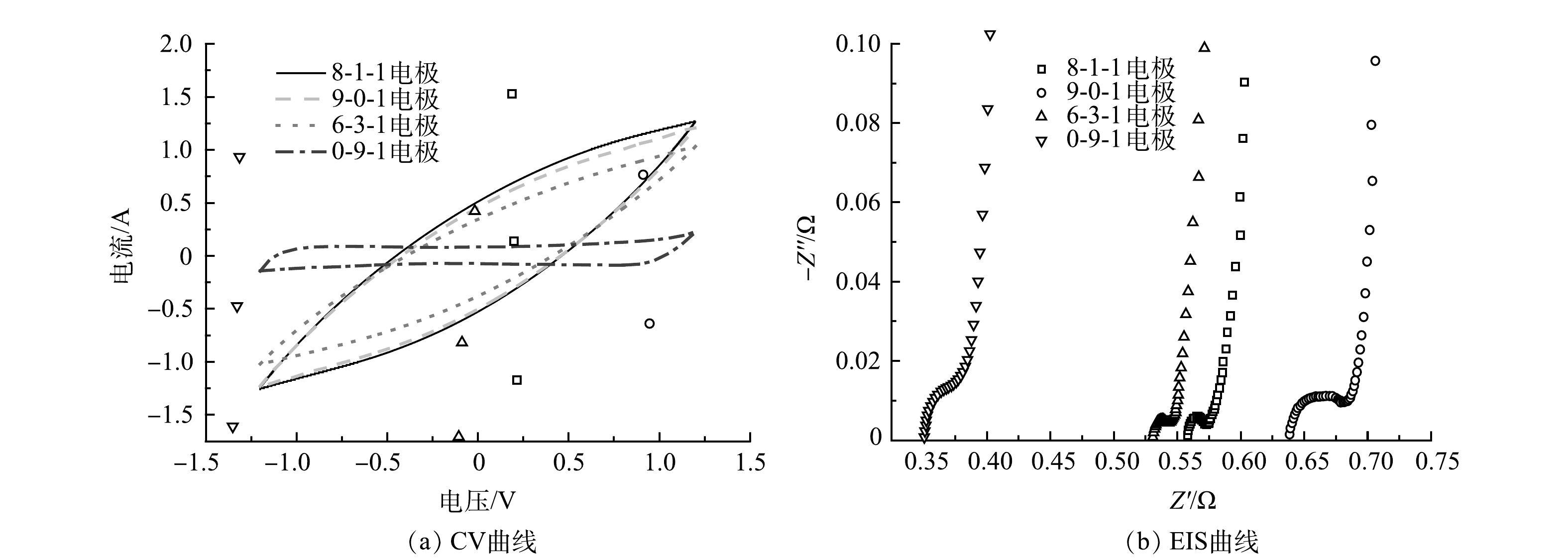
采用循环伏安法(CV)和电化学阻抗测试(EIS)对电极片进行电化学表征。测试过程在三电极体系(玻碳电极作为工作电极,饱和甘汞电极作为参比电极,铂电极作为对电极)的密闭型电解池(电解液为1 mol·L−1的NaCl溶液)中进行。CV的扫描电压为−1.2~1.2 V,扫速50 mV·s−1。EIS的扫描频率为0.1~105 Hz。CV曲线的比电容根据式(6)进行计算。
式中:C为比电容,F·g−1;a为扫速,mV·s−1;
Va 和Vc 分别为低电位截止值和高电位截止值,V。 -
为了进一步研究电极对离子的吸附过程,利用热力学和动力学对不同组分电极进行模拟。Langmuir和Freundlich等温吸附方程[15-16]可用式(7)和式(8)表示,准一级和准二级动力学模型如式(9)和式(10)所示。
式中:Ce为平衡浓度,g·L−1;qe是平衡时吸附的离子量,mg·g−1;qm为最大吸附量,mg·g−1;KL是与吸附有关的Langmuir常数;KF是与吸附剂相对吸附容量有关的Freundlich常数;1/n(在0~1)称为Freundlich系数,表示吸附质的吸附趋势。
式中:qt表示时间为t时的吸附量,mg·g−1;K1表示一级吸附速率常数,h;K2表示二级吸附速率常数,g·(mg·h)−1。
-
1)电极材料质量比的影响。不同材料质量比对电极脱盐性能的影响结果如图3所示。由图3(a)和图3(b)可以看出,在相同时间内,8-1-1电极的脱盐速度最快,且电导率下降幅度最大。由此计算的吸附容量可高达10.13 mg·g−1。由图3(c)可以看出,4种电极的电荷效率均保持在90%左右,且脱盐率随着AC占比的下降而降低。其原因为,AC具有大的比表面积和丰富的孔径,在脱盐过程中提供了主要的吸附位点[8]。由图3(d)中可以看出,8-1-1电极的电极片同样具有最优的单位能量脱盐量和脱盐速度。当电极中没有AC时,即0-9-1电极片的脱盐速度(0.084 μmol·(cm2·min)−1)仅为8-1-1电极片脱盐速度(0.443 μmol·(cm2·min)−1)的20%。其原因为,当CB作为单独电极材料时,其比表面积小,所能提供的吸附位点很少[17],导致其吸附容量低。综上所述,具有最佳脱盐性能的电极片为8-1-1。
2)加电电压的影响。由图4(a)和图4(b)可以看出,出水电导率受加电电压的影响较大。随着运行电压的增大,MCDI装置中静电场力增强,有利于离子的迁移,1.2 V时的脱盐率是0.6 V的5.56倍。然而,当电压超过1.23 V时会引起水的电解,产生氢气和氧气[18]。电解产生的气体会对离子迁移有干扰作用,同时电解过程发生的各类副反应会消耗电荷,导致相比于电压为1.8 V时,1.2 V下的装置具有更高的吸附容量、电荷效率和脱盐效率(图4(b)、图4(c))。由图4(d)可以看出,随着加电电压的升高,其单位能量脱盐量也会降低。虽然施加0.6 V电压时系统单位能量脱盐量高,但1.2 V时的脱盐速度0.44 μmol·(cm2·min)−1和吸附容量10.13 mg·g−1要比0.6 V时的0.079 μmol·(cm2·min)−1和1.81 mg·g−1分别高出5.57倍和5.60倍。综合比较,1.2 V为最佳的加电电压。
3)进水流速的影响。由图5(a)可以看出,出水电导率均呈现先下降的趋势[19],随着脱盐过程的不断进行,电极脱盐能力逐渐变弱,电导率随之上升。加电脱盐10 min后,施加相反电压进行解吸,出水电导率会逐渐恢复到初始值。我们还可以清楚的观察到电极解吸回初始电导率的时间随流速的降低而增加。由图5(b)和图5(c)可以看出,进水流速为4 mL·min−1时,电导率下降最快,此时具有最大的吸附容量、脱盐率和电荷效率;而进水流速为1 mL·min−1时,其脱盐率和电荷效率较高,但流速低导致盐水的水力停留时间长,1 mL·min−1的水力停留时间(0.477 min)是4 mL·min−1(0.119 min)的4倍,会限制MCDI的处理能力[20]。综合图5(d)所示的单位能量脱盐量和脱盐速度,当进水流速为4 mL·min−1时,MCDI系统具有最优的脱盐性能。
-
由图6(a)可以看出,当进水盐质量浓度为0.2、0.4、0.6 g·L−1时,电导率下降值分别为249.7、406.7和633.1 μS·cm−1。这表明随着进水浓度的增加,脱盐率上升,电极的吸附容量也随之增加。如图6(b)所示,当进水盐质量浓度为0.6 g·L−1时,电极片的吸附容量最高为13.48 mg·g−1。其原因是,增大进水盐浓度可使进水的电阻下降,系统脱盐电流也会变大[21-22],且电极和溶液之间形成更高的盐离子浓度梯度,降低了盐离子的扩散阻力,从而提升了电极的吸附容量[23]。由图6(c)可以看出,脱盐率随进水浓度的增加而降低。这是因为当离子浓度超过一定值时,随着进水中离子浓度的增加,进水中增加的离子数远大于出水中吸附去除的离子数,致使脱盐率降低[24]。同时,在电极与溶液界面形成的双电层厚度与脱盐溶液浓度成反比,当溶液浓度较高时,双电层较薄,电极材料几乎不能有效地保持电荷[25],这也会导致高进水盐浓度下MCDI的脱盐率下降。由图6(d)可以看出,脱盐速度随进水盐浓度的增加而增加。当初始盐质量浓度为0.6 g·L−1时,其脱盐速度为0.59 μmol·(cm2·min)−1。而单位能量脱盐量随着初始盐浓度的增加而稍有降低。进水盐浓度增加会使吸附容量增大,从而系统脱盐速度更快,但电极区域没有足够的吸附位点,导致一部分离子无法吸附在电极中,致使单位能量脱盐量有所降低。单位能量脱盐量和脱盐率是表征MCDI装置耗能和脱盐效果的2个重要指标,综合考虑以上2个指标可认为,当进水盐质量浓度为0.4 g·L−1时,MCDI系统具有最优的脱盐效果。
-
电极的CV曲线如图7(a)所示,平滑曲线的矩形伏安图代表了完美的双电层电容[26]。由式(6)计算可得,8-1-1、9-0-1、6-3-1和0-9-1电极的比电容分别为43.0、39.1、30.1、10.3 F·g−1。8-1-1电极片比电容最高,可归因于合适的AC与CB配比及AC良好的孔隙结构和CB优异的导电性质[8]。由图7(b)可以看出,几种电极的EIS曲线均分别在高频区和低频区呈现一个半圆和一条直线。其中高频半圆的直径代表电荷转移电阻(Rct),低频斜线的斜率对应于warburg电阻(Zw)引起的离子在电解质中的扩散阻力[27-28]。曲线与X轴的交点显示了装置整体的接触电阻,由图7(b)可以看出,0-9-1电极片的接触电阻比其他电极片电阻小很多,且8-1-1、9-0-1、6-3-1和0-9-1电极片的Rct分别为0.576、0.652、0.545和0.387 Ω。上述结果说明CB的加入使电荷转移电阻降低,这主要归因于CB具有优异的导电性,可在AC间提供电子迁移通道。但CB比表面积小,尤其内比表面积比例低,导致其可为离子提供的吸附位点少,而AC比表面积大且孔径丰富,能为离子储存提供很高的电容,所以AC和CB作为电极组分可优势互补,合适的配比能进一步提升系统的脱盐性能。
-
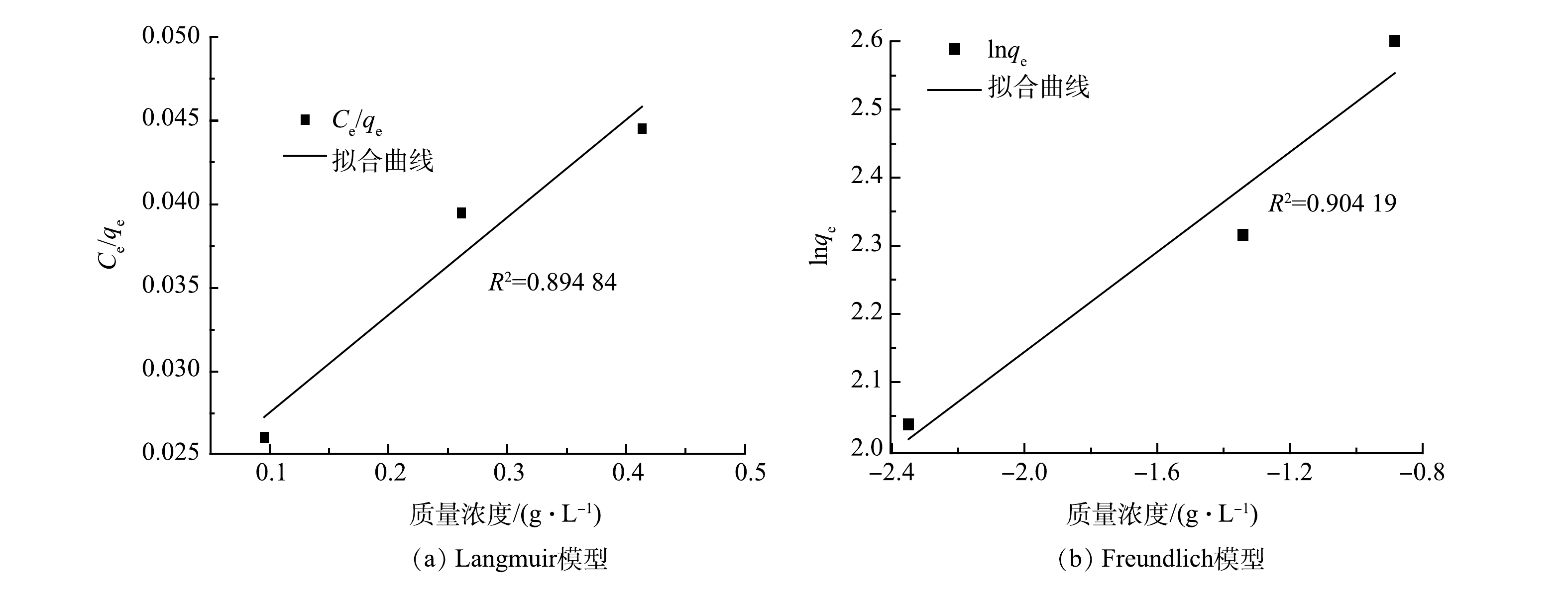
本研究利用Langmuir和Freundlich吸附等温方程来拟合在不同进水盐浓度下电吸附脱盐过程,结果如图8和表1所示。Langmuir等温吸附方程为单层吸附,吸附质间没有相互作用。Freundlich等温吸附方程描述了吸附剂表面的非均匀能量分布和被吸附物种之间的相互作用,其中吸附可以扩展到离子在电极表面的多层覆盖[8]。由图8和表1拟合得到的可决系数R2可以看出,Freundlich吸附等温方程可以更好的描述电吸附脱盐过程。这也说明离子在电极表面吸附位点上为多层吸附。
MCDI的电吸附动力学过程采用准一级和准二级动力学模型进行拟合,拟合结果如表2所示。准一级动力学模型吸附速率主要受物理吸附机理控制,可视为一个物理吸附过程;准二级动力学模型吸附速率受化学吸附的控制,这种化学吸附涉及到吸附剂与吸附质之间的电子共用和电子转移[29]。由表2中R2值可以看出,实验数据更符合准二级动力学模型。
-
1)在进水氯化钠质量浓度为0.4 g·L−1时,控制活性炭、炭黑及聚四氟乙烯质量比为8∶1∶1、运行电压为1.2 V、进水流速为4 mL·min−1,此时MCDI具有最佳脱盐性能,其脱盐率为34.45%,电荷效率为95.27%,吸附容量为10.13 mg·g−1,脱盐速率为0.44 μmol·(cm2·min)−1,单位能量脱盐量为8.23 μmol·J−1。
2)在CV扫速为50 mV·s−1时,8-1-1电极片具有最高的比电容,为43.0 F·g−1;同时,8-1-1电极片中AC提供了较多的电吸附位点,而添加适量的CB有利于降低电荷转移电阻,使其具有更优异的电吸附脱盐性能。
3)电吸附脱盐过程符合Freundlich吸附等温方程和准二级动力学方程,表明电吸附脱盐过程为多分子层吸附且以化学吸附为主。
活性炭与炭黑混合电极的脱盐性能及相关工艺参数的优化
Optimization of membrane capacitive deionization performance of activated carbon and carbon black mixed electrode
-
摘要: 混合电极中各组分的质量比是影响膜电容去离子(membrane capacitive deionization, MCDI)系统脱盐性能的重要因素。重点研究了混合电极中活性材料(活性炭)、导电剂(炭黑)和粘结剂(聚四氟乙烯)3种组分的质量比对MCDI系统脱盐性能的影响,并优化了工艺参数。实验结果表明,在进水氯化钠质量浓度为0.4 g·L−1时,控制活性炭、炭黑及聚四氟乙烯的质量比为8∶1∶1、运行电压为1.2 V,进水流速为4 mL·min−1,MCDI系统具有较优异的脱盐性能,其吸附容量和脱盐速度分别为10.13 mg·g−1和0.44 μmol·(cm2·min)−1,电荷效率和单位能量脱盐量可分别达95.27%和8.23 μmol·J−1;而且,增大进水中氯化钠浓度会进一步提升MCDI系统的吸附容量和脱盐速度,但其脱盐率会有所降低。吸附热力学和动力学拟合结果表明,此混合电极材料脱盐过程分别符合Freundlich吸附等温方程和准二级动力学方程。Abstract: The mass ratio of each component in the mixed electrode is an important factor for the desalination performance of membrane capacitor deionization (MCDI). In this study, the effects of the mass ratio of active ingredient (activated carbon), conductive agent (carbon black) and binder (polytetrafluoroethylene) on the desalination performance of MCDI were investigated, and the operation parameters were optimized. The experimental results showed that the MCDI system had a good desalination performance when the influent sodium chloride concentration was 0.4 g·L−1, the mass ratio of activated carbon, carbon black and polytetrafluoron was 8∶1∶1, the operating voltage was 1.2 V and the influent flow rate was 4 mL·min−1. The adsorption capacity and desalination rate were 10.13 mg·g−1 and 0.44 μmol·cm−2·min−1, respectively, charge efficiency and unit energy desalination capacity could reach 95.27% and 8.23 μmol·J−1, respectively. Moreover, the increase of the influent sodium chloride concentration could further improve the adsorption capacity and desalination rate of MCDI system, but the desalination rate decreased. The fitting results of adsorption thermodynamics and kinetics showed that the desalination process of the mixed electrode material conforms to Freundlich adsorption isothermal equation and second-order kinetic equation, respectively.
-
Key words:
- mixed electrode /
- MCDI /
- parameter optimization /
- dynamics /
- thermodynamics
-
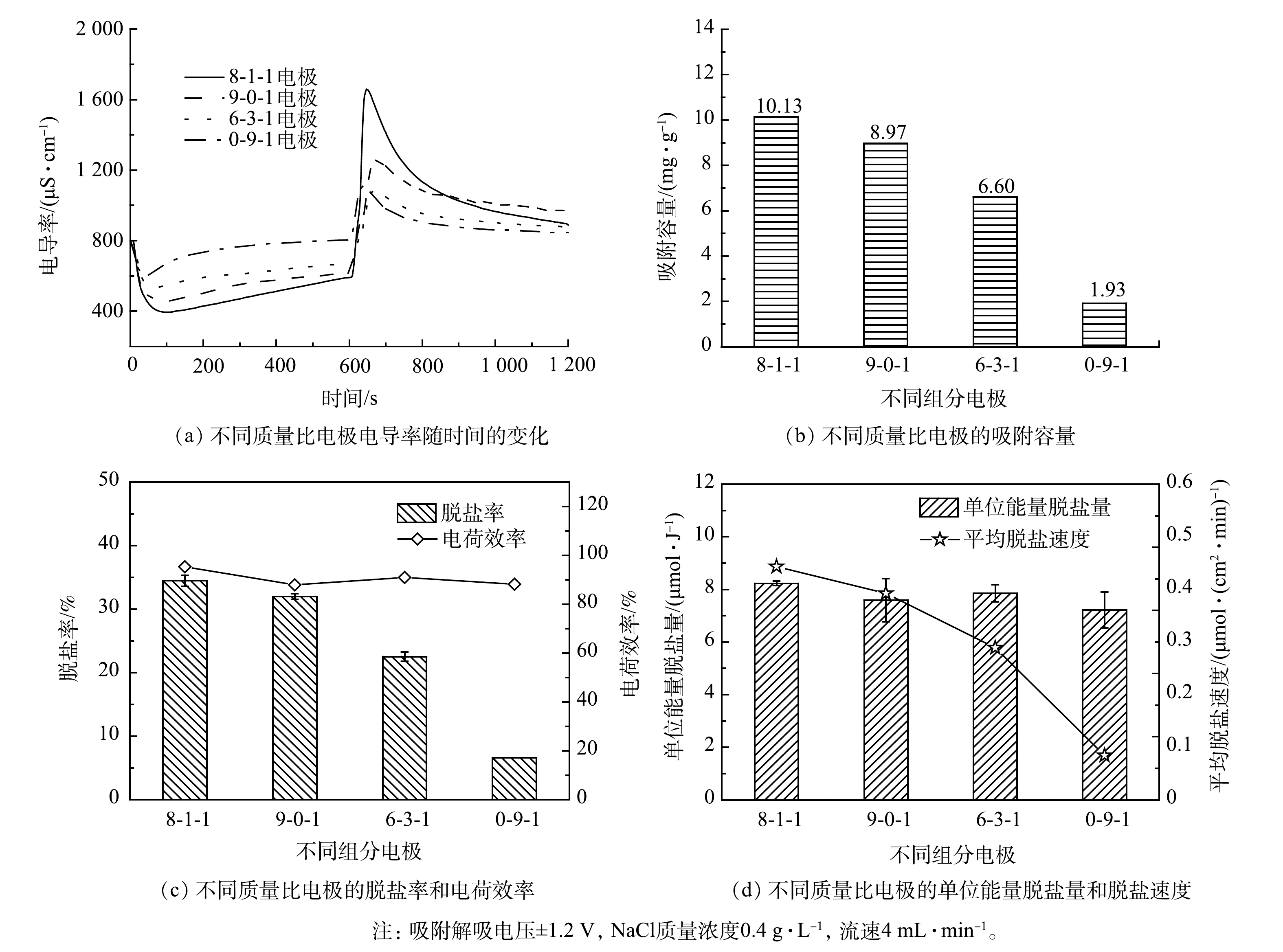
图 3 不同质量比电极的脱盐性能
Figure 3. Desalination performance of electrodes with different mass ratios
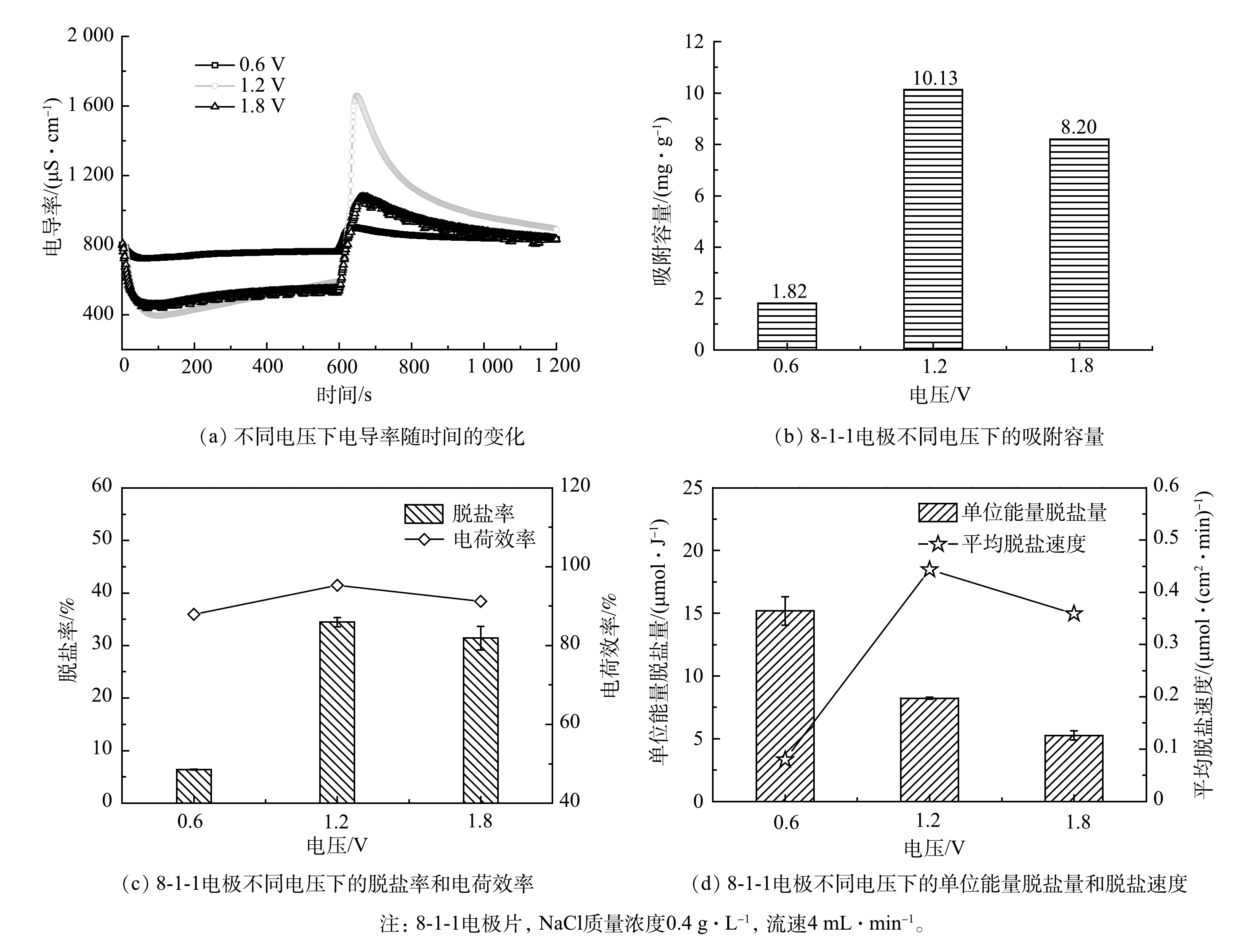
图 4 8-1-1电极在不同电压下的脱盐性能
Figure 4. Desalination performance of 8-1-1 electrode sheet at different voltages
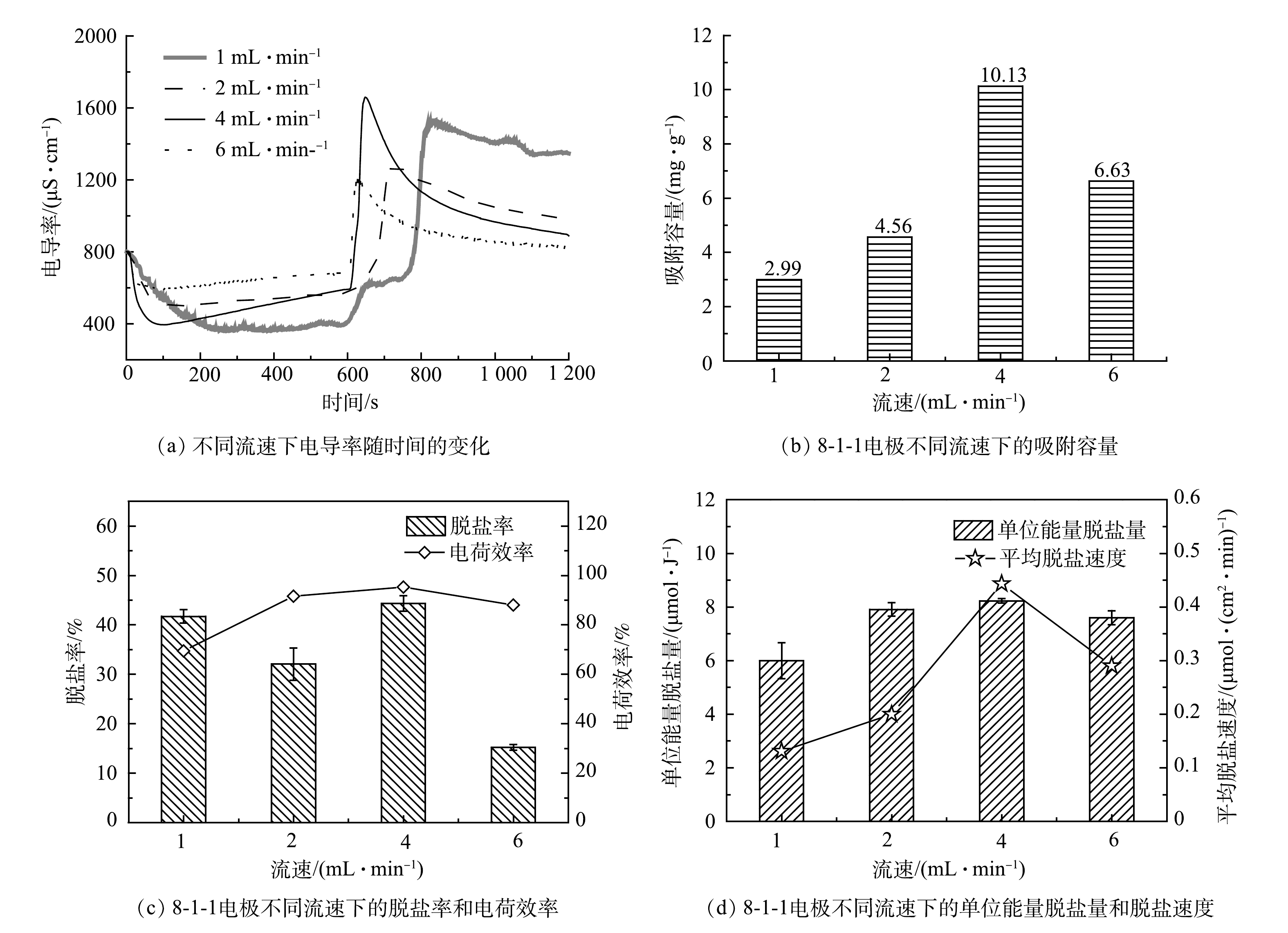
图 5 8-1-1电极在不同流速下的脱盐性能
Figure 5. Desalination performance of 8-1-1 electrode sheet at different flow rates
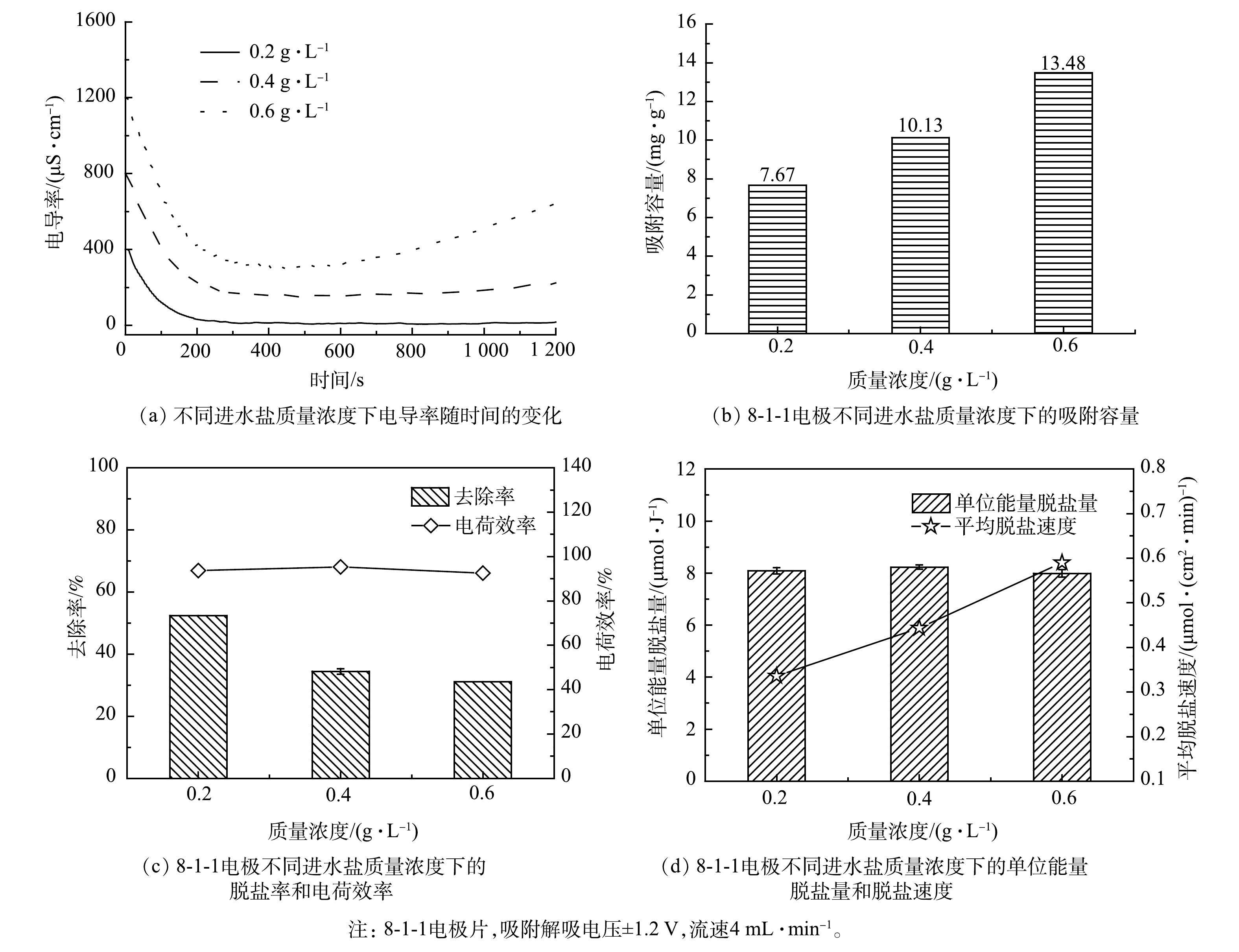
图 6 8-1-1电极在不同浓度下的脱盐性能
Figure 6. Desalination performance of 8-1-1 electrode sheet at different concentrations
表 1 8-1-1电极片吸附盐离子的等温吸附参数
Table 1. Isothermal adsorption parameters of 8-1-1 electrodes to salt ions
Langmuir模型 Freundlich模型 qm/(mg·g−1) KL R2 n KF R2 17.14 0.372 0.895 2.729 17.760 0.904  下载: 导出CSV
下载: 导出CSV
表 2 不同条件的准一级和准二级动力学模型拟合数据
Table 2. First order and second order dynamic model fitting data under different conditions
序号 一级动力学模型 二级动力学模型 K1 R2 K2 R2 1 0.005 18 0.933 0.018 36 0.996 2 0.004 21 0.961 0.015 10 0.994 3 0.004 09 0.961 0.011 72 0.997 4 0.006 97 0.998 0.012 61 0.990 5 0.003 59 0.970 0.003 20 0.999 6 0.003 86 0.958 0.013 78 0.997 7 0.004 48 0.892 0.001 60 0.969 8 0.003 79 0.951 0.004 60 0.985 9 0.003 96 0.968 0.012 00 0.998 10 0.001 41 0.980 0.001 00 0.994 11 0.001 83 0.968 0.001 61 0.996 12 0.002 19 0.961 0.001 00 0.980 13 0.005 18 0.933 0.018 36 0.996 14 0.004 21 0.962 0.015 10 0.994 注: 1~4分别为8-1-1、9-0-1、6-3-1、0-9-1; 5~7分别为0.6、1.2、1.8 V;序号8~11分别为1、2、4、6 mL·min−1;序号12~14分别为不同进水盐质量浓度0.2、0.4、0.6 g·L−1。
下载: 导出CSV
-
[1] SUSS M, PORADA S, SUN X, et al. Water desalination via capacitive deionization: What is it and what can we expect from it?[J]. Energy & Environmental Science, 2015, 8(8): 2296-2319. [2] Ma J X, HE C, HE D, et al. Analysis of capacitive and electrodialytic contributions to water desalination by flow-electrode CDI[J]. Water Research, 2018, 144: 296-303. doi: 10.1016/j.watres.2018.07.049 [3] XING W L, LIANG J, TANG W W, et al. Perchlorate removal from brackish water by capacitive deionization: Experimental and theoretical investigations[J]. Chemical Engineering Journal, 2019, 361: 209-218. doi: 10.1016/j.cej.2018.12.074 [4] SHANNON M A, BOHN P W, ELIMELECH M, et al. Science and technology for water purification in the coming decades[J]. Nature, 2009, 452: 337-346. [5] TAN C, HE C, TANG W W, et al. Integration of photovoltaic energy supply with membrane capacitive deionization (MCDI) for salt removal from brackish waters[J]. Water Research, 2018, 147(15): 276-286. [6] 赵飞, 苑志华, 钟鹭斌, 等. 电容去离子技术及其电极材料研究进展[J]. 水处理技术, 2016, 42(5): 38-44. [7] THAMILSELVAN A, NESARAJ A S, NOEL M. Review on carbon-based electrode materials for application in capacitive deionization process[J]. International Journal of Environmental Science and Technology, 2016, 13(12): 2961-2976. doi: 10.1007/s13762-016-1061-9 [8] YANG S J, KIM T, LEE K, et al. Solvent evaporation mediated preparation of hierarchically porous metal organic framework-derived carbon with controllable and accessible large-scale porosity[J]. Carbon, 2014, 71: 294-302. doi: 10.1016/j.carbon.2014.01.056 [9] HARO M, RASINES G, MACIAS C, et al. Stability of a carbon gel electrode when used for the electro-assisted removal of ions from brackish water[J]. Carbon, 2011, 49(12): 3723-3730. doi: 10.1016/j.carbon.2011.05.001 [10] WIMALASIRI Y, MOSSAD M, ZOU L. Thermodynamics and kinetics of ammonium ions by graphene laminate electrodes in capacitive deionization[J]. Desalination, 2015, 357: 178-188. doi: 10.1016/j.desal.2014.11.015 [11] XU S, WANG T H, WANG C F, et al. The effect of crystal phase of manganese oxide on the capacitive deionization of simple electrolytes[J]. Science of the Total Environment, 2019, 675: 31-40. doi: 10.1016/j.scitotenv.2019.04.172 [12] HUYSKENS C, HELSEN J, HAAN A D. Capacitive deionization for water treatment: Screening of key performance parameters and comparison of performance for different ions[J]. Desalination, 2013, 328: 8-16. doi: 10.1016/j.desal.2013.07.002 [13] MOSSAD M, ZOU L. A study of the capacitive deionization performance under various operational conditions[J]. Journal Hazardous Materials, 2012, 213: 491-497. [14] ZHAO R, SATPRADIT O, RIJNAARTS H, et al. Optimization of salt adsorption rate in membrane capacitive deionization[J]. Water Research, 2013, 47(5): 1941-1952. doi: 10.1016/j.watres.2013.01.025 [15] DING Z B, XU X T, LI Y Q et al. Significantly improved stability of hybrid capacitive deionization using nickel hexacyanoferrate/reduced graphene oxide cathode at low voltage operation - ScienceDirect[J] Desalination, 2019, 468: 114078-114087. [16] MOSSAD M, ZOU L. A study of the capacitive deionization performance under various operational conditions[J]. Journal Hazardous Materials, 2012, 213-214: 491-497. doi: 10.1016/j.jhazmat.2012.02.036 [17] 杨宏艳, 张卫珂, 葛坤, 等. 流动性电极电容去离子技术的脱盐性能研究[J]. 环境污染与防治, 2017, 39(8): 911-919. [18] KIM Y H, TANG K, CHANG J, et al. Potential limits of capacitive deionization and membrane capacitive deionization for water electrolysis[J]. Separation Science and Technology, 2019, 54(13): 2112-2125. doi: 10.1080/01496395.2019.1608243 [19] LIANG P, SUN X, BIAN Y, et al. Optimized desalination performance of high voltage flow-electrode capacitive deionization by adding carbon black in flow-electrode[J]. Desalination, 2017, 420(5): 63-69. [20] OREN Y. Capacitive deionization (CDI) for desalination and water treatment-past, present and future: A review[J]. Desalination, 2008, 228(1-3): 10-29. doi: 10.1016/j.desal.2007.08.005 [21] SHIN Y, DONG C D, HUANG Y H, et al. Electro-sorption of ammonium ion onto nickel foam supported highly microporous activated carbon prepared from agricultural residues (dried Luffa cylindrica)- ScienceDirect[J]. Science of the Total Environment, 2019, 673: 296-305. doi: 10.1016/j.scitotenv.2019.04.066 [22] AMP N G T, RITTERSUPA/SUP U. Influence of concentration of supporting electrolyte on electrochemistry of redox systems on multi-walled carbon nanotubes[J]. Physics and Chemistry of Liquids, 2012, 50(5): 661-668. doi: 10.1080/00319104.2012.663496 [23] LI H, LIANG S, LI J, et al. The capacitive deionization behavior of a carbon nanotube and reduced graphene oxide composite[J]. Journal of Materials Chemistry A, 2013, 1(21): 6335-6341. doi: 10.1039/c3ta10681k [24] XU B, XU X, GAO H, et al. Electro-enhanced adsorption of ammonium ions by effective graphene-based electrode in capacitive deionization[J]. Separation and Purification Technology, 2020, 250: 117243. doi: 10.1016/j.seppur.2020.117243 [25] ZHANG X, XIE K, GAO J, et al. Highly pore-expanded benzidine-functionalized graphene framework for enhanced capacitive deionization[J]. Desalination, 2018, 445: 149-158. doi: 10.1016/j.desal.2018.08.001 [26] JOLANTA N, DOROTA G F, KRZYSZTOF N, et al. The effect of supporting electrolyte concentration on zinc electrodeposition kinetics from methimazole solutions[J]. Electroanalysis, 2019, 31(6): 1141-1149. doi: 10.1002/elan.201800852 [27] WANG C, SONG H, ZHANG Q, et al. Parameter optimization based on capacitive deionization for highly efficient desalination of domestic wastewater biotreated effluent and the fouled electrode regeneration[J]. Desalination, 2015, 365: 407-415. doi: 10.1016/j.desal.2015.03.025 [28] NIE C, PAN L, LIU Y, et al. Electrophoretic deposition of carbon nanotubes-polyacrylic acid composite film electrode for capacitive deionization[J]. Electrochemical Acta, 2012, 66: 06-109. [29] YAN W, LUO H, HOU W, et al. Adsorption of hexavalent chromium from aqueous solutions by graphene modified with cetyltrimethylammonium bromide[J]. Journal of Colloid & Interface Science, 2013, 394(1): 183-191. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4983
- HTML全文浏览数: 4983
- PDF下载数: 89
- 施引文献: 0











