




 下载:
下载:








-
易挥发的剧毒氯代烃有机物1,2-二氯乙烷(1,2-dichloroethane,简称1,2-DCA)具有致癌、致畸、致突变效应,是土壤和地下水环境中常见有机污染物之一[1]。因其溶解性高、性质稳定,导致其在土壤和地下水中环境的降解难度大。因此,研究和探讨1,2-二氯乙烷在地下水中的降解具有重要意义。
地下水有机污染的主要修复方法包括抽出处理[2-3]、气相抽提[4]、可渗透反应墙[5]以及生物修复[6]等,但这些方法在处理成本、去除率及运行周期等方面尚存在不足之处。原位化学氧化技术虽然在污染地下水修复方面还处于研究发展阶段,但已表现出良好的处理效果[7-8]。碱活化过硫酸盐原位修复地下水是其中一种被广泛应用的技术。有研究[9]表明,过硫酸盐氧化体系中存在硫酸盐自由基(
SO−⋅4 )和羟基自由基(·OH),其对有机污染物的降解效果随pH变化而不同。在pH>8.5的条件下,SO−⋅4 能与OH−反应并促进体系中·OH的生成,体系中的部分有机污染物可由·OH氧化降解[10-11]。MA等[12]选择活化过硫酸盐氧化苯酚作为研究对象,在碱性条件下苯酚的氧化速率有所增加,pH升高可显著降低化学反应活化能,降解苯酚时过硫酸盐的有效碱性活化pH阈值最小约为11。增大修复药剂在地下水含水层中的有效影响半径是地下水原位修复技术工程化应用的关键之一。修复药剂在含水层的运移受含水层自身水文地质条件和地下水渗流场影响较大。水动力控制法是利用井群系统,通过抽水或向含水层注水,人为地改变地下水的水力梯度,进而影响含水层的地下水渗流特征。在进行原位化学氧化修复时,通过水动力调控可以影响修复药剂的扩散和运移,从而间接影响地下水中污染物降解效果。
目前,针对氯代烃污染地下水原位化学修复的研究主要是室内氧化批实验以及土柱或砂箱模拟实验,因其存在尺度效应而影响了该技术的推广应用。本研究中,以某有机污染场地污染含水层为实验对象,采用水动力控制法强化原位化学氧化技术修复受1,2-二氯乙烷污染含水层,分析了碱活化过硫酸盐降解目标污染物的效果,探讨实验过程中目标污染物及地下水参数的变化规律,以期为该技术修复污染地下水的工程化应用提供参考。
全文HTML


-
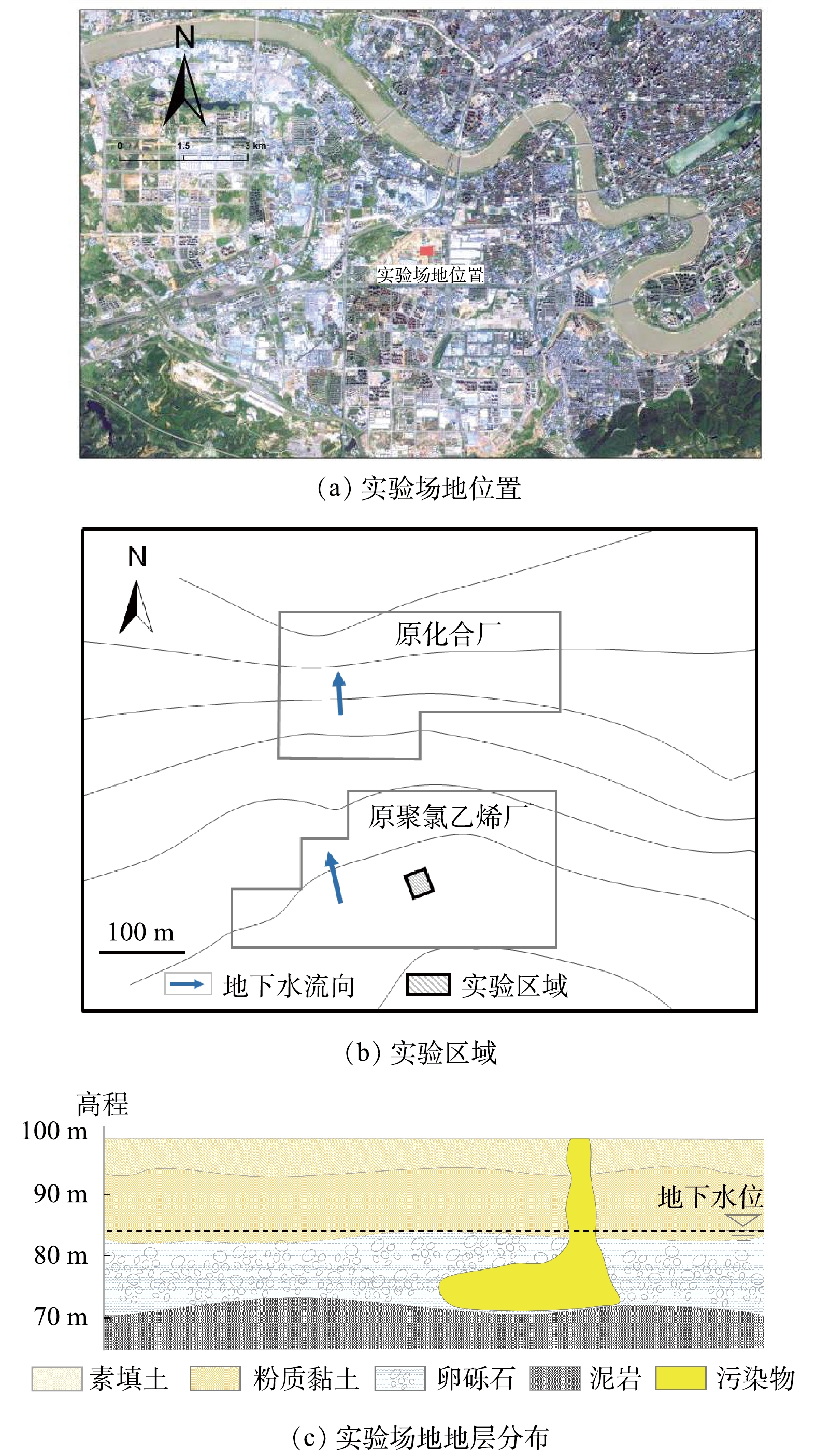
本研究中,以某化工厂停产搬迁后的遗留有机污染场地含水层为实验对象,场地位置如图1(a)所示,实验区域如图1(b)所示。场地地层结构主要由素填土、粉质黏土、圆砾和泥岩组成(图1(c)),各层厚度分别为0~6.00、7.00~15.00、10.00~29.50 m,泥岩层未穿透,地下水位埋深11.48~17.00 m。实验区域位于污染区域内,地下水主要赋存于卵砾石潜水含水层中,实验中修复的目标含水层位于地下15.00~29.00 m,含水层中的主要污染物为1,2-二氯乙烷。
-
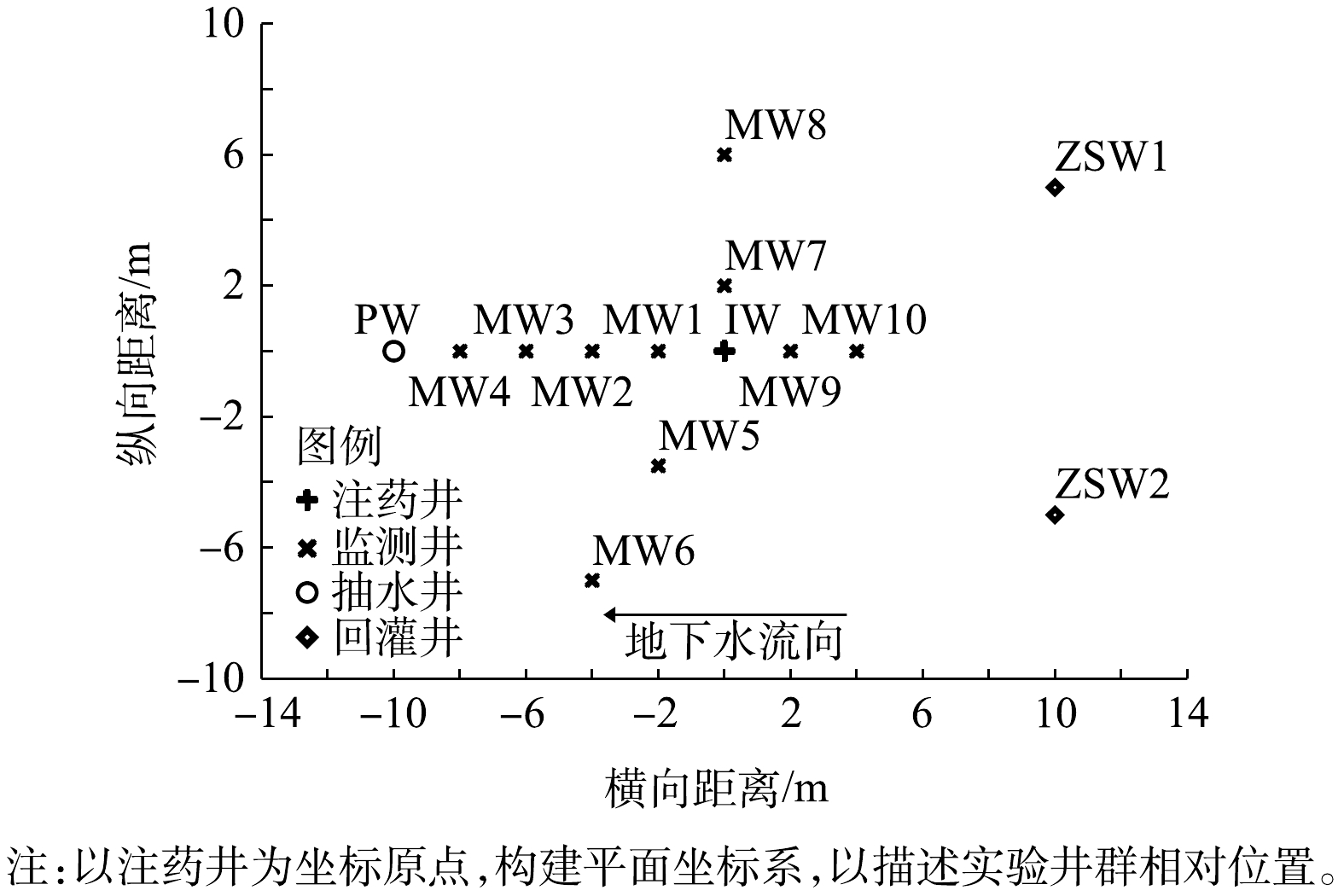
实验分为3个阶段进行:第1阶段为水文地质调查阶段,在实验区内开展抽水实验和示踪实验获取水文地质参数,并在实验区周围完善实验井群(图2)布置;第2阶段为修复实验阶段,开展地下水水动力控制、监控修复前地下水监测井中污染物浓度,将过硫酸钠和氢氧化钠注入目标含水层;第3阶段为采样监测阶段,在注药完成后,采集实验区域内地下水样品,检测和分析目标污染物含量及相应的理化参数。
1)水文地质实验。水文地质实验分为抽水实验和示踪实验。抽水实验为单孔完整井稳定流抽水实验[13],实验过程中使用秒表、电磁流量计记录抽水过程中的时间间隔和流量值。实验现场无其他观测井,将抽水井PW直接作为观测井处理,使用水位仪记录抽水井水位变化。实验过程持续9 h,抽水稳定流量为7 m3·h−1,最大降深为2.30 m。
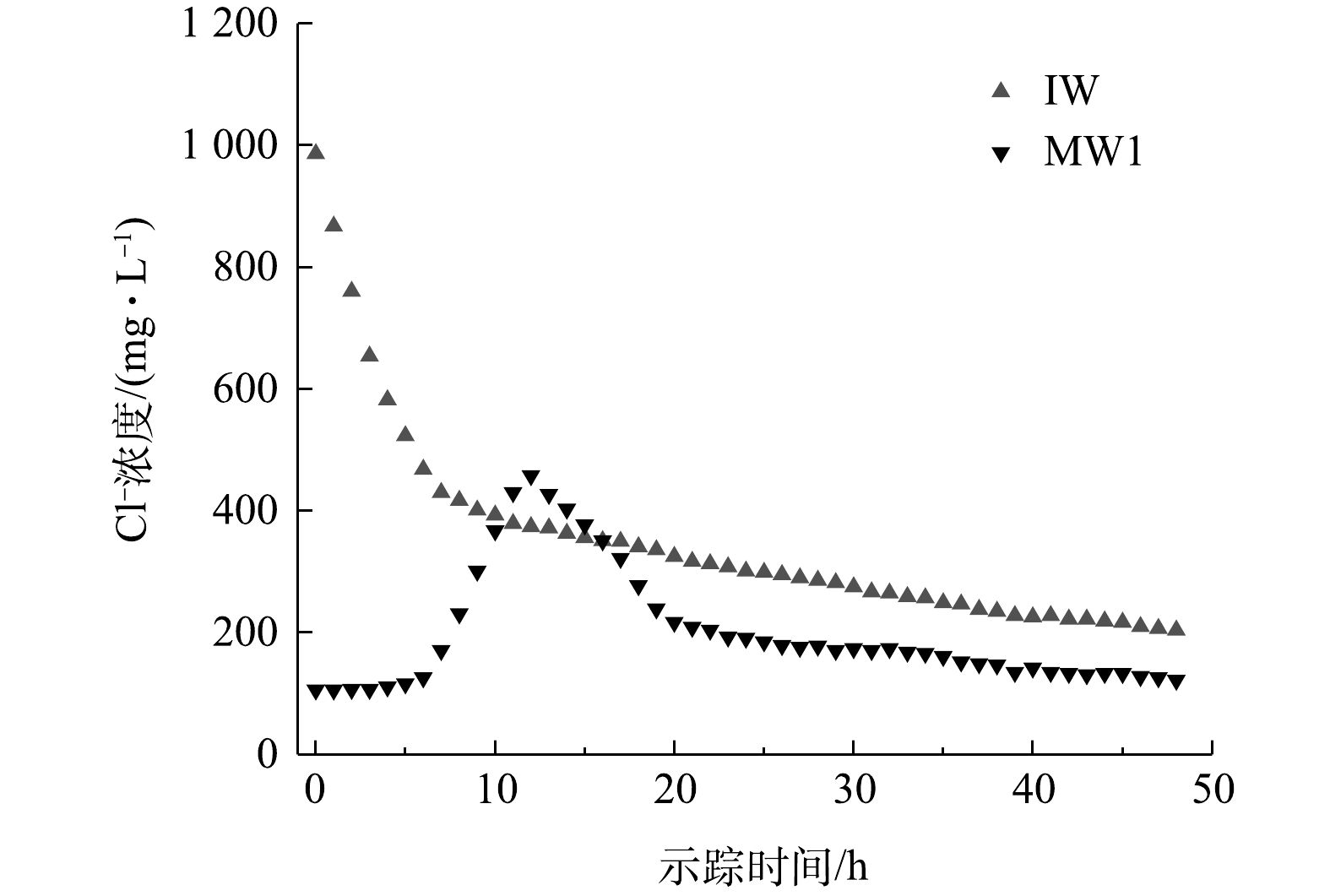
示踪实验采用配制的饱和NaCl溶液作为示踪剂,实验前开启抽水泵抽水,待观测井的水位降深达到稳定后开始进行实验。实验过程中,抽水井PW定流量抽水,抽水流量为7 m3·h−1。示踪剂投放前,先对示踪剂投源井IW和观测井MW1处于稳定水位时的地下水Cl−浓度进行测定。经测定,实验区地下水Cl−背景浓度为104~110 mg·L−1。实验所用示踪剂溶液由质量为250 kg NaCl配制而成,一次性持续注入,并将示踪剂投放时间作为弥散实验开始的时间。投放示踪剂后,平均每隔1 h在示踪剂投源井IW和观测井MW1取样监测Cl−浓度。现场实验的延续时间,主要是根据水样中Cl−质量浓度随时间变化曲线而决定的。当所取水样中的示踪剂离子质量浓度从背景值达到峰值并逐渐降低到起始背景质量浓度值,然后再稳定一段时后终止实验。本次实验中示踪剂注入时间为抽水实验进行9 h后,共持续48 h。
2)原位化学氧化修复。实验中采用的氧化剂为工业过硫酸钠(含量99%),活化剂为氢氧化钠(液碱,含量30%)。在注入地下水含水层前需将过硫酸钠配制成浓度适合的流体状药剂。根据以往应用经验、实验室小试数据及场地的实际污染程度,确定本次实验期间过硫酸钠溶液注入浓度为300 kg·m−3,实验期间共注入过硫酸钠15 t和氢氧化钠12 m3。
注药前24 h,开启抽水井PW进行抽水,并回灌至注水井ZSW1和ZSW2,抽、注水流量为7 m3·h−1,通过抽水和注水控制实验区域内地下水水力梯度。注药时,开启注药泵,向注药井IW内注入过硫酸钠溶液和液碱。过硫酸钠溶液为持续性恒流量注入,注入流量为2 m3·h−1;液碱为间歇性注入,注入流量为0.5 m3·h−1,注入时间和间歇时间均为20 min。注药结束后,停止抽水和回灌,水动力控制结束。
3)修复效果监测。药剂注入工作完成后,从监测井中采集地下水样品进行分析测试。地下水监测项目主要包括1,2-二氯乙烷含量和地下水的理化性质。采用便携气囊式低流量采样泵(QED MP50,美国)慢速洗井和采样,在监测井水位下2、8和14 m处取样,取平均值作为监测井中样品浓度(Mean ± SEM)。有机样品使用40 mL棕色玻璃瓶在4 ℃保存,无机样品使用250 mL塑料瓶保存。
地下水pH、溶解氧、氧化还原电位、电导率采用HACH Q300HD便捷式水质分析仪现场测定。地下水样品中Cl−含量采用硝酸银滴定法测定。地下水样品中硫酸盐含量采用离子色谱法检测,检测仪器为离子色谱仪(Aquion,美国),具有电导检测器和抑制器,电导检测器进样体积200 μL,使用阴离子分离柱,流速1.2 mL·min−1,柱温25 ℃,碳酸盐淋洗液Na2CO3为0.6 mmol·L−1,NaHCO3为0.6 mmol·L−1。地下水样品中1,2-二氯乙烷含量采用吹扫捕集/气相色谱-质谱法检测,检测仪器为气相色谱-质谱联用仪(Agilent,美国),配备DB-624极性石英毛细管色谱柱,AQUATek 100吹扫捕集进样器(Agilent,美国);进样口温度260 ℃,分流比10∶1,柱流量1 mL·min−1,传输线温度230 ℃;色谱柱升温程序:初始柱温40 ℃,保持3 min,以20 ℃·min−1升至200 ℃,再以10 ℃·min−1升至250 ℃,保持3 min,载气为高纯氦气(99.999%);电离能量69.9 eV,离子源温度230 ℃,四级杆温度150 ℃,采集模式为SIM模式,溶剂延迟3 min;定量方法采用峰面积外标法。
1.1. 实验区概况
1.2. 实验方法
-
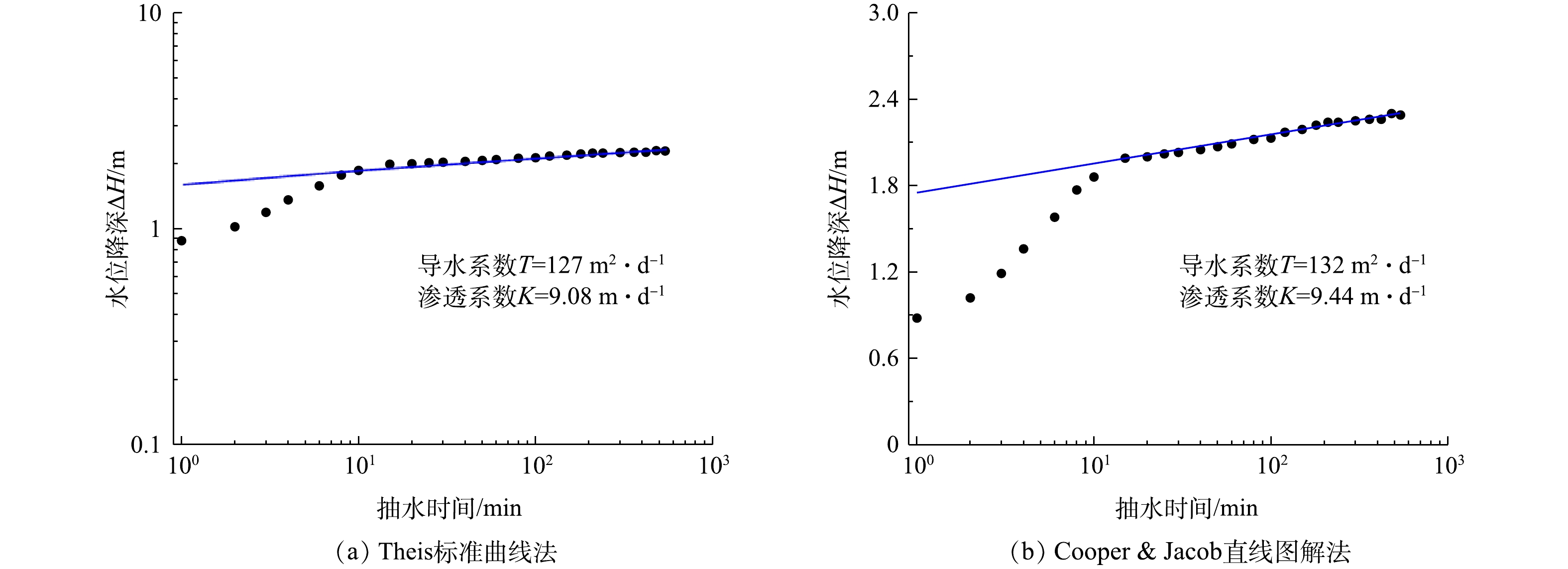
1)抽水实验结果与分析。抽水实验是野外求取含水层参数的重要方法,NEUMAN等利用抽水实验求取重力释水滞后条件下含水层参数[14]。本次抽水实验中无观测井,以抽水井实验数据为基础求解含水层参数,因此,在数据处理过程中需要对抽水井井储[15]及表皮效应[16]的影响进行修正。由于抽水前期数据受井损和水泵扬程等影响流量变化较快,故需对前期变化较快数据进行筛选,利用 Aquifer Test中Theis模型求解其对抽水井PW井抽水数据处理,运用Theis曲线拟合及Cooper & Jacob线性拟合分别求解含水层参数,拟合结果如图3所示。
Dupuit公式法是水文地质调查过程中常用的求取含水层参数(式(1)~式(3))的方法,薛禹群[17]对以稳定流为基础的Dupuit公式和影响半径问题有详细的阐述。
式中:K为渗透系数,m·d−1;Q为抽水井涌水量,m3·d−1;s为抽水井稳定时水位降深值,m;R为影响半径,m;r为抽水井半径,m;H为潜水含水层的厚度,m;T为导水系数,m2·d−1。
本研究中采用试算法求解。在求解过程中,由于Dupuit所利用的是最终达到“稳定”阶段的水位数据,因此,抽水流量采用最终稳定流量计算。抽水井井径为0.13 m,稳定抽水流量为168 m3·d−1,含水层厚度为14 m,最大降深为2.30 m,经试算法求解得到抽水影响半径为39.08 m,含水层渗透系数为5.15 m·d−1,导水系数为72 m2·d−1。
对于Aquifer Test中Theis标准曲线拟合法求解,可有效利用所有数据,但在求解过程中随意性较大。对于Dupuit公式法求解,方法较为成熟,其要求抽水实验时间长,但在求解过程中需关注最终稳定流量和水位降深,前期数据利用效率低。综合运用不同的含水层参数求解方法,得到实验区域含水层参数(平均值),结果表明,渗透系数为7.89 m·d−1,导水系数为101 m2·d−1。
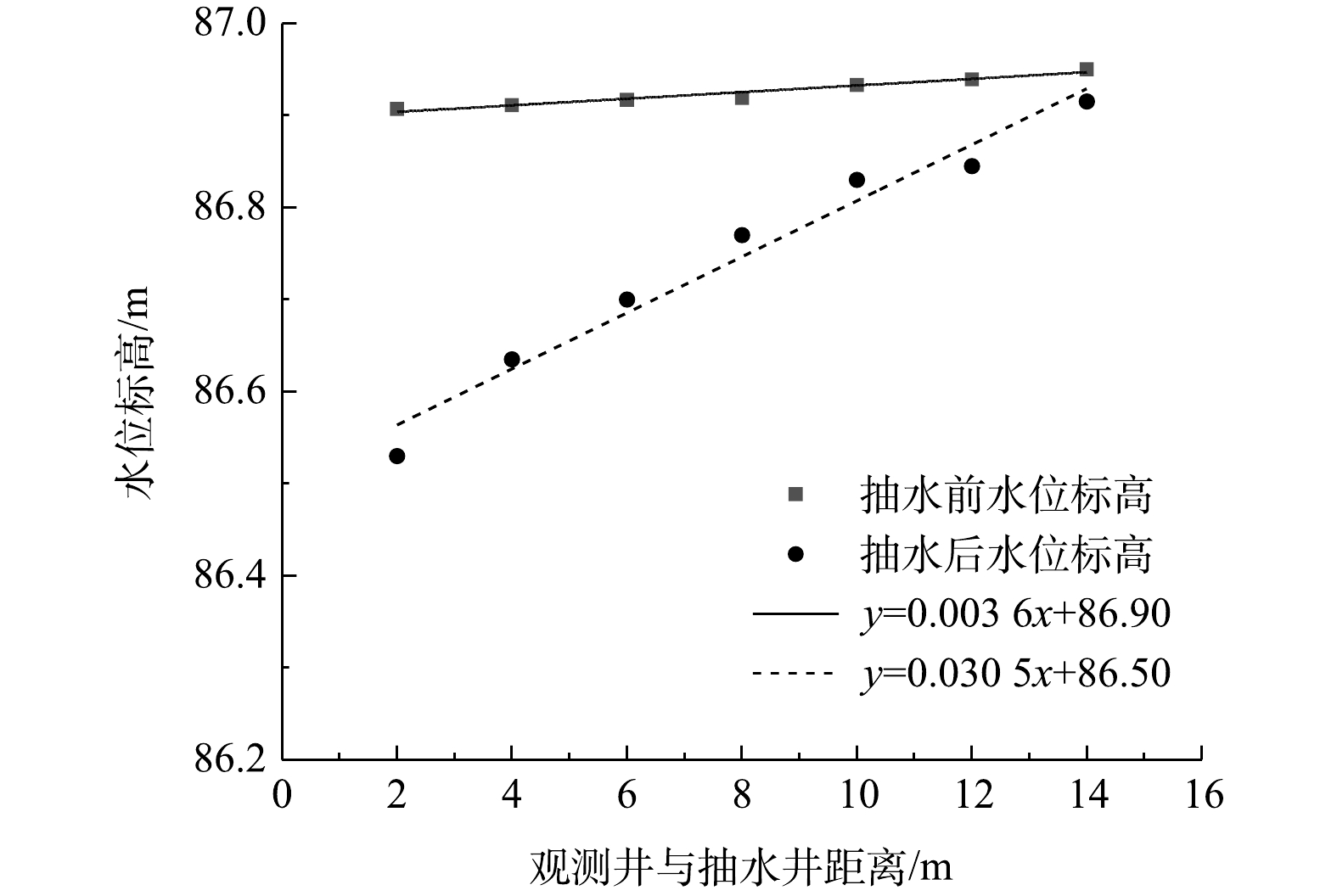
2)场地水动力弥散特征。抽水实验前后实验区域观测井水位标高如图4所示,自然流场下地下水流向由南向北,水力坡度为0.36%,持续抽水9 h后,地下水流场趋于稳定,场地水力坡度为3.05%,通过稳定流抽水可有效调控实验区域地下水渗流场。
实验区域潜水含水层示踪弥散实验开始于抽水井PW抽水9 h后,示踪实验历时48 h,Cl−浓度随时间变化过程见图5。观测井MW1中Cl−浓度在实验开始后4 h开始升高,在12 h后达到峰值458 mg·L−1,之后逐渐下降。
以示踪剂投源井IW为坐标原点,地下水流速主方向为x轴正方向,垂直于地下水流速方向为y轴正方向,观测井位于x轴上。向IW井中注入示踪剂NaCl,Cl−在含水层中的运移过程主要受机械弥散作用,分子扩散作用的影响可忽略不计,其运移过程可简化为一维稳定流场中的二维弥散问题,位于坐标为(x,y)点处观测井中的Cl−浓度随时间变化过程的解析式如式(4)所示。
式中:C(x, y, t)为t时刻区域任意点处示踪剂的浓度,mg·L−1;M为单位含水层厚度上目标离子瞬时投放质量,kg;n为含水层有效孔隙率,u为地下水流速,m·d−1;αL、αT为含水层纵、横向弥散度,m。
由图5所示的示踪剂投源井IW和观测井MW1中Cl−浓度与时间的关系,以式(4)对观测井MW1中Cl−浓度进行拟合,可得αL=0.89 m、u=3.85 m·d−1。根据前人的研究结果及经验判断,场地的横向弥散度αT大致为纵向弥散度αL的1/10[18],可得横向弥散度αT的经验推断值为0.089 m。
-
在注入过硫酸钠和氢氧化钠后,不同监测井中1,2-二氯乙烷降解规律存在明显距离-速率效应。注药后,距离注药井较近区域(4 m内)地下水中的污染物浓度呈快速下降趋势,距离注药井较远区域(6~8 m)地下水中的污染物浓度呈缓慢下降趋势。距离注药井2 m处监测井(MW1、MW7和MW9)和距离注药井4 m处监测井(MW2、MW5和MW10)中1,2-二氯乙烷浓度的变化情况如图6所示。MW1、MW7和MW9中1,2-二氯乙烷初始浓度分别为2.31、3.18和3.32 mg·L−1,在注药后地下水中1,2-二氯乙烷快速下降,注药后28 d,3个监测井中的1,2-二氯乙烷均被完全降解(低于检出限1.4 μg·L−1);注药后58 d,3个监测井中1,2-二氯乙烷浓度开始回升,到实验结束时,3个监测井中的1,2-二氯乙烷浓度分别为1.91、3.18和1.22 mg·L−1。MW2、MW5和MW10中1,2-二氯乙烷初始浓度为4.17、3.13和3.67 mg·L−1,在注药后地下水中1,2-二氯乙烷浓度快速下降;注药后28 d,3个监测井中的1,2-二氯乙烷浓度分别为0.28、0.44和0.35 mg·L−1,与初始浓度相比,分别降低了94.00%、85.86%和86.70%。与2 m处监测井相似,MW2、MW5和MW10中1,2-二氯乙烷浓度均在实验期间出现下降后回升,到实验结束时,3个监测井中的1,2-二氯乙烷浓度分别为3.46、4.43和1.10 mg·L−1。
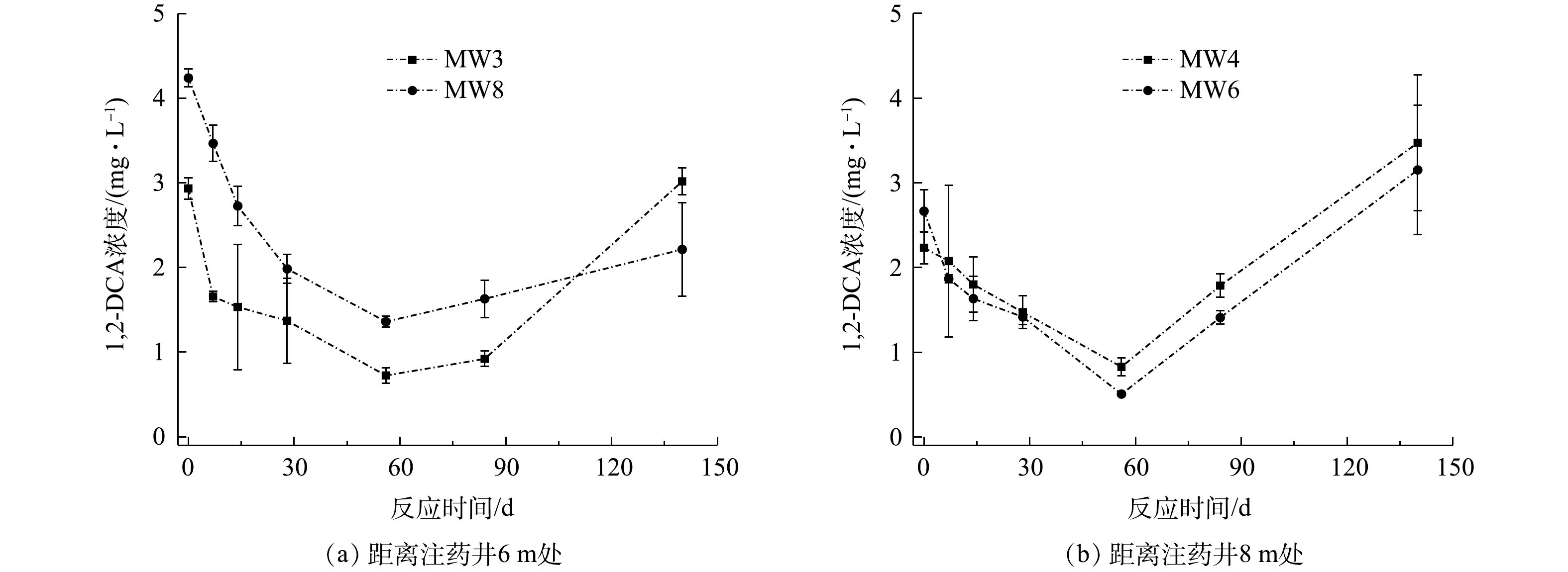
距离注药井6 m处监测井(MW3和MW8)和距离注药井8 m处监测井(MW4和MW6)中1,2-二氯乙烷浓度变化情况如图7所示。MW3、MW8、MW4和MW6中1,2-二氯乙烷初始浓度为2.93、4.24、2.23和2.67 mg·L−1,在注药后56 d内,监测井中1,2-二氯乙烷浓度稳定下降,在第56天,4个监测井中1,2-二氯乙烷浓度下降至最低值,分别为0.72、1.36、0.83和0.51 mg·L−1,与初始浓度相比,分别下降了53.34%、53.22%、33.98%和46.83%。注药84 d后,监测井中观测到1,2-二氯乙烷浓度开始回升,到实验结束时,MW3、MW8、MW4和MW6中1,2-二氯乙烷浓度分别为3.02、2.21、3.47和3.15 mg·L−1。
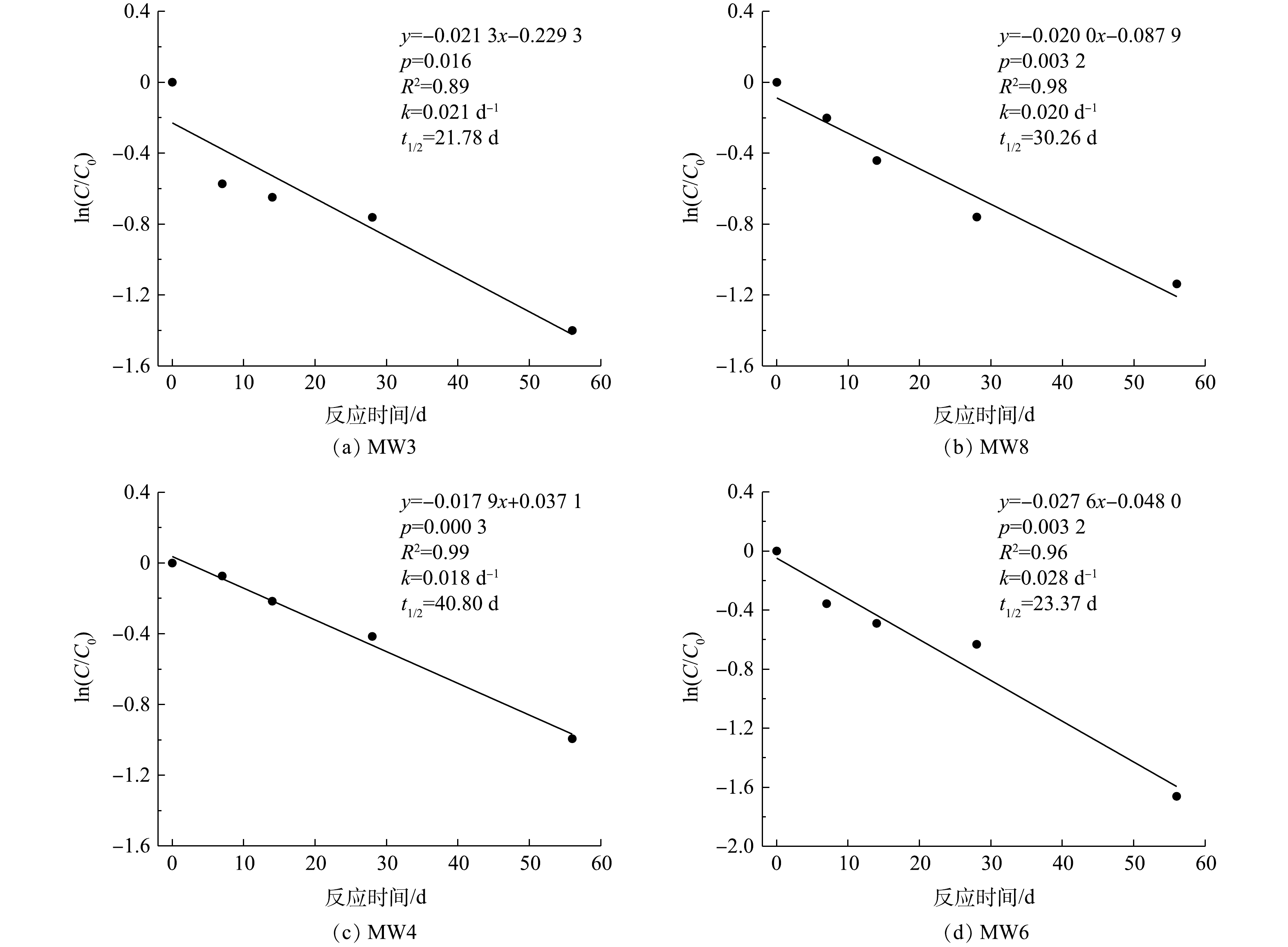
在注药后56 d内,对MW3、MW8、MW4和MW6中1,2-二氯乙烷浓度与其初始浓度比值的自然对数值-时间进行拟合,由图8可知,ln(C/C0)-t呈线性关系,地下水中1,2-二氯乙烷降解过程遵循准一级动力学方程(式(5))。
式中,k为反应速率常数,d−1;C为t时刻的反应物浓度,mg·L−1;C0为反应物初始浓度,mg·L−1;t为反应时间,d;t1/2为半衰期,d。
分析实验期间1,2-二氯乙烷浓度随时间变化规律,求解得到各监测井中1,2-二氯乙烷的反应速率常数和半衰期(平均值),碱活化过硫酸盐降解1,2-二氯乙烷反应速率常数为0.022 d−1,半衰期为29 d。
-
注入修复药剂后,实验区域含水层中1,2-二氯乙烷浓度分布变化情况如图9所示。注药后第7天,仅MW1、MW9和注药井修复达标(浓度低于场地修复目标值0.15 mg·L−1即为达标);第14天,MW1、MW2、MW5、MW9和注药井修复达标。注药后0~28 d,实验区域内地下水1,2-二氯乙烷污染物浓度总体呈下降趋势,实验区域内1,2-二氯乙烷浓度修复达标区域面积逐渐增大,这表明碱活化过硫酸盐对受1,2-二氯乙烷污染含水层有较好的修复效果。实验28 d后,实验区域地下水中1,2-二氯乙烷浓度开始回升,达标区域开始减小。其原因在于,实验区间注入修复药剂有限,当氧化药剂被完全消耗后,周边含水层未降解的污染物由于分子扩散作用向达标区补给,使修复达标区域的含水层重新被污染。
修复药剂在含水层中的迁移和扩散受含水层自身的非均质性和地下水的渗流特征控制,抽取地下水时也会对地下水流场产生严重干扰,从而间接影响修复药剂迁移[19]。本实验区域含水层为卵砾石层,渗透系数为7.89 m·d−1,透水性较好,修复药剂注入期间,受抽水的机械弥散作用影响,修复药剂随地下水渗流方向运移,污染物修复达标区域逐渐变为以注药点为中心,长轴方向与地下水渗流方向一致的近似椭圆形。但污染物修复达标范围有限,这是由于地下水水力坡度(0.36%~3.05%)较小以及修复药剂总量相对较少造成的[20]。
-
在注入氧化剂和活化剂后,地下水中硫酸盐浓度变化情况如图10(a)所示(图中列举了地下水主流向上监测井中硫酸盐浓度的变化)。在实验前,实验区域硫酸盐浓度为30.30~47.70 mg·L−1。MW1和MW2在注药后前28 d内,硫酸盐浓度持续上升,最高分别达到425 mg·L−1和408 mg·L−1;第28天后,硫酸盐浓度持续稳定下降,实验结束时,恢复至原浓度水平。MW3和MW4在注药后的第7天,硫酸盐浓度出现明显增长,分别达到102 mg·L−1和75.70 mg·L−1,与MW1和MW2中硫酸盐浓度峰值相比较小,之后呈下降趋势。过硫酸盐氧化过程中会产生了大量的硫酸盐,硫酸盐在过硫酸盐被完全消耗前会持续增加,在过硫酸盐被完全消耗之后,硫酸盐浓度呈下降趋势,这也反映了化学氧化作用的剧烈程度。这与吴圣华等[21]在采用过硫酸盐氧化处理汽油BTEX污染地下水时所得的结果类似。
碱活化过硫酸盐体系会引起地下水pH的变化。图10(b)所示是实验过程中地下水pH的变化情况。在注入氧化剂和活化剂前,实验区域地下水pH为4.93~5.57。MW1和MW2在注药后第7天,pH分别上升至10.90和12.08,为碱活化过硫酸盐创造较适宜的pH环境,之后总体呈下降趋势。MW3和MW4在注药后的28 d内pH有小幅度下降。这可能是由于实验期间注入过硫酸盐总量大于氢氧化钠总量,在水动力控制条件下,过硫酸盐自然扩散和随水力控制运移范围大于氢氧化钠,造成距离注药井相对较远的MW3和MW4中pH有一定程度下降。有研究[22]表明,过硫酸钠投加可对地下水环境造成一定的酸化效应。随着过硫酸盐消耗及周边地下水侧向补给,在实验结束时,监测井中pH恢复至原水平。LIANG等[23]认为,过硫酸盐的氧化作用较缓慢且温和,虽然反应过程中土壤pH会下降至2~4,但随着过硫酸盐的继续消耗,pH会逐渐回升,二次污染可控,故使用过硫酸盐原位修复有机污染土壤和地下水具有独特的优势和较好的应用前景。
过硫酸钠的化学氧化机理是:其溶解后形成的过硫酸盐
S2O82− 具有强氧化性,可以降解氯代烃、苯系物等污染物,并且在一定活性催化剂的作用下(如碱、Fe2+等)会释放出SO−⋅4 ,具有更强的氧化性[24]。从过硫酸钠的化学氧化机理可以看出,化学氧化过程中产生了大量的硫酸盐,会在一定范围内形成硫酸盐富集区域。在本研究中,采用碱活化过硫酸盐修复污染地下水中1,2-二氯乙烷,实验初期和中期(前56 d内),在注药井周边4 m以内出现高浓度的硫酸盐,浓度值高于《地下水环境质量标准》(GB/T 14848-2017)中IV类水标准350 mg·L−1。有研究[25]表明,对于土壤和地下水含水介质中硫酸盐的代谢和降解,硫酸盐还原菌起到了重要的作用,这类微生物能够在厌氧条件下利用硫酸盐作为电子受体进行厌氧呼吸,在厌氧呼吸过程中,SO2−4 被还原成了S2−并产生能量以提供微生物增长。在实验后期(56~140 d),地下水中硫酸盐浓度逐渐下降,最终恢复到实验前的初始浓度水平,这可能是由于硫酸盐还原菌代谢降解硫酸盐所致。
2.1. 实验区的水文地质参数
2.2. 1,2-二氯乙烷降解规律及动力学分析
2.3. 地下水修复效果
2.4. 地下水中硫酸盐浓度和pH的变化



-
1)实验区含水层渗透系数为7.89 m·d−1,导水系数为101 m2·d−1,在一维稳定流场中二维弥散条件下,含水层纵、横向弥散度αL和αT分别为0.89 m和0.089 m,地下水流速为3.85 m·d−1,水动力条件明显优于自然状态,通过水动力控制法干扰地下水流场可有效控制修复药剂在含水层中的扩散速度和影响范围。
2)注药后实验区污染物浓度整体呈下降趋势,在第14天,注药井4 m以内1,2-二氯乙烷浓度低于检出限,药剂效果在含水层中可保持28 d。碱活化过硫酸盐降解1,2-二氯乙烷反应速率常数为0.022 d−1,半衰期为29 d。在实验后期,因周边未修复区污染物向修复区扩散、补给,导致实验区地下水中1,2-二氯乙烷浓度出现回升。
3)实验过程中,实验区地下水中硫酸盐浓度先升高后下降,在140 d内恢复至原浓度水平,对实验场地二次污染影响较小。碱活化过硫酸盐在氯代烃类污染场地修复中具有广阔的应用前景。
